

Tuberculosis (TB) is the leading cause of death from a single infectious agent. The emergence of drug resistance in strains of M. tuberculosis, the etiologic agent of TB, emphasizes the need to identify new targets and antimicrobial agents.
KEYWORDS: tuberculosis, Mycobacterium tuberculosis, conditional mutant, acyltransferase, glycolipid, mycobacterium
ABSTRACT
Mycobacterium tuberculosis comprises an unusual cell envelope dominated by unique lipids and glycans that provides a permeability barrier against hydrophilic drugs and is central for its survival and virulence. Phosphatidyl-myo-inositol mannosides (PIMs) are glycolipids considered to be not only key structural components of the cell envelope but also the precursors of lipomannan (LM) and lipoarabinomannan (LAM), important lipoglycans implicated in host-pathogen interactions. Here, we focus on PatA, a membrane-associated acyltransferase that transfers a palmitoyl moiety from palmitoyl coenzyme A (palmitoyl-CoA) to the 6-position of the mannose ring linked to the 2-position of inositol in PIM1/PIM2. We validate that the function of PatA is vital for M. tuberculosis in vitro and in vivo. We constructed a patA conditional mutant and showed that silencing patA is bactericidal in batch cultures. This phenotype was associated with significantly reduced levels of Ac1PIM2, an important structural component of the mycobacterial inner membrane. The requirement of PatA for viability was also demonstrated during macrophage infection and in a mouse model of infection, where a dramatic decrease in viable counts was observed upon silencing of the patA gene. This is reminiscent of the behavior of PimA, the mannosyltransferase that initiates the PIM pathway, also found to be essential for M. tuberculosis growth in vitro and in vivo. Altogether, the experimental data highlight the significance of the early steps of the PIM biosynthetic pathway for M. tuberculosis physiology and reveal that PatA is a novel target for drug discovery programs against this major human pathogen.
IMPORTANCE Tuberculosis (TB) is the leading cause of death from a single infectious agent. The emergence of drug resistance in strains of M. tuberculosis, the etiologic agent of TB, emphasizes the need to identify new targets and antimicrobial agents. The mycobacterial cell envelope is a major factor in this intrinsic drug resistance. Here, we have focused on the biosynthesis of PIMs, key virulence factors and important components of the cell envelope. Specifically, we have determined that PatA, the acyltransferase responsible for the first acylation step of the PIM synthesis pathway, is essential in M. tuberculosis. These results highlight the importance of early steps of the PIM biosynthetic pathway for mycobacterial physiology and the suitability of PatA as a potential new drug target.
INTRODUCTION
Mycobacterium tuberculosis, the etiologic agent of tuberculosis (TB), is the second most deadly infectious agent in the world after HIV. In 2018, there were about 10 million new cases and 1.2 million deaths from TB, with an estimated one-quarter of the human population carrying a latent infection (1–4). First-line treatment for active, drug-susceptible TB, requires a standard administration of four drugs during a period of 6 months, including isoniazid, rifampicin, ethambutol, and pyrazinamide (5). Multidrug-resistant TB (MDR-TB) and extensively drug-resistant TB (XDR-TB) emerge mainly as a result of poor compliance during short-course chemotherapy (6). Specifically, MDR-TB is a form of TB caused by bacteria that do not respond to isoniazid and rifampicin. Although MDR-TB is curable, it requires expensive and toxic second-line drugs and an extensive chemotherapy of up to 2 years. XDR-TB is a more serious form of MDR-TB caused by bacteria that do not respond to the most effective second-line anti-TB drugs, often leaving patients without any further treatment options (6). MDR-TB and XDR-TB remain a public health crisis and a health security threat, making the discovery of new targets and novel anti-TB drugs with bactericidal mechanisms different from those of currently available agents an urgent need (7). The cell envelope of M. tuberculosis exhibits a unique architecture comprised of sugars and lipids of exceptional chemical structure, providing a hydrophobic impermeable barrier against many commonly used antibiotics and allowing the bacteria to survive in extremely hostile intracellular conditions. Due to its essential role in the biology of M. tuberculosis, the cell envelope represents a highly attractive target for the discovery of novel anti-TB drugs (8–10). In that sense, several key medicines used in current TB therapy inhibit enzymes involved in cell envelope biosynthesis.
The phosphatidyl-myo-inositol mannosides (PIMs) are glycolipids found in abundant quantities in the inner and outer membranes of the cell envelope of Mycobacterium spp. and a few other actinomycetes (Fig. 1) (11–13). PIMs are based on a phosphatidyl-myo-inositol (PI) anchor and can contain one to six mannose residues and up to four acyl chains. The tri- and tetra-acylated phosphatidyl-myo-inositol dimannosides (Ac1PIM2 and Ac2PIM2, respectively) are considered both metabolic end products and intermediates in the biosynthesis of the tri- and tetra-phosphatidyl-myo-inositol hexamannosides (Ac1PIM6 and Ac2PIM6, respectively), lipomannan (LM), and lipoarabinomannan (LAM) (11, 12, 14). PIMs, LM, and LAM play an essential role as structural components of the mycobacterial cell envelope but are also important molecules implicated in host-pathogen interactions in the course of TB and leprosy (11, 13, 15–18). The biosynthesis of PIMs takes place on both sides of the mycobacteria inner membrane (Fig. 1) (12, 13). The biosynthesis of PIMs is initiated on the cytoplasmic side by the mannosyltransferase PimA (Rv2610c; the Rv numbers of the nomenclature based on the M. tuberculosis H37Rv genome are included hereafter), which catalyzes the transfer of a mannose residue from GDP-mannose (GDP-Man) to the 2-position of the myo-inositol ring of PI to produce phosphatidyl-myo-inositol monomannoside (PIM1 [19, 20]). The second step is mediated by the mannosyltransferase PimB (Rv2188c), which transfers a mannose residue from GDP-Man to the 6-position of the myo-inositol ring of PIM1, to produce phosphatidyl-myo-inositol dimannoside (20–23). The acyltransferase PatA catalyzes the transfer of a palmitoyl moiety from palmitoyl coenzyme A (palmitoyl-CoA) to PIM1 and PIM2 at the 6-position of the mannose ring transferred by PimA to form Ac1PIM1 and Ac1PIM2, respectively (24–26). The following two pathways were originally proposed for the biosynthesis of Ac1PIM2: (i) PI is mannosylated by the consecutive action of PimA and PimB to form PIM2, which is further acylated to form Ac1PIM2, and (ii) PIM1 is acylated by PatA to form Ac1PIM1 and then mannosylated by PimB to form Ac1PIM2. The experimental data indicate that both pathways might coexist in mycobacteria, although the sequence of events PI→PIM1→PIM2→Ac1PIM2 is favored (20, 27, 28). Ac1PIM2 can be further acylated on position 3 of the myo-inositol ring to form Ac2PIM2 (20, 28). This acyltransferase as well as most of the mannosyltransferases involved in the biosynthesis of higher forms of PIMs still remains to be identified (11–13, 29). Strikingly, the following four enzymes participating in the early steps of the PIM pathway were found to be essential in vitro in Mycobacterium smegmatis: the phosphatidylinositol phosphate synthase PIP (Rv2612c), PimA, PimB, and PatA (19, 26, 30, 31). From a drug discovery perspective, the essential character of the enzymes involved in the early steps of PIM biosynthesis and their restriction to mycobacteria and a few other actinomycetes highlight their interest as novel targets for TB drug discovery (12, 13).
FIG 1.

The PIMs from M. tuberculosis. (A) The chemical structure of PIMs. The major PIM species have two (PIM2) or six (PIM6) mannose residues with different degrees of acylation. (B) The current model of PIM biosynthesis. The currently accepted model proposes that PimA and PimB catalyze the two first consecutive mannosylation steps in the cytoplasmic face of the plasma membrane. The identity and location of the enzyme(s) responsible for the third and fourth mannosylation steps, as well as the existence of a flippase, are under debate. The fifth mannosylation step is catalyzed by PimE in the periplasmic face of the plasma membrane. The identity of the enzyme responsible for the sixth mannosylation step is still unknown. PIMs can be also acylated by the action of two acyltransferases: PatA catalyzes the transfer of an acyl chain to the mannose ring transferred by PimA at the cytosolic face of the plasma membrane, whereas an unknown acyltransferase catalyzes the transfer of an acyl chain to the inositol ring of the glycolipid. In the inset, the acylation steps on Ac2PIM2 are shown as an example. (C) PatA transfers a palmitoyl moiety from palmitoyl-CoA to the 6-position of the mannose ring linked to 2-position of inositol in PIM1/PIM2.
Here, we focus on PatA (2.3.1.265), an enzyme that belongs to an important family of acyltransferases, including HtrB and MsbB, involved in the last steps of Kdo2-lipid A biosynthesis in Gram-negative bacteria (32). We previously provided the very first crystal structures of PatA in the presence of (i) its naturally occurring acyl donor palmitoyl group, (ii) a nonhydrolyzable palmitoyl-CoA analog, and (iii) the 6-O-palmitoyl-α-d-mannopyranoside product, unraveling the acyl donor and acceptor binding mechanisms (32, 33). In this study, we carefully investigated the essentiality of PatA from M. tuberculosis in vitro and in vivo, in a mouse model of TB, in order to evaluate its potential for drug discovery purposes.
RESULTS
Silencing of patA in axenic cultures results in bacterial death.
To probe the essentiality of PatA, a conditional knockdown (cKD) mutant for the patA gene (rv2611c) (TB489) was constructed in M. tuberculosis H37Rv as described in Materials and Methods. Since patA is part of a predicted operon including pimA (rv2610c) and rv2609c (19, 20, 34), to avoid artifacts due to polar effects, both genes were provided in trans on pFRA247, a pMV261-derived plasmid expressing the two genes from the constitutive promoter Phsp60. In the resulting strain, named TB506.1, expression of patA was expected to be downregulated following the addition of anhydrotetracycline (ATc) to the culture medium.
As shown in Fig. 2A, when serial dilutions of a TB506.1 suspension were spotted on Middlebrook 7H10 containing 500 ng/ml ATc, bacterial growth was inhibited, confirming the predicted essentiality of patA in vitro. The visible residual growth corresponds to spots containing the largest number of bacteria, suggesting that bacteria must go through some generations before PatA is titrated down and the phenotype becomes detectable. As expected, the growth of TB38, an M. tuberculosis H37Rv derivative containing pFRA61 (carrying the TetR/PipOFF) on the chromosome, was unaffected by the presence of the drug at this concentration (see Fig. S2 in the supplemental material). A similar experiment was performed growing the bacteria in Middlebrook 7H9 liquid medium with or without ATc (500 ng/ml). As depicted in Fig. 2B, two passages in fresh ATc-containing medium were necessary to observe bacterial growth inhibition, confirming that some time was necessary to titrate down PatA after the downregulation of its structural gene. We also followed the viability of the culture of the patA conditional mutant grown in the presence of ATc after its growth curve diverged from that of the culture grown without ATc. As visible in Fig. 2B, despite the optical density of the culture with ATc remaining stable, the number of viable cells decreased, suggesting that PatA depletion is bactericidal.
FIG 2.

patA is an essential gene in M. tuberculosis. (A) Serial dilutions of log-phase cultures of the conditional mutant TB506.1 were spotted on Middlebrook 7H10 plates with or without 500 ng/ml ATc. (B) Growth curves of the conditional knockdown TB506.1 in the presence (triangles) or absence (circles) of 500 ng/ml ATc; 1:10 dilutions of the cultures in fresh medium with or without ATc (dashed arrows) were performed until the growth of the culture with ATc stopped. Gray bars represent the number of CFU per milliliter in the culture with ATc after its growth arrested. The results represent the average of three independent experiments. *, P < 0.5; **, P < 0.05.
To demonstrate that the observed phenotype was indeed due to the downregulation of patA and not to the failure to complement pimA (rv2609c) with pFRA247, this plasmid was transformed in a previously characterized pimA (rv2609c) conditional mutant TB99 (34) to obtain TB513. In this case, pFRA247 was able to complement the downregulation of pimA, as the presence of ATc did not affect the growth of this complemented strain (see Fig. S3 in the supplemental material). Further, to confirm expression of pimA in the patA mutant, we performed a side-by-side reverse transcriptase PCR (RT-PCR) analysis of both pimA and patA in TB99 and TB506.1 strains. We found that levels of pimA expression were comparable in the two strains in the absence of ATc, while in the presence of ATc, pimA was exclusively repressed in TB99 and patA was exclusively repressed in TB506.1 (see Fig. S4 in the supplemental material). Importantly, it is worth noting that the mRNA levels of rv2612c remained constant in strains TB99 and TB506.1 (Fig. S4).
Production of PIM is greatly reduced in patA-depleted mycobacteria.
The effect of patA gene silencing on PIM biosynthesis was examined by one-dimensional and two-dimensional (2D) thin-layer chromatography (TLC) analysis of the lipids extracted from M. tuberculosis TB38 and TB506.1 cells collected at different time points (0, 4, and 12 days) in medium supplemented with 500 ng/ml ATc. Lipid identification was performed by calculating the retention factor (Rf) value, defined as the distance travelled by a given compound divided by the distance traveled by the solvent (35), and comparing to previous reports using the same experimental setup. As expected, the TB38 strain showed a similar lipid profile during the time course (Fig. 3A). In contrast in the TB506.1 strain, we could observe a reduction in lipid bands I, II, and III, starting from the origin (O), corresponding to Ac1PIM6, Ac2PIM6, and Ac1PIM2, respectively (Fig. 3A) (19), and an increase in the lipid band assigned to PI relative to the TB38 strain (Fig. 3A) (36).
FIG 3.

Loss of Ac1PIM2 in the PatA-depleted strain. (A) Lipids were extracted from M. tuberculosis TB38 and TB506.1 grown in the presence of 500 ng ml−1 ATc at different time points (0, 4, and 12 days), separated by TLC in the solvent CHCl3-CH3OH-concentrated NH4OH-H2O (65:25:0.5:4) and visualized with α-naphthol. (B) Alternatively, samples from day 12 were separated by 2D-TLC using the same solvent system as in panel A for dimension 1 and chloroform, acetic acid, methanol, and water (50:60:2.3:3) for dimension 2 and visualized as in panel A. Data are representative of two independent experiments with similar results. At the indicated days, cultures were submitted to serial dilutions in fresh medium with ATc in order to visualize a progressive decrease in the Ac1PIM2 levels.
We performed 2D-TLC analysis of the same samples and corroborated that production of Ac1PIM6, Ac2PIM6, and Ac1PIM2 lipid species is affected in the TB506.1 strain relative to the TB38 strain (Fig. 3B). This was particularly evident in the case of Ac1PIM2, which is absent in the mutant and is considered an end product of the PIM pathway (12, 13, 18). Altogether, this profile is consistent with the biochemical reaction catalyzed by PatA, which transfers a palmitoyl moiety from palmitoyl-CoA to the 6 position of the mannose ring linked to the 2 position of myo-inositol in PIM1 or PIM2, leading to the synthesis of Ac1PIM1/Ac1PIM2. It is evident that depletion of PatA caused a block in the biosynthesis of PIMs, resulting in severe changes in the composition of the mycobacterial cell membrane, which correlates with the loss of viability observed.
patA is essential for growth in macrophages.
To validate the essentiality of patA during intracellular growth on macrophages, we infected THP-1-derived macrophages with the patA conditional mutant TB506.1 and the parental strain TB38, in the presence or absence or ATc, and determined viable counts 2, 4, and 6 days after infection. Both TB38 parental strain and TB506.1 retained their infective capacity in the absence of ATc and beside the presence of TetR and Pip regulators. Conversely, only the TB506.1 strain showed reduced viability on the presence of ATc being significantly reduced at day 4 and 6 after infection (Fig. 4). Importantly, no difference in cell viability was observed between treatments (data not shown). These results indicate the essentiality of patA for macrophage infection.
FIG 4.
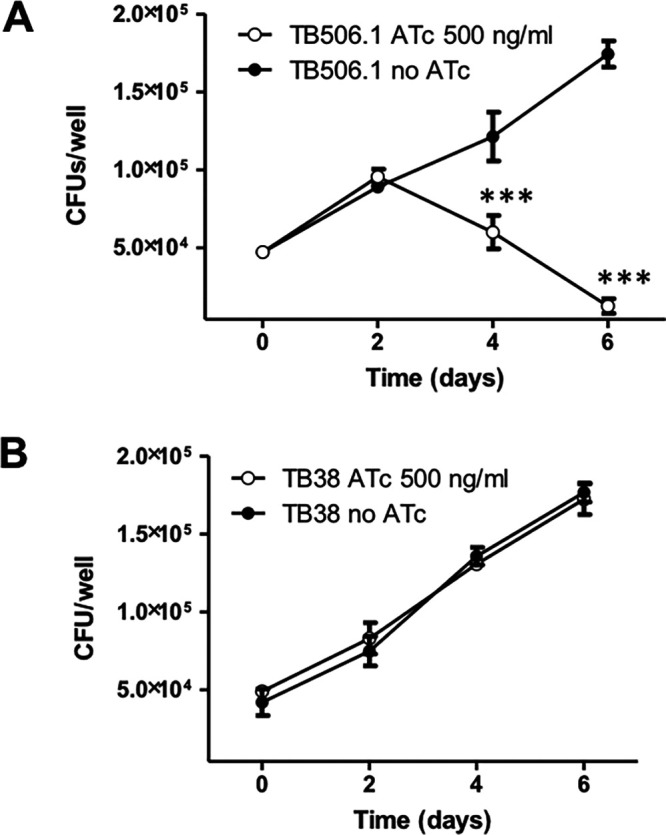
Growth curves of TB506.1 and of the parental strain TB38 in THP-1-derived macrophages. The patA conditional mutant TB506.1 and the parental strain TB38 were used to infect THP-1-derived macrophages. While TB506.1 was able to replicate intracellularly only in the absence of ATc (A), TB38 was able to divide regardless of the presence of ATc in the cell culture medium (B). Filled circles, no ATc; open circles, 500 ng/ml ATc added to the culture medium. Data are representative of two independent experiments with similar results. ***, P < 0.001 one-way ANOVA with Tukey posttest.
patA is essential for growth in mouse.
To assess the essentiality of patA in vivo, C57BL/6J mice were intranasally infected with the conditional mutant strain TB506.1. Silencing of patA was achieved by supplying animals with water with 5% sucrose containing doxycycline starting on the day of the infection. Control groups did not received doxycycline, allowing normal expression of patA. As depicted in Fig. 5, we observed that M. tuberculosis H37Rv sustained infection in the presence and in the absence of doxycycline, 4 and 8 weeks after challenge. In the same line, the TB506.1 mutant in the absence of doxycycline reached similar rates of bacterial replication during the acute (4 weeks) and chronic (8 weeks) phases of infection. In contrast, TB506.1 manifested a dramatic reduction in CFU at 4 and 8 weeks after infection in the presence of doxycycline, indicating the essential nature of patA during both the acute and chronic phases of infection. This significant trend of reduction in cell viability for TB506.1 in the presence of doxycycline was observed in both lungs and spleen. Altogether, these findings clearly highlight the essential role played by PatA during M. tuberculosis infection.
FIG 5.

Validation of patA essentiality in the mouse model of infection. The graph illustrates the bacterial loads in the lungs (A) and spleens (B) of C57BL/6JOlaHsd mice infected with TB506.1. Silencing of patA was achieved by supplying animals with water supplemented with 5% sucrose containing doxycycline. Results represent the mean value and error for 5 mice per group and time point. **, P < 0.01; ***, P < 0.001 one-way ANOVA with Tukey posttest.
DISCUSSION
The gene patA is predicted to be an essential gene for in vitro growth of H37Rv according to analysis of saturated Himar1 transposon libraries (31, 37, 38). The aim of this work is to demonstrate the essentiality of patA during in vitro growth and experimental macrophage and mouse infections. These results strongly suggest that the membrane acyltransferase PatA is a potential drug target in M. tuberculosis according to its essentiality. We did this by exploiting a repressible promoter system based on the TetR and Pip regulators (34, 39). The patA gene is part of a cluster of genes potentially organized as a single transcriptional unit (12, 26) involved in the early steps of PIM biosynthesis. The upstream gene encodes the phosphatidylinositol phosphate synthase PIP (Rv2612c) (rv2612c, PIP) (30, 40–42), whereas the downstream open reading frame (ORF) encodes a putative GDP-mannose hydrolase (rv2609c) and the phosphatidyl-myo-inositol mannosyltransferase PimA (rv2610c) (19, 20). Although rv2609c was predicted to be nonessential (37), we decided to provide this ORF and that of the pimA gene in trans on a replicative plasmid to the TB506.1 strain in order to counteract potential polar effects caused by the single recombination event in the patA gene. In this regard, we are confident that the described phenotype is therefore due to the lack of the patA gene product only.
When grown on solid medium, the conditional mutant was clearly able to replicate only in the absence of ATc, and the same phenotype was evident also in liquid medium. Moreover, to observe an effect during growth in liquid culture, cells had to undergo two passages. This suggests that the conditional mutant accumulation of both PatA and PIMs allows cell growth for several rounds of replication after patA expression is repressed. This phenomenon has already been observed for other conditional mutants generated using the same TetR/PipOFF system and could be due to the higher strength of the ptr promoter compared to that of the original promoter of the downregulated gene causing its overexpression in the absence of ATc (43, 44). The sharp decrease of bacterial viability following patA repression resembled that observed after repression of pimA and expanded the essentiality to the early steps of the PIM biosynthetic pathway (12, 18). Biochemical analyses of the lipids after patA repression showed a clear depletion of PIMs and a consequent accumulation of the PIMs precursor PI, with no effect on the remaining pools of lipids, further supporting the essential role of PatA in this biochemical pathway.
The observed accumulation of PI and depletion of PIMs in the TB506.1 strain upon treatment with ATc seem to be deleterious during experimental infection of both macrophages and mice. We observed a rapid decline in viable counts 2 days after macrophage infection, indicating that alteration of the PIM biosynthesis pathway is critical during the initial stages of infection. Although macrophages represent a starting point at the time of interrogating the role of patA during infection, our results suggest that M. tuberculosis needs to keep the integrity of the early steps of PIM biosynthesis for optimal intracellular replication. To test the role of this gene during a more relevant complex infection scenario, we infected C57BL/6 mice with TB506.1 and control strains and provided water supplemented with or without doxycycline. The dramatic and significant decrease in the number of CFU in lung and spleens both at 4 and 8 weeks after infection for the TB506.1 strain in the presence of doxycycline strongly supports that the absence of patA leads to loss of bacterial cell viability. Although we do not know the exact mechanism by which silencing of patA might have accelerated the process of lung clearance, it is possible that some cell envelope instability due to altered lipid composition renders M. tuberculosis more susceptible to innate and adaptive immune responses.
Ac2PIM2 was found to be the most abundant lipid group in the inner membrane (IM), accounting for up to 42% (by weight) of all of the lipids in the IM extract (16, 44–47). Our TLC analysis indicates the complete disappearance of Ac1PIM2 and the reduction of Ac1PIM6 and Ac2PIM6 glycolipid species in the TB506.1 strain relative to the TB38 strain (Fig. 3B). The accumulation of PI in the M. tuberculosis patA-deficient mutant is somehow expected as well as the accumulation of PIM1 and/or PIM2. Indeed, Kordulakova and colleagues have demonstrated the accumulation of PIM1 and PIM2 glycolipids in an M. smegmatis patA-deficient mutant (26). The lack of the palmitoyl residue in Ac2PIM2 glycolipid can certainly modify the properties of the mycobacterial cell envelope (48). In that sense, the M. smegmatis patA mutant could only be grown in the presence of a high concentration of NaCl (15 g/liter) and in the absence of Tween 80. The sodium salts might protect the weakened membrane by osmotic pressure or by neutralizing the surface charges of the membrane. Importantly, the mRNA levels of rv2612c remained constant in the two strains, TB99 and TB506.1, ruling out any defect in the PI biosynthesis. What seems interesting is the fact that, together with the complete disappearance of Ac1PIM2 and the reduction of Ac1PIM6 and Ac2PIM6, the levels of Ac2PIM2 remain unchanged. According to Bansal-Mutalik and colleagues, Ac2PIM2 is the major component of the internal leaflet of the mycobacterial inner membrane (45). One possible explanation for these results is the presence of compensatory acyltransferase activities/specificities that could contribute to generate and maintain the Ac2PIM2 levels (26). Taken together, the experimental data suggest that Ac1PIM2 and Ac2PIM2 primarily play a structural role in the mycobacterial cell envelope (49). The alteration in the relative abundance of lipids/glycolipids leads to structural defects in the mycobacterial cell envelope, explaining the observed severe growth defects. Therefore, patA is another potential target whose depletion causes severe consequences in M. tuberculosis cell viability, similar to what has been reported for other mycobacterial genes (34, 50).
Our long-term goal is the discovery of novel anti-TB drugs targeting the early steps of PIM biosynthesis. From a drug discovery perspective, the essential character of PIM biosynthetic enzymes and their restriction to mycobacteria and a few other actinomycetes emphasizes their interest as novel targets for anti-TB chemotherapeutic agents (Fig. 6). The following four enzymes of the PIM pathway were found to be essential in M. smegmatis and/or M. tuberculosis: (i) in vitro, PIP (Rv2612c [30]), PimA (Rv2610c [19, 34]), PimB (Rv2188c [20]), and PatA (Rv2611c [26; this study]); and (ii) in vivo in M. tuberculosis, PimA (34) and PatA (this study). Interestingly, the crystal structures of PimA, PimB, and PatA have been reported (32, 33, 51–53). PimA and PimB belong to the GT-B family of glycosyltransferases, consisting of two Rossmann fold domains with a deep fissure at the interface that includes the catalytic center (54–58). As a consequence, the occurrence of an important interdomain movement has been well established in GT-B enzymes (27, 59). Specifically, PimA is an outstanding example of GT-B flexibility and intrinsic dynamic properties for the study of GT-B enzymes. The crystal structures of the unliganded form of PimA and that of the GDP-Man complex revealed the local reshuffling of secondary structure elements within the flexible segment (residues 118 to 163) in the N-terminal domain (52). Very recently, we identified four functionally relevant states of PimA that coexist in dynamic equilibrium in solution undergoing conformational exchange on timescales from milliseconds to seconds. Specifically, fold switching is a slow process, on the order of seconds, whereas domain motions occur simultaneously but are substantially faster, on the order of milliseconds (60). We hypothesize that such conformational flexibility has impaired identification of potent inhibitors against PimA using in silico and in vitro target-based drug screening approaches. In contrast, the crystal structures of PatA in the presence of (i) its naturally occurring acyl donor palmitoyl group, (ii) a nonhydrolyzable palmitoyl-CoA analog, and (iii) the 6-O-palmitoyl-α-d-mannopyranoside product revealed clear defined grooves that participate in the association of the substrates and products. We hypothesize that the presence of clear and deep pockets might facilitate the in silico and in vitro screenings for the discovery of potent inhibitors against the enzyme.
FIG 6.

Essentiality of the early steps in PIM biosynthesis. Two pathways were originally proposed for the biosynthesis of Ac1PIM2 in the cytoplasmic phase of the mycobacterial inner membrane as follows: (i) PI is mannosylated to form PIM2 by the consecutive action of the GT-B glycosyltransferases PimA and PimB, respectively, and PIM2 is then acylated by PatA to form Ac1PIM2; and (ii) PI is mannosylated by PimA to form PIM1 and then acylated by PatA to form Ac1PIM1, which in turn is mannosylated by PimB to form Ac1PIM2. In vitro experimental evidence indicates that although both pathways might coexist in mycobacteria, the sequence of events PI→PIM1→PIM2→Ac1PIM2 is favored (20, 28). The production and purification of high yields of PimA, PimB, and PatA, along with the determination of their three-dimensional structure, place us in an unprecedented position to initiate a drug discovery program focusing on those enzymes (32, 33, 51–53).
Taken together, these data clearly show the essentiality of PatA for M. tuberculosis growth and survival in axenic cultures and during macrophage or mouse infection, making it a potential candidate for in vitro target-based drug screening approaches. Finally, the modulation of PimA or PatA expression by the use of the conditional mutant could find application in target-based whole-cell screening assays for the identification of compounds targeting PimA or PatA or other cellular processes that are part of the same pathway.
MATERIALS AND METHODS
Bacterial strains and media.
M. tuberculosis strain H37Rv was grown at 37°C in Middlebrook 7H9 or 7H10 (Difco) supplemented with 0.05% (vol/vol) Tween 80 (Sigma-Aldrich), 0.2% (vol/vol) glycerol (Sigma-Aldrich), and 10% ADN (2% glucose, 5% bovine serum albumin, 0.85% NaCl). For cloning procedures, Escherichia coli strain DH5α was grown in Luria-Bertani medium (LB). When required, hygromycin (Roche) was used at a final concentration of 50 μg/ml for M. tuberculosis and 150 μg/ml for E. coli. Streptomycin and kanamycin (Sigma-Aldrich) were used at a concentration of 20 μg/ml. Anhydrotetracycline (Sigma-Aldrich) was used at a concentration of 500 ng/ml.
Construction of the M. tuberculosis patA conditional mutant.
The patA (rv2611c) conditional mutant was constructed in M. tuberculosis H37Rv using the TetR/PipOFF repressible promoter system (39, 43). In this system, the gene of interest is placed under the transcriptional control of the repressible promoter Pptr; then, the gene encoding its repressor (Pip) is introduced in the chromosome together with the gene encoding the repressor TetR. Since the gene encoding Pip is transcribed from a TetR-dependent promoter, addition of anhydrotetracycline (ATc) to the culture medium leads to its induction and consequent repression of the gene of interest (39).
To place patA under Pptr transcriptional control, we constructed a suicide plasmid cloning the first 620 bp of the patA gene (PCR amplified using primers RP1862bis, 5′-CCAATGCATATTGCCGGCCTTAAGGGCT-3′, and RP1889, 5′-AGGCCTCAGACCACTCGGTTGTTCCT-3′) downstream Pptr in pFRA50 (39), conferring resistance to hygromycin. Ten micrograms of the resulting plasmid (pFRA238) was then introduced by electroporation into M. tuberculosis H37Rv, and transformants were selected on 7H10 plated containing hygromycin (see Fig. S1A in the supplemental material). The correct integration of the suicide plasmid by insertional duplication was confirmed by PCR, using an upper primer in the Pptr region (RP1908, 5′-CGACCTGACCGCGGAGAAAT-3′) and a lower primer external to the homology region used for recombination (RP1909, 5′-CAGCAATGTGTGGGCAGCAA-3′) (Fig. S1B). The resulting strain (TB498) was finally electroporated with the integrative plasmid pFRA61.1, containing the TetR-PipOFF system, conferring resistance to streptomycin to obtain TB499. Since patA might be cotranscribed with its downstream genes rv2609c and rv2610 (pimA), we decided to provide these genes in trans expressed from a constitutive promoter so that addition of ATc would lead to the repression of only patA transcription. For this purpose, pimA and rv2609c were amplified from the H37Rv genome using the primers RP1890, 5′-AGGCCTCTCGGATCGGCATGATTTGTC-3′, and RP1891, 5′-GTCGACCGACTTTTCTAAGCGGTCCA-3′, and then cloned downstream Phsp60 in the replicative plasmid pMV261, conferring resistance to kanamycin. The resulting plasmid, named pFRA247, was than introduced in the conditional mutant TB499 to obtain TB506.1 (Table 1).
TABLE 1.
M. tuberculosis strains used in this study
| Strain name | Parental strain | Description | Reference or source |
|---|---|---|---|
| TB38 | H37Rv | TetR/PipOFF system | 39 |
| TB498 | H37Rv | PptrpatA_pimA_rv2609c | This paper |
| TB499 | TB498 | PptrpatA_pimA_rv2609c, TetR/PipOFF system | This paper |
| TB506.1 | TB499 | TB499(pFRA247) (pMV261-derived plasmid expressing pimA_rv2609c under Phsp60) | This paper |
| TB99 | H37Rv | PptrpimA_rv2609c, TetR/PipOFF system | 34 |
| TB513 | TB99 | TB99(pFRA247) | This paper |
Characterization of the patA conditional mutant.
To characterize the growth curves of the bacterial strains, mid-log cultures were diluted up to a theoretical optical density at 540 nm (OD540) of 0.06 in Middlebrook 7H9 albumin-dextrose-catalase (ADC) with or without 500 ng/ml ATc and incubated without shaking at 37°C. OD540 was recorded at different time points to obtain the growth curves. To characterize the growth of the bacterial strains on solid medium, 5 μl of 10-fold serial dilutions of a mid-log culture grown in Middlebrook 7H9 ADC was spotted on Middlebrook 7H10 plates with or without 500 ng/ml ATc. Plates were than incubated at 37°C until growth was visible.
Extraction of lipids from the patA-deficient M. tuberculosis mutant.
M. tuberculosis TB506.1 was grown at 37°C in standing Middlebrook 7H9 supplemented with 500 ng/ml ATc. The bacteria were diluted every 120 h three times in fresh ATc-containing medium and collected before each dilution. Cells were harvested by centrifugation at 3,500 rpm for 5 min, and pellets were heat inactivated at 100°C for 1 h and used for lipid extraction. A culture of M. tuberculosis TB506.1 grown without ATc served as a control. Five milliliters 2:1 (vol/vol) chloroform-methanol was added to the cell pellet and incubated at room temperature (RT) for 12 h with constant stirring. The organic extract was separated by centrifugation (1,000 × g, 5 min, RT) and decantation. The obtained pellet was extracted with 5 ml 2:1 (vol/vol) chloroform-methanol for 1 h at RT with constant stirring and separated under the same conditions. The combined two organic extracts were dried under a stream of nitrogen gas at RT and subjected to a biphasic Folch wash with 2.1 ml of 4:2:1 (by volume) water-chloroform-methanol mixture. Finally, the hydrophilic phase was removed, and the organic phase was saved and dried under a nitrogen gas stream.
Lipid profile characterization by thin-layer chromatography.
The dried organic phase was resuspended with 4 μl of 2:1 (vol/vol) chloroform-methanol per 1 mg of dry weight. Fourteen microliters of each sample was applied on 60 F254 silica gel plates (Merck), and lipid components were separated by TLC using chloroform, methanol, ammonium hydroxide, and water (65:25:0.5:4) as a mobile phase. To develop plates, we repeated the process of pulverizing the staining solution and heating the plate at 100°C 4 to 5 times. To visualize the different glycolipids of the lipid mixture, we used α-naphthol staining, which contains 10.5 ml 15% ethanolic solution of 1-naphthol, 6.5 ml 97% sulfuric acid, 40.5 ml ethanol, and 4 ml water. Alternatively, total lipids were separated by 2D-TLC using chloroform, methanol, ammonium hydroxide, and water (65:25:0.5:4) as a mobile phase 1, and chloroform, acetic acid, methanol, and water (50:60:2.3:3) as mobile phase 2.
RNA extraction and quantitative reverse transcription-PCR.
RNA extraction and quantitative reverse transcription-PCR were performed as previously described using the primers shown in Table S1 in the supplemental material (61). The sigA mRNA was used as internal invariant control for data normalization (36). RNA samples that had not been reverse transcribed were included in all experiments to exclude significant DNA contamination. For each sample, melting curves were performed to confirm the purity of the products.
Infection of macrophages.
THP-1 macrophages (American Type Culture Collection) were grown in suspension at 37°C in 5% CO2 in bicarbonate-buffered RPMI (Gibco) supplemented with 10% (vol/vol) fetal bovine serum (FBS; Gibco), 50 μmol/liter β-mercaptoethanol, and 50 μg/ml gentamicin to a density of about 0.5 × 106 cells/ml (34). To induce differentiation of monocytes into macrophages, cells were seeded in 96-well plates and stimulated with 50 ng/ml phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich). After 24 h, PMA was removed, and cells were infected with M. tuberculosis H37Rv and TB506.1 at a multiplicity of infection of 1:10 for 90 min as previously described (62). Extracellular bacteria were further removed by washing with warm phosphate-buffered saline (PBS), and fresh medium supplemented with 200 ng/ml ATc was added. Macrophages were lysed at 0 h and 2, 4, and 6 days after infection, and extracts were plated to determine viable bacteria.
Intranasal infection of mice.
The mice used in the experiment were 7-week-old female C57BL/6J purchased from Envigo. Forty mice were challenged via the intranasal route with 103 CFU of M. tuberculosis H37Rv or TB506.1 suspended in 15 μl sterile PBS. For this procedure, mice were anesthetized with 75 mg/kg of ketamine and 1 mg/kg of medetomidine and reversed by 1 mg/kg of atipamezole. Drinking water was supplemented with 1 mg/ml of doxycycline (Sigma-Aldrich; D9891) and 5% sucrose. Control groups without doxycycline received drinking water with 5% sucrose. At day 1, week 4, and week 8 after infection, mice were euthanized in a CO2 chamber, and lungs and spleen were collected for bacterial load estimation. The enumeration of viable CFU was performed by plating appropriate lung homogenate dilutions on Middlebrook 7H11 plates for M. tuberculosis H37Rv and Middlebrook 7H11 plates containing 50 μg/ml hygromycin and 20 μg/ml of streptomycin for TB506.1. The animal experiment was performed at the biosafety level 3 (BSL3) animal facilities of NEIKER Institute (Derio, Spain), having received institutional approval. All institutional and ethical guidelines for animal care and experimentation were adhered to with permits provided by “Diputacion Foral de Bizkaia 15/2017 and 20/2019.” The NEIKER Animal Ethics Committee, registered with the Government of Spain (registration no. ES489010006099), approved all animal experimental protocols and usage.
Statistics.
Statistical analyses were performed in GraphPad Prism 5.0. Two-way analysis of variance (ANOVA) with Tukey posttest was used to evaluate paired replicates. P values of less than 0.05 were considered significant.
Supplementary Material
ACKNOWLEDGMENTS
This project was supported by grants from MINECO/FEDER EU contracts BFU2016-77427-C2-2-R, BFU2017-92223-EXP, PID2019-105649RB-I00; Severo Ochoa Excellence Accreditation SEV-2016-0644; the Basque Government contract KK-2019/00076; and NIH R01AI149297 (to M.E.G.). R.P.-R. is further a “Ramon y Cajal” fellow from the Spanish Ministry of Economy and Competitiveness. R.P.-R. acknowledges support by MINECO/FEDER EU contract SAF2016–77433-R and PID2019-110240RB-I00.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Zumla A, Raviglione M, Hafner R, von Reyn CF. 2013. Tuberculosis. N Engl J Med 368:745–755. doi: 10.1056/NEJMra1200894. [DOI] [PubMed] [Google Scholar]
- 2.Guinn KM, Rubin EJ. 2017. Tuberculosis: just the FAQs. mBio 8:e01910-17. doi: 10.1128/mBio.01910-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Getahun H, Matteelli A, Chaisson RE, Raviglione M. 2015. Latent Mycobacterium tuberculosis infection. N Engl J Med 372:2127–2135. doi: 10.1056/NEJMra1405427. [DOI] [PubMed] [Google Scholar]
- 4.World Health Organization. 2019. Global tuberculosis report 2019. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 5.Horsburgh CR, Jr, Barry CE, III, Lange C. 2015. Treatment of tuberculosis. N Engl J Med 373:2149–2160. doi: 10.1056/NEJMra1413919. [DOI] [PubMed] [Google Scholar]
- 6.Dheda K, Gumbo T, Gandhi NR, Murray M, Theron G, Udwadia Z, Migliori GB, Warren R. 2014. Global control of tuberculosis: from extensively drug-resistant to untreatable tuberculosis. Lancet Respir Med 2:321–338. doi: 10.1016/S2213-2600(14)70031-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Libardo MD, Boshoff HI, Barry CE, III. 2018. The present state of the tuberculosis drug development pipeline. Curr Opin Pharmacol 42:81–94. doi: 10.1016/j.coph.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jackson M, McNeil MR, Brennan PJ. 2013. Progress in targeting cell envelope biogenesis in Mycobacterium tuberculosis. Future Microbiol 8:855–875. doi: 10.2217/fmb.13.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dulberger CL, Rubin EJ, Boutte CC. 2020. The mycobacterial cell envelope—a moving target. Nat Rev Microbiol 18:47–59. doi: 10.1038/s41579-019-0273-7. [DOI] [PubMed] [Google Scholar]
- 10.Johnson EO, LaVerriere E, Office E, Stanley M, Meyer E, Kawate T, Gomez JE, Audette RE, Bandyopadhyay N, Betancourt N, Delano K, Da Silva I, Davis J, Gallo C, Gardner M, Golas AJ, Guinn KM, Kennedy S, Korn R, McConnell JA, Moss CE, Murphy KC, Nietupski RM, Papavinasasundaram KG, Pinkham JT, Pino PA, Proulx MK, Ruecker N, Song N, Thompson M, Trujillo C, Wakabayashi S, Wallach JB, Watson C, Ioerger TR, Lander ES, Hubbard BK, Serrano-Wu MH, Ehrt S, Fitzgerald M, Rubin EJ, Sassetti CM, Schnappinger D, Hung DT. 2019. Large-scale chemical-genetics yields new M. tuberculosis inhibitor classes. Nature 571:72–78. doi: 10.1038/s41586-019-1315-z. [DOI] [PubMed] [Google Scholar]
- 11.Angala SK, Belardinelli JM, Huc-Claustre E, Wheat WH, Jackson M. 2014. The cell envelope glycoconjugates of Mycobacterium tuberculosis. Crit Rev Biochem Mol Biol 49:361–399. doi: 10.3109/10409238.2014.925420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guerin ME, Kordulakova J, Alzari PM, Brennan PJ, Jackson M. 2010. Molecular basis of phosphatidyl-myo-inositol mannoside biosynthesis and regulation in mycobacteria. J Biol Chem 285:33577–33583. doi: 10.1074/jbc.R110.168328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sancho-Vaello E, Albesa-Jove D, Rodrigo-Unzueta A, Guerin ME. 2017. Structural basis of phosphatidyl-myo-inositol mannosides biosynthesis in mycobacteria. Biochim Biophys Acta Mol Cell Biol Lipids 1862:1355–1367. doi: 10.1016/j.bbalip.2016.11.002. [DOI] [PubMed] [Google Scholar]
- 14.Khoo KH, Dell A, Morris HR, Brennan PJ, Chatterjee D. 1995. Structural definition of acylated phosphatidylinositol mannosides from Mycobacterium tuberculosis: definition of a common anchor for lipomannan and lipoarabinomannan. Glycobiology 5:117–127. doi: 10.1093/glycob/5.1.117. [DOI] [PubMed] [Google Scholar]
- 15.Jankute M, Cox JA, Harrison J, Besra GS. 2015. Assembly of the mycobacterial cell wall. Annu Rev Microbiol 69:405–423. doi: 10.1146/annurev-micro-091014-104121. [DOI] [PubMed] [Google Scholar]
- 16.Haites RE, Morita YS, McConville MJ, Billman-Jacobe H. 2005. Function of phosphatidylinositol in mycobacteria. J Biol Chem 280:10981–10987. doi: 10.1074/jbc.M413443200. [DOI] [PubMed] [Google Scholar]
- 17.Torrelles JB, Schlesinger LS. 2010. Diversity in Mycobacterium tuberculosis mannosylated cell wall determinants impacts adaptation to the host. Tuberculosis (Edinb) 90:84–93. doi: 10.1016/j.tube.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morita YS, Fukuda T, Sena CB, Yamaryo-Botte Y, McConville MJ, Kinoshita T. 2011. Inositol lipid metabolism in mycobacteria: biosynthesis and regulatory mechanisms. Biochim Biophys Acta 1810:630–641. doi: 10.1016/j.bbagen.2011.03.017. [DOI] [PubMed] [Google Scholar]
- 19.Kordulakova J, Gilleron M, Mikusova K, Puzo G, Brennan PJ, Gicquel B, Jackson M. 2002. Definition of the first mannosylation step in phosphatidylinositol mannoside synthesis. PimA is essential for growth of mycobacteria. J Biol Chem 277:31335–31344. doi: 10.1074/jbc.M204060200. [DOI] [PubMed] [Google Scholar]
- 20.Guerin ME, Kaur D, Somashekar BS, Gibbs S, Gest P, Chatterjee D, Brennan PJ, Jackson M. 2009. New insights into the early steps of phosphatidylinositol mannoside biosynthesis in mycobacteria: PimB' is an essential enzyme of Mycobacterium smegmatis. J Biol Chem 284:25687–25696. doi: 10.1074/jbc.M109.030593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lea-Smith DJ, Martin KL, Pyke JS, Tull D, McConville MJ, Coppel RL, Crellin PK. 2008. Analysis of a new mannosyltransferase required for the synthesis of phosphatidylinositol mannosides and lipoarbinomannan reveals two lipomannan pools in Corynebacterineae. J Biol Chem 283:6773–6782. doi: 10.1074/jbc.M707139200. [DOI] [PubMed] [Google Scholar]
- 22.Mishra AK, Batt S, Krumbach K, Eggeling L, Besra GS. 2009. Characterization of the Corynebacterium glutamicum ΔpimB' ΔmgtA double deletion mutant and the role of Mycobacterium tuberculosis orthologues Rv2188c and Rv0557 in glycolipid biosynthesis. J Bacteriol 191:4465–4472. doi: 10.1128/JB.01729-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Torrelles JB, DesJardin LE, MacNeil J, Kaufman TM, Kutzbach B, Knaup R, McCarthy TR, Gurcha SS, Besra GS, Clegg S, Schlesinger LS. 2009. Inactivation of Mycobacterium tuberculosis mannosyltransferase pimB reduces the cell wall lipoarabinomannan and lipomannan content and increases the rate of bacterial-induced human macrophage cell death. Glycobiology 19:743–755. doi: 10.1093/glycob/cwp042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brennan P, Ballou CE. 1967. Biosynthesis of mannophosphoinositides by Mycobacterium phlei. The family of dimannophosphoinositides. J Biol Chem 242:3046–3056. doi: 10.1016/S0021-9258(18)95931-4. [DOI] [PubMed] [Google Scholar]
- 25.Brennan P, Ballou CE. 1968. Biosynthesis of mannophosphoinositides by Mycobacterium phlei. Enzymatic acylation of the dimannophosphoinositides. J Biol Chem 243:2975–2984. doi: 10.1016/S0021-9258(18)93368-5. [DOI] [PubMed] [Google Scholar]
- 26.Kordulakova J, Gilleron M, Puzo G, Brennan PJ, Gicquel B, Mikusova K, Jackson M. 2003. Identification of the required acyltransferase step in the biosynthesis of the phosphatidylinositol mannosides of mycobacterium species. J Biol Chem 278:36285–36295. doi: 10.1074/jbc.M303639200. [DOI] [PubMed] [Google Scholar]
- 27.Guerin ME, Schaeffer F, Chaffotte A, Gest P, Giganti D, Kordulakova J, van der Woerd M, Jackson M, Alzari PM. 2009. Substrate-induced conformational changes in the essential peripheral membrane-associated mannosyltransferase PimA from mycobacteria: implications for catalysis. J Biol Chem 284:21613–21625. doi: 10.1074/jbc.M109.003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Svetlikova Z, Barath P, Jackson M, Kordulakova J, Mikusova K. 2014. Purification and characterization of the acyltransferase involved in biosynthesis of the major mycobacterial cell envelope glycolipid—monoacylated phosphatidylinositol dimannoside. Protein Expr Purif 100:33–39. doi: 10.1016/j.pep.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berg S, Kaur D, Jackson M, Brennan PJ. 2007. The glycosyltransferases of Mycobacterium tuberculosis—roles in the synthesis of arabinogalactan, lipoarabinomannan, and other glycoconjugates. Glycobiology 17:35–56. doi: 10.1093/glycob/cwm010. [DOI] [PubMed] [Google Scholar]
- 30.Jackson M, Crick DC, Brennan PJ. 2000. Phosphatidylinositol is an essential phospholipid of mycobacteria. J Biol Chem 275:30092–30099. doi: 10.1074/jbc.M004658200. [DOI] [PubMed] [Google Scholar]
- 31.DeJesus MA, Gerrick ER, Xu W, Park SW, Long JE, Boutte CC, Rubin EJ, Schnappinger D, Ehrt S, Fortune SM, Sassetti CM, Ioerger TR. 2017. Comprehensive essentiality analysis of the Mycobacterium tuberculosis genome via saturating transposon mutagenesis. mBio 8:e02133-16. doi: 10.1128/mBio.02133-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Albesa-Jove D, Svetlikova Z, Tersa M, Sancho-Vaello E, Carreras-Gonzalez A, Bonnet P, Arrasate P, Eguskiza A, Angala SK, Cifuente JO, Kordulakova J, Jackson M, Mikusova K, Guerin ME. 2016. Structural basis for selective recognition of acyl chains by the membrane-associated acyltransferase PatA. Nat Commun 7:10906. doi: 10.1038/ncomms10906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tersa M, Raich L, Albesa-Jove D, Trastoy B, Prandi J, Gilleron M, Rovira C, Guerin ME. 2018. The molecular mechanism of substrate recognition and catalysis of the membrane acyltransferase PatA from mycobacteria. ACS Chem Biol 13:131–140. doi: 10.1021/acschembio.7b00578. [DOI] [PubMed] [Google Scholar]
- 34.Boldrin F, Ventura M, Degiacomi G, Ravishankar S, Sala C, Svetlikova Z, Ambady A, Dhar N, Kordulakova J, Zhang M, Serafini A, Vishwas KG, Vishwas VG, Kolly GS, Kumar N, Palù G, Guerin ME, Mikusova K, Cole ST, Manganelli R. 2014. The phosphatidyl-myo-inositol mannosyltransferase PimA is essential for Mycobacterium tuberculosis growth in vitro and in vivo. J Bacteriol 196:3441–3451. doi: 10.1128/JB.01346-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Geiss F. 1987. Fundamentals of thin-layer chromatography (planar chromatography). Heidelberg, Basel, New York. [Google Scholar]
- 36.Manganelli R, Dubnau E, Tyagi S, Kramer FR, Smith I. 1999. Differential expression of 10 sigma factor genes in Mycobacterium tuberculosis. Mol Microbiol 31:715–724. doi: 10.1046/j.1365-2958.1999.01212.x. [DOI] [PubMed] [Google Scholar]
- 37.Sassetti CM, Boyd DH, Rubin EJ. 2003. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol 48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 38.Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, Sassetti CM. 2011. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog 7:e1002251. doi: 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boldrin F, Casonato S, Dainese E, Sala C, Dhar N, Palu G, Riccardi G, Cole ST, Manganelli R. 2010. Development of a repressible mycobacterial promoter system based on two transcriptional repressors. Nucleic Acids Res 38:e134. doi: 10.1093/nar/gkq235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salman M, Lonsdale JT, Besra GS, Brennan PJ. 1999. Phosphatidylinositol synthesis in mycobacteria. Biochim Biophys Acta 1436:437–450. doi: 10.1016/s0005-2760(98)00151-9. [DOI] [PubMed] [Google Scholar]
- 41.Morii H, Ogawa M, Fukuda K, Taniguchi H, Koga Y. 2010. A revised biosynthetic pathway for phosphatidylinositol in Mycobacteria. J Biochem 148:593–602. doi: 10.1093/jb/mvq093. [DOI] [PubMed] [Google Scholar]
- 42.Morii H, Okauchi T, Nomiya H, Ogawa M, Fukuda K, Taniguchi H. 2013. Studies of inositol 1-phosphate analogues as inhibitors of the phosphatidylinositol phosphate synthase in mycobacteria. J Biochem 153:257–266. doi: 10.1093/jb/mvs141. [DOI] [PubMed] [Google Scholar]
- 43.Boldrin F, Degiacomi G, Serafini A, Kolly GS, Ventura M, Sala C, Provvedi R, Palu G, Cole ST, Manganelli R. 2018. Promoter mutagenesis for fine-tuning expression of essential genes in Mycobacterium tuberculosis. Microb Biotechnol 11:238–247. doi: 10.1111/1751-7915.12875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taneja R, Malik U, Khuller GK. 1979. Effect of growth temperature on the lipid composition of Mycobacterium smegmatis ATCC 607. J Gen Microbiol 113:413–416. doi: 10.1099/00221287-113-2-413. [DOI] [PubMed] [Google Scholar]
- 45.Bansal-Mutalik R, Nikaido H. 2014. Mycobacterial outer membrane is a lipid bilayer and the inner membrane is unusually rich in diacyl phosphatidylinositol dimannosides. Proc Natl Acad Sci U S A 111:4958–4963. doi: 10.1073/pnas.1403078111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morita YS, Patterson JH, Billman-Jacobe H, McConville MJ. 2004. Biosynthesis of mycobacterial phosphatidylinositol mannosides. Biochem J 378:589–597. doi: 10.1042/BJ20031372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morita YS, Velasquez R, Taig E, Waller RF, Patterson JH, Tull D, Williams SJ, Billman-Jacobe H, McConville MJ. 2005. Compartmentalization of lipid biosynthesis in mycobacteria. J Biol Chem 280:21645–21652. doi: 10.1074/jbc.M414181200. [DOI] [PubMed] [Google Scholar]
- 48.Larrouy-Maumus G, Marino LB, Madduri AV, Ragan TJ, Hunt DM, Bassano L, Gutierrez MG, Moody DB, Pavan FR, de Carvalho LP. 2016. Cell-envelope remodeling as a determinant of phenotypic antibacterial tolerance in Mycobacterium tuberculosis. ACS Infect Dis 2:352–360. doi: 10.1021/acsinfecdis.5b00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matsumoto K. 1997. Phosphatidylserine synthase from bacteria. Biochim Biophys Acta 1348:214–227. doi: 10.1016/s0005-2760(97)00110-0. [DOI] [PubMed] [Google Scholar]
- 50.Evans JC, Trujillo C, Wang Z, Eoh H, Ehrt S, Schnappinger D, Boshoff HI, Rhee KY, Barry CE, III, Mizrahi V. 2016. Validation of CoaBC as a bactericidal target in the coenzyme A pathway of Mycobacterium tuberculosis. ACS Infect Dis 2:958–968. doi: 10.1021/acsinfecdis.6b00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guerin ME, Kordulakova J, Schaeffer F, Svetlikova Z, Buschiazzo A, Giganti D, Gicquel B, Mikusova K, Jackson M, Alzari PM. 2007. Molecular recognition and interfacial catalysis by the essential phosphatidylinositol mannosyltransferase PimA from mycobacteria. J Biol Chem 282:20705–20714. doi: 10.1074/jbc.M702087200. [DOI] [PubMed] [Google Scholar]
- 52.Giganti D, Albesa-Jove D, Urresti S, Rodrigo-Unzueta A, Martinez MA, Comino N, Barilone N, Bellinzoni M, Chenal A, Guerin ME, Alzari PM. 2015. Secondary structure reshuffling modulates glycosyltransferase function at the membrane. Nat Chem Biol 11:16–18. doi: 10.1038/nchembio.1694. [DOI] [PubMed] [Google Scholar]
- 53.Batt SM, Jabeen T, Mishra AK, Veerapen N, Krumbach K, Eggeling L, Besra GS, Futterer K. 2010. Acceptor substrate discrimination in phosphatidyl-myo-inositol mannoside synthesis: structural and mutational analysis of mannosyltransferase Corynebacterium glutamicum PimB'. J Biol Chem 285:37741–37752. doi: 10.1074/jbc.M110.165407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lairson LL, Henrissat B, Davies GJ, Withers SG. 2008. Glycosyltransferases: structures, functions, and mechanisms. Annu Rev Biochem 77:521–555. doi: 10.1146/annurev.biochem.76.061005.092322. [DOI] [PubMed] [Google Scholar]
- 55.Albesa-Jove D, Giganti D, Jackson M, Alzari PM, Guerin ME. 2014. Structure-function relationships of membrane-associated GT-B glycosyltransferases. Glycobiology 24:108–124. doi: 10.1093/glycob/cwt101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Albesa-Jove D, Guerin ME. 2016. The conformational plasticity of glycosyltransferases. Curr Opin Struct Biol 40:23–32. doi: 10.1016/j.sbi.2016.07.007. [DOI] [PubMed] [Google Scholar]
- 57.Moremen KW, Haltiwanger RS. 2019. Emerging structural insights into glycosyltransferase-mediated synthesis of glycans. Nat Chem Biol 15:853–864. doi: 10.1038/s41589-019-0350-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rodrigo-Unzueta A, Ghirardello M, Urresti S, Delso I, Giganti D, Anso I, Trastoy B, Comino N, Tersa M, D'Angelo C, Cifuente JO, Marina A, Liebau J, Mäler L, Chenal A, Albesa-Jové D, Merino P, Guerin ME. 2020. Dissecting the structural and chemical determinants of the “open-to-closed” motion in the mannosyltransferase PimA from mycobacteria. Biochemistry 59:2934–2945. doi: 10.1021/acs.biochem.0c00376. [DOI] [PubMed] [Google Scholar]
- 59.Giganti D, Alegre-Cebollada J, Urresti S, Albesa-Jove D, Rodrigo-Unzueta A, Comino N, Kachala M, Lopez-Fernandez S, Svergun DI, Fernandez JM, Guerin ME. 2013. Conformational plasticity of the essential membrane-associated mannosyltransferase PimA from mycobacteria. J Biol Chem 288:29797–29808. doi: 10.1074/jbc.M113.462705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liebau J, Tersa M, Trastoy B, Patrick J, Rodrigo-Unzueta A, Corzana F, Sparrman T, Guerin ME, Mäler L. 2020. Unveiling the activation dynamics of a fold-switch bacterial glycosyltransferase by 19F NMR. J Biol Chem 295:9868–9878. doi: 10.1074/jbc.RA120.014162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Macįg A, Dainese E, Rodriguez GM, Milano A, Provvedi R, Pasca MR, Smith I, Palù G, Riccardi G, Manganelli R. 2007. Global analysis of the Mycobacterium tuberculosis Zur (FurB) regulon. J Bacteriol 189:730–740. doi: 10.1128/JB.01190-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Manganelli R, Voskuil MI, Schoolnik GK, Smith I. 2001. The Mycobacterium tuberculosis ECF sigma factor σE: role in global gene expression and survival in macrophages. Mol Microbiol 41:423–437. doi: 10.1046/j.1365-2958.2001.02525.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.