

Abstract
Purpose
The ectodermal dysplasias (EDs) constitute a group of disorders characterized by abnormalities in two or more ectodermal derivatives, including skin, hair, teeth, and sweat glands. The purpose of the current study was to evaluate ocular manifestations in pediatric patients with ED.
Methods
Retrospective case series including consecutive ED subjects who were treated in the ophthalmology department at the Children’s Hospital of Philadelphia over a 12-year period (2009–2020). Main Outcome Measures were ocular and ocular adnexal abnormalities.
Results
Thirty subjects were included: 20 males (67%), mean age of 4.5 years (range 0.3–18). Patients with different subtypes were included, with the hypohidrotic ED and ectrodactyly-ectodermal dysplasia-clefting variants being most prevalent. Most common findings were: lacrimal drainage obstruction in 12 (40%) including punctal agenesis in 10 (33%), refractive errors in 13 (43%) and amblyopia in 6 (20%). A new finding of eyelid ptosis or eyelash ptosis was demonstrated in 11 subjects (37%), mostly associated with TP63 or EDA1 genes variants.
Conclusion
Ectodermal dysplasias are associated with various ocular pathologies and amblyopia in the pediatric population. We report a possible genetic association between lash ptosis and EDA1 gene, and eyelid ptosis and TP63 or EDA1 genes variants.
Keywords: Ectodermal dysplasia, Ptosis, Lash ptosis, EDA1, TP63, Ectrodactyly-ectodermal dysplasia-clefting, EEC, Ankyloblepharon-ectodermal defects-cleft lip/palate, AEC
Introduction
Ectodermal dysplasias (ED) are genetic conditions affecting the development and/or homeostasis of two or more ectodermal derivatives, including skin, hair, teeth, nails, and sweat glands [1]. They constitute a large and diverse group of over 200 disorders, heterogeneous in their genetic causes and clinical phenotypes, with a variable range of reported prevalence [1–6]. The anomalies affecting the epidermis and epidermal appendages are extremely variable; many are associated with malformations in other organs and systems, thus management usually requires a multidisciplinary approach [4].
Several EDs were reported to have ocular abnormalities; however, few large cohort studies focus on ocular manifestations of these conditions [1, 4–13]. The purpose of the current study was to describe the ocular phenotype including new findings in a large cohort of pediatric subjects with various ED disorders.
Methods
Retrospective analysis of all consecutive ED subjects who were treated at the ophthalmology department in the Children’s Hospital of Philadelphia in a 12-year period (2009–2020) was performed. Data collected included demographics, clinical photographs, symptoms, diagnosis, genetics, and management. Statistical analysis was carried out using Microsoft Excel (Microsoft, Redmond, WA, USA). The described research adhered to the tenets of the Declaration of Helsinki. The study and data accumulation were carried out with approval from the Institutional Review Board (IRB).
Results
Thirty subjects were studied: 20 male (67%) and 10 females (33%), with a mean age of 4.5 years at first visit (range 0.3 to 18 years), and a mean follow up time of 4.0 years (range 0–17.8). Diagnoses were as follows: hypohidrotic ED in 8 patients (HED) (27% of all patients), hidrotic ED (n = 2/30, 7%), ectrodactyly-ectodermal dysplasia-clefting (EEC) (n = 8/30, 27%), ankyloblepharon-ectodermal defects-cleft lip/palate (AEC) (n = 3/30, 10%), Rapp-Hodgkin ED (n = 2/30, 7%), Marshall syndrome (n = 2/30, 7%), unspecified ED (n = 2/30, 7%), ED with immunodeficiency (n = 1/30, 3%), tricho-dento-osseous syndrome (n = 1/30, 3%), and oculo-ectodermal syndrome (n = 1/30, 3%).
The most common presenting symptoms were tearing (14 subjects, 47%), and photophobia (6 subjects, 20%). Visual acuity was appropriate for age in each eye for 24 children (n = 24/30, 80%). Thirteen subjects (n = 13/30, 43%) had refractive errors requiring spectacles. Six children (n = 6/30, 20%) had amblyopia with a visual acuity in the range of 20/25-20/600. Among those children, 4 (n = 4/30, 13%) had strabismus.
The most common finding was lacrimal drainage obstruction (n = 12/30, 40%), and punctal agenesis was found in 10 of these 12 subjects (n = 10/30, 33%); these were most prevalent in EEC cases (Fig. 1). Additional findings were: dry eye (n = 4/30, 13%), blepharitis (n = 3/30, 10%), allergic conjunctivitis (n = 3/30, 10%), cataracts (n = 2/30, 7%), distichiasis with trichiasis and recurrent corneal abrasions (n = 2/30, 7%) (Fig. 2). Posterior embryotoxon, posterior pole osteomas, and bilateral peripapillary colobomas were observed in a patient with oculo-ectodermal syndrome. A patient with Marshall syndrome had shallow orbits and hypertelorism on clinical impression and increased axial length, giving the appearance of lid retraction and proptosis (Fig. 3).
Fig. 1.
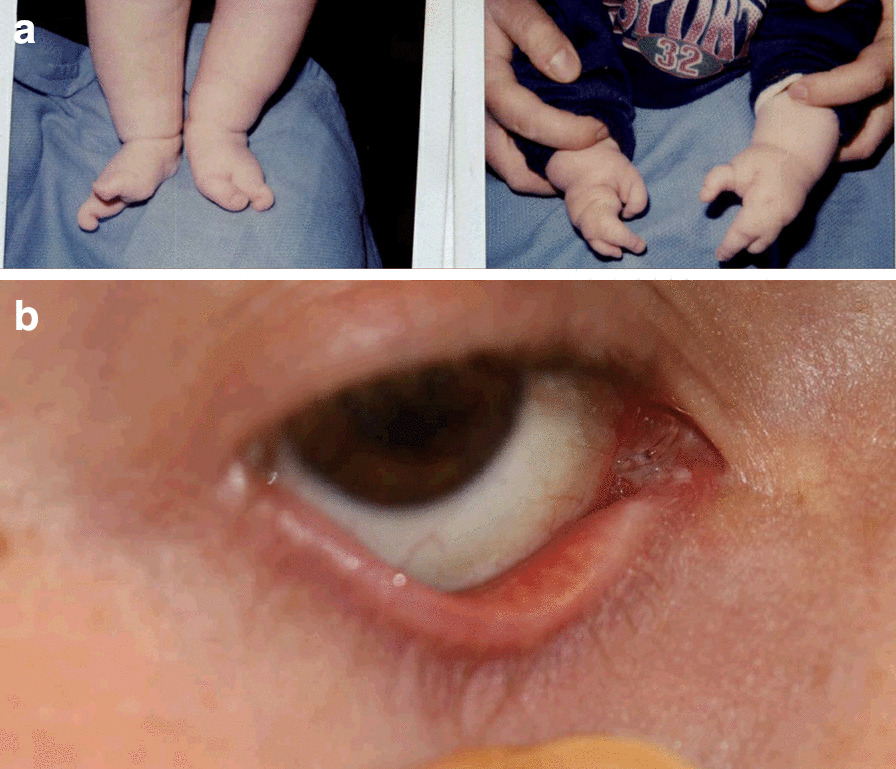
A male patient with Ectrodactyly-ectodermal dysplasia-clefting (EEC) syndrome. He had constant epiphora due to right sided punctal agenesis that resolved with CJDCR at the age of 18 years. Systemic disorders included cleft lip and palate, syndactyly, dental abnormalities, midface hypoplasia, and hearing disorders. a The patient at infancy, showing ectrodactyly. b Same patient, at age 18 years, showing absent lower punctum on the right side (punctal agenesis), and a Lester-Jones tube
Fig. 2.
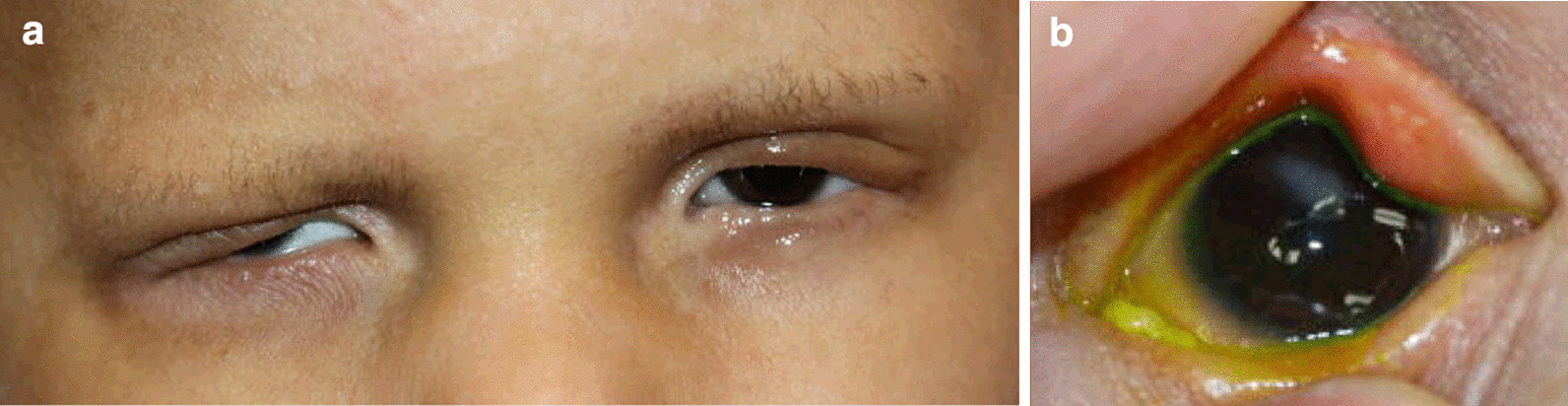
A 13-year-old male patient with Ankyloblepharon-ectodermal defects-cleft lip/palate (AEC) syndrome. The subject was extremely photophobic due to distichiasis and trichiasis (a) causing recurrent corneal erosions and subsequent right eye corneal scarring (b)
Fig. 3.
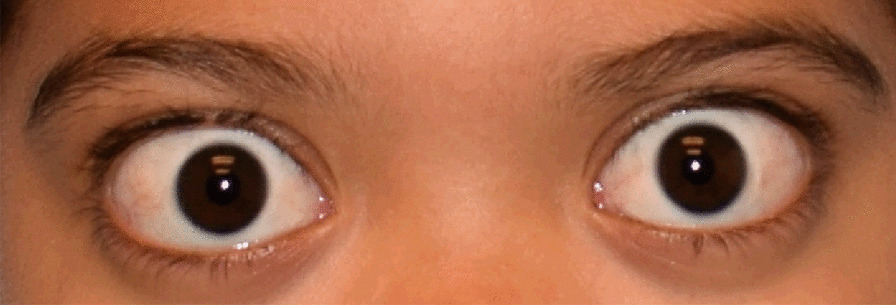
A 10-year-old male with Marshall syndrome, with high myopia (− 20D), shallow orbits and hypertelorism. The patient developed bilateral posterior subcapsular cataracts, had PPV lensectomies with prophylactic peripheral retinal laser ablation at the age of 4 years, and was left aphakic. Final visual acuity at age 10 was 20/60 OD, 20/80 OS
Twelve subjects (40%) required surgical intervention. Lacrimal drainage surgery was performed in 7 (23%) subjects, including dacryocystorhinostomy (DCR) in 4 (13%) or conjunctivodacryocystorhinostomy (CJDCR) with Jones tube placement in 3 (10%). Eyelid surgery was performed in 6 children (20%). This involved the repair of trichiasis (2), ankyloblepharon (2), ptosis (1), and lesion biopsies (1). Bilateral cataract extraction was performed in one child with Marshall syndrome. Demographics of study population, clinical presentations, and surgical interventions are summarized in Table 1.
Table 1.
Demographics, clinical presentations, and surgical interventions of 30 pediatric subjects with Ectodermal Dysplasia
| All subtypes (total) | Unspecified ED, hidrotic and hypohidrotic | EEC | AEC | Rapp-Hodgkin ED | Marshall’s syndrome | HED with immunodeficiency | Tricho-dento-osseous syndrome | Oculo-ectodermal syndrome | |
|---|---|---|---|---|---|---|---|---|---|
| Number of subjects | 30 | 12 | 8 | 3 | 2 | 2 | 1 | 1 | 1 |
| Gender (M:F) | 20:10 | 8:4 | 6:2 | 3:0 | 0:2 | 2:0 | 1:0 | 0:1 | 0:1 |
| Age at first visit, years | 4.5 | 5.4 | 3 | 4.4 | 0.5 | 2.7, 0.4 | 3 | 9.5 | 12.5 |
| Genetic variant (n of tests) | EDA1 (6) | TP63 (3) | TP63 (2) | TP63 (2) | COL11-A1 (1) | NEMO (1) | DLX3 & COL1A1 (1) | KRAS (1) | |
| Symptoms (n of subjects, %) | |||||||||
| Tearing/discharge | 14, 47% | 4, 33% | 7, 87% | 1, 33 | 1 | 1 | – | – | – |
| Photophobia | 6, 20% | 2, 17% | 1, 14% | 2, 67% | 1 | – | – | – | – |
| Diagnoses (n of subjects, %) | |||||||||
| Lacrimal disorders | 12, 40% | 2, 17% | 7, 87% | 2, 67% | 1 | – | – | – | – |
| Punctal agenesis | 10, 33% | 2, 17% | 6, 62% | 1, 33% | 1 | – | – | – | – |
| Dry eye | 4, 13% | 1, 8% | 1, 14% | 1, 33% | 1 | – | – | – | – |
| Strabismus | 4, 13% | 1, 8% | – | 1, 33% | – | – | – | – | – |
| Blepharitis | 3, 10% | 1, 8% | 2, 29% | – | – | – | – | – | – |
| With MGD | 2, 6.7% | 1, 8% | 1, 14% | – | – | – | – | – | – |
| Allergic conjunctivitis | 3, 10% | 2, 17% | – | 1, 33% | – | – | – | – | – |
| Distichiasis and trichiasis | 2, 6.7% | – | – | 2, 67% | – | – | – | – | – |
| Cataract | 2, 6.7% | 1, 8% | – | – | – | 1 | – | – | – |
| Ptosis/lash ptosis | 11,37% | 5, 42% | 3, 43% | 2, 67% | – | – | – | – | 1 |
| Isolated lash ptosis | 3, 10% | 2, 17% | 1, 14% | – | – | – | – | – | – |
| Isolated ptosis | 4, 13% | 1, 8% | – | 2, 67% | – | – | – | – | 1 |
| Combined ptosis & lash ptosis | 4, 13% | 2, 17% | 2, 29% | – | – | – | – | – | – |
| Spectacles | 13, 43% | 5, 42% | 3, 43% | 2, 67% | – | 1 | – | 1 | 1 |
| Amblyopia | 6, 20% | 2, 17% | 1, 14% | 1, 33% | – | 1 | – | – | 1 |
| Other | 1, 3% | – | – | – | – | – | – | – | 1a |
| Surgical intervention (n of subjects, %) | 12, 40% | 1, 8% | 6, 75% | 3, 100% | 1 | 1 | – | – | 1 |
| Lacrimal repairs | 7, 23% | – | 5, 62% | 1, 33% | 1 | – | – | – | – |
| Trichiasis repair | 2, 6.7% | – | – | 2, 67% | – | – | – | – | – |
| Ankyloblepharon repair | 2, 6.7% | – | – | 2, 67% | – | – | – | – | – |
| Ptosis repair | 1, 3% | – | – | – | – | – | – | – | 1 |
| Cataract extraction | 1, 3% | – | – | – | – | 1 | – | – | - |
| Other | 2, 3% | 1, 8%b | - | - | - | 1 | - | - | 1c |
ED ectodermal dysplasia, HED hypohidrotic ED, EEC ectrodactyly-ectodermal dysplasia-clefting syndrome, AEC ankyloblepharon-ectodermal defects-cleft lip/palate syndrome, MGD meibomian gland dysfunction
aPosterior embryotoxon, posterior pole osteomas, and bilateral peripapillary colobomas
bExamination under anesthesia and punctal plugs
cOcular surface lesions biopsies
Eyelid abnormalities
Lash ptosis or ptosis were present in clinical photographs of 11 subjects (n = 11/30, 37%) at a mean age of 10 years. Four subjects had both entities, 3 had isolated lash ptosis, and 4 had isolated ptosis. Among the seven subjects with lash ptosis, genetic testing was available for three HED subjects, and were all positive for pathogenic EDA1 gene variants. Among the subjects with primary eyelid ptosis, genetic testing was available for two subjects; one had HED with confirmed pathogenic EDA1 gene variant (Fig. 4), and the other had AEC with confirmed TP63 pathogenic variant.
Fig. 4.
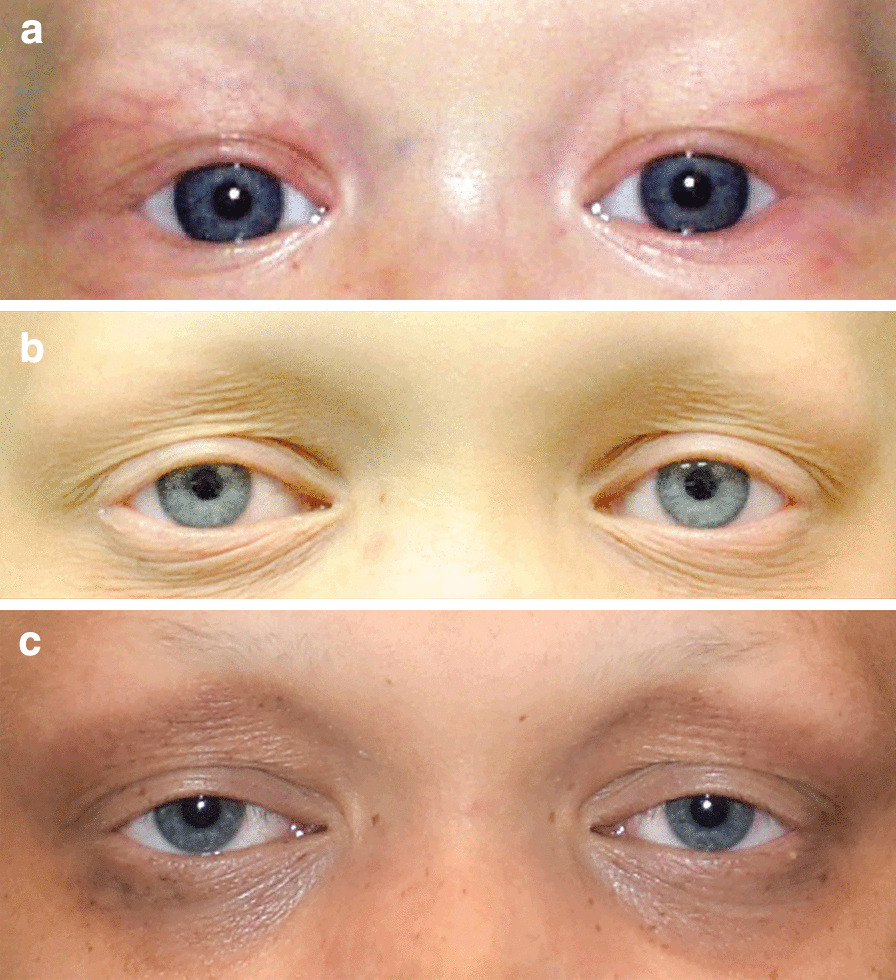
A male subject with hypohidrotic Ectodermal Dysplasia (HED) and EDA1 gene variant, demonstrating bilateral acquired ptosis and lash ptosis. Note the typical ED presentation with light and sparse eyelashes and eyebrow hair (hypotrichosis), and progressive periocular skin changes. a The patient at age 5 months, with normal eyelid position. b Age 10, demonstrating mild bilateral ptosis, lash ptosis, and periocular skin changes. c Age 14, with progression of his bilateral ptosis, lash ptosis, and periocular pigmentary skin changes
Discussion
Ectodermal dysplasias are genetically determined developmental defects of tissues of ectodermal origin, heterogeneous in their genetic causes and clinical phenotypes. During embryonic development, the ectoderm gives rise to the epidermis, the central and peripheral nervous system, the placodes, and neural crest cells [14]. Disruption of these tissue and cell types causes the clinical spectrum characteristic of this group of disorders. Known ocular findings in EDs include scant eyebrows and eyelashes, lacrimal gland hypoplasia, dry eyes, and blepharitis [2, 15].
Several case series focused on ocular findings in ED [6, 11–13, 16]. In the largest series, published in 2004, Kaercher [6] described a case series of 36 subjects with confirmed ED which included 30 subjects with HED, EEC syndrome (3), AEC syndrome (2), and Gorlin–Goltz syndrome (1). The author found a high percentage of dry eye (94%), eyebrows diminution (94%), lashes alterations (92%), and meibomian gland alterations (95%) detected by meibomianoscopy, and concluded that meibomian gland alterations are a reliable ocular sign of ED.
The second-largest series was published by Keklikci et al. [13] in 2014 and described 24 ED subjects at a mean age of 15.8 years (range 3–45), including HED (21 subjects) and EEC (2). Eighteen subjects (75%) suffered from ocular complaints related to the ocular surface. Eleven subjects (46%) reported irritation, tearing, epiphora, photophobia, redness, and recurrent inflammations of the lids; these were attributed to dry eye. The authors concluded that ocular complaints, particularly dry eye symptoms, may increase as age advances.
These series, as well as others, emphasized the ocular surface findings in ED, including various keratopathies, meibomian gland disorders, and dry eye [6, 12, 17, 18]. In our series, tearing secondary to lacrimal disorders including punctal agenesis was the most prevalent symptom, especially in TP63-related disorders (EEC, AEC, RHS), followed by photophobia secondary to dry eye, trichiasis, blepharitis, and corneal disorders.
Less than 20% of the subjects were diagnosed with dry eye, blepharitis or meibomian gland abnormalities, and no corneal dystrophies were diagnosed. These differences could be explained by the younger age of our subjects, making the history information, ocular examination and auxiliary tests more challenging. In addition, as some of these disorders develop or progress with age, these changes would be expected to be more prevalent in adulthood [13].
Oculo-ectodermal syndrome (OES)
OES, also named Toriello Lacassie Droste syndrome, was first described by Toriello et al. [19] in 1993. About 20 cases have been reported in the literature [20, 21]. It is caused by somatic variants in the KRAS gene on chromosome [12, 21]. It is characterized by epibulbar dermoids and cutis aplasia congenital [22]. Phenotypic expression is highly variable, and various other abnormalities have occasionally been reported, including growth failure, lymphedema, cardiovascular defects, neurodevelopmental symptoms, non-ossifying fibromas of the long bones, giant cell granulomas of the jaws, arachnoid cysts in the brain, seizure disorder, hyperpigmented nevi, and rhabdomyosarcoma [20, 23].
In addition to unilateral or bilateral epibulbar dermoids, ocular anomalies such as upper eyelid skin tags, corneal opacities, hyperopia and astigmatism, strabismus, and microphthalmia can be present [21, 22, 24]. Gardner and Viljeon [25] described an affected patient with a small optic disc on the right and a large optic disc with abnormal retinal pigmentation on the left. Boppudi et al. [21] described a case with deeply set eyes and a narrow intercanthal distance, while Toriello [19] described a child with chorioretinal atrophy, prominent eyes and strabismus.
In addition to epibulbar dermoids, our subject’s ocular history included posterior embryotoxon, posterior pole osteomas, and bilateral peripapillary colobomas. These findings have not been previously described in patients with OES. Interestingly, optic nerve coloboma has been described in Encephalo‐cranio‐cutaneous lipomatosis (ECCL) [26], which supports the notion that OES may be a mild variant of ECCL [22]; furthermore, that supports an association between optic coloboma and OES.
Eyelid malposition associated with ectodermal dysplasias
It is difficult to strictly classify the origin of individual eyelid structures into an ectodermal or mesenchymal origin [27]. Generally, the surface ectoderm gives rise to the conjunctiva, skin epithelium, hair follicles, Zeis glands, glands of Moll, and meibomian glands; the Levator aponeurosis, being of neural crest origin, is another ectodermal derivative [27]. The tarsal plate, levator muscle, orbicularis muscle, orbital septum, and tarsal muscle of Müller develop from the mesenchyme [27].
It has been advocated that gene expression in the EDs is not limited to the ectoderm and that there is a concomitant effect on developing mesenchymal structures [4, 5]. The involvement of both ectodermal and mesodermal structures in the pathogenesis of these disorders may explain the various eyelid and eyelashes abnormalities demonstrated in this series, including the high portion (37%) of eyelid ptosis and/or lash ptosis.
Ptosis
Ptosis has been anecdotally described in ED. Examples include Jackson and Barr (1978) [28] who described 2 sisters with ED and ptosis. Zanolli et al. [29] described ED with signs of both ectodermal and mesodermal dysplasia, associated with ptosis. Ptosis was also described in a pediatric case of Goltz syndrome, a rare ED subtype, but this was concomitant with bilateral microcorneas, microphthalmos, and iris colobomas [30]. A paper stated that ptosis is associated with AEC, but no relevant reference was recognized [2]. Salinas et al. [31] suggested ptosis as one of the manifestations of Rapp-Hodgkin syndrome.
Our findings suggest that the rate of ptosis in ED subjects, especially those with TP63 and EDA1 gene variants, is higher than previously demonstrated, and that such genetic associations might exist. The ptosis was noted to be acquired rather than congenital (Fig. 4). Progressive decrease in anterior lamellar tone or levator dehiscence may be the cause of eyelid malposition in our series. Actual entropion due to tarsoconjunctival contraction was not noted, however, in this series.
Lash ptosis
A unique observation was the high rate of lash ptosis, especially given the infrequency of this observation in the pediatric population. As previously discussed, eyelash disorders are associated with EDs, including sparse and thin lashes, pseudodistichiasis, and trichiasis [6, 32] However, lash ptosis was not previously described in ED subjects. This subtle diagnosis can be easily overlooked, especially with the HED pediatric subjects with their sparse, light eyelashes. We report a possible genetic association between the EDA1 gene and lash ptosis.
In summary, EDs are associated with various ocular and ocular adnexal abnormalities. In this series, several new findings were observed, including increased risk of amblyopia in most subtypes, as well as eyelid ptosis and lash ptosis in subtypes with TP63 or EDA1 gene variants. In addition, trichiasis may be more common in AEC than previously described, and posterior pole osteomas and peripapillary colobomas may be found with OES. These various findings mandate early ophthalmic evaluation in this unique group of children.
Acknowledgements
None.
Authors' contributions
All authors contributed to design, data collection and analysis, writing, and review. All authors read and approved the final manuscript.
Funding
None.
Availability of data and materials
Data available on request.
Declarations
Ethics approval and consent to participate
The study and data accumulation were carried out with approval from the Institutional Review Board (IRB). Data collected in a retrospective manner.
Consent for publication
Not applicable.
Competing interests
None.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Wright JT, Fete M, Schneider H, et al. Ectodermal dysplasias: Classification and organization by phenotype, genotype and molecular pathway. Am J Med Genet A. 2019;179:442–447. doi: 10.1002/ajmg.a.61045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jen M, Nallasamy S. Ocular manifestations of genetic skin disorders. Clin Dermatol. 2016;34:242–275. doi: 10.1016/j.clindermatol.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 3.Ansari A, Pillarisetty LS. Embryology, Ectoderm. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2020. Available at: http://www.ncbi.nlm.nih.gov/books/NBK539836/ [Accessed June 1, 2020]. [PubMed]
- 4.Priolo M. Ectodermal dysplasias: an overview and update of clinical and molecular-functional mechanisms. Am J Med Genet A. 2009;149A:2003–2013. doi: 10.1002/ajmg.a.32804. [DOI] [PubMed] [Google Scholar]
- 5.Itin PH, Fistarol SK. Ectodermal dysplasias. Am J Med Genet C Semin Med Genet. 2004;131C:45–51. doi: 10.1002/ajmg.c.30033. [DOI] [PubMed] [Google Scholar]
- 6.Kaercher T. Ocular symptoms and signs in patients with ectodermal dysplasia syndromes. Graefes Arch Clin Exp Ophthalmol. 2004;242:495–500. doi: 10.1007/s00417-004-0868-0. [DOI] [PubMed] [Google Scholar]
- 7.Nguyen-Nielsen M, Skovbo S, Svaneby D, et al. The prevalence of X-linked hypohidrotic ectodermal dysplasia (XLHED) in Denmark, 1995–2010. Eur J Med Genet. 2013;56:236–242. doi: 10.1016/j.ejmg.2013.01.012. [DOI] [PubMed] [Google Scholar]
- 8.Pinheiro M, Freire-Maia N. Ectodermal dysplasias: a clinical classification and a causal review. Am J Med Genet. 1994;53:153–162. doi: 10.1002/ajmg.1320530207. [DOI] [PubMed] [Google Scholar]
- 9.Fete M, vanBokhoven H, Clements SE, et al. International research symposium on ankyloblepharon-ectodermal defects-cleft lip/palate (AEC) syndrome. Am J Med Genet A. 2009;149A:1885–1893. doi: 10.1002/ajmg.a.32761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lamartine J. Towards a new classification of ectodermal dysplasias. Clin Exp Dermatol. 2003;28:351–355. doi: 10.1046/j.1365-2230.2003.01319.x. [DOI] [PubMed] [Google Scholar]
- 11.Chandravanshi SL. Hypohidrotic ectodermal dysplasia: a case report. Orbit 2019:1–4. [DOI] [PubMed]
- 12.Mawhorter LG, Ruttum MS, Koenig SB. Keratopathy in a family with the ectrodactyly-ectodermal dysplasia-clefting syndrome. Ophthalmology. 1985;92:1427–1431. doi: 10.1016/s0161-6420(85)33847-2. [DOI] [PubMed] [Google Scholar]
- 13.Keklikci U, Yavuz I, Tunik S, et al. Ophthalmic manifestations in patients with ectodermal dysplasia syndromes. Adv Clin Exp Med. 2014;23:605–610. doi: 10.17219/acem/37235. [DOI] [PubMed] [Google Scholar]
- 14.Wright JE, Sullivan TJ, Garner A, et al. Orbital venous anomalies. Ophthalmology. 1997;104:905–913. doi: 10.1016/s0161-6420(97)30208-5. [DOI] [PubMed] [Google Scholar]
- 15.McNab AA, Potts MJ, Welham RA. The EEC syndrome and its ocular manifestations. Br J Ophthalmol. 1989;73:261–264. doi: 10.1136/bjo.73.4.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tyagi P, Tyagi V, Hashim AA. Ocular and non-ocular manifestations of hypohidrotic ectodermal dysplasia. BMJ Case Rep 2011;2011. [DOI] [PMC free article] [PubMed]
- 17.Messmer EM, Kenyon KR, Rittinger O, et al. Ocular manifestations of keratitis-ichthyosis-deafness (KID) syndrome. Ophthalmology. 2005;112:e1–6. doi: 10.1016/j.ophtha.2004.07.034. [DOI] [PubMed] [Google Scholar]
- 18.Mondino BJ, Bath PE, Foos RY, et al. Absent meibomian glands in the ectrodactyly, ectodermal dysplasia, cleft lip-palate syndrome. Am J Ophthalmol. 1984;97:496–500. doi: 10.1016/s0002-9394(14)76134-3. [DOI] [PubMed] [Google Scholar]
- 19.Toriello HV, Lacassie Y, Droste P, Higgins JV. Provisionally unique syndrome of ocular and ectodermal defects in two unrelated boys. Am J Med Genet. 1993;45:764–766. doi: 10.1002/ajmg.1320450620. [DOI] [PubMed] [Google Scholar]
- 20.Figueiras DDA, Leal DMDCB, Kozmhinsky V. Oculoectodermal syndrome: twentieth described case with new manifestations. Ann Bras Dermatol. 2016;91:160–162. doi: 10.1590/abd1806-4841.20164409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boppudi S, Bögershausen N, Hove HB, et al. Specific mosaic KRAS mutations affecting codon 146 cause oculoectodermal syndrome and encephalocraniocutaneous lipomatosis. Clin Genet. 2016;90:334–342. doi: 10.1111/cge.12775. [DOI] [PubMed] [Google Scholar]
- 22.Ardinger HH, Horii KA, Begleiter ML. Expanding the phenotype of oculoectodermal syndrome: possible relationship to encephalocraniocutaneous lipomatosis. Am J Med Genet A. 2007;143A:2959–2962. doi: 10.1002/ajmg.a.31969. [DOI] [PubMed] [Google Scholar]
- 23.Habib F, Elsaid MF, Salem KY, et al. Oculo-ectodermal syndrome: a case report and further delineation of the syndrome. Qatar Med J. 2014;2014:114–122. doi: 10.5339/qmj.2014.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hunter AGW. Oculocerebrocutaneous and encephalocraniocutaneous lipomatosis syndromes: blind men and an elephant or separate syndromes? Am J Med Genet A. 2006;140:709–726. doi: 10.1002/ajmg.a.31149. [DOI] [PubMed] [Google Scholar]
- 25.Gardner J, Viljoen D. Aplasia cutis congenita with epibulbar dermoids: further evidence for syndromic identity of the ocular ectodermal syndrome. Am J Med Genet. 1994;53:317–320. doi: 10.1002/ajmg.1320530403. [DOI] [PubMed] [Google Scholar]
- 26.Chacon-Camacho OF, Lopez-Moreno D, Morales-Sanchez MA, et al. Expansion of the phenotypic spectrum and description of molecular findings in a cohort of patients with oculocutaneous mosaic RASopathies. Mol Genet Genom Med. 2019;7:e625. doi: 10.1002/mgg3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tawfik HA, Abdulhafez MH, Fouad YA, Dutton JJ. Embryologic and fetal development of the human eyelid. Ophthal Plast Reconstr Surg. 2016;32:407–414. doi: 10.1097/IOP.0000000000000702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jackson LG, Barr MA. Conductive deafness with ptosis and skeletal malformations in sibs: a probably autosomal recessive disorder. Birth Defects Orig Artic Ser. 1978;14:199–204. [PubMed] [Google Scholar]
- 29.Zannolli R, Inchingolo G, Serracca L, et al. Ectodermal dysplasia syndrome with eyebrow alopecia, ptosis, strabismus, nystagmus, joint laxity, cerebellar ataxia, and osteopenia. Am J Med Genet. 2002;113:111–113. doi: 10.1002/ajmg.10713. [DOI] [PubMed] [Google Scholar]
- 30.Rishi E, Sodhi P, Swaminathan M, Rishi P. Goltz syndrome: Primary diagnosis by an ophthalmologist. Indian J Ophthalmol. 2019;67:1467–1468. doi: 10.4103/ijo.IJO_724_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salinas CF, Montes GM. Rapp-Hodgkin syndrome: observations on ten cases and characteristic hair changes (pili canaliculi) Birth Defects Orig Artic Ser. 1988;24:149–168. [PubMed] [Google Scholar]
- 32.Howard GR, Wilson ME. Pseudodistichiasis as a manifestation of anhidrotic ectodermal dysplasia. J Pediatr Ophthalmol Strabismus. 1993;30:204–205. doi: 10.3928/0191-3913-19930501-17. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data available on request.