

Abstract
Purpose
Long intergenic non-coding RNAs (lincRNAs) are increasingly recognized as important regulators for pathogenesis and/or prognosis of breast cancer, including triple-negative breast cancer (TNBC) subtype. However, few previous studies used RNA-sequencing (RNA-Seq) technology, and none included an independent replication.
Methods
To systematically evaluate the association between expression of lincRNAs and TNBC survival, we examined lincRNA expression profiles in TNBC tissues using RNA-Seq data for 200 TNBC patients from the Shanghai Breast Cancer Survival Study (SBCSS) and Southern Community Cohort Study (SCCS).
Results
Twenty-five lincRNAs were found to be associated with overall survival (P < 0.05 and no significant heterogeneity across studies at Q statistic P > 0.1), and 61 lincRNAs were associated with disease-free survival (DFS). Among these, two lincRNAs (LINC01270 and LINC00449) were significantly associated with both worse overall survival and DFS and were expressed at significantly higher levels in tumor tissues compared with adjacent normal breast tissues (log2[Fold Change] > 0.5 and FDR < 0.05). We further evaluated the potential functions of LINC01270 and LINC00449 using in vitro functional experiments and found that siRNA-mediated knockdown of LINC01270 and LINC00449 expression significantly decreased cell viability, colony formation and cell migration ability in TNBC cells (P < 0.05).
Conclusions
Evidence from observational studies and in vitro experiments indicates that LINC00449 and LINC01270 may be prognostic biomarkers for TNBC.
Keywords: Triple-negative breast cancer, LINCRNA, Survival, RNA-Seq, In vitro experiments
Introduction
Long non-coding RNAs (lncRNAs) are a large class of non-coding regulatory RNAs with a length greater than 200 nucleotides (nt). LncRNAs can be classified by their genomic locations, including independent transcription units (long intergenic non-coding RNAs, lincRNAs) [1], regions upstream of protein-coding genes (promoter upstream transcripts, PROMPTs) [2] and enhancers (enhancer RNAs). LincRNAs play important roles in chromosome remodeling [3], immune responses [4] and cancer progression [5].
As the most common cancer among women worldwide, breast cancer has a generally favorable outcome; its survival rate has improved substantially over the last three decades, owing to advances in early detection and improvements in treatment [6]. However, outcomes for triple-negative breast cancer (TNBC), a subtype of breast cancer lacking expression of estrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER2), are still unsatisfactory, with a higher risk of early relapse and mortality compared to other breast cancer subtypes [7]. Recently, several lincRNAs such as MALAT1 [8], XIST [9] and NEAT1 [10] have been reported as potential biomarkers for breast cancer, including TNBC progression. Studies have applied RNA-sequencing (RNA-Seq) to evaluate the expressions or derive expression signatures of lincRNAs in predicting breast cancer outcomes [11, 12]. However, none of these studies have specifically focused on investigating the association between lincRNA expression and TNBC survival, and none included an independent replication.
Presented in this report is a comprehensive evaluation of the association between lincRNA expressions and TNBC survival using RNA-Seq data from 200 TNBC patients from two independent studies, the Shanghai Breast Cancer Survival Study (SBCSS) and Southern Community Cohort Study (SCCS). Furthermore, we carried out in vitro studies (cell viability, colony formation, cell migration and invasion assays) to investigate whether the lincRNAs identified from our observational studies can affect cell functions by short interfering RNA (siRNA)-silenced knockdown in the MDA-MB-231 TNBC cell line.
Methods
Study populations
The Shanghai Breast Cancer Survival Study (SBCSS), initiated in 2002, is a longitudinal study of 5042 breast cancer patients [13]. Patients were identified from a population-based tumor registry and recruited to the study approximately 6 months after cancer diagnosis. In-person interviews collected information on demographics, cancer diagnosis and treatment, lifestyle factors, quality of life and comorbidities. Patients were followed-up via in-person surveys at 1.5, 3, 5 and 10 years post-diagnosis to collect data on clinical information, recurrence, survival status and causes of death. Record linkages with a tumor registry follow-up database and vital statistics registry were also conducted to supplement information on survival status. Medical records were obtained to verify cancer diagnosis and collect information on initial cancer treatment, tumor ER/PR status and breast cancer stage. HER2 status was obtained either from the hospital record or measured in the Vanderbilt Molecular Epidemiology Laboratory [13, 14]. Tumor sections were collected for 4036 patients (85%); of these, 525 (10%) were TNBC samples. RNA-Seq data were generated from 150 TNBC cases and included in the current study.
The Southern Community Cohort Study (SCCS) recruited 85,806 participants, aged 40–79 years, between 2002 and 2009 from 12 southeastern states in the US [15]. Approximately two-thirds of participants were African Americans, and 60% were women. Information on demographics, lifestyle factors, comorbidities and other exposures was collected at baseline and updated in follow-up surveys. Annual linkages of the cohort with the 12-state cancer registries covering the SCCS catchment area are performed to identify incident breast cancer cases and collect information on tumor ER/PR/HER2 status. Mortality information is obtained via annual linkage with the National Death Index (NDI) databases. Collection of medical records and breast tumor tissues began in 2010. Information on breast cancer histology, pathology, stage and grade, including ER/PR/ HER2 status, was obtained from these cancer registries. Sixty-nine TNBC cases, with tissue samples, were included in an RNA-Seq study.
We excluded from the current study patients with stage 0 or stage IV diseases (SBCSS: N = 0; SCCS: N = 19), resulting in a total of 200 TNBC cases from the SBCSS and SCCS for the current study.
Gene expression profiling and data processing
SBCSS and SCCS data
Hematoxylin and eosin (H&E) slides were reviewed by a study pathologist, and tumor tissues were dissected from one to five unstained formalin-fixed paraffin-embedded (FFPE) tissue sections to ensure that samples contained > 80% tumor cells for total RNA extraction [16]. Total RNA was extracted and purified using the Qiagen miRNeasy FFPE Kit. The quantity and quality of RNA samples extracted from tumor tissue FFPE sections were checked by NanoDrop (E260, E260/E280 ratio, spectrum 220–320 nm) and by separation on an Agilent BioAnalyzer. The RNase H method was used for rRNA depletion [16]. Illumina TruSeq RNA Sample Prep Kit v2 was used to prepare a sequencing library, and HiSeq 2000 was used for sequencing. Each sample was sequenced paired-end at a read length of 100 bp. A minimum of 10 M reads was obtained for each sample. All RNA-Seq data were processed following the mRNA analysis pipeline from The Cancer Genome Atlas (TCGA) Genomic Data Commons (GDC) [17]. The STAR two-pass method was used for raw data alignment to the human reference genome (hg38) [18]. GENCODE v22 was used for coding gene and non-coding RNA annotation in the human genome [19]. Gene expression levels were measured using Fragments Per Kilobase of transcript per Million mapped reads (FPKM).
All gene expression data from the SBCSS and SCCS were log2-transformed after excluding unexpressed genes from over half the samples (median FPKM = 0). Quantile normalization was performed for all samples to standardize expression levels to the same scale. Only lincRNAs annotated in GENCODE v22 were investigated in this study (N = 7656).
Intrinsic molecular breast cancer subtypes (luminal A, luminal B, basal-like, HER2-enriched and normal-like) were determined following Giovanni’s strategy using the PAM50 classifier [20, 21] and implemented using the R package genefu [22]. In order to make our sample subtype distribution more resemble the online reference, which included all subtypes of breast cancer [20, 21], we included RNA-Seq data for non-TNBC cases in the SBCSS (N = 160) and SCCS (N = 386) in deriving subtypes using the PAM50 classifier.
TCGA data
RNA-Seq data from breast cancer patients in TCGA, publicly available from the NCI GDC data portal [23], were used to perform differential gene expression analysis between tumor and adjacent normal tissues. HTSeq-Count data were downloaded as gene expression profiles for TCGA-BRCA patients and prepared for differential gene expression analysis by the R package TCGAbiolinks [24].
In vitro studies
Breast cancer cell line
The human TNBC cell line, MDA-MB-231, was purchased from American Type Culture Collection (ATCC) and cultured in Dulbecco’s Modified Eagle Medium (DMEM, Gibco), supplemented with 100 IU/mL penicillin–streptomycin (Gibco) and 10% fetal bovine serum (Gibco) at 37 °C in a humidified atmosphere with 5% CO2.
Gene expression levels in MDA-MB-231 cell line
Total RNA was isolated from MDA-MB-231 cells using the miRNeasy Mini Kit (Qiagen) according to the manufacturer’s protocol. Complementary DNA (cDNA) was synthesized using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). Quantitative real-time PCR (qRT-PCR) was performed using DNA primers in a CFX384 Touch Real-Time PCR Detection System (Bio-Rad) with Luna Universal qPCR Master Mix (New England BioLabs). Relative lincRNA expression was calculated using the ΔΔCt method [25] and normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Primer sequences for each lincRNA and GAPDH are listed in Supplementary Table 1.
Short interfering RNA (siRNA) silencing
Following transfection optimization for MDA-MB-231 cells, reverse transfections of siRNAs targeting our genes of interest (GOI) were performed on 5000 cells per well in 100 μL culture media, without antibiotics, in a 96-well plate (Costar). Two Silencer™ Select Pre-Designed siRNAs targeting LINC00449 (siRNA ID: n486205, n503412) and LINC01270 (siRNA ID: n503492, n503493) were purchased from Thermo Fisher Scientific. Positive control siRNA (AllStars Hs Cell Death Control siRNA, Qiagen) and a Silencer™ Select Negative Control siRNA (NC siRNA, Thermo Fisher Scientific) were used as controls to confirm transfection efficiencies for each experiment. RNAiMAX (Life Technologies) transfection reagent was used for siRNA delivery in Opti-MEM reduced serum media (Gibco). Validation of efficient siRNA knockdown of gene expression was assessed 48 h post-transfection by qRT-PCR and normalized to cells transfected with the NC siRNA. Target sequences for each siRNA are listed in Supplementary Table 2.
Cell viability assay
Cell viability was determined using alamarBlue (Thermo Fisher Scientific), as previously described [26]. On Day 5, following 96 h of transfection, 10 μL alamarBlue was added to wells containing siRNA-transfected cells (1:10 dilution), incubated for 2 to 6 h, and fluorescence (ex570nm/ em585nm) was measured using BioTek Synergy HT plate reader. Relative cell viability was normalized to the NC siRNA. Results represent data obtained from three independent experiments.
Colony formation assay
For colony formation assays, MDA-MB-231 cells were transfected with siRNAs in 96-well plates, as previously described [26]. After 16 h, siRNA-transfected cells were reseeded in 6-well plates with a density of 2000 cells per well in 2 mL antibiotic-free culture media and allowed to proliferate for 11 to 14 days. Colonies consisting of ≥ 50 cells were fixed with 10% neutral buffered formalin for 30 min, stained with crystal violet (0.1% w/v in H2O, Sigma-Aldrich), scanned and counted using ImageJ software (NIH). Colony counts were normalized to the NC siRNA group and expressed as percent (%) of negative control. Three independent experiments were performed for siRNAs targeting lincRNAs.
Cell migration and invasion assays
Cell migration and invasion assays were performed in 24-well plates with 8 μm pore size chamber inserts (Millipore), as previously described [27]. The invasion assay used coated Matrigel (Corning, 1:10 dilution) in the upper chamber, simulating characteristics of the extracellular matrix, and the migration assay used chamber inserts only. MDA-MB-231 cells were transfected with LINC00449 and LINC01270 siRNAs and NC siRNA for 16 h. Then, siRNA-transfected cells were reseeded into the upper chamber with 100 μL serum-free medium (6 × 104 cells/well). A total of 750 μL medium containing 10% FBS was placed in the lower chamber as a chemoattractant. After incubation at 37 °C for 48 h, cells adhering to the lower surface membrane of the upper chamber were fixed in 10% neutral buffered formalin for 30 min, followed by staining with 0.1% crystal violet (Sigma-Aldrich), and then subjected to microscopic inspection. Five visual fields of each insert were randomly chosen. Crystal violet was then eluted using 33% acetic acid and quantified by measuring the absorbance at 590 nm with BioTek Synergy HT plate reader. Relative migrated and invaded cells were normalized to the NC siRNA group and expressed as percent (%) of negative control. Results represent data obtained from three independent experiments.
Data analysis
Cox proportional hazards were used for multivariable modeling adjusting for age at diagnosis, TNM stage, race and PAM50 subtypes. Data analyses were carried out separately in the SBCSS and SCCS, and a fixed-effect meta-analysis was performed to summarize the results from the two studies. We used TCGA data to evaluate differential gene expression analysis between tumor and adjacent breast normal tissues with DESeq 2 [28]. All statistical analyses were performed in R 3.6.0.
For in vitro studies, statistical differences between groups were evaluated using GraphPad Prism 8.0 statistical software. All experiments were performed in triplicate and repeated at least three times. The results were presented as the mean ± SD. One-way analysis of variance (ANOVA), followed by Dunnett’s multiple comparison test, was used to compare the differences between siRNA-mediated lincRNA knockdown cells and negative control cells. P values < 0.05 were considered significant.
Results
A total of 200 patients (SBCSS: 150 and SCCS: 50) with stage I, II and III TNBC were included in this study. Demographic and clinical characteristics of study participants are presented in Table 1. Median follow-up periods were 145 months for the SBCSS (range 8–187 months) and 135 months for the SCCS (range 24–174 months). Basal-like is the major subtype, accounting for 62% and 84% in SBCSS and SCCS samples, respectively.
Table 1.
Clinicopathological characteristics of triple-negative breast cancer patients of the SBCSS and SCCS
| SBCSS | SCCS | TCGA | |
|---|---|---|---|
| No | 150 | 50 | 110 |
| Age (mean (SD)) | 54.9 (12.1) | 58.3 (8.8) | 54.8 (12.1) |
| Race | |||
| Asian | 150 (100.0) | 0 | 8 (7.3) |
| African American | 0 | 37 (74.0) | 31 (28.2) |
| European American | 0 | 13 (26.0) | 71 (64.5) |
| Stage | |||
| Stage I | 27 (18.0) | 10 (20.0) | 19 (17.3) |
| Stage II | 90 (60.0) | 28 (56.0) | 72 (65.5) |
| Stage III | 33 (22.0) | 12 (24.0) | 19 (17.3) |
| PAM50 | |||
| Luminal A | 13 (8.7) | 1 (2.0) | 1 (0.9) |
| Luminal B | 9 (6.0) | 0 (0.0) | 2 (1.8) |
| Basal-like | 93 (62.0) | 42 (84.0) | 96 (87.3) |
| HER2-Enriched | 22 (14.7) | 4 (8.0) | 8 (7.3) |
| Normal-like | 13 (8.7) | 3 (6.0) | 3 (2.7) |
We first evaluated each individual lincRNA for its association with overall survival using multivariate Cox proportional hazards model. Within the 7656 lincRNAs annotated in GENCODE v22, there were 1727 lincRNAs expressed (median FPKM > 0) in SBCSS data and 2487 expressed in SCCS data. Of them, 1711 lincRNAs were expressed in both datasets. After adjusting for age at diagnosis, TNM stage, race (in the SCCS only) and PAM50 subtypes, we found that 220 lincRNAs in the SBCSS and 239 lincRNAs in the SCCS were associated with overall survival at a significance level of P < 0.05. There were 39 lincRNAs significantly associated with overall survival in both the SBCSS and SCCS, 33 of which showed consistent directionality of the association in both studies. In the meta-analysis using the fixed-effect model, we found that 25 lincRNAs showed no significant heterogeneity across studies (Q statistic P > 0.1) and were significantly associated with overall survival (Supplementary Table 3). Using TCGA data, we further found that 3 of the 25 lincRNAs (LINC00449, LINC01270 and LINC01910) were expressed significantly higher in tumor tissues compared with adjacent normal tissues (log2[Fold Change] > 0.5 and FDR < 0.05; Table 2) and were associated with lower overall survival.
Table 2.
Results of overall survival, disease-free survival and gene differential expression analyses for LINC00449, LINC01270 and LINC01910
| ENSEMBL ID | Gene Symbol | SBCSS (OS) | SCCS (OS) | Meta Analysis (OS) | SBCSS (DFS) | Diff Exp (Tumor Vs Normal) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| HR | P Value | HR | P Value | Q Stat | P Value | HR | P Value | log2(FC) | FDR | ||
| ENSG00000203441 | LINC00449 | 1.55 (1.2 – 2.1) | 0.004 | 2.65 (1.3 – 5.4) | 0.008 | 0.181 | <0.001 | 1.63 (1.2 – 2.2) | 0.002 | 0.52 | <0.001 |
| ENSG00000203999 | LINC01270 | 1.55 (1 – 2.4) | 0.041 | 2.41 (1.2 – 5) | 0.017 | 0.305 | 0.003 | 1.59 (1.1 – 2.4) | 0.026 | 0.75 | <0.001 |
| ENSG00000266278 | LINC01910 | 1.48 (1 – 2.1) | 0.031 | 2.36 (1 – 5.3) | 0.038 | 0.303 | 0.005 | 1.37 (0.97 – 1.9) | 0.074 | 1.04 | <0.001 |
In addition, we investigated the association between individual lincRNA and breast cancer disease-free survival (DFS) in the SBCSS using the same method and analysis filtration criteria as for overall survival (DFS information was unavailable in the SCCS). After adjusting for age at diagnosis, TNM stage, race and PAM50 subtypes, we identified that 226 lincRNAs were significantly associated with DFS at a significance level of P < 0.05. Of these 226 lincRNAs, 61 were significantly higher expressed in tumor tissues compared with adjacent normal tissues within TCGA data (log2[Fold Change] > 0.5 and FDR < 0.05; Supplementary Table 4) and were associated with lower DFS. From the above analyses, we selected two lincRNAs, LINC00449 and LINC01270, that were significantly associated with both worse overall survival and DFS in TNBC patients, as well as having significantly higher expression in tumor tissues than adjacent normal breast tissue for further functional evaluation.
In vitro functional assays using TNBC cell line MDA-MB-231
To evaluate the potential functions of LINC00449 and LINC01270, we quantitatively explored LINC00449 and LINC01270 expression in TNBC cell line by using total RNA isolated from MDA-MB-231 cells. As shown in Figure S1A, LINC00449 and LINC01270 expression were detected by qRT-PCR analysis in MDA-MB-231 cells, as mean ΔCt values for LINC00449 and LINC01270 were 12.7 and 10.7 normalized to GAPDH. We further conducted functional assays via knockdown experiments in MDA-MB-231 cells. We used two siRNAs for each lincRNA silencer. Knockdown efficiencies of all siRNAs were validated by qRT-PCR (Figure S1B).
Cell viability and colony formation were then investigated. We used alamarBlue to investigate cell viability and quantified the relative cell viability after knocking down lincRNAs compared with NC siRNA (Fig. 1A). Results showed that, compared with the negative control cells, knocking down LINC01270 and LINC00449 expression significantly decreased MDA-MB-231 cell viability, as mean cell viabilities were 86.9% (siLINC00449–02), 74.2% (siLINC01270–01) and 44.0% (siLINC01270–02) compared with 100% for the NC siRNA (Fig. 1A). However, siLINC00449–01 for silencing LINC00449 expression did not show any effect.
Fig. 1.
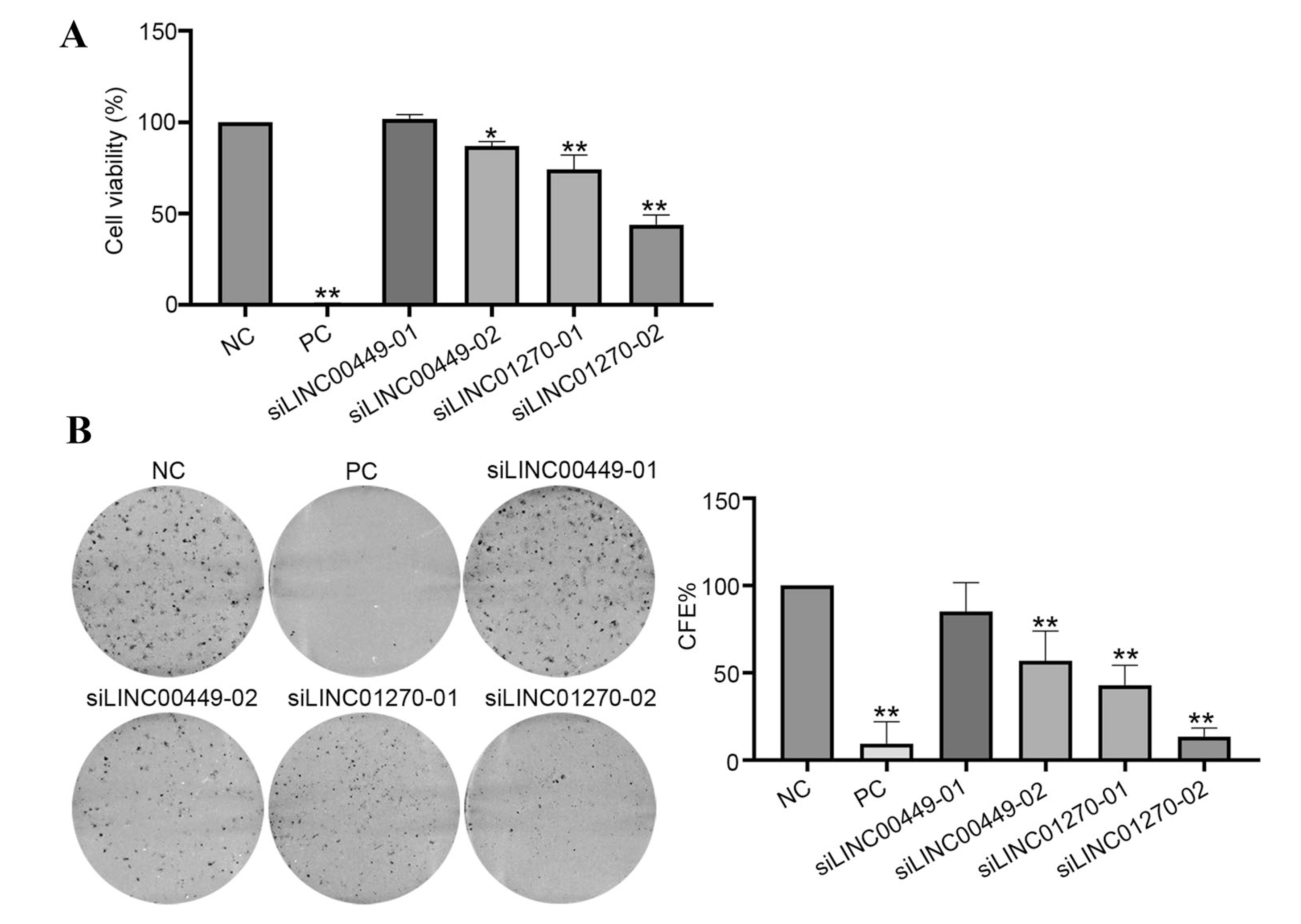
Effect on cell viability and colony formation efficiency by silencing lincRNA expressions in MDA-MB-231 cells. A Cell viability. MDA-MB-231 cells were transfected with indicated siRNAs for 96 h. Cell viability was determined using alamarBlue reagent. Percent relative viability was calculated as (mean lincRNA siRNA value/ mean NC siRNA value) × 100. Data were presented as mean ± SD from three independent experiments. B Colony formation efficiency. MDA-MB-231 cells were transfected with indicated siRNAs, and then reseeded after 16 h for colony formation (CF) assay. After 11–14 days, colonies were fixed with 10% neutral buffered formalin, stained with crystal violet, scanned and counted by ImageJ software. Colony formation efficiency (CFE) was normalized to NC siRNA group and expressed as percent (%) of control. All the experiments were repeated three times independently and presented as mean ± SD. P values were determined by one-way ANOVA followed by Dunnett’s multiple comparisons test: *P value < 0.05, **P value < 0.01. NC: negative control siRNA; PC: cell death positive control siRNA
We further investigated the effects of silencing these lincRNAs on colony forming ability to evaluate the longer-term effects of lincRNA silencing on cell proliferation. Results indicated that knocking down of LINC01270 and LINC00449 showed significant inhibition on colony formation ability in MDA-MB-231 cells compared with negative control cells (Fig. 1B), with the mean colony formation efficiencies being 85.1% (siLINC00449–01), 56.8% (siLINC00449–02), 42.9% (siLINC01270–01) and 13.5% (siLINC01270–02) (Fig. 1B).
The effects of LINC00449 and LINC01270 on cell migration and invasion were further evaluated in MDA-MB-231 cells. Results indicated that silencing LINC01270 and LINC00449 resulted in decreased cell migration ability in MDA-MB-231 cells compared with negative control cells, with the mean ratio of the migrated cell number being 49.3% (siLINC00449–02) and 45.0% (siLINC01270–02). Furthermore, knocking down of LINC00449 (siLINC00449–02) decreased cell invasion by about 51% relative to the negative control in MDA-MB-231 cells. We did not observe a significant effect of LINC01270 on cell invasion in MDA-MB-231 cells (Fig. 2).
Fig. 2.
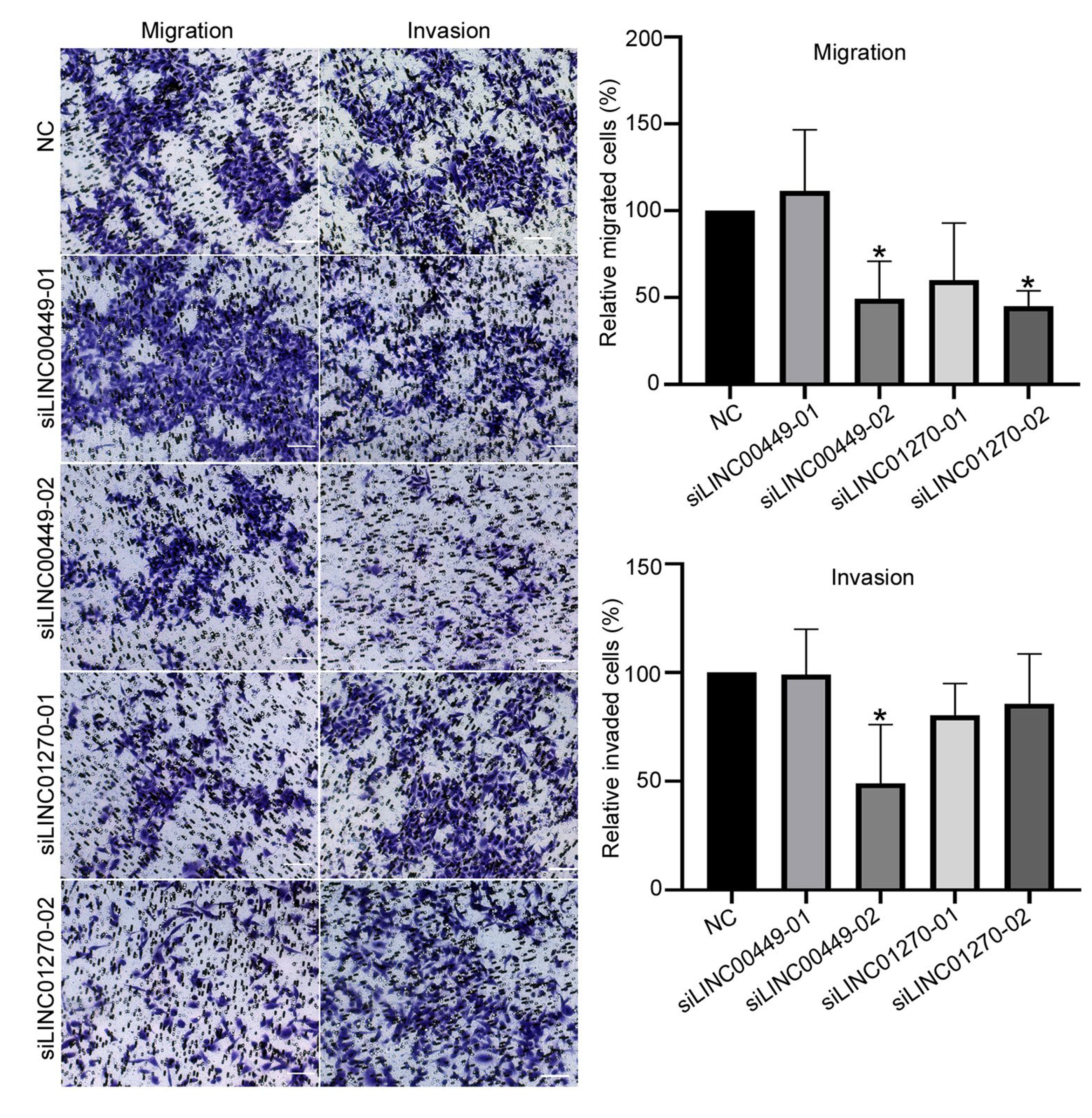
Effect on cell migration and invasion ability in MDA-MB-231 cells by silencing lincRNA expressions. MDA-MB-231 cells were transfected with indicated siRNAs and negative control (NC) siRNA, respectively, and then reseeded after 16 h in upper chamber for the transwell assay. After 48 h, the cells adhering to the lower surface of the membrane on the upper chamber were fixed with 10% neutral buffered formalin, stained with crystal violet, and then subjected to microscopic inspection. Five visual fields of each insert were randomly chosen. Then crystal violet was eluted using 33% acetic acid and quantified by measuring the absorbance at 590 nm. Relative migrated and invaded cells were normalized to the NC siRNA group and expressed as percent (%) of control. All the experiments were repeated three times independently and presented as mean ± SD. P values were determined by one-way ANOVA followed by Dunnett’s multiple comparisons test: *P value < 0.05. NC: negative control siRNA
Discussion
LincRNAs have been increasingly recognized as important regulators of pathogenesis and/or prognosis for a wide range of cancers, although few studies have included a validation study. In the current study, we first evaluated the association between lincRNA expression in tumor tissues and breast cancer survival among 200 TNBC patients from two independent studies, the SBCSS and SCCS. We identified two lincRNAs (LINC01270 and LINC00449) that were significantly associated with worse overall survival and DFS in TNBC patients from the SBCSS and SCCS and were expressed significantly higher in tumor tissues than adjacent normal breast tissues in TCGA cases. We carried out in vitro functional experiments and showed that siRNA-mediated knockdown of LINC01270 and LINC00449 significantly decreased cell viability, colony formation and cell migration ability in TNBC MDA-MB-231 cells. Furthermore, knocking down of LINC00449 significantly inhibited cell invasion in MDA-MB-231 cells. Overall, evidence from both observational studies and in vitro results suggest that high expression of LINC00449 and LINC01270 may be biomarkers for worse prognosis for TNBC.
No previous studies have investigated the roles of LINC00449 and LINC01270 on TNBC prognosis, although one study reported that LINC01270 was a risk biomarker for lung adenocarcinoma in TCGA samples [29]. LINC01270 is located between CEBPB and PTPN1, both of which have critical roles in breast cancer progression [30, 31]. LINC00449 is located in the intergenic region of gene TM9SF2, a member of the transmembrane 9 superfamily, which may play a role in small molecule transport or act as an ion channel [32]. A previous study identified TM9SF2 as a candidate for a cell surface marker common to breast cancer [33]. We performed additional differential gene expression analysis between tumor and adjacent normal tissues using TCGA data for all the cancer types, and found these two lincRNAs were overexpressed in cancer tissues than their adjacent normal tissue data in many cancer types (Supplementary Table 5). These results suggest a possible common biological role of these lincRNAs in carcinogenesis across multiple cancers. Further studies are warranted to investigate the biological mechanisms underlying the associations of LINC00449 and LINC01270 with TNBC progression.
We used two siRNAs to knockdown gene expression levels in in vitro assays. Results for LINC01270 were consistent between the two siRNAs. For LINC00449, the siLINC00449–01 only had moderate inhibitory effects on colony formation but did not have any effect on other cell functional assays, although this siRNA knocked down LINC00449 expression by ~ 60%. It is possible that the effect of siLINC00449–01 may be more pronounced in long-term incubation, and there may be reverse effects by this siRNA treatment. It is also possible that siLINC00449–01 may have unintended “off-target” effects that may silence other genes which affect cell functions [34, 35]. Further studies using the stable silencing method are warranted. In addition, studies are needed to use stable knockdown technology and orthotopic xenograft models to confirm the functions and regulatory mechanisms of LINC01270 and LINC00449 in TNBC.
Several lincRNAs, including MALAT1 [8], XIST [9] and NEAT1 [10], have been previously reported to be associated with TNBC prognosis. However, none of these lincRNAs were significantly associated with overall survival or DFS in our study. Differences in patient population, sample size, type of tumor sample, sequencing method and chance finding could have, individually or jointly, contributed to the discrepancies.
The major strength of our study is its utilizing samples and data from two independent epidemiological studies as well as TCGA, plus its inclusion of in vitro functional studies for validation. The ethnic differences between participants of the SBCSS and SCCS allowed a cross-population validation of lincRNAs that are associated with TNBC prognosis, minimizing chance findings or biases. On the other hand, the ethnic differences between the two studies, and the small sample size of each study, prohibited us from identifying race-specific TNBC prognosis-associated lincRNAs. Our study also has other limitations. First, RNA-Seq data from the SBCSS and SCCS were generated from FFPE samples rather than fresh frozen tissues. Thus, gene expressions of some lincRNAs, particularly those with low expression levels, may have not been reliably measured, which may have led to false-negative results. Second, as mentioned above, the sample size of our studies was small, particularly for the SCCS, which comprised the statistical power of our study. Last, only overall survival data were available in the SCCS, so we could not perform the analysis with DFS.
Conclusions
In this comprehensive study, we found that lincRNAs (LINC01270 and LINC00449) were associated with prognosis for TNBC in observational studies and cellular functions related to cancer progression in in vitro functional analyses. These results suggest that LINC00449 and LINC01270 may serve as prognostic biomarkers for TNBC patients.
Supplementary Material
Acknowledgements
We thank Regina Courtney for her help with RNA sample preparation and Dr. Mary Shannon Byers for assistance with editing and manuscript preparation.
Acknowledgments
Aunding
This study was supported by grants from the US Department of Defense (DOD) Breast Cancer Research Program (DAMD 17–02-1–0607 to X.-O. Shu) and the National Institutes of Health (NIH; R01 CA118229 to X.-O. Shu, P50CA098131 to C. Arteaga, U01CA202979 to W. J. Blot and W. Zheng). Sample preparation was conducted at the Survey and Biospecimen Shared Resources, which is supported in part by the Vanderbilt-Ingram Cancer Center (P30CA068485).
Footnotes
Electronic supplementary material The online version of this article (https://doi.org/10.1007/s10549-020-06021-6) contains supplementary material, which is available to authorized users.
Compliance with ethical standards
Conflict of interest None of the authors have a conflict of interest.
Ethical approval The study protocol was approved by the institutional review boards of Vanderbilt University and the Shanghai Municipal Center for Disease Control and Prevention.
Informed consent All participants provided written informed consent.
Publisher’s Note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Data availability
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
References
- 1.Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, Cabili MN, Jaenisch R Mikkelsen TS, Jacks T, Hacohen N, Bernstein BE, Kellis M, Regev A, Rinn JL, Lander ES (2009) Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458(7235):223–227. 10.1038/nature07672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Preker P, Nielsen J, Kammler S, Lykke-Andersen S, Christensen MS, Mapendano CK, Schierup MH, Jensen TH (2008) RNA exosome depletion reveals transcription upstream of active human promoters. Science 322(5909):1851–1854. 10.1126/science.1164096 [DOI] [PubMed] [Google Scholar]
- 3.Han P, Chang CP (2015) Long non-coding RNA and chromatin remodeling. RNA Biol 12(10):1094–1098. 10.1080/15476286.2015.1063770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hadjicharalambous MR, Lindsay MA (2019) Long non-coding RNAs and the innate immune response. Noncoding RNA 5(2):34. 10.3390/ncrna5020034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huarte M, Rinn JL (2010) Large non-coding RNAs: missing links in cancer? Hum Mol Genet 19(R2):R152–161. 10.1093/hmg/ddq353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68(6):394–424. 10.3322/caac.21492 [DOI] [PubMed] [Google Scholar]
- 7.Anders C, Carey LA (2008) Understanding and treating triple-negative breast cancer. Oncology 22(11):1233–1239 (discussion 1239–1240, 1243) [PMC free article] [PubMed] [Google Scholar]
- 8.Kim J, Piao HL, Kim BJ, Yao F, Han Z, Wang Y, Xiao Z, Siverly AN, Lawhon SE, Ton BN, Lee H, Zhou Z, Gan B, Nakagawa S, Ellis MJ, Liang H, Hung MC, You MJ, Sun Y, Ma L (2018) Long noncoding RNA MALAT1 suppresses breast cancer metastasis. Nat Genet 50(12):1705–1715. 10.1038/s41588-018-0252-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xing F, Liu Y, Wu SY, Wu K, Sharma S, Mo YY, Feng J, Sanders S, Jin G, Singh R, Vidi PA, Tyagi A, Chan MD, Ruiz J, Debinski W, Pasche BC, Lo HW, Metheny-Barlow LJ, D’Agostino RB Jr, Watabe K (2018) Loss of XIST in breast cancer Activates MSN-c-met and reprograms microglia via exosomal miRNA to promote brain metastasis. Cancer Res 78(15):4316–4330. 10.1158/0008-5472.CAN-18-1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shin VY, Chen J, Cheuk IW, Siu MT, Ho CW, Wang X, Jin H, Kwong A (2019) Long non-coding RNA NEAT1 confers oncogenic role in triple-negative breast cancer through modulating chemoresistance and cancer stemness. Cell Death Dis 10(4):270. 10.1038/s41419-019-1513-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ding X, Zhu L, Ji T, Zhang X, Wang F, Gan S, Zhao M, Yang H (2014) Long intergenic non-coding RNAs (LincRNAs) identified by RNA-seq in breast cancer. PLoS ONE 9(8):e103270. 10.1371/journal.pone.0103270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Wagner EK, Guo X, May I, Cai Q, Zheng W, He C, Long J (2016) Long intergenic non-coding RNA expression signature in human breast cancer. Sci Rep 6:37821. 10.1038/srep37821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baglia ML, Cai Q, Zheng Y, Wu J, Su Y, Ye F, Bao PP, Cai H, Zhao Z, Balko J, Zheng W, Lu W, Shu XO (2014) Dual specificity phosphatase 4 gene expression in association with triple-negative breast cancer outcome. Breast Cancer Res Treat 148(1):211–220. 10.1007/s10549-014-3127-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Su Y, Zheng Y, Zheng W, Gu K, Chen Z, Li G, Cai Q, Lu W, Shu XO (2011) Distinct distribution and prognostic significance of molecular subtypes of breast cancer in Chinese women: a population-based cohort study. BMC Cancer 11:292. 10.1186/1471-2407-11-292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Signorello LB, Hargreaves MK, Steinwandel MD, Zheng W, Cai Q, Schlundt DG, Buchowski MS, Arnold CW, McLaughlin JK, Blot WJ (2005) Southern community cohort study: establishing a cohort to investigate health disparities. J Natl Med Assoc 97(7):972–979 [PMC free article] [PubMed] [Google Scholar]
- 16.Guo Y, Wu J, Zhao S, Ye F, Su Y, Clark T, Sheng Q, Lehmann B, Shu XO, Cai Q (2016) RNA sequencing of formalin-fixed, paraffin-embedded specimens for gene expression quantification and data mining. Int J Genomics 2016:9837310. 10.1155/2016/9837310034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.TCGA GDC mRNA analysis pipeline. https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/
- 18.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29(1):15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, Barnes I, Berry A, Bignell A, Carbonell Sala S, Chrast J, Cunningham F, Di Domenico T, Donaldson S, Fiddes IT, Garcia Giron C, Gonzalez JM, Grego T, Hardy M, Hourlier T, Hunt T, Izuogu OG, Lagarde J, Martin FJ, Martinez L, Mohanan S, Muir P, Navarro FCP, Parker A, Pei B, Pozo F, Ruffier M, Schmitt BM, Stapleton E, Suner MM, Sycheva I, Uszczynska-Ratajczak B, Xu J, Yates A, Zerbino D, Zhang Y, Aken B, Choudhary JS, Gerstein M, Guigo R, Hubbard TJP, Kellis M, Paten B, Reymond A, Tress ML, Flicek P (2019) GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res 47(D1):D766–D773. 10.1093/nar/gky955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, Davies S, Fauron C, He X, Hu Z, Quackenbush JF, Stijleman IJ, Palazzo J, Marron JS, Nobel AB, Mardis E, Nielsen TO, Ellis MJ, Perou CM, Bernard PS (2009) Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 27(8):1160–1167. 10.1200/JCO.2008.18.1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, Zhang H, McLellan M, Yau C, Kandoth C, Bowlby R, Shen H, Hayat S, Fieldhouse R, Lester SC, Tse GM, Factor RE, Collins LC, Allison KH, Chen YY, Jensen K, Johnson NB, Oesterreich S, Mills GB, Cherniack AD, Robertson G, Benz C, Sander C, Laird PW, Hoadley KA, King TA, Network TR, Perou CM (2015) Comprehensive molecular portraits of invasive lobular breast cancer. Cell 163(2):506–519. 10.1016/j.cell.2015.09.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gendoo DM, Ratanasirigulchai N, Schroder MS, Pare L, Parker JS, Prat A, Haibe-Kains B (2016) Genefu: an R/Bioconductor package for computation of gene expression-based signatures in breast cancer. Bioinformatics 32(7):1097–1099. 10.1093/bioinformatics/btv693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.NCI GDC data portal. https://portal.gdc.cancer.gov/
- 24.Mounir M, Lucchetta M, Silva TC, Olsen C, Bontempi G, Chen X, Noushmehr H, Colaprico A, Papaleo E (2019) New functionalities in the TCGAbiolinks package for the study and integration of cancer data from GDC and GTEx. PLoS Comput Biol 15(3):e1006701. 10.1371/journal.pcbi.1006701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25(4):402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 26.Wu L, Wang J, Cai Q, Cavazos TB, Emami NC, Long J, Shu XO, Lu Y, Guo X, Bauer JA, Pasaniuc B, Penney KL, Freedman ML, Kote-Jarai Z, Witte JS, Haiman CA, Eeles RA, Zheng W, the Practical CBPCCPC (2019) Identification of novel susceptibility loci and genes for prostate cancer risk: a transcriptome-wide association study in over 140,000 European descendants. Cancer Res 79(13):3192–3204. 10.1158/0008-5472.CAN-18-3536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu H, Liu Y, Shu XO, Cai Q (2016) MiR-374a suppresses lung adenocarcinoma cell proliferation and invasion by targeting TGFA gene expression. Carcinogenesis 37(6):567–575. 10.1093/carcin/bgw038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15(12):550. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y, Fu J, Wang Z, Lv Z, Fan Z, Lei T (2019) Screening key lncRNAs for human lung adenocarcinoma based on machine learning and weighted gene co-expression network analysis. Cancer Biomark 25(4):313–324. 10.3233/CBM-190225 [DOI] [PubMed] [Google Scholar]
- 30.Zahnow CA (2009) CCAAT/enhancer-binding protein beta: its role in breast cancer and associations with receptor tyrosine kinases. Expert Rev Mol Med 11:e12. 10.1017/S1462399409001033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bentires-Alj M, Neel BG (2007) Protein-tyrosine phosphatase 1B is required for HER2/Neu-induced breast cancer. Cancer Res 67(6):2420–2424. 10.1158/0008-5472.CAN-06-4610 [DOI] [PubMed] [Google Scholar]
- 32.Schimmoller F, Diaz E, Muhlbauer B, Pfeffer SR (1998) Characterization of a 76 kDa endosomal, multispanning membrane protein that is highly conserved throughout evolution. Gene 216(2):311–318. 10.1016/s0378-1119(98)00349-7 [DOI] [PubMed] [Google Scholar]
- 33.Abou-Sharieha S, Sugii Y, Tuoya YuD, Chen L, Tokutaka H, Seno M (2009) Identification of TM9SF2 as a candidate of the cell surface marker common to breast carcinoma cells. Chin J Clin Oncol 6:1 [Google Scholar]
- 34.Riba A, Emmenlauer M, Chen A, Sigoillot F, Cong F, Dehio C, Jenkins J, Zavolan M (2017) Explicit modeling of siRNA-dependent on- and off-target repression improves the interpretation of screening results. Cell Syst 4(2):182–193. 10.1016/j.cels.2017.01.011 [DOI] [PubMed] [Google Scholar]
- 35.Jackson AL, Burchard J, Schelter J, Chau BN, Cleary M, Lim L, Linsley PS (2006) Widespread siRNA “off-target” transcript silencing mediated by seed region sequence complementarity. RNA 12(7):1179–1187. 10.1261/rna.25706 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.