

Abstract
Precision Medicine (PM) is an innovative approach that, by relying on large populations’ datasets, patients’ genetics and characteristics, and advanced technologies, aims at improving risk stratification and at identifying patient-specific management through targeted diagnostic and therapeutic strategies. Cardiac channelopathies are being progressively involved in the evolution brought by PM and some of them are benefiting from these novel approaches, especially the long QT syndrome. Here, we have explored the main layers that should be considered when developing a PM approach for cardiac channelopathies, with a focus on modern in vitro strategies based on patient-specific human-induced pluripotent stem cells and on in silico models. PM is where scientists and clinicians must meet and integrate their expertise to improve medical care in an innovative way but without losing common sense. We have indeed tried to provide the cardiologist’s point of view by comparing state-of-the-art techniques and approaches, including revolutionary discoveries, to current practice. This point matters because the new approaches may, or may not, exceed the efficacy and safety of established therapies. Thus, our own eagerness to implement the most recent translational strategies for cardiac channelopathies must be tempered by an objective assessment to verify whether the PM approaches are indeed making a difference for the patients. We believe that PM may shape the diagnosis and treatment of cardiac channelopathies for years to come. Nonetheless, its potential superiority over standard therapies should be constantly monitored and assessed before translating intellectually rewarding new discoveries into clinical practice.
Keywords: Precision Medicine, Cardiac arrhythmias, Cardiac channelopathies, Genetics, Long QT syndrome, Pluripotent stem cells
Introduction
Medicine evolves steadily but sometimes new ideas or discoveries lead to either sudden turns or abrupt jumps forward. It happened with the discovery of blood typing and with the realization that invisible forms of life identifiable by a microscope could cause fatal infections. What followed were the introduction of safe blood transfusions and of specific antibiotics against different types of bacteria. Progressively, these highly selective approaches favoured the development of the term ‘Precision Medicine’ (PM) (still often used interchangeably with the older term ‘Personalized Medicine’1), which gained favour because it is objectively attractive and also conveys the reassuring feeling that doctors have therapies that are just ‘right for us’.
Since a few years, this concept is impacting not only the research on inherited cardiac diseases but also their clinical management,2 and here, we discuss the current role and potential future development of PM for the management of arrhythmogenic disorders of genetic origin. While considering in principle all of them, our focus will be on the long QT syndrome (LQTS) because it is the best characterized inherited arrhythmia and the one on which we have long-term experience.3 , 4
As defined by the Precision Medicine Initiative,5 , 6 ‘Precision Medicine is an emerging approach for disease treatment and prevention that takes into account individual variability in genes, environment, and lifestyle for each person’. The main purpose of this strategy is to predict treatment and preventive actions, which could be more effective for the individual patient or, more realistically, for groups of patients with common characteristics. This approach is in contrast to the current one-size-fits-all, in which disease treatment and prevention strategies are developed based on the results of randomized trials, without consideration for individual differences. To put it more simply and less elegantly, PM is the opposite of an implantable cardioverter defibrillator (ICD).
We will discuss not only how PM can help to better understand the mechanisms underlying cardiac channelopathies and to improve risk stratification but also which tools are being developed for the identification of patient-specific therapies, including the repurposing of drugs already approved for other uses, a strategy increasingly pursued. Due to space limits, we will not discuss another potentially important aspect of PM, namely gene therapy, which is under progressive development.
While presenting the available approaches and admitting our fascination for PM, especially for its amazing potential to penetrate uncharted territory and our enthusiasm for its intellectual rewards, we will also attempt to honestly recognize its limits—when present—and to preserve common sense when comparing fancy vs. run-of-the-mill approaches. Indeed, besides admiring conceptual elegance, scientists have the responsibility of assessing, as objectively as possible, the superiority of one approach vs. the other.
Cardiac channelopathies
The discovery of the first three genes responsible for LQTS in 1995 and 19967–9 had a major impact on the diagnosis and treatment of cardiac arrhythmias of genetic origin. It paved the way to the awareness that genetic variants can produce significant functional alterations in clinical electrophysiology and that, conversely, they could represent potential targets for novel therapies. Ever since, the identification and study of novel disease-causing genes grew exponentially and opened the genetic era for cardiac arrhythmias.
As most of the genes responsible for several inherited arrhythmic diseases encode for ion channels or accessory proteins involved in the excitation–contraction coupling, these disorders are called cardiac channelopathies. The most relevant are LQTS, catecholaminergic polymorphic ventricular tachycardia, Brugada syndrome, and short QT syndrome.2
Being mostly monogenic diseases that manifest early in life, channelopathies represent an ideal model for PM studies since background genetic heterogeneity and the influence of complex environmental and socio-economic factors have generally a low weight in the phenotypic traits of the disease. There are several layers worth considering for the development of PM, all important and with notable intersections (Figure 1). Some of these are discussed in detail below.
Figure 1.

Precision medicine layers. Layers that constitute a Precision Medicine pipeline for a dynamic patient risk stratification.
Clinical data
Population data collected from large cohorts of patients are the starting point for PM approaches. As the low prevalence of cardiac channelopaties hinders the availability of large sample sizes, the establishment of international registries has allowed the collection and the analysis of clinical data from centres worldwide resulting in significant progress.10–12
For instance, a study on ∼700 patients all with arrhythmic events and known genotype has documented that triggers of arrhythmias in LQTS are gene specific.13 LQT1 patients are at risk especially during sympathetic activation, e.g. during physical activity or emotional stress, since they have an impaired slow delayed-rectifier potassium current (IKs) and therefore their QT interval does not shorten as it should during heart rate increases. By contrast, LQT2 and LQT3 patients, whose IKs levels are normal, are at low risk during physical activity. Indeed, whereas 99% of syncope while swimming occur among LQT1 patients, 80% of those triggered by a sudden noise occur among LQT2 patients13; in LQT3 arrhythmias mostly occur during rest/sleep.13 This population-based observation now allows to suspect the genotype simply on the basis of the medical history, before the results of the genetic test, and to implement gene-specific management14 as outlined further down in this section.
The current understanding of LQTS has already allowed clinical decisions that could very well be regarded as aspects of PM, not fancy but effective and dictated by common sense. Two examples are relevant.
When in 1995 it was realized that one form of LQTS (i.e. LQT3) was caused by an excess of inward sodium current, we proposed to test in these patients the sodium channel blocker mexiletine to verify whether or not the QT interval would have shortened.15 Our approach was successful and mexiletine for LQT3 patients is now part of standard therapy.16 , 17 This was the very first gene-specific therapy for LQTS and an early example of PM.
When in 2001 it became evident that the most dangerous time of the day for LQT2 patients was the awakening,13 we decided to shift nadolol administration from the traditional once a day in the morning to twice a day, morning and evening, to ensure an adequate plasma concentration in early morning hours while still asleep.14 This is a simple form of PM, but it is clinically meaningful and shows that understanding the specific, almost ‘private’, aspects of channelopathies impacts on management.
Conversely, it is fair to recognize that the main therapeutic tools used for LQTS, namely β-blockers, left cardiac sympathetic denervation, and ICDs, while being conceptually rather gross interventions, are actually quite effective and have saved the lives of most LQTS patients. As we explore the growing potential of PM using increasingly refined methodologies, the fact that we already have very effective therapies should not be underestimated. To put things in a proper perspective, it might be useful to compare the management of LQTS before and after 1995–1996 (the big divide being represented by the identification of the first three major genes for LQTS).7–9 Until then, the management for patients affected by LQTS, according to the diagnostic criteria in use at that time,18 was essentially the same: β-blockers for all and for those still at risk either left cardiac sympathetic denervation or an ICD.19 , 20 That was it. Genetics, more than bringing a true revolution led to a major refinement of our management. It became clear, based on the genotype–phenotype correlation mentioned above,13 that LQT1 patients should limit strenuous exercise and that it would be wise for them to swim only in the presence of an adult (one able to swim!).2 Similarly, for LQT2 patients, it was recommended to keep the K+ plasma levels at no less than 4 mEq/L, to avoid sleep disruption in the post-partum period and sudden noise while asleep. For LQT3 patients, at risk especially during night-time, we recommended sharing the room with an adult or having an audio surveillance system in the room to alert adults in case of gasping noises.21 For them,15 as mentioned above, and more recently also for some LQT2 patients,22 mexiletine was proven useful to shorten QTc. It has also become evident that LQT1 males who, while off therapy, remain without symptoms until age 25 are at extremely low risk of a first cardiac event and may not need therapy, if they so prefer. As to recommendations for exercise and competitive sports, there is uniformity in regarding LQT2 and LQT3 as at lower risk compared to LQT1, but there are differences between the American (more liberal) and the European (less liberal) views.2 , 23
These changes in management, which may well be labelled as PM, represent not only an undeniable progress in the way we treat and protect our LQTS patients from life-threatening arrhythmias but also a major reward for the efforts to never stop in the search of novel insights into the mechanisms underlying this disease, which constitutes, better than any other, the link between molecular genetics and clinical medicine.4
Genetics
Genetics have a pivotal role in the discovery of new disease-causing genes or variants associated with a specific disease phenotype. Seventeen genes, responsible for or associated with LQTS, have been so far identified.2 The three main ones, KCNQ1 (LQT1), KCNH2 (LQT2) and SCN5A (LQT3), account for ∼75% of the clinically defined cases of LQTS, while the minor genes contribute an additional 5% together. About 20% of LQTS cases still do not find a genetic characterization and represent an important opportunity for in-depth diagnostic use of personalized approaches.24
Next-generation sequencing (NGS) technology provides the genome sequence data in hours. The large datasets generated by the NGS technology led to an exponential increase in the amounts of genes and variants associated with specific clinical phenotype but have also caused the proliferation of functionally uncharacterized variants, which represent a non-negligible portion of all genotype-positive patients for a specific disease. For these ‘variants of uncertain significance’ (VUS), the evidence in support of their pathogenicity is lacking or controversial.25 In our clinical databases, ∼19% of the variants found in LQT1 and ∼33% of those found in LQT2 patients are classified as VUS (Figure 2).
Figure 2.
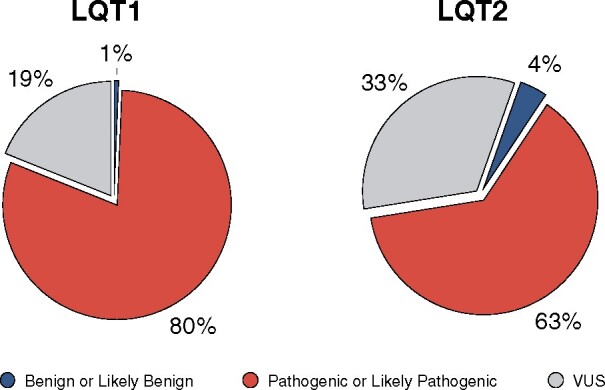
Variants of Unknown Significance. Percentage of variants classified as VUS in our clinical LQT1 and LQT2 patient database. N = LQT1: 153 variants, 29 VUS, 777 subjects; LQT2: 152 variants, 50 VUS, 563 subjects.
Improvements in diagnostic technologies and the increase in population data worldwide have contributed significantly to the reappraisal and reclassification of genes and variants previously associated with certain disease phenotypes. By strictly revising the criteria with which genes were considered as associated with an LQTS phenotype, Adler et al.26 recently observed that the genotype–phenotype associations behind many genes that were originally identified as disease causing for LQTS were instead limited or disputed for disease causation. Some genes were reclassified due to intrinsic limitations of the ‘candidate gene’ approach in comparison to more modern and robust genome-wide methods. Another issue included the underestimation of the frequency of the variant in the general population; this led to the conclusion that certain variants, with unknown frequencies at the time of their identification, were common in the general population and even more common in some ethnicities; thus, the disease phenotype was very unlikely to be caused by an autosomal dominant inheritance. Besides the specific case of LQTS, this study indicates that a PM approach to diagnosis and therapy must include a prompt re-evaluation of the interpretation of genetic findings that parallels progress in knowledge and technology.
In addition to variants in coding regions, for which ancillary data such as transcript sequence, protein structure, or functional analyses may facilitate the inference of disease causality, it is necessary to highlight the importance of regulatory variants in non-coding regions of the genome. While current knowledge of the architecture and functional role of regulatory elements in the human genome is scanty, there is a plethora of evidence suggesting that intergenic regulatory elements and non-coding RNAs27 , 28 may play a significant role in both disease causality and disease expression for monogenic disorders.29 The advent of state-of-the-art genome sequencing technologies will allow functional genomics to explore the contribution of regulatory genetic elements to ultimately explain the incomplete penetrance and variable expressivity observed in LQTS patients.2 , 30
On a cautionary side, it is important to avoid over-interpretation of genetic findings and assumptions with limited evidence to avoid misdiagnosis and potential harm to patients.31 Accordingly, it is crucial to identify potentially disease-causing variants and to validate their pathogenicity with high-fidelity in vitro technologies that can circumvent the need of large populations of patients.
In vitro characterization of variants
High-throughput approaches based on heterologous systems are a widespread method to acquire information on the pathogenicity of putative disease-causing variants identified with large yield sequencing platforms.32 Robotic and automated pipelines may allow the rapid electrophysiological characterization of hundreds of variants,32 significantly contributing to their functional classification and to a more efficient patient risk stratification. However, albeit these high-throughput pipelines should be routinely implemented, it remains evident that the in vitro electrophysiological effects of a variant do not yet necessarily translate into predictable clinical effects for various reasons. First, a variant may have different functionality in immortalized cell lines (e.g. HEK293) transfected with plasmids than in adult cardiomyocytes; this may be due to cell-specific post-translational modifications of ion channels or to different expression levels of accessory subunits or regulatory proteins. The presence of splicing variants further complicates genotype–phenotype correlations and requires additional in vitro studies to identify and characterize the altered transcript and the altered splicing mechanisms prior to any electrophysiological study. However, this additional complexity does not preclude a proper functional characterization, and the putative function of splicing variants can still be captured with an electrophysiological analysis in heterologous systems.33 , 34 Second, the contribution of genetic modifiers may be concealed while approaching the disease characterization in heterologous systems, potentially leading to wrong assumptions of variant pathogenicity that may have relevant clinical implications.35–37 Third, the contribution of patient’s genetic background and external factors still cannot be recapitulated by heterologous systems. For instance, the KCNQ1-A341V variant shows a mild in vitro phenotype when expressed in heterologous systems but its in vivo phenotype is very severe.38 , 39 Conversely, the KCNQ1-Y111C trafficking variant exhibits a severe loss of function in heterologous system while patients have a low risk of cardiac events,40 a condition that might be potentially explained by the concomitant presence of common variants in genes related to protein (ion channel) turnover.41 Correlating the clinical effect of each variant with their functional in vitro role is a possible first step to identify all the variants that may have outlier or abnormal genotype–phenotype correlations. However, more physiological cellular models capable to mimic the genetic background in which the variants exert their effect are required for advanced mechanistic studies.
Patient-specific induced pluripotent stem cell-derived cardiomyocytes
Human induced pluripotent stem cells (iPSCs) were first generated in 2007 by transforming dermal fibroblasts using retroviruses encoding reprogramming genes.42 Shortly thereafter, other groups generated human iPSCs by reprogramming different somatic cell sources including mononuclear cells isolated from peripheral blood,43 a non-invasive method offering clear advantages for the recruitment of patients, especially children.
Human iPSCs can be differentiated into cardiomyocytes (iPSC-CMs).44–46 Differentiation efficiency has profoundly increased over time and current protocols generate highly enriched populations of cardiomyocytes with the minimal characteristics required to study channelopathies.47–50 As a result, applications and diseases modelled with the iPSC technology have skyrocketed and have validated these cellular models as important preclinical tools for identifying novel mechanisms of disease51–57 and for drug screening purposes.58 , 59 iPSC-CMs fairly faithfully summarize ‘clinical trial on a plate’ (population level) and even ‘patient on a plate’ (individual level) conditions, thus representing a good model for the implementation of PM to better identify and manage LQTS and other cardiac channelopathies.60
The first study describing LQT1 modelling showed that patient-derived iPSC-CMs summarize the typical electrophysiological features of the disease such as prolongation of action potential duration, abnormal response to the stimulation of catecholamine prevented by β-blockers, and altered activation and deactivation kinetics of IKs.61 Besides the three main LQTS types,3 multiple studies further demonstrated the ability of iPSC-CMs to model minor subtypes, including calmodulinopathies.62–66 Correlation between in vitro parameters recorded with Multi-Electrode Arrays (MEAs)67 and the ECG seem to confirm the potential for iPSC-CMs to model some clinically relevant features in vitro,68 although large-scale comparisons of electrophysiological parameters from the same donor have still to be generated.
The iPSC-CMs allow researchers to use cells that share ethnicity, gender, and genetic background with the donor, boosting the capabilities to observe and describe patient-specific responses in a human cellular environment comprehensive of the patient’s genetic background. For this reason, disease lines should be matched for gender and ethnicity to perform proper comparisons. The age of the donor should also be considered as somatic cells from older patients have accumulated a larger number of spontaneous mutations that may become relevant after reprogramming.69 iPSCs derived from relatives, who partially share the genetic background, may help in comparative studies to minimize confounding factors derived from donor-specific genetic background differences that still represent the largest source of genetic variation.70 Studying multiple cell lines harbouring the same disease-causing mutations should also contribute to dilute potential confounding factors arising from experimental procedures, clonal variability, random variant accumulation, and in vitro selective pressure. A candidate approach to tackle the functional effect of rare genetic variants would ideally involve multiple laboratories in the generation of different iPSC lines for each single variant to triangulate the VUS effect across different centres, cell platforms, and differentiation methods. In this context, genome editing technologies have become a powerful tool to discriminate the effect of VUSs for monogenic disorders as cardiac channelopathies.71 These approaches integrate two main strategies: (i) insert the variant in a bona fide wild-type line to verify whether the variant triggers a disease phenotype or (ii) correct the variant in patient-specific iPSC lines to rescue the disease phenotype. This is also particularly relevant for tackling the intrinsic phenotypic variability exhibited by iPSC-CMs from different lines or donors,72 and in both approaches each genome edited line represents the proper control for comparison when validating the pathogenicity of a variant.41 , 63 , 64 , 73 , 74 Operators should also work blindly when investigating VUS pathogenicity to limit bias due to stochastic variations in the data. A remarkable contribution to minimize such variability may come from high-throughput robotics and automated liquid handling platforms, now capable of performing complex tasks as cell culture, line derivation, differentiation,75 , 76 as well as complex functional characterizations.77
Such technological advances have also made possible to use the iPSC technology for drug discovery and screening78 purposes, where new or already marketed drugs are being used to target specific mechanisms underlying different channelopathies. This strategy has the potential to significantly impact the PM concept and promising results are already available.78–83 Some of these early examples will be discussed later, in sections dedicated to drug screening and repurposing.
How to use iPSC-CMs to improve current Precision Medicine strategies
Having said that iPSC technology will help push forward several aspects linked with personalized choices, we also underline that a robust PM strategy should rely more on extensive population data since putting all hopes and efforts in downstream individual patient-specific approaches cannot represent the magic bullet for several reasons. Specifically, it is technically and financially unsustainable at the present state of technology to generate personalized cell lines for each single patient; feature-specific or cohort-specific approaches are instead more realistic. To this end, it is crucial to gather patients in clusters stratified by major factors as age, gender, ethnicity, variant type, and clinical features. The contribution of cell banks will guarantee methodological consistency and proper quality controls. Nevertheless, a personalized approach (i.e. one iPSC line per patient) should not always be discarded since it might be useful for specific situations, for instance, as a filter to evaluate which subjects will likely respond to specific pharmacological treatments.84 , 85
Use of iPSC-CMs to identify and characterize genetic modifiers
Personalised approaches to study LQTS can also foster the discovery of subtle regulatory mechanisms that would have hardly been captured with large population studies. The incomplete penetrance of LQTS manifests in individuals affected by identical disease-causing mutations having a variable expressivity of the disease phenotype.30 Combinations of patient-specific iPSC-CMs, genetics, and genome editing have already allowed the identification of putative modifier genes involved in the phenotype of LQT141 and LQT2.73 Both studies started from LQTS families in which patients showed a different disease severity despite the presence of an identical disease-causing mutation. Subject-specific iPSC-CMs reproduced the variable phenotype severity of their donors in vitro on action potential duration, field potential duration (a gross in vitro equivalent of the QT interval) and ion current levels. Whole exome sequencing allowed the precise identification of variants putatively regulating genotype–phenotype discordance. The correction of these variants by CRISPR/Cas9 genome editing either rescued73 or unmasked41 the LQTS disease phenotype, respectively, confirming the damaging and protective role of these variants. The data were further confirmed in small cohorts of patients harbouring the variants but large population studies will now be required to potentially extend these results to other LQTS variants or subtypes. The identification of factors influencing disease severity contributes to a more precise risk stratification system that could impact clinical management (Figure 3).
Figure 3.

Modifier genes. Schematic representation of the mechanism of action of a putative protective modifier gene indirectly altering the electrophysiology and protecting the patient from cardiac events.
In addition to these small-scale and patient-specific approaches, factors capable of shaping disease severity may potentially be discovered through genome-wide association studies (GWASs). Despite the requirement of large populations of patients, GWAS-based approaches can identify loci harbouring genetic variants affecting the QT interval duration in the general population and may provide a reliable starting point for more patient-specific analyses on specific subpopulations of patients. This was the case for two common variants in the nitric oxide synthase 1 adaptor protein (NOS1AP) gene, previously identified as a modulator of QT interval in the general population through a GWAS,86 which was later associated with an increased risk of life-threatening arrhythmias in an LQT1 founder population87 and whose mechanism of action was characterized using patient-specific iPSC-CMs.88 We foresee that combined improvements in the identification of these modifiers and in their downstream functional characterization might lead to more precise risk stratification and clinical management of cardiac channelopathies (Figure 4).
Figure 4.

Precision Medicine Gap. Schematic representation of the journey of a patient diagnosed with symptoms that suggest the presence of a cardiac channelopathy. Once the diagnosis is validated, the patient blood is sent for sequencing with results returned within weeks. From that patient’s blood it is possible to generate iPSCs, correct the genetic defect with gene editing and obtain functional cardiomyocytes within a few months. After these key steps have been made, it is then possible to start specific drug testing on patient-derived cells to attempt a more personalised therapy. This entire process could require more than one year, during which time a buffer therapy has to be implemented to guarantee the care of the patient.We call this period the ‘Precision Medicine gap’: it represents a time in which PM approaches cannot yet be implemented. We anticipate that this gap will be progressively shortened as further progress will be made.
Drug toxicity
Perhaps the most intuitive application of iPSC-CMs is their use for the safety assessment of preclinical drug candidates.72 , 89 Traditional experimental models to assess cardiac toxicity in the preclinical phase of drug development rely on heterologous systems, costly and often inconclusive animal models or more recently on computational approaches. More specifically, the risk of developing ventricular arrhythmias, in particular Torsades de Pointes (TdP), is tested in vitro by measuring IKr blockade.90 However, this parameter alone is not a robust predictor of TdP or of ventricular arrhythmias in general and the attrition rate of drugs during development raises questions on the reliability of these approaches.91 Indeed, even though some of these systems can be partially automated and performed at high throughput with excellent yields,92–94 their predictive value is often low, and this has resulted in the withdrawal of drugs not only in advanced phases of development but also after having entered the market.95 , 96
Thus, an easily accessible human-based cellular model, potentially with a higher translational relevance, has attracted basic scientists, pharmaceutical industries and regulatory agencies. An almost complete set of endogenous cardiac ion channels and the ability to potentially perform high-throughput assays make this platform very attractive for the early stages of drug development where medicinal chemistry require guidance.97 , 98 The suitability of iPSC-CMs for the prediction of arrhythmic cardiotoxicity is currently being investigated also by large consortia as part of the Comprehensive in vitro Proarrhythmia Assay (CiPA) initiative, meant to revise tests for hERG blockade and possibly replace thorough QT studies to more accurately predict the arrhythmogenic risk of compounds, to reduce the rate of false positives.99–101
To date, the area in which platforms based on iPSC-CMs have been most tested to study drug toxicity in the field of cardiac arrhythmias is that of drug-induced LQTS (diLQTS).102 Even though, strictly speaking, diLQTS is not a channelopathy, we have shown that 30% of patients developing diLQTS carry LQTS disease-causing mutations.102 A remarkable example of diLQTS modelling has been recently reported.63 The investigators first shortlisted healthy volunteers based on their QT prolonging potential exhibited 3 h after a sotalol challenge in a clinical trial (clinicaltrials.gov, NCT01338441). Then, they selected the most extreme phenotypes of low responders, e.g. individuals with an absent or modest QT prolongation after sotalol, and high responders, who did exhibit important QT prolongation after the challenge. They generated subject-specific iPSC-CMs and measured in vitro their pharmacological response using MEA. Despite some variability in ion channel expression (hERG) or drug response, the majority of cell lines clustered according to their in vivo response to sotalol, demonstrating a remarkable correlation between clinical and in vitro data. A similar approach can also be used to screen drug toxicity of new or repurposed drugs in iPSC-CMs derived from patients with LQTS or other types of channelopathies (see below).
Limitations of approaches based on iPSC-CMs
Despite the great enthusiasm for this cellular model, there are still important limitations that may partly impact their use for clinically relevant approaches. The reprogramming of somatic cells into iPSCs alters some of the epigenetic modifications103 and the effects of stimuli that have been accumulated during lifetime by the subject. This has the unintended consequence of removing the contribution of environmental factors as physical activity, diet or concurrent pharmacological therapies in the exacerbation of the disease phenotype. This may have a minor importance for inherited channelopathies with premature onset, but the potential contribution of environmental factors on the long-term progression of the disease should be considered when investigating the severity of genetic variants.
Another possible limitation is that iPSC-CMs generally show immature characteristics in morphology, cytoskeletal proteins, and ion channel expression and organization. The iPSC-CMs described so far have characteristics more similar to CMs of foetal origin than to mature adult CMs.48 Since the majority of LQTS mutations do generate clear cut phenotypes as a result of variants whose phenotypic consequence is often manifested at young age, it would be logical to hypothesize that the maturation level currently reached by the iPSC-CM technology could be sufficient to reproduce most of the LQTS phenotypes in vitro. However, the cell maturation level might be critical when investigating mutations that require a fully organized and functional excitation–contraction coupling apparatus, or when the mutation phenotype is shaped by VUS, modifier genes or for proteins which expression levels or isoforms vary with age.104 Strategies to tackle and partially overcome this issue include the use of long-term cultures105 or miRNA,106–108 the development of more effective maturation media,109 , 110 the use of electrical stimulation,111 or stretch112 during differentiation. Numerically modelled IK1 injected into iPSC-CMs by dynamic clamp, an advanced state-of-the-art approach, has been successfully used to overcome issues derived by the low expression of this ion current in isolated iPSC-CMs.41 , 62 , 113 , 114 Another important limitation of standard iPSC-CMs cultures is that they lack the complexity of cardiac tissue. For this reason, tridimensional multicellular models such as engineered heart tissues,115–117 cell sheets,118 or microtissues constituted of two or three defined cell types119 , 120 have been developed, with good results also in terms of cell maturation. The generation of organ-on-a-chip platforms will contribute to further refine the predictability and clinical relevance of this technology.121
Computational approaches
Computational approaches have introduced important breakthroughs in the way experiments are performed and data are analysed and interpreted. Technology has advanced to the point of becoming a key aspect of medicine, with the potential to improve diagnostic accuracy, risk stratification, and drug safety. These methods integrate data generated by clinical, in vitro and in silico sources and use them for predictions, such as the potentially cardiotoxic effect of a drug treatment. They are widely used in safety pharmacology and are a pillar of the CiPA initiative.122 Recent examples demonstrated that these approaches may even have a superior accuracy than animal models to detect cardiotoxicity caused by proarrhythmic drugs123 and future technological advancements will certainly contribute to further refine these models.124
In the context of PM, one of the most mature concept proposed so far is the digital twin,125 which aims to acquire and combine data from multiple sources as clinical records, imaging reports, -omics, and in vitro experimental data to build mathematical models capable to extract relevant parameters for clinicians and generate new knowledge. This concept has emerged with some notable examples126–128 in the field of cardiology and could be further refined to embrace also early-onset diseases as congenital channelopathies, with population data, genomics and functional in vitro evaluations guiding risk stratification and clinical pharmacological treatments. However, it is important to include diverse genetic backgrounds, ethnicities, and genders while training models and classifying variants, since a wrong enrichment of specific groups in the training dataset may skew predictions, and thus clinical decisions, towards unnecessary therapy or inaction, both with potentially negative consequences.129
In this regard, the interpretation of ECG features with neural networks may allow a substantially improved diagnostic accuracy, with the possibility to use the power of large datasets containing hundreds of thousands of annotated ECG records to train algorithms and detect features specific to the main channelopathies.130 Since ECG interpretation may be considered a statistical classification problem, thus requiring large ‘training’ data sets, the accuracy and precision of these models will likely increase with their use, and this can provide great benefits to small hospitals with little experience in cardiac channelopathies. Some studies already begun with collaborations between large hospitals and industry (e.g. NCT04441892 or NCT02412709 on clinicaltrials.gov); moreover, these technologies have proven their efficacy in enhancing risk stratification for acute coronary syndrome,131 to identify the patients who could be at higher risk of developing the disease,127 and for the classification of rhythm disturbances to reduce the rate of misdiagnoses in ECG interpretations.128 Caution should still be put in the output generated by neural networks as the criteria used by these algorithms for classification are concealed and the data interpretation in these early phases of development should still be further validated.132 Nevertheless, these promising results for PM appear particularly exciting when coupled with the widespread use of wearable technologies allowing ECG monitoring without the need of healthcare specialists.133 , 134
When dreams meet reality
Despite all these promising and fascinating attempts, it is important to note that so far only a tiny fraction of the progress from the described in vitro and in silico strategies have truly benefitted the patients. The main question is how to handle patient-specific tests that may take months or years to be implemented after the first diagnostic hypothesis is made (Figure 5). The widespread diffusion of high-throughput genetic testing has been coupled, especially in small centres or in developing economies, with insufficient resources to analyse data, functionally validate the output, and extrapolate meaningful information to improve patient’s care. Even though it is acceptable for patients to receive the results of their genetic testing within weeks, at the present stage of technology, it is clearly unfeasible to introduce long-term strategies based on patient-specific cellular models for the majority of our patients. High-throughput platforms often encounter the bottleneck of a low-throughput functional biology.
Figure 5.
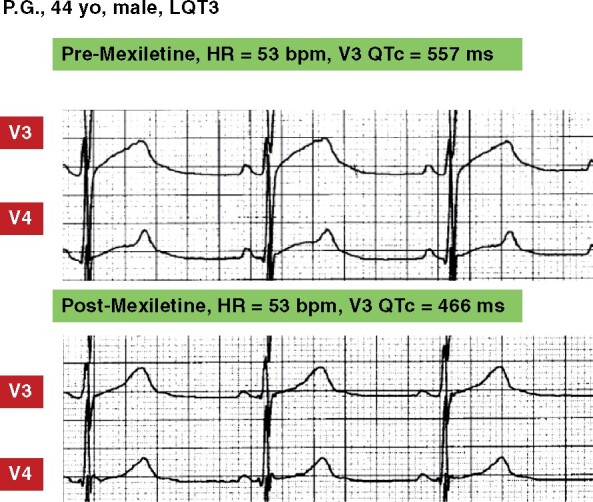
Mexiletine effect. This LQT3 patient (SCN5A-p.M1320V), already on therapy with nadolol (0.28 mg/kg/die limited by bradycardia), continued to show a major QT interval prolongation with a very abnormal T wave. Following mexiletine (5.6 mg/kg/die), both QTc and the T wave morphology became close to normal. The Holter tracings were recorded while the patient was in chronic therapy; the changes observed within 2 h from the acute oral administration were similar and prompted the addition of mexiletine to the therapy. The patient continues to remain asymptomatic.
As previously mentioned, variant-specific therapeutic strategies may be implemented and the power of high-throughput, robotic functional screenings for the biophysical characterization of variants will significantly shorten the time required to obtain the results, and their implementation may be considered alongside with a thorough genetic screening. iPSC-CMs will not offer useful interpretations of clinical realities to guide clinical practice until shared efforts among groups worldwide will be made to develop and predict, in large heterogeneous cohorts, the functional effects of individual variables on disease traits and drug responses. These efforts will require comprehensive consortia and an extensive protocol automation75 and standardization similar to what has been done within the CiPA initiative122 and others.135
Despite all these limitations, the progress in the understanding of the mechanisms underlying channelopathies has already led to new tools to identify responders to a specific therapy, to proposals for novel targeted therapies, and to attempts for drug repurposing. However, each novel PM strategy should improve on current therapies and would need to survive a careful cost–benefit analysis before becoming clinically meaningful. What follows is a series of a few recent representative ground-breaking examples of PM that, despite the novelty of their approach and scientific quality, still have to overcome a number of obstacles before becoming truly useful to patients.
New tools to identify responders
The pioneering 1995 report on the efficacy of mexiletine to shorten QTc in LQT3 patients15 was endorsed by guidelines16 , 17 and confirmed by widespread clinical experience.136 However, it soon became evident that not all LQT3 patients shorten their QTc with mexiletine. It was recently shown that there is a blunted response to mexiletine when mutations affect the Domain III voltage sensing domain of NaV1.5 (DIII-VSD) and that this peculiarity can be exploited to predict whether or not mexiletine will shorten QTc.137 Since the method uses the voltage dependence of DIII-VSD to properly predict the functional response in patients, this must be a known prerequisite for each specific SCN5A variant. However, as biophysical characterization is available only for a minority of variants, it follows that this elegant procedure would often take months to get an answer. By contrast, what now happens clinically is simple and rapid: the patient is given half of the daily dose orally and his/her QTc is monitored continuously for 2 h; within that time mexiletine reaches its therapeutic plasma concentration and if the QTc shortens it is immediately evident (Figure 5). The rapidity, safety, and clarity of the results with the acute oral drug testing method are such that a complex and time-consuming method does not appear very practical with the current state of technology and knowledge on SCN5A variants. Nevertheless, the pool of biophysically characterized SCN5A variants will eventually grow and allow these refined and prompt predictions to be clinically useful.
Novel therapies
Disease-specific iPSC models may also facilitate the identification and optimization of compounds capable to rescue the disease phenotype. A logical in vitro strategy to correct the disease phenotype caused by impaired IKs (LQT1, LQT5) or IKr (LQT2, LQT6) is to pharmacologically enhance these two key repolarizing currents. PM approaches have been attempted to correct the disease phenotype on patient-specific iPSC-CMs by using pharmacological activators, i.e. small molecules able to alter the biophysical properties of KV7.1 or KV11.1 and enhance IKs 138 , 139 or IKr.52 , 78 , 140–142 Despite the remarkable shortening of field potential duration and action potential duration that has been obtained in vitro with some of them,78 , 142 a clinical translation appears premature and requires caution since many of these compounds have a very narrow therapeutic window. In addition, it has been suggested that some of them could exert proarrhythmic effects because of an over- or inaccurate correction of the LQTS phenotype.142 The identification of the correct dose considering the biophysics of each specific gene variant is needed. This additional step is likely to encounter difficulties before this approach could prove to be useful in the daily practice, given the safety and efficacy of β-blockers for most LQTS patients.
By using human iPSC-CMs derived from a SCN5A-1795insD+/− mutation carrier, it was possible to validate the effects of GS967, a new potent and selective inhibitor of INaL, on repolarization abnormalities, resulting in antiarrhythmic effects with no deleterious consequences on cardiac conduction.81 Importantly, the in vitro results were confirmed in knock-in mice harbouring the homologous mutation, demonstrating that iPSC-CMs can provide reliable disease models. Very recently, a chemical refinement of the antiarrhythmic drug mexiletine was obtained via high-throughput screening on iPSC-CMs derived from LQT3 patients, leading to analogues with increased potency and selectivity for inhibiting INaL across a panel of 7 LQT3 variants.143 The authors hypothesize that these mexiletine analogues can be exploited as mechanistic probes and for clinical development.
Drug repurposing
Drug repurposing represents an alternative approach to tailor drug responses to patient needs. By relying on drugs already approved for clinical use, this strategy has the advantage of reducing time and costs related to safety trials and drug approval processes and, if proved effective, offers results readily applicable to the patients. The same approach is currently used and is being optimized by industry with the objective of integrating preclinical drug testing on iPSC-CMs Large-scale assays are being developed with single candidate healthy iPSC lines (bona fide wild-type genomes), with the aid, when needed, of gene editing technologies.
The majority of LQT2 mutations cause a defect in hERG trafficking.144 The identification of a drug capable of correcting the trafficking defect could therefore have potential positive effects for these patients. Mehta et al.145 reported that Lumacaftor, a drug acting on protein trafficking and successfully used for the treatment of cystic fibrosis,146 can restore the trafficking of hERGs in iPSC-CMs derived from LQT2 patients carrying Class 2 mutations and can shorten the corrected field potential duration. This effect was Class-specific as it was absent in iPSC-CMs from LQT2 patients with Class 1 mutations (hERG protein synthesis).145 We moved from the laboratory to the clinic and performed a first translational study in two of the patients whose iPSC-CMs responded to Lumacaftor in Mehta’s study, having considered that Lumacaftor is already approved for clinical use with a good safety profile.147 We documented QTc shortening in both patients, and a substantial increase in the time during which the QTc values were in the lower tertile of the values at baseline. However, the magnitude of QTc shortening (∼30 ms) was lower than we had expected based on the in vitro results.147 Very recently, O’Hare et al.148 confirmed, in similar experimental conditions, that Lumacaftor rescues hERG trafficking in three LQT2 patient-derived iPSCs. The action potential shortening was observed in the two cell lines characterized by a pure trafficking defect, while it was prolonged in a cell line with co-existing trafficking and gating defects. While Lumacaftor may or may not turn out to be sufficiently useful for LQT2 patients with trafficking defect (a clinical trial—clinicaltrials.gov NCT04581408—is ongoing and should provide definitive answers), our combined experimental and clinical studies have shown that iPSC-CMs represent a valid tool for drug-repurposing strategies.149
Conclusions
PM is an important evolution of traditional medicine and the rapidity of its progress is impressive. It is exciting and stimulating also because its challenge is intellectually rewarding. It has the potential to significantly impact our societies and healthcare systems, by reducing the social and economic burden associated with inadequate prevention strategies, and patients alike, by offering tailored therapies of improved efficacy. To acquire sufficiently large datasets from in vivo and in vitro patient-specific strategies to train models, validate predictions, and implement on a large scale the various systems just described will require time, but this is the way by which science goes forward.
Nevertheless, PM should not be considered as an entirely new path but an ‘evolution’, which means a progress made without the need to jettison the huge amount of practical experience and knowledge accumulated over the years. In this review, we have tried to preserve the thread linking the more advanced technological approaches to the insights derived by relatively simple medical observations emerged by the time-honoured principle that understanding the mechanisms is, most of the time, the key for an effective therapy. We recognize the almost irresistible attraction of complex novelties, which we embrace, but at the same time we equally respect what has been learnt by those cardiologists who have fruitfully merged knowledge and clinical experience. This consideration should guide us when we have to critically assess whether new approaches to therapy, besides their novelty and elegance, are indeed of greater benefit to our true stakeholders, the patients.
Acknowledgements
The authors are grateful to Pinuccia De Tomasi, BS, for expert editorial support, and to Maria-Christina Kotta, MSc, PhD, for constructive suggestions on genetic aspects.
Funding
This work was supported by: Leducq Foundation for Cardiovascular Research [18CVD05] ‘Towards Precision Medicine with Human iPSCs for Cardiac Channelopathies’ to P.J.S. and M.G.; ‘Grant Dipartimenti d’Eccellenza 2018–2022’, Department of Molecular Medicine (Pavia University) to M.G.; Marie Skłodowska-Curie Individual Fellowship (H2020-MSCA-IF-2017 grant agreement No. 795209) to L.S.; and Fondazione CARIPLO, ‘Biomedical Research Conducted by Young Researchers’, grant No. 2019-1691 to L.S.
Conflict of interest: none declared.
Contributor Information
Massimiliano Gnecchi, Department of Cardiothoracic and Vascular Sciences—Coronary Care Unit and Laboratory of Clinical and Experimental Cardiology, Fondazione IRCCS Policlinico San Matteo, Viale Golgi 19, 27100 Pavia, Italy; Department of Molecular Medicine, Unit of Cardiology, University of Pavia, Viale Golgi 19, 27100 Pavia, Italy; Department of Medicine, University of Cape Town, J-Floor, Old Main Building, Groote Schuur Hospital, Observatory, 7925 Cape Town, South Africa.
Luca Sala, Istituto Auxologico Italiano IRCCS, Center for Cardiac Arrhythmias of Genetic Origin and Laboratory of Cardiovascular Genetics, Via Pier Lombardo 22 - 20135 Milan, Italy.
Peter J Schwartz, Istituto Auxologico Italiano IRCCS, Center for Cardiac Arrhythmias of Genetic Origin and Laboratory of Cardiovascular Genetics, Via Pier Lombardo 22 - 20135 Milan, Italy.
References
- 1.National Research Council, Division on Earth and Life Studies, Board on Life Sciences, Committee on a Framework for Developing a New Taxonomy of Disease. Toward Precision Medicine: Building a Knowledge Network for Biomedical Research and a New Taxonomy of Disease. Washington, DC: National Academies Press; 2012. [PubMed] [Google Scholar]
- 2. Schwartz PJ, Ackerman MJ, Antzelevitch C, Bezzina CR, Borggrefe M, Cuneo BF, Wilde AAM. Inherited cardiac arrhythmias. Nat Rev Dis Primers 2020;6:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sala L, Gnecchi M, Schwartz PJ. Long QT syndrome modelling with cardiomyocytes derived from human-induced pluripotent stem cells. Arrhythm Electrophysiol Rev 2019;8:105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schwartz PJ. 1970-2020: 50 years of research on the long QT syndrome-from almost zero knowledge to precision medicine. Eur Heart J 2020. doi:10.1093/eurheartj/ehaa769. [DOI] [PubMed] [Google Scholar]
- 5. Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med 2015;372:793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ashley EA. The precision medicine initiative: a new national effort. JAMA 2015;313:2119–2120. [DOI] [PubMed] [Google Scholar]
- 7. Curran ME, Splawski I, Timothy KW, Vincen GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 1995;80:795–803. [DOI] [PubMed] [Google Scholar]
- 8. Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, Moss AJ, Towbin JA, Keating MT. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 1995;80:805–811. [DOI] [PubMed] [Google Scholar]
- 9. Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, Shen J, Timothy KW, Vincent GM, De Jager T, Schwartz PJ, Towbin JA, Moss AJ, Atkinson DL, Landes GM, Connors TD, Keating MT. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet 1996;12:17–23. [DOI] [PubMed] [Google Scholar]
- 10. Moss AJ, Schwartz PJ. 25th anniversary of the International Long-QT Syndrome Registry: an ongoing quest to uncover the secrets of long-QT syndrome. Circulation 2005;111:1199–1201. [DOI] [PubMed] [Google Scholar]
- 11. Giustetto C, Schimpf R, Mazzanti A, Scrocco C, Maury P, Anttonen O, Probst V, Blanc J-J, Sbragia P, Dalmasso P, Borggrefe M, Gaita F. Long-term follow-up of patients with short QT syndrome. J Am Coll Cardiol 2011;58:587–595. [DOI] [PubMed] [Google Scholar]
- 12. Crotti L, Spazzolini C, Boczek NJ, Jimenez JJ, Makita N, Tester D, Etheridge SP, Beckmann BM, Papagiannis J, Webster GR, Lahrouchi N, Bezzina C, Horie M, Nyegaard M, Bennhagen R, Hamilton RM, Wilde A, George AL, Ackerman MJ, Schwartz PJ. International Calmodulinopathy Registry (ICaMR). Circulation 2016;134:A14840. [Google Scholar]
- 13. Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, Denjoy I, Guicheney P, Breithardt G, Keating MT, Towbin JA, Beggs AH, Brink P, Wilde AA, Toivonen L, Zareba W, Robinson JL, Timothy KW, Corfield V, Wattanasirichaigoon D, Corbett C, Haverkamp W, Schulze-Bahr E, Lehmann MH, Schwartz K, Coumel P, Bloise R. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation 2001;103:89–95. [DOI] [PubMed] [Google Scholar]
- 14. Schwartz PJ, Ackerman MJ. The long QT syndrome: a transatlantic clinical approach to diagnosis and therapy. Eur Heart J 2013;34:3109–3116. [DOI] [PubMed] [Google Scholar]
- 15. Schwartz PJ, Priori SG, Locati EH, Napolitano C, Cantù F, Towbin JA, Keating MT, Hammoude H, Brown AM, Chen LS, Colatsky TJ. Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate. Implications for gene-specific therapy. Circulation 1995;92:3381–3386. [DOI] [PubMed] [Google Scholar]
- 16. Al-Khatib SM, Stevenson WG, Ackerman MJ, Bryant WJ, Callans DJ, Curtis AB, Deal BJ, Dickfeld T, Field ME, Fonarow GC, Gillis AM, Granger CB, Hammill SC, Hlatky MA, Joglar JA, Kay GN, Matlock DD, Myerburg RJ, Page RL. 2017 AHA/ACC/HRS Guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. Circulation 2018;138:e272–e391. [DOI] [PubMed] [Google Scholar]
- 17. Priori SG, Blomström-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, Elliott PM, Fitzsimons D, Hatala R, Hindricks G, Kirchhof P, Kjeldsen K, Kuck K-H, Hernandez-Madrid A, Nikolaou N, Norekvål TM, Spaulding C, Van Veldhuisen DJ. Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC) Endorsed by Association for European Paediatric and Congenital Cardiology (AEPC). Europace 2015;17:1601–1687. [DOI] [PubMed] [Google Scholar]
- 18. Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome. An update. Circulation 1993;88:782–784. [DOI] [PubMed] [Google Scholar]
- 19. Schwartz PJ. Idiopathic long QT syndrome: progress and questions. Am Heart J Elsevier 1985;109:399–411. [DOI] [PubMed] [Google Scholar]
- 20. Schwartz PJ, Locati EH, Moss AJ, Crampton RS, Trazzi R, Ruberti U. Left cardiac sympathetic denervation in the therapy of congenital long QT syndrome—a Worldwide Report. Circulation 1991;84:503–511. [DOI] [PubMed] [Google Scholar]
- 21. Schwartz PJ. Management of long QT syndrome. Nat Clin Pract Cardiovasc Med 2005;2:346–351. [DOI] [PubMed] [Google Scholar]
- 22. Bos JM, Crotti L, Rohatgi RK, Castelletti S, Dagradi F, Schwartz PJ, Ackerman MJ. Mexiletine shortens the QT interval in patients with potassium channel-mediated type 2 long QT syndrome. Circ Arrhythm Electrophysiol 2019;12:e007280. [DOI] [PubMed] [Google Scholar]
- 23. Heidbuchel H, Arbelo E, D’Ascenzi F, Borjesson M, Boveda S, Castelletti S, Miljoen H, Mont L, Niebauer J, Papadakis M, Pelliccia A, Saenen J, de la Garza MS, Schwartz PJ, Sharma S, Zeppenfeld K, Corrado D. Recommendations for participation in leisure-time physical activity and competitive sports of patients with arrhythmias and potentially arrhythmogenic conditions. Part 2: ventricular arrhythmias, channelopathies, and implantable defibrillators . Europace 2020. doi:10.1093/europace/euaa106. [DOI] [PubMed] [Google Scholar]
- 24. Ingles J, Semsarian C. Time to rethink the genetic architecture of long QT syndrome. Circulation 2020;141:440–443. [DOI] [PubMed] [Google Scholar]
- 25. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, on behalf of the ACMG Laboratory Quality Assurance Committee Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Adler A, Novelli V, Amin AS, Abiusi E, Care M, Nannenberg EA, Feilotter H, Amenta S, Mazza D, Bikker H, Sturm AC, Garcia J, Ackerman MJ, Hershberger RE, Perez MV, Zareba W, Ware JS, Wilde AAM, Gollob MH. An International, multicentered, evidence-based reappraisal of genes reported to cause congenital long QT syndrome. Circulation 2020;141:418–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cheng X, Waghulde H, Mell B, Morgan EE, Pruett-Miller SM, Joe B. Positional cloning of quantitative trait nucleotides for blood pressure and cardiac QT-interval by targeted CRISPR/Cas9 editing of a novel long non-coding RNA. PLoS Genet 2017;13:e1006961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jiang Y, Du W, Chu Q, Qin Y, Tuguzbaeva G, Wang H, Li A, Li G, Li Y, Chai L, Yue E, Sun X, Wang Z, Pavlov V, Yang B, Bai Y. Downregulation of long non-coding RNA Kcnq1ot1: an important mechanism of arsenic trioxide-induced long QT syndrome. Cell Physiol Biochem 2018;45:192–202. [DOI] [PubMed] [Google Scholar]
- 29. Li X, Montgomery SB. Detection and impact of rare regulatory variants in human disease. Front Genet 2013;4:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Priori SG, Napolitano C, Schwartz PJ. Low penetrance in the long-QT syndrome: clinical impact. Circulation 1999;99:529–533. [DOI] [PubMed] [Google Scholar]
- 31. Gollob MH. Gene discovery: from biological plausibility to genetic evidence supporting disease causation. Heart Rhythm 2019;16:1707–1709. [DOI] [PubMed] [Google Scholar]
- 32. Vanoye CG, Desai RR, Fabre KL, Gallagher SL, Potet F, DeKeyser JM, Macaya D, Meiler J, Sanders CR, George AL. High-throughput functional evaluation of KCNQ1 decrypts variants of unknown significance. Circ Genom Precis Med 2018;11:e002345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gong Q, Zhang L, Moss AJ, Vincent GM, Ackerman MJ, Robinson JC, Jones MA, Tester DJ, Zhou Z. A splice site mutation in hERG leads to cryptic splicing in human long QT syndrome. J Mol Cell Cardiol 2008;44:502–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tsuji-Wakisaka K, Akao M, Ishii TM, Ashihara T, Makiyama T, Ohno S, Toyoda F, Dochi K, Matsuura H, Horie M. Identification and functional characterization of KCNQ1 mutations around the exon 7-intron 7 junction affecting the splicing process. Biochim Biophys Acta 2011;1812:1452–1459. [DOI] [PubMed] [Google Scholar]
- 35. Watanabe H, Yang T, Stroud DM, Lowe JS, Harris L, Atack TC, Wang DW, Hipkens SB, Leake B, Hall L, Kupershmidt S, Chopra N, Magnuson MA, Tanabe N, Knollmann BC, George AL, Roden DM. Striking in vivo phenotype of a disease-associated human SCN5A mutation producing minimal changes in vitro. Circulation 2011;124:1001–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schwartz PJ, Crotti L, George AL. Modifier genes for sudden cardiac death. Eur Heart J 2018;39:3925–3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. de la Roche J, Angsutararux P, Kempf H, Janan M, Bolesani E, Thiemann S, Wojciechowski D, Coffee M, Franke A, Schwanke K, Leffler A, Luanpitpong S, Issaragrisil S, Fischer M, Zweigerdt R. Comparing human iPSC-cardiomyocytes versus HEK293T cells unveils disease-causing effects of Brugada mutation A735V of NaV1.5 sodium channels. Sci Rep 2019;9:11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brink PA, Crotti L, Corfield V, Goosen A, Durrheim G, Hedley P, Heradien M, Geldenhuys G, Vanoli E, Bacchini S, Spazzolini C, Lundquist AL, Roden DM, George AL, Schwartz PJ. Phenotypic variability and unusual clinical severity of congenital long-QT syndrome in a founder population. Circulation 2005;112:2602–2610. [DOI] [PubMed] [Google Scholar]
- 39. Crotti L, Spazzolini C, Schwartz PJ, Shimizu W, Denjoy I, Schulze-Bahr E, Zaklyazminskaya EV, Swan H, Ackerman MJ, Moss AJ, Wilde AAM, Horie M, Brink PA, Insolia R, De Ferrari GM, Crimi G. The common long-QT syndrome mutation KCNQ1/A341V causes unusually severe clinical manifestations in patients with different ethnic backgrounds: toward a mutation-specific risk stratification. Circulation 2007;116:2366–2375. [DOI] [PubMed] [Google Scholar]
- 40. Winbo A, Diamant U-B, Stattin E-L, Jensen SM, Rydberg A. Low incidence of sudden cardiac death in a Swedish Y111C type 1 long-QT syndrome population. Circ Cardiovasc Genet 2009;2:558–564. [DOI] [PubMed] [Google Scholar]
- 41. Lee Y-K, Sala L, Mura M, Rocchetti M, Pedrazzini M, Ran X, Mak TSH, Crotti L, Sham PC, Torre E, Zaza A, Schwartz PJ, Tse H-F, Gnecchi M. MTMR4 SNVs modulate ion channel degradation and clinical severity in congenital long QT syndrome: insights in the mechanism of action of protective modifier genes. Cardiovasc Res 2021;117:767–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007;131:861–872. [DOI] [PubMed] [Google Scholar]
- 43. Staerk J, Dawlaty MM, Gao Q, Maetzel D, Hanna J, Sommer CA, Mostoslavsky G, Jaenisch R. Reprogramming of human peripheral blood cells to induced pluripotent stem cells. Cell Stem Cell 2010;7:20–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zwi L, Caspi O, Arbel G, Huber I, Gepstein A, Park I-H, Gepstein L. Cardiomyocyte differentiation of human induced pluripotent stem cells. Circulation 2009;120:1513–1523. [DOI] [PubMed] [Google Scholar]
- 45. Lian X, Hsiao C, Wilson G, Zhu K, Hazeltine LB, Azarin SM, Raval KK, Zhang J, Kamp TJ, Palecek SP. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc Natl Acad Sci USA 2012;109:E1848–E1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mura M, Ginevrino M, Zappatore R, Pisano F, Boni M, Castelletti S, Crotti L, Valente EM, Schwartz PJ, Gnecchi M. Generation of the human induced pluripotent stem cell (hiPSC) line PSMi003-A from a patient affected by an autosomal recessive form of Long QT Syndrome type 1. Stem Cell Res 2018;29:170–173. [DOI] [PubMed] [Google Scholar]
- 47. Berg C V D, Okawa S, Chuva de Sousa Lopes SM, Iperen L. V, Passier R, Braam SR, Tertoolen LG, Sol A. D, Davis RP, Mummery CL. Transcriptome of human foetal heart compared with cardiomyocytes from pluripotent stem cells. Development 2015;142:3231–3238. [DOI] [PubMed] [Google Scholar]
- 48. Veerman CC, Kosmidis G, Mummery CL, Casini S, Verkerk AO, Bellin M. Immaturity of human stem-cell-derived cardiomyocytes in culture: fatal flaw or soluble problem? Stem Cells Dev 2015;24:1035–1052. [DOI] [PubMed] [Google Scholar]
- 49. Gnecchi M, Stefanello M, Mura M. Induced pluripotent stem cell technology: toward the future of cardiac arrhythmias. Int J Cardiol 2017;237:49–52. [DOI] [PubMed] [Google Scholar]
- 50. L van den B, Brandão KO, Yiangou L, Mol MPH, Grandela C, Mummery CL, Verkerk AO, Davis RP. Cryopreservation of human pluripotent stem cell-derived cardiomyocytes is not detrimental to their molecular and functional properties. Stem Cell Res 2020;43:101698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Itzhaki I, Maizels L, Huber I, Zwi-Dantsis L, Caspi O, Winterstern A, Feldman O, Gepstein A, Arbel G, Hammerman H, Boulos M, Gepstein L. Modelling the long QT syndrome with induced pluripotent stem cells. Nature 2011;471:225–229. [DOI] [PubMed] [Google Scholar]
- 52. Matsa E, Rajamohan D, Dick E, Young L, Mellor I, Staniforth A, Denning C. Drug evaluation in cardiomyocytes derived from human induced pluripotent stem cells carrying a long QT syndrome type 2 mutation. Eur Heart J 2011;32:952–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gnecchi M, Schwartz PJ. The unstoppable attraction for induced pluripotent stem cells: are they the magic bullet for modeling inherited arrhythmogenic diseases? J Am Coll Cardiol 2012;60:1001–1004. [DOI] [PubMed] [Google Scholar]
- 54. Bellin M, Casini S, Davis RP, D'Aniello C, Haas J, Ward-van Oostwaard D, Tertoolen LGJ, Jung CB, Elliott DA, Welling A, Laugwitz K-L, Moretti A, Mummery CL. Isogenic human pluripotent stem cell pairs reveal the role of a KCNH2 mutation in long-QT syndrome. EMBO J 2013;32:3161–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhang M, D’Aniello C, Verkerk AO, Wrobel E, Frank S, Ward-van Oostwaard D, Piccini I, Freund C, Rao J, Seebohm G, Atsma DE, Schulze-Bahr E, Mummery CL, Greber B, Bellin M. Recessive cardiac phenotypes in induced pluripotent stem cell models of Jervell and Lange-Nielsen syndrome: disease mechanisms and pharmacological rescue. Proc Natl Acad Sci USA 2014;111:E5383–E5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mura M, Mehta A, Ramachandra CJ, Zappatore R, Pisano F, Ciuffreda MC, Barbaccia V, Crotti L, Schwartz PJ, Shim W, Gnecchi M. The KCNH2-IVS9-28A/G mutation causes aberrant isoform expression and hERG trafficking defect in cardiomyocytes derived from patients affected by long QT syndrome type 2. Int J Cardiol 2017;240:367–371. [DOI] [PubMed] [Google Scholar]
- 57. Schwartz PJ, Sala L. Precision vs traditional medicine. Clinical questions trigger progress in basic science: a favor not always returned. Circ Res 2019;124:459–461. [DOI] [PubMed] [Google Scholar]
- 58. Del Álamo JC, Lemons D, Serrano R, Savchenko A, Cerignoli F, Bodmer R, Mercola M. High throughput physiological screening of iPSC-derived cardiomyocytes for drug development. Biochim Biophys Acta 2016;1863:1717–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Elitt MS, Barbar L, Tesar PJ. Drug screening for human genetic diseases using iPSC models. Hum Mol Genet 2018;27:R89–R98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pang L, Sager P, Yang X, Shi H, Sannajust F, Brock M, Wu JC, Abi-Gerges N, Lyn-Cook B, Berridge BR, Stockbridge N. Workshop Report: FDA Workshop on improving cardiotoxicity assessment with human-relevant platforms. Circ Res 2019;125:855–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Moretti A, Bellin M, Welling A, Jung CB, Lam JT, Bott-Flügel L, Dorn T, Goedel A, Höhnke C, Hofmann F, Seyfarth M, Sinnecker D, Schömig A, Laugwitz K-L. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med 2010;363:1397–1409. [DOI] [PubMed] [Google Scholar]
- 62. Rocchetti M, Sala L, Dreizehnter L, Crotti L, Sinnecker D, Mura M, Pane LS, Altomare C, Torre E, Mostacciuolo G, Severi S, Porta A, De Ferrari GM, George AL Jr, Schwartz PJ, Gnecchi M, Moretti A, Zaza A. Elucidating arrhythmogenic mechanisms of long-QT syndrome CALM1-F142L mutation in patient-specific induced pluripotent stem cell-derived cardiomyocytes. Cardiovasc Res 2017;113:531–541. [DOI] [PubMed] [Google Scholar]
- 63. Yamamoto Y, Makiyama T, Harita T, Sasaki K, Wuriyanghai Y, Hayano M, Nishiuchi S, Kohjitani H, Hirose S, Chen J, Yokoi F, Ishikawa T, Ohno S, Chonabayashi K, Motomura H, Yoshida Y, Horie M, Makita N, Kimura T. Allele-specific ablation rescues electrophysiological abnormalities in a human iPS cell model of long-QT syndrome with a CALM2 mutation. Hum Mol Genet 2017;26:1670–1677. [DOI] [PubMed] [Google Scholar]
- 64. Limpitikul WB, Dick IE, Tester DJ, Boczek NJ, Limphong P, Yang W, Choi MH, Babich J, DiSilvestre D, Kanter RJ, Tomaselli GF, Ackerman MJ, Yue DT. A precision medicine approach to the rescue of function on malignant calmodulinopathic long-QT syndrome. Circ Res 2017;120:39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kotta MC, Sala L, Ghidoni A, Badone B, Ronchi C, Parati G, Zaza A, Crotti L. Calmodulinopathy: a novel, life-threatening clinical entity affecting the young. Front Cardiovasc Med 2018;5:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Badone B, Ronchi C, Kotta MC, Sala L, Ghidoni A, Crotti L, Zaza A. Calmodulinopathy: functional effects of CALM mutations and their relationship with clinical phenotypes. Front Cardiovasc Med 2018;5:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sala L, Ward-van Oostwaard D, Tertoolen LGJ, Mummery CL, Bellin M. Electrophysiological analysis of human pluripotent stem cell-derived cardiomyocytes (hPSC-CMs) using multi-electrode arrays (MEAs). J Vis Exp 2017;123:55587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kim J, Shah D, Potapov I, Latukka J, Aalto-Setälä K, Räsänen E. Scaling and correlation properties of RR and QT intervals at the cellular level. Sci Rep 2019;9:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lo Sardo V, Ferguson W, Erikson GA, Topol EJ, Baldwin KK, Torkamani A. Influence of donor age on induced pluripotent stem cells. Nat Biotechnol 2017;35:69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kyttälä A, Moraghebi R, Valensisi C, Kettunen J, Andrus C, Pasumarthy KK, Nakanishi M, Nishimura K, Ohtaka M, Weltner J, Van Handel B, Parkkonen O, Sinisalo J, Jalanko A, Hawkins RD, Woods N-B, Otonkoski T, Trokovic R. Genetic variability overrides the impact of parental cell type and determines iPSC differentiation potential. Stem Cell Rep 2016;6:200–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Garg P, Oikonomopoulos A, Chen H, Li Y, Lam CK, Sallam K, Perez M, Lux RL, Sanguinetti MC, Wu JC. Genome editing of induced pluripotent stem cells to decipher cardiac channelopathy variant. J Am Coll Cardiol 2018;72:62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sala L, Bellin M, Mummery CL. Integrating cardiomyocytes from human pluripotent stem cells in safety pharmacology: has the time come? Br J Pharmacol 2017;174:3749–3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chai S, Wan X, Ramirez-Navarro A, Tesar PJ, Kaufman ES, Ficker E, George AL, Deschênes I. Physiological genomics identifies genetic modifiers of long QT syndrome type 2 severity. J Clin Invest 2018;128:1043–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chavali NV, Kryshtal DO, Parikh SS, Wang L, Glazer AM, Blackwell DJ, Kroncke BM, Shoemaker MB, Knollmann BC. Patient-independent human induced pluripotent stem cell model: a new tool for rapid determination of genetic variant pathogenicity in long QT syndrome. Heart Rhythm 2019;16:1686–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Paull D, Sevilla A, Zhou H, Hahn AK, Kim H, Napolitano C, Tsankov A, Shang L, Krumholz K, Jagadeesan P, Woodard CM, Sun B, Vilboux T, Zimmer M, Forero E, Moroziewicz DN, Martinez H, Malicdan MCV, Weiss KA, Vensand LB, Dusenberry CR, Polus H, Sy KTL, Kahler DJ, Gahl WA, Solomon SL, Chang S, Meissner A, Eggan K, Noggle SA. Automated, high-throughput derivation, characterization and differentiation of induced pluripotent stem cells. Nat Methods 2015;12:885–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Conway MK, Gerger MJ, Balay EE, O’Connell R, Hanson S, Daily NJ, Wakatsuki T. Scalable 96-well plate based iPSC culture and production using a robotic liquid handling system. J Vis Exp 2015;2015:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Suk H-J, van Welie I, Kodandaramaiah SB, Allen B, Forest CR, Boyden ES. Closed-loop real-time imaging enables fully automated cell-targeted patch-clamp neural recording in vivo. Neuron 2017;96:244–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sala L, Yu Z, Ward-van Oostwaard D, Veldhoven J. V, Moretti A, Laugwitz K-L, Mummery CL, IJzerman AP, Bellin M. A new hERG allosteric modulator rescues genetic and drug-induced long-QT syndrome phenotypes in cardiomyocytes from isogenic pairs of patient induced pluripotent stem cells. EMBO Mol Med 2016;8:1065–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Sasaki K, Makiyama T, Yoshida Y, Wuriyanghai Y, Kamakura T, Nishiuchi S, Hayano M, Harita T, Yamamoto Y, Kohjitani H, Hirose S, Chen J, Kawamura M, Ohno S, Itoh H, Takeuchi A, Matsuoka S, Miura M, Sumitomo N, Horie M, Yamanaka S, Kimura T. Patient-specific human induced pluripotent stem cell model assessed with electrical pacing validates S107 as a potential therapeutic agent for catecholaminergic polymorphic ventricular tachycardia. PLoS One 2016;11:e0164795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Maizels L, Huber I, Arbel G, Tijsen AJ, Gepstein A, Khoury A, Gepstein L. Patient-specific drug screening using a human induced pluripotent stem cell model of catecholaminergic polymorphic ventricular tachycardia type 2. Circ Arrhythm Electrophysiol 2017;10:e004725. [DOI] [PubMed] [Google Scholar]
- 81. Portero V, Casini S, Hoekstra M, Verkerk AO, Mengarelli I, Belardinelli L, Rajamani S, Wilde AAM, Bezzina CR, Veldkamp MW, Remme CA. Anti-arrhythmic potential of the late sodium current inhibitor GS-458967 in murine Scn5a-1798insD+/− and human SCN5A-1795insD+/− iPSC-derived cardiomyocytes. Cardiovasc Res 2017;113:829–838. [DOI] [PubMed] [Google Scholar]
- 82. Song L, Park S-HE, Isseroff Y, Morikawa K, Yazawa M. Inhibition of CDK5 alleviates the cardiac phenotypes in Timothy syndrome. Stem Cell Rep 2017;9:50–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. El Battrawy I, Lan H, Cyganek L, Zhao Z, Li X, Buljubasic F, Lang S, Yücel G, Sattler K, Zimmermann W-H, Utikal J, Wieland T, Ravens U, Borggrefe M, Zhou XB, Akin I. Modeling short QT syndrome using human-induced pluripotent stem cell-derived cardiomyocytes. J Am Heart Assoc 2018;7:e007394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Burridge PW, Li YF, Matsa E, Wu H, Ong S-G, Sharma A, Holmström A, Chang AC, Coronado MJ, Ebert AD, Knowles JW, Telli ML, Witteles RM, Blau HM, Bernstein D, Altman RB, Wu JC. Human induced pluripotent stem cell-derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat Med 2016;22:547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Stillitano F, Hansen J, Kong CW, Karakikes I, Funck-Brentano C, Geng L, Scott S, Reynier S, Wu M, Valogne Y, Desseaux C, Salem JE, Jeziorowska D, Zahr N, Li R, Iyengar R, Hajjar RJ, Hulot JS. Modeling susceptibility to drug-induced long QT with a panel of subject-specific induced pluripotent stem cells. Elife 2017;6:1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Arking DE, Pfeufer A, Post W, Kao WHL, Newton-Cheh C, Ikeda M, West K, Kashuk C, Akyol M, Perz S, Jalilzadeh S, Illig T, Gieger C, Guo C-Y, Larson MG, Wichmann HE, Marbán E, O'Donnell CJ, Hirschhorn JN, Kääb S, Spooner PM, Meitinger T, Chakravarti A. A common genetic variant in the NOS1 regulator NOS1AP modulates cardiac repolarization. Nat Genet 2006;38:644–651. [DOI] [PubMed] [Google Scholar]
- 87. Crotti L, Monti MC, Insolia R, Peljto A, Goosen A, Brink PA, Greenberg DA, Schwartz PJ, George AL. NOS1AP is a genetic modifier of the long-QT syndrome. Circulation 2009;120:1657–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ronchi C, Bernardi J, Mura M, Stefanello M, Badone B, Rocchetti M, Crotti L, Brink P, Schwartz PJ, Gnecchi M, Zaza A. NOS1AP polymorphisms reduce NOS1 activity and interact with prolonged repolarization in arrhythmogenesis. Cardiovasc Res 2021;117:472–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Braam SR, Tertoolen L, A van de S, Meyer T, Passier R, Mummery CL. Prediction of drug-induced cardiotoxicity using human embryonic stem cell-derived cardiomyocytes. Stem Cell Res 2010;4:107–116. [DOI] [PubMed] [Google Scholar]
- 90. Bowes J, Brown AJ, Hamon J, Jarolimek W, Sridhar A, Waldron G, Whitebread S. Reducing safety-related drug attrition: the use of in vitro pharmacological profiling. Nat Rev Drug Discov 2012;11:909–922. [DOI] [PubMed] [Google Scholar]
- 91. Gintant G, Sager PT, Stockbridge N. Evolution of strategies to improve preclinical cardiac safety testing. Nat Rev Drug Discov 2016;15:457–471. [DOI] [PubMed] [Google Scholar]
- 92. Willumsen NJ, Bech M, Olesen SP, Jensen BS, Korsgaard MPG, Christophersen P. High throughput electrophysiology: new perspectives for ion channel drug discovery. Recept Channels 2003;9:3–12. [PubMed] [Google Scholar]
- 93. Dunlop J, Bowlby M, Peri R, Vasilyev D, Arias R. High-throughput electrophysiology: an emerging paradigm for ion-channel screening and physiology. Nat Rev Drug Discov 2008;7:358–368. [DOI] [PubMed] [Google Scholar]
- 94. Obergrussberger A, Goetze TA, Brinkwirth N, Becker N, Friis S, Rapedius M, Haarmann C, Rinke-Weiß I, Stölzle-Feix S, Brüggemann A, George M, Fertig N. An update on the advancing high-throughput screening techniques for patch clamp-based ion channel screens: implications for drug discovery. Expert Opin Drug Discov 2018;13:269–277. [DOI] [PubMed] [Google Scholar]
- 95. Ninan W, Ninan B, Wertheimer AI. Withdrawing drugs in the U.S. versus other countries. Inov Pharm 2012;3:1–5.22844651 [Google Scholar]
- 96. Waring MJ, Arrowsmith J, Leach AR, Leeson PD, Mandrell S, Owen RM, Pairaudeau G, Pennie WD, Pickett SD, Wang J, Wallace O, Weir A. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat Rev Drug Discov 2015;14:475–486. [DOI] [PubMed] [Google Scholar]
- 97. Liang P, Lan F, Lee AS, Gong T, Sanchez-Freire V, Wang Y, Diecke S, Sallam K, Knowles JW, Wang PJ, Nguyen PK, Bers DM, Robbins RC, Wu JC. Drug screening using a library of human induced pluripotent stem cell–derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity. Circulation 2013;127:1677–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Matsa E, Burridge PW, Yu K-H, Ahrens JH, Termglinchan V, Wu H, Liu C, Shukla P, Sayed N, Churko JM, Shao N, Woo NA, Chao AS, Gold JD, Karakikes I, Snyder MP, Wu JC. Transcriptome profiling of patient-specific human ipsc-cardiomyocytes predicts individual drug safety and efficacy responses in vitro. Cell Stem Cell 2016;19:311–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Blinova K, Stohlman J, Vicente J, Chan D, Johannesen L, Hortigon-Vinagre MP, Zamora V, Smith G, Crumb WJ, Pang L, Lyn-Cook B, Ross J, Brock M, Chvatal S, Millard D, Galeotti L, Stockbridge N, Strauss DG. Comprehensive translational assessment of human-induced pluripotent stem cell derived cardiomyocytes for evaluating drug-induced arrhythmias. Toxicol Sci 2017;155:234–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ando H, Yoshinaga T, Yamamoto W, Asakura K, Uda T, Taniguchi T, Ojima A, Shinkyo R, Kikuchi K, Osada T, Hayashi S, Kasai C, Miyamoto N, Tashibu H, Yamazaki D, Sugiyama A, Kanda Y, Sawada K, Sekino Y. A new paradigm for drug-induced torsadogenic risk assessment using human iPS cell-derived cardiomyocytes. J Pharmacol Toxicol Methods 2017;84:111–127. [DOI] [PubMed] [Google Scholar]
- 101. Huang H, Pugsley MK, Fermini B, Curtis MJ, Koerner J, Accardi M, Authier S. Cardiac voltage-gated ion channels in safety pharmacology: review of the landscape leading to the CiPA initiative. J Pharmacol Toxicol Methods 2017;87:11–23. [DOI] [PubMed] [Google Scholar]
- 102. Itoh H, Crotti L, Aiba T, Spazzolini C, Denjoy I, Fressart V, Hayashi K, Nakajima T, Ohno S, Makiyama T, Wu J, Hasegawa K, Mastantuono E, Dagradi F, Pedrazzini M, Yamagishi M, Berthet M, Murakami Y, Shimizu W, Guicheney P, Schwartz PJ, Horie M. The genetics underlying acquired long QT syndrome: impact for genetic screening. Eur Heart J 2016;37:1456–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Kim K, Doi A, Wen B, Ng K, Zhao R, Cahan P, Kim J, Aryee MJ, Ji H, Ehrlich LIR, Yabuuchi A, Takeuchi A, Cunniff KC, Hongguang H, Mckinney-Freeman S, Naveiras O, Yoon TJ, Irizarry RA, Jung N, Seita J, Hanna J, Murakami P, Jaenisch R, Weissleder R, Orkin SH, Weissman IL, Feinberg AP, Daley GQ. Epigenetic memory in induced pluripotent stem cells. Nature 2010;467:285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Veerman CC, Mengarelli I, Lodder EM, Kosmidis G, Bellin M, Zhang M, Dittmann S, Guan K, Wilde AAM, Schulze-Bahr E, Greber B, Bezzina CR, Verkerk AO. Switch from fetal to adult SCN5A isoform in human induced pluripotent stem cell-derived cardiomyocytes unmasks the cellular phenotype of a conduction disease-causing mutation. J Am Heart Assoc 2017;6:e005135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Kamakura T, Makiyama T, Sasaki K, Yoshida Y, Wuriyanghai Y, Chen J, Hattori T, Ohno S, Kita T, Horie M, Yamanaka S, Kimura T. Ultrastructural maturation of human-induced pluripotent stem cell-derived cardiomyocytes in a long-term culture. Circ J 2013;77:1307–1314. [DOI] [PubMed] [Google Scholar]
- 106. Pisano F, Altomare C, Cervio E, Barile L, Rocchetti M, Ciuffreda MC, Malpasso G, Copes F, Mura M, Danieli P, Viarengo G, Zaza A, Gnecchi M. Combination of miRNA499 and miRNA133 exerts a synergic effect on cardiac differentiation. Stem Cells 2015;33:1187–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Gnecchi M, Pisano F, Bariani R. microRNA and cardiac regeneration. Adv Exp Med Biol 2015;887:119–141. [DOI] [PubMed] [Google Scholar]
- 108. White MC, Pang L, Yang X. MicroRNA-mediated maturation of human pluripotent stem cell-derived cardiomyocytes: towards a better model for cardiotoxicity? Food Chem Toxicol 2016;98:17–24. [DOI] [PubMed] [Google Scholar]
- 109. Parikh SS, Blackwell DJ, Gomez-Hurtado N, Frisk M, Wang L, Kim K, Dahl CP, Fiane A, Tønnessen T, Kryshtal DO, Louch WE, Knollmann BC. Thyroid and glucocorticoid hormones promote functional t-tubule development in human-induced pluripotent stem cell-derived cardiomyocytes. Circ Res 2017;121:1323–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Feyen DAM, McKeithan WL, Bruyneel AAN, Spiering S, Hörmann L, Ulmer B, Zhang H, Briganti F, Schweizer M, Hegyi B, Liao Z, Pölönen R-P, Ginsburg KS, Lam CK, Serrano R, Wahlquist C, Kreymerman A, Vu M, Amatya PL, Behrens CS, Ranjbarvaziri S, Maas RGC, Greenhaw M, Bernstein D, Wu JC, Bers DM, Eschenhagen T, Metallo CM, Mercola M. Metabolic maturation media improve physiological function of human iPSC-derived cardiomyocytes. Cell Rep 2020;32:107925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Ronaldson-Bouchard K, Ma SP, Yeager K, Chen T, Song L, Sirabella D, Morikawa K, Teles D, Yazawa M, Vunjak-Novakovic G. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 2018;556:239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Abilez OJ, Tzatzalos E, Yang H, Zhao M-T, Jung G, Zöllner AM, Tiburcy M, Riegler J, Matsa E, Shukla P, Zhuge Y, Chour T, Chen VC, Burridge PW, Karakikes I, Kuhl E, Bernstein D, Couture LA, Gold JD, Zimmermann WH, Wu JC. Passive stretch induces structural and functional maturation of engineered heart muscle as predicted by computational modeling. Stem Cells 2018;36:265–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Wilders R. Dynamic clamp: a powerful tool in cardiac electrophysiology. J Physiol 2006;576:349–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Meijer van Putten RME, Mengarelli I, Guan K, Zegers JG, Ginneken A. V, Verkerk AO, Wilders R. Ion channelopathies in human induced pluripotent stem cell derived cardiomyocytes: a dynamic clamp study with virtual IK1. Front Physiol 2015;6:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Eschenhagen T, Eder A, Vollert I, Hansen A. Physiological aspects of cardiac tissue engineering. Am J Physiol Heart Circ Physiol 2012;303:H133–H143. [DOI] [PubMed] [Google Scholar]
- 116. Nunes SS, Miklas JW, Liu J, Aschar-Sobbi R, Xiao Y, Zhang B, Jiang J, Massé S, Gagliardi M, Hsieh A, Thavandiran N, Laflamme MA, Nanthakumar K, Gross GJ, Backx PH, Keller G, Radisic M. Biowire: a platform for maturation of human pluripotent stem cell-derived cardiomyocytes. Nat Methods 2013;10:781–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Turnbull IC, Karakikes I, Serrao GW, Backeris P, Lee J-J, Xie C, Senyei G, Gordon RE, Li RA, Akar FG, Hajjar RJ, Hulot J-S, Costa KD. Advancing functional engineered cardiac tissues toward a preclinical model of human myocardium. FASEB J 2014;28:644–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Shaheen N, Shiti A, Huber I, Shinnawi R, Arbel G, Gepstein A, Setter N, Goldfracht I, Gruber A, Chorna SV, Gepstein L. human induced pluripotent stem cell-derived cardiac cell sheets expressing genetically encoded voltage indicator for pharmacological and arrhythmia studies. Stem Cell Rep 2018;10:1879–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Giacomelli E, Bellin M, Sala L, van Meer BJ, Tertoolen LGJ, Orlova VV, Mummery CL. Three-dimensional cardiac microtissues composed of cardiomyocytes and endothelial cells co-differentiated from human pluripotent stem cells. Development 2017;144:1008–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Giacomelli E, Meraviglia V, Campostrini G, Cochrane A, Cao X, Helden R. V, Krotenberg Garcia A, Mircea M, Kostidis S, Davis RP, Meer B. V, Jost CR, Koster AJ, Mei H, Míguez DG, Mulder AA, Ledesma-Terrón M, Pompilio G, Sala L, Salvatori DCF, Slieker RC, Sommariva E, Vries A. D, Giera M, Semrau S, Tertoolen LGJ, Orlova VV, Bellin M, Mummery CL. Human-iPSC-derived cardiac stromal cells enhance maturation in 3D cardiac microtissues and reveal non-cardiomyocyte contributions to heart disease. Cell Stem Cell 2020;26:862–879.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Berg A. V D, Mummery CL, Passier R, Meer A. V D. Personalised organs-on-chips: functional testing for precision medicine. Lab Chip 2019;19:198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Fermini B, Hancox J,A, Gerges N, Bridgland-Taylor M, Chaudhary KW, Colatsky T, Correll K, Crumb W, Damiano B, Erdemli G, Gintant G, Imredy J, Koerner J, Kramer J, Levesque P, Li Z, Lindqvist A, Obejero-Paz CA, Rampe D, Sawada K, Strauss DG, Vandenberg JI. A new perspective in the field of cardiac safety testing through the comprehensive in vitro proarrhythmia assay paradigm. J Biomol Screen 2016;21:1–11. [DOI] [PubMed] [Google Scholar]
- 123. Passini E, Britton OJ, Lu HR, Rohrbacher J, Hermans AN, Gallacher DJ, Greig RJH, Bueno-Orovio A, Rodriguez B. Human in silico drug trials demonstrate higher accuracy than animal models in predicting clinical pro-arrhythmic cardiotoxicity. Front Physiol 2017;8:15–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Paci M, Passini E, Klimas A, Severi S, Hyttinen J, Rodriguez B, Entcheva E. All-optical electrophysiology refines populations of in silico human iPSC-CMs for drug evaluation. Biophys J 2020;118:2596–2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Corral-Acero J, Margara F, Marciniak M, Rodero C, Loncaric F, Feng Y, Gilbert A, Fernandes JF, Bukhari HA, Wajdan A, Martinez MV, Santos MS, Shamohammdi M, Luo H, Westphal P, Leeson P, DiAchille P, Gurev V, Mayr M, Geris L, Pathmanathan P, Morrison T, Cornelussen R, Prinzen F, Delhaas T, Doltra A, Sitges M, Vigmond EJ, Zacur E, Grau V, Rodriguez B, Remme EW, Niederer S, Mortier P, McLeod K, Potse M, Pueyo E, Bueno-Orovio A, Lamata P. The ‘Digital Twin’ to enable the vision of precision cardiology. Eur Heart J 2020;41:4556–4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Dey D, Slomka PJ, Leeson P, Comaniciu D, Shrestha S, Sengupta PP, Marwick TH. Artificial intelligence in cardiovascular imaging. J Am Coll Cardiol 2019;73:1317–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Attia ZI, Kapa S, Lopez-Jimenez F, McKie PM, Ladewig DJ, Satam G, Pellikka PA, Enriquez-Sarano M, Noseworthy PA, Munger TM, Asirvatham SJ, Scott CG, Carter RE, Friedman PA. Screening for cardiac contractile dysfunction using an artificial intelligence-enabled electrocardiogram. Nat Med 2019;25:70–74. [DOI] [PubMed] [Google Scholar]
- 128. Hannun AY, Rajpurkar P, Haghpanahi M, Tison GH, Bourn C, Turakhia MP, Ng AY. Cardiologist-level arrhythmia detection and classification in ambulatory electrocardiograms using a deep neural network. Nat Med 2019;25:65–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Manrai AK, Funke BH, Rehm HL, Olesen MS, Maron BA, Szolovits P, Margulies DM, Loscalzo J, Kohane IS. Genetic misdiagnoses and the potential for health disparities. N Engl J Med 2016;375:655–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Saqué V, Vaglio M, Funck-Brentano C, Kilani M, Bourron O, Hartemann A, Badilini F, Salem J-E. Fast, accurate and easy-to-teach QT interval assessment: the triplicate concatenation method. Arch Cardiovasc Dis 2017;110:475–481. [DOI] [PubMed] [Google Scholar]
- 131. Goto S, Kimura M, Katsumata Y, Goto S, Kamatani T, Ichihara G, Ko S, Sasaki J, Fukuda K, Sano M. Artificial intelligence to predict needs for urgent revascularization from 12-leads electrocardiography in emergency patients. PLoS One 2019;14:e0210103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Mincholé A, Rodriguez B. Artificial intelligence for the electrocardiogram. Nat Med 2019;25:22–23. [DOI] [PubMed] [Google Scholar]
- 133. Castelletti S, Dagradi F, Goulene K, Danza AI, Baldi E, Stramba-Badiale M, Schwartz PJ. A wearable remote monitoring system for the identification of subjects with a prolonged QT interval or at risk for drug-induced long QT syndrome. Int J Cardiol 2018;266:89–94. [DOI] [PubMed] [Google Scholar]
- 134. Perez MV, Mahaffey KW, Hedlin H, Rumsfeld JS, Garcia A, Ferris T, Balasubramanian V, Russo AM, Rajmane A, Cheung L, Hung G, Lee J, Kowey P, Talati N, Nag D, Gummidipundi SE, Beatty A, Hills MT, Desai S, Granger CB, Desai M, Turakhia MP. Large-scale assessment of a smartwatch to identify atrial fibrillation. N Engl J Med 2019;381:1909–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Blinova K, Dang Q, Millard D, Smith G, Pierson J, Guo L, Brock M, Lu HR, Kraushaar U, Zeng H, Shi H, Zhang X, Sawada K, Osada T, Kanda Y, Sekino Y, Pang L, Feaster TK, Kettenhofen R, Stockbridge N, Strauss DG, Gintant G. International multisite study of human-induced pluripotent stem cell-derived cardiomyocytes for drug proarrhythmic potential assessment. Cell Rep 2018;24:3582–3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Mazzanti A, Maragna R, Faragli A, Monteforte N, Bloise R, Memmi M, Novelli V, Baiardi P, Bagnardi V, Etheridge SP, Napolitano C, Priori SG. Gene-specific therapy with mexiletine reduces arrhythmic events in patients with long QT syndrome type 3. J Am Coll Cardiol 2016;67:1053–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Zhu W, Mazzanti A, Voelker TL, Hou P, Moreno JD, Angsutararux P, Naegle KM, Priori SG, Silva JR. Predicting patient response to the antiarrhythmic mexiletine based on genetic variation: personalized medicine for long QT syndrome. Circ Res 2019;124:539–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Ma D, Wei H, Lu J, Huang D, Liu Z, Loh LJ, Islam O, Liew R, Shim W, Cook SA. Characterization of a novel KCNQ1 mutation for type 1 long QT syndrome and assessment of the therapeutic potential of a novel IKs activator using patient-specific induced pluripotent stem cell-derived cardiomyocytes. Stem Cell Res Ther 2015;6:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Matschke V, Piccini I, Schubert J, Wrobel E, Lang F, Matschke J, Amedonu E, Meuth SG, Strünker T, Strutz-Seebohm N, Greber B, Scherkenbeck J, Seebohm G. The natural plant product Rottlerin activates Kv7.1/KCNE1 channels. Cell Physiol Biochem 2016;40:1549–1558. [DOI] [PubMed] [Google Scholar]
- 140. Duncan G, Firth K, George V, Hoang MD, Staniforth A, Smith G, Denning C. Drug-mediated shortening of action potentials in LQTS2 human induced pluripotent stem cell-derived cardiomyocytes. Stem Cells Dev 2017;26:1695–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Kopljar I, Lu HR, Van Ammel K, Otava M, Tekle F, Teisman A, Gallacher DJ. Development of a human ipsc cardiomyocyte-based scoring system for cardiac hazard identification in early drug safety de-risking. Stem Cell Rep 2018;11:1365–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Perry MD, Ng C-A, Mangala MM, Ng TYM, Hines AD, Liang W, Xu MJO, Hill AP, Vandenberg JI. Pharmacological activation of IKr in models of long QT type 2 risks overcorrection of repolarization. Cardiovasc Res 2020;116:1434–1445. [DOI] [PubMed] [Google Scholar]
- 143. McKeithan WL, Feyen DAM, Bruyneel AAN, Okolotowicz KJ, Ryan DA, Sampson KJ, Potet F, Savchenko A, Gómez-Galeno J, Vu M, Serrano R, George AL Jr, Kass RS, Cashman JR, Mercola M. Reengineering an antiarrhythmic drug using patient hiPSC cardiomyocytes to improve therapeutic potential and reduce toxicity. Cell Stem Cell 2020;27:813–821.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Anderson CL, Delisle BP, Anson BD, Kilby JA, Will ML, Tester DJ, Gong Q, Zhou Z, Ackerman MJ, January CT. Most LQT2 mutations reduce Kv11.1 (hERG) current by a Class 2 (trafficking-deficient) mechanism. Circulation 2006;113:365–373. [DOI] [PubMed] [Google Scholar]
- 145. Mehta A Ramachandra C J A Singh P Chitre A Lua C H Mura M Crotti L Wong P Schwartz P J Gnecchi M Shim W. Identification of a targeted and testable antiarrhythmic therapy for long-QT syndrome type 2 using a patient-specific cellular model. European Heart Journal 2018;39:1446–1455. 10.1093/eurheartj/ehx394 [DOI] [PubMed] [Google Scholar]
- 146. Boyle MP, Bell SC, Konstan MW, McColley SA, Rowe SM, Rietschel E, Huang X, Waltz D, Patel NR, Rodman D; VX09-809-102 study group. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet Respir Med 2014;2:527–538. [DOI] [PubMed] [Google Scholar]
- 147. Schwartz PJ, Gnecchi M, Dagradi F, Castelletti S, Parati G, Spazzolini C, Sala L, Crotti L. From patient-specific induced pluripotent stem cells to clinical translation in long QT syndrome type 2. Eur Heart J 2019;40:1832–1836. [DOI] [PubMed] [Google Scholar]
- 148. O’Hare BJ, Kim CSJ, Hamrick SK, Ye D, Tester DJ, Ackerman MJ. Promise and potential peril with Lumacaftor for the trafficking defective type 2 long-QT syndrome-causative variants, p.G604S, p.N633S, and p.R685P, using patient-specific re-engineered cardiomyocytes. Circ Genom Prec Med 2020;13:466–475. [DOI] [PubMed] [Google Scholar]
- 149. Wu JC, Garg P, Yoshida Y, Yamanaka S, Gepstein L, Hulot J-S, Knollmann BC, Schwartz PJ. Towards precision medicine with human iPSCs for cardiac channelopathies. Circ Res 2019;125:653–658. [DOI] [PMC free article] [PubMed] [Google Scholar]