

Summary
Glycolysis plays a central role in organismal metabolism, but its quantitative inputs across mammalian tissues remain unclear. Here we use 13C-tracing in mice to quantify glycolytic intermediate sources: circulating glucose, intra-tissue glycogen, and circulating gluconeogenic precursors. Circulating glucose is the main source of circulating lactate, the primary end product of tissue glycolysis. Yet, circulating glucose highly labels glycolytic intermediates in only a few tissues: blood, spleen, diaphragm and soleus muscle. Most glycolytic intermediates in the bulk of body tissue, including liver and quadriceps muscle, come instead from glycogen. Gluconeogenesis contributes less but also broadly to glycolytic intermediates, and its flux persists with physiologic feeding (but not hyperinsulinemic clamp). Instead of suppressing gluconeogenesis, feeding activates oxidation of circulating glucose and lactate to maintain glucose homeostasis. Thus, the bulk of the body slowly breaks down internally stored glycogen while select tissues rapidly catabolize circulating glucose to lactate for oxidation throughout the body.
eTOC blurb
Glucose consumption by glycolysis is key for maintaining euglycemia. Here, TeSlaa et al. quantify the sources of glycolytic intermediates in mammalian tissues, finding evidence for specialized highly glycolytic cell populations, including red muscle, that produce most circulating lactate.
Graphical Abstract
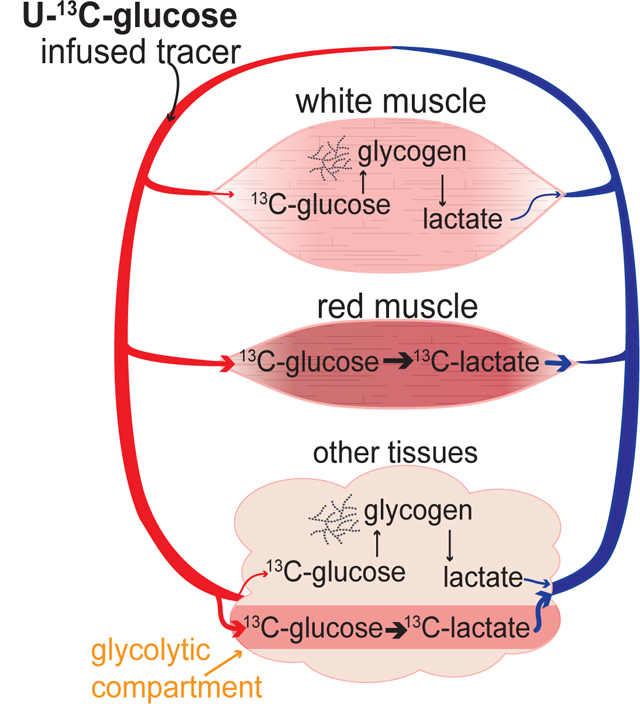
Introduction
Glucose has long been considered the most important circulating energy precursor in mammals. Its catabolic pathway, glycolysis, directly makes two ATP molecules per molecule of glucose. Glycolysis also produces NADH and pyruvate, which can be oxidized in mitochondria to make additional ATP. When mitochondrial respiration is impaired, pyruvate is instead reduced by NADH to lactate, which cells secrete as a waste product. Mammals, however, do not net excrete substantial lactate. Instead, lactate released by one cell is used as fuel for other cells, with most tissues of the body taking in circulating lactate and metabolizing it into pyruvate and downstream TCA intermediates (Brooks, 2018; Faubert et al., 2017; Hui et al., 2017).
Uptake of circulating lactate provides a route for cells to generate pyruvate and energy from carbohydrate without consuming glucose or running glycolysis. Thus, glucose uptake and glycolysis may not be universal features of mammalian cells. What cell types or tissues directly use circulating glucose? In cells that do not rely on glucose, where do glycolytic intermediates come from?
Glucose uptake into tissues has been extensively studied (Baron et al., 1988; DeFronzo et al., 1989; Gerich, 1993; James et al., 1985a); (Gruetter et al., 1994; Taylor et al., 1992). 18F-FDG imaging and isotope labeled 2DG uptake assays have revealed high uptake in brain, transformed tissue, the heart, and some muscles (Büsing et al., 2013; James et al., 1985a; Wetter et al., 1999), yet uptake of glucose or its analogs does not necessarily correlate with glucose catabolism via glycolysis (Barrio et al., 2019; Bauckneht et al., 2020; Kernstine et al., 2020). Accordingly, it is important to directly measure tissue glycolytic intermediate labeling. But, in large part due to detection challenges for glycolytic intermediates, recent reports utilizing stable isotope tracing in vivo have focused mostly on TCA cycle metabolites (Courtney et al., 2018; Davidson et al., 2016; Faubert et al., 2017; Hui et al., 2017).
In addition to glucose, there are several other potential contributors to tissue glycolytic intermediate pools including glycogen breakdown and gluconeogenesis (Shulman and Rothman, 2017). Ultimately, glycolysis, gluconeogenesis, and glycogen synthesis and breakdown work in concert to maintain not only tissue glycolytic intermediates, but also circulating glucose homeostasis. Defective glucose homeostasis is the hallmark of type II diabetes. Accordingly, understanding the in vivo activity of these pathways is of great medical importance.
Here we utilize intravenous infusion of 13C-labeled metabolites, including glucose and gluconeogenic precursors, in mice to investigate the source of glycolytic intermediates. By conducting both direct 13C-glucose infusion and 13C-glucose pulse chase, we are able to dissect the contributions of glucose and glycogen. We find that circulating glucose is a minority contributor to glycolytic intermediates in many tissues and the bulk body as a whole. Both glycogen breakdown and gluconeogenesis contribute to glycolytic intermediates across a strikingly wide range of tissues. Despite this, glucose is the largest source of circulating lactate, indicating rapid glycolytic flux driven by circulating glucose somewhere in the body. We propose that this rapid glycolytic flux, which plays a central role in circulating glucose clearance, localizes to tissues where circulating glucose strongly labels glycolytic intermediates, including red muscle.
Results
Low labeling of glycolytic intermediates from glucose
To explore the direct catabolism of circulating glucose through glycolysis, we intravenously infused U-13C-glucose and measured its contribution to tissue glycolytic metabolites (Figure 1A-B). Infusions were carried out over the final portion of an 8 h fast, with the infusion rate selected to achieve ~15% circulating glucose labeling. Serum collected throughout the infusion and tissues collected at the end of the infusion were analyzed by LC-MS (Figure 1A).
Figure 1. Circulating glucose supplies only a fraction of glycolytic intermediates.

(A) Schematic of experimental procedures. [U-13C]glucose is infused into jugular vein catheterized mice. At the end of the infusion, serum and tissue are collected for LC-MS analysis.
(B) Schematic of glycolysis showing hexose phosphate (hp), fructose-1,6-bisphosphate (fbp), and 3-phosphoglycerate (3pg).
(C) Serum glucose and lactate labeling from [U-13C]glucose infusion (n=5 mice). Data is fit with a single exponential.
(D) Normalized labeling of circulating lactate when sampled from the tail (n=10) versus from a carotid artery catheter (n=16). P-value calculated by unpaired t-test; **p=0.0021.
(E) Labeling in arterial serum glucose, arterial serum lactate (n=16 from 1D), and tissue glycolytic intermediates at the end of 2.5 h fasted [U-13C]glucose infusions. n=4 for most tissues except gastroc (n=2) and diaphragm (n=9). Tissue abbreviations are quadriceps fermoris muscle (quad), gastrocnemius muscle (gastroc), diaphragm (diaph), small intestine (small int), gonadal white adipose tissue (gwat), inguinal white adipose tissue (iwat), and brown adipose tissue (bat).
Mean ± SEM. Replicates indicate number of mice in which measurements were made.
The infusion of glucose results in rapid labeling of circulating lactate suggesting robust whole-body glycolysis (Figure 1C). When normalized to the labeling of circulating glucose, approximately 50% of lactate is labeled within the first ninety minutes of infusion (Figure S1A). Labeling of lactate was lower in blood collected by carotid artery catheter when compared to blood collected from the tail, a site commonly sampled during intravenous infusions (Figure 1D). This indicates active catabolism of glucose to lactate locally within the tail.
In many tissues, the labeling observed in tissue glycolytic intermediates was strikingly lower than in circulating lactate. While labeling was detectable for glucose-6 phosphate (which, together with any analytically unresolved isomers, we refer to as hexose-phosphate), labeling was often below the limit of quantitation for other intermediates. To improve our detection, we developed an LC-MS method employing selected ion monitoring (SIM) scans for two relatively abundant glycolytic metabolites: fructose-1,6-bisphosphate and 3-phosphoglycerate, which in combination with hexose-phosphate allowed measurements in upper, mid, and lower glycolysis (Table S1; Figure S1B).
The resulting improved measurement confirmed that there is only a small contribution of circulating glucose to tissue glycolytic intermediates, especially in tissues known to have important glycogen pools including liver and quadriceps, a classical example of white muscle rich in type IIB (fast-twitch) fibers and low in mitochondria. This paucity of labeling was persistent even with 8 h glucose infusions (Figure S1F-H). In contrast, red blood cells, spleen, soleus, and diaphragm display glycolytic intermediate labeling as high as circulating lactate labeling (Figure 1E). Red blood cells are known to be glycolytic (Rose and Warms, 1966), but measurements of their lactate production rate ex vivo imply that they only account for approximately 2.3 nmol min−1 g body weight−1 of lactate production, roughly 1% of whole-body lactate turnover (Figure S1C-E). The soleus and diaphragm are muscles rich in type I (slow oxidative) fibers and type IIA (fast oxidative) fibers with a bright red appearance due to high mitochondrial content. These muscles stand out for particularly high usage of circulating glucose to support glycolysis.
Feeding modestly increases the fraction of glycolysis from glucose
Circulating glucose homeostasis is maintained after a meal by insulin-induced glucose uptake into tissue. To determine if feeding induces a large shift in the in fraction of glycolytic intermediates that label from circulating glucose, we infused mice with U-13C-glucose after refeeding previously fasted mice (Figure 2A). The labeling of circulating lactate from glucose increases modestly with refeeding, representing an increase in whole-body lactate production from circulating glucose (Figure 2B). In tissues, there is a small increase in labeling in the quadricep muscles and the liver in the refed state when compared with fasting. Some tissues have notably lower labeling in fructose-1,6-bisphosphate when compared to hexose phosphate and 3-phosphoglycerate, including adipose tissue and the lung. These tissues may contain certain cells with high fructose-1,6-bisphosphate and low glucose contribution to glycolysis, and other cells with low fructose-1,6-bisphosphate and high glucose contribution to glycolysis. Overall, glycolytic intermediates in many tissues remain less than 50% labeled from circulating glucose in both the fasted and refed state (Figure 1E and S2A).
Figure 2. Hyperinsulinemia increases glucose use in glycolysis more than physiologic feeding.

(A) Illustration of 2.5 h fasted and refed [U-13C]glucose infusion.
(B) Normalized labeling in arterial serum lactate after 2.5 h infusion in fasted (n=16 from Figure 1B) and refed (n=6) states. P-value calculated by unpaired t-test; *p=0.015.
(C) Normalized labeling from 2.5 h [U-13C]glucose infusion of glycolytic intermediates hexose-phosphate (hp), fructose-1,6-bisphosphate (fbp), and 3-phosphoglycerate (3pg) in the quadriceps muscles, the soleus, and the liver for fasted (n = 4 from Figure 1E) or refed (n=6 for liver and quad, n=5 for soleus) mice. P-values calculated by two-way ANOVA; *p<0.05
(D) Illustration of hyperinsulinemic-euglycemic clamp with additional [U-13C]glucose tracer. Mice infused with either 2.4 or 0.8 mU min−1 kg−1 insulin, variable glucose to maintain euglycemia (required only in 2.5 mU min−1 kg−1 insulin group), and tracer amounts of stable isotope labeled glucose. Red blood cells were also infused to maintain hematocrit to allow for repeated blood sampling.
(E) Insulin levels measured by ELISA in different states: fasted (n=10), refed (n=7), 0.8 mU min−1 kg−1 insulin infusion (n=5), or 2.5 mU min−1 kg−1 insulin infusion (n=6). P-value calculated with one-way ANOVA; ****p<0.0001
(F) Normalized label in arterial serum lactate after infusion of either 0.8 (n=2) or 2.5 (n=3) mU min−1 kg−1 of insulin with [U-13C]glucose.
(G) Normalized labeling of glycolytic intermediates hexose-phosphate (hp), fructose-1,6-bisphosphate (fbp), and 3-phosphoglycerate (3pg) in the quadriceps muscles, the soleus, and the liver after infusion of either 0.8 (n=2) or 2.5 (n=3) mU min−1 kg−1 of insulin with [U-13C]glucose. P-value calculated with two-way ANOVA; ***p=0.0005, ****p<0.0001.
Mean ± SEM. Replicates indicate number of mice in which measurements were made.
Hyperinsulinemic clamp induces glucose use in glycolysis
Numerous studies have utilized infusion of supraphysiologic insulin to study insulin action, with infusion of glucose to maintain euglycemia, a technique termed the hyperinsulinemic-euglycemic clamp (Ayala et al., 2011; Ayala et al., 2006). We performed clamp studies to determine if hyperinsulinemia could increase the labeling of tissue glycolytic intermediates from circulating glucose. For comparison, a separate group of mice were infused with a lower concentration of insulin (0.8 mU min−1 kg−1 vs 2.5 mU min−1 kg−1) that is more physiologic (Figure 2E and S2B-C). Hyperinsulinemia results in a large increase in the labeling of circulating lactate from glucose (Figure 2F). With the exception of liver, glycolytic intermediate labeling was also increased (Figure 2G). This confirms that sustained high levels of insulin induce the direct catabolism of circulating glucose via glycolysis, while other substrates make glycolytic intermediates under physiologic feeding and fasting.
Widespread glycolytic intermediate production from glycogen
A potential source of glycolytic intermediates is glycogen. Glycogen is predominantly synthesized during feeding and has slower turnover than circulating metabolites. We therefore hypothesized that prolonged glucose infusions that spanned a feeding period would label glycogen stores and thereby increase labeling in glycolytic intermediates. Consistent with this, extending the U-13C-glucose infusion duration to 24 h markedly increased serum lactate and tissue glycolytic intermediate labeling, e.g., from approximately 14% to about 50% in quadriceps and from 16% to 40% in liver (Figure S1G,H).
To better distinguish glycolytic intermediate labeling from circulating versus stored glucose, we pursued a strategy of U-13C-glucose pulse-chase infusions. The pulse is a 16 h infusion of U-13C-glucose overnight (during the time when mice eat most liberally) which is designed to label the glycogen pool in tissues. After the pulse, the infusion is stopped, food is removed, and the mice are fasted for 8 h (Figure 3A). During this 8h “chase,” labeling in serum glucose declines, but labeling of glycogen is maintained (Figure 3B and S3A).
Figure 3. Glycogen supplies glycolytic intermediates in all tissues.

(A) Schematic of pulse-chase experiment. Mice were infused with [U-13C]glucose for 16 h with the goal of labeling glycogen. Next an 8 h chase was performed in which the infusion was stopped during 8 h of fasting to allow circulating metabolite labeling to decrease.
(B) Arterial serum glucose and lactate labeling over the course of the pulse-chase experiments. Combined data during the infusion is fit with a single exponential. Labeling in liver glycogen collected at the end of the pulse-chase is also displayed. Mean ± SEM; n=4 mice for most time points except serum at 20 and 22 h (n=2), serum at 24 h (n=9), liver glycogen at 20 h (n=5) and liver glycogen at 24 h (n=8). Error bars for some data points are too small to be visible.
(C-D) Labeling in serum glucose and lactate and tissue glycolytic intermediates normalized to glycogen labeling in the liver (C) or the quadriceps (D) at the end of the pulse-chase experiment. Expected labeling from circulating glucose is calculated based on the 2.5 h glucose infusion data from Figure 1 as described by the displayed equation; n=6 mice.
(E) Labeling in glycolytic intermediates at the end of the pulse chase experiment normalized to glycogen labeling measured in each tissue. n vary by tissue; n=4 mice for most tissues; n=6 for liver, quad, heart, and small intestine; n=5 for soleus and diaphragm, n=3 for iwat, bat and gastroc, and n=2 for brain.
Mean ± SEM. Replicates indicate number of mice in which measurements were made.
This pulse-chase strategy effectively highlights the importance of glycogen as a glycolytic input. If there were no glycogen contribution to glycolytic intermediates, then one could predict tissue glycolytic intermediate labeling during the chase from circulating glucose labeling. For example, relative to liver glycogen labeling, circulating glucose labeling during the chase is 50%. Based on the 2.5 h 13C-glucose infusion experiments, circulating glucose contributes only 16% of liver glycolytic intermediates (Figure 1E). Thus, we would expect liver glycolytic intermediate labeling of only 8% relative to liver glycogen. In contrast, the observed labeling is roughly 7-fold higher (> 50%), reflecting a major local contribution from liver glycogen (Figure 3C). Similarly, while circulating glucose contributes minimally to quadriceps glycolytic intermediates, in the pulse-chase experiment, glycolytic intermediate labeling relative to quadriceps glycogen is 80% (Figure 3D). Except for spleen, soleus, and diaphragm muscle, the labeling of glycolytic intermediate after pulse-chase is substantial in most tissues (Figure 3E, Figure S3B). Therefore, our experiments reveal an unanticipated widespread contribution from glycogen to glycolysis.
A potential concern in these experiments is that the major contribution of glycogen, as opposed to circulating glucose, may be due to acute stress on the animals. Glycogen breakdown, or glycogenolysis, can be stimulated by epinephrine (Deibert and Defronzo, 1980; Sutherland and Wosilait, 1956). To determine if the low contribution of circulating glucose to glycolytic intermediate is due to a stress response, we infused glucose after treating mice with propranolol, a beta-adrenergic receptor blocker (Juhlin-Dannfelt et al., 1982). Treatment with propranolol did not increase the labeling of glycolytic intermediate from circulating glucose (Figure S3C). Thus, in fasted mice, glycogen appears to be a major physiological source of glycolytic intermediates.
Glycogen contribution to glycolysis in the fed state
We also examined the fed state contribution of glycogen, via pulse-chase experiments in which the chase ends during feeding (Figure S4A). As feeding suppresses production of circulating glucose from glycogen, at the end of the fed chase, the labeling of circulating glucose is almost zero, and any labeling observed in glycolytic intermediates presumably comes from glycogen. While only a small fraction of the glycolytic intermediate pool labeled in the liver at the end of the chase (Figure S4B), extensive labeling was seen in quadriceps (Figure S4C). Substantial labeling was also observed in many other tissues, indicating a meaningful contribution of glycogen breakdown in tissues of fed mice (Figure S4D).
Gluconeogenic contribution to glycolytic intermediates
Another potential source of glycolytic intermediates is from the reverse flux of circulating substrates via gluconeogenesis. In bona fide gluconeogenic organs such as the liver and kidney, in which glucose-6-phosphatase is expressed, gluconeogenic flux can produce circulating glucose (Cori, 1981; Felig et al., 1970; Krebs, 1964) (Felig et al., 1970). A partial gluconeogenic pathway may also contribute to glycolytic intermediates in organs not classically considered gluconeogenic. Therefore, we quantified the contribution of gluconeogenesis to circulating glucose and tissue glycolytic intermediates by infusing 13C-labeled gluconeogenic precursors: lactate, alanine, glycerol, and glutamine (Figure 4A).
Figure 4. Gluconeogenic substrates broadly contribute to glycolytic intermediates in mammalian tissues.

(A) Schematic of the entry points of measured substrates into glycolysis.
(B-E) Labeling in circulating metabolites (lactate, glucose, alanine, glycerol, and glutamine) and tissue glycolytic intermediates after 2.5 h fasted infusion (normalized to enrichment of the tracer in serum). Each panel represents data from a different tracer: [U-13C]lactate (B), [U-13C]alanine (C), [U-13C]glycerol (D), and [U-13C]glutamine (E). Mean ± SEM; n=4 mice in all cases except f in [U-13C]lactate infusion where n=10 mice for serum measurements and n=8 for liver and kidney measurements.
In fasted mice, we observe a substantial contribution of lactate (40%) and glycerol (33%), and to a lesser extent (~ 10%) of alanine and glutamine, to circulating glucose (Figure 4B-E). Interestingly, we observed substantial glycolytic intermediate labeling not only in classical gluconeogenic tissues (liver, kidney), but also in most other organs. This presumably involves lactate uptake, entry into the TCA cycle via pyruvate carboxylase, and subsequent conversion of oxaloacetate into glycolytic intermediates via phosphoenolpyruvate carboxykinase (PEPCK), whose mitochondrial isoform is widely expressed across tissues (Stark and Kibbey, 2014). Consistent with this pathway, in most tissues, we observed substantial TCA labeling (including M+3 malate, reflective of pyruvate carboxylase flux; Figure S5A-B) and greater labeling in 3-phosphoglycerate (3pg) than in glycolytic intermediates closer to glucose. We also observe some correspondence between tissue TCA fuel preferences and gluconeogenic substrate preferences, e.g., pancreas preferentially uses glutamine for both, running a partial gluconeogenic pathway that largely terminates at fructose-1,6-bisphosphate (Figure 4E, S5C). Thus, most mammalian tissues engage in at least a partial gluconeogenic pathway that produces glycolytic intermediates.
Even in the classical gluconeogenic organs, labeling of glycolytic intermediates was not particularly high (~ 20%). Importantly, nearly twice as much labeling from infused lactate was observed in circulating glucose than in either liver or kidney glycolytic intermediates (Figure 4B). This suggests that the bulk of gluconeogenic flux in liver and kidney occurs in a subset of cells, with the other cells having lower gluconeogenic flux and therefore a lesser contribution of lactate to glycolytic metabolites. Such heterogeneity is consistent with the idea of zonation of the liver (Ben-Moshe et al., 2019; Cheng et al., 2018; Halpern et al., 2017; Katz, 1992). In the kidney, it is consistent with gluconeogenic activity localizing to the cortex, with the medulla instead consuming glucose (Gerich et al., 2001).
Gluconeogenesis in the fed state
We also performed gluconeogenic substrate infusions in the fed state. After refeeding, infusion of 13C-lactate leads to about 16% labeling of circulating glucose, less than half that observed in the fasted state, but nevertheless substantial. Analysis of tissue glycolytic intermediates revealed a persistent large contribution of lactate (Figure S6A). In contrast, fed-state infusions of 13C-alanine, 13C-glycerol, and 13C-glutamine resulted in limited tissue glycolytic intermediate labeling (Figure S6B-D). We also measured the labeling in liver glycogen, finding that, in the fed state, lactate contributes to hepatic glycogen production (Figure S6E) reflecting glycogen synthesis both directly from glucose and indirectly through gluconeogenesis (Moore et al., 1991; O’Doherty et al., 2000).
Hyperinsulemic clamp but not feeding blocks gluconeogenesis
We observed a substantial contribution from gluconeogenic substrates to liver and kidney glycolytic intermediates in both the fasted and fed states. Accordingly, we sought to quantify endogenous glucose production fluxes in the fed and fasted state. While individual tracing experiments reveal the overall contribution from the infused 13C-nutrient to downstream metabolites, there may be intervening paths. For example, when labeling is observed in circulating glucose from infused 13C- lactate, it can be either from lactate-driven gluconeogenesis within that tissue or from circulating alanine that was produced from lactate in another tissue in the body. To resolve such crosstalk, we used data from each infusion to determine how much each substrate labels each other potential substrate, using the pulse-chase experiments to reveal contributions from glycogen. The resulting labeling matrix can be applied to solve for each substrate’s direct contribution (i.e. contribution without passing through another circulating metabolite) to a given metabolite pool (Figure 5A) (Hui et al., 2020; Hui et al., 2017)).
Figure 5. Gluconeogenesis persists in the physiologic fed state.

(A) Illustration of substrate interconversions used to calculate the direct contributions to circulating glucose
(B) Direct substrate contributions to circulating glucose production flux. The total height of each bar is reflective of total glucose turnover, calculated by the . n ≥ 3 mice for each nutrient and condition.
(C) Illustration of hyperinsulinemic-euglycemic clamps performed in combination with U-13C-lactate infusion. Mice infused with either 2.4 or 0.8 mU min−1 kg−1 insulin, variable glucose to maintain euglycemia (required only in 2.5 mU min−1 kg−1 insulin group), and tracer amounts of stable isotope labeled lactate. Red blood cells were also infused to maintain hematocrit allowing for repeated blood sampling.
(D) Normalized labeling in arterial serum glucose after the indicated 13C-lactate infusions; n vary by condition; fasted (n=10 mice from Figure 4B); fed (n=7 mice); insulin infusions (n=3 mice).
(E) Normalized labeling of liver glycolytic intermediates hexose-phosphate (hp, n=3 mice), fructose-1,6-bisphosphate (fbp, n=2), and 3-phosphoglycerate (3pg, n=2) during U-13C-lactate infusion in combination with either 0.8 or 2.5 mU min−1 kg−1 of insulin with variable glucose.
Mean ± SEM.
Analysis of the direct sources of fasting circulating glucose revealed a gluconeogenesis contribution (spread across substrates) of 69% and a glycogenolysis contribution of 28%. The glycogen contribution disappeared with feeding. The gluconeogenesis contribution decreased as a fraction (Figure S6F), but due to increased total glucose turnover with feeding, absolute gluconeogenic flux was similar across the fasted and fed states (Figure 5B).
This was accompanied by a shift in gluconeogenic precursor usage, from glycerol (whose contribution decreased, consistent with insulin inhibition of lipolysis) to lactate, with the flux from circulating lactate to circulating glucose trending up (Figure 5B). Thus, the Cori cycle is active throughout fed and fasted cycles.
We reasoned that the higher insulin levels induced by clamp (Hatting et al., 2018), as compared to feeding, might suppress gluconeogenesis. Therefore, we performed hyperinsulinemic-euglycemic clamps (2.5 mU min−1 kg−1 insulin) in combination with U-13C-lactate infusion, finding essentially complete suppression of gluconeogenesis (Figure 5C, D). Labeling of 3-phosphoglycerate and fructose-1,6-bisophosphate in the liver persists during hyperinsulinemia, while labeling of hexose phosphate is inhibited, suggesting that insulin acts to inhibit fructose-1,6-bisphosphatase (Figure 5E). Collectively, these studies show that hyperinsulinemic euglycemic clamp, but not physiological insulin induced by feeding, suppresses lactate-driven gluconeogenesis.
Carbohydrate oxidation matches glucose turnover in fasting and feeding
Because we found minimal differences in endogenous glucose production between fasting and feeding, we reasoned that consumption pathways must be important for maintaining euglycemia (Figure 6A). Glucose turnover increases more than two-fold after feeding due to the large influx of glucose from food, and this requires a compensatory increase in glucose consumption (Figure 6B). Utilizing published indirect calorimetry data (Hui et al., 2020), we found that the amount of carbohydrate that is burned between fasting and feeding matches the changes in glucose turnover observed in these states (Figure 6C). This suggests that, at least in rodents refed during their active dark cycle, carbohydrate oxidation is the main way of clearing incoming dietary glucose.
Figure 6. Carbohydrate is primarily cleared through oxidation.

(A) Schematic indicating the measured fates of glucose.
(B) Glucose turnover in the fasted (n=9 mice) and fed states (n=5 mice) from Figure 5B.
(C) The rate of carbohydrate oxidation measured by indirect calorimetry (n=6 mice per condition) calculated based on data from (Hui et al., 2020).
(D) Carbohydrate consumption fluxes.
(E) Fluxes of carbohydrate oxidative measured independently by stable isotope tracing (as consumption flux not flowing into other metabolites) or by indirect calorimetry. (F) Model summarizing carbohydrate production and consumption fluxes in fasting and feeding. The model is calculated from metabolite interconverting fluxes determined by isotope labeled glucose, lactate, glycerol, alanine, and glutamine infusions in each of these conditions. Fluxes <50 nmol C min−1 g body weight−1 are not displayed.
Mean ± SEM.
There is literature evidence for the importance also of other glucose consuming pathways: glycogen synthesis and de novo lipogenesis (Irimia et al., 2010; Solinas et al., 2015). We quantified glycogen levels in tissues to calculate net glycogen synthesis rates. To quantify the rate of de novo lipogenesis, we performed infusion of D2O (Hellerstein et al., 1996; Zhang et al., 2017). When compared to carbohydrate oxidation, glycogen synthesis only accounted for a small fraction of whole-body carbohydrate consumption. Similarly, de novo lipogenesis is negligible in comparison to carbohydrate consumption by oxidation (Figure 6D). Therefore, in the tested circumstance in mice, oxidation is the quantitatively most important means of carbohydrate consumption.
A whole-body model of carbohydrate metabolism
Using observations of circulating metabolite labeling, without relying on tissue metabolite measurements, we can use mass and isotope balancing to obtain a pseudosteady-state model of glucose and lactate production and consumption fluxes (Methods S1). Such modeling does not consider which tissues or cell subtypes are responsible for particular fluxes, but provides an overview of whole-body metabolic activity, integrating the results from the different tracer experiments. We assume that unmeasured glucose and lactate effluxes (not making glycogen or other metabolites) reflect terminal oxidation. The resulting oxidation rates, determined independent of any calorimetry data, match the amount of carbohydrate oxidation measured with calorimetry (Figure 6E-F). This model emphasizes the high flux conversion of glucose to circulating lactate in both the fasted and the fed states, coupled with circulating lactate uptake and oxidation.
Compartmentalized flux from circulating glucose into circulating lactate
To bring tissue specificity to our quantitative analysis, we sought to determine the direct sources of tissue glycolytic intermediates and circulating lactate. Similar to our analysis of the source of circulating glucose, we utilized the infusion data from each potential substrate to determine the direct contribution of each to glycolysis. This analysis revealed that, in the fasted state, all tissues made glycolytic intermediates from glycogen, with the highest glycogen contribution in quadriceps muscles, heart, and liver. Circulating glucose also contributes broadly, with its contribution greatest in soleus (Figure 7A). Gluconeogenic substrates also make direct contributions to glycolytic intermediates, with lactate heavily used in liver and quadriceps, glycerol in kidney and adipose, and amino acids in pancreas.
Figure 7. Select tissues convert circulating glucose to circulating lactate, bypassing the bulk of whole-body glycolytic intermediates.

(A) Direct nutrient contributions to production of glycolytic intermediates in each tissue.
(B) Direct nutrient contributions to production of circulating lactate. The total height of each bar represents total lactate turnover.
(C) Illustration showing the amount of whole-body glycolytic intermediates that come from circulating glucose or glycogen, compared to the amount of circulating lactate that comes from each of these sources.
Mean ± SEM; n ≥ 3 mice for each nutrient and condition.
Feeding decreases the contribution of glycogen to glycolytic intermediates in many tissues, most markedly in the liver, and leads to greater contribution from circulating glucose in heart and brown adipose tissue (BAT). In quadriceps muscles, however, even in the fed state, only a small amount of glucose directly contributes to glycolytic intermediates (Figure 7A). As skeletal muscles are known to uptake glucose in response to insulin (James et al., 1985a), this suggests that glucose is first assimilated into the quadriceps’ glycogen pool before being used in glycolysis.
To gain a whole-body perspective on the source of glycolytic intermediates, we summed the contributions in different tissues which were weighted for their contribution to body mass. Largely due to the high mass of white muscle, glycogen was the dominant overall source of glycolytic intermediates.
We then performed similar analysis for circulating lactate. In the fasted state, approximately 43% of circulating lactate (205 nmol min−1 g−1) comes directly from circulating glucose, while about 20% (97 nmol min−1 g−1) comes from glycogen (Figure 7B). Upon feeding, the total production flux of lactate increases, with a shift toward increased percent contribution from glucose (64% or 419 nmol min−1 g−1) versus glycogen (18% or 116 nmol min−1 g−1) (Figure 7B), indicating persistent glycogen breakdown after eating, which aligns with the dominant contribution of glycogen to white muscle glycolytic intermediates even in the fed state. Overall, we observe a striking dichotomy between the source of whole-body glycolytic intermediates (glycogen) and lactate (circulating glucose), reflecting compartmentation of the flux between circulating glucose and circulating lactate (Figure 7C).
Discussion
Here we quantified the relative contribution of circulating nutrients and internally stored glycogen to glycolytic intermediate pools within tissues and to circulating glucose and lactate. This analysis revealed a striking discrepancy between the sources of circulating carbohydrate, glucose and lactate, and the sources of tissue glycolytic intermediates. In the fasted state, the greatest source of tissue glycolytic intermediates is glycogen, yet the largest source of circulating lactate is circulating glucose. This discrepancy argues for functional compartmentalization of glycolysis in mammals: most flux from circulating glucose to circulating lactate occurs in specialized locale, while most glycolytic intermediates reside in other compartments that excrete lactate relatively slowly in the sedentary state and are fed primarily by glycogen not glucose.
The glucose-driven, highly glycolytic compartment could be organized at the level of a subcellular compartment, cell type, or tissue. For example, even though glucose labels glycolytic intermediates in intestine to a lesser extent than it labels circulating lactate, there may well be cell types (or subcellular compartments) within the intestine that avidly convert circulating glucose to circulating lactate.
The lactate secreted by the glucose-driven, highly glycolytic compartment plays a central role in whole body energy production, as circulating lactate—produced originally from glucose—serves as a major TCA substrate (Hui et al., 2017; Liu et al., 2020)). The predominance of lactate as a direct TCA substrate need not reflect its net consumption, but rather may reflect rapid exchange between tissue pyruvate and circulating lactate (Jang et al., 2019; Liu et al., 2020; Rabinowitz and Enerbäck, 2020). Such exchange enables the equilibration of the cytosolic redox balance with circulating lactate and pyruvate (Corkey and Deeney, 2020; Goodman et al., 2020; Patgiri et al., 2020). The high magnitude of these reversible fluxes also ensures the capacity for net shuttling of three-carbon units between tissues and cell types (Brooks, 2018), likely from relatively rare highly glycolytic cells to other widely distributed cell types.
Interestingly, among tissues, different muscle types were on the opposite ends of the spectrum in terms of use of circulating glucose versus glycogen for glycolysis. Circulating glucose most strongly labels glycolytic intermediates in red muscle (soleus and diaphragm). In contrast, it makes the smallest contribution in quadriceps, a white muscle.
Muscle termed red or oxidative can be comprised of two different myofiber types: I and IIA, both with high oxidative capacity and mitochondrial density (Augusto et al., 2004). In mice, soleus is a mixture of both, while the diaphragm in mice is majority type IIA fibers (Greising et al., 2015). In contrast, the quadriceps in mice is majority type IIB fast-twitch fibers and is termed a white or glycolytic muscle. Interestingly, while referred to as “oxidative,” type I and type IIA fibers were previously shown to have high glucose uptake both basally and in response to insulin (Henriksen et al., 1991; James et al., 1985a; James et al., 1985b). Our data argue that they are major contributors to whole body glycolytic flux, converting circulating glucose to circulating lactate.
While flux from circulating glucose to circulating lactate localizes to specific tissues and/or cell types, glycogen metabolism proved to be a ubiquitous source of tissue glycolytic intermediates. This broad-based glycogenolysis renders each individual tissue capable of glycolytic energy production without drawing on the circulating glucose pool, partially dissociating circulating glucose homeostasis from tissue fuel requirements. It also makes glucose readily available for use by each tissue internally, even if circulation is impaired, e.g., due to a wound.
We further found that gluconeogenesis—at least a partial pathway from the TCA to glycolytic intermediates—occurs throughout the body. Strikingly, both glycolytic intermediate production via gluconeogenesis and complete gluconeogenic flux to produce circulating glucose persist in fed animals. A potential physiological benefit of fed-state gluconeogenesis is conversion of nutrients that cannot be readily stored (e.g. amino acids, lactate) into glucose and eventually glycogen. Understanding the physiological significance of this metabolic activity is an important future objective, as is determining its role in impaired regulation of circulating glucose in diabetes.
Muscle is a primary site of insulin resistance in diabetes, and our results suggest that red muscles play a central role in direct catabolism of circulating glucose (DeFronzo and Tripathy, 2009; Kelley et al., 2002; Rothman et al., 1995). Interestingly, there are shifts in fiber composition toward increased fast-twitch fiber types in obesity and type II diabetes (He et al., 2001; MÅrin et al., 1994; Oberbach et al., 2006; Stuart et al., 2013; Tanner et al., 2002). Given the greater direct use of circulating glucose for glycolysis in type I and type IIa fibers, as opposed to type IIb fibers, it is tempting to speculate that such fiber type switching contributes to insulin resistance. Further investigation into glucose handling in different muscle fiber types holds promise for advancing understanding and treatment of diabetes.
Limitations of the Study
The present study is limited to mice on a standard laboratory rodent diet, a high carbohydrate diet. While mice are omnivores like humans, they are much smaller, with higher surface-to-volume ratio, heat generation needs, and metabolic rate. In addition, mice generally have a higher of fraction fast-twitch muscle fibers than other mammals (Augusto et al., 2004). In the present study, the mice did not exercise. Exercise can induce both muscle glucose uptake and glycogen breakdown (Fueger et al., 2004; James et al., 1985b; Jensen and Richter, 2012). In addition, because fasted and feeding took place during their typical light and dark periods, the effects of feeding cannot be decoupled from the diurnal cycle.
In terms of measurement limitations, although our tracer experiments covered circulating glucose, major circulating gluconeogenic substrates, and glycogen, they did not explore all potential glycolytic intermediate precursors. For example, glycolytic intermediates could be derived locally from sources other than glycogen, such as glycerol from lipolysis or amino acids from protein breakdown. We did not probe such precursors. Perhaps for this reason, our measurements failed to account for a substantial fraction of glycolytic intermediates in several tissues. In addition, our analysis of glycogen contribution to glycolytic intermediates treats glycogen as a homogenous, well-mixed pool and does not account for the structure of glycogen, in which the labeling may be more concentrated on the outer layers of the glycogen molecule. Finally, and most fundamentally, our measurements were limited to ground whole tissues and therefore represent average glycolytic intermediate labeling within the tissue. A key implication of this study, based on comparing these tissue measurements to circulating metabolite labeling, is that glycolytic flux and gluconeogenic flux can be concentrated spatially within tissues, likely within particular cell types. Accordingly, direct measurement of glycolytic intermediate labeling in different cell types – by sorting or imaging – is a critical future objective.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Joshua Rabinowitz (joshr@princeton.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
El-Maven (Elucidata) and AccuCor Isotope Natural Abundance Correction used for the analysis of LC-MS data is available on GitHub (https://github.com/lparsons/accucor). Code used for direct contribution calculations was developed in MATLAB (Matworks) as is available as Supplemental Code S1. Isotope labeling data generated during this study is included as Supplemental Data S1.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse studies followed protocols approved by the Princeton University Animal Care and Use Committee. Animals were housed on a normal light cycle (8AM-8PM) and fed a standard rodent chow (PicoLab Rodent 20 5053, St. Louis, MO). Jugular vein catheterization was performed on 10–24 week old male C57BL/6N male mice (Charles River Laboratories). Aseptic surgery was performed to place a catheter in the right jugular vein and to connect the catheter to a vascular access button implanted under the skin on the back of the mouse. For arterial sampling, an additional catheter was implanted in the left carotid artery and connected to a two-channel vascular access button. Catheters and vascular access buttons (VABs) were bought from Instech Labs. Mice were allowed to recover from jugular vein catheterization surgery for at least 5 days before experimentation. For intravenous infusions, 13C-metabolites were prepared in saline at following concentrations: 0.2 M (fasted) and 0.8 M (fed) U-13C-glucose, 0.2 M (fasted) and 0.3 M (fed) 13C-alanine, 100 mM U-13C-glutamine, and 100 mM 13C-glycerol. Sodium 13C-lactate was diluted to 5% w/w in water. The infusion setup (Instech Laboratories) included a swivel and tether to allow the mouse to move around the cage freely. Infusion rate was set to 0.1 μl min−1 g−1. Tail blood was collected by tail snip (~10 μl) while arterial blood was collected through the carotid artery catheter. In both cases blood was directly collected into blood collection tubes with clotting factor (Sarstedt 16.442.100). Blood samples were stored on ice and then centrifuged at 16,000 × g for 10 minutes at 4°C to get serum samples. Tissue harvest was performed at the end of the infusion after euthanasia either by cervical dislocation or by intravenous administration of 70 mg/kg pentobarbital. Tissues were quickly dissected, clamped with a pre-cooled Wollenberger clamp, and dropped in liquid nitrogen. Propranolol was administered at a dosage of 50 mg/kg by oral gavage of 5 μl/g of 10 mg/ml propranolol (Sigma) in 0.1% Tween 80, 0.5% Hydroxypropyl-β-cyclodextrin (MP Biomedicals, Cat #153540).
METHOD DETAILS
Hyperinsulinemic-euglycemic clamp
Hyperinsulinemic-euglycemic clamps were performed according to standard procedure with the addition of stable isotope tracers. Carotid artery and jugular vein catheters were placed at least five days before procedure. On the day of the procedure mice were fasted at 8 am (ZT0). At approximately 11:30 am, a primed infusion of either [U-13C]glucose (0.2 M in saline at 1 μl min−1 g−1 body weight−1 for 2 minutes, then 0.1 μl min−1 g−1 body weight−1) or [U-13C]lactate (5% v/v Na·[U-13C]lactate in water at 1 μl min−1 g−1 body weight−1 for 2 minutes, then 0.1 μl min−1 g−1 body weight−1) was begun through jugular vein catheter to measure Fcirc values. Serum samples of 20 and 50 μl, respectively, were collected through the carotid artery catheter after 75 and 85 min of infusion. After 90 minutes, the insulin clamp was begun and by infusion of 2.5 mU min−1 kg−1 insulin (Humulin, Eli Lily), variable glucose (50% dextrose w/v or 2 M dextrose + 0.8 M [U-13C]glucose), and red blood cell mixture (1:1 saline washed blood cells:10 U/ml heparinized saline) at 3.5 μl min−1. When [U-13C]lactate was a tracer, it was additionally infused from a separate pump (5% v/v Na·[U13C]lactate in water at 0.1 μl min−1 g−1 body weight−1 ). Euglycemia was maintained (120–150 mg/dl) by measuring glucose every ten minutes with a glucometer and adjusting the glucose infusion rate as necessary. The insulin clamp was carried out for 2 h with blood sample collected every 10 minutes (20–50 μl) for the last 1 h. For tissue collection, 70 mg/kg pentobarbital was administered through a y-connector to the jugular catheter.
Serum Metabolite Extraction
Serum (2 μl) was added to 68 μl of −80°C 100% methanol, vortexed, and put on dry ice for at least 5 minutes. The resulting extract was centrifuged at 16,000 × g for 10 minutes at 4°C and supernatant was mixed 1:1 with 80% methanol. After centrifugation again at 16,000 × g for 10 minutes at 4°C, supernatant was transferred to tubes for LC-MS analysis.
Tissue Metabolite Extraction
Frozen tissue was ground by a Cyromill at cryogenic temperature (Retsch, Newtown, PA). Ground tissue was then weighed (~20 mg) and mixed with −20°C 40:40:20 methanol:acetonitrile:water with 0.5% formic acid (extraction solvent) at a concentration of 25mg/ml. Samples were briefly vortexed before neutralizing with 8 μl of 15% ammonium bicarbonate per 100 μl of extraction solvent. Extract was then vortexed and centrifuged twice at 16,000 × g for 20 minutes at 4°C before the final supernatant was transferred to LC-MS tubes for analysis.
Metabolite Measurement by LC-MS
A quadrupole-orbitrap mass spectrometer (Q Exactive, Thermo Fisher Scientific, San Jose, CA) operating in negative mode was coupled to hydrophobic interaction chromatography (HILIC) via electrospray ionization. Scans were performed from m/z 70 to 1000 at 1 Hz and 70,000 resolution. LC separation was on a XBridge BEH Amide column (2.1 mm × 150 mm × 2.5 μm particle size, 130 Å pore size; Water, Milford, MA) using a gradient of solvent A (20 mM ammonium acetate, 20 mM ammounium hydroxide in 95:5 water:acetonitrile, pH 9.45) and solvent B (acetonitrile). Flow rate was 150 μl/min. The LC gradient was: 0 min, 85% B; 2 min, 85% B; 3 min, 80% B; 5 min, 80% B; 6 min, 75% B; 7 min, 75% B; 8 min, 70% B; 9 min, 70% B; 10 min, 50% B; 12 min, 50% B; 13 min, 25% B; 16 min, 25% B; 18 min, 0% B; 23 min, 0% B; 24 min, 85% B. Autosampler temperature was 5°C, and injection volume was 5–10 μl for serum samples and 15 μl for tissue samples.
For improved detection of fructose-1,6-bisphosphate and 3-phosphoglycerate, selected ion monitoring (SIM) scans were added. For 3-phosphoglycerate and fructose-1,6-bisphosphate, scans were performed from 180–190 m/z and 336–350 m/z, respectively, from 13–15 minutes of the 25-minute gradient run with 70,000 resolution and maximum IT of 500 ms and AGC target of 3e6.
Data were analyzed using the MAVEN or El-MAVEN (Elucidata) software (Melamud et al., 2010). For tracer experiments, isotope labeling was corrected for natural 13C abundance (Su et al., 2017).
Ex vivo blood cell culture
Mice fed ad lib were anesthetized using ketamine/xylazine, and blood was collected via cardiac puncture into a heparinized syringe through a 25G needle. Blood was incubated at 37 °C with constant agitation. 50 μL aliquots of whole blood were taken over the course of 6 hours and extracted with 200 μL ice cold LC-MS MeOH, vortexed, and placed on dry ice for at least 30 minutes to produce an extract with a final composition of 80% MeOH. Metabolite extracts were spun at 4 °C for 15 minutes and the supernatant was transferred to a fresh tube. The supernatant was dried under a N2 gas and re-suspended in 60 μL ultrapure MilliQ water. Glucose and lactate concentrations were determined using the YSI 2900 biochemistry analyzer. Calculations assume 40% of blood volume is RBCs and that the blood content of the mouse is 0.07 ml/g (Hui et al., 2020).
Glycogen Hydrolysis and Measurement by LC-MS
Frozen cryomill ground tissue was weighed out (~10 mg) and mixed with 2 M hydrochloric acid (10 μl/mg of tissue). Glycogen acid hydrolysis was facilitated by placing in 80°C water bath for 2 hours. Samples were then neutralized with 12 μl/mg saturated ammonium bicarbonate before extracting with 88 μl/mg 50:50 methanol:acetonitrile. For brain samples, glycogen was digested with amyloglucosidase (Sigma). Cryomill ground brain samples were first suspended 1 mg/μl in 100mM sodium acetate buffer pH 4.5 and boiled for 5 minutes. 25 μl of sample were mixed with 25 μl of 20 mg/ml amyloglucosidase in the same 100mM sodium acetate buffer pH 4.5. Enzyme and tissue mix were incubated at 37°C for 4 hours and then extracted with 200 25 μl of 50:50 MeOH/ACN. All samples (both acid and enzyme digested) were centrifuged at 16,000 × g and supernatant transferred to LC-MS tube for analysis. All samples were run side-by-side with a tissue sample that was directly extracted (no hydrolysis) with 110 μl/mg 40:40:20 methanol:acetonitrile:water to measure free glucose. Samples were analyzed as described in the Metabolite Measurement by LC-MS section except scans were performed from m/z 178 to 188 and the resolution was 140,000. The LC gradient was: 0 min, 60% B; 1 min, 60% B; 1.5 min, 50% B; 3 min, 50% B; 4 min, 25% B; 6 min, 25% B; 7 min, 0% B; 7.5 min, 70% B; 10 min, 70% B. Injection volume was 10 μl. To subtract out free glucose, ion counts for non-hydrolyzed samples were subtracted from those of hydrolyzed samples to get the values for glycogen.
The concentration of glycogen at different time points was then used to calculate the net glycogen synthesis flux.
Direct Contribution Calculations
For metabolite pools, the mass isotopomer distribution (after natural isotope correction) was used to calculate the fractional carbon-atom labeling given by:
where n is the number of C atoms in the metabolite, i is the different mass isotopolgues (m + 0, m + 1 etc.) and m is the abundance of the given isotopolgoue as previously described(Fendt et al., 2013).
Normalized labeling is defined as the fractional labeling of the metabolite of interest divided by the fractional labeling of the tracer (e.g. of circulating glucose in a standard glucose infusion experiment). For pulse-chase experiments, glycogen labeling in the tissue of interest is used as the tracer labeling to calculate the normalized labeling of glycolytic intermediates.
The direct contribution of circulating nutrients and glycogen to tissue glycolytic intermediate, circulating glucose, and circulating lactate was calculated as described previously1. The logic is as follows: for each infusion condition, the measured labeling in a given metabolite pool is the sum of the direct contribution from the tracer and contribution from other substrates that acquire labeling from the tracer (these other substrates are sometimes referred to as secondary tracers). By performing independent infusions to probe each secondary tracer, we can solve for the direct contributions of each potential substrate to a given metabolite pool. For example, for glucose and glycogen, we can write two equations:
where LGI←glc represents the labeling observed in glycolytic intermediates from 2.5 h glucose infusion, Lglc←glc represents the labeling in glucose from the 2.5 h infusion of glucose, fGI←glc represents the direct contribution of circulating glucose to glycolytic intermediates, Lglycogen←glc represents the labeling in glycogen from 2.5 h infusion of glucose, fGI←glc represents the direct contribution of tissue glycogen to glycolytic intermediates, LGI←glycogen represents the labeling observed in glycolytic intermediates from the pulse-chase experiment, Lglc←glycogen represents the labeling in circulating glucose from the pulse-chase experiment, and Lglycogen←glycogen represents the labeling in glycogen from the pulse-chase experiment. As all of the L values are empirically measured, we can determine the f values by linear algebra (system of 2 equations with 2 unknowns). When expanding to more tracers (1..n), we can represent this in matrix format:
Both of the matrix on the left and the vector on the right are empirically measured. By matrix inversion, we can thereby determine the f vector containing the direct contributions. The same approach was applied to calculate the direct contributions to circulating lactate and glucose. Calculations were performed in MATLAB using constrained linear optimization solver “lsqlin”, where, the constraints are fi>=0 and sum(f)<=1. Errors of the output f were obtained by Monte Carlo simulations. We randomly sample each measured element L (in both the matrix and the vector) multiple times (50), assuming a normal distribution with the experimentally measured standard deviation. The linear solver was run for each randomly sampled set of L, from which the errors of f were determined. To quantify the fraction of body tissue that comes from each source, direct contributions were multiplied by tissues glycolytic intermediate concentrations from Table S1 and published tissue weight values(Brown et al., 1997).
Circulatory turnover flux measurements
To measure the circulatory turnover flux of a circulating metabolite, we infused uniformly 13C labeled form of the metabolite. At pseudo-steady state, we measure the mass isotope distribution of the metabolite in serum. The circulatory turnover flux, Fcirc, and carbon atom circulatory turnover flux, , were calculated as previously described4,5. The fraction of the metabolite that is fully labeled, Lm+n (e.g. m+6 of glucose), is used to calculated the circulatory turnover flux, Fcirc:
where R is the infusion rate of the labeled tracer. To determine the carbon-atom circulatory turnover flux we instead use the fractional labeling of the tracer, L.
Calculation of circulating metabolite interconversion fluxes
We calculated the direct contribution of all of our tracers (circulating glucose, lactate, alanine, glycerol, glutamine, and stored glycogen) to each other. Using this information and the carbon atom circulatory flux (), we can calculate the fluxes converting one circulating metabolite into another (interconversion fluxes), as described in detail by our group recently (Hui et al., 2020; Hui et al., 2017) and in Supplemental Methods S1.
To do this, we first calculate the total flux through a circulating nutrient k, Jcirc,k, and then multiply it by the direct contribution values (as fractions) to obtain the direct contributing fluxes (in units of nmol carbon/min/g). The total flux through a circulating nutrient is related to its , but somewhat greater if some of the influxes are themselves labeled. To determine the Jcirc,k, we first determine the fraction of influx that is unlabeled of the given metabolite, k, (ck) and use this constant to calculate the total circulatory flux through the metabolite:
Incorporation of interconversion fluxes into whole-body model of carbohydrate metabolism
See Methods S1 in the supplemental information.
Quantification of de novo lipogenesis
To quantify the rate of de novo lipogenesis in fasted and fed states, we infused 20% D2O saline intravenously into mice in 12 h intervals either from 8am to 8pm (ZT0-ZT12) or from 8pm to 8am (ZT12-ZT0). When D2O is infused, the D2O enrichment in serum and tissue is not at steady state but increasing linearly with time. The final serum enrichment (p1) is a linear function of time:
Rapid H-D exchange of tissue NADPH and D2O results in quick equilibrium between these pools24. NAPDH labeling (p2), therefore, also linearly increases with time:
Because synthesis of one palmitate molecule requires 14 H from NADPH and 7 H from H2O, the expected deuterium per palmitate (E) will be:
Define
f = the average de novo lipogenesis flux per hour (gram/hour)
D = measured average deuterium per fatty acid
C = total fat amount
For an infusion of duration t hours, the deuterium amount incorporated into new synthesized fatty acid is:
Newly synthesized palmitate (F) in t = 12 h is given by:
Values were then converted to nmol glucosyl units per hour where one 2-carbon unite equals 0.5 molecules of glucose.
Quantification of carbohydrate oxidation
Indirect calorimetry data recently published from our laboratory was used for the calculation of carbohydrate oxidation rates in fasting and fed states4. To calculate the approximate amount of VO2 consumption that comes from burning of carbohydrate, VO2 consumption and CO2 production rates were converted into units of nmol min−1 g body weight−1. For fasting values, we used the values spanning the last 2.5 h of an 8 h fast, which corresponds with our infusion experiments. Fed data was taken from the first 2.5 h after the lights were turned off which corresponds with our fed infusions data. Because more oxygen is consumed for fat oxidation than carbohydrate, we can calculate the amount of carbon used for oxygen consumption from carbohydrate, VO2carb, and from fat, VO2fat. To do this we solved the following equations:
Insulin measurement
Serum insulin concentrations were measured with Insulin ELISA kit (Crystal Chem) following the manufacturer’s instructions.
Quantification and Statistical Analysis
Data are presented as means with error bars representing the standard error of the mean. T-test and one-way or 2-way ANOVA tests with correction for multiple comparisons were utilized to calculate p-values. p-value of less than 0.05 was considered to be significant. Prism (GraphPad) was used for statistical analysis.
Supplementary Material
Document S1. Figures S1-S7 and Table S1.
Methods S1. Carbohydrate fluxes – rates of interconversion and oxidation and Table S2-S5. Related to Figure 6 and STAR Methods.
Code S1. Code used for calculation of direct contribution of circulating nutrients to metabolite pools. Related to Figure 7 and STAR Mehtods.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| D-glucose (U-13C6, 99%) | Cambridge Isotope Laboratories | CLM-1396 |
| Sodium D-lactate (13C3, 98%) 20% w/w in H2O | Cambridge Isotope Laboratories | CLM-10768 |
| L-Alanine (13C3, 99%) | Cambridge Isotope Laboratories | CLM-2184-H |
| Glycerol (13C3, 99%) | Cambridge Isotope Laboratories | CLM-1510 |
| L-Glutamine (13C5, 99%) | Cambridge Isotope Laboratories | CLM-1822-H |
| Propranolol Hydrochloride | Sigma-Aldrich | 1576005 |
| Heparin Sodium | Fisher Scientific | H19 |
| Humulin 100U/ml | Thermo Fisher | NC1415864 |
| Deuterium Oxide | Cambridge Isotope Laboratories | DLM-6-PK |
| Glycerokinase | Sigma | 6278 |
| Experimental Models: Cell Lines | ||
| Human: A5469 | ATCC | CCL-185 |
| Human: HCT 116 | ATCC | CCL-247 |
| Human: MIA PaCa-2 | ATCC | CRL-1420 |
| Human: PANC-1 | ATCC | CRL-1469 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57Bl/6N | Charles River Laboratories | C57Bl/6 |
| Software and Algorithms | ||
| MATLAB software | MathWorks | |
| Prism software | GraphPad | |
| MAVEN software | Princeton University | http://genomics-pubs.princeton.edu/mzroll/index.php |
| El-MAVEN software | Elucidata | https://elucidatainc.github.io/ElMaven/Downloads/ |
| Accucor | GitHub | https://github.com/lparsons/accucor |
| Biorender | Biorender | biorender.com |
| Others | ||
| XBridge BEH Amide XP column | Waters | 176002889 |
| Insulin ELISA Kit | Crystal Chem | 90080 |
| Mouse Jugular Vein Catheter | Instech Laboratories | C20PU-MJV1301 |
| Mouse Carotid Artery Catheter | Instech Laboratories | C10PU-MCA1459 |
| Mouse Vascular Access Button, 1-channel | Instech Laboratories | VABM1B/25 |
| Mouse Vascular Access Button, 2-channel | Instech laboratories | VABM2B/25R25 |
Highlights.
Most circulating lactate comes from circulating glucose
But most tissue glycolytic intermediates come from glycogen
This discrepancy reflects compartmentalization of glycolysis
Glycolytic flux is concentrated in red muscle and other specialized cell populations
Acknowledgements
T.T. is supported by the NIH grant F32DK118856. S.H. is supported by the NIH grant R00DK117066. This work was supported by NIH Pioneer award 1DP1DK113643, Diabetes Research Center grant P30 DK019525, and the Paul G. Allen Family Foundation grant 0034665. We thank members of the Rabinowitz lab for scientific discussions. Schematics were created with Biorender.com.
Footnotes
Declaration of Interests
J.D.R. is an advisor and stockholder in Colorado Research Partners; a paid consultant of Pfizer; a founder and stockholder in Toran Therapeutics; and inventor of patents held by Princeton University.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Augusto V, Padovani CR, and Rocha Campos GE (2004). Skeletal muscle fiber types in C57Bl6J mice. pp. 89–94. [Google Scholar]
- Ayala JE, Bracy DP, Malabanan C, James FD, Ansari T, Fueger PT, McGuinness OP, and Wasserman DH (2011). Hyperinsulinemic-euglycemic clamps in conscious, unrestrained mice. JoVE (Journal of Visualized Experiments), e3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala JE, Bracy DP, McGuinness OP, and Wasserman DH (2006). Considerations in the Design of Hyperinsulinemic-Euglycemic Clamps in the Conscious Mouse. Diabetes 55, 390. [DOI] [PubMed] [Google Scholar]
- Baron AD, Brechtel G, Wallace P, and Edelman SV (1988). Rates and tissue sites of non-insulin- and insulin-mediated glucose uptake in humans. American Journal of Physiology-Endocrinology and Metabolism 255, E769–E774. [DOI] [PubMed] [Google Scholar]
- Barrio JR, Satyamurthy N, Huang S-C, Scafoglio C, Yu A, Alavi A, and Krohn KA (2019). Does 2-FDG-PET Accurately Reflect Quantitative In vivo Glucose Utilization? Journal of Nuclear Medicine, jnumed.119.237446-jnumed.237119.237446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauckneht M, Cossu V, Castellani P, Piccioli P, Orengo AM, Emionite L, Di Giulio F, Donegani MI, Miceli A, Raffa S, et al. (2020). FDG uptake tracks the oxidative damage in diabetic skeletal muscle: An experimental study. Molecular Metabolism 31, 98–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Moshe S, Shapira Y, Moor AE, Manco R, Veg T, Bahar Halpern K, and Itzkovitz S. (2019). Spatial sorting enables comprehensive characterization of liver zonation. Nature Metabolism 1, 899–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks GA (2018). The Science and Translation of Lactate Shuttle Theory. (Cell Press; ), pp. 757–785. [DOI] [PubMed] [Google Scholar]
- Brown RP, Delp MD, Lindstedt SL, Rhomberg LR, and Beliles RP (1997). Physiological parameter values for physiologically based pharmacokinetic models. Toxicology and Industrial Health 13, 407–484. [DOI] [PubMed] [Google Scholar]
- Büsing KA, Schönberg SO, Brade J, and Wasser K. (2013). Impact of blood glucose, diabetes, insulin, and obesity on standardized uptake values in tumors and healthy organs on 18F-FDG PET/CT. Nuclear Medicine and Biology 40, 206–213. [DOI] [PubMed] [Google Scholar]
- Cheng X, Kim SY, Okamoto H, Xin Y, Yancopoulos GD, Murphy AJ, and Gromada J. (2018). Glucagon contributes to liver zonation. Proceedings of the National Academy of Sciences 115, E4111–E4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cori CF (1981). The Glucose–Lactic Acid Cycle and Gluconeogenesis. In Current Topics in Cellular Regulation. Estabrook RW, and Srere P, eds. (Academic Press; ), pp. 377–387. [PubMed] [Google Scholar]
- Corkey BE, and Deeney JT (2020). The Redox Communication Network as a Regulator of Metabolism. Frontiers in Physiology 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney KD, Bezwada D, Mashimo T, Pichumani K, Vemireddy V, Funk AM, Wimberly J, McNeil SS, Kapur P, Lotan Y, et al. (2018). Isotope Tracing of Human Clear Cell Renal Cell Carcinomas Demonstrates Suppressed Glucose Oxidation In Vivo. Cell Metabolism 28, 793–800.e792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson Shawn M., Papagiannakopoulos T, Olenchock Benjamin A., Heyman Julia E., Keibler Mark A., Luengo A, Bauer Matthew R., Jha Abhishek K., O’Brien James P., Pierce Kerry A., et al. (2016). Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metabolism 23, 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFronzo RA, Ferrannini E, and Simonson DC (1989). Fasting hyperglycemia in non-insulin-dependent diabetes mellitus: Contributions of excessive hepatic glucose production and impaired tissue glucose uptake. Metabolism 38, 387–395. [DOI] [PubMed] [Google Scholar]
- DeFronzo RA, and Tripathy D. (2009). Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. (American Diabetes Association), pp. S157–S163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deibert DC, and Defronzo RA (1980). Epinephrine-induced Insulin Resistance in Man. The Journal of Clinical Investigation 65, 717–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, Yang C, Do QN, Doucette S, Burguete D, et al. (2017). Lactate Metabolism in Human Lung Tumors. Cell 171, 358–371.e359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felig P, Pozefsk T, Marlis E, and Cahill GF (1970). Alanine: Key Role in Gluconeogenesis. Science 167, 1003–1004. [DOI] [PubMed] [Google Scholar]
- Fendt SM, Bell EL, Keibler MA, Davidson SM, Wirth GJ, Fiske B, Mayers JR, Schwab M, Bellinger G, Csibi A, et al. (2013). Metformin decreases glucose oxidation and increases the dependency of prostate cancer cells on reductive glutamine metabolism. Cancer Research 73, 4429–4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fueger PT, Hess HS, Posey KA, Bracy DP, Pencek RR, Charron MJ, and Wasserman DH (2004). Control of exercise-stimulated muscle glucose uptake by GLUT4 is dependent on glucose phosphorylation capacity in the conscious mouse. Journal of Biological Chemistry 279, 50956–50961. [DOI] [PubMed] [Google Scholar]
- Gerich JE (1993). Control of glycaemia. Baillière’s Clinical Endocrinology and Metabolism 7, 551–586. [DOI] [PubMed] [Google Scholar]
- Gerich JE, Meyer C, Woerle HJ, and Stumvoll M. (2001). Renal Gluconeogenesis. Its importance in human glucose homeostasis 24, 382–391. [DOI] [PubMed] [Google Scholar]
- Goodman RP, Markhard AL, Shah H, Sharma R, Skinner OS, Clish CB, Deik A, Patgiri A, Hsu Y-HH, Masia R, et al. (2020). Hepatic NADH reductive stress underlies common variation in metabolic traits. Nature 583, 122–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greising SM, Medina-Martínez JS, Vasdev AK, Sieck GC, and Mantilla CB (2015). Analysis of muscle fiber clustering in the diaphragm muscle of sarcopenic mice. Muscle and Nerve 52, 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruetter R, Magnusson I, Rothman DL, Avison MJ, Shulman RG, and Shulman GI (1994). Validation of 13C NMR measurements of liver glycogen in vivo. Magnetic Resonance in Medicine 31, 583–588. [DOI] [PubMed] [Google Scholar]
- Halpern KB, Shenhav R, Matcovitch-Natan O, Tóth B, Lemze D, Golan M, Massasa EE, Baydatch S, Landen S, Moor AE, et al. (2017). Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature 542, 352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatting M, Tavares CDJ, Sharabi K, Rines AK, and Puigserver P. (2018). Insulin regulation of gluconeogenesis. Ann N Y Acad Sci 1411, 21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Watkins S, and Kelley DE (2001). Skeletal Muscle Lipid Content and Oxidative Enzyme Activity in Relation to Muscle Fiber Type in Type 2 Diabetes and Obesity. Diabetes 50, 817–823. [DOI] [PubMed] [Google Scholar]
- Hellerstein MK, Schwarz J-M, and Neese RA (1996). Regulation of hepatic de novo lipogenesis in humans. Annual review of nutrition 16, 523–557. [DOI] [PubMed] [Google Scholar]
- Henriksen EJ, Rodnick KJ, Mondon CE, James DE, and Holloszy JO (1991). Effect of denervation or unweighting on GLUT-4 protein in rat soleus muscle. Journal of Applied Physiology 70, 2322–2327. [DOI] [PubMed] [Google Scholar]
- Hui S, Cowan AJ, Zeng X, Yang L, TeSlaa T, Li X, Bartman C, Zhang Z, Jang C, Wang L, et al. (2020). Quantitative Fluxomics of Circulating Metabolites. Cell Metabolism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui S., Ghergurovich JM., Morscher RJ., Jang C., Teng X., Lu W., Esparza LA., Reya T., Le Z., Yanxiang Guo J., et al. (2017). Glucose feeds the TCA cycle via circulating lactate. Nature 551, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irimia JM, Meyer CM, Peper CL, Zhai L, Bock CB, Previs SF, McGuinness OP, DePaoli-Roach A, and Roach PJ (2010). Impaired Glucose Tolerance and Predisposition to the Fasted State in Liver Glycogen Synthase Knock-out Mice. Journal of Biological Chemistry 285, 12851–12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James DE, Jenkins AB, and Kraegen EW (1985a). Heterogeneity of insulin action in individual muscles in vivo: euglycemic clamp studies in rats. American Journal of Physiology-Endocrinology and Metabolism 248, E567–E574. [DOI] [PubMed] [Google Scholar]
- James DE, Kraegen EW, and Chisholm DJ (1985b). Effects of exercise training on in vivo insulin action in individual tissues of the rat. The Journal of clinical investigation 76, 657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang C, Hui S, Zeng X, Cowan AJ, Wang L, Chen L, Morscher RJ, Reyes J, Frezza C, Hwang HY, et al. (2019). Metabolite Exchange between Mammalian Organs Quantified in Pigs. Cell Metabolism 30, 594–606.e593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen TE, and Richter EA (2012). Regulation of glucose and glycogen metabolism during and after exercise. (John Wiley & Sons, Ltd; ), pp. 1069–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhlin-Dannfelt A, Terblanche S, Fell R, Young J, and Holloszy J. (1982). Effects of beta-adrenergic receptor blockade on glycogenolysis during exercise. Journal of Applied Physiology 53, 549–554. [DOI] [PubMed] [Google Scholar]
- Katz NR (1992). Metabolic Heterogeneity of Hepatocytes across the Liver Acinus. The Journal of Nutrition 122, 843–849. [DOI] [PubMed] [Google Scholar]
- Kelley DE, He J, Menshikova EV, and Ritov VB (2002). Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 51, 2944–2950. [DOI] [PubMed] [Google Scholar]
- Kernstine KH, Faubert B, Do QN, Rogers TJ, Hensley CT, Cai L, Torrealba J, Oliver D, Wachsmann JW, Lenkinski RE, et al. (2020). Does Tumor FDG-PET Avidity Represent Enhanced Glycolytic Metabolism in Non-Small Cell Lung Cancer? The Annals of Thoracic Surgery 109, 1019–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs HA (1964). The Croonian Lecture, 1963 Gluconeogenesis. Proceedings of the Royal Society of London. Series B. Biological Sciences 159, 545–564. [DOI] [PubMed] [Google Scholar]
- Liu S, Dai Z, Cooper DE, Kirsch DG, and Locasale JW (2020). Quantitative Analysis of the Physiological Contributions of Glucose to the TCA Cycle. Cell Metabolism 32, 619–628.e621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MÅrin P., Andersson B., Krotkiewski M., and Björntorp P. (1994). Muscle Fiber Composition and Capillary Density in Women and Men With NIDDM. Diabetes Care 17, 382–386. [DOI] [PubMed] [Google Scholar]
- Melamud E, Vastag L, and Rabinowitz JD (2010). Metabolomic Analysis and Visualization Engine for LC−MS Data. Analytical Chemistry 82, 9818–9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MC, Cherrington AD, Cline G, Pagliassotti MJ, Jones EM, Neal DW, Badet C, and Shulman GI (1991). Sources of carbon for hepatic glycogen synthesis in the conscious dog. The Journal of Clinical Investigation 88, 578–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Doherty RM, Jensen PB, Anderson P, Jones JG, Berman HK, Kearney D, and Newgard CB (2000). Activation of direct and indirect pathways of glycogen synthesis by hepatic overexpression of protein targeting to glycogen. The Journal of Clinical Investigation 105, 479–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberbach A, Bossenz Y, Lehmann S, Niebauer J, Adams V, Paschke R, Schön MR, Blüher M, and Punkt K. (2006). Altered Fiber Distribution and Fiber-Specific Glycolytic and Oxidative Enzyme Activity in Skeletal Muscle of Patients With Type 2 Diabetes. Diabetes Care 29, 895–900. [DOI] [PubMed] [Google Scholar]
- Patgiri A, Skinner OS, Miyazaki Y, Schleifer G, Marutani E, Shah H, Sharma R, Goodman RP, To T-L, Robert Bao X, et al. (2020). An engineered enzyme that targets circulating lactate to alleviate intracellular NADH:NAD+ imbalance. Nature Biotechnology 38, 309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowitz JD, and Enerbäck S. (2020). Lactate: the ugly duckling of energy metabolism. Nature Metabolism 2, 566–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose IA, and Warms JVB (1966). Control of Glycolysis in the Human Red Blood Cell. Journal of Biological Chemistry 241, 4848–4854. [PubMed] [Google Scholar]
- Rothman DL, Magnusson I, Cline G, Gerard D, Kahn CR, Shulman RG, and Shulman GI (1995). Decreased muscle glucose transport/phosphorylation is an early defect in the pathogenesis of non-insulin-dependent diabetes mellitus. Proceedings of the National Academy of Sciences of the United States of America 92, 983–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman RG, and Rothman DL (2017). The Glycogen Shunt Maintains Glycolytic Homeostasis and the Warburg Effect in Cancer. Trends in Cancer 3, 761–767. [DOI] [PubMed] [Google Scholar]
- Solinas G, Borén J, and Dulloo AG (2015). De novo lipogenesis in metabolic homeostasis: More friend than foe? Molecular Metabolism 4, 367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark R, and Kibbey RG (2014). The mitochondrial isoform of phosphoenolpyruvate carboxykinase (PEPCK-M) and glucose homeostasis: has it been overlooked? Biochim Biophys Acta 1840, 1313–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart CA, McCurry MP, Marino A, South MA, Howell MEA, Layne AS, Ramsey MW, and Stone MH (2013). Slow-twitch fiber proportion in skeletal muscle correlates with insulin responsiveness. Journal of Clinical Endocrinology and Metabolism 98, 2027–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X, Lu W, and Rabinowitz JD (2017). Metabolite Spectral Accuracy on Orbitraps. Analytical Chemistry 89, 5940–5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland EW, and Wosilait WD (1956). The relationship of epinephrine and glucagon to liver phosphorylase. J. biol. Chem 218, 459–468. [PubMed] [Google Scholar]
- Tanner CJ, Barakat HA, Dohm GL, Pories WJ, MacDonald KG, Cunningham PRG, Swanson MS, and Houmard JA (2002). Muscle fiber type is associated with obesity and weight loss. American Journal of Physiology-Endocrinology and Metabolism 282, E1191–E1196. [DOI] [PubMed] [Google Scholar]
- Taylor R, Price TB, Rothman DL, Shulman RG, and Shulman GI (1992). Validation of 13c nmr measurement of human skeletal muscle glycogen by direct biochemical assay of needle biopsy samples. Magnetic Resonance in Medicine 27, 13–20. [DOI] [PubMed] [Google Scholar]
- Wetter TJ, Gazdag AC, Dean DJ, and Cartee GD (1999). Effect of calorie restriction on in vivo glucose metabolism by individual tissues in rats. American Journal of Physiology-Endocrinology and Metabolism 276, E728–E738. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Chen L, Liu L, Su X, and Rabinowitz JD (2017). Chemical Basis for Deuterium Labeling of Fat and NADPH. Journal of the American Chemical Society 139, 14368–14371. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1. Figures S1-S7 and Table S1.
Methods S1. Carbohydrate fluxes – rates of interconversion and oxidation and Table S2-S5. Related to Figure 6 and STAR Methods.
Code S1. Code used for calculation of direct contribution of circulating nutrients to metabolite pools. Related to Figure 7 and STAR Mehtods.
Data Availability Statement
El-Maven (Elucidata) and AccuCor Isotope Natural Abundance Correction used for the analysis of LC-MS data is available on GitHub (https://github.com/lparsons/accucor). Code used for direct contribution calculations was developed in MATLAB (Matworks) as is available as Supplemental Code S1. Isotope labeling data generated during this study is included as Supplemental Data S1.