

Abstract
Introduction
We investigated the frequency, neuropathology, and phenotypic characteristics of spastic paraplegia (SP) that precedes dementia in presenilin 1 (PSEN1) related familial Alzheimer's disease (AD).
Methods
We performed whole exome sequencing (WES) in 60 probands with hereditary spastic paraplegia (HSP) phenotype that was negative for variants in known HSP‐related genes. Where PSEN1 mutation was identified, brain biopsy was performed. We investigated the link between HSP and AD with PSEN1 in silico pathway analysis and measured in vivo the stability of PSEN1 mutant γ‐secretase.
Results
We identified a PSEN1 variant (p.Thr291Pro) in an individual presenting with pure SP at 30 years of age. Three years later, SP was associated with severe, fast cognitive decline and amyloid deposition with diffuse cortical plaques on brain biopsy. Biochemical analysis of p.Thr291Pro PSEN1 revealed that although the mutation does not alter active γ‐secretase reconstitution, it destabilizes γ‐secretase‐amyloid precursor protein (APP)/amyloid beta (Aβn) interactions during proteolysis, enhancing the production of longer Aβ peptides. We then extended our analysis to all 226 PSEN1 pathogenic variants reported and show that 7.5% were associated with pure SP onset followed by cognitive decline later in the disease. We found that PSEN1 cases manifesting initially as SP have a later age of onset, are associated with mutations located beyond codon 200, and showed larger diffuse, cored plaques, amyloid‐ring arteries, and severe CAA.
Discussion
We show that pure SP can precede dementia onset in PSEN1‐related familial AD. We recommend PSEN1 genetic testing in patients presenting with SP with no variants in known HSP‐related genes, particularly when associated with a family history of cognitive decline.
Keywords: Alzheimer's disease, dementia, hereditary spastic paraplegia, HSP, presenilin, PSEN1, spastic gait, spastic paraparesis, spastic paraplegia
1. BACKGROUND
Alzheimer's disease (AD) is the major cause of dementia worldwide, presenting with memory loss and behavioral disturbances as the core clinical features. 1 Around 4.7 million individuals 65 years of age or older are estimated to carry a diagnosis of AD dementia in the United States alone. 2 By 2050, AD cases are expected to reach up to 13.8 million in the United States and 130 million worldwide. 3 Neuropathologically, intracellular neurofibrillary tangles of abnormally phosphorylated tau protein and extracellular amyloid beta (Aβ) plaques are the hallmarks of AD. 4 The majority of AD cases are sporadic, with a late onset (≥65 years). Mutations in presenilin 1 (PSEN1) (OMIM: 104311), presenilin 2 (PSEN2) (OMIM: 600759) and the amyloid precursor protein APP (OMIM: 104760) cause familial forms with early onset disease. 1 , 4 , 5 Familial AD is characterized by memory and behavioral disturbances similar to sporadic AD. Nevertheless, atypical phenotypes associated with spastic paraplegia (SP) have been described in familial AD (FAD), especially in PSEN1 mutations, the most common genetic cause of AD. 1 , 4 , 5 , 6 , 7 , 8
Systematic review: The authors reviewed the literature using traditional (eg, PubMed) sources, meeting abstracts, and presentations. Analysis of all 226 PSEN1 pathogenic variants reported to date showed that 7.5% presented as pure spastic paraplegia (SP) followed by cognitive decline, and PSEN1 cases manifesting initially as SP have a later age of onset and are associated with mutations located beyond codon 200. These mutations were associated with cortical cotton wool plaques (CWP) on neuropathological examination.
Interpretation: We show that pure spastic paraplegia can precede the onset of dementia in PSEN1‐related familial Alzheimer's disease (AD) and that clinical features of SP correlate with the position of pathogenic mutations. We recommend PSEN1 genetic testing in patients presenting with SP negative for variants in known hereditary SP (HSP)–related genes, particularly in the presence of a family history of cognitive decline.
Future directions: This study highlights the clinical overlap between spastic paraplegia and PSEN1‐related Alzheimer's disease. Early screening of PSEN1 mutations would alleviate the need for further invasive testing, such as brain biopsy. Unraveling the phenotypic heterogeneity within PSEN1 mutation carriers could provide new potential targets for clinical trials of patients with familial AD.
Spastic paraplegia (or SP) is a common feature of many genetic neurological diseases. It is the defining clinical feature of a group of diseases collectively known as hereditary spastic paraplegias (HSPs). 9 , 10 HSP can present as pure (defined by isolated lower limb spasticity) or complicated by other neurological manifestations such as cognitive impairment, ataxia, or peripheral neuropathy. 11 All types of inheritance have been reported in HSPs.
In 1997, mutations in PSEN1 were originally identified as a cause of FAD complicated by SP. 12 , 13 , 14 PSEN1, PSEN2, and APP encode the catalytic subunit—presenilin1/2(PSEN1/2)—of the γ‐secretase protease complex and its substrate, the amyloid precursor protein (APP). γ‐Secretase performs sequential cleavages on the transmembrane domain of APP and many other type 1 transmembrane proteins (eg, LIN‐12/Notch proteins). 15 The sequential γ‐secretase–mediated processing of APP generates amyloid beta (Aβ) peptides of different lengths and has been studied in detail. 8 , 15 Several reports suggest that other γ‐secretase substrates are processed by the protease in a sequential manner, but the precise number and position of cuts are not known. 15
We have reported previously that PSEN1 AD‐linked mutations consistently lead to the release of relatively longer Aβs. 16 , 17 It is important to note that the enhanced release of longer peptides is caused by the destabilization of γ‐secretase‐APP interactions during its sequential processing. 18 Of note, the degree of destabilization of the enzyme‐substrate complexes had been shown to correlate with the age at AD onset. 18
The AD‐linked PSEN1 mutations associated with SP share characteristic neuropathological findings; one that is shared by all cases is cotton wool plaques (CWPs). CWPs, not typically observed in sporadic AD, represent large non‐cored plaques without significant neuritic dystrophy. 19 , 20 An expanding number of families with AD complicated by SP has been subsequently reported, many of them displaying characteristic CWP. 13 Very rarely, SP can even precede the development of cognitive decline in these patients, sometimes by several years. 13 In the era of next‐generation DNA sequencing and emerging disease‐modifying therapies for familial and sporadic AD, early provision of a definite genetic diagnosis for these patients becomes extremely important.
Here, we investigated the frequency, neuropathological findings, and phenotypic characteristics of spastic paraplegia (SP) that precedes the onset of dementia in PSEN1‐related familial AD.
2. METHODS
2.1. Subjects
In this retrospective study we recruited 60 Greek HSP index cases referred to a tertiary neurology department under ethics‐approved research protocols (UCLH:04/N034) with informed consent. The probands had slowly progressive SP as the first or most prominent manifestation. Acquired and metabolic causes of SP were excluded with extensive autoimmune, metabolic, and neuroimaging investigations, including brain and spine magnetic resonance imaging (MRI), cerebrospinal fluid (CSF) analysis, evoked potentials studies, nerve conduction and electromyography (EMG), electroencephalography (EEG), and testing for HIV, syphilis, serum vitamin B12, and vitamin E.
2.2. Clinical phenotyping
Comprehensive clinical phenotyping was performed by neurogenetics specialists in Athens (Neurogenetics Unit, UOA) during the period from 2012 to 2019. The Ashworth scale was used to quantify the grade of spasticity, and the Spastic Paraplegia Rating Scale (SPRS) to evaluate the severity of SP. The Montreal Cognitive Assessment (MoCA) was used for cognitive assessment.
2.3. Neuropathology
Biopsy specimens were fixed overnight in formalin (4%) and routinely processed into liquid paraffin. The sections were cut at 4 μm thickness with a microtome (Microm, Heidelberg, Germany) and stained with hematoxylin and eosin (H&E) and Congo red. Immunohistochemical staining was performed using an automated staining apparatus (Ventana Benchmark, Roche, Mannheim, Germany) and an antibody directed against Aβ (Clone 4G8; Covance, Emeryville, California, USA) according to the manufacturer's protocol.
2.4. Genetic analysis
DNA was extracted from peripheral blood. All cases were investigated with whole exome sequencing (WES). WES and alignments were carried out at Macrogen (Macrogen Inc, Seoul, South Korea). Exome capture was performed using SureSelect Human All Exon V4 enrichment kit (Agilent Technologies, Santa Clara, CA, USA) and sequenced on an Illumina HiSeq4000 instrument (Illumina, San Diego, CA, USA) generating 100‐bp paired‐end reads. Reads alignment using the hg19 human genome reference, variant calling and annotation were performed as described previously. 21 All cases were screened and were negative for known HSP‐related genes.
The analysis was then extended to include all coding genes. Only variants with MAF of < 0.01 in 1000 Genomes Project, 22 NHLBI GO Exome Sequencing, and gnomAD 23 and predicted to be likely pathogenic or known pathogenic, in genes associated with neurological phenotype, were considered as likely causal variants. The PSEN1 variant was confirmed using bi‐directional Sanger Sequencing using the following primers: 5′‐GGAAGACTGGCGATTTGTGT‐3′ and 5′‐CAGTGACCCTGAAAAATCAAGA‐3′.
2.5. Generation of stable cell lines
Mouse embryonic fibroblasts (MEFs) stably expressing wild‐type or the p.Thr291Pro human PSEN1 were generated as previously reported. 24 Briefly, PSEN1/PSEN2 dKO MEFs were cultured in Dulbecco's modified Eagle's medium (DMEM)/F‐12 (Life Technologies) containing 10% fetal bovine serum (FBS) (Life Technologies). The cells were transduced with retroviruses, carrying pMSCVpuro plasmids encoding wild‐type (WT) or p.Thr291Pro PSEN1, using a replication‐defective recombinant retroviral expression system (Clontech). Post transduction the culture medium was supplemented with 5 μg/mL puromycin (Sigma‐Aldrich) to select clones stably expressing PSEN, which were further maintained in DMEM/F‐12, 10% FBS, and 3 μg/mL puromycin. NCT and PSEN1 expression levels and the reconstitution of the functional GSEC complexes in the different cell lines were analyzed by SDS‐PAGE/western blot. GSEC inhibitor X (565771) was purchased from Calbiochem and anti‐NCT (612290) from BD Biosciences.
The following antibodies were used in the study: anti‐FLAG M2 (F3165) from Sigma‐Aldrich, anti‐PSEN‐CTF (MAB5232) from Merck, horseradish peroxidase (HRP)–conjugated anti‐mouse (1721011), anti‐rabbit IgG (1721019) from Bio‐Rad, and anti‐PEN‐2 (B126.2). 25
2.6. In vitro thermo‐activity assays using solubilized γ‐secretase
MEF microsomal fractions were solubilized in 1% CHAPSO as described previously. 16 A total of 5 μg of the solubilized material was used in in vitro thermo‐activity assays with purified APPC99‐3xFLAG (1.5μM final) (as described 18 ) in 50 mM PIPES, pH 7.0, 0.25 M sucrose, 1 mM EGTA, 1x proteinase inhibitors (Roche), 0.1% phosphatidylcholine, and 2.5% DMSO (or 10 μM Inhibitor X). The activity assays were performed over a temperature gradient ranging from 37°C to 65°C (similar to 18 ). Briefly, enzyme mixes (containing all components except the substrate) and substrate dilutions were pre‐incubated separately at the indicated temperatures for 10 minutes. After pre‐incubation, the substrate was added to the enzyme mix and proteolysis proceeded for 4 hours. AICD production was assessed by western blot analysis with M2 FLAG antibody, and Aβ generation was quantified by MSD multiplex ELISA as described previously. 18 The ELISA capture antibodies (JRF AB038 for Aβ38, JRF/cAb40/28 for Aβ40 and JRF/cAb42/26 for Aβ42) and the detection antibody (JRF/AbN/25 raised against the N‐terminus of Aβ) were obtained through collaboration with Janssen Pharmaceutica NV (Beerse, Belgium).
2.7. Analysis of all reported PSEN1 pathogenic variants
We expanded our analysis into the link between SP and PSEN1‐related AD by investigating the frequency of SP in cases with previously reported PSEN1 pathogenic variants. Literature searches were conducted on MEDLINE (PubMed), Scopus, clinicaltrials.gov, and database of AD mutations (http://www.molgen.ua.ac.be/ADMutations) using several key words and their combinations, up to December 2019. Search terms included: hereditary spastic paraplegia, spastic paraplegia, familial Alzheimer's disease, and PSEN1. References from the selected articles were also thoroughly screened for other pertinent articles. Additional searches were performed for specific topics (eg, neuropathology of Alzheimer's disease, cotton wool plaques). Studies reporting mutations in PSEN1, presenting initially as SP, were further analyzed. During the screening of the abstracts/full texts, the publications that were not relevant to this review were removed. No language or publication period restrictions were applied to the initial searches. However, only English‐speaking publications were further studied in detail.
2.8. Pathway analysis
We assessed the biological functions of HSP‐causative genes as members of a putative interactome, that is, a protein network jointly mediating several intracellular processes. Expanded details of functional enrichment analyses and in silico knockdown of PSEN1 are described in Supplementary e‐1.
3. RESULTS
3.1. PSEN1 p.Thr291Pro variant presenting with spastic paraplegia several years before the onset of cognitive decline
From screening a cohort of 60 patients with symptoms of HSP we identified one individual presenting with pure HSP phenotype followed by the development of severe cognitive decline several years later. The patient in his thirties (individual II‐1, Figure 1A) with an unremarkable past medical history presented with a 1‐year history of progressive gait disturbance. Initial neurological examination revealed a mild spastic gait with pyramidal syndrome (brisk reflexes, bilateral upgoing plantar reflexes, ankle clonus, and increased tone with an Ashworth score of 1 [of 4]). Cognitive function was within normal limits on initial examination (Montreal Cognitive Assessment [MoCA] 30/30). Brain and spine MRI in the proband revealed no specific findings. His mother had an onset of SP in her thirties and no cognitive impairment reported at onset. She became bedridden with severe cognitive impairment and died before age 40. No further family history was available, because she was adopted.
FIGURE 1.
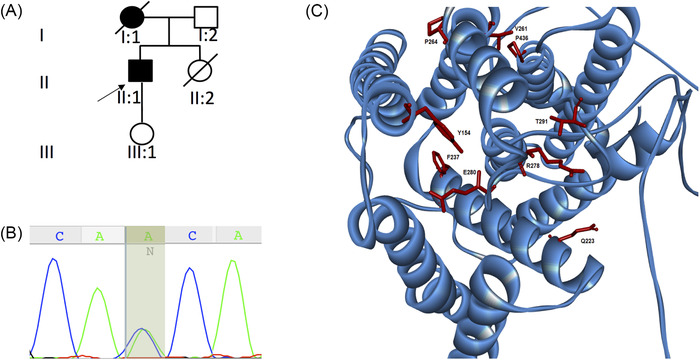
Genetic and protein analysis for the PSEN1 mutation–associated spastic paraplegia. A, Pedigree of the family reported in this study. B, Sanger sequencing confirmation of the proband highlighting the c.871A > C (p.Thr291Pro) mutation. C, Localization of PSEN1 mutations associated with spastic paraparesis as presenting phenotype. PSEN1 protein has nine transmembrane domains. Pathogenic substitutes in the p.Thr291Pro and other reported mutations are shown
HSP gene panel and multiplex ligation‐dependent probe amplification (MLPA) for ‐ copy number variants in SPG4, the most frequent cause of pure HSP, were negative. The spasticity progressed, and at 1‐year follow‐up the patient presented with a severe spastic gait but no additional clinical features. Memory disturbances were first noted at ≈2.5 years following the onset of spasticity and prompted a re‐evaluation of the initial diagnosis. A repeat brain MRI did not show any abnormal findings; therefore, a brain biopsy was performed to exclude a spongiform encephalopathy. Of interest, the only findings on brain biopsy were Aβ‐A4‐deposition within the vessel walls with several diffuse cortical CWPs. Three years following symptom onset, he had severe worsening of his cognitive abilities (MoCA20/30, MMSE25/30) with further motor deterioration. At last examination, 4.5 years into the disease (at 37.5 years), the patient was wheelchair‐bound with severe SP, walking only a few steps with bilateral support. SP rating scale was 48/52 and MoCA was 14/30.
WES in the proband (II‐1) revealed the heterozygous variant c.871A > C (p.Thr291Pro) in PSEN1 (NM_000021.3), confirmed by Sanger sequencing (Figure 1B). The c.871A > C (p.Thr291Pro) is a known pathogenic missense mutation located in exon 9. 26 The p.Thr291Pro has an anti‐parallel β‐sheet location, which, according to recent structural data, interacts directly with the C‐terminal domain of the substrates (APP and Notch). 27 , 28
3.2. The p.Thr291Pro variant leading to generation of relatively longer Aβ peptides from APP
To investigate whether the pathogenic variant identified in our cohort, the p.Thr291Pro PSEN1 substitution, destabilizes the interactions between γ‐secretase and APP leading to altered Aβ production, we analyzed the activities and thermostabilities of WT and mutant γ‐secretase complexes in vitro. To this end, we rescued the expression of human WT and p.Thr291Pro mutant PSEN1s in PSEN1/2‐deficient mouse embryonic fibroblasts (MEFs) and analyzed the effects of the mutation on γ‐secretase complex reconstitution and activity. The WT and mutant PSEN1 proteins both restored γ‐secretase complex levels, as indicated by the levels of NTF‐ and CTF‐PSEN fragments (Figure 2A). We next assessed the effects of the p.Thr291Pro substitution on the generation of Aβ peptides in an in vitro γ‐secretase activity assay. The analysis showed increased production of Aβ42, relative to the shorter Aβ40/38 peptides by the mutant protease when compared to the WT. The increased production of Aβ42, relative to the shorter Aβ40/38, is a hallmark of familial AD‐linked PSEN1 mutations. Analysis of the p.Thr291Pro PSEN1 in thermoactivity assays confirmed that this substitution destabilizes “enzyme‐substrate” interactions at the level of both total activity (AICD generation) as well as Aβ production (Figure 2B‐2C). This is indicated by shifts in the thermoactivity profiles for AICD and Aβ peptide production, respectively. The lower stability of enzyme‐substrate interactions formed by the p.Thr291Pro PSEN1 γ‐secretase is in line with the reported effects of AD‐linked PSEN1 variants on APP processing. 16 , 17 , 18 , 29 We point at this effect as central to the pathogenesis of AD‐linked PSEN1 variants.
FIGURE 2.
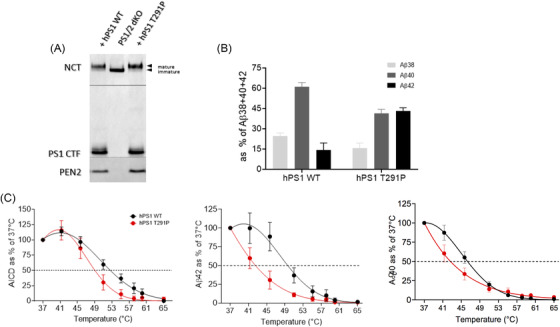
AD‐linked p.Thr291Pro PSEN1 destabilizes γ‐secretase‐APPc99/Aβ interactions resulting in generation of longer Aβ peptides. A, Representative SDS‐PAGE/western blot analysis of CHAPSO‐solubilized membranes from dKO PS1/PS2 mouse embryonic fibroblasts (MEFs) stably expressing wild‐type (WT) or mutant p.Thr291Pro hPS1. The presence of mature, glycosylated NCT, C‐terminal fragment of the endoproteolyzed PS1 and PEN2 (compared to dKO PS1/PS2 cells) indicates that p.Thr291Pro hPS1 reconstitutes active γ‐secretase complexes. Arrowheads indicate the position of molecular weight markers. B, Aβ profiles generated by solubilized membranes prepared from stable MEF PS1/2 dKO cell lines expressing WT or p.Thr291Pro hPS1 γ‐secretase in in vitro activity assays with purified APPC99‐3xFLAG. Increase in the production of Aβ42 at the expense of the shorter Aβ38 and Aβ40 indicates pathogenic impairment of γ‐secretase processivity of APPC99 by the AD‐linked p.Thr291Pro hPS1 mutation. N = 3 ± SD. C, In vitro thermoactivity data confirm that mutant p.Thr291Pro hPS1 destabilizes γ‐secretase‐APPC99/Aβ interactions, when compared to the WT complex, at the level of total activity (AICD generation) as well as Aβ peptide production, inducing premature dissociation of long Aβ peptides. N ≥ 3 ± SD
3.3. Spastic paraplegia phenotype in PSEN1‐related AD
Given the unusual presentation of pure SP preceding cognitive impairment by several years, we reviewed the clinical phenotype of all known PSEN1 pathogenic variants reported to date. We found that 13.7% of reported pathogenic variants were associated with SP (31 of 226 pathogenic and likely pathogenic variants reported in PSEN1) (http://www.molgen.ua.ac.be/ADMutations). Of these, including the variant reported here, 17 (7.5%) have initially presented with pure SP, followed by cognitive decline months or years later (Table 1, Figure 1C). The average age at onset in the group of patients presenting initially with pure SP was 40.5 years old, and the majority of these variants were located in or after exon 8. The average age at onset in the PSEN1 mutation carriers manifesting only with dementia was 37.3 years old and SP was, in most of these cases, prominent only after some years of cognitive impairment (Supplementary e‐2). The majority of patients with this phenotype had mutations that clustered in exons 4 and 5 1 .
TABLE 1.
All published PSEN1 mutations presenting with pure SP at onset
| Mutations | Exon | Families (n) | Age of onsetSP (years) | Neuropathological findings | Presenting symptom | Study |
|---|---|---|---|---|---|---|
| p.Ile83_Met84del | EX4 | 1 | 34‐38 | CWP | SP | Houlden et al. (2000) |
| p.Tyr154Asn | EX5 | 1 | 37 | – | SP | Hattori et al. (2004) |
| p.Phe237Ile | EX7 | 1 | 31 | – | SP | Sodeyama et al. (2001) |
| p.Gln223Arg | EX7 | 1 | 35 | CWP, neuritic plaques, neurofibrillary tangles, neuropil threads | SP | Saint‐Aubert et al. (2013) |
| p.Val261Phe | EX8 | 1 | 38 | CWP | SP | Farlow et al (2000) |
| p.Pro264Leu | EX8 | 1 | 54 | – | SP | Jacquemont et al. (2002) |
| p.Pro264Leu | EX8 | 1 | – | – | SP | Dumanchin et al. (2006) |
| p.Pro264Leu | EX8 | 1 | – | CWP | SP | Wallon et al. (2012) |
| p.Arg278Thr | EX8 | 1 | 34 | – | SP | Kwok et al. (1997) |
| p.Arg278Lys | EX8 | 1 | 41‐45 | – | SP |
Assini et al. (2003) |
| p.Glu280Gly | EX8 | 1 | 40‐43 | Amyloid plaques, CWP | SP, myoclonus, ataxia | O'Riordan et al. (2002) |
| p.Glu280Gly | EX8 | 1 | 47 | Amyloid plaques, CWP, tau positive neurofibrillary tangles | SP | Sinha et al. (2013) |
| Skipping of entire exon 9 | – | 2 | 45‐55 |
CWP Neurofibrillary tangles, amyloid plaques |
SP |
Crook et al. (1998) Brooks et al. (2003) |
| p.Thr291Pro | EX9 | 2 | 32 | CWP | SP | This study |
| p.Thr291Pro | EX9 | 1 | 33 | – | SP | Dumanchin et al. (2006) |
| p.Val391Gly | EX11 | 1 | 23 | – | SP | Lou et al. (2017) |
| p.Pro436Gly | EX12 | 1 | 20‐42 | CWP | SP | Houlden et al. (2000) |
Legend: SP, spastic paraparesis; CWP, cotton‐wool plaques; –, not available; EX, exon.
The novel PSEN1 case (p.Thr291Pro) identified in this study is also listed.
3.4. Hereditary spastic paraplegia pathways including PSEN1 act as a true interactome
Given the occurrence of SP phenotype in the early course of PSEN1‐related AD in over 10% of all reported cases, we investigated the overlap between HSP and PSEN1 dysfunction using gene set enrichment analysis (GSEA) and in silico knockdown of PSEN1 from the resulting network (Supplementary e‐1). HSP‐associated genes were significantly enriched for gene ontology terms and pathways involving neurogenesis, axonal development, and CNS myelination, indicating a point of divergence from the rest of the interactome. It is important to note that the retrieval of significantly enriched pathways (particularly by examining the gene list containing PSEN1) has indicated that the HSP‐causative genes function as a true interactome. Following PSEN1 knockdown, the pathways “neurotrophin signaling” and “Wnt signaling” were eliminated. In canonical (β‐catenin dependent) Wnt signaling, presenilins maintain β‐catenin stability, allowing the translocation to the nucleus and the subsequent expression of Wnt target genes, promoting neuronal growth, synaptogenesis, and plasticity. Much like the perturbed Wnt signaling disrupts adult neurogenesis and plasticity in AD, deleterious alterations have also been reported as upstream events in neurotrophin cascades 30 (such as BDNF signaling). In vivo animal models of PSEN1 knockdown have demonstrated the conditional depletion of BDNF levels, corresponding to the pathway silencing denoted in our in silico experiment.
3.5. Neuropathological findings in PSEN1 variants presenting with spastic paraplegia show distinct features from sporadic AD: Case report and extended analysis
Although the PSEN1 c.871A > C was reported previously in familial AD, its neuropathological expression has not been previously described. The brain biopsy in individual II‐1 from this study showed amyloid deposition within the vessel walls with diffuse cortical plaques, with the characteristic morphology of CWPs (Figure 3). Neuropathological studies of 24 mutations of PSEN1‐related familial AD associated with SP were identified in the literature. CWPs were identified in 21 of 24 studies, amyloid plaques in 10 of 24, and cerebral amyloid angiopathy (CAA) in 3 of 24. Different amounts of CAA in different mutations were also observed. In mutations located before codon 200, many diffuse cored plaques, a few white matter plaques, and mild to moderate CAA were identified. In comparison, mutations beyond codon 200 showed larger diffuse, cored plaques surrounding with amyloid‐rings arteries, with severe CAA (Table 1).
FIGURE 3.
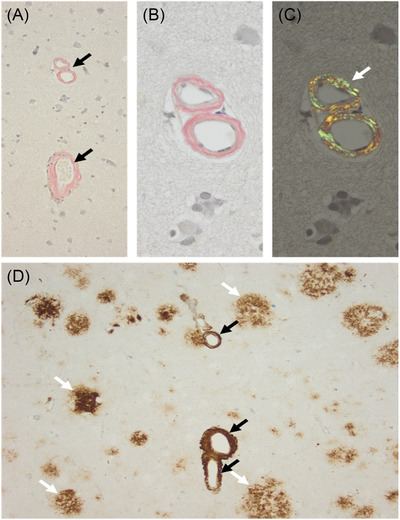
Neuropathological study in individual II‐1 from this study (c.871A ≥ C, p.Thr291Pro). A, Several vessels with thickened vessels showing congophilia (black arrows; Congo Red staining). B, Higher magnification of A with congophilia of blood vessels (B) and after polarization of the same area showing typical apple‐green color (white arrow in C). D, Beta‐A4‐amyloid immunohistochemistry confirms amyloid deposition within the vessel walls (black arrows) and in addition several diffuse cortical plaques (white arrows), with the characteristic morphology of cotton‐wool plaques
4. DISCUSSION
In this study, we assessed the clinical, molecular, and neuropathological features of AD‐linked PSEN1 mutations presenting with pure HSP‐like phenotype several years before the onset of cognitive decline. Our analysis of all reported AD‐linked PSEN1 cases showed that 7.5% of all known pathogenic PSEN1 pathogenic variants presented initially as SP. Furthermore, 13.7% of PSEN1 pathogenic variants were associated with SP at some point during the disease, suggesting that this phenotype is not infrequent and should prompt inclusion of PSEN1 variants in the differential diagnosis of patients presenting with SP.
We show that atypical presentations including SP are mostly associated with PSEN1 variants located beyond exon 8. Pathogenic variants located before codon 200 in exon 8, affecting the first hydrophilic loop of PSEN1, usually present with an earlier age of onset and pure dementia phenotype, 1 whereas the PSEN1 cases manifesting initially as SP have a later age at onset, in the same range with HSP patients, making the differential diagnosis of familial dementia challenging.
Our study, the first neuropathological description of AD caused by p.Thr291Pro PSEN1 variant, shows Aβ‐A4‐deposition within the vessel walls with several diffuse CWPs. We cannot comment further as the patient is alive, and no full autopsy report is available. Neuropathological studies of PSEN1 families presenting with SP identified CWPs, consisting of congophilic cores and dystrophic neurites, which were not observed in sporadic AD, 19 , 20 but are seen in other familial AD cases, such as PSEN2 or APP‐related. CWPs consist mainly of amino‐terminally truncated forms of Aβ peptide, starting after amino acid 11, where the alternative cleavage site is located. 13 PSEN1 mutations with CWPs produce high levels of the Aβ peptide ending at amino acid 42 or 43. 13 CWPs are found mainly in the frontal motor cortices. However, abnormal tau accumulation in the primary motor cortex was reported recently in a PSEN1 patient with SP. 31 Neurofibrillary tangles (NFTs) were identified in several cases carrying PSEN1 variants and manifesting SP. 26 , 32 However, not all PSEN1‐related cases associated with SP showed the presence of overt tangles. 33 On neuropathology examination, PSEN1 mutations located beyond codon 200, as in the case presented here, have more extensive CAA than mutations located before codon 200. 34
Pathway analysis in HSP‐related genes and PSEN1 showed perturbed “neurotrophin signaling” and “Wnt signaling” when PSEN1 is knocked down. Both these pathways have been associated with the pathogenesis of AD, mediating both neuronal morphogenesis and mature CNS neuroprotection. Furthermore, the roles mediated by these pathways include motor neuron development, phenotype diversification, and survival. 30 , 35 Conversely, loss of canonical signaling in each of these pathways has been implicated in the pathophysiology of ALS. 36 Restoration or de novo targeting of these pathways, in turn, has been shown to either halt neuron death or promote proliferation. 33 Taken together, these studies suggest that a knockdown in either or both of these pathways could promote neurodegeneration and synaptopathy, and that the arrest of these aberrant processes could occur if canonical signaling were restored. Thus pathways and molecular functions mediated by this interactome would further outline potentially targetable pathophysiological mechanisms.
The frequent co‐occurrence of PSEN1 variants and spasticity might thereby be driven by shared vulnerability of corticospinal tract axons and cortical neurons toward disturbances of the same molecular pathways and the effect of specific mutations affecting γ‐secretase activity in a way that alters its catalytic action on some other substrates particularly important for motor neurons. Over 80 different γ‐secretase substrates have been identified 37 and a growing number of additional proteins cleaved by presenilin/γ‐secretase continue to be discovered. Most of these proteins are type‐I transmembrane proteins involved in vital signaling functions regulating cell fate, adhesion, migration, neurite outgrowth, or synaptogenesis. It is notable that the HSP networks analyses highlight very similar molecular disease mechanisms including cellular transport, nucleotide metabolism, and synapse and axon development. 38 Furthermore, some presenilin γ‐secretase substrates have already been linked with neurodegenerative conditions associated with spasticity and/or motor neuron involvement such as colony‐stimulating factor‐1 (CSF1R), 39 , 40 , 41 netrin 1 receptor (DCC), 42 , 43 receptor tyrosine‐protein kinase erbB‐4 (ERBB4), 44 and very low density lipoprotein receptor (VLDLR). 45 , 46
Two distinct pathogenic pathways emerge in PSEN1‐related disease. First, the typical AD type presenting initially with dementia and characterized by neuritic plaques on brain biopsy, and the atypical course presenting initially with SP followed by cognitive decline and characterized by CWP on neuropathology. 20 A “threshold effect” was speculated (which most PSEN1 mutations fail to exceed) as well as a differential effect on Aβ42 production. 20 , 26 Some mutations are reported to cause a nearly complete loss of γ‐secretase activity, suggesting that pathogenic PSEN1 mutations could also cause a general loss of presenilin function. 47 However, the great majority of familial AD patients are heterozygous for PSEN1 mutations and the studies analyzing γ‐secretase activity in post‐mortem brain samples of different PSEN1‐related familial AD show that the WT allele not only contributes to the global brain γ‐secretase endopeptidase activity but in fact compensates for potential mutant‐associated impairments, at least on APP processing. 17 Mutant‐induced impairments on the γ‐secretase‐processing of particular substrates cannot be excluded.
Aβ42/Αβ40 ratio and increased percentage of N‐terminal truncated species are also speculated to contribute to these phenotypic differences, 48 suggesting the existence of critical areas of the protein. For example, mutations in exons 4 and 5 are reported to cause, particularly, early onset disease. Without affecting the endopeptidase function of γ‐secretase, these mutations located in the extracellular loop change an important allosteric core that alters the Aβ profiles generated through carboxypeptidase‐like activity of γ‐secretase. 24 Our analyses show that the identified p.Thr291Pro PSEN1 mutation does not impair the assembly of γ‐secretase, or its activation through autoendoproteolysis into an active protease. The mutation, however, destabilizes the γ‐secretase‐APP/Aβ interactions, leading to premature dissociation of longer Aβ peptides. This destabilizing effect has been shown to characterize AD‐linked PSEN1 mutations and correlates with AD onset. 18
Compared to PSEN1‐associated SP, HSP usually has a slow progression and does not shorten patients’ life expectancy; therefore, a definite molecular diagnosis of familial PSEN1‐related disease has major implications for adequate counseling, healthcare provision, and access to treatments and services. We highlight the urgency to screen these patients for PSEN1 mutations, especially in the age of emerging anti‐amyloid disease‐modifying therapy. A delay in the treatment of familial AD, often by years, because of an initial presentation as HSP, could have a significant negative impact on the patient and their family. Therefore, we recommend that the mutations in PSEN1 should be considered in the genetic work‐up of all patients presenting with SP, especially when associated with cognitive impairment or in those with a family history of early onset dementia; and that, consequently, PSEN1 should be included in HSP diagnostic gene panels.
Our study highlights the clinical overlap between SP and PSEN1‐related AD. We show that over 7% of PSEN1 mutations present with pure HSP phenotype preceding onset of dementia by several years. These mutations were associated with cortical CWPs on neuropathological examination. Early screening of PSEN1 mutations would alleviate the need for further invasive testing, such as brain biopsy. Unraveling the phenotypic heterogeneity within PSEN1 mutation carriers could provide new potential targets for clinical trials of patients with familial AD.
CONFLICTS OF INTEREST
The authors declare no competing conflict of interest. M. Breza has received a research grant from European Academy of Neurology (EAN) and travel grants from Genesis Pharma, Teva‐ Specifar, Pfizer, Sanofi‐Genzyme. G. Koutsis has received research grants from Genesis Pharma and Teva; and consultation fees, advisory boards, and honoraria from Sanofi‐Genzyme, Genesis Pharma, Teva, Novartis, Roche, Merck, Pfizer, Specifar, and Integris Pharma. G. Karadima has received research grants from Pfizer and advisory board remuneration from Roche.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
We would like to thank the participants and their families for their essential help with this work. This work was supported by The Wellcome Trust (Synaptopathies strategic award (104033)), a Wellcome Trust Multi‐User Equipment Grant, Muscular Dystrophy UK (MD UK), MDC USA and The Medical Research Council (MRC UK International Centre and project grants), and the Spastic Paraplegia Foundation. Marianthi Breza is supported by a research grant from European Academy of Neurology (EAN). Georgios Koutsis would acknowledge support from Genesis Pharma (grant no. 13044) special account for research grants for the National and Kapodistrian University of Athens. John Hardy is supported by Medical Research Council (MR/N026004/1), Wellcome Trust Hardy (award number 202903/Z/16/Z), Dolby Family Fund, and by the UK Dementia Research Institute. Maria Szaruga and Lucía Chávez‐Gutiérrez would like to acknowledge support from Stichting Alzheimer Onderzoek (SAO), the Fund for Scientific Research, Flanders, and the Vlaams Institute voor Biotechnologie (VIB).
Chelban V, Breza M, Szaruga M, et al. Spastic paraplegia preceding PSEN1‐related familial Alzheimer's disease. Alzheimer's Dement. 2021;13:e12186. 10.1002/dad2.12186
Viorica Chelban, Marianthi Breza, Henry Houlden, and Georgios Koutsis authors contributed equally this study.
REFERENCES
- 1. Ryan NS, Nicholas JM, Weston PSJ, et al. Clinical phenotype and genetic associations in autosomal dominant familial Alzheimer's disease: a case series. Lancet Neurol. 2016;15(13):1326‐1335. [DOI] [PubMed] [Google Scholar]
- 2. Hebert LE, Weuve J, Scherr PA, Evans DA. Alzheimer disease in the United States (2010‐2050) estimated using the 2010 census. Neurology. 2013;80(19):1778‐1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. 2020 Alzheimer's disease facts and figures. Alzheimers Dement, 2020. [DOI] [PubMed] [Google Scholar]
- 4. Olsson B, Lautner R, Andreasson U, et al. CSF and blood biomarkers for the diagnosis of Alzheimer's disease: a systematic review and meta‐analysis. Lancet Neurol. 2016;15(7):673‐684. [DOI] [PubMed] [Google Scholar]
- 5. Finckh U, Kuschel C, Anagnostouli M, et al. Novel mutations and repeated findings of mutations in familial Alzheimer disease. Neurogenetics. 2005;6(2):85‐89. [DOI] [PubMed] [Google Scholar]
- 6. Wasco W, Pettingell WP, Jondro PD, et al. Familial Alzheimer's chromosome 14 mutations. Nat Med. 1995;1(9):848. [DOI] [PubMed] [Google Scholar]
- 7. Gallo M, Frangipane F, Cupidi C, et al. The novel PSEN1 M84V mutation associated to frontal dysexecutive syndrome, spastic paraparesis, and cerebellar atrophy in a dominant Alzheimer's disease family. Neurobiol Aging. 2017;56:213.e7‐213.e12.e12. [DOI] [PubMed] [Google Scholar]
- 8. Soosman SK, Joseph‐Mathurin N, Braskie MN, et al. Widespread white matter and conduction defects in PSEN1‐related spastic paraparesis. Neurobiol Aging. 2016;47:201‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lo Giudice T, Lombardi F, Santorelli FM, Kawarai T, Orlacchio A. Hereditary spastic paraplegia: clinical‐genetic characteristics and evolving molecular mechanisms. Exp Neurol. 2014;261:518‐539. [DOI] [PubMed] [Google Scholar]
- 10. Chelban V, Patel N, Vandrovcova J, et al. Mutations in NKX6‐2 cause progressive spastic ataxia and hypomyelination. Am J Hum Genet. 2017;100(6):969‐977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kara E, Tucci A, Manzoni C, et al. Genetic and phenotypic characterization of complex hereditary spastic paraplegia. Brain. 2016;139(Pt 7):1904‐1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kwok JBJ, Taddei K, Hallupp M, et al. Two novel (M233T and R278T) presenilin‐1 mutations in early‐onset Alzheimer's disease pedigrees and preliminary evidence for association of presenilin‐1 mutations with a novel phenotype. Neuroreport. 1997;8(6):1537‐1542. [DOI] [PubMed] [Google Scholar]
- 13. Karlstrom H, et al. Variable phenotype of Alzheimer's disease with spastic paraparesis. J Neurochem. 2008;104(3):573‐583. [DOI] [PubMed] [Google Scholar]
- 14. Montojo CA, Congdon E, Hwang L, et al. Neural mechanisms of response inhibition and impulsivity in 22q11.2 deletion carriers and idiopathic attention deficit hyperactivity disorder. Neuroimage Clin. 2015;9:310‐321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Steiner H, Fukumori A, Tagami S, Okochi M. Making the final cut: pathogenic amyloid‐beta peptide generation by gamma‐secretase. Cell Stress. 2018;2(11):292‐310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chávez‐Gutiérrez L, Bammens L, Benilova I, et al. The mechanism of gamma‐Secretase dysfunction in familial Alzheimer disease. Embo J. 2012;31(10):2261‐2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Szaruga M, Veugelen S, Benurwar M, et al. Qualitative changes in human gamma‐secretase underlie familial Alzheimer's disease. J Exp Med. 2015;212(12):2003‐2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Szaruga M, Munteanu B, Lismont S, et al. Alzheimer's‐causing mutations shift abeta length by destabilizing gamma‐secretase‐abetan interactions. Cell. 2017;170(3):443‐456.e14. [DOI] [PubMed] [Google Scholar]
- 19. Crook R, Verkkoniemi A, Perez‐Tur J, et al. A variant of Alzheimer's disease with spastic paraparesis and unusual plaques due to deletion of exon 9 of presenilin 1. Nat Med. 1998;4(4):452‐455. [DOI] [PubMed] [Google Scholar]
- 20. Houlden H, Baker M, Mcgowan E, et al. Variant Alzheimer's disease with spastic paraparesis and cotton wool plaques is caused by PS‐1 mutations that lead to exceptionally high amyloid‐beta concentrations. Ann Neurol. 2000;48(5):806‐808. [PubMed] [Google Scholar]
- 21. Chelban V, Carecchio M, Rea G, et al. MYORG‐related disease is associated with central pontine calcifications and atypical parkinsonism. Neurol Genet. 2020;6(2):e399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Auton A, Brooks LD, Durbin RM, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein‐coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285‐291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Petit D, Hitzenberger M, Lismont S, et al. Extracellular interface between APP and Nicastrin regulates Abeta length and response to gamma‐secretase modulators. Embo j. 2019;38(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bammens L, Chávez‐Gutiérrez L, Tolia A, Zwijsen An, De Strooper B. Functional and topological analysis of Pen‐2, the fourth subunit of the gamma‐secretase complex. J Biol Chem. 2011;286(14):12271‐12282. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26. Dumanchin C, Tournier I, Martin C, et al. Biological effects of four PSEN1 gene mutations causing Alzheimer disease with spastic paraparesis and cotton wool plaques. Hum Mutat. 2006;27(10):1063. [DOI] [PubMed] [Google Scholar]
- 27. Zhou R, Yang G, Guo X, Zhou Q, Lei J, Shi Y. Recognition of the amyloid precursor protein by human gamma‐secretase. Science. 2019(6428):363. [DOI] [PubMed] [Google Scholar]
- 28. Yang G, Zhou R, Zhou Q, et al. Structural basis of Notch recognition by human gamma‐secretase. Nature. 2019;565(7738):192‐197. [DOI] [PubMed] [Google Scholar]
- 29. Fernandez MA, Klutkowski JA, Freret T, Wolfe MS. Alzheimer presenilin‐1 mutations dramatically reduce trimming of long amyloid beta‐peptides (Abeta) by gamma‐secretase to increase 42‐to‐40‐residue Abeta. J Biol Chem. 2014;289(45):31043‐31052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nordström U, Maier E, Jessell TM, Edlund T. An early role for WNT signaling in specifying neural patterns of Cdx and Hox gene expression and motor neuron subtype identity. PLoS Biol. 2006;4(8):e252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lyoo CH, Cho H, Choi JY, et al. Tau accumulation in primary motor cortex of variant Alzheimer's disease with spastic paraparesis. J Alzheimers Dis. 2016;51(3):671‐675. [DOI] [PubMed] [Google Scholar]
- 32. Sodeyama N. Very early onset Alzheimer's disease with spastic paraparesis associated with a novel presenilin 1 mutation (Phe237Ile). J Neurol Neurosurg Psychiatry. 2001;71(4):556‐557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Smith MJ, Kwok JBJ, Mclean CA, et al. Variable phenotype of Alzheimer's disease with spastic paraparesis. Ann Neurol. 2001;49(1):125‐129. [DOI] [PubMed] [Google Scholar]
- 34. Mann DMA, Pickering‐Brown SM, Takeuchi A, Iwatsubo T. Amyloid angiopathy and variability in amyloid beta deposition is determined by mutation position in presenilin‐1‐linked Alzheimer's disease. Am J Pathol. 2001;158(6):2165‐2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Noelanders R, Vleminckx K. How Wnt signaling builds the brain: bridging development and disease. Neuroscientist. 2017;23(3):314‐329. [DOI] [PubMed] [Google Scholar]
- 36. González‐Fernández C, Mancuso R, Del Valle J, Navarro X, Rodríguez FJ. Wnt signaling alteration in the spinal cord of amyotrophic lateral sclerosis transgenic mice: special focus on Frizzled‐5 cellular expression pattern. PLoS One. 2016;11(5):e0155867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Haapasalo A, Kovacs DM. The many substrates of presenilin/gamma‐secretase. J Alzheimers Dis. 2011;25(1):3‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Novarino G, Fenstermaker AG, Zaki MS, et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science. 2014;343(6170):506‐511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Swerdlow RH, Miller BB, Lopes MBS, et al. Autosomal dominant subcortical gliosis presenting as frontotemporal dementia. Neurology. 2009;72(3):260‐267. [DOI] [PubMed] [Google Scholar]
- 40. Rademakers R, Baker M, Nicholson AM, et al. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat Genet. 2011;44(2):200‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nicholson AM, Baker MC, Finch NA, et al. CSF1R mutations link POLD and HDLS as a single disease entity. Neurology. 2013;80(11):1033‐1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Srour M, Riviere JB, Pham JMT, et al. Mutations in DCC cause congenital mirror movements. Science. 2010;328(5978):592. [DOI] [PubMed] [Google Scholar]
- 43. Jamuar SS, Schmitz‐Abe K, D'gama AM, et al. Biallelic mutations in human DCC cause developmental split‐brain syndrome. Nat Genet. 2017;49(4):606‐612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Takahashi Y, Fukuda Y, Yoshimura J, et al. ERBB4 mutations that disrupt the neuregulin‐ErbB4 pathway cause amyotrophic lateral sclerosis type 19. Am J Hum Genet. 2013;93(5):900‐905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dixon‐Salazar TJ, Silhavy JL, Udpa N, et al. Exome sequencing can improve diagnosis and alter patient management. Sci Transl Med. 2012;4(138):138ra78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Boycott KM, Flavelle S, Bureau A, et al. Homozygous deletion of the very low density lipoprotein receptor gene causes autosomal recessive cerebellar hypoplasia with cerebral gyral simplification. Am J Hum Genet. 2005;77(3):477‐483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Heilig EA, Xia W, Shen J, Kelleher RJ. A presenilin‐1 mutation identified in familial Alzheimer disease with cotton wool plaques causes a nearly complete loss of gamma‐secretase activity. J Biol Chem. 2010;285(29):22350‐22359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mastrangelo P, Mathews PM, Chishti MA, et al. Dissociated phenotypes in presenilin transgenic mice define functionally distinct gamma‐secretases. Proc Natl Acad Sci U S A. 2005;102(25):8972‐8977. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information