

Abstract
High-throughput Plasmodium genomic data is increasingly useful in assessing prevalence of clinically important mutations and malaria transmission patterns. Understanding parasite diversity is important for identification of specific human or parasite populations that can be targeted by control programs, and to monitor the spread of mutations associated with drug resistance. An up-to-date understanding of regional parasite population dynamics is also critical to monitor the impact of control efforts. However, this data is largely absent from high-burden nations in Africa, and to date, no such analysis has been conducted for malaria parasites in Tanzania country-wide. To this end, over 1,000 P. falciparum clinical isolates were collected in 2017 from 13 sites in seven administrative regions across Tanzania, and parasites were genotyped at 1,800 variable positions genome-wide using molecular inversion probes. Population structure was detectable among Tanzanian P. falciparum parasites, roughly separating parasites from the northern and southern districts and identifying genetically admixed populations in the north. Isolates from nearby districts were more likely to be genetically related compared to parasites sampled from more distant districts. Known drug resistance mutations were seen at increased frequency in northern districts (including two infections carrying pfk13-R561H), and additional variants with undetermined significance for antimalarial resistance also varied by geography. Malaria Indicator Survey (2017) data corresponded with genetic findings, including average region-level complexity-of-infection and malaria prevalence estimates. The parasite populations identified here provide important information on extant spatial patterns of genetic diversity of Tanzanian parasites, to which future surveys of genetic relatedness can be compared.
Keywords: malaria, Plasmodium falciparum, population structure, molecular inversion probes, isolation-by-distance, drug resistance, Tanzania
INTRODUCTION
Understanding how pathogen populations change over space and time provides key information for public health surveillance efforts. Infectious diseases can have complex spatial patterns influenced by geographic barriers and host movement (Real et al. 2005; Vora, Burke, and Cummings 2008; Takehisa et al. 2009). Genetic data is increasingly used as a tool for infectious disease epidemiology and ecology for explaining heterogeneity in disease prevalence, and to identify how genetic structure forms in pathogen populations. Such data is also extremely useful for complex disease systems such as vector-borne diseases (Hemming-Schroeder et al. 2018), where interactions of host, vector, and pathogen are often affected by unique environmental variables (Kent et al. 2007; Massaro, Kondor, and Ratti 2019; Barbu et al. 2013), and where pathogen movement is mediated by both vector (Bataille et al. 2009; Egizi et al. 2016) and host (Wesolowski et al. 2012; Dalziel, Pourbohloul, and Ellner 2013) mobility.
Human malaria, caused by infections with members of the genus Plasmodium, is one example of a disease with complex transmission dynamics influenced by both the Anopheline vector (Thomson et al. 1995; Huestis et al. 2019) and human host mobility (Wesolowski et al. 2012; Guerra et al. 2019). While global malaria morbidity and mortality has decreased over the past two decades, malaria remains entrenched in key areas. Currently, 70% of the world’s malaria cases occur in 11 countries (“World Malaria Report” 2019). These “high burden, high impact” nations have recently become the subject of more focused malaria control efforts. Shifting control priorities potentially require new tools to help monitor and inform public health programs, and could benefit from the recent advances in integrating genomic and epidemiologic data in malaria research (Wesolowski et al. 2018; S. K. Tessema et al. 2019).
Understanding parasite dynamics underlying malaria in high transmission regions is critical for ongoing malaria elimination initiatives in sub-Saharan Africa. Monitoring parasite population structure and genomic signatures of selection can offer insights on the impact that interventions have on parasite populations (Daniels et al. 2015), the spread of mutations associated with drug resistance (Hamilton et al. 2019), and can identify transmission patterns reflecting human movement (S. Tessema et al. 2019). Recently, high-throughput genomics have elucidated important geographic (Miotto et al. 2013; Takala-Harrison et al. 2015) and temporal (Amato et al. 2018; Cerqueira et al. 2017) fluctuations of parasite populations, as well as fine-scale relatedness across short distances (Taylor et al. 2017), in Southeast Asia. While these analyses have shown within-country dynamics in regions of relatively low transmission, it is unclear how these insights translate to high transmission settings of Africa, home to 10/11 “high burden” countries. Also lacking are thorough baseline genetic characterizations of circulating parasites in these regions, to which subsequent analyses can be compared. Several studies have managed to identify between-country genetic signatures utilizing whole genome sequence data across sub-Saharan Africa (Amambua-Ngwa et al. 2012; Ocholla et al. 2014; Duffy et al. 2018; Amambua-Ngwa et al. 2019), but a lack of thorough within-country sampling and reliance on whole genome sequencing (which can be cost-prohibitive for large surveys) has limited further exploration of population genetics in Africa.
Recently, molecular inversion probes (MIPs) have been adapted for high-throughput characterization of Plasmodium falciparum clinical infections; these assays allow for the rapid characterization of tens to thousands of positions across the genome. MIPs have successfully been used to track the spread of drug resistance mutations (Aydemir et al. 2018; Deutsch-Feldman et al. 2019; Mensah et al. 2020) in several African countries and to describe parasite population structure within the Democratic Republic of Congo (a high burden nation) (Verity et al. 2020). Expanding MIPs to explore these trends in other high transmission settings in Africa is an efficient way to establish contemporary genetic trends, and can provide ongoing surveillance of circulating parasite populations. Such characterization would provide control programs with baseline (and ongoing) surveillance data to evaluate public health interventions in high-transmission areas of sub-Saharan Africa that are pivoting to elimination efforts (Dalmat et al. 2019; Wesolowski et al. 2018).
Tanzania is classified as a high burden, high transmission setting, with almost the entire population at risk for malaria (“World Malaria Report” 2019). However, malaria transmission is heterogeneous across the country (Figure 1) (Hagenlocher and Castro 2015; Ministry of Health, Community Development, Gender, Elderly and Children (MoHCDGEC) [Tanzania Mainland] et al. 2017; Thawer et al. 2020), with regions of both high and low transmission. Given these patterns, the genetics of underlying parasite populations may be similarly heterogeneous. Additionally, while several studies have investigated patterns of drug resistance markers across the country (Gesase et al. 2009; Kavishe et al. 2016; Ngondi et al. 2017), there have been no large-scale analyses of within-Tanzania parasite population structure using genome-wide genetic markers to provide additional context to these findings.
Figure 1:
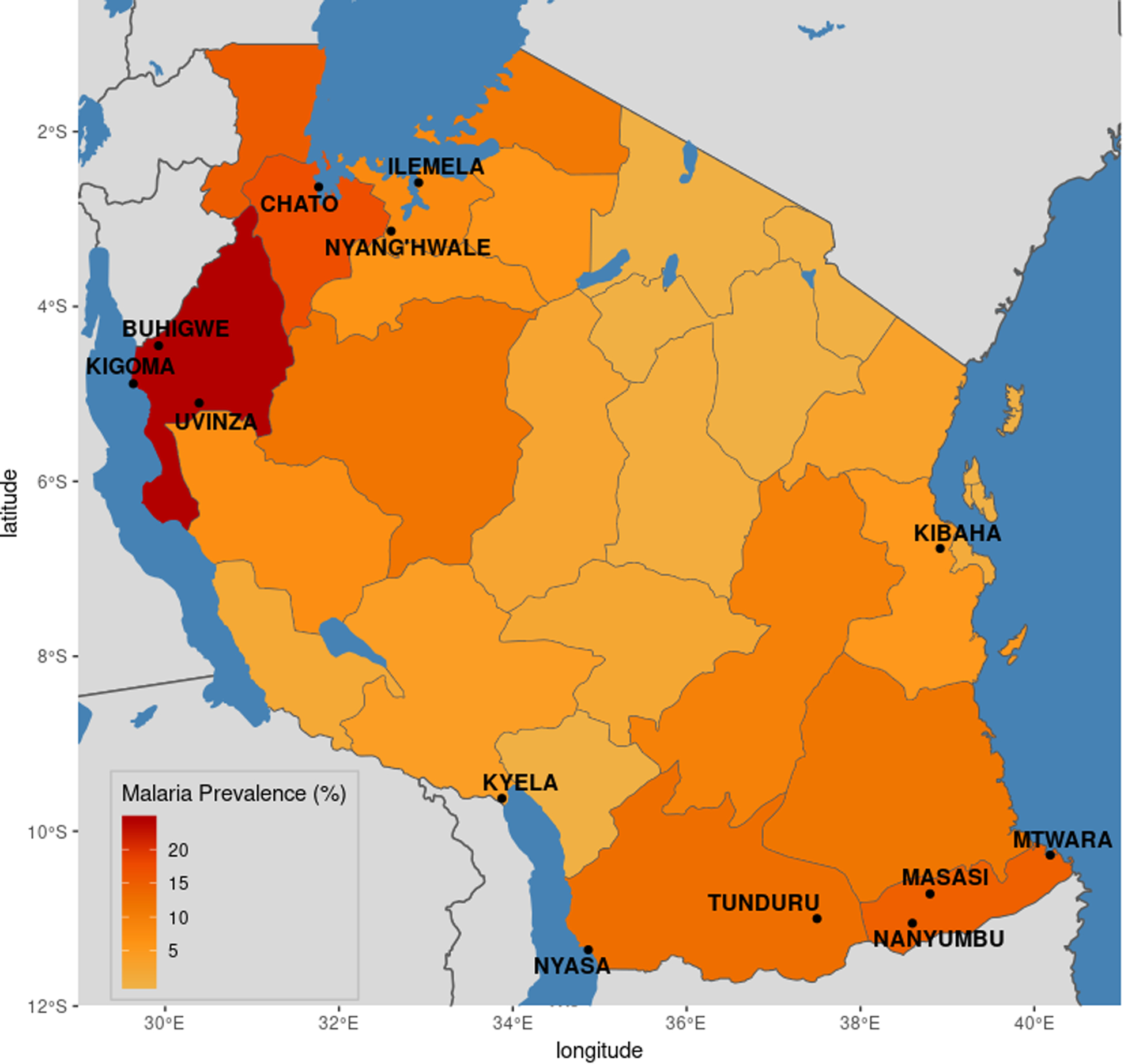
Region-level malaria prevalence in Tanzania. Malaria prevalence in children under five years of age at the regional level, as measured by rapid diagnostic tests (RDTs), was calculated using data from the 2017 Malaria Indicator Survey (MIS). Districts in which sampling occurred for the current study are labeled.
In this work, over 1,000 P. falciparum infections collected across Tanzania in 2017 were genotyped using MIP panels. Using data generated at ~1.8k genome-wide positions, as well as known or putative drug resistance loci, a current picture of parasite dynamics within the country is described. Results show that parasite populations differ genetically across the country, and that this differentiation is likely driven by a combination of geographical distance and selection due to past or ongoing antimalarial drug use.
METHODS
Sample collection:
1,232 samples from 13 districts in 7 administrative regions of Tanzania (Figure 1) were available from previously conducted cross-sectional surveys and drug efficacy studies. These districts were drawn from regions capturing the variability of transmission that occurs in malaria-endemic regions of Tanzania, with malaria prevalence ranging from ~5% (Mbeya region) to ~25% (Kigoma). For the cross-sectional studies, 552 samples were collected between July and November 2017 from eight districts (Chato, Nyan’ghwale, Buhigwe, Uvinza, Nyasa, Tunduru, Nanyumbu, and Mtwara) with historically high transmission (Chacky et al. 2018; Ministry of Health, Community Development, Gender, Elderly and Children (MoHCDGEC) [Tanzania Mainland] et al. 2017). All individuals in 120 households from two villages selected from each district were tested for malaria, representing both symptomatic and asymptomatic malaria cases in adults and children (Chiduo et al. 2020). Parasitemia (microscopy) was available for these subjects. The remaining 680 samples came from five national malaria control program (NMCP) sentinel sites (Ilemela, Kigoma, Kyela, Masasi, and Kibaha) where two in vivo therapeutic efficacy studies (TES) were conducted in 2017. The first TES assessed the efficacy and safety of artesunate-amodiaquine and dihydroartemisinin-piperaquine in enrolled symptomatic children between 6 months and 10 years of age (Mandara et al. 2019); the second evaluated artemether-lumefantrine (Ishengoma et al. In preparation). Ethical clearance for the above studies was obtained from the Medical Research Coordinating Committee (MRCC) of the National Institute for Medical Research (NIMR-MRCC) in Tanzania. Analyses utilizing parasite genomes from deidentified samples were deemed nonhuman subjects research at Brown University and the University of North Carolina Chapel Hill.
Parasite Genotyping and Variant Calling:
DNA was extracted from dried blood spots using QIAmp DNA Blood kits per manufacturer’s protocols (Qiagen, Hilden, Germany). Extracted DNA underwent capture and sequencing using two MIP panels previously described (Aydemir et al. 2018). The first panel targeted 1,834 single nucleotide polymorphisms (SNPs) across the genome shown to be phylogeographically informative for sub-Saharan African parasite populations, as well as putatively neutral SNPs, based on the publicly available Pf3k P. falciparum Community project from the MalariaGEN Consortium (The Pf3K Project 2016). The second MIP panel targeted SNPs known or suspected to be associated with drug resistance. Libraries were sequenced on a Illumina Nextseq 500 using 150 bp paired end sequencing for the genome-wide panel, and on an Illumina MiSeq using 250 bp paired-end reads for the drug resistance panel, at Brown University ([dataset]Moser et al., Genbank).
Variant calling was conducted as previously described (Aydemir et al. 2018; Verity et al. 2020). Briefly, sequences were reconstructed by stitching together mate-pair reads, and then clustering sequences by 1) their unique molecular identifiers (UMIs) and 2) with the qluster algorithm from SeekDeep (Hathaway et al. 2018). Each reconstructed sequence was assigned a unique MIP-microhaplotype ID, so that SNPs occurring on the same probe could be linked together. Reconstructed sequences were used to identify SNPS through pairwise alignment of each sequence to the 3D7 reference genome using LastZ (Harris 2007). Variants were subset to the original SNP positions targeted by the MIPs to minimize calls from PCR or sequencing error, and SNPs with poor sequence quality (Illumina phred score < 20) were removed. Non-biallelic SNPs in the genome-wide panel were removed across all samples. After these steps, samples and SNPs were filtered to remove those that had greater than 90% missing genotypes (across variants or samples).
Population Genetic Analyses:
Several approaches were used to detect genetic population structure in the sample set. Principal component analysis (PCA) was conducted with the prcomp function in R (v3.6) (R Core Team 2019). Discriminatory analysis of principal components (DAPC) was done with the dapc function from the adegenet package (Jombart and Ahmed 2011). To determine an appropriate number of PCs to retain for DAPC (so as to avoid over-fitting), alpha score optimization was used. Additionally, the program Admixture (Alexander, Novembre, and Lange 2009) was used to test K values of 1–10 (10 replicates each). The cross-validation (CV) value from the replicate with the highest log-likelihood value for each K was plotted, and the K with the lowest CV value was chosen as the final K. For sample-level analyses using admixture results, samples were assigned to K using the within-sample admixture proportion estimates.
SNPs associated with population structure were identified with two approaches. First, a random forest approach using the randomForest package in R (Liaw and Wiener 2002) was used to identify SNPs (predictor variables) associated with two response variables: 1) geographic categories (defined broadly as north vs south) and K subpopulation (K1 vs K2). Kibaha was removed from this approach for simplicity, given its role as the sole district of eastern Tanzania in the dataset. 10,000 trees and 200 features (SNPs) at each split; these values were optimized observing the out-of-bag (OOB) error over different parameter values. Receiver-operator curves (ROCs) were constructed from the geographic and K models to assess the performance of each model. Secondly, loading values from the above PCAs were taken to identify SNPs which contribute the most to the first three PCs. SNPs identified through both the random forest and PCA approach were then compared.
To investigate the relationship between genetic relatedness and geographic distance (as measured by greater circle distance using district administrative center), the inbreeding_mle function of the mipanalyzer R package was used to calculate the inbreeding coefficient (F) between sample pairs (Verity et al. 2020). In order to avoid artificially high inbreeding coefficients for samples with high amounts of missing genotypes, this analysis was restricted to samples with < 50% missing genotypes. As polyclonality (multiple strains within the same infection) is common in high-transmission settings of sub-Saharan Africa, several approaches were used to handle loci where more than one allele was detected. For PCA-based approaches (PCA, DAPC), the within-sample allele frequency (WSAF), defined as the alternative allele UMI count divided by total UMI coverage of the site within the sample, was used. This value ranged from zero to one, with zero values representing sites that only found the reference allele at that site, and values of one representing sites that found only the alternate allele at that site. For all other analyses (admixture, random forest, inbreeding coefficients), the major allele (the allele represented by > 50% of the UMIs) at heterozygous positions was used.
Drug resistance analysis:
To increase confidence in the genotyping calls used for reporting SNPs associated with drug resistance, if the alternate allele was supported by only a single UMI, or if the WSAF of the alternate allele was less than 1%, the genotype at that position in that sample was considered to only have the reference allele at that position; otherwise the sample was considered positive for the alternate allele. The prevalence of SNPs in known or suspected drug resistance genes was then calculated for target SNP positions from the drug-resistance MIP panel at the sample level. Samples with missing genotypes were excluded from the denominator in prevalence calculations. To explore haplotype information, SNPs that were detected by the same probe were grouped within each sample, using a unique MIP-microhaplotype ID given to each reconstructed sequence (see Variant Calling above), allowing for the examination of within-sample haplotypes for positions covered by the same probe within both monoclonal and polyclonal samples.
Epidemiologic Trends:
To identify epidemiologic correlations with genetic findings from the above analyses, data from the 2017 Malaria Indicator Survey (MIS) from Tanzania was downloaded from the Demographic Health Survey (DHS) website using the rdhs R package (Watson, FitzJohn, and Eaton 2019). Malaria prevalence and antimalarial use in children under five years of age was calculated using survey weights with the survey package in R (version 3.34). Because the study population presented here included individuals over five years of age, prevalence estimates were also obtained from previously conducted studies within the study districts that included older subjects (Abt Associates 2017). The number of strains per infection (complexity of infection, or COI) was calculated using the categorical method of THE REAL McCOIL (Chang et al. 2017) with the filtered genome-wide variant calls.
RESULTS
Genotyping success:
Samples were taken from 13 administrative districts in 7 administrative regions of Tanzania with different malaria transmission intensities, with region prevalence ranging from ~5–25% (Figure 1). Initial results showed higher levels of missingness in some of the northern districts (Table S1). While low parasitemia was one contributor to this loss, examining data by experiment showed signs of inefficient capture on several plates (Figures S1 & S2). This failure appeared to be largely responsible for increased missingness in certain districts and not necessarily due to inherent characteristics of parasites from those regions. Additionally, different patterns of missingness by district were seen for both the genome-wide and drug-resistance MIP panel (Figure S1), further evidence of experimental failure. Due to the above plate failure, a high proportion of samples (> 80%) from Chato and Nyang’hwale (both in the northern district of Geita) were lost (Table S1). However, 64 (76.5%) samples from Ilemela (also in the northern part of Tanzania) were successfully genotyped, offering representation of parasites from that region in the below analysis.
Population structure:
After variant calling and filtering, 742 and 934 samples (from the genome-wide and drug resistance panels, respectively) from 12 districts were retained for analysis (Table S1). High levels of missingness were observed in samples from Nyang’hwale, and were removed (Figure S1 & S2, Supplemental Text). After excluding these samples, 737 samples from 12 districts and 1,617 variable biallelic SNPs originally targeted by the MIPs were used to explore population structure in Tanzania. Principal component analysis (PCA) showed separation between the northwest and southern regions of the country, with samples from Kibaha in the east intermingling with samples from both regions (Figure S3). Discriminatory PCA (DAPC) analysis using the first 112 components explaining 46% variation of the original PCA (Figure S4) highlighted additional separation of samples by their district-level origin (Figure 2), particularly separating samples from Ilemela in the north from Kigoma and Buhigwe in the northwest. Admixture analysis identified two populations within the sample set (Figure S5); one population almost solely represented samples from the northwestern districts, while the other represented samples from all districts (Figure 3A). Samples from the northwestern districts and Kibaha in the east also had signs of increased admixture, as compared to samples from southern regions.
Figure 2:

Separation of Tanzanian P. falciparum samples by geographic region using discriminatory analysis of principal components (DAPC). DAPC analysis using the first 112 components of a principal component analysis (PCA) is shown (explaining 46% of the variation in the original PCA). Each dot is a sample colored by its geographic origin based on the 12 districts included in the analysis (inset map). A minimum spanning tree, constructed from genetic distances between groups, is overlaid on the plot (black dashed line).
Figure 3:
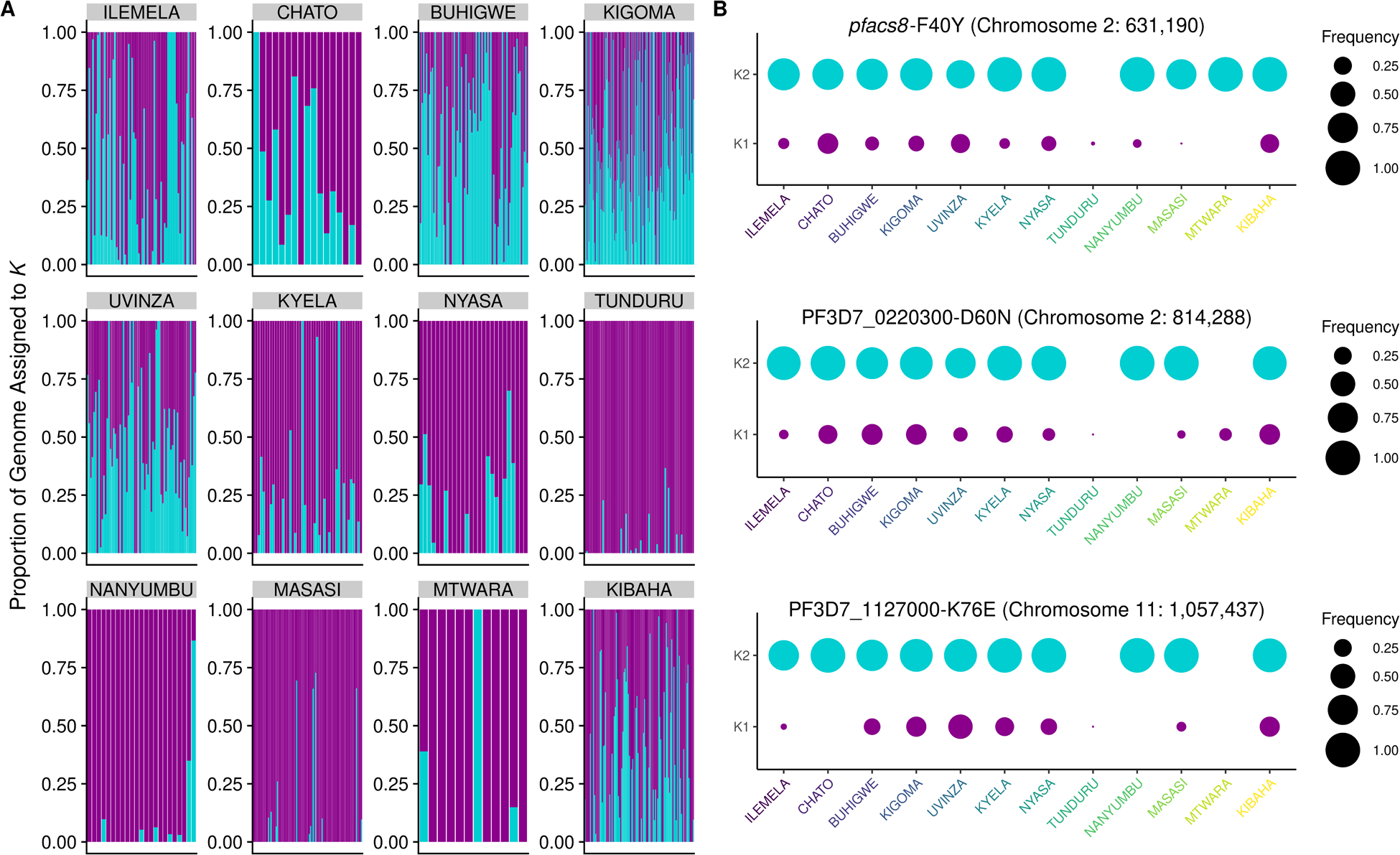
Tanzanian parasite population structure and associated variants. A. Admixture analysis using 737 samples and 1,614 genome-wide SNPs revealed two populations, K1 (purple) and K2 (turquoise) across the 12 districts included in the analysis. B. Frequency of key SNPs that were associated with population structure by random forest (Figure S8) and PCA (Figures S9), stratified by K subpopulation. Area of the circle represents the frequency of the alternate allele at that position within each subpopulation, and districts labels colored as in Figure 2 (from north, to northwest, to south, to east).
As the genetic relatedness of parasites from the above analyses, particularly the PCA-based approaches, appeared to be best represented by a gradient across the sites (versus distinct subpopulations), we estimated genetic relatedness between each sample-pair as the inbreeding coefficient F, which is the probability that any two samples are related by descent, and geographic distance between sample pairs, using only those samples that had at least 50% of targeted SNPs successfully genotyped (n=515). The majority of comparisons showed little to no genetic relatedness (Figure S6), as expected in diverse populations of parasites. However, a small number of sample pairs (n=8 comprising of 16 unique samples) had inbreeding coefficients exceeding 0.90; these highly related samples only occurred between parasites within the same districts (Ilemela, Buhigwe, Kigoma, Uvinza, and Kibaha). Dates of collection were available for seven of the eight pairs; three pairs were collected on the same day (Buhigwe and Uvinza comparisons), and the remaining four pairs were collected 1 (Kigoma), 2 (Kigoma), 9 (Kigoma), and 16 (Kibaha) days apart. After averaging coefficients in bins of increasing geographic distance, an inverse relationship was observed between genetic relatedness and geographic distance (Figure 4A). Variation in the amount of genetic sharing was also observed by site. Sample comparisons from southern district pairs (from within the same district and between different districts) appeared to show slightly more inbreeding than other district-level comparisons (Figure 4B, Figure S7); additionally, comparisons involving Kibaha did not always track with distance (Figure S7).
Figure 4:
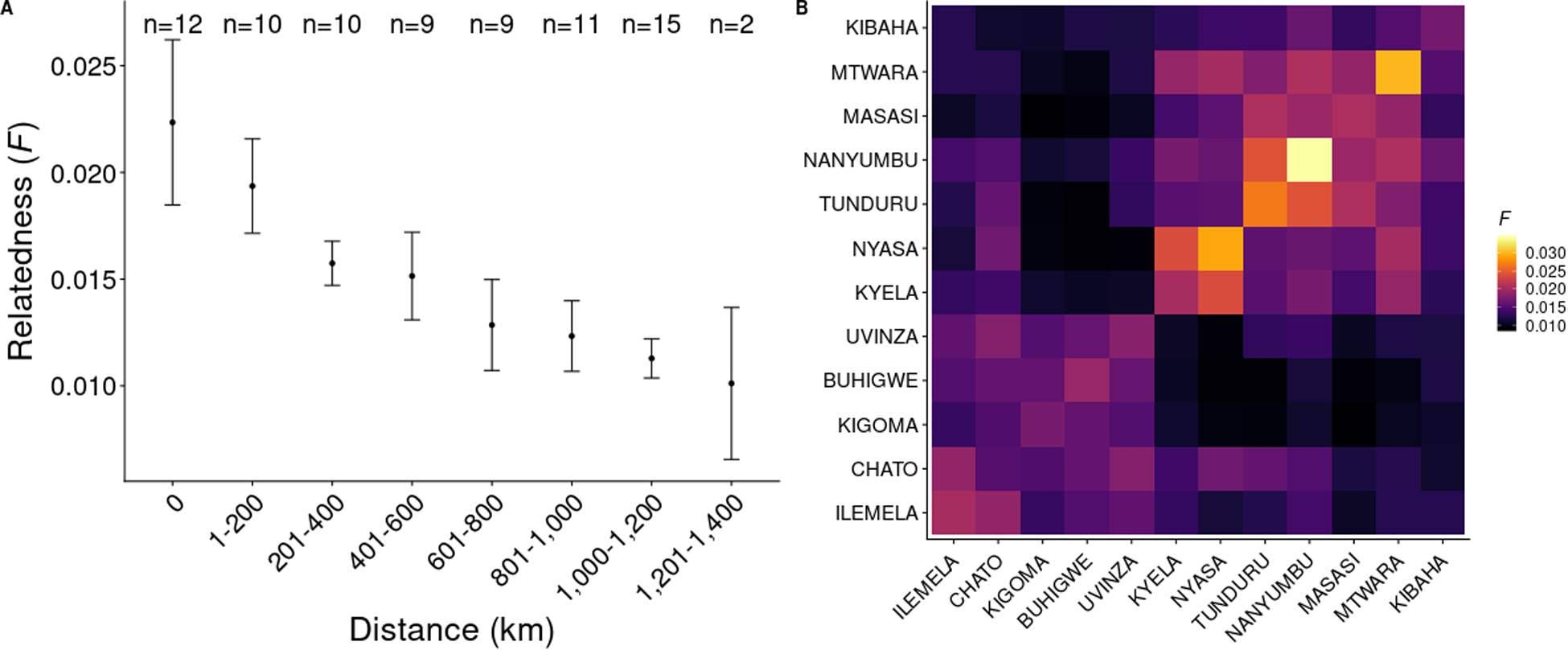
Genetic relatedness by geographic distance. A. Relatedness (as measured by the inbreeding coefficient F) binned by geographic distance (greater circle distance). An average-of-averages approach (average of all district-level average F) was used to avoid district comparisons with more samples contributing more to a bin. Bars represent 95% confidence intervals. The number of district-level pairwise comparisons in each bin is shown at the top of the plot (for example, there are 12 districts included in this analysis, so the number of same-district comparisons in n=12 for bin size 0 km). B. Heatmap of averaged F between districts. Districts are arranged by geographic location (north and northwest, to south and southeast, to east).
Variants associated with population structure:
To better understand the genetic drivers of the spatial distribution of parasites, variants associated with the observed population structure were identified by a random forest analysis, using SNPs as features to predict geographic (north vs south) or genetic (K1 vs K2) populations (Figure S8). (To simplify the classification procedure, samples from Kibaha were excluded from this analysis). While the random forest model predicting genetic populations had better predictive performance (AUC=0.96) than the model predicting geographic populations (AUC: 0. 87), overlap was seen in regions of the genome containing SNPs best predicting population categories. While individually these variants carried very little predictive power towards classification methods, the top hits detected on chromosomes 2 and 11 also showed relatively high contributions towards PC1 of the PCA (Figure S9). Chromosome 2 hits were in an acyl-CoA synthetase (ACS8; PF3D7_0215300) and a Plasmodium exported protein of unknown function (PF3D7_0220300). pfacs8-F40Y showed particular separation by geography, not only being more prevalent in the northwestern regions of Tanzania, but also occurring more frequently in samples whose genome more likely represented the K2 population identified in the above admixture analysis (Figure 3B). Similar results were seen for several SNPs in PF3D7_0220300 (D60N and 146T) and in the chromosome 11 hit PF3D7_1127000 (K76E).
SNPs associated with drug resistance have been previously identified as contributors to population structure in sub-Saharan Africa (Amambua-Ngwa et al. 2018, 2019; Verity et al. 2020). While drug resistance loci were not among the top hits of the above random forest analysis, SNPs in a region upstream of pfdhps contributed to PC2 and PC3. Additionally, SNPs in a coding region downstream of pfcrt (pfcg2, a gene often in linkage with pfcrt (Cooper et al. 2005) also contributed to PC3 (Figure S9).
Drug Resistance Patterns in Tanzania:
After variant calling and filtering, 934 samples at 32 target SNP positions in 12 genes associated with drug resistance were successfully genotyped (Table S1). Mutations associated with sulfadoxine-pyrimethamine (SP) resistance in both the pfdhfr and pfdhps gene were at high frequencies independent of location, such as pfdhps-K540E (Supplemental Dataset 1). However, other mutations showed differential frequencies across geographic regions. The pfcrt mutation K76T was elevated in the north and northwestern districts (Figure 5A). Similarly, the A581G mutation in pfdhps was observed in the northwest of the country (particularly in Kigoma, Buhigwe, Uvinza) but was absent from the southern districts (Figure 5B). Haplotypes for the pfdhps mutations using variants detected on the same MIP further highlighted this geographic separation, with samples containing both K540E and A581G mutations overwhelming occurring in the northern districts (Table 1).
Figure 5:
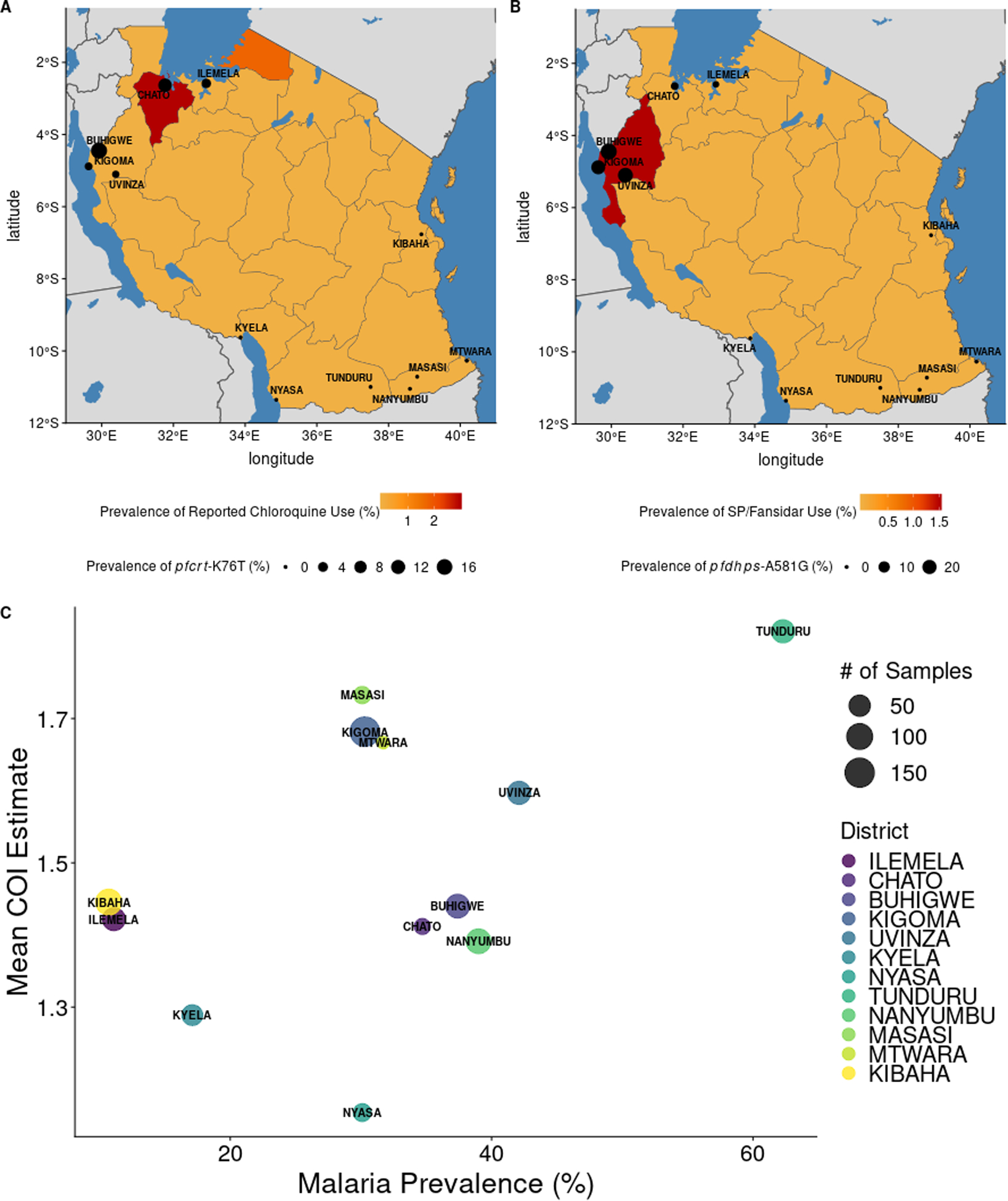
Patterns of antimalarial use and malaria transmission with genetic measures. Data from the 2017 Malaria Indicator Survey (MIS) was assessed for correlation with genetic measures from this study. A. District level prevalences of pfcrt-K76T are presented on region-level estimates of chloroquine use among children under five years of age. Mutation frequencies for A. and B. can be found in Supplemental Dataset 1. B. District level prevalences of pfdhps-A581G are presented on region-level estimates of SP/Fansidar use among children under five years of age. C. The relationship between P. falciparum prevalence, as measured by rapid diagnostic tests (RDTs) in school-age children (Abt Associates, 2017), and average COI estimate, at the district level. Each point is a district, and the size of the district indicates how many samples were in each district.
Table 1:
pfdhps haplotypes for mutations within the same MIP probe (K540E, A581G, and A613S), by geographic region (North: Ilemela, Chato, Kigoma, Buhigwe, and Uvinza; South & East: all others).
| Pfdhps Mutation Combination | Geographic Region | Total | |
|---|---|---|---|
| North | South & East | ||
| A613 Only | 3 (42.9%) | 4 (57.1%) | 7 |
| A581G Only | 15 (100%) | 0 (0%) | 15 |
| K540E Only | 290 (58%) | 210 (42%) | 500 |
| K540E + A581G | 57 (91.9%) | 5 (8.1%) | 62 |
| K540E + A613S | 0 (0%) | 1 (100%) | 1 |
Historically, mutations in pfk13 that have been confirmed or associated with causing artemisinin resistance have been observed at low frequency (or not at all) in sub-Saharan Africa. None of the following mutations associated (or confirmed) with delayed parasite clearance phenotypes were observed in this study population: F446I, P553L, N458Y, M476I C580Y, Y493H, R539T, 543T, P441L, G538V, G449A, V568G, C469F, P574L, A481V, F673I, P527H, A675V, and N537I. However, two samples carried the R561H mutation (Supplemental Dataset 1). These alleles had good UMI support (36/36 and 74/170 UMI support). Both were from the districts in the north and northwest of the country (Chato and Buhigwe).
Correlation of Demographic Patterns with Genetic Signals:
Higher levels of mutations associated with both SP and chloroquine were observed in north and northwestern districts (Supplemental Dataset 1), despite the replacement of these drugs with artemisinin combination therapies (ACTs) in 2006 (Ministry of Health and Social Welfare. In: A. Mwita, F. Molten, (Eds.) 2006). To determine if the frequencies of drug resistance mutations reported here correlated with ongoing drug use, reported antimalarial use was queried from the 2017 Malaria Indicator Survey (MIS) data. Reported use of chloroquine, quinine, and SP/Fansidar were all less than 5% (although only ~20% of participants responded to these queries) (Figure 5A–B). However, some overlap was observed with regions reporting SP/Fansidar and chloroquine use, and districts with >5% prevalence of pfdhps-A581G and pfcrt-K76T.
Tanzania is considered a high-transmission setting; however, malaria prevalence is heterogeneous across the country, with varying levels of prevalence across districts (Figure 1). This is also reflected in our genetic data. Complexity of infection (COI) estimates varied by region, with averaged district COI estimates ranging from 1.15 to 1.82. The proportion of samples that were polyclonal (COI > 1) ranged from 11.5% to 50%, with max COI ranging from 3–15. District-level malaria prevalence estimates were collated from previously conducted studies estimating malaria burden in the study districts, as well as from the 2017 MIS data. As expected, districts with higher average COI estimates also tended to have higher malaria prevalence (Figure 5C). This trend held regardless of the source of malaria prevalence estimates (Figure S10).
DISCUSSION
Genomic data of P. falciparum infections in malaria endemic regions can provide key information for implementation and control efforts. In addition to monitoring drug resistance mutations, parasite population genetics are currently being used to characterize important contributions of human mobility to parasite spread (Chang et al. 2019; S. Tessema et al. 2019); such data can allow for the identification of regions and populations to be targeted by control methods. Genomic data has also been used to assess the impacts of interventions (Daniels et al. 2015), and in the future could be used to detect unexpected population shifts after mass drug administration campaigns (the emergence of known and novel drug resistance mutations) and large-scale vaccine feasibility studies (the emergence of vaccine-resistant parasite populations). However, for such studies to be successful, it is important that the background variation in standing parasite populations is well described. To address these points in Tanzania, this paper explored patterns of parasite population structure and diversity in combination with drug resistance mutation frequencies.
While two main parasite populations were observed (roughly north and south), patterns representing gradients of genetic relatedness over geographic distance were identified. The lack of sample pairs from different districts with high genetic relatedness indicates that, in this dataset, there is little detectable direct contribution by very recent human movement to parasite population genetics in this region. However, several signals may still reflect broad patterns in human migration, such as the greater genetic relatedness among samples from Tunduru and surrounding districts. A major highway (A19) directly connects Tunduru to Nanyumbu, Masasi and, eventually, Mtwara. Kyela and Nyasa comparisons also showed greater amounts of inbreeding; both of these districts border Lake Nyasa, and may reflect increased human movement upon waterways. While some southern districts had small sample sizes for this analysis (Nanyumbu, Mtwara Nyasa), high inbreeding was still observed within and between other southern districts which were represented by 40 or more samples (Tunduru, Masasi). The increased sharing relative to that seen in regions of similar distance in the north could be due to more admixed populations driving greater genetic distance. However, stratification of the analysis by K1 and K2 subpopulations did not show an expected increase in relatedness (data not shown).
By leveraging previously conducted MIS data (collected at temporally equivalent timeframes as the samples included in the present study), this analysis found a correlation between transmission intensity and COI. This relationship is important to document for each malaria-endemic regions of interest, as previous studies have reported no relationship with COI and malaria prevalence (Koepfli et al. 2018). This is in contrast to other previously published work (Chang et al. 2017; Bei et al. 2018; Verity et al. 2020), including recent work in the Democratic Republic of the Congo with MIPs, showing higher COIs in areas with higher transmission. Overall, discrepancies between studies of transmission intensity and COI could reflect region-specific transmission dynamics, but could also reflect the methodological differences used to assess COI. This manuscript and others which have detected a relationship between malaria transmission and COI have used genome-wide SNP approaches, in contrast to approaches relying on the detection of size differences of amplified fragments of a small number of genes. The COI results also highlight the ability of MIPs to detect temporal changes in transmission. A decrease in average COI was observed between the samples from this study (2017) and from Tanzanian samples collected in 2015 that were genotyped in the same manner (Verity et al. 2020), corresponding to the overall decrease in malaria transmission in Tanzania during that period (“World Malaria Report” 2018).
The genomic results also provide context for previously published work exploring drug resistance mutation patterns in Tanzania. High prevalence of certain mutations across Tanzania, such as dhps-K540E mutations, and the higher prevalence of dhps-A581G mutations in regions of northern Tanzania compared to southern regions, have been previously described (Kavishe et al. 2016; Ngondi et al. 2017). Contextually, high frequencies of both dhps-A581G and pfcrt-K76T have also been observed in nations bordering the northwestern regions of Tanzania (Verity et al. 2020), and may be a reflection of historical expansion of drug-resistance parasites in this region of Africa. However, while reported drug use in 2017 was low for SP and chloroquine, it was nonetheless reported in the Tanzanian districts where relatively higher mutation frequencies were observed for SNPs associated with resistance to these antimalarials. Further studies investigating drug use in these regions would be important to tease out any contributions that current inappropriate antimalarial use may have on maintaining or further spreading drug resistance mutations among these parasite populations. It could be that fitness costs associated with mutations in pfcrt (Ord et al. 2007; Osman et al. 2007; Mharakurwa et al. 2013) and dhps (Osman et al. 2007) could result in reversion to the wild-type in areas that removed the corresponding drug pressure sooner than other regions, negating the necessity of ongoing drug use in the northern regions. Alternatively, use of antimalarials with similar resistance mechanisms (such as amodiaquine) could slow reversion to wild type populations. Finally, recent reports of pfk13 R561H mutations in southeast Tanzania and Rwanda (Uwimana et al. 2020; Bwire et al. 2020) highlight the need for continuous monitoring of pfk13 mutations in sub-Saharan Africa. While only two samples were positive for R561H (both collected through the cross-sectional surveys), this may imply that this mutation has been circulating in the region since 2017.
Several variants found to associate with population structure here may be useful for future molecular epidemiology studies identifying importation of parasite genotypes between districts in Tanzania. While none of these variants have been definitively tied to antimalarial use, several variants identified here have been reported in studies of resistance. Allele frequencies of pfacs8 have been shown to differ in Malawi, where SP was used for longer periods of time compared to other African nations (Ravenhall et al. 2016). In addition, the chromosome 2 region identified here also contributed to signals seen in population structure analyses from other African nations, including the Democratic Republic of Congo (Verity et al. 2020). Finally, PF3D7_0220300 has been reported to be upregulated (along with pfacs8) in studies investigating the response of Plasmodium parasites to dihydroartemisinin exposure (Shaw et al. 2015). The regions in which these genes fall have also been identified as being under selection across space and time in the Gambia (Amambua-Ngwa et al. 2018). Given drug resistance loci often represent some of the strongest population signals (Amambua-Ngwa et al. 2019; Verity et al. 2020), these variants require future study, particularly in regards to their current distribution across sub-Saharan Africa.
Efficient genotyping with MIPs allowed for hundreds of infections in Tanzania to be genotyped. The lower per-sample cost compared to whole genome sequencing makes these MIP panels amenable to large population surveys with denser sampling, potentially improving the understanding of parasite populations as sampling strategies can become more rigorous and complex. While the use of previously conducted studies allowed for representation of geographically disparate regions of the country, the TES studies (which contained infections from symptomatic individuals < 10 years of age) do not broadly sample different ages and disease severities. While it is generally not expected that there are different subpopulations across age or disease groups (consistent with a eukaryotic pathogen undergoing frequent sexual recombination), it is possible that the full extent of the parasite population was not captured. Thus, TES studies may not be representative of the parasite population circulating in these districts, and may possibly underestimate genetic diversity if infections in younger, symptomatic individuals are different than older, asymptomatic infections. However, only the east of the country (Kibaha) was represented by samples only from TES; all other regions are represented by both TES and cross-sectional surveys. Additionally, while MIPs have been shown to be robust to fluctuations in frequencies of strains within polyclonal infections down to 29 parasites/μL (Aydemir et al. 2018), this does imply that MIPs may not recover a representative sampling of the low-density infections present in this sample set, and higher levels of missingness did occur in samples with lower parasite densities. This is in part due to MIPS being dependent on a capture step, which compared to standard PCR amplicon sequencing is impacted more by low DNA concentrations or degraded DNA (Aydemir et al. 2018). However, this capture step allows for the minimization of errors due to the incorporation of UMIs, and MIPs are more sensitive and less expensive than other methods such as whole genome sequencing. MIP panels are also adaptable; additional probes can be easily designed and added to existing panels to capture new targets of interest as surveillance efforts expand to new regions.
The molecular inversion probe approach, coupling population-based surveys with an efficient method of genotyping, allowed for the identification of population structure country-wide in Tanzania, using fewer markers compared to whole genome sequencing, and therefore could be useful tools for monitoring parasite populations over time in this region. As Tanzania continues to make strides in malaria control, including advancing the pre-elimination area on the Zanzibar Archipelago, surveillance of genetic changes of Tanzanian parasite population will be critical for monitoring and optimizing interventions, ensuring elimination success.
Supplementary Material
ACKNOWLEDGEMENTS
This research was funded by the National Institutes of Health (grant numbers R01AI121558 and R01AI139520 K24AI134990). The cross sectional study and TES of artesunate-amodiaquine were funded by the Global Fund to fight Tuberculosis, HIV and Malaria through the National Malaria Control Program/Ministry of Health. The TES of artemether-lumefantrine was funded by the US President’s Malaria Initiative through the Boresha Afya/Jhpiego project. The study drugs, filter papers and technical support for TES were provided by the World Health Organization through the Country Office in Tanzania and headquarters in Geneva, Switzerland.
Authors greatly appreciate the contribution of the research scientists and assistants who took part in the sample collection surveys and TES: William Makunde, Method Segeja, Seth Misago, Daniel Challe, Gasper Lugela, Ezekiel Malecela, Juma Tupa, August Nyaki, Zakayo Nzella, John Masimba, Benson Swai, Fides Mumburi, Francis Chambo, Tilaus Gustav, Masunga Malimi, Juma Akida, Paul Martine, Mwanaidi Mtui, Caroline Minja, Richard Makono, Filbert Francis, Gineson Nkya, Richard Malisa, Raphael Charles, Respigi Kiwango, Lifoba Mosoud, Stumai George, Ali Idris, Ibrahim Materego, Neema Barua, and Michael Makange, Isolide Sylvester, Edwin Liheluka, Cloud Tesha, Emmanuel Chagoha, Happyness Jeremiah, Yahya Derua, Bernard Malongo, Martin Zuakuu, Adam Mgaya, Hashim Ally, Heri Bakari, Alex John, Neema Richard, Rukia Ahmed, Israel Msangi, Shabani Shabani, Joyce Geho, Praygod Swai, Neema Mwangoka, Simon Lugoye, Barnabas Muganda, Hosea Nchana, Claris Ossere, Sofian Mdegela, Moses Sarya, Rosemary Batunika, Jaffari Ramadhani, Charles Chacha, Mary Mwacha, Elinzuu Nicodemu, Juliana Joseph, Stumai George, Mzubwa Paul, Nerbert Mwasote, Caroline Minja, Samwel Bushukatale, Katamapahe Ndimbo, Masudi Ngamaley, Elisha Mg’andile, Hemed Suleiman, Neema Nziku, Peter Kapelanga, Sebastian Kobelo, Eulalia Mwageni, Alex Godwin, Ulrick Mosha, Devotha Mlelwa, Elfrida Kilumbo, and Ruth Mwakyombe. Technical, administrative and logistic support was provided by the finance department at Tanga Centre (Lydia Lugomola, Derick Maira, Joseph Said and Selemani Mandia) and NMCP (Abdallah Kajuna and Judith Kirama); drivers (Seth Nguhu, Francis Mkongo, Saidi Maivaji, Silvester Msamila, Eliwasiri Mmbaga, Thomas Semdoe, Athumani Simba and Ally Mshana) and others (Robert Mhilu, Salome Ngoda, Elfrida Mosha, Haruna Mrisho, Beatrice Semng’indo and Rehema Mtibusa) are highly appreciated. Special appreciation goes to the President’s Office Local Government and Regional Authorities and the village, district and regional authorities, particularly village Leaders, District Executive Directors (DEDs), District Medical Officers (DMOs) and District Malaria Focal Persons (DMFPs) for supporting the research teams during the entire study period. We also thank the Director General of NIMR for granting a permission to publish this paper.
Footnotes
DATA AVAILABILITY
Raw sequencing reads generated through this project have been deposited into the NCBI SRA (Bioprojects PRJNA631258 and PRJNA631263). Code to recreate analyses and figures presented in this manuscript can be found at https://github.com/kmogroethe/Mns-Tz-2017-PopGen-MIPs.
CONFLICTS OF INTEREST & DISCLAIMERS
The authors have no conflict of interest to declare. EJR is affiliated with the CDC; the findings and conclusions in this report are those of the author(s) and do not necessarily represent the official position of the Centers for Disease Control and Prevention/the Agency for Toxic Substances and Disease Registry. RN is a staff member of the World Health Organization and MW is a recently retired staff of the World Health Organization. They alone are responsible for the views expressed in this publication, which do not necessarily represent the decisions, policy or views of the World Health Organization.
REFERENCES
- Abt Associates. 2017. “PMI | Africa IRS (AIRS) Project Indoor Residual Spraying (IRS 2) Task Order Six, 2017 Tanzania End of Spray Report.” Bethesda, MD. https://www.pmi.gov/docs/default-source/default-document-library/implementing-partner-reports/tanzania-2017-end-of-spray-report.pdf?sfvrsn=4. [Google Scholar]
- Alexander David H., Novembre John, and Lange Kenneth. 2009. “Fast Model-Based Estimation of Ancestry in Unrelated Individuals.” Genome Research 19 (9): 1655–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amambua-Ngwa Alfred, Amenga-Etego Lucas, Kamau Edwin, Amato Roberto, Ghansah Anita, Golassa Lemu, Randrianarivelojosia Milijaona, et al. 2019. “Major Subpopulations of Plasmodium Falciparum in Sub-Saharan Africa.” Science 365 (6455): 813–16. [DOI] [PubMed] [Google Scholar]
- Amambua-Ngwa Alfred, Jeffries David, Amato Roberto, Worwui Archibald, Karim Mane, Ceesay Sukai, Nyang Haddy, et al. 2018. “Consistent Signatures of Selection from Genomic Analysis of Pairs of Temporal and Spatial Plasmodium Falciparum Populations from The Gambia.” Scientific Reports 8 (1): 9687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amambua-Ngwa Alfred, Tetteh Kevin K. A., Manske Magnus, Gomez-Escobar Natalia, Stewart Lindsay B., Deerhake M. Elizabeth, Cheeseman Ian H., et al. 2012. “Population Genomic Scan for Candidate Signatures of Balancing Selection to Guide Antigen Characterization in Malaria Parasites.” PLoS Genetics 8 (11): e1002992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amato Roberto, Pearson Richard D., Almagro-Garcia Jacob, Amaratunga Chanaki, Lim Pharath, Suon Seila, Sreng Sokunthea, et al. 2018. “Origins of the Current Outbreak of Multidrug-Resistant Malaria in Southeast Asia: A Retrospective Genetic Study.” The Lancet Infectious Diseases 18 (3): 337–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aydemir Ozkan, Janko Mark, Hathaway Nick J., Verity Robert, Kashamuka Mwandagalirwa Melchior, Tshefu Antoinette K., Tessema Sofonias K., et al. 2018. “Drug-Resistance and Population Structure of Plasmodium Falciparum Across the Democratic Republic of Congo Using High-Throughput Molecular Inversion Probes.” The Journal of Infectious Diseases. 10.1093/infdis/jiy223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbu Corentin M., Hong Andrew, Manne Jennifer M., Small Dylan S., Quintanilla Calderón Javier E., Sethuraman Karthik, Quispe-Machaca Víctor, et al. 2013. “The Effects of City Streets on an Urban Disease Vector.” PLoS Computational Biology 9 (1): e1002801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bataille Arnaud, Cunningham Andrew A., Cedeño Virna, Cruz Marilyn, Eastwood Gillian, Fonseca Dina M., Causton Charlotte E., et al. 2009. “Evidence for Regular Ongoing Introductions of Mosquito Disease Vectors into the Galápagos Islands.” Proceedings of the Royal Society B: Biological Sciences 276 (1674): 3769–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bei Amy K., Niang Makhtar, Deme Awa B., Daniels Rachel F., Sarr Fatoumata D., Sokhna Cheikh, Talla Cheikh, et al. 2018. “Dramatic Changes in Malaria Population Genetic Complexity in Dielmo and Ndiop, Senegal, Revealed Using Genomic Surveillance.” The Journal of Infectious Diseases 217 (4): 622–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bwire George M., Ngasala Billy, Mikomangwa Wigilya P., Kilonzi Manase, and Kamuhabwa Appolinary A. R.. 2020. “Detection of Mutations Associated with Artemisinin Resistance at K13-Propeller Gene and a near Complete Return of Chloroquine Susceptible Falciparum Malaria in Southeast of Tanzania.” Scientific Reports 10 (1): 3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerqueira Gustavo C., Cheeseman Ian H., Schaffner Steve F., Nair Shalini, McDew-White Marina, Pyae Phyo Aung, Ashley Elizabeth A., et al. 2017. “Longitudinal Genomic Surveillance of Plasmodium Falciparum Malaria Parasites Reveals Complex Genomic Architecture of Emerging Artemisinin Resistance.” Genome Biology 18: 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacky Frank, Runge Manuela, Rumisha Susan F., Machafuko Pendael, Chaki Prosper, Massaga Julius J., Mohamed Ally, et al. 2018. “Nationwide School Malaria Parasitaemia Survey in Public Primary Schools, the United Republic of Tanzania.” Malaria Journal 17 (1): 452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Hsiao Han, Wesolowski Amy, Sinha Ipsita, Jacob Christopher G., Mahmud Ayesha, Uddin Didar, Ibna Zaman Sazid, et al. 2019. “Mapping Imported Malaria in Bangladesh Using Parasite Genetic and Human Mobility Data.” ELife 8 (April). 10.7554/eLife.43481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Hsiao Han, Worby Colin J., Yeka Adoke, Nankabirwa Joaniter, Kamya Moses R., Staedke Sarah G., Dorsey Grant, et al. 2017. “THE REAL McCOIL: A Method for the Concurrent Estimation of the Complexity of Infection and SNP Allele Frequency for Malaria Parasites.” PLoS Computational Biology 13 (1): e1005348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiduo Mercy G., Mandara Celine I., Rumisha Susan F., Chacky Frank, Francis Filbert T., Mmbando Bruno P., Seth Misago D., et al. 2020. “Assessing the Intrinsic and Extrinsic Drivers and Targeting the Observed Resilience of Malaria in Northwestern and Southern Tanzania: A Protocol for a Cross-Sectional Exploratory Study.” Infectious Diseases (except HIV/AIDS). medRxiv 10.1101/2020.05.05.20091330. [DOI] [Google Scholar]
- Cooper Roland A., Papakrivos Janni, Lane Kristin D., Fujioka Hisashi, Lingelbach Klaus, and Wellems Thomas E.. 2005. “PfCG2, a Plasmodium Falciparum Protein Peripherally Associated with the Parasitophorous Vacuolar Membrane, Is Expressed in the Period of Maximum Hemoglobin Uptake and Digestion by Trophozoites.” Molecular and Biochemical Parasitology 144 (2): 167–76. [DOI] [PubMed] [Google Scholar]
- Dalmat Ronit, Naughton Brienna, Kwan-Gett Tao Sheng, Slyker Jennifer, and Stuckey Erin M.. 2019. “Use Cases for Genetic Epidemiology in Malaria Elimination.” Malaria Journal 18 (1): 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalziel Benjamin D., Pourbohloul Babak, and Ellner Stephen P.. 2013. “Human Mobility Patterns Predict Divergent Epidemic Dynamics among Cities.” Proceedings. Biological Sciences / The Royal Society 280 (1766): 20130763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels Rachel F., Schaffner Stephen F., Wenger Edward A., Proctor Joshua L., Chang Hsiao-Han, Wong Wesley, Baro Nicholas, et al. 2015. “Modeling Malaria Genomics Reveals Transmission Decline and Rebound in Senegal.” Proceedings of the National Academy of Sciences of the United States of America 112 (22): 7067–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch-Feldman Molly, Aydemir Ozkan, Carrel Margaret, Brazeau Nicholas F., Bhatt Samir, Bailey Jeffrey A., Kashamuka Melchior, et al. 2019. “The Changing Landscape of Plasmodium Falciparum Drug Resistance in the Democratic Republic of Congo.” BMC Infectious Diseases 19 (1): 872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy Craig W., Amambua-Ngwa Alfred, Ahouidi Ambroise D., Diakite Mahamadou, Awandare Gordon A., Ba Hampate, Tarr Sarah J., et al. 2018. “Multi-Population Genomic Analysis of Malaria Parasites Indicates Local Selection and Differentiation at the Gdv1 Locus Regulating Sexual Development.” Scientific Reports 8 (1): 15763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egizi Andrea, Kiser Jay, Abadam Charles, and Fonseca Dina M.. 2016. “The Hitchhiker’s Guide to Becoming Invasive: Exotic Mosquitoes Spread across a US State by Human Transport Not Autonomous Flight.” Molecular Ecology 25 (13): 3033–47. [DOI] [PubMed] [Google Scholar]
- Gesase Samwel, Gosling Roly D., Hashim Ramadhan, Ord Rosalynn, Naidoo Inbarani, Madebe Rashid, Mosha Jacklin F., et al. 2009. “High Resistance of Plasmodium Falciparum to Sulphadoxine/Pyrimethamine in Northern Tanzania and the Emergence of Dhps Resistance Mutation at Codon 581.” PloS One 4 (2): e4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra Carlos A., Kang Su Yun, Citron Daniel T., Hergott Dianna E. B., Perry Megan, Smith Jordan, Phiri Wonder P., et al. 2019. “Human Mobility Patterns and Malaria Importation on Bioko Island.” Nature Communications 10 (1): 2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagenlocher Michael, and Castro Marcia C.. 2015. “Mapping Malaria Risk and Vulnerability in the United Republic of Tanzania: A Spatial Explicit Model.” Population Health Metrics 13 (1). 10.1186/s12963-015-0036-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton William L., Amato Roberto, van der Pluijm Rob W., Jacob Christopher G., Hong Quang Huynh, Thuy-Nhien Nguyen Thanh, Tinh Hien Tran, et al. 2019. “Evolution and Expansion of Multidrug-Resistant Malaria in Southeast Asia: A Genomic Epidemiology Study.” The Lancet Infectious Diseases 19 (9): 943–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris Robert S. 2007. “Improved Pairwise Alignment of Genomic DNA.” PhD, Pennsylvania State University.http://www.bx.psu.edu/~rsharris/rsharris_phd_thesis_2007.pdf. [Google Scholar]
- Hathaway Nicholas J., Parobek Christian M., Juliano Jonathan J., and Bailey Jeffrey A.. 2018. “SeekDeep: Single-Base Resolution de Novo Clustering for Amplicon Deep Sequencing.” Nucleic Acids Research. 10.1093/nar/gkx1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemming-Schroeder Elizabeth, Lo Eugenia, Salazar Cynthia, Puente Sandie, and Yan Guiyun. 2018. “Landscape Genetics: A Toolbox for Studying Vector-Borne Diseases.” Frontiers in Ecology and Evolution 6: 21. [Google Scholar]
- Huestis Diana L., Dao Adama, Diallo Moussa, Sanogo Zana L., Samake Djibril, Yaro Alpha S., Ousman Yossi, et al. 2019. “Windborne Long-Distance Migration of Malaria Mosquitoes in the Sahel.” Nature 574 (7778): 404–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jombart Thibaut, and Ahmed Ismaïl. 2011. “Adegenet 1.3–1: New Tools for the Analysis of Genome-Wide SNP Data.” Bioinformatics 27 (21): 3070–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavishe Reginald A., Kaaya Robert D., Nag Sidsel, Krogsgaard Camilla, Ginsbak Notland Jakob, Kavishe Adellaida A., Ishengoma Deus, Roper Cally, and Alifrangis Michael. 2016. “Molecular Monitoring of Plasmodium Falciparum Super-Resistance to Sulfadoxine–Pyrimethamine in Tanzania.” Malaria Journal 15 (1): 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent Rebekah J., Thuma Philip E., Mharakurwa Sungano, and Norris Douglas E.. 2007. “Seasonality, Blood Feeding Behavior, and Transmission of Plasmodium Falciparum by Anopheles Arabiensis after an Extended Drought in Southern Zambia.” The American Journal of Tropical Medicine and Hygiene 76 (2): 267–74. [PMC free article] [PubMed] [Google Scholar]
- Koepfli Cristian, Waltmann Andreea, Ome-Kaius Maria, Robinson Leanne J., and Mueller Ivo. 2018. “Multiplicity of Infection Is a Poor Predictor of Village-Level Plasmodium Vivax and P. Falciparum Population Prevalence in the Southwest Pacific.” Open Forum Infectious Diseases 5 (11): ofy240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaw A, and Wiener M. 2002. “Classification and Regression by RandomForest.” R News. https://www.researchgate.net/profile/Andy_Liaw/publication/228451484_Classification_and_Regression_by_RandomForest/links/53fb24cc0cf20a45497047ab/Classification-and-Regression-by-RandomForest.pdf. [Google Scholar]
- Mandara Celine I., Francis Filbert, Chiduo Mercy G., Ngasala Billy, Mandike Renata, Mkude Sigsbert, Chacky Frank, et al. 2019. “High Cure Rates and Tolerability of Artesunate-Amodiaquine and Dihydroartemisinin-Piperaquine for the Treatment of Uncomplicated Falciparum Malaria in Kibaha and Kigoma, Tanzania.” Malaria Journal 18 (1): 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massaro Emanuele, Kondor Daniel, and Ratti Carlo. 2019. “Assessing the Interplay between Human Mobility and Mosquito Borne Diseases in Urban Environments.” Scientific Reports 9 (1): 16911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mensah Benedicta A., Aydemir Ozkan, Myers-Hansen James L., Opoku Millicent, Hathaway Nicholas J., Marsh Patrick W., Anto Francis, Bailey Jeffrey, Abuaku Benjamin, and Ghansah Anita. 2020. “Antimalarial Drug Resistance Profiling of Plasmodium Falciparum Infections in Ghana Using Molecular Inversion Probes and Next-Generation Sequencing.” Antimicrobial Agents and Chemotherapy 64 (4). 10.1128/AAC.01423-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mharakurwa Sungano, Sialumano Mavis, Liu Kun, Scott Alan, and Thuma Philip. 2013. “Selection for Chloroquine-Sensitive Plasmodium Falciparum by Wild Anopheles Arabiensis in Southern Zambia.” Malaria Journal 12 (December): 453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ministry of Health and Social Welfare. In: Mwita A, Molten F, (Eds.). 2006. “National Guidelines for Malaria Diagnosis and Treatment.” United Republic of Tanzania. http://apps.who.int/medicinedocs/documents/s19271en/s19271en.pdf. [Google Scholar]
- Ministry of Health, Community Development, Gender, Elderly and Children (MoHCDGEC) [Tanzania Mainland], Ministry of Health (MoH) [Zanzibar], National Bureau of Statistics (NBS), Office of the Chief Government Statistician (OCGS), and ICF. 2017. “Tanzania Malaria Indicator Survey 2017.” Dar es Salaam, Tanzania, and Rockville, Maryland, USA: MoHCDGEC, MoH, NBS, OCGS, and ICF. https://www.dhsprogram.com/pubs/pdf/MIS31/MIS31.pdf. [Google Scholar]
- Miotto Olivo, Almagro-Garcia Jacob, Manske Magnus, MacInnis Bronwyn, Campino Susana, Rockett Kirk A., Amaratunga Chanaki, et al. 2013. “Multiple Populations of Artemisinin-Resistant Plasmodium Falciparum in Cambodia.” Nature Genetics 45 (6): 648–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [dataset]Moser KA Madebe RA, Aydemir O, Chiduo MG, Mandara CI, Rumisha SF, Chaky F et al. 2020. Describing the current status of Plasmodium falciparum population structure and drug resistance within mainland Tanzania using molecular inversion probes. GenBank https://www.ncbi.nlm.nih.gov/bioproject/PRJNA631258, https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA631263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngondi Jeremiah M., Ishengoma Deus S., Doctor Stephanie M., Thwai Kyaw L., Keeler Corinna, Mkude Sigsbert, Munishi Oresto M., et al. 2017. “Surveillance for Sulfadoxine-Pyrimethamine Resistant Malaria Parasites in the Lake and Southern Zones, Tanzania, Using Pooling and next-Generation Sequencing.” Malaria Journal 16 (1): 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocholla Harold, Preston Mark D., Mipando Mwapatsa, Jensen Anja T. R., Campino Susana, MacInnis Bronwyn, Alcock Daniel, et al. 2014. “Whole-Genome Scans Provide Evidence of Adaptive Evolution in Malawian Plasmodium Falciparum Isolates.” The Journal of Infectious Diseases 210 (12): 1991–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ord Rosalynn, Alexander Neal, Dunyo Sam, Hallett Rachel, Jawara Musa, Targett Geoffrey, Drakeley Christopher J., and Sutherland Colin J.. 2007. “Seasonal Carriage of Pfcrt and Pfmdr1 Alleles in Gambian Plasmodium Falciparum Imply Reduced Fitness of Chloroquine-Resistant Parasites.” The Journal of Infectious Diseases 196 (11): 1613–19. [DOI] [PubMed] [Google Scholar]
- Osman Maha E., Mockenhaupt Frank P., Bienzle Ulrich, Elbashir Mustafa I., and Giha Hayder A.. 2007. “Field-Based Evidence for Linkage of Mutations Associated with Chloroquine (Pfcrt/Pfmdr1) and Sulfadoxine-Pyrimethamine (Pfdhfr/Pfdhps) Resistance and for the Fitness Cost of Multiple Mutations in P. Falciparum.” Infection, Genetics and Evolution: Journal of Molecular Epidemiology and Evolutionary Genetics in Infectious Diseases 7 (1): 52–59. [DOI] [PubMed] [Google Scholar]
- R Core Team. 2019. “R: A Language and Environment for Statistical Computing.” Vienna, Austria: R Foundation for Statistical Computing. https://www.R-project.org/. [Google Scholar]
- Ravenhall Matt, Diez Benavente Ernest, Mipando Mwapatsa, Jensen Anja T. R., Sutherland Colin J., Roper Cally, Sepúlveda Nuno, et al. 2016. “Characterizing the Impact of Sustained Sulfadoxine/Pyrimethamine Use upon the Plasmodium Falciparum Population in Malawi.” Malaria Journal 15 (1): 575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Real Leslie A., Henderson J. Caroline, Biek Roman, Snaman Jennifer, Lambert Jack Tracy, Childs James E., Stahl Eli, Waller Lance, Tinline Rowland, and Nadin-Davis Susan. 2005. “Unifying the Spatial Population Dynamics and Molecular Evolution of Epidemic Rabies Virus.” Proceedings of the National Academy of Sciences of the United States of America 102 (34): 12107–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw Philip J., Chaotheing Sastra, Kaewprommal Pavita, Piriyapongsa Jittima, Wongsombat Chayaphat, Suwannakitti Nattida, Koonyosying Pongpisid, Uthaipibull Chairat, Yuthavong Yongyuth, and Kamchonwongpaisan Sumalee. 2015. “Plasmodium Parasites Mount an Arrest Response to Dihydroartemisinin, as Revealed by Whole Transcriptome Shotgun Sequencing (RNA-Seq) and Microarray Study.” BMC Genomics 16 (October): 830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takala-Harrison Shannon, Jacob Christopher G., Arze Cesar, Cummings Michael P., Silva Joana C., Dondorp Arjen M., Fukuda Mark M., et al. 2015. “Independent Emergence of Artemisinin Resistance Mutations Among Plasmodium Falciparum in Southeast Asia.” The Journal of Infectious Diseases 211 (5): 670–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takehisa Jun, Kraus Matthias H., Ayouba Ahidjo, Bailes Elizabeth, Van Heuverswyn Fran, Decker Julie M., Li Yingying, et al. 2009. “Origin and Biology of Simian Immunodeficiency Virus in Wild-Living Western Gorillas.” Journal of Virology 83 (4): 1635–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor Aimee R., Schaffner Stephen F., Cerqueira Gustavo C., Nkhoma Standwell C., Anderson Timothy J. C., Sriprawat Kanlaya, Pyae Phyo Aung, Nosten François, Neafsey Daniel E., and Buckee Caroline O.. 2017. “Quantifying Connectivity between Local Plasmodium Falciparum Malaria Parasite Populations Using Identity by Descent.” PLoS Genetics 13 (10): e1007065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessema Sofonias K., Raman Jaishree, Duffy Craig W., Ishengoma Deus S., Amambua-Ngwa Alfred, and Greenhouse Bryan. 2019. “Applying Next-Generation Sequencing to Track Falciparum Malaria in Sub-Saharan Africa.” Malaria Journal 18 (1): 268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessema Sofonias, Wesolowski Amy, Chen Anna, Murphy Maxwell, Wilheim Jordan, Mupiri Anna-Rosa, Ruktanonchai Nick W., et al. 2019. “Using Parasite Genetic and Human Mobility Data to Infer Local and Cross-Border Malaria Connectivity in Southern Africa.” ELife 8 (April). 10.7554/eLife.43510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thawer Sumaiyya G., Chacky Frank, Runge Manuela, Reaves Erik, Mandike Renata, Lazaro Samwel, Mkude Sigsbert, et al. 2020. “Sub-National Stratification of Malaria Risk in Mainland Tanzania: A Simplified Assembly of Survey and Routine Data.” Malaria Journal 19 (1): 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Pf3K Project. 2016. “Pf3k Pilot Data Release 5.” https://malariagen.net/data/pf3k-5.
- Thomson MC, Connor SJ, Quiñones ML, Jawara M, Todd J, and Greenwood BM. 1995. “Movement of Anopheles Gambiae s.l. Malaria Vectors between Villages in The Gambia.” Medical and Veterinary Entomology 9 (4): 413–19. [DOI] [PubMed] [Google Scholar]
- Uwimana Aline, Legrand Eric, Stokes Barbara H., Jean-Louis Mangala Ndikumana, Warsame Marian, Umulisa Noella, Ngamije Daniel, et al. 2020. “Emergence and Clonal Expansion of in Vitro Artemisinin-Resistant Plasmodium Falciparum Kelch13 R561H Mutant Parasites in Rwanda.” Nature Medicine, August. 10.1038/s41591-020-1005-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verity Robert, Aydemir Ozkan, Brazeau Nicholas F., Watson Oliver J., Hathaway Nicholas J., Kashamuka Mwandagalirwa Melchior, Marsh Patrick W., et al. 2020. “The Impact of Antimalarial Resistance on the Genetic Structure of Plasmodium Falciparum in the DRC.” Nature Communications 11 (1): 2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vora A, Burke DS, and Cummings DAT. 2008. “The Impact of a Physical Geographic Barrier on the Dynamics of Measles.” Epidemiology and Infection 136 (5): 713–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson Oliver J., FitzJohn Rich, and Eaton Jeffrey W.. 2019. “Rdhs: An R Package to Interact with The Demographic and Health Surveys (DHS) Program Datasets.” Wellcome Open Research 4 (June): 103. [Google Scholar]
- Wesolowski Amy, Eagle Nathan, Tatem Andrew J., Smith David L., Noor Abdisalan M., Snow Robert W., and Buckee Caroline O.. 2012. “Quantifying the Impact of Human Mobility on Malaria.” Science 338 (6104): 267–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesolowski Amy, Taylor Aimee R., Chang Hsiao-Han, Verity Robert, Tessema Sofonias, Bailey Jeffrey A., Perkins T. Alex, Neafsey Daniel E., Greenhouse Bryan, and Buckee Caroline O.. 2018. “Mapping Malaria by Combining Parasite Genomic and Epidemiologic Data.” BMC Medicine 16 (1): 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- “World Malaria Report.” 2018. Geneva: World Health Organization. [Google Scholar]
- “World Malaria Report.” 2019. Geneva: World Health Organization. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.