

Abstract
Blood vessels support tumours by providing nutrients and oxygen, while also acting as conduits for the dissemination of cancer1. Here we use mouse models of breast and lung cancer to investigate whether endothelial cells also have active ‘instructive’ roles in the dissemination of cancer. We purified genetically tagged endothelial ribosomes and their associated transcripts from highly and poorly metastatic tumours. Deep sequencing revealed that metastatic tumours induced expression of the axon-guidance gene Slit2 in endothelium, establishing differential expression between the endothelial (high Slit2 expression) and tumoural (low Slit2 expression) compartments. Endothelial-derived SLIT2 protein and its receptor ROBO1 promoted the migration of cancer cells towards endothelial cells and intravasation. Deleting endothelial Slit2 suppressed metastatic dissemination in mouse models of breast and lung cancer. Conversely, deletion of tumoural Slit2 enhanced metastatic progression. We identified double-stranded RNA derived from tumour cells as an upstream signal that induces expression of endothelial SLIT2 by acting on the RNA-sensing receptor TLR3. Accordingly, a set of endogenous retroviral element RNAs were upregulated in metastatic cells and detected extracellularly. Thus, cancer cells co-opt innate RNA sensing to induce a chemotactic signalling pathway in endothelium that drives intravasation and metastasis. These findings reveal that endothelial cells have a direct instructive role in driving metastatic dissemination, and demonstrate that a single gene (Slit2) can promote or suppress cancer progression depending on its cellular source.
Highly metastatic cells exhibit an enhanced capacity to recruit endothelial cells into the primary tumour site2–4. Moreover, it has previously been shown that endothelial cells release ‘angiocrine’ growth factors and cytokines that are detected by haematopoietic stem cells and epithelial cells5–9. These observations suggest that endothelial cells might provide molecular signals that are detected by tumour cells and that could consequently promote metastasis2.
SLIT2 induced in the metastatic endothelium
To identify a potential endothelial-derived factor that may promote metastasis, we used a systematic approach that integrated in vivo Cre-mediated ribosomal tagging (RiboTag)10 in endothelial cells with affinity purification of endothelial ribosome-bound messenger RNAs (mRNAs) followed by deep sequencing. The axon-guidance gene Slit2 was the top secreted factor that was upregulated in the vasculature of highly metastatic mouse melanoma B16F10 tumours relative to vessels of less-metastatic isogenic B16F0 tumours (Fig. 1a, b). Quantitative real-time PCR (qPCR) of ribosome-bound mRNAs isolated from the endothelial cells of tumours in RiboTag mice validated these findings (Fig. 1c). Immunofluorescent staining for SLIT2 and the endothelial marker endomucin in B16F0, B16F10 and the isogenic mouse mammary tumour lines 67NR (nonmetastatic) and 4T1 (highly metastatic) revealed increased SLIT2 expression within the primary tumour blood vessels of the highly metastatic 4T1 and B16F10 lines, relative to the tumour blood vessels of the poorly metastatic 67NR and B16F0 lines (Fig. 1d, e). Conditioned medium from highly metastatic 4T1 cells was sufficient to induce SLIT2 expression in mouse lung endothelial cells, as detected by immunofluorescent staining (Fig. 1f) and qPCR (Extended Data Fig. 1a, b). Thus, highly metastatic breast and melanoma cells induce SLIT2 expression in endothelial cells.
Fig. 1 |. Highly metastatic tumours induce SLIT2 expression in endothelial cells.
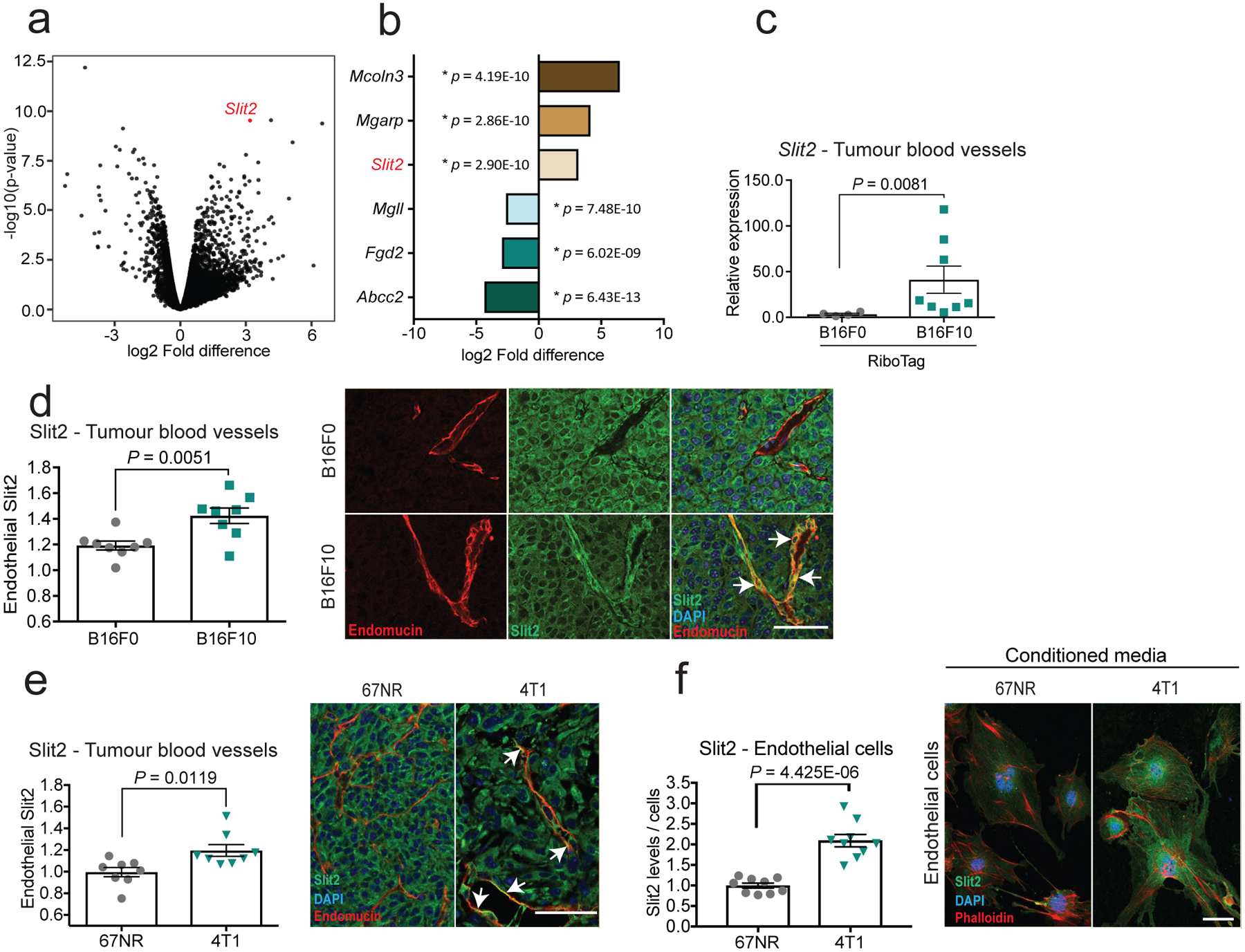
The RiboTag model and endothelial-specific (CDH5) Cre-mediated recombination were used to immunopurify haemagglutinin (HA)-tagged RPL22 ribosomal protein and associated transcripts for sequencing. a, b, Volcano plot (a) and bar chart (b) show log2-transformed fold differences in endothelial gene expression between highly metastatic B16F10 (n = 7) and poorly metastatic B16F0 (n = 5) tumours. Two-sided Wald tests. c, d, Dot plots depict Slit2 expression in tumour blood vessels determined by quantitative real-time PCR (c) (B16F0, n = 4; B16F10, n = 8; two-sided Mann–Whitney test), and fluorescent intensities of SLIT2 expression in tumour blood vessels (d) in highly metastatic B16F10 tumours (n = 8) compared to poorly metastatic B16F0 tumours (n = 8). Unpaired two-tailed Student’s t-test. Mean ± s.e.m. Representative immunofluorescent images (right) depict SLIT2 (green) and endomucin (red) expression, and DAPI staining (blue). Arrows indicate co-localization of SLIT2 and endomucin in blood vessels. e, Dot plots depict fluorescent intensities of SLIT2 protein immunostaining in tumour blood vessels in highly metastatic 4T1 (n = 8) and nonmetastatic 67NR (n = 8 unpaired two-tailed Student’s t-test) isogenic mammary tumours (e) and in mouse lung endothelial cells treated with conditioned medium from 67NR (n = 9) and 4T1 cells (f) (n = 9) (two-tailed Student’s t-test) cells. Representative images as in d. Data are mean ± s.e.m.
Endothelial SLIT2 drives metastasis
We used an inducible knockout model using Cdh5(PAC)-creERT211 miceto drive endothelial-specific deletion of Slit212 (hereafter referred to as ecSLIT2 knockout). Endothelial SLIT2 inactivation was confirmed at the RNA and protein levels by qPCR and western blotting of lung endothelial cells, respectively (Fig. 2a, b). Furthermore, immunofluorescent staining of tumour sections for SLIT2 and endomucin confirmed SLIT2 deletion in tumour blood vessels (Fig. 2c).
Fig. 2 |. Endothelial-specific deletion of SLIT2 suppresses metastasis in several models of metastatic breast and lung cancer.
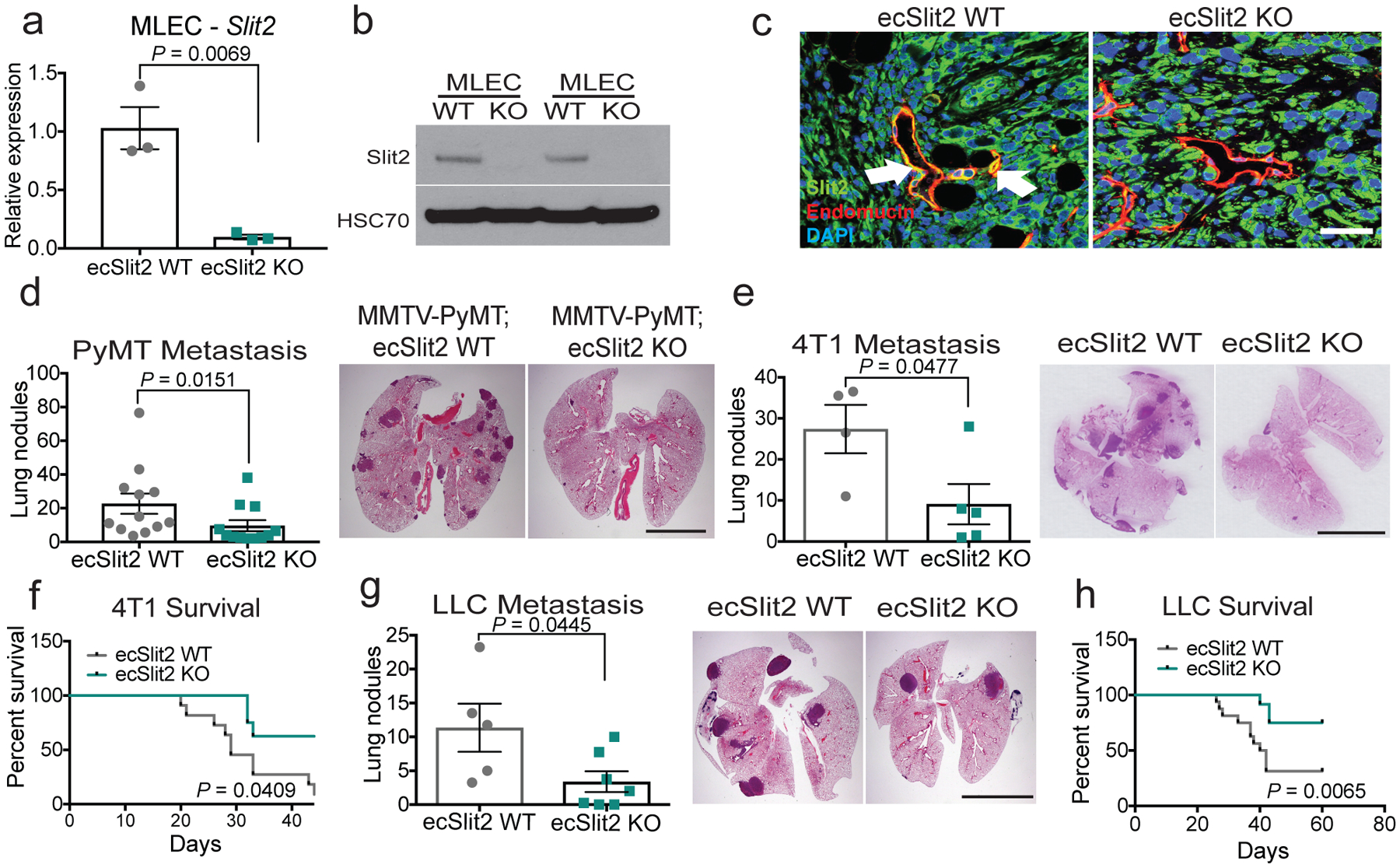
a, b, Slit2 expression by qPCR (mean Slit2 expression in ecSLIT2 knockout (KO) relative to wild type (WT) ± s.e.m; n = 3; two-tailed Student’s t-test) (a), and SLIT2 expression by western blot for ecSLIT2-knockout and wild-type mouse lung endothelial cells (MLECs). HSC70 was used as a loading control. c, Representative immunofluorescent images depict SLIT2 (green) and endomucin (red) expression, and DAPI staining (blue), in wild-type (left) and ecSLIT2-knockout (right) mice bearing 4T1 tumours. Arrows indicate co-localization of SLIT2 and endomucin in blood vessels. Three independent experiments. Scale bar, 100 μm. d, e, g, Dot plots represent numbers of metastatic lung nodules per mouse, from MMTV-PyMT ecSLIT2 wild-type (n = 12) and MMTV-PyMT ecSLIT2-knockout (n = 12) mice (two-tailed Mann–Whitney test) (d); 4T1-bearing wild-type (n = 4) and ecSLIT2-knockout (n = 5) mice (unpaired two-tailed Student’s t-test) (e); and Lewis-lung-carcinoma (LLC)-bearing wild-type (n = 5) and ecSLIT2-knockout (n = 7) mice (two-tailed Mann–Whitney test) (g). Representative haematoxylin and eosin (H&E) images of lungs are shown (right). f, h, Kaplan–Meier curves comparing post-surgical survival after primary tumour resection of 4T1-bearing wild-type (grey) (n = 11) and ecSLIT2-knockout (green) (n = 8) mice (Gehan–Breslow–Wilcoxon test) (f) and LLC-bearing wild-type (grey) (n = 16) and ecSLIT2-knockout (green) (n = 12) mice (Gehan–Breslow–Wilcoxon test) (h). In all H&E images, scale bars are 1 cm. Data are mean ± s.e.m.
Vascular Slit2 deletion in the genetically initiated MMTV-PyMT mammary tumour mouse model (which expresses polyoma virus middle T antigen (PyMT) under control of mouse mammary tumour virus (MMTV) substantially reduced the formation of lung metastasis, without impairing primary tumour growth or angiogenesis (Fig. 2d, Extended Data Fig. 2a, d, g, h). Furthermore, in a different model, primary 4T1 mammary tumours growing in ecSLIT2-knockout mice displayed no significant impairment in growth rate (Extended Data Fig. 2b) or angiogenesis (Extended Data Fig. 2e). However, ecSLIT2-knockout mice containing 4T1 tumours developed significantly fewer metastases than did wild-type littermate controls, and ecSLIT2-knockout mice exhibited increased survival upon primary tumour resection relative to wild-type controls (Fig. 2e, f). Injection of cancer cells directly into the venous circulation—which bypasses the primary tumour site—did not significantly affect metastatic colonization or survival in ecSLIT2-knockout mice relative to wild-type littermate controls (Extended Data Fig. 3a–f). We observed outcomes similar to those of the 4T1 model when using the Lewis lung carcinoma model (Fig. 2g, h, Extended Data Fig. 2c, f). These observations reveal that endothelial SLIT2 promotes metastasis in both syngeneic breast and lung cancer models and in a genetically induced model of breast cancer. Importantly, and consistent with a lack of impaired primary tumour growth in these models, 4T1 tumours in ecSLIT2-knockout mice displayed normal blood vessel density, perfusion and permeability (Extended Data Fig. 2i, j), which suggests that conditional Slit2 deletion within the tumour blood vessels of adult mice does not impair angiogenesis or the vascular function required to sustain primary tumour growth. Moreover, these findings suggest that endothelial SLIT2 may promote intravasation by tumour cells from the primary tumour site.
Endothelial SLIT2 drives intravasation
To better understand the mechanism by which endothelial SLIT2 promotes cancer progression, circulating tumour cells that express luciferase–ZsGreen were isolated from the blood of wild-type and ecSLIT2-knockout mice that bore 4T1 mammary tumours. Bioluminescence quantification of circulating tumour cells revealed that endothelial Slit2 deletion significantly reduced intravasation by tumour cells (Fig. 3a). Depending on context, SLIT2 has previously been shown to act as both a chemoattractant and chemorepellent, instructing axon guidance or the migration of neuronal progenitor cells in the developing nervous system13,14, as well as tissue morphogenesis15. We hypothesized that endothelial SLIT2 may recruit tumour cells towards blood vessels and, consequently, facilitate intravasation by tumour cells. We thus determined whether recombinant SLIT2 could promote the migration of tumour cells. Using a transwell migration assay, we exposed B16F10 and 4T1 tumour cells to increasing concentrations of recombinant mouse SLIT2. Both cell lines displayed an enhanced migration towards increasing concentrations of mouse SLIT2 (Fig. 3b). Next, endothelial cells that overexpress SLIT2 or ecSLIT2-knockout cells were plated in the lower chamber of a transwell assay and 4T1 tumour cells were plated in the upper chamber. Endothelial cells that overexpress SLIT2 induced greater transwell migration of tumour cells (Fig. 3c). By contrast, SLIT2-deficient endothelial cells and endothelial cells that express only the C-terminal fragment of SLIT216 exhibited a reduced capacity to induce the transwell migration of tumour cells (Fig. 3c, Extended Data Fig. 4a, b, e, f). These observations reveal that SLIT2 produced by endothelial cells can act as a recruitment signal to promote the migration of cancer cells.
Fig. 3 |. Endothelial SLIT2 promotes intravasation and migration of tumour cells via tumoural ROBO1.
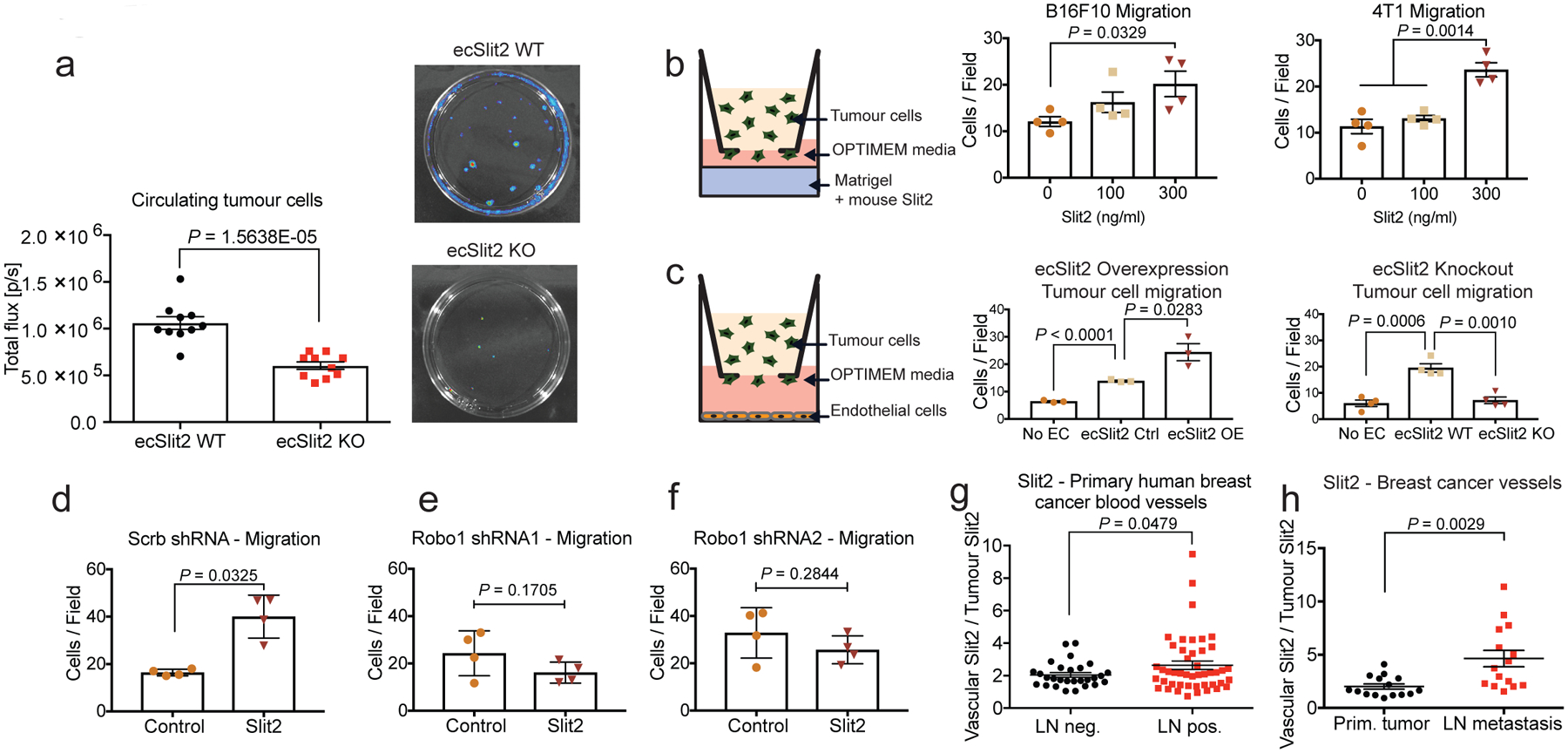
a, Left, dot plot depicts bioluminescence quantification of luciferase-positive circulating tumour cell colonies isolated from whole blood of wild-type (n = 10) and ecSLIT2-knockout (n = 10) mice. Unpaired two-tailed Student’s t-test. Right, representative images of luciferase-positive colonies cultured from blood. b, Schematic (left) and dot plot quantification (right) of quantified B16F10 and 4T1 tumour cells per optical field (10× magnification) that migrated across the transwell towards increasing concentrations of recombinant SLIT2. B16F10 and 4T1 migration, n = 4 for each condition. Two-tailed Student’s t-test. c, Schematic (left) and dot plot quantification (right) of 4T1 tumour cells per optical field that migrated across the transwell towards (middle and right) no endothelial cells (EC) (n = 3), wild-type endothelial cells (n = 3) and SLIT2-overexpressing (OE) endothelial cells (n = 3). Bar chart (right) depicts the same migration assay with no endothelial cells (n = 4), wild-type endothelial cells (n = 4) or ecSLIT2-knockout endothelial cells (n = 4). Two-tailed Student’s t-test. d–f, Dot plots represent number of control or Robo1 shRNA-depleted B16F10 tumour cells that migrated towards 300 ng ml−1 recombinant mouse SLIT2 per optical field (10× magnification). d, Scrambled shRNA control (control, n = 4; SLIT2, n = 4). e, Robo1 shRNA hairpin 1 (control, n = 4; SLIT2, n = 4). f, Robo1 shRNA hairpin 2 (control, n = 4; SLIT2, n = 4). Two-tailed Student’s t-test. g, h, Dot plot represents NIH tissue microarray of blood vessels of primary human breast cancer (g) or Abcam tissue array of breast cancer vessels (h) with vascular or tumoural SLIT2 fluorescence intensity of lymph-node (LN)-negative (n = 28) and -positive (n = 46) primary breast cancers (g) (unpaired one-tailed Student’s t-test) or primary (n = 15) or lymph-node-metastatic (n = 15) cancers (h) (two-tailed Student’s t-test). Data are mean ± s.e.m.
To define how ecSLIT2 promotes the migration of tumour cells, we used short-hairpin RNAs (shRNAs) to knock down Robo1—the predominant SLIT2 receptor across the cell lines studied herein. The depletion of ROBO1 in B16F10 tumour cells reduced the migration of cancer cells towards a gradient of recombinant SLIT2 (Fig. 3d–f), consistent with previously described promigratory and cancer-promoting roles for ROBO receptors17. Consistent with a prometastatic role, expression of Robo1 was higher in metastatic 4T1 cells relative to nonmetastatic 67NR cells and in metastatic 4T1 cells relative to parental 4T1 cells (Extended Data Fig. 5a, b). These findings implicate ROBO1 as a tumoural receptor that responds to endothelial-derived SLIT2 to mediate the migration of cancer cells towards the endothelium and intravasation.
We next analysed endothelial and tumoural expression of SLIT2 protein in tissue microarrays of human breast cancer from the Cooperative Human Tissue Network (https://www.chtn.org/). Higher-stage primary breast tumours that exhibit lymph node metastases (and confer reduced survival outcomes) expressed significantly higher endothelial SLIT2 relative to lower-stage (lymph-node-negative) primary tumours (Fig. 3g). Additionally, in an independent, commercially available tissue microarray for which primary tumour and metastatic lymph node samples were available, endothelial expression of SLIT2 protein was significantly higher in the endothelium of lymph node metastases relative to the endothelium of primary tumours (Fig. 3h). Higher endothelial expression of SLIT2 in 20 patient-derived xenografts from patients with breast cancer also tended be associated with reduced survival of the patient (Extended Data Fig. 5c). Moreover, analysis of mRNA-sequencing data from breast tumours from patients and the matched circulating tumour cells18 revealed reduced SLIT2 expression levels in circulating tumour cells relative to the associated primary tumours (Extended Data Fig. 5d). Consistent with these findings, reduced SLIT2 expression in publicly available gene-expression data from 1,660 breast tumours was significantly associated with a higher likelihood of relapse (Extended Data Fig. 5e), and higher ROBO1 expression in 3,951 breast cancers was also associated with reduced relapse-free survival (Extended Data Fig. 5f). Additionally, ROBO1 depletion in human MDA-MB-231 breast cancer cells reduced orthotopic metastasis in NOD-SCID-gamma (NSG) mice (Extended Data Fig. 10a–d). These findings support a model in which enhanced expression of endothelial SLIT2 relative to tumoural SLIT2 acts upon tumoural ROBO1 to drive cancer metastasis.
Tumour RNA drives endothelial SLIT2 trough TLR3
To identify the tumour-derived factor(s) that induce SLIT2 in endothelial cells, we treated endothelial cells with conditioned medium from highly metastatic 4T1 cells. Conditioned medium from 4T1 cells was filtered with different pore sizes; we observed that the flow-through from the 10-kDa filtration did not induce Slit2 upregulation in endothelial cells, as assessed by qPCR (Fig. 4a). Filtration with a 10-kDa membrane would exclude proteins larger than 10 kDa as well as nucleic acids. Consistent with the involvement of nucleic acids, treatment of the 4T1 conditioned medium with RNase A (Fig. 4b)—but not DNase I (Extended Data Fig. 1d)—abrogated SLIT2 induction, which suggests that tumour-derived RNA may induce endothelial SLIT2. Consistent with this hypothesis, heat inactivation of the conditioned medium also abolished SLIT2 induction in endothelial cells (Extended Data Fig. 1e).
Fig. 4 |. dsRNA secreted by metastatic cells activates endothelial TLR3 to induce SLIT2 expression.
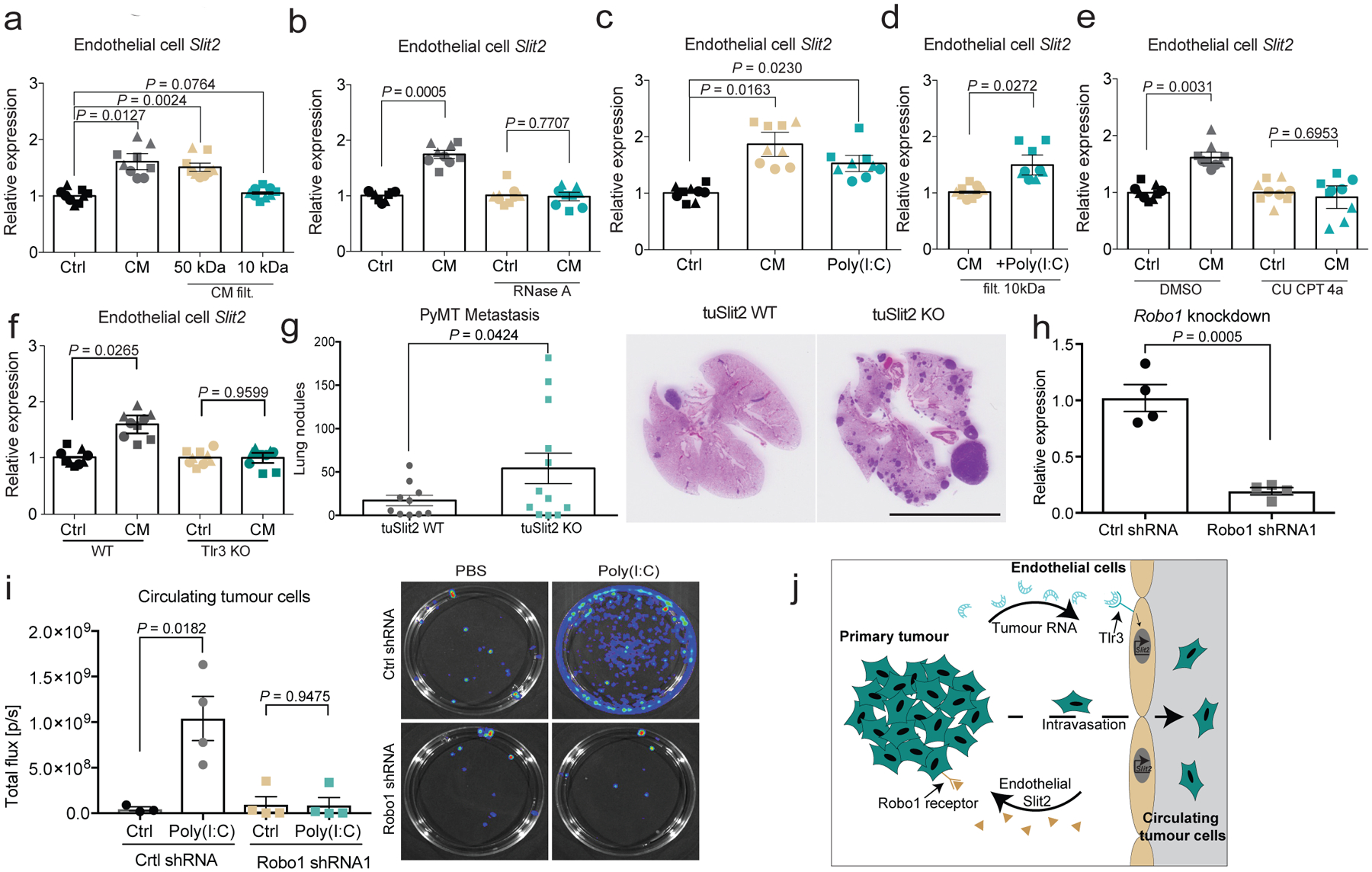
a, b, Dot plots represent Slit2 mRNA expression by qPCR in endothelial cells exposed to 4T1 conditioned medium (CM) (n = 3), basal OPT medium (control, n = 3), and 4T1 conditioned medium that had undergone 10-kDa (n = 3) or 50-kDa (n = 3) filtration (a) or RNase A treatment (25 μg ml−1) (n = 3) (b). Two-tailed Student’s t-test. c, d, Same as a, except that poly(I:C) (2.5 μg ml−1) was added to basal medium (control, n = 3; conditioned medium, n = 3; poly(I:C), n = 3) (two-tailed Student’s t-test) (c) or to 10-kDa-filtered 4T1 conditioned medium (conditioned medium, n = 3; +poly(I:C), n = 3) (d). One-tailed Student’s t-test. e, Same as a, except dimethyl sulfoxide (DMSO) (control, n = 3; conditioned medium, n = 3) or CU CPT 4a (control, n = 3; conditioned medium, n = 3) was added. Two-tailed Student’s t-test. f, Dot plot depicting Slit2 mRNA levels (by qPCR) induced by diluted 4T1 conditioned medium (1:8) in either wild-type endothelial cells (control, n = 3; conditioned medium, n = 3) or TLR3-knockout endothelial cells (control, n = 3; conditioned medium, n = 3). Two-tailed Student’s t-test. a–f, Biological replicates, each replicate represented by different symbol. g, Dot plot represents the number of metastatic lung nodules per mouse (left) and representative images of MMTV-PyMT-driven tumours (right) in tuSLIT2-knockout (n = 12) and wild-type (n = 10) mice. One-tailed Student’s t-test. Scale bar, 1 cm. h, Dot plot represents ROBO1 expression in 4T1-Luc-zsGreen cells expressing control shRNA (control shRNA, n = 4) or Robo1 shRNA (n = 4). Two-tailed Student’s t-test. i, Dot plot represents bioluminescence signal of whole-blood-derived circulating tumour cell colonies from tumours expressing control or Robo1 shRNA in NSG mice that were injected intravenously with 25 μg poly(I:C) or phosphate-buffered saline (PBS). Control shRNA: control, n = 3; poly(I:C), n = 4. Robo1 shRNA1: control, n = 4; poly(I:C), n = 4. Two-tailed Student’s t-test. Representative images (right) of bioluminescence imaging of plates containing circulating tumour cell colonies. j, Proposed model. Highly metastatic tumour cells release dsRNA, which is detected by endothelial-cell TLR3 RNA-sensing receptors—leading to endothelial SLIT2 induction. Endothelial SLIT2 acts on tumoural ROBO1 receptors to drive tumour-cell migration towards vessels, which facilitates intravasation by tumour cells and, consequently, metastatic dissemination. Data are mean ± s.e.m.
We next observed that the addition of the synthetic double-stranded RNA (dsRNA) analogue polyinosinic:polycytidylic acid (poly(I:C)) to basal medium (Fig. 4c) or to the 10-kDa conditioned-medium filtrate induced endothelial SLIT2 expression to a degree similar to that observed with 4T1 conditioned medium (Fig. 4d). These observations suggest that dsRNA released by tumour cells may upregulate Slit2 expression in endothelial cells. In mammalian cells, Toll-like receptor 3 (TLR3) is a major receptor for dsRNA19. Consistent with this, pharmacological inhibition of TLR3 with the TLR3-specific antagonist CU CPT 4a, as well as genetic inactivation of Tlr3, suppressed SLIT2 induction in endothelial cells by conditioned medium from highly metastatic cells (Fig. 4e, f, Extended Data Fig. 4c, d). Endocytosis has previously been shown to be important for TLR3 signalling20,21. Consistent with this, pharmacological inhibition of endocytosis with dynasore suppressed the induction of endothelial SLIT2 (Extended Data Fig. 1c). Tlr3-knockout endothelial cells also displayed reduced phosphorylation of ERK1 and ERK2, which has been previously implicated in TLR3 downstream signalling22 (Extended Data Fig. 1f). Moreover, treatment of the 4T1 conditioned medium with RNase A also impaired the phosphorylation of ERK1 and ERK2 in endothelial cells (Extended Data Fig. 1g). Additionally, Tlr3 deletion in the host impaired intravasation by tumour cells (Extended Data Fig. 6a, b). Importantly, activation of TLR9 with two different concentrations of the TLR9 synthetic ligand CpG oligodeoxynucleotide did not affect Slit2 expression in endothelial cells (Extended Data Fig. 1h–j). These findings reveal that endothelial TLR3 detects extracellular RNA from highly metastatic tumours and induces SLIT2.
Tumoural SLIT2 represses metastasis
Tumoural Slit2 silencing through promoter hypermethylation or allelic deletions have previously been reported23,24, which suggests a tumour-suppressive role for tumoural SLIT2. Paradoxically, the SLIT receptor ROBO1 has previously been reported to become overexpressed in some cancers, which suggests a tumour-promoting role for this pathway17,25. Promoter hypermethylation and allelic deletions of Slit2 in tumours have been difficult to reconcile with the neurodevelopmental roles of SLIT proteins in promoting cell migration, and a tractable model for how SLIT signalling affects cancer progression has not emerged. Analysis of methylation of the Slit2 promoter and Slit2 expression data in publicly available datasets from the MethHC database26 revealed that the Slit2 promoter is significantly more methylated in breast tumours relative to normal mammary-gland tissue (Extended Data Fig. 7a). Highly metastatic 4T1 cells expressed reduced Slit2 relative to nonmetastatic 67NR cells (Extended Data Fig. 7b), and treatment of 4T1 cells with the demethylating agent 5-azacytidine induced Slit2 expression—consistent with methylation-induced repression (Extended Data Fig. 7c). Moreover, both Slit2 pre-mRNA and genomic copy number were reduced in highly metastatic 4T1 cells (Extended Data Fig. 7d, e). Collectively, our data reconcile seemingly contradictory past clinical and pathological observations, and support a model in which enhanced endothelial expression of SLIT2 relative to tumoural expression of SLIT2 drives cancer metastasis. A major prediction of this model is that genetic inactivation of Slit2 in the tumoural compartment would promote metastasis—in stark contrast to endothelial inactivation of SLIT2, which reduced metastasis. To directly test this, we genetically inactivated Slit2 in the tumour compartment by driving Cre recombinase expression in mammary glands of Slit2-floxed MMTV-PyMT mice (hereafter referred to as tuSLIT2-knockout). SLIT2 inactivation in the tumour compartment substantially enhanced metastatic progression without affecting primary tumour growth or angiogenesis (Fig. 4g, Extended Data Fig. 2k–m). Deletion of tumoural Slit2 did not affect tumour cell apoptosis or the expression of other SLIT2-related factors such as netrin 1, SDF1 or MCP1 (Extended Data Fig. 8a–g). Consistent with observations on in vivo metastasis, depletion of tumoural SLIT2 enhanced tumour-cell migration towards exogenous recombinant SLIT2 (Extended Data Fig. 4g, h). These data are consistent with publicly available gene-expression data (from https://kmplot.com/analysis/) that reveal a significant association between reduced tumoural expression of Slit2 and a worse prognosis in human breast cancer (Extended Data Fig. 5e). Moreover, western blot analysis of endothelial-cell and tumour-cell lysates revealed higher levels of SLIT2 protein in endothelial cells (Extended Data Fig. 4b). Previous studies have implicated SLIT2 in the development of the mammary gland27–29. Our observations in mouse models of cancer reveal that the same gene (that is, Slit2) can act as a driver or suppressor of the metastatic progression of breast cancer depending on the compartment within which it is expressed—endothelial versus tumoural.
Our findings reveal a model in which RNA released from highly metastatic tumour cells activates an innate immune RNA-sensing pathway in endothelial cells, inducing SLIT2 upregulation via TLR3 (Fig. 4j). Because TLR3 is a sensor of dsRNA, we searched for a dsRNA source that becomes elevated in highly metastatic cells. Immunofluorescence quantification using the dsRNA-binding monoclonal antibody J2 revealed higher levels of dsRNA in several highly metastatic tumours relative to less-metastatic isogenic counterparts (Extended Data Fig. 9a, b). Additionally, more cell-free RNA was detected in the conditioned medium of highly metastatic cells relative to isogenic less-metastatic cells as well as in the plasma of mice that bear highly metastatic 4T1 tumours (Extended Data Fig. 1k–m).
Endogenous retroviral elements (ERVs) represent a potential source of endogenous dsRNA. RNA sequencing (RNA-seq) revealed significantly higher expression of annotated ERVs in highly metastatic breast and melanoma cancer cells relative to poorly metastatic parental populations (Extended Data Fig. 9c, d). dsRNA can be highly stable in solution. Consistently, we observed even greater differential levels of ERVs in the conditioned medium of highly metastatic cells relative to poorly metastatic cells (Extdened Data Fig. 9e). To determine whether we could detect ERVs as dsRNA species in highly metastatic cells, we pulled down tumoural dsRNA using the J2 antibody and detected multiple ERV species (Extended Data Fig. 9f). These findings reveal that highly metastatic breast and melanoma cells contain and secrete higher levels of endogenous dsRNA species, which contribute to their enhanced capacity to activate TLR3-dependent induction of endothelial SLIT2.
These findings have clinical implications, as TLR3 agonists such as poly(I:C) are being tested in clinical trials as a means of activating the innate immune system. Our experiments suggest that the efficacy of such approaches may be impaired by the unintended consequence of driving metastatic dissemination in the neo-adjuvant setting. To directly investigate this possibility, we tested the effect of the commonly used clinical trial adjuvant poly(I:C) on intravasation by cancer cells. To extricate the effect of poly(I:C) on intravasation from its known downstream antitumour adaptive immune effects, we performed this study in immune-deficient mice. Treatment of NSG mice that bore 4T1 primary breast tumours with poly(I:C) substantially enhanced intravasation in a tumoural ROBO1-dependent manner, as assessed by quantification of circulating tumour cells (Fig. 4h, i, Extended Data Fig. 6f, g). Treatment with poly(I:C) increased vascular SLIT2 expression in tumours without further increasing tumour vascular leakage (Extdended Data Fig. 6c–e). These findings reveal a detrimental role for TLR3 agonism in the dissemination of cancer and suggest that combining poly(I:C) treatment with the inhibition of the endothelial SLIT2–ROBO1 axis may enhance the beneficial effect of TLR3 agonism in the clinic.
Discussion
Links between cancer inflammation and pathogen- or danger-associated molecular pattern-detection mechanisms have previously been reported30–31. Moreover, it has previously been shown that cancer cells can activate inflammatory pathways in response to stromal RNA32 and multiple cancers have been found to release RNA33. Additionally, oncogenic transformation has been associated with increased ERV expression34,35. RNA stress has also been observed in cardiovascular disease and vascular inflammation occurs in sepsis36,37. Our experiments reveal what we believe to be the first demonstration that endothelial cells can detect tumoural RNA, become activated and instructively promote metastatic progression. This endothelial RNA stress response probably evolved as a means of detecting pathogenic microorganisms33,36,37. By co-opting this response, cancer cells selectively exploit a feature of the inflammatory response to drive progression38. Our findings also reveal a link between TLR3 activation by extracellular RNA and endothelial SLIT2. Although metastatic cell RNA activated TLR3 in these models, other RNA sensors may mediate tumoural endothelial activation in other cancers. Our findings thus uncover molecular alterations that drive metastatic progression that could perhaps aid in the earlier diagnosis and potentially treatment of high-risk cancers.
METHODS
Exact values of significance are indicated in all figures. No statistical methods were used to predetermine sample size. The experiments were not randomized and investigators were not blinded to allocation during experiments and outcome assessment, except for certain microscopic imaging assessments, where the experimentalist was blinded to allocation.
Endothelial and tumour cell culture
All cancer cells were propagated as previously described39. Primary mouse lung endothelial cells and immortalized mouse lung endothelial cells (PyMT cells) were cultured in coated 75-cm2 tissue culture treated flasks (Falcon) or 10-cm tissue culture dishes (Falcon) in MLEC medium40. The 67NR and 4T07 cell lines were provided by W. P. Schiemann. The 4T1, B16F0, B16F10 and LLC lines were obtained from the ATCC41,42. Tumour cells were cultured in uncoated tissue culture dishes in D10F medium (880 ml 1× DMEM, + 4,5 g/l d-glucose, + l-glutamine, + 110 mg/l sodium pyruvate (Gibco)), 100 ml fetal bovine serum (Sigma), 10 ml penicillin–streptomycin (Gibco) and 3 ml amphotericin B (Lonza). Lungs from wild-type C57BL/6J, BalbC and Cdh5(PAC)-creERT2;Slit2fl/fl mice were used to isolate MLECs, as previously described40. After a negative sort with rat anti-CD16/CD32 (BD Biosciences 553141), cells were immortalized via polyoma middle T (PyMT) antigen viral transduction by incubating them on 2 consecutive days for 4 h each with supernatant from GgP+E packaging cells43. Cells were grown in MLEC medium and supplemented with 500 nM 4-hydroxytamoxifen (Sigma) for Cdh5(PAC)-creERT2;Slit2fl/fl mice. Two positive sorts using rat anti-ICAM2 (BD Biosciences 553326) and sheep anti-rat IgG magnetic beads (Dynabeads) were performed as previously described40. Tamoxifen-treated endothelial cells isolated from Cdh5(PAC)-creERT2;Slit2fl/fl mice were used to generate SLIT2-depleted endothelial cells (ecSLIT2-knockout), and Cre-negative litter mates yielded wild-type endothelial cells (wild type).
Conditioned medium treatment of endothelial cells
Conditioned medium was generated by plating 1 × 106 tumour cells (67NR or 4T1) in 10-cm dishes. After allowing 8 h for cell attachment, cells were washed twice with low serum, basal medium-Opti-MEM (Gibco) and incubated in 15 ml of Opti-MEM for 12 h. Conditioned medium was collected and spun down for 5 min at 424g (1,500 rpm). Sixty thousand immortalized endothelial cells were plated in a 12-well plate. After 24 h in culture, cells were washed twice in Opti-MEM and 1 ml of conditioned medium or Opti-MEM (negative control) was added. After 12 h incubation, total RNA was extracted (Norgen total RNA kit). 4T1 conditioned medium was either used directly or filtered (50 kDa or 10 kDa) (Amicon Ultra-15). Additionally, conditioned medium or basal Opti-MEM was treated with DNase I (10 μg/ml) (Worthington), RNase A (25 μg/ml) (Ambion AM2271) for 2 h at 37 °C before addition to endothelial cells. Alternatively, basal Opti-MEM or filtered conditioned medium were supplemented with synthetic dsRNA–poly(I:C) (Sigma) (2.5 μg/ml). Heat inactivation of conditioned medium or Opti-MEM was done at 95 °C for 10 min. CU CPT 4a (Tocris 4843) was used at the final concentration of 27 μM. CU CPT 4a was added to Opti-MEM or 4T1 conditioned medium and endothelial cells were treated as described. Dynasore hydrate (Sigma D7693) was supplemented to conditioned medium or Opti-MEM basal medium (5 μM) and the same concentration of DMSO was used as negative control. Synthetic dsRNA–CpG oligodeoxynucleotide (ODN) (Invivogen ODN 1585) was diluted in Opti-MEM (1×), to 2.5 μg/ml and 12.5 μg/ml and endothelial cells were treated as described. All conditioned medium experiments were conducted in biological triplicates.
Mouse studies
All mouse work was performed in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) at Rockefeller University. Wild-type C57BL/6J mice were obtained from Jackson Laboratory and wild-type BALB/c (BALB/cAnNCrl) mice were obtained from Charles River Laboratories. Slit2-floxed mice were obtained from A. Chedotal12. The endothelial-specific inducible Cre line Cdh5(PAC)-creERT2 was obtained from R. Adams11. Cdh5(PAC)-creERT2;Slit2-floxed mice were crossed for at least five generations with pure wild-type BALB/c or pure C57BL/6J mice and then inter-crossed to obtain Cdh5(PAC)-CreERT2;Slit2-floxed mice. Rpl22-floxed (RiboTag) mice were obtained from Jackson Laboratory10. Cdh5(PAC)-creERT2 mice were crossed with Rpl22HA/HA (RiboTag) mice to generate Cdh5(PAC)-creERT2;Rpl22fl/fl/HA mice. Cdh5(PAC)-creERT2;Slit2-floxed C57BL6 mice were crossed with MMTV-PyMT mice44 to generate Cdh5(PAC)-CreERT2;Slit2-floxed;MMTV-PyMT mice. MMTV-Cre mice45 were obtained from Jackson Laboratory and crossed with the Slit2fl/fl (Slit2 gene gets deleted not SLIT2 protein) mice to generate the Slit2 deletion in the tumoural compartment (tuSLIT2 knockout) in MMTV-PyMT mice. NSG and TLR3-knockout mice were purchased from Jackson Laboratory (NSG, stock number 5557; TLR3-knockout, stock number 5217). Genotyping primers and expected product sizes are listed in the Supplementary Table 1.
Immunoprecipitation of polysomes and RiboTag profiling
Upon tamoxifen treatment of Cdh5(PAC)-creERT2 mice, Cre recombinase becomes active in endothelial cells and recombines loxP sites, which flank exon 4 of Rpl22, replacing it with an HA-tagged exon 4. To activate Cre, mice were injected intraperitoneally with 150 μl of tamoxifen diluted in corn oil (10 mg/ml) for 2 consecutive days. Concomitantly, mouse chow was replaced with tamoxifen-supplemented food (250 mg/kg) (Envigo TD.130856). After 7 days, mice were injected subcutaneously with either 100,000 B16F0 or B16F10 cells diluted 1:1 in PBS and reduced growth factor matrigel (BD Biosciences). Tumours were grown for 12 days and mice were killed. Tumours were resected and placed in a Petri dish on ice. After separating a piece (about 200 mg) of tumour with a scalpel, the sample was weighed and homogenized with a dounce (10% w/v) in homogenization buffer (50 mM Tris, pH 7.5, 100 mM KCl, 12 mM MgCl2, 1% NP-40, 1 mM DTT, 200 U/ml Promega RNasin, 1 mg/ml heparin, 100 μg/ml cyclohexamide, Sigma protease inhibitor mixture). One hundred μl of conjugated anti-HA magnetic beads (Dynabeads, Invitrogen) was added to an Eppendorf tube and washed once in homogenization buffer on a magnetic rack (Invitrogen). Samples were spun down at 10,000g for 10 min and supernatants (400 μl) were added directly to antibody-coupled magnetic beads and rotated overnight at 4 °C. After 16 h, samples were placed on magnet and the supernatant was collected. The pellets were washed 3 times for 5 min in high salt buffer (50 mM Tris, pH 7.5, 300 mM KCl, 12 mM MgCl2, 1% NP-40, 1 mM DTT, 100 μg/ml cyclohexamide). To isolate RNA, 350 μl of Qiagen RLT buffer (supplemented with 2-mercaptoethanol) was added to anti-HA magnetic beads (polysomes). Total RNA extraction was performed according to manufacturer’s instructions using an RNeasy Micro Plus kit (Qiagen) and quantified using a NanoDrop.
RNA-seq
The RiboTag system was used to isolate ribosome-bound mRNA (n = 5 B16F0 tumours, n = 7 B16F10 tumours) and prepared for high-throughput sequencing. Ribosomal RNA (rRNA) depletion was performed using Ribo-Zero rRNA Removal kit (Epicentre RZH110424). Fifty ng of rRNA depleted sample was used for amplification and labelled with the ScriptSeq RNA-Seq Library Preparation kit (Epicentre–SSV21124). Multiplexed sequencing was performed with ScriptSeq Index PCR primers (Epicentre RSBC10948) during library preparation. TruSeq Stranded Total RNA Library Prep (Illumina) was used to generate RNA-seq libraries from conditioned medium cell-free RNA. For multiplex sequencing, TruSeq RNA Single Index Set A and Set B were used (Illumina). RNA-seq libraries were quantified using a Bioanalyzer (Agilent). Pooled samples were sequenced on a Illumina HiSeq 2500 (1 × 50 bp) in two independent batches. As a part of our RNA-seq pipeline, the quality of the FASTQ files were checked with FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and adaptor sequences were trimmed with cutadapt v.1.16. Reads were aligned to the mm10 mouse genome with STAR v.2.2.1. Count files were generated with featureCounts v.1.6.0 and imported to R v.3.3.2. EdgeR was used to perform differential expression analysis and P value correction (batch information was included in the design). For ERV analysis, reads were quality-trimmed and subjected to adaptor removal (cutadapt -v.2.3, -q 15 -a AGATCGGAAGAGCACACGTCT). The resulting reads were then mapped to the mouse genome (build mm10) using BWA (v.0.7, bwa mem) with default parameters. The aligned reads were then filtered on the basis of the following criteria: (i) number of clipped positions over the length of the read, and (ii) the edit distance over the number of matching positions (both ratios were required to be <2%). The filtered reads were then assigned to mouse ERVs (build mm10), downloaded from UCSC genome browser, using featureCounts v.1.6.0. The resulting count table was then analysed using DESeq2 (v.1.22). The library sizes were set using counts from all genes (mm10) before differential ERV expression analysis. To analyse SLIT2 expression in circulating tumour cells of patients with cancer, RNA-seq data from a prior study were used (circulating tumour cell (CTC) RNA-seq from n = 16 patients, relative to tumour RNA-seq available for n = 12 of the same patients)18. Processed read counts were downloaded from Gene Expression Omnibus (GEO) (GSE111842) and combined into a count table. DESeq2 (v.1.22) was used to generate a normalized count matrix. The normalized count for SLIT2 across all samples was used to compare its expression in blood, tumour and CTC samples.
Human breast cancer samples
Tissue microarrays from primary breast cancers were provided by the NCI Cooperative Human Tissue Network. Cancer Diagnosis Program (CDP) Breast Cancer Progression Tissue Microarrays (Clinical Progression) were de-waxed, rehydrated, blocked in 10% goat serum, incubated in rabbit anti-SLIT2 antibody (Proteintech 20217–1-AP) and mouse anti-human CD31 (Westernblot, NCL-L-CD31–607) in 0.5% goat serum in PBS overnight at 4 °C. After washing three times in PBS, samples were incubated with anti-mouse Alexa Fluor 555 and anti-rabbit Alexa Fluor 488 (Thermo Fisher Scientific) both diluted 1:100 in 0.5% goat serum in PBS, washed in PBS and incubated with DAPI (2.5 μg/ml, Roche 10236276001) diluted in PBS. Primary and metastatic tumour vessel SLIT2 expression was assessed from the breast cancer tissue array (Abcam, TMA AB178118) using the immunostaining protocol described in ‘Immunostaining of mouse tumours and lung sections’. Images were acquired with a Leica Confocal sp8 microscope. Blood vessel SLIT2 expression quantification was computationally calculated using CellProfiler (Broad Institute). The association between tumoural SLIT2 expression levels and relapse-free survival was obtained using the kmplot database46. Patient-derived xenograft (PDX) sections from patients with breast cancer were obtained from A. L. Welm47,48. PDX sections were immunostained for SLIT2 and endomucin (endothelial marker) as described in ‘Immunostaining of mouse tumours and lung sections’. ImageJ software was used to quantify colocalization of SLIT2 within the PDX vessels.
Immunostaining of mouse tumours and lung sections
Sections from fixed mouse tumours were dewaxed and rehydrated in descending concentrations of ethanol and microwaved in antigen retrieving solution (AntigenPlus Buffer pH10; EMD Millipore, 71290–3) for 20 min. After blocking the samples in 5% goat serum (Sigma, G9023) diluted in PBS for 1 h at room temperature, sections were stained for endothelial cells using rat anti-endomucin (V.7C7; Santa Cruz, SC-65495) and rabbit anti-SLIT2 antibody (Proteintech 20217–1-AP) diluted 1:100 in 0.5% goat serum in PBS overnight at 4 °C. After washing three times in PBS, samples were incubated with anti-rabbit Alexa Fluor 555 and anti-rat Alexa Fluor 488 (Thermo Fisher Scientific) both diluted 1:100 in 0.5% goat serum in PBS, washed in PBS and incubated with DAPI (2.5 μg/ml, Roche 10236276001) diluted in PBS. For SLIT2 immunostaining of zsGreen 4T1 tumours, anti-rabbit Alexa Fluor 647 (Thermo Fisher Scientific) secondary antibody was used. Confocal microscopy was used to acquire images (LSM 880, Zeis) and colocalization of SLIT2 and endomucin or levels of SLIT2 in tumour vessels were quantified using ImageJ. For detection and quantification of lung metastases an anti-PyMT antibody (Novus Biologicals, NB100–2749) was used. The entire lung section image was acquired with a RS-G4 scanning confocal microscope (Caliber I.D.) and the area of individual metastases was calculated with Fiji. For each tumour, the average area for individual metastases was calculated. Lesions were considered to represent either micro- or macrometastases if they were smaller or larger than the mean, respectively. This protocol was also performed for the remaining immunostainings. The following antibodies were used: rabbit anti-cleaved caspase 3 (Cell Signaling, 9661S), mouse anti-MCP1 (ThermoFisher Scientific MA5–17040), chicken anti-netrin 1 (Novus Biologicals, NB100–1605), rabbit anti-SDF1 (Proteintech, 17402–1-AP) and mouse J2 antibody (Scicons, 10010200). Control IgG from the same species was used as a negative control. For mouse antibodies, blocking solution was supplemented with mouse on mouse blocking reagent according to manufacturer instructions (Vector Labs, MKB-2213). Metastatic nodules were quantified under the microscope as areas greater than 2,000 μm2.
Tumour growth, metastasis and circulating tumour cell assays
Cdh5(PAC)-creERT2;Slit2-floxed;MMTV-PyMT mice developed mammary gland tumours at the age of 11 ± 2 weeks. Lungs of tamoxifen-treated mice were resected at 23 weeks of age. Lung nodule numbers in each mouse were quantified as the average number of lung metastases detected under 10× objective of an optical microscope of two H&E-stained sections with a 100-μm interval. Eight-to-ten-week-old female Cdh5(PAC)-creERT2;Slit2-floxed or Slit2-floxed littermates (BALB/c) were treated with tamoxifen as described in ‘Immunoprecipitation of polysomes and RiboTag profiling’, to generate ecSLIT2-knockout and wild-type mice. One hundred thousand 4T1 cells (ATCC, mycoplasma-free) were resuspended in 100 μl of a 1:1 mixture of PBS and reduced growth factor Matrigel (Corning) and injected into the fourth fat pad on the right side. Tumour volumes were estimated using the formula: volume = large diameter × short diameter2 × 0.52. Tumours were surgically resected at an average of 200 mm3 in volume. Eight-to-ten -week-old male and female Cdh5(PAC)-CreERT2;Slit2-floxed or Slit2-floxed littermates (C57BL/6J) were treated with tamoxifen as described in ‘Immunoprecipitation of polysomes and RiboTag profiling’ and injected subcutaneously in flanks with 100,000 LLC cells (ATCC; mycoplasma-free) re-suspended in 100 μl of a 1:1 mixture of PBS and reduced growth factor Matrigel (Corning). Tumour volumes were estimated using the formula described above and were surgically resected when reaching an average of 150 mm3 in volume. For survival studies in both 4T1 and LLC metastasis assays, mice were checked twice a day and euthanized upon any sign of illness or distress. The date of death was assumed to be the following day. For metastasis quantification, mice were euthanized 19 days after surgical resection of 4T1 fat pad tumours and 20 days after surgical resection of LLC subcutaneous tumours. The lungs were sectioned and stained with H&E at two different depths. The number of lung nodules per section was quantified using a 10× magnification objective in an optical microscope. To detect circulating tumour cells, 4T1 cells (ATCC; mycoplasma-free) were transduced with lentivirus to express luciferase and zsGreen (pHIV-Luc-ZsGreen was a gift from B. Welm (Addgene plasmid no. 39196)). Forty-eight h after transduction, zsGreen-positive 4T1 cells were sorted using a FACSAria cell sorter. One hundred thousand 4T1-Luc-zsGreen cells were injected into the fat pad of 8–10 week-old female wild-type mice and ecSLIT2-knockout littermates. The tumours were grown for 32 days, at which point mice were terminally anaesthetized with 400 μl of ketamine–xylazine (17.8 ml sterile water (Corning), 2 ml ketamine (Henry Schein), 200 μl xylazine (100 ng/ml, Sigma)), and whole blood of mice was collected through cardiac puncture with a 27 G needle and 1 ml syringe containing 50 μl of 0.5M EDTA (Quality Biological 351-027-101). Total blood cells were separated from plasma by centrifugation at 2,000g for 5 min at 4 °C. After discarding the supernatant, the cell pellet was resuspended in 1 ml erythrocyte lysis buffer (Buffer EL-QIAGEN) and incubated for 20 min at room temperature. After another centrifugation step at 400g for 8.5 min at room temperature, the supernatant was discarded, and cells were resuspended again in 1 ml of EL buffer and incubated for 5 min before spinning down one more time for 8.5 min at 400g. Cell pellets were then washed once in 1 ml of D10F, spun down for 8.5 min at 400g, resuspended in D10F and plated on a 10-cm tissue culture dish. Plates were imaged using an IVIS machine 7 days after collection of blood to detect luciferase-positive colonies derived from CTCs. For this purpose, D10F medium was replaced with a luciferin solution (0.1667 mg/ml D-luciferin (Perkin Elmer P/N122796) diluted in PBS with Ca2+ and Mg2+). Luminescence of individual tissue culture plates was measured using an IVIS spectrum machine (PerkinElmer). CTCs were also quantified in synthetic dsRNA–poly(I:C)-treated NSG mice. Eight-to-ten-week-old aged-matched female NSG mice were fat-pad-injected with 4T1-Luc-zsGreen cells. After allowing tumours to grow for 15 days, the mice were injected daily with either PBS or poly(I:C) (25 μg diluted in 100 μl PBS) intravenously in the tail vein for 7 consecutive days. Whole blood was collected 2 h after the last injection and quantification of CTCs performed as described. Lung Metastatic 4T1 cells were isolated after homogenizing the lungs with collagenase (as described in ‘Endothelial and tumour cell culture’) of wild-type BALB/c mice with 500-mm3 fat-pad 4T1 tumours. MDA-MB-231 breast cancer cells obtained from ATCC were transduced with a triple reporter lentiviral vector49. Two independent Robo1 shRNAs were used. Two hundred thousand cells were injected bilaterally into mammary fat pads of NSG female mice. Tumours were surgically resected at 300 mm3 and lung luminescence was subsequently measured using an IVIS machine.
Genotyping of transgenic mouse lines
Genotyping of various mouse colonies was performed after extracting DNA from an ear skin biopsy using the PCR primers annotated in the Supplementary Table 1.
Tumour migration assays and endothelial and tumour cell chemotaxis assays
Twenty-four-well plate wells (Falcon) were covered with 250 μl of growth factor reduced Matrigel (Corning) containing different concentrations of recombinant mouse N-terminal and C-terminal fragments of SLIT2 (SLIT-N and SLIT2-C, respectively) (100 μg/ml in PBS) (R&D Systems): 0 ng/ml, 100 ng/ml and 300 ng/ml. After polymerizing the Matrigel for 30 min at 37 °C, 250 μl of Opti-MEM medium (Gibco) was added on top of the SLIT2–Matrigel mixture. Fifty thousand serum-starved B16F10 or 4T1 cells were plated on the top of 3.0-μm pore size transwell inserts (high pore density polyester track-etched membranes (Falcon)). After incubating the cells for 20 h at 37 °C, nonmigrated cells on top of membranes were removed with cotton swabs. The remaining cells were fixed in 4% paraformaldehyde (Alfa Aesar) for 20 min at room temperature and washed in PBS. Membranes were detached from the insert and mounted on microscope slides (Fisher Scientific) with mounting medium containing DAPI (Vector). Fluorescent images were taken with an Axiovert 40 CFL microscope (Zeiss). DAPI-positive nuclei were counted to quantify cell migration. To perform endothelial and tumour cell chemotaxis assays, 50,000 SLIT2-overexpressing (pCMV3-Slit2-FLAG Sino Biological) and empty-vector-control endothelial cells (pCMV3-untagged-NCV Sino Biological) were plated in 4 wells of a 24-well plate (Falcon). The cells were incubated for 24 h in MLEC medium. Matrigel invasion chambers (8.0 μm PET membranes) (Corning) were placed after changing the culture medium to Opti-MEM (Gibco). Fifty thousand serum-starved 4T1 cells in Opti-MEM were seeded on top of inserts. Cells were incubated at 37 °C for 20 h. PET membranes were isolated and stained with DAPI. Migrated cells were quantified by counting DAPI stained nuclei per optical field of view using an Axiovert 40 CFL microscope (Zeiss). All conditions were tested in quadruplicates or triplicates.
Western blotting
Protein lysates from MLECs were prepared with ice-cold RIPA buffer supplemented with protease and phosphatase inhibitors (Roche). Thirty μg of protein lysates were separated using SDS–polyacrylamide gel electrophoresis and transferred to a PVDF membrane (Immobilion-P, Millipore, IPVH00010). After blocking the membranes in 5% milk in TBST (1× TBS (Cell Signalling); 0.1% Tween20 (Sigma)), the membranes were incubated overnight at 4 °C with either rabbit anti-SLIT2 antibody (Abcam- ab134166) diluted 1:1,000 in 5% BSA (Sigma) or mouse anti-HSC 70 antibody (Santa Cruz Biotechnology - B-6) diluted 1:5,000 in 5% milk in TBST. Anti-Flag (Cell Signaling, 2368S), anti-SLIT2 (Proteintech, 20217–1-AP), p-p44/42 MAPK (pERK1/2) (Cell Signaling, 4370S) and p44/42 MAPK (ERK1 and ERK2) (Cell Signaling 4695S).
Primary antibodies were incubated in 5% BSA in TBST overnight at 4 °C. After washing the blots 3 times for 15 min each in TBST, the membranes were incubated with HRP-conjugated goat anti-rabbit IgG (H+L) or HRP-conjugated goat anti-mouse IgG (H+L) secondary antibody (Invitrogen). Finally, the membranes were incubated with ECL western blot substrate (Thermo Scientific) for 1 min. X-Ray film (Fujifilm) was exposed to the western blot membranes (30 s, 1 and 3 min) and developed with a film processor (SRX-101A, Konica Minolta).
Quantification of phosphorylated ERK1 and ERK2, was performed by measurement of densitometry of phosphorylated ERK1 and ERK2 bands relative to total ERK1 and ERK2 using ImageJ software.
Generation of lentivirus, knockdown cells and overexpressing cells
Generation of lentivirus-mediated knockdown cells was performed as previously described39. shRNA (pLKO.1-puro vector - Sigma) vectors were used to generate lentivirus to knock down ROBO1 or SLIT2. HEK293 cells (ATCC) were incubated with 12 μg of pLKO.1-puro shRNA vector (Sigma), 4 μg pRSV-Rev (Cell Biolabs), 4 μg pCgpV (Cell Biolabs), 4 μg pCMV-VSV-G (Cell Biolabs) and 30 μl lipofectamin 2000 reagent (Invitrogen) diluted in 5 ml of Opti-MEM medium (Gibco). Medium was changed to antibiotic-free D10F after 5 h and once again after 24 h. Medium containing the shRNA-packaged virus was collected and spun down for 5 min at 524g (1,500 rpm), filtered (0.45-μm filter, Pall) and supplemented with polybrene (10 μg/ml) (Speciality Media) before adding to the cells. 4T1 cells were grown to a confluence of 80% and incubated in shRNA lentivirus medium for 24 h. Forty-eight h after transfection, medium was changed to D10F, supplemented with 2 μg/ml of puromycin (Gibco). The following shRNA sequences were cloned into pLKO.1-puro vectors: mouse Robo1 shRNA no. 1: CCG GGC CGA AGG AAT ATG GCA GAA ACT CGA GTT TCT GCC ATA TTC CTT CGG CTT TTT G (Sigma); shRNA no. 2: CCG GCC AGT TAG ATT CTC ACG GAA ACT CGA GTT TCC GTG AGA ATC TAA CTG GTT TTT G (Sigma); human ROBO1 shRNA no. 1: CCG GTG ACA CAT GAC GCC AGA TAA ACT CGA GTT TAT CTG GCG TCA TGT GTC ATT TTT G (Sigma); shRNA no. 2: CCG GGC AGA CAA AGA GAA CAA GCA ACT CGA GTT GCT TGT TCT CTT TGT CTG CTT TTT G; mouse Slit2 shRNA no. 1: CCG GCT TGA CCA TGT TGG ACT AAT TCT CGA GAA TTA GTC CAA CAT GGT CAA GTT TTT G (Sigma) shRNA no. 2: CCG GGC CTT GTC ACA CTT AGC GAT TCT CGA GAA TCG CTA AGT GTG ACA AGG CTT TTT G (Sigma); scrambled shRNA (control): CCG GCA ACA AGA TGA AGA GCA CCA ACT CGA GTT GGT GCT CTT CAT CTT GTT GTT TTT G (Sigma). SLIT2 overexpression was obtained by transfecting either PyMT immortalized mouse lung endothelial cells or B16F10 cells with the plasmids: pCMV3-Slit2-FLAG (Sino Biological) or empty-vector control pCMV3-untagged-NCV (Sino Biological). Five μg of each plasmid was diluted in 1.5 ml of Opti-MEM (Gibco); 30 μl of lipofectamin 2000 reagent (Invitrogen) in 1.5 ml of Opti-MEM. After incubating each solution for 5 min at room temperature, both solutions were combined and incubated for an additional 20 min. The cells were then washed 4 times in Opti-MEM before adding the plasmid and lipofectamine solution together with an additional 5 ml of Opti-MEM. After 5 h incubation, the transfection solution was replaced with normal MLEC medium.
Adenoviral transduction of mouse lung endothelial cells
ecSLIT2-knockout endothelial cells were isolated and immortalized as described in ‘Endothelial and tumour cell culture’. Adenoviral particles were donated by K. J. Sevensson and generated from pAd/CMV/V5-DEST vectors as previously described16. Adenovirus containing full-length SLIT2, C-terminal SLIT2 and LacZ (negative control) were diluted in 15 ml of MLEC medium and added to 1 × 106 SLIT2-knockout endothelial cells (500 × 106 viral particles, multiplicity of infection of 500). Expression of the different constructs was confirmed after 48 h and transduced cells were used for migration assays (as described in ‘Tumour migration assays and endothelial and tumour cell chemotaxis assay’) between 48 and 72 h from initial viral transduction.
Blood vessel density, perfusion and permeability in mouse tumour
To assess blood vessel density, mouse tumours (B16F10, LLC, 4T1 and MMTV-PyMT tumours) were fixed in 4% PFA diluted in PBS overnight at room temperature and then transferred to 70% ethanol. Tumour sections were immunostained for endothelial cells using rat anti-endomucin (V.7C7; Santa Cruz, SC-65495) diluted 1:100 in 0.5% goat serum in PBS overnight at 4 °C, washed 3 times in PBS and incubated with anti-rat Alexa 555 (Thermo Fisher Scientific) for 1 h at room temperature. After washing once in PBS, tumour sections were incubated in 3 μg/ml of DAPI in PBS for 10 min, washed two more times in PBS and mounted using ProLong Gold (Invitrogen). Blood vessel density was quantified by measuring endomucin-stained areas relative to DAPI-positive areas using ImageJ software. For blood vessel perfusion and permeability studies, wild-type or ecSLIT2-knockout mice were injected with 100,000 4T1 cells in the fat pad and the tumours were allowed to grow until they reached an average of 200 mm3. For analysis of blood vessel perfusion, 10 min before being killed the mice were injected in the tail vein with 100 μl of PE–PECAM antibody (BioLegend). To assess blood vessel permeability, mice were injected with Hoechst (100 μl of 4 mg/ml) (Thermo Fisher Scientific) via the tail vein 1 min before euthanasia. Tumours were isolated and snap-frozen. Cryosections were briefly thawed and then immersed in −20 °C acetone for 10 min. After rehydrating the tissue sections in PBS, samples were mounted with coverslips using ProLong Gold antifade reagent (Invitrogen). Blood vessel perfusion was quantified as the ratio of tumour blood vessels perfused with PE–PECAM over total blood vessel stained with rat anti-mouse PECAM (CD31) antibody (BD Pharmingen, 550274) and anti-rat Alexa 488 secondary antibody (Invitrogen). ImageJ software was used to quantify the areas of Hoechst-positive nuclei normalized to perfused vessels as a measurement of vessel permeability.
qPCR
Total RNA Purification Kit (Norgen Biotek) was used to extract RNA from cells growing on tissue culture dishes. cDNA was generated by using SuperScipt III First-Strand Synthesis System for PCR with reverse transcription (RT–PCR) (Invitrogen). The qPCR was conducted in 384-well PCR microplates (Axygen) in a 7900HT Fast Real-Time PCR System (ThermoFisher). cDNA samples were analysed in quadruplicates. Each well contained a total volume of 10 μl, including 0.5 μl cDNA (concentration 200–600 ng/μl), 2.0 μl primers (2.5 mM), 5.0 μl Fast SYBR Green Master Mix (ThermoFisher) and 2.5 μl DEPC-treated water (Ambion). The reaction started with a denaturation step at 95 °C for 20 s followed by 40 cycles of denaturation at 95 °C for 1 s and annealing and elongation at 60 °C for 20 s. The fluorescence was measured at the end of each cycle. Measured data was analysed using the comparative CT method (ΔΔCT method).
Mouse Gapdh (Integrated DNA Technologies), forward: AACTTTGGCATTGTGGAAGG; reverse: ACACATTGGGGGTAGGAACA; human GAPDH, forward: AGCCACATCGCTCAGACA; reverse: GCCCAATACGACCAAATCC; mouse Slit2, forward: AGCACCATCGAGAGGGGAG; reverse: GATCAAGCCGGTAGAGCTTCG; exon 8 Slit2, forward: CTGGCTCTCAGACTGGCTTC, reverse: TCTCGTTTTTGAACCTCTGCT; mouse Robo1, forward: CCTTCAGACCTGATCGTCTCC; reverse: TGAGCGCGGGTCATCTTTG; human ROBO1, forward: CTTACACCCGTAAAAGTGACGC; reverse: TGGTCTCTCTAAGACAGTCAGC.
For analysis of mouse Slit2 primers (mSlit2Exon3F AGGGGAGCATTCCAGGATCT and mSlit2Intron3R AGCCACCACGAAACTGAAGA) were designed for the border of exon 3 with the downstream intron.
Copy-number assay
Genomic DNA (gDNA) was extracted from cells growing in vitro using the DNeasy Blood and Tissue Kit (Qiagen, 69506). The manufacturer protocol was followed including an RNAase treatment step with 4 μl of 100 mg/ml RNase A (Qiagen, 19101). Two μl of gDNA samples (diluted in 5 ng/μl in DEPC-treated water) were added to the reaction mixture: 5.5 μl TaqMan Genotyping Master Mix (ThermoFisher Scientific, 4371353); 0.55 μl TaqMan Copy Number Assay (ThermoFisher Scientific Slit2 PN4400291,): 0.55 μl TaqMan Copy Number Reference Assay (Mouse Tfrc, ThermoFisher Scientific 4458370); 2.2 μl DEPC-treated water. A standard real-time PCR protocol was used (95 °C for 10 min, 40 cycles of 15 s at 95 °C and 60 s at 60 °C) performed with a QuantStudio 5 System (ThermoFisher Scientific). Samples were denatured at 95 °C for 10 min, followed by 40 cycles of 15 s denaturation at 95 °C and annealing plus elongation for 60 s at 60 °C. Copy number analysis was performed with the CopyCaller software (Version 2.1, ThermoFisher Scientific) with a manual CT threshold of 0.2 and auto baseline option selected.
5-Azacytidine treatment of tumour cells
4T1 tumour cells were incubated for 72 h in D10F supplemented with 5,000 μM 5-azacytidine (Sigma A2385) or the same volume of DMSO as negative control. Cells were changed to D10F without DMSO or 5-azacytidine for 9 days before RNA extraction. cDNA synthesis and qPCR for SLIT2 were performed.
Cell-free RNA conditioned medium and in plasma
B16F0 and B16F10, and 67NR and 4T1, cells were plated and incubated in Opti-MEM (Gibco) as described in ‘Conditioned medium treatment of endothelial cells’. Ten ml of the medium was concentrated into a final volume of 300 μl using 10-kDa filtration system (Amicon Ultra-15). Total RNA was isolated (Norgen plasma/Serum RNA Purification Mini kit). RNA was quantified by measuring the samples on a Nanodrop instrument and normalizing the concentration to the number of cells.
For cell-free RNA isolation from the plasma, BALB/c (BALB/cAnNCrl) mice were implanted with either 67NR or 4T1 tumours in the mammary fat pad as described in ‘Tumour growth, metastasis and circulating tumour cells assays’. When the tumours reached 600 mm3, the whole blood was collected with a 27G needle in a 1-ml syringe containing 50 μl of 0.5M EDTA, through cardiac puncture. The blood was then spun down 1,500g for 10 min and the plasma was collected. Two hundred μl of plasma was used for total RNA extraction (Norgen plasma/Serum RNA Purification Mini kit). RNA was quantified using a Bioanalyzer 2100 (Agilent).
RNA immunoprecipitation using J2 antibody
Immunoprecipitation of dsRNA with a dsRNA-specific J2 antibody was adapted from previously published work50. In brief, breast cancer cells (67NR or 4T1) were lysed with NP-40 Lysis Buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM EDTA, and 0.5% Igepal CA-630) supplemented with cOmplete EDTA-free protease inhibitor (Roche). Cell lysates were passed through a G26 needle before centrifugation at 20,000g at 4 °C for 10 min. Ten per cent of the supernatant (input) was set aside for RNA isolation using TRIzol-LS and Direct-zol Miniprep kit (Zymo Research). The rest of the supernatant was diluted fivefold using NET-2 buffer (50 mM Tris-HCl, pH 7.5, 1 mM supplemented with and 0.5% Igepal CA-630) and supplemented with 10 U of Turbo DNase (Ambion) and 10 mM of MgCl2. Protein A Dynabeads that were coupled with J2 or mouse IgG2a isotype-specific control antibody were added to the supernatant and rotated at 4 °C for 2 h. The beads were washed twice with NET-2 buffer, then twice with HSWB (50 mM Tris-HCl, pH 7.5, 750 mM NaCl, 1 mM EDTA, 1% Igepal CA-630, 0.5% sodium deoxycholate, 0.1% SDS) and another two times with NET-2 buffer. After all liquids were drained from the beads, RNAs were isolated from the beads using TRIzol and RNA Clean & Concentrator-5 (Zymo Research). Both the input and J2-immunoprecipitated RNA were depleted of rRNAs using NEBNext rRNA Depletion Kit (Human/Mouse/Rat) before they were used for library construction using NEBNext Ultra II Directional RNA Library Prep Kit. Constructed libraries were paired-end sequenced for 75 cycles using NextSeq 500.
Statistics and reproducibility
Results are presented in dot plots, with dots representing individual values, and in bar charts that depict average values ± s.e.m. The number of samples for each group was chosen on the basis of the expected levels of variation and consistency. Experiments were performed in a blinded fashion. Unless otherwise stated, statistical significance was assessed by a two-tailed Student’s t-test or Mantel–Cox test for mouse survival analysis with P value < 0.05 being considered statistically significant. All studies were performed at least twice, and all repeats were successful.
Ethical regulations
All experiments were performed in compliance with all relevant ethical regulations. All mouse experiments were performed under the supervision and approval of the IACUC at Rockefeller University. Mice were euthanized before individual tumours reached a maximum volume of 1,500 mm3 (IACUC-approved protocol limit), or whenever displaying any signs of pain or distress. Human breast cancer samples were de-identified and obtained with signed informed consent from patients and approval of the ethical committee.
Extended Data
Extended Data Fig. 1 |. Endothelial cells upregulate SLIT2 upon treatment with conditioned medium from highly metastatic 4T1 cells.
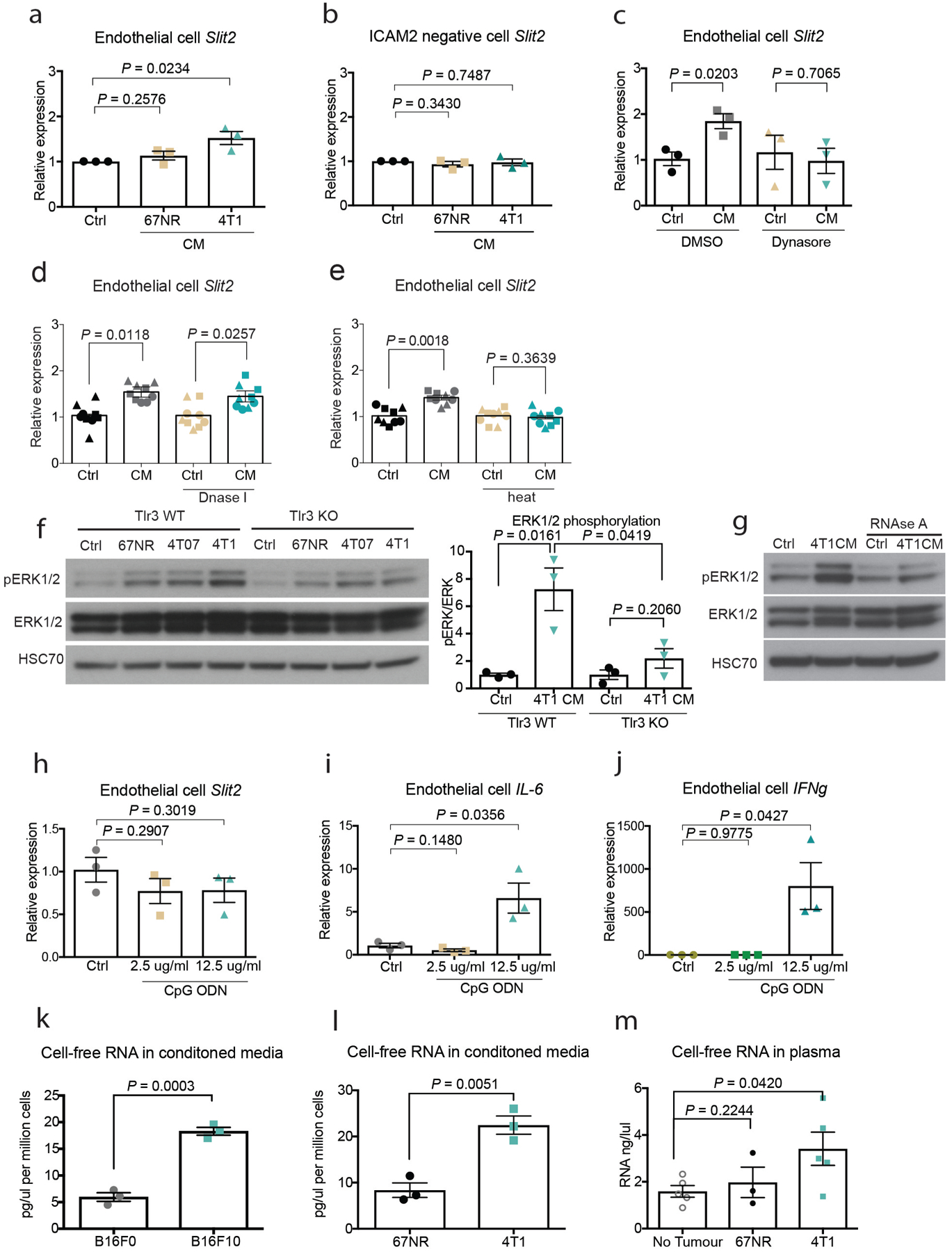
a, Primary MLECs (ICAM2-positive) upregulate SLIT2 when treated with conditioned medium derived from 4T1 cells (n = 3). Dot plot represents Slit2 mRNA levels measured by qPCR for each biological replicate with mean ± s.e.m. Two-tailed Student’s t-test. b, Primary nonendothelial cells (ICAM2-negative) from the lung do not upregulate SLIT2 upon treatment with 4T1 conditioned medium (n = 3). Dot plot represents Slit2 mRNA levels measured by qPCR for each biological replicate with mean ± s.e.m. Two-tailed Student’s t-test. c, Treatment of endothelial cells with 5 μM dynasore inhibits SLIT2 expression upon treatment with conditioned medium from 4T1 cells (n = 3). Dot plot represents Slit2 mRNA levels measured by qPCR for each biological replicate with mean ± s.e.m. Two-tailed Student’s t-test. d, e, Dot plots represent Slit2 mRNA expression by qPCR in endothelial cells exposed to 4T1 conditioned medium treated with (e) DNase I (10 μg/ml; n = 3), and (d) heat treatment (95 °C, 10 min; n = 3). Data are mean ± s.e.m. Two-tailed Student’s t-test. f, TLR3 wild-type (Tlr3 WT) and TLR3-knockout (Tlr3 KO) endothelial cells were treated with conditioned medium from 67NR, 4T07 and 4T1 cells. Western blot analysis revealed that wild-type endothelial cells display increased phosphorylation of ERK1 and ERK2 upon treatment with the conditioned medium from highly metastatic 4T1 cells. TLR3-knockout endothelial cells displayed reduced phosphorylation of ERK1 and ERK2 relative to wild-type controls. Dot plot displays densitometry quantification for three independent experiments. Two-tailed Student’s t-test. g, RNase A treatment of the 4T1 conditioned medium blunted endothelial phosphorylation of ERK1 and ERK2. h, Supplementation of basal medium with synthetic TLR9 ligand (CpG ODN, 2.5 or 12.5 μg/ml) did not induce endothelial SLIT2 upregulation (n = 3). Dot plot represents Slit2 levels measured by qPCR for each biological replicate with mean ± s.e.m. Two-tailed Student’s t-test. i, j, Supplementation of basal medium with synthetic TLR9 ligand (CpG ODN, 2.5 or 12.5 μg/ml) induced (i) endothelial Il6 (n = 3) and (j) Ifng mRNA expression (n = 3). Dot plot represents Il6 and Ifng levels measured by qPCR for each biological replicate with mean ± s.e.m. Two-tailed Student’s t-test. k, l, Quantification of RNA isolated from conditioned medium of (k) B16F0 (n = 3) and B16F10 cells (n = 3) and (l) 67NR (n = 3) and 4T1 cells (n = 3). Dot plot represents RNA concentrations detected in conditioned medium normalized by the cell number with mean ± s.e.m. Two-tailed Student’s t-test. m, RNA detection in plasma isolated from mice with 67NR (n = 3) and 4T1 (n = 5) mammary gland tumours. Tumour-free mice (n = 5) were used as a negative control. Increased concentrations of RNA were detected in the plasma of mice with the metastatic 4T1 tumours. Dot plot represents the RNA concentrations detected in the plasma of each mouse, either with no tumour or with 67NR and 4T1 tumours. Two-tailed Student’s t-test.
Extended Data Fig. 2 |. Endothelial SLIT2 deletion does not impair primary tumour growth and angiogenesis.
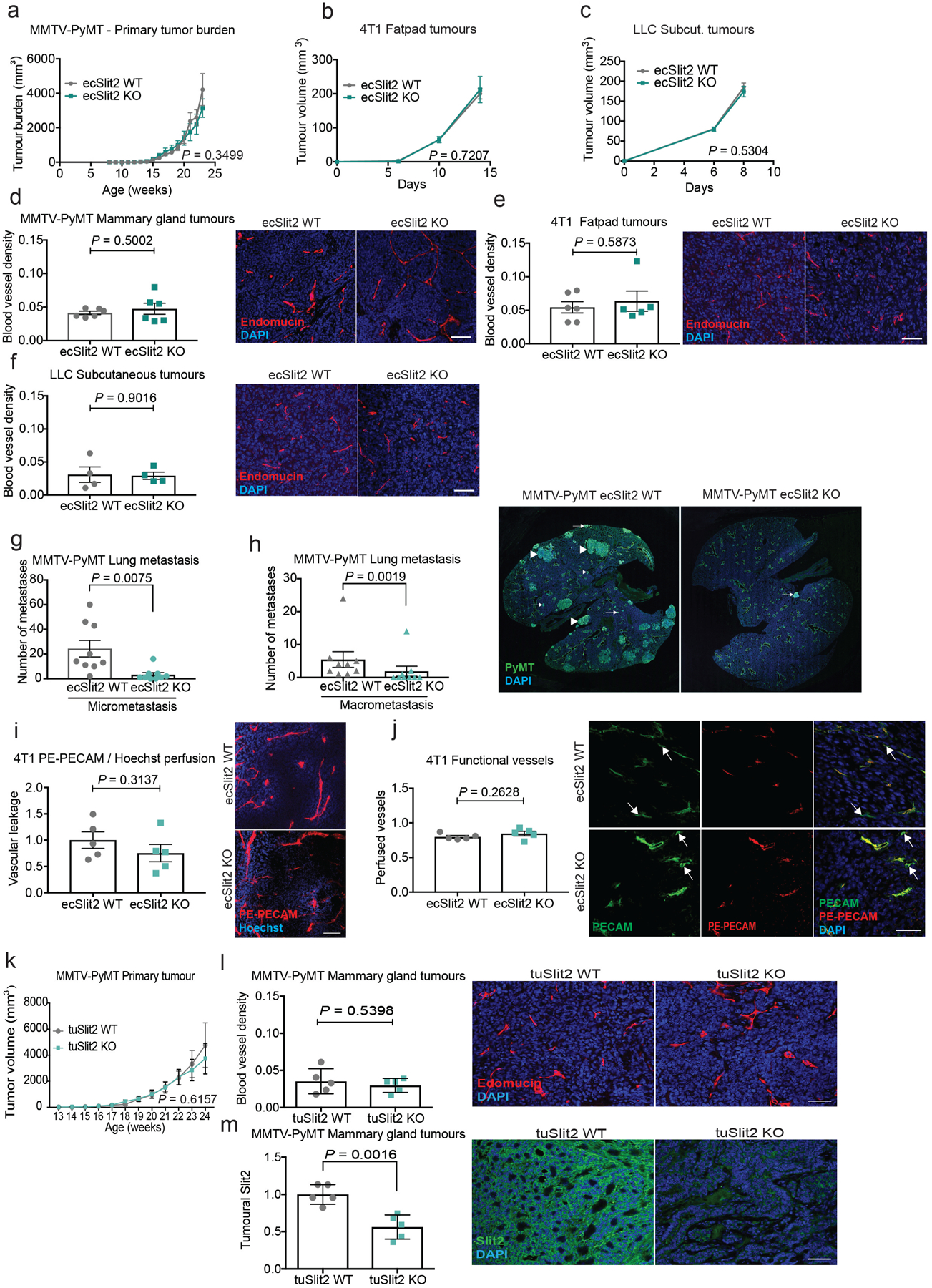
a–c, Tumour growth rates (left) for (a) spontaneous MMTV-PyMT mammary gland tumours (total tumour burden) in wild-type (n = 8) and ecSLIT2-knockout mice (n = 7), (b) orthotopic 4T1 mammary tumours in wild-type (n = 11) and ecSLIT2-knockout mice (n = 8), and (c) subcutaneous LLC tumours in wild-type (n = 22) and ecSLIT2-knockout mice (n = 19). Mean tumour volume ± s.e.m. for each time point. Two-tailed t-test for last time point. d, Mammary gland tumours from tamoxifen-treated Cdh5(PAC)-creERT2;Slit2-floxed;MMTV-PyMT (ecSLIT2-knockout) or CreERT2-negative Slit2-floxed;MMTV-PyMT (ecSLIT2 wild-type) mice were sectioned and stained for endomucin. No significant difference in blood vessel density was observed between tumours growing in wild-type and ecSLIT2-knockout mice. Each dot represents the average of endomucin area relative to total DAPI area in sections for each tumour, measured with ImageJ. Mean ± s.e.m. ecSLIT2 wild type, n = 6; ecSLIT2 knockout, n = 6. Scale bar, 50 μm. Two-tailed Student’s t-test. e, The 4T1 tumour sections were stained for endomucin. No difference in vessel density was observed between tumours from wild-type and ecSLIT2-knockout mice. Dot plot depicts endomucin area relative to DAPI area for each tumour, quantified by ImageJ. Mean ± s.e.m. ecSLIT2 wild type, n = 6; ecSLIT2 knockout, n = 5; Scale bar, 50 μm. Two-tailed Student’s t-test. f, LLC tumour sections were stained for endomucin. No difference in blood vessel density was observed between tumours growing in ecSLIT2-knockout and wild-type mice. Mean ± s.e.m. ecSLIT2 wild type, n = 4; ecSLIT2 knockout, n = 4. Scale bar, 50 μm. Two-tailed Student’s t-test. g, h, Immunofluorescence staining for PyMT in lung sections of MMTV-PyMT ecSLIT2 wild type or ecSLIT2-knockout mice reveals reduction in both micrometastasis (g) and macrometastasis (h). Dot plot displays the number of lung nodules per mouse, divided into micrometastases or macrometastases. ecSLIT2 wild type, n = 9; ecSLIT2 knockout, n = 9. Data are mean ± s.e.m. Two-tailed Mann–Whitney test. Arrowheads indicate macrometastasis and arrows indicate micrometastasis. i, Wild-type and ecSLIT2-knockout mice bearing 4T1 primary tumours were intravenously injected with PE–PECAM antibody and Hoechst. The 4T1 tumour sections were prepared, and vessel permeability was quantified. Representative images of tumour sections showing Hoechst nuclear staining and perfused PE–PECAM vessels. Scale bar, 50 μm. Dot plot represents the mean ratio of Hoechst signal relative to PE–PECAM signal ± s.e.m.; ecSLIT2 wild type, n = 5; ecSLIT2 knockout, n = 5. j, Tumour sections from wild-type and ecSLIT2-knockout mice bearing 4T1 primary tumours were injected via tail vein with PE–PECAM antibody and stained for PECAM to quantify the proportion of perfused vessels relative to total tumour vessels. Representative images of tumour sections showing PE–PECAM perfused vessels (functional vessels) relative to total vessels stained with PECAM. White arrows indicate nonperfused blood vessels. Scale bar, 50 μm. Bar chart represents the mean ratio of Hoechst relative to endomucin staining ± s.e.m. ecSLIT2 wild type, n = 5; ecSLIT2 knockout, n = 5. i, j, Two-tailed Student’s t-test. k, Tumour growth rates for the MMTV-PyMT tumours in tuSLIT2-knockout (n = 12) or wild-type (n = 10) control mice. Tumour burden was calculated by adding individual tumours in each mouse. Data are mean ± s.e.m. Two-tailed t-test for last time point. l, Blood vessel density was measured by immunostaining for endomucin in sections of mammary gland tumours from MMTV-PyMT mice (tuSLIT2 wild type or tuSLIT2 knockout). Bar chart represents the average endomucin area relative to DAPI area ± s.e.m. Scale bar, 50 μm. tuSLIT2 wild type, n = 5; tuSLIT2 knockout, n = 5. m, Tumoural SLIT2 deletion was confirmed by immunostaining of tumours for SLIT2. Fluorescent quantification revealed a significant reduction in SLIT2 levels in tuSLIT2-knockout tumours. Bar chart with each dot representing the average of fluorescent quantification of different tumour sections for each mouse ± s.e.m. tuSLIT2 wild type, n = 5; tuSLIT2 knockout, n = 5. l, m, Two-tailed Student’s t-test. Scale bar, 50 μm.
Extended Data Fig. 3 |. Endothelial SLIT2 deletion does not affect metastatic colonization upon tail vein injection.
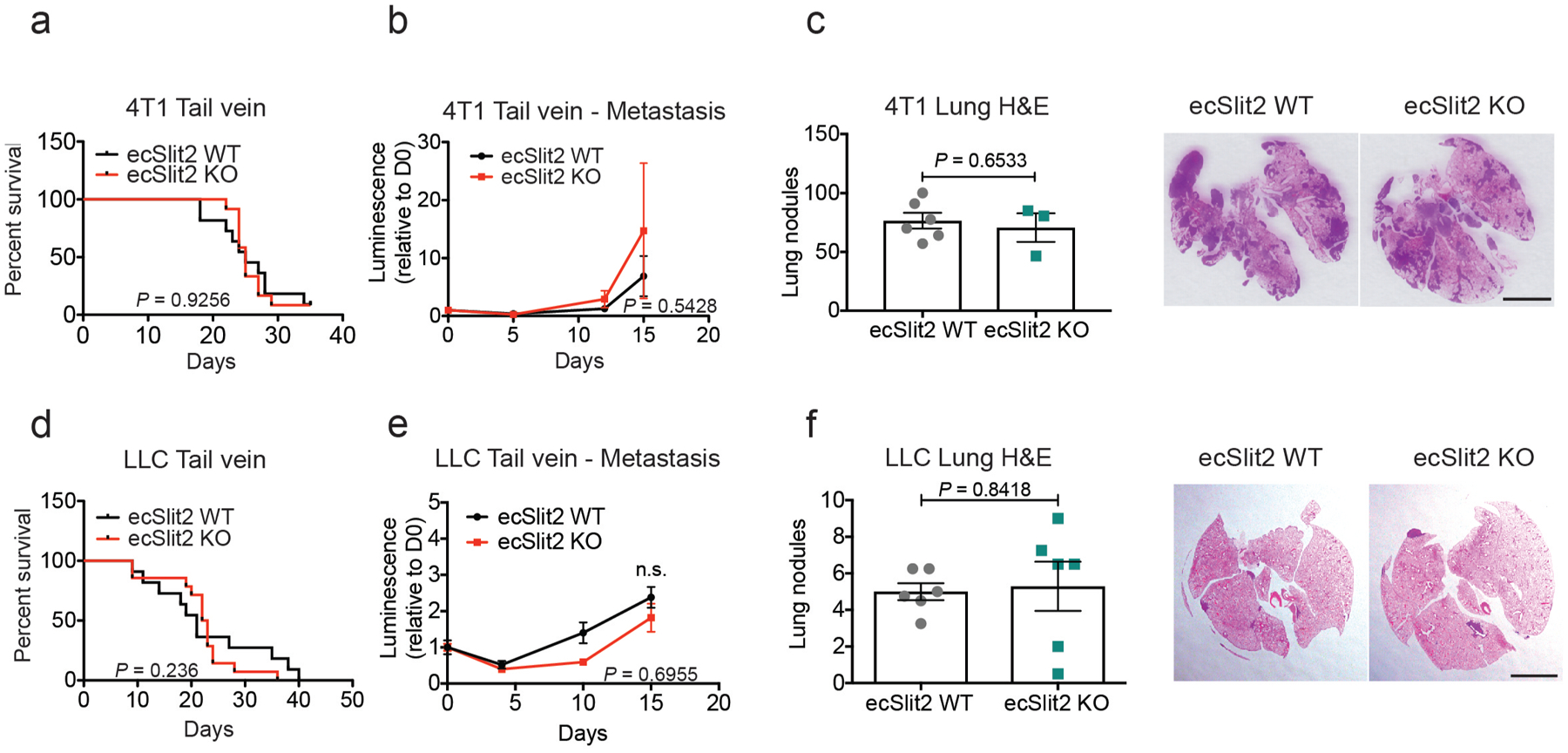
a, 4T1 cells were injected intravenously into the tail veins of ecSLIT2-knockout or wild-type littermate controls. Survival is depicted as the number of days until each mouse was euthanized owing to metastatic disease. ecSLIT2 wild type, n = 11; ecSLIT2 knockout, n = 12. log-rank (Mantel–Cox) test. b, Metastatic burden was measured by quantification of mean luminescence relative to day 0 ± s.e.m. ecSLIT2 wild type, n = 11; ecSLIT2 knockout, n = 12. Two-tailed Student’s t-test. c, H&E-stained lung sections were used for quantification of lung nodules 17 d after injection of cells. Dot plot represents the average number of lung nodules per mouse ± s.e.m. ecSLIT2 wild type, n = 6; ecSLIT2 knockout, n = 3. Scale bar, 0.5 cm. Two-tailed Student’s t-test. d, LLC cells were injected into the tail veins of wild-type or ecSLIT2-knockout littermate controls. Survival is depicted as in a. ecSLIT2 wild type, n = 11; ecSLIT2 knockout, n = 14. log-rank (Mantel–Cox) test. e, Metastatic burden was measured by mean bioluminescence quantification relative to day 0 ± s.e.m. ecSLIT2 wild type, n = 6, ecSLIT2 knockout, n = 6. Two-tailed Student’s t-test. f, H&E-stained lung sections revealed no significant difference in lung nodule numbers between groups, 15 d after injection. Data are mean ± s.e.m. ecSLIT2 wild type, n = 6; ecSLIT2 knockout, n = 6. Scale bar, 0.5 cm. Two-tailed Student’s t-test.
Extended Data Fig. 4 |. Time course of endothelial SLIT2 upregulation upon conditioned medium treatment.
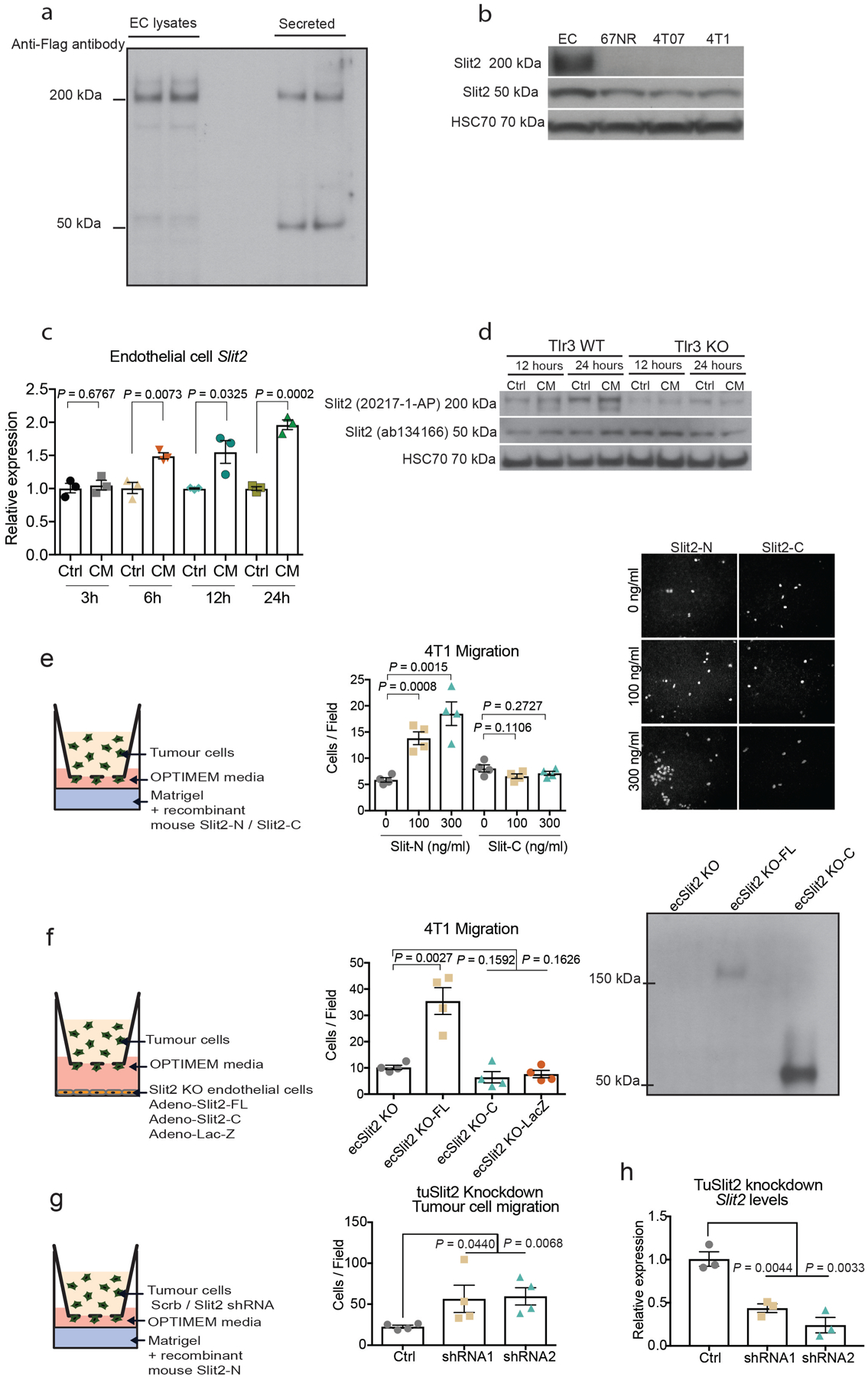
C-terminal SLIT2 fragment is insufficient to promote 4T1 tumour cell migration. a, Endothelial cells overexpressing SLIT2-C–Flag were used to generate conditioned medium or lysed for protein extraction. Anti-Flag antibody was used to detect SLIT2 in either cell lysates or secreted SLIT2 in the conditioned medium. b, Western blot for SLIT2 to detect the full-length SLIT2 or its C-terminal fragment. HSC70 was used as a loading control. c, Endothelial cells were treated with conditioned medium from 4T1 cells for 3, 6, 12 and 24 h, and SLIT2 levels were assessed. Dot plot represents Slit2 mRNA levels for each biological replicate with mean ± s.e.m. n = 3 for each group. Two-tailed Student’s t-test. d, Western blot of SLIT2 protein upon treatment of wild-type endothelial cells or TLR3-knockout endothelial cells with conditioned medium from 4T1 cells or basal medium (control). e, The 4T1 tumour cells displayed enhanced migration towards increasing concentrations of recombinant SLIT2-N, but not SLIT2-C. Dot plot represents the number of migrated cells per optical field of view (10× objective) with mean ± s.e.m. Panel displays representative images from migrated cells (10× objective). n = 4 for each group. Two-tailed Student’s t-test. f, Adenoviral expression of full-length SLIT2 in ecSLIT2-knockout endothelial cells promoted tumour cell migration in a transwell assay while adenoviral expression of C-terminal SLIT2 (ecSLIT2 KO-C) or LacZ (ecSLIT2 KO-LacZ) did not enhance tumour cell migration. Mean ± s.e.m. Two-tailed Student’s t-test. Western blot with antibody against C-terminal region of SLIT2 detected full-length and C-terminal forms of SLIT2 in ecSLIT2-knockout endothelial cells transduced with adenovirus. n = 4 for each condition. g, SLIT2 depletion in 4T1 tumour cells using two independent shRNAs increased migration of tumour cells towards endothelial cells in a transwell migration assay. Dot plot represents the number of migrated 4T1 cells per optical field of view (10× objective) with mean ± s.e.m. P value between control and shRNA no. 1 and no. 2 corresponds to a one-tailed Student’s t-test. n = 4 for each condition. h, SLIT2 expression in 4T1 cells transduced with either scrambled shRNA or two independent shRNAs targeting SLIT2. Dot plot represents Slit2 mRNA levels for each biological replicate with mean ± s.e.m. Two-tailed Student’s t-test. n = 3 for each condition.
Extended Data Fig. 5 |. Endothelial and tumoural SLIT2 and tumoural ROBO1 levels are associated with cancer progression in patients with breast cancer.
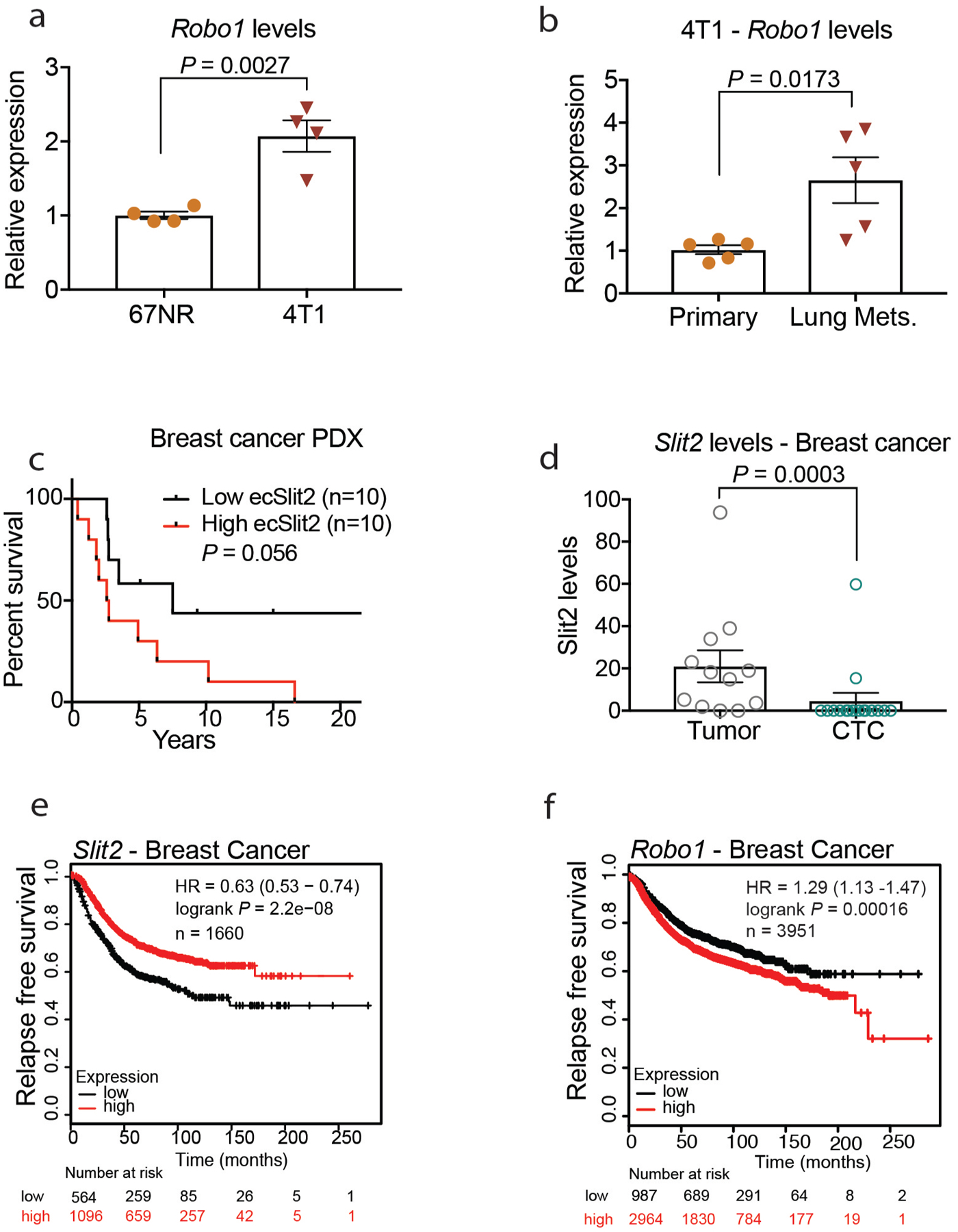
a, b, Dot plots represent relative Robo1 mRNA expression by qPCR in mouse metastatic 4T1 (n = 4) and nonmetastatic 67NR cells (n = 4) (a) and parental 4T1 cells (primary n = 5) and 4T1-derived lung metastases (n = 5) (b). a, b Two-tailed Student’s t-test. c, SLIT2 quantification in endomucin-positive vessels of PDX tumours from patients with breast cancer revealed that high levels of endothelial SLIT2 correlate with worse prognosis. Survival data for the patients from whom the PDXs were isolated were stratified into high and low endothelial SLIT2 expression. High ecSLIT2 expression, n = 10 patients; low ecSLIT2 expression, n = 10 patients. log-rank (Mantel–Cox) test. d, RNA-seq data analysis from primary tumours and CTCs of patients with breast cancer from Gene Expression Omnibus GSE11184218 revealed reduced or undetectable SLIT2 expression in CTCs from patients with stage II or III breast cancer when compared with SLIT2 levels of matched primary tumours. Dot plot represents the mean SLIT2 levels ± s.e.m. Primary tumour samples, n = 12; CTC samples, n = 16. Two-tailed Mann–Whitney test. e, f, Kaplan–Meier survival analysis for SLIT2 (e) and ROBO1 (f) expression in human breast tumours generated using KMPLOT, with auto select best cut-off selected. SLIT2, n = 1,660; ROBO1, n = 3,951. log-rank (Mantel–Cox) test.
Extended Data Fig. 6 |. TLR3 knockout in the tumour stroma reduces tumour cell intravasation, whereas synthetic dsRNA induces intravasation by tumour cells.
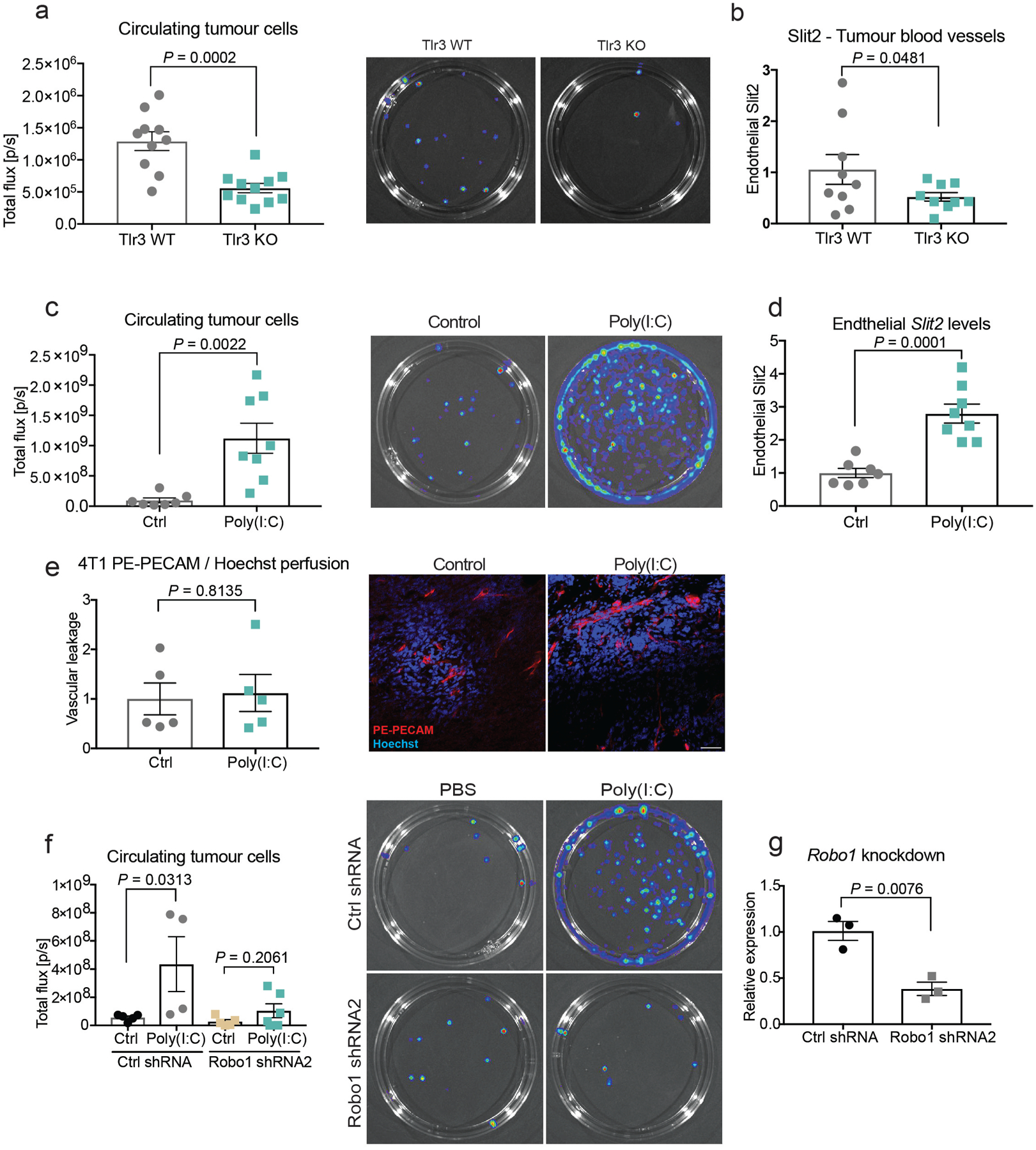
a, 4T1-Luc-zsGreen tumours were established in the mammary fat pads of wild-type and TLR3-knockout mice. Circulating tumour cells were isolated from the whole blood of mice, and quantified by identifying luciferase-positive colonies. Dot plot represents measured bioluminescence (photons s−1). Representative images of luciferase-positive colonies growing on 10-cm tissue culture dishes. TLR3 wild type, n = 10; TLR3 knockout, n = 11. b, Quantification of SLIT2 expression in the blood vessels of 4T1 mammary tumours in either wild-type or TLR3-knockout mice. Dot plot represents mean fluorescence intensities of SLIT2 in endomucin-positive vessels of 4T1 tumours ± s.e.m. TLR3 wild type, n = 9; TLR3 knockout, n = 9 tumours. a, b, Data are mean ± s.e.m. Two-tailed Student’s t-test. c, Injection of poly(I:C) (25 μg) into NSG mice promoted intravasation by tumour cells, measured by quantification of circulating tumour cells through detection of luminescence (photons s−1) from luciferase-positive colonies. Dot plot with each dot representing measured bioluminescence (photons s−1), for the whole-blood-derived colonies for each mouse. Control group (ctrl), n = 7; poly(I:C), n = 8. Representative images of luciferase-positive colonies growing on a 10-cm tissue culture dish. d, ImageJ quantification of immunofluorescent SLIT2 staining that co-localized with endomucin-positive vessels in 4T1 tumours injected with either PBS (control) or poly(I:C). Dot plot represents fluorescent intensities of SLIT2 in the vasculature of 4T1 tumours ± s.e.m. Control, n = 7; poly(I:C), n = 8 tumours. e, PE–PECAM antibody and Hoechst perfusion did not reveal changes in vascular permeability by poly(I:C) treatment. Representative images of tumour sections showing Hoechst nuclear staining and perfused PE–PECAM vessels. Scale bar, 50 μm. Bar chart represents the average ratio of Hoechst signal relative to PE–PECAM signal normalized to the control group ± s.e.m.; n = 5 tumours for each group. c–e, Data are mean ± s.e.m. Two-tailed Student’s t-test. f, Robo1 knockdown in tumour cells with a second shRNA (Robo1 shRNA no. 2) inhibited poly(I:C)-induced intravasation. Dot plot with each dot representing measured bioluminescence (photons s−1), for the whole-blood-derived luciferase-positive colonies for each mouse with mean ± s.e.m. Control shRNA: control, n = 5; poly(I:C), n = 4; Robo1 shRNA no. 2: control, n = 5; poly(I:C), n = 6. One-tailed Student’s t-test. g, ROBO1 expression in 4T1-Luc-zsGreen cells transduced with either scrambled shRNA (control shRNA) or shRNA no. 2 targeting Robo1. Dot plot represents Robo1 mRNA levels for each replicate with mean ± s.e.m. Control shRNA, n = 3; Robo1 shRNA no. 2, n = 3. Two-tailed Student’s t-test.
Extended Data Fig. 7 |. SLIT2 promoter is hypermethylated in breast cancer in humans.
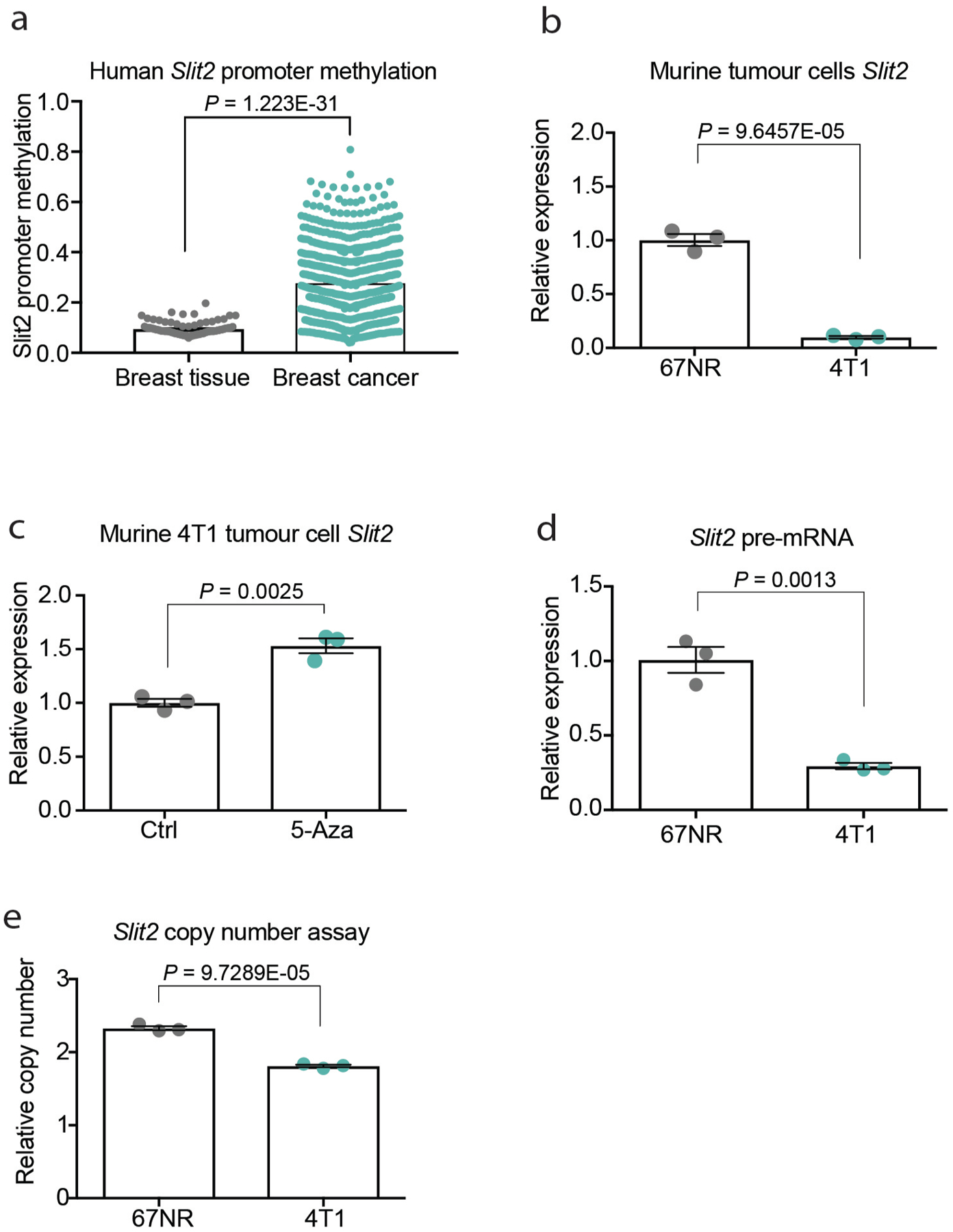
a, SLIT2 promoter methylation in normal breast tissues and invasive breast carcinomas, reproduced from the Human Cancer database (MethHC)26. Dot plot represents the mean SLIT2 promoter methylation ± s.e.m. Breast tissue, n = 92; Breast cancer, n = 735. b, Slit2 expression by real-time qPCR in 67NR and 4T1 tumour cells. Dot plot represents Slit2 mRNA levels for each biological replicate with mean ± s.e.m. 67NR, n = 3; 4T1, n = 3. c, Treatment of 4T1 tumour cells with the demethylating agent 5-azacytidine (5-aza) upregulated SLIT2 expression. Dot plot represents Slit2 mRNA levels for each biological replicate with mean ± s.e.m. Control group (ctrl) n = 3; 5-aza group, n = 3. d, Pre-mRNA levels were measured by real-time qPCR using primers that span the exon–intron junction. Dot plot represents pre-mRNA Slit2 levels for each biological replicate (n = 3) with mean ± s.e.m. e, Genomic copy number for Slit2 in 67NR and 4T1 tumour cells relative to mouse lung endothelial cells (MLEC). Dot plot represents Slit2 copy number (TaqMan assay) for each replicate (n = 3) with mean ± s.e.m. a–e, Two-tailed Student’s t-test.
Extended Data Fig. 8 |. Tumoural SLIT2 deletion does not significantly affect apoptosis and expression of SLIT2-related cytokines.
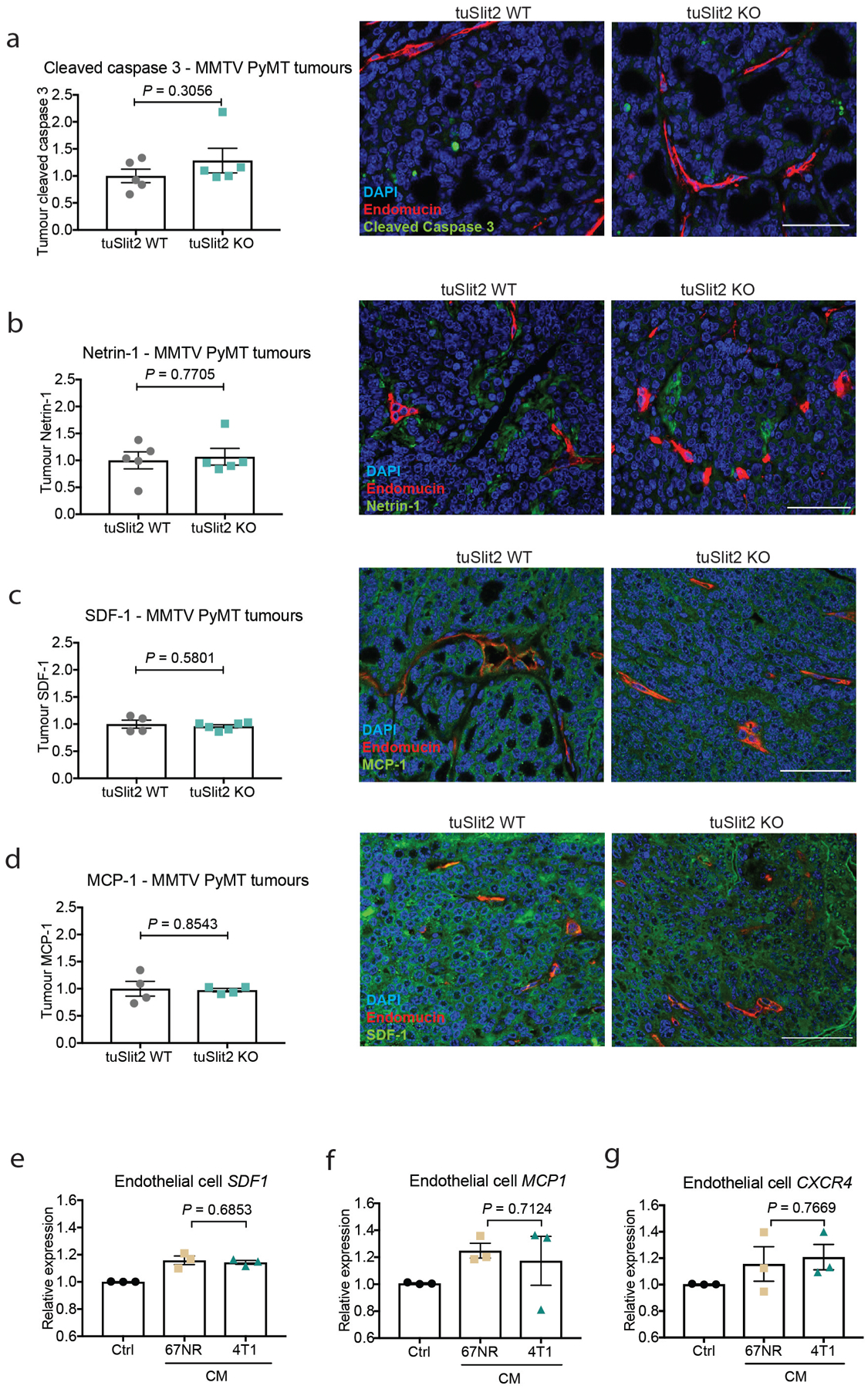
a, No difference in cleaved caspase 3 staining in MMTV-PyMT tumour sections of wild-type versus ecSLIT2-knockout mice. Dot plot represents fluorescent intensity of cleaved caspase 3 in tumour sections. Mean ± s.e.m. tuSLIT2 wild type, n = 5; tuSLIT2 knockout, n = 5 tumours. Scale bar, 50 μm. b, Deletion of SLIT2 in the tumour compartment of MMTV-PyMT tumours did not significantly alter tumour netrin 1 expression. Dot plot represents fluorescent intensity of netrin 1 in tumour sections. Mean ± s.e.m. tuSLIT2 wild type, n = 5; tuSLIT2 knockout, n = 5 tumours. Scale bar, 50 μm. c, Deletion of SLIT2 in the tumour compartment of MMTV-PyMT tumours did not significantly affect tumour SDF1 expression; Dot plot represents fluorescent intensity of SDF1 in tumour sections ± s.e.m. tuSLIT2 wild type, n = 4; tuSLIT2 knockout, n = 6 tumours. Scale bar, 50 μm. d, Deletion of SLIT2 in the tumour compartment of MMTV-PyMT tumours did not significantly change tumour MCP1 expression; Dot plot represents fluorescent intensity of MCP1 in tumour sections ± s.e.m. tuSLIT2 wild type, n = 4; tuSLIT2 knockout, n = 4 tumours. Scale bar, 50 μm. e, Sdf1 (also known as Cxcl12) expression was measured by qPCR in endothelial cells treated with conditioned medium from the poorly metastatic 67NR or highly metastatic 4T1 cells, or with basal medium (control). Bar chart represents the mean expression levels of SDF1 ± s.e.m. Each group, n = 3. f, Mcp1 (also known as Mcpt1) expression was measured by qPCR in endothelial cells treated with conditioned medium from poorly metastatic 67NR or highly metastatic 4T1 cells, or basal medium (control). Bar chart depicts the mean expression of MCP1 ± s.e.m. Each group, n = 3. g, Cxcr4 expression was assessed by qPCR in endothelial cells treated with conditioned medium from poorly metastatic 67NR or highly metastatic 4T1 cells, or basal medium (control). Bar chart represents the mean expression values of CXCR4 ± s.e.m. Each group, n = 3. a–g, Two-tailed Student’s t-test.
Extended Data Fig. 9 |. Increased detection of dsRNA and ERVs in highly metastatic tumours.
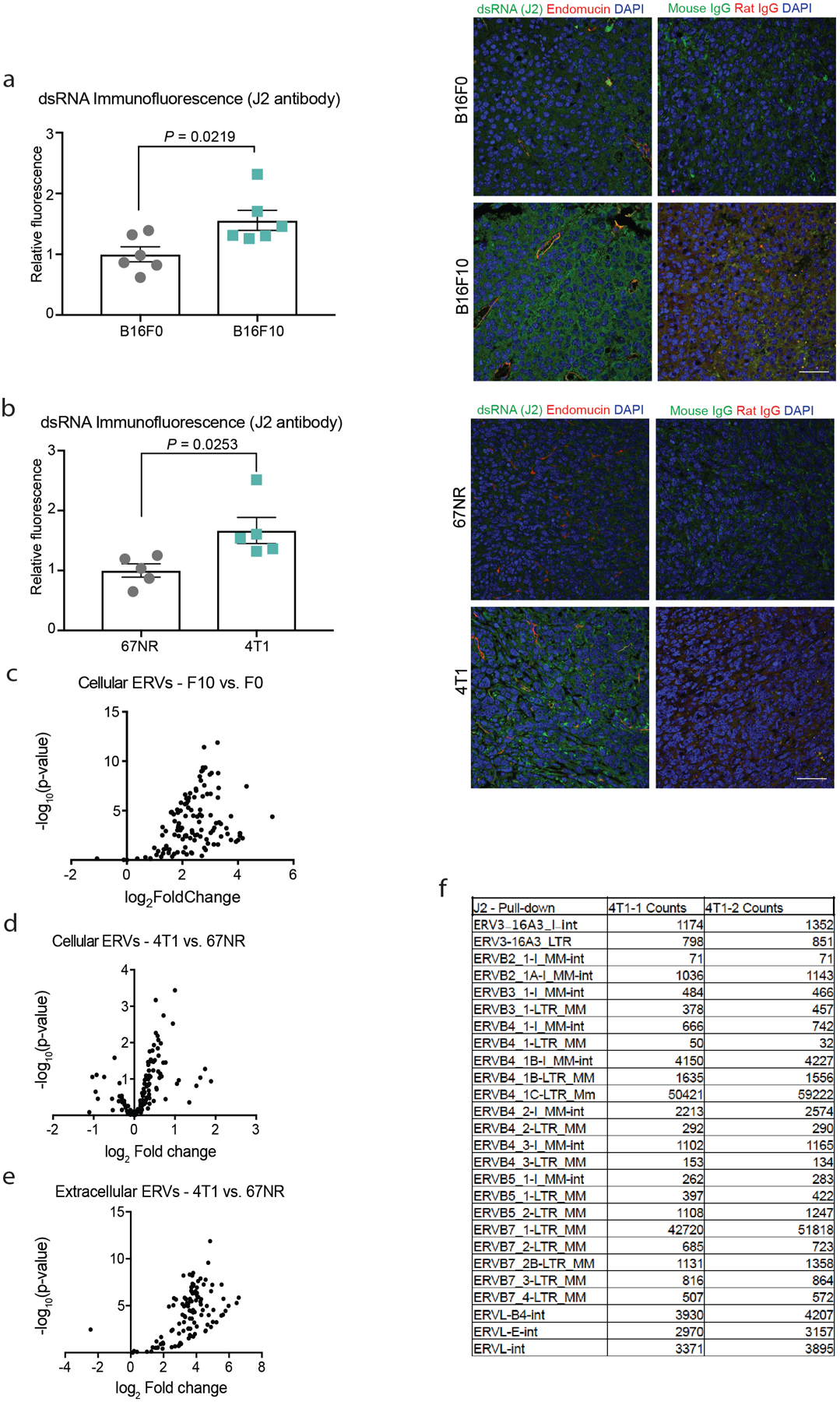
dsRNA was detected by immunostaining of tumours with the J2 antibody. a, b, Fluorescent quantification revealed a significant increase in dsRNA signal in highly metastatic B16F10 relative to B16F0 tumours (a) and in metastatic 4T1 relative to nonmetastatic 67NR tumours (b). Bar chart with each dot representing the mean relative fluorescent quantification of different tumour sections for each tumour normalized to the low-metastatic B16F0 or 67NR tumours ± s.e.m. B16F0, n = 6; B16F10, n = 6; 67NR, n = 5; 4T1, n = 5. Representative images are shown for the immunostaining of dsRNA (J2), endomucin and DAPI in tumour sections of B16F0 and B16F10, and 67NR and 4T1, tumours. Scale bar, 50μm. a, b, Two-tailed Student’s t-test. c, d, Volcano plot displays the log2-transformed fold differences in expression of ERVs between B16F10 relative to B16F0 cells (c) as well as 4T1 relative to 67NR cells (d). Out of 12,332 annotations, 123 ERV sequences were detected in our RNA-seq libraries. n = 3 biological replicates. e, Cell-free RNA was isolated from the conditioned medium of 67NR and 4T1 cell cultures, and RNA-seq libraries were generated for analysis of the aforementioned 123 ERVs. Volcano plot displays the log2-transformed fold differences in detected ERVs in the supernatant of 4T1 cells relative to 67NR cells. n = 3 biological replicates. f, Pull-down of dsRNA with the J2 antibody followed by RNA-seq detected ERVs secreted by 4T1 cells. Table shows the total counts of ERV reads detected for two immunoprecipitation replicates (4T1–1 and 4T1–2).
Extended Data Fig. 10 |. ROBO1 knockdown in human MDA cells reduces orthotopic lung metastasis.
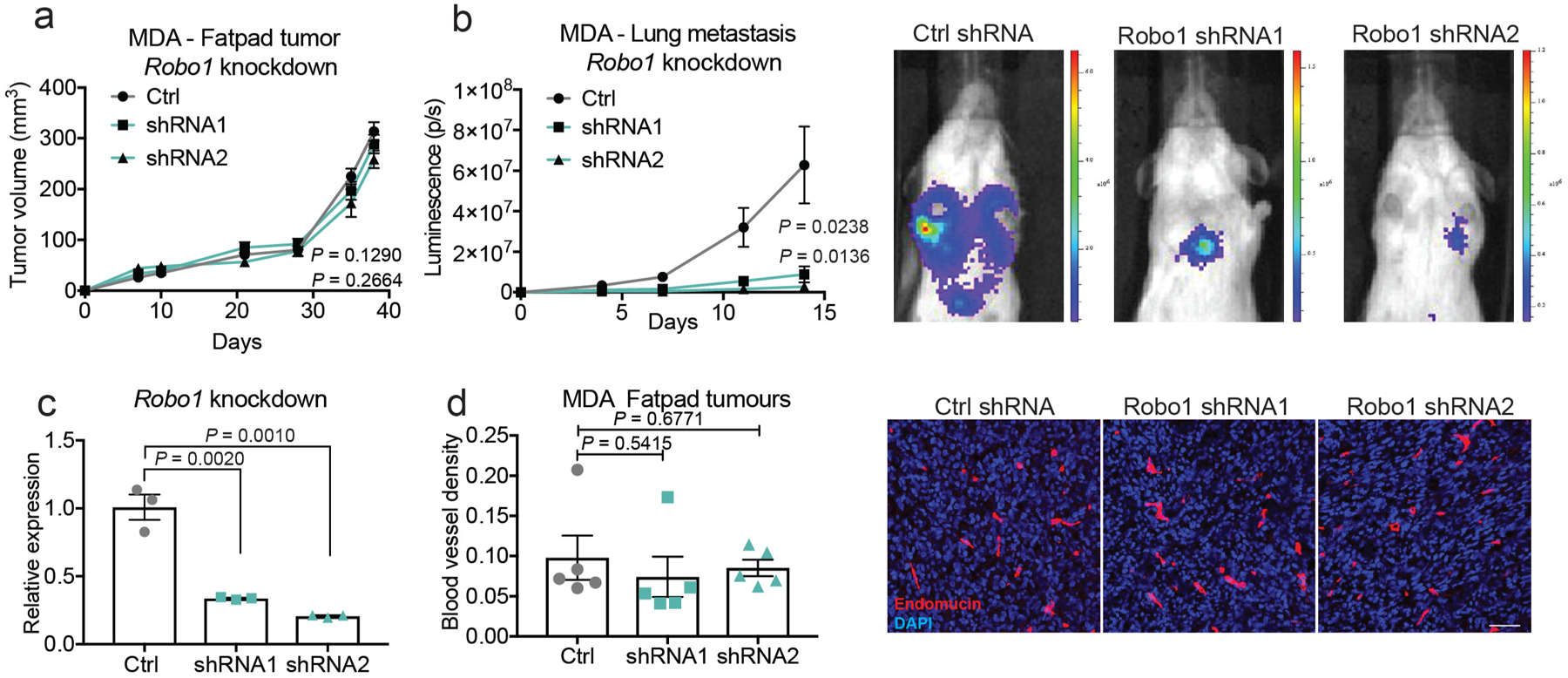
a, Two independent shRNAs were used to knockdown ROBO1 in human breast cancer MDA cells. Both ROBO1 knockdown (shRNA no. 1 and shRNA no. 2) and control shRNA (control) cells were injected into the mammary fat pads of NSG mice. Tumours were measured at days 7, 10, 21, 28, 35 and 38, and surgically resected on day 38. P values for the last time point (control–shRNA no. 1 and control–shRNA no. 2) are shown. Two-tailed Student’s t-test. b, Lung metastasis was assessed by bioluminescence imaging (photons per second). n = 5 for each group. P values for the last time point (control–shRNA no. 1 and control–shRNA no. 2) are shown. Two-tailed Student’s t-test. Representative images of lung bioluminescence are shown with colour scale (photons per s per cm2 per sr) for each independent group (ctrl and shRNA no. 1 or no. 2) c, Robo1 knockdown levels were confirmed by qPCR (n = 3). Two-tailed Student’s t-test. d, Blood vessel density was assessed by staining tumour sections with endomucin and DAPI. No significant difference in blood vessel density was detected between the control and Robo1-knockdown tumour groups (shRNA no. 1 or no. 2). Dot plot represents endomucin area relative to DAPI area for each tumour, quantified using ImageJ. Mean ± s.e.m. n = 5 each group. Two-tailed Student’s t-test. Scale bar, 50 μm.
Supplementary Material
Acknowledgements
We thank Veena Padmanaban, Lisa Noble, Dennis Hsu, Doowon Huh, Ryan Moy, Serkan Belkaya, Baris Boyraz, and Daniel Mucida for thoughtful comments on previous versions of the manuscript. We also thank Rockefeller University resource centers: Svetlana Mazel and Stanka Semova of the flow cytometry resource center, Connie Zhao from the genomics resource center, Vaughn Francis from the Comparative bioscience center and veterinary staff for animal husbandry and care, and Alison North, Christina Pyrgaki and staff of the Bio-Imaging resource facility. We thank Prof. Sandra J. Gendler from Mayo Clinic Arizona for providing MMTV-PyMT mice, Allain Chedotal for generation and generous distribution of the Slit2 floxed mice, Marc Tessier-Lavigne for kindly transferring Slit2 floxed mice, and Prof. Ralf Adams for kindly providing the Cdh5(PAC)-CreERT2 mice. Additionally, we would like to thank Katrin Svensson for generously providing the Slit2 adenoviral particles. T.M., K.W., M.S., M.M. and J.Y.K. are members of the German Academic Scholarship Foundation (Studienstiftung des deutschen Volkes). K.W. and S.R. were awarded a fellowship from Boehringer Ingelheim Foundation. B.T. was supported by the Lucy Lee Chiles Fellowship (HFCR-15-06-04) from the Hope Funds for Cancer Research. This work was supported by grants from the National Institutes of Health (RO1-CA236954-01A1 to S.F.T.; RO1-GM123977 to H.G.) and the Department of Defense Collaborative Scholars and Innovators award (W81-XWH-12-1-0301). S.F.T. was also supported by a Faculty Scholars grant from the Howard Hughes Medical Institute, the Breast Cancer Research Foundation award, the Black Family Foundation, and the Reem-Kayden award.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
RNA-seq data have been deposited in the NCBI GEO accession GSE145319. Other data generated are available from the corresponding author upon reasonable request.
Data availability
Code generated is available from the author upon reasonable request.
Readers are welcome to comment on the online version of this article at www.nature.com/nature.
The authors declare no competing financial interests.
References
- 1.Strilic B et al. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature 536, 215–218, doi: 10.1038/nature19076 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Png KJ, Halberg N, Yoshida M & Tavazoie SF A microRNA regulon that mediates endothelial recruitment and metastasis by cancer cells. Nature 481, 190–194, doi: 10.1038/nature10661 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Pencheva N et al. Convergent multi-miRNA targeting of ApoE drives LRP1/LRP8-dependent melanoma metastasis and angiogenesis. Cell 151, 1068–1082, doi: 10.1016/j.cell.2012.10.028 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pencheva N & Tavazoie SF Control of metastatic progression by microRNA regulatory networks. Nat Cell Biol 15, 546–554, doi: 10.1038/ncb2769 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rafii S, Butler JM & Ding BS Angiocrine functions of organ-specific endothelial cells. Nature 529, 316–325, doi: 10.1038/nature17040 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kobayashi H et al. Angiocrine factors from Akt-activated endothelial cells balance self-renewal and differentiation of haematopoietic stem cells. Nat Cell Biol 12, 1046–1056, doi: 10.1038/ncb2108 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ding BS et al. Endothelial-derived angiocrine signals induce and sustain regenerative lung alveolarization. Cell 147, 539–553, doi: 10.1016/j.cell.2011.10.003 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ding BS et al. Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration. Nature 468, 310–315, doi: 10.1038/nature09493 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tavora B et al. Endothelial-cell FAK targeting sensitizes tumours to DNAdamaging therapy. Nature 514, 112–116, doi: 10.1038/nature13541 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanz E et al. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc Natl Acad Sci U S A 106, 13939–13944, doi: 10.1073/pnas.0907143106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 465, 483–486, doi: 10.1038/nature09002 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Gibson DA et al. Dendrite self-avoidance requires cell-autonomous slit/robo signaling in cerebellar purkinje cells. Neuron 81, 1040–1056, doi: 10.1016/j.neuron.2014.01.009 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brose K et al. Slit proteins bind Robo receptors and have an evolutionarily conserved role in repulsive axon guidance. Cell 96, 795–806 (1999). [DOI] [PubMed] [Google Scholar]
- 14.Wang KH et al. Biochemical purification of a mammalian slit protein as a positive regulator of sensory axon elongation and branching. Cell 96, 771–784 (1999). [DOI] [PubMed] [Google Scholar]
- 15.Strickland P, Shin GC, Plump A, Tessier-Lavigne M & Hinck L Slit2 and netrin 1 act synergistically as adhesive cues to generate tubular bi-layers during ductal morphogenesis. Development 133, 823–832, doi: 10.1242/dev.02261 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Svensson KJ et al. A Secreted Slit2 Fragment Regulates Adipose Tissue Thermogenesis and Metabolic Function. Cell Metab 23, 454–466, doi: 10.1016/j.cmet.2016.01.008 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ballard MS & Hinck L A roundabout way to cancer. Adv Cancer Res 114, 187–235, doi: 10.1016/B978-0-12-386503-8.00005-3 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lang JE et al. RNA-Seq of Circulating Tumor Cells in Stage II-III Breast Cancer. Ann Surg Oncol 25, 2261–2270, doi: 10.1245/s10434-018-6540-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gantier MP & Williams BR The response of mammalian cells to double-stranded RNA. Cytokine Growth Factor Rev 18, 363–371, doi: 10.1016/j.cytogfr.2007.06.016 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnsen IB et al. Toll-like receptor 3 associates with c-Src tyrosine kinase on endosomes to initiate antiviral signaling. EMBO J 25, 3335–3346, doi: 10.1038/sj.emboj.7601222 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Itoh K, Watanabe A, Funami K, Seya T & Matsumoto M The clathrin-mediated endocytic pathway participates in dsRNA-induced IFN-beta production. J Immunol 181, 5522–5529, doi: 10.4049/jimmunol.181.8.5522 (2008). [DOI] [PubMed] [Google Scholar]
- 22.Kawasaki T & Kawai T Toll-like receptor signaling pathways. Front Immunol 5, 461, doi: 10.3389/fimmu.2014.00461 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shivapurkar N et al. Multiple regions of chromosome 4 demonstrating allelic losses in breast carcinomas. Cancer Res 59, 3576–3580 (1999). [PubMed] [Google Scholar]
- 24.Dallol A et al. Frequent epigenetic inactivation of the SLIT2 gene in gliomas. Oncogene 22, 4611–4616, doi: 10.1038/sj.onc.1206687 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Grone J et al. Robo1/Robo4: differential expression of angiogenic markers in colorectal cancer. Oncol Rep 15, 1437–1443 (2006). [PubMed] [Google Scholar]
- 26.Huang WY et al. MethHC: a database of DNA methylation and gene expression in human cancer. Nucleic Acids Res 43, D856–861, doi: 10.1093/nar/gku1151 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Macias H et al. SLIT/ROBO1 signaling suppresses mammary branching morphogenesis by limiting basal cell number. Dev Cell 20, 827–840, doi: 10.1016/j.devcel.2011.05.012 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Macias H & Hinck L Mammary gland development. Wiley Interdiscip Rev Dev Biol 1, 533–557, doi: 10.1002/wdev.35 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marlow R et al. SLITs suppress tumor growth in vivo by silencing Sdf1/Cxcr4 within breast epithelium. Cancer Res 68, 7819–7827, doi: 10.1158/0008-5472.CAN-08-1357 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Escamilla-Tilch M et al. The interplay between pathogen-associated and danger-associated molecular patterns: an inflammatory code in cancer? Immunol Cell Biol 91, 601–610, doi: 10.1038/icb.2013.58 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Bakhoum SF et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472, doi: 10.1038/nature25432 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nabet BY et al. Exosome RNA Unshielding Couples Stromal Activation to Pattern Recognition Receptor Signaling in Cancer. Cell 170, 352–366 e313, doi: 10.1016/j.cell.2017.06.031 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Redzic JS, Balaj L, van der Vos KE & Breakefield XO Extracellular RNA mediates and marks cancer progression. Semin Cancer Biol 28, 14–23, doi: 10.1016/j.semcancer.2014.04.010 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rooney MS, Shukla SA, Wu CJ, Getz G & H acohen N Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160, 48–61, doi: 10.1016/j.cell.2014.12.033 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kassiotis G Endogenous retroviruses and the development of cancer. J Immunol 192, 1343–1349, doi: 10.4049/jimmunol.1302972 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zernecke A & Preissner KT Extracellular Ribonucleic Acids (RNA) Enter the Stage in Cardiovascular Disease. Circ Res 118, 469–479, doi: 10.1161/CIRCRESAHA.115.307961 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Khakpour S, Wilhelmsen K & Hellman J Vascular endothelial cell Toll-like receptor pathways in sepsis. Innate Immun 21, 827–846, doi: 10.1177/1753425915606525 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Mantovani A, Allavena P, Sica A & Balkwill F Cancer-related inflammation. Nature 454, 436–444, doi: 10.1038/nature07205 (2008). [DOI] [PubMed] [Google Scholar]
Methods References
- 39.Tavazoie SF et al. Endogenous human microRNAs that suppress breast cancer metastasis. Nature 451, 147–U143, doi:Doi 10.1038/Nature06487 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reynolds LE & Hodivala-Dilke KM Primary mouse endothelial cell culture for assays of angiogenesis. Methods Mol Med 120, 503–509 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Aslakson CJ & Miller FR Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res 52, 1399–1405 (1992). [PubMed] [Google Scholar]
- 42.Fidler IJ Biological behavior of malignant melanoma cells correlated to their survival in vivo. Cancer Res 35, 218–224 (1975). [PubMed] [Google Scholar]
- 43.May T et al. Establishment of murine cell lines by constitutive and conditional immortalization. J Biotechnol 120, 99–110, doi: 10.1016/j.jbiotec.2005.03.027 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Guy CT, Cardiff RD & Muller WJ Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol 12, 954–961 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wagner KU et al. Cre-mediated gene deletion in the mammary gland. Nucleic Acids Res 25, 4323–4330 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lanczky A et al. miRpower: a web-tool to validate survival-associated miRNAs utilizing expression data from 2178 breast cancer patients. Breast Cancer Res Treat 160, 439–446, doi: 10.1007/s10549-016-4013-7 (2016). [DOI] [PubMed] [Google Scholar]
- 47.DeRose YS et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat Med 17, 1514–1520, doi: 10.1038/nm.2454 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sikora MJ et al. Invasive lobular carcinoma cell lines are characterized by unique estrogen-mediated gene expression patterns and altered tamoxifen response. Cancer Res 74, 1463–1474, doi: 10.1158/0008-5472.CAN-13-2779 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ponomarev V et al. A novel triple-modality reporter gene for whole-body fluorescent, bioluminescent, and nuclear noninvasive imaging. Eur J Nucl Med Mol Imaging 31, 740–751, doi: 10.1007/s00259-003-1441-5 (2004). [DOI] [PubMed] [Google Scholar]
- 50.Dhir A et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 560, 238–242, doi: 10.1038/s41586-018-0363-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.