

Abstract
The histone acetyltransferase MOF (KAT8) is mainly involved in the acetylation of histone H4 at lysine 16 (H4K16) and some non‐histone proteins. The MOF expression level is significantly reduced in many cancers, however the biological function of MOF and its underlying mechanism are still elusive in hepatocellular carcinoma (HCC). Estrogen receptor α (ERα) has been considered as a tumor suppressor in HCC. Here, we demonstrated that MOF expression is significantly reduced in HCC samples, and is positively correlated with that of ERα. MOF interacts with ERα, and participates in acetylation of ERα at K266, K268, K299, thereby inhibiting ERα ubiquitination to maintain the stability of ERα. In addition, MOF participates in the upregulation of ERα‐mediated transactivation. Depletion of MOF significantly promotes cell growth, migration, and invasion in HCC cell lines. Taken together, our results provide new insights to understand the mechanism underlying the modulation function of MOF on ERα action in HCC, suggesting that MOF might be a potential therapeutic target for HCC.
Keywords: acetylation, estrogen receptor α, hepatocellular carcinoma, MOF, tumor suppression
In summary, we have demonstrated that MOF as a crucial histone acetylase participates in the acetylation of ERα, thereby inhibiting the ubiquitination of ERα to stabilize ERα protein in HCC. MOF upregulates ERα‐induced transactivation. MOF depletion promotes the cell proliferation, migration, and invasion in HCC cells.
Abbreviations
- AFP
alpha‐fetoprotein
- CHIP
carboxyl terminus of hsc70‐interacting protein
- ChIP
chromatin immunoprecipitation
- Co‐IP
co‐immunoprecipitation
- E2
17β‐estradiol
- ERα
estrogen receptor α
- ERβ
estrogen receptor β
- EtOH
ethanol
- HAT
histone acetyltransferase
- HCC
hepatocellular carcinoma
- IHC
immunohistochemical
- MDM2
murine double minute 2
- MOF (KAT8)
males absent on the first (lysine acetyltransferase 8)
- MYST
moz‐ybf2/sas3‐sas2‐tip60
- qRT‐PCR
quantitative real‐time PCR
- SHP
small heterodimer partner
- SMAD7
SMAD Family Member 7
1. INTRODUCTION
Primary liver cancers include HCC (75%‐85% of cases) and intrahepatic cholangiocarcinoma (10%‐15% of cases) and other rare types. There are approximately 841 000 new cases and 782 000 deaths each year. In most parts of the world, the incidence and mortality for men are 2 to 3 times higher than for women. 1 The incidence of HCC increases in postmenopausal women, and estrogen therapy can suppress this phenomenon. 2 , 3 The prognosis for women with HCC is better than that for men. 4 Currently, only sorafenib and lenvatinib have been approved for first‐line treatment of advanced, unresected HCC, but they only produce a small degree of survival benefit, 5 , 6 therefore new therapeutic targets need to be identified to improve current HCC treatments.
Estrogens play a biological role mainly by binding to ERα and estrogen receptor β (ERβ). ERα is a member of the nuclear receptor steroid superfamily, and its activity is affected by ligands and co‐regulators (including co‐activators and co‐inhibitors). 7 , 8 Studies have shown that ERα expression in HCC is significantly lower than that in normal liver tissue. 9 The combination of ERα and SP1 mediates apoptosis in Hep3B cells. 10 ERα inhibits invasion and metastasis by targeting MTA1 and regulating NF‐κB and MMP‐9 in liver cancer. 9 , 11 ERα improves fatty liver progression by upregulating the transcription of SHP. 12 , 13 A previous study showed that Erbin promotes the association between ERα and CHIP, which is an E3 ligase, thereby increasing ERα ubiquitination and degradation to promote HCC tumorigenesis. 14 Therefore, the ERα signaling pathway plays a crucial protective role in HCC progression.
MOF, also named as lysine acetyltransferase 8 (KAT8), a member of MYST (Moz, Ybf2/Sas3, Sas2, Tip60) family, which has a highly conserved histone acetyltransferase (HATs) domain in a variety of species. 15 , 16 MOF is acetylated in vivo and in vitro. Acetylation is limited in conserved MYST domains (C2HC zinc fingers and HAT), of which the K274 residue is the major self‐acetylation site. 17 In addition to histone acetylation modifications, MOF has even been found to acetylate non‐histone proteins such as P53, 18 FASN, 19 LSD1, 20 NoRC 21 and IRF3. 22
Abnormal expression of MOF in several tumor tissues has been previously reported. 23 , 24 MOF expression is reduced in HCC and the lower expression of MOF is correlated with the poor prognosis of HCC. 25 However, the molecular mechanism of MOF in HCC progression still needs to be clarified.
In this study, our results showed that the expression of MOF is significantly decreased in HCC tissue samples. Interestingly, we examined that the expression of MOF in HCC is positively correlated with that of ERα. Our results demonstrated that the acetylation activity of MOF is required for downregulation of ERα ubiquitination to maintain the stability of ERα. In addition, MOF acting as a novel co‐activator of ERα enhances the endogenous expression of ERα target genes. In the response of estrogen, MOF or ERα is recruited to the promoter region of ERα target gene. Functionally, MOF depletion significantly promotes cell growth, migration, and invasion in HCC cell lines. Collectively, our study provides new insights to understand the mechanism underlying the modulation of function of MOF on ERα action in HCC, indicating that MOF might be a potential target for HCC treatment.
2. MATERIALS AND METHODS
2.1. Antibodies
The FLAG‐MOF plasmid was purchased from Sinobiological (Cat: HG13797‐NF). The FLAG‐MOF‐K274R mutant was cloned into the PCMV‐Flag vector. The ERα‐K266R, ERα‐K268R, ERα‐K299R, ERα‐K302R, ERα‐K303R, and ERα‐K302/303R mutants were cloned into the pcDNA3‐HA vector. Final constructs were verified using DNA sequencing. The expression plasmid for human ERα (pSG5‐ERα) and pGL‐ERE‐AdML reporter plasmid carrying 3 consensus estrogen response elements (3 × ERE) were kindly provided by Dr. Shigeki Kato.
The antibodies used in this study were: anti‐MOF (Bethyl laboratories # A300‐992A‐2), anti‐acetylated‐lysine (AcK) (Cell Signaling Technology # 9441), anti‐ERα (Cell Signaling Technology # 8644), anti‐SMAD7 (Sigma # SAB4200346), anti‐SHP(NR0B2) (ZEN BIO # 501836), anti‐GAPDH (Shanghai Kangchen # KC5G4), anti‐FLAG and anti‐HIS (GNI), anti‐rabbit/mouse (ABclonal), anti‐IgG (Santa # sc‐2025).
2.2. Cell culture, siRNA transfection, and lentivirus infection
Detailed experimental procedures for this section are described in Supporting Information. The sequences for siRNAs are described in Supporting Information
2.3. Dual luciferase reporter assay and quantitative real‐time PCR (qPCR)
Detailed experimental procedures for this section are described in Supporting Information. The primer sequences used to detect mRNA expression are listed in Table S1.
2.4. Co‐immunoprecipitation and Immunofluorescence
Detailed experimental procedures for this section are described in Supporting Information.
2.5. Chromatin immunoprecipitation (ChIP) assay
Detailed experimental procedures for this section are described in Supporting Information. Primer sequences are listed in Table S2.
2.6. Colony formation assay, cell growth, cell migration, invasion experiments, and lipid drop formation experiment
Detailed experimental procedures for this section are described in Supporting Information.
2.7. Xenograft tumor growth
In total, 1 × 106 cells were injected subcutaneously in mice. Tumor diameter were measured every week using an electronic caliper. Tumor volume (cubic millimeters) was calculated as volume = (short diameter)2 × long diameter/2. 26 At 4 wk later, tumor‐bearing mice were killed following the policy for the humane treatment of animals. All procedures for animal experiments were carried out in compliance with ethical regulations approved by the Animal Ethics Committee of China Medical University.
2.8. Collection of clinical HCC tissue samples and IHC analysis
Detailed experimental procedures for this section are described in Supporting Information.
2.9. Statistical analysis
Statistical analysis for this study was performed using the SPSS (v.22.0) statistical software program. Overall survival curves were plotted based on the Kaplan‐Meier method with the log‐rank test applied for comparison. For qPCR and luciferase assay, two‐sided Student t test was used to determine significant differences. *P < .05; **P < .01; ***P < .001. For clinical data analysis, chi‐square test was adopted. For the correlation between MOF and ERα, the Pearson correlation coefficient was calculated.
3. RESULTS
3.1. Expression of MOF is significantly reduced in HCC samples and is positively correlated with that of ERα
MOF belonging to the MYST family is reported to participate in suppression of HCC growth, 25 however the molecular mechanisms underlying the biological function of MOF in HCC progression are largely unknown. On the Kaplan‐Meier plotter website we analyzed the data from 364 patients with HCC; the results demonstrated that regarding short‐term overall survival (0‐60 mo) of patients with lower expression of MOF there was a poor prognosis (Figure 1A). In addition, the patients with HCC with high MOF expression, regardless of hepatitis virus infection, showed a better prognosis compared with those with low expression (Figure S1). In addition, we downloaded and analyzed the GSE25097, GSE50579, and GSE22405 datasets deposited in the Gene Expression Omnibus (GEO) database, these 3 datasets showed that MOF mRNA expression was reduced in HCC compared with that in normal liver tissue (Figure 1B). To evaluate MOF expression in HCC, we performed immunohistochemical staining on clinical samples from 145 clinical HCC tissues and 48 matched adjacent noncancerous tissues. The results demonstrated that higher expression of MOF was significantly associated with good differentiation, while no significant association was observed between MOF expression and age, gender, alpha‐fetoprotein (AFP) expression or HBV infection (Figure 1C, and Tables 1 and 2). Unexpectedly, when detecting protein expression in 33 pairs of fresh HCC tissues and the adjacent noncancerous tissues using western blot, we found that MOF expression in 22 HCC samples (66.67%) and ERα expression in 18 HCC samples (54.54%) was significantly reduced in HCC tissues (Figure 1D,E). Moreover, expression of MOF was positively correlated with that of ERα, which is a putative tumor suppressor in HCC (r 2 = .4255, P = 4.00E‐04), providing evidence for the possibility of positive regulation between MOF and ERα (Figure 1F). Taken together, these results indicated that MOF expression was significantly reduced in HCC and may be a potential biomarker for HCC prognosis. MOF expression was positively correlated with that of ERα.
FIGURE 1.
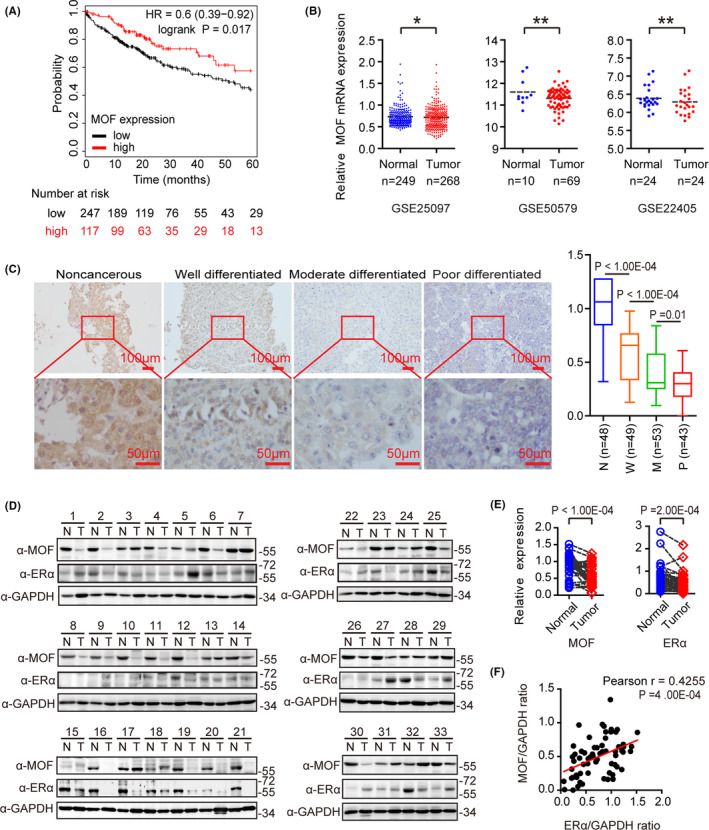
MOF is lower expressed in hepatocellular carcinoma and positively correlated with ERα. A, The overall survival curve generated from the Kaplan‐Meier plotter website shows the contribution of MOF to 364 liver cancer patients. B, MOF mRNA expression from GSE25097, GSE50579, GSE22405 datasets was analyzed. C, MOF expression of different degrees of differentiation was shown by immunohistochemical staining in clinical specimens. N: noncancerous liver tissue; W: well differentiated HCC; M: moderately differentiated HCC; P: moderately differentiated HCC; magnification: ×10×40; scale bars: 100 μm/50 μm. Mann‐Whitney U test was used. D, Protein expression of MOF or ERα in 33 pairs of HCC (T) and adjacent non‐tumor tissues (N) was analyzed using western blot. E, Using the gray‐scale expression of MOF or ERα in Figure 1D and GAPDH as a reference, a two‐tailed Student t test was used for statistical analysis. F, There is a positive correlation between MOF and ERα protein expression. The expression levels of MOF and ERα in Figure 1D were quantified by densitometry, standardized with GAPDH, and analyzed by Pearson correlation. In the above experiments, *P < .05, **P < .01, and ***P < .001
TABLE 1.
MOF expression in HCC and adjacent noncancerous liver tissues
| Group | Case | MOF expression | P‐value a | |
|---|---|---|---|---|
| n = 193 | Low | High | ||
| HCC | 145 | 93 | 52 | <.001 |
| Noncancerous liver tissues | 48 | 5 | 43 | |
Chi‐square test.
TABLE 2.
Relationship between MOF expression and clinicopathological characteristics
| Group | Case | MOF expression | P‐value a | |
|---|---|---|---|---|
| n = 145 (%) | Low | High | ||
| 93 | 52 | |||
| Gender | ||||
| Male | 109 (75.2%) | 68 | 41 | .444 |
| Female | 36 (24.8%) | 25 | 11 | |
| Age (years) | ||||
| <55 | 76 (52.4%) | 49 | 27 | .93 |
| ≥55 | 69 (47.6%) | 44 | 25 | |
| Pathological grades | ||||
| Well | 49 (33.8%) | 22 | 27 | .002** |
| Moderate | 53 (36.6%) | 38 | 15 | |
| Poor | 43 (29.6%) | 33 | 10 | |
| AFP | ||||
| Negative | 49 (33.8%) | 35 | 14 | .191 |
| Positive | 96 (66.2%) | 58 | 38 | |
| HBV infection | ||||
| Negative | 53 (36.6%) | 37 | 16 | .28 |
| Positive | 92 (63.4%) | 56 | 36 | |
Bold values represent P < .05.
Chi‐square test.
3.2. MOF interacts with ERα and stabilizes ERα protein
Our results in this study have shown that the reduced expression of MOF was positively correlated with that of ERα in clinical HCC samples. To further evaluate the modulation function of MOF on ERα, Co‐IP experiments were first performed to find a physical interaction between MOF and ERα in HCCLM3 and Huh7 (Figure 2A‐C). Immunofluorescence confocal experiments showed that MOF and ERα mainly co‐localized to the nucleus in the presence of E2 in HCCLM3 and Huh7 (Figures 2D and S2A). Interestingly, our results also demonstrated that ectopic expression of MOF increased ERα protein levels (Figure 2A). qPCR experiments did not show any influence of MOF on ERα mRNA levels in Huh7 and HCCLM3 cell lines (Figures 2E and S2B), we therefore further performed western blotting experiments with transfection FLAG‐MOF plasmid. The results showed that MOF increased ERα protein levels in a dose‐dependent manner in HEK293 and HCCLM3 cell lines (Figure 2F). Conversely, MOF depletion markedly decreased ERα expression in HCCLM3 and Huh7 cells (Figure 2G). We further treated HCCLM3 and Huh7 cells with the protein synthesis inhibitor cycloheximide (CHX) to examine the influence of MOF on ERα stability. The results demonstrated that ERα degradation was decreased with MOF overexpression (Figure 2H,I). However, in the presence of the proteasome inhibitor MG132, the function of MOF on maintenance of ERα stability was significantly impaired (Figure 2J,K). Our results suggested that MOF interacted with ERα to participate in maintenance of ERα stability.
FIGURE 2.
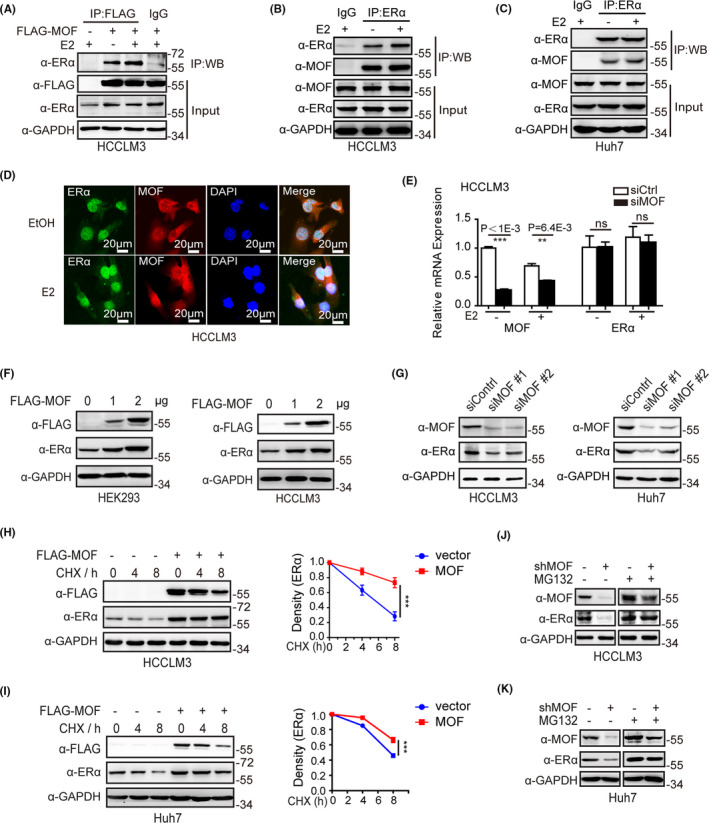
MOF interacts with ERα to be involved in ERα protein stability. A‐C, MOF interacts with ERα. Co‐IP experiment is performed with the indicated antibodies. D, MOF is co‐localized with ERα. Immunofluorescence confocal analysis was performed with ethanol vehicle or 10−7 M E2 treatment. E, MOF has no effect on the ERα mRNA level. F, MOF overexpression increases the ERα protein level in a dose‐dependent manner. G, MOF depletion decreases the expression of ERα. H, I, MOF overexpression decreased ERα degradation. HCCLM3 and Huh7 were treated with 10 mg/mL cycloheximide (CHX) for the indicated time points, followed by western blotting analysis. J, K, MOF depletion decreases the expression of ERα. The cell lysates were subjected to western blot analysis with indicated antibodies with MG132 (5 μM) for 6 h
3.3. MOF acetylates ERα to maintain ERα stability by inhibition of ERα polyubiquitination
Previous studies have reported that MOF participates in acetylation of P53, FASN, and MOF itself. 17 , 18 , 19 Having shown that MOF interacts with ERα and stabilizes ERα protein expression, we therefore turned to examine whether MOF was involved in ERα acetylation. Co‐IP results showed that ectopic expression of MOF participated in ERα acetylation, while MOF depletion markedly decreased ERα acetylation level in HCCLM3 and Huh7 cells (Figure 3A,B). In addition, we also observed that ERα acetylation level was reduced in HCC samples, and that the lower acetylation level of ERα was positively correlated with MOF expression (Figure S3). Therefore, we tried to detect the exact sites of ERα acetylated by MOF. It has been reported that 5 lysine sites on ERα can be acetylated by p300, including K266, K268, K299, K302, and K303. 27 We further constructed several ERα mutants substituting K266, K268, K299, K302, K303, and K302/K303 with arginine (R) to make acetylation‐resistant mimics. The acetylation assay results showed that acetylation levels of ERα mutants (K266R, K268R, K299R) were significantly reduced compared with the ERα wild‐type acetylation levels mediated by MOF (Figure 3C), indicated that K266, K268 and K299 on ERα were possible acetylation sites of ERα mediated by MOF.
FIGURE 3.
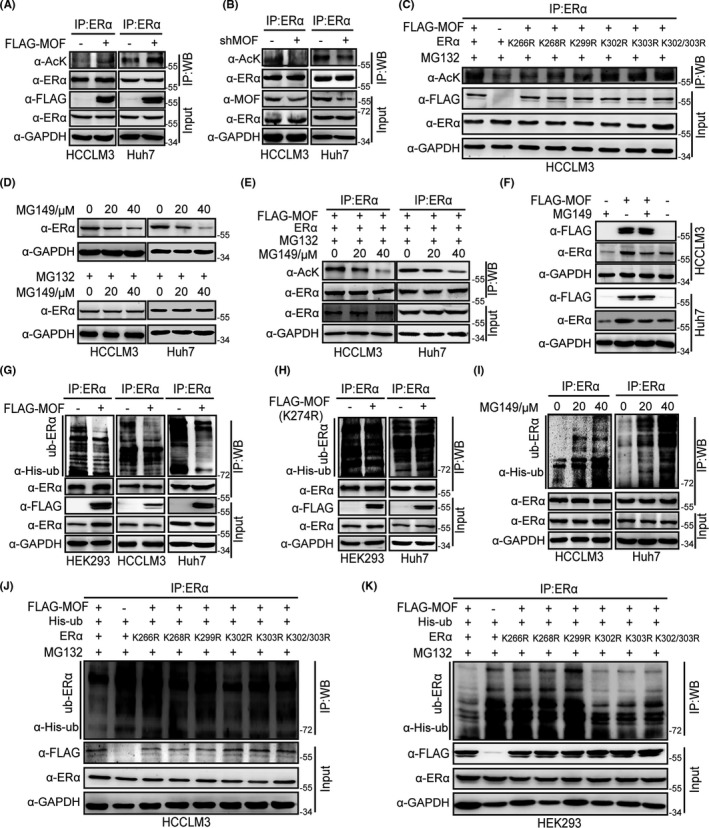
MOF participates in ERα acetylation to maintain ERα stability by alteration of ERα ubiquitination. A, B, MOF participates in acetylation of ERα. HCCLM3 and Huh7 cells were transfected with ERα and FLAG‐MOF (A) or shMOF (B). After incubation with TSA (5 μM) and 5 mM nicotinamide (5 μM) for 12 h, and MG132 (5 μM) for 6 h, cell lysates were subjected to IP assays with ERα antibody, western blot detected with indicated antibodies. C, MOF acetylates the K266, K268, and K299 sites of ERα. D, MG149 reduces ERα protein expression in a dose‐dependent manner. E, MG149 inhibits ERα acetylation induced by MOF. F, MG149 prevents MOF‐mediated ERα stability. G, MOF weakens the polyubiquitination of ERα in HEK293, HCCLM3, and Huh7 cells. H, Loss of function of MOF acetylase activity by FLAG‐MOF mutant (FLAG‐MOF K274R) does not affect ERα polyubiquitination. I, MOF inhibitor MG149 increases ERα polyubiquitination level. J, K, ERα mutants (K266R, K268R, K299R) carrying loss of function of ERα acetylation reverses the effect of MOF on attenuation of ERα polyubiquitination in HCCLM3 and HEK293 cells
MG149 is a potent histone acetyltransferase inhibitor with an IC50 of 74 µM for Tip60 and 47 µM for MOF. Therefore, we selected MG149 (47 µM) as an MOF inhibitor to examine whether MOF‐mediated ERα acetylation promoted ERα protein stability. We first detected ERα protein levels by treatment of MG149 with or without the proteasome inhibitor MG132, the results showed that MG149 promoted ERα degradation, while MG132 abolished the effect of MG149 on ERα (Figure 3D). Moreover, acetylation assays with the MG149 treatment were performed, the results demonstrated that MG149 clearly inhibited ERα acetylation by MOF in HCCLM3 and Huh7 cells (Figure 3E). In addition, as shown in Figure 3F, our results showed that MG149 prevented MOF‐mediated increase of ERα expression. The above results suggested that ERα acetylation by MOF promoted ERα protein stability, and MOF‐mediated maintenance of ERα stability might be related to ubiquitination. Next, we analyzed how MOF influenced ERα stability by alteration of ubiquitination. Our results showed that wild‐type MOF overexpression weakened the polyubiquitination of ERα in HEK293, HCCLM3, and Huh7 cells (Figure 3G). However, the enzyme active site mutation plasmid FLAG‐MOF (K274R) did not affect the polyubiquitination of ERα (Figure 3H). Moreover, MOF inhibitor MG149 increased the polyubiquitination level of ERα (Figure 3I). ERα mutants (K266R, K268R, and K299R) abolished the effect of MOF on ERα ubiquitination (Figure 3J,K). Taken together, these results suggest that MOF acetylates ERα at K266, K268, K299 sites to maintain ERα stability by alteration of ERα ubiquitination.
3.4. MOF upregulates ERα‐mediated transactivation
ERα acting as an important transcription factor plays a key role in suppression of HCC progression. Our results have also demonstrated that MOF is involved in the maintenance of ERα stability. Therefore, it was critical to investigate the effect of MOF on ERα transcriptional activity. We performed a series of dual luciferase reporting experiments. The results showed that MOF overexpression significantly upregulated ERα‐mediated transactivation in HCCLM3 and Huh7 cells (Figure 4A,B), and MOF knockdown inhibited ERα‐mediated transactivation (Figure 4C). Furthermore, we found that MOF upregulated ERα‐mediated transactivation with or without E2. ERα contains an amino‐terminal ligand‐independent transcription activation AF1 domain and a carboxy‐terminal ligand‐dependent transcription activation AF2 domain. 28 To examine if the specific activation transcription domain of ERα was modulated by MOF, the truncated ERα harboring the ERα AF1 and ERα AF2 reporter plasmid was subjected to a double luciferase reporting experiment. The results demonstrated that MOF enhanced ERα AF1‐mediated transactivation and, in the response of E2, MOF enhanced ERα AF2‐mediated transactivation (Figure 4D). Taken together, these results demonstrated that MOF acts as a novel co‐activator of ERα to upregulates ERα‐mediated transactivation.
FIGURE 4.
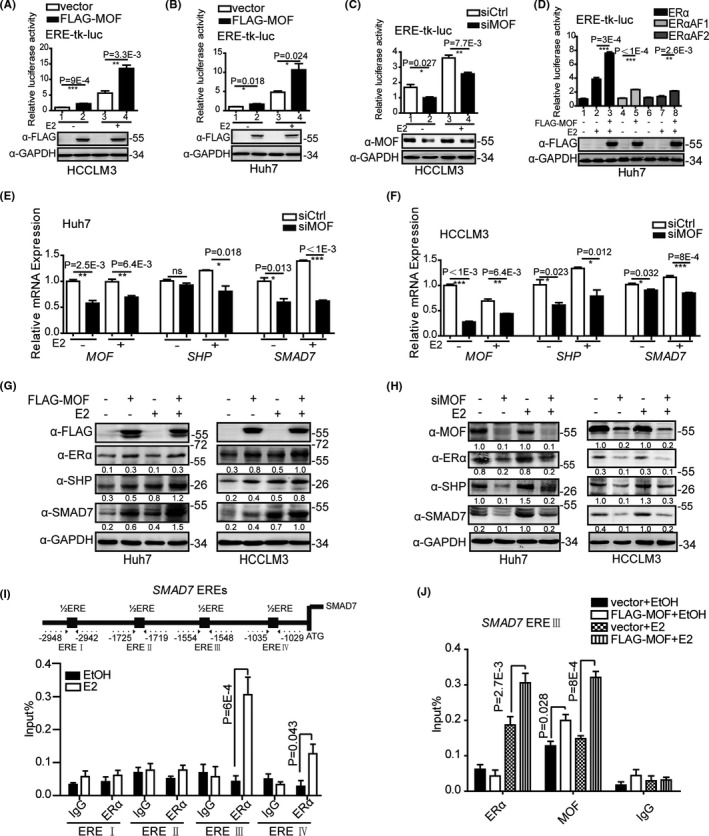
MOF enhances ERα‐mediated transactivation in HCC cells. A, B, MOF overexpression promotes ERα‐mediated transactivation. C, MOF depletion represses ERα‐mediated transactivation. D, MOF overexpression promotes ERαAF1 and ERαAF2‐mediated transactivation. E, F, MOF depletion attenuated the mRNA expression of ERα target genes. G, MOF overexpression enhanced the protein expression of ERα target genes. H, MOF depletion attenuated the protein expression of ERα target genes. I, ERα was recruited mainly in SMAD7‐ERE III. J, MOF overexpression enhanced the recruitment of ERα on SMAD7‐ERE III. A two‐tailed Student t test was used for statistical analysis, and error bars represent mean ± SD (*P < .05, **P < .01, ***P < .001)
To further investigate the role of MOF in endogenous ERα target genes, we detected the target gene expression of ERα in HCCLM3 and Huh7 with MOF knockdown or overexpression. The results showed that MOF depletion reduced mRNA expression of endogenous ERα target genes such as SHP, 12 , 13 and SMAD family member 7 (SMAD7) 29 in Huh7 and HCCLM3 cells (Figure 4E,F). MOF overexpression increased the protein levels of SHP and SMAD7. MOF depletion reduced the protein levels of SHP and SMAD7 in Huh7 and HCCLM3cells (Figure 4G,H). At the same time, the results showed that MOF overexpression increased ERα protein expression, and MOF depletion reduced ERα protein expression (Figure 4G,H), which was consistent with our preceding conclusion that MOF stabilizes ERα protein. The above results indicated that MOF is involved in upregulating the expression of ERα target genes.
Considering that MOF co‐activates ERα‐mediated transcription regulation, ChIP analysis was further performed to examine whether MOF is recruited to the ERα binding regions of the ERα target gene. First, we found four half‐ERE sites in the SMAD7 promoter region. ChIP analysis results showed that ERα was recruited mainly at the ERE III site and a small amount was recruited at the ERE IV site (Figure 4I). Next, ChIP analysis was further performed at SMAD7 ERE III site. ChIP analysis results showed that in the response of E2, MOF or ERα was recruited to the ERE III site of the SMAD7 promoter region. Moreover, the ectopic expression of MOF increased ERα recruitment on SMAD7 ERE Ⅲ (Figure 4J). MOF depletion reduced ERα recruitment on SMAD7 ERE Ⅲ (Figure S4). These results suggested that MOF upregulates ERα‐mediated transactivation.
3.5. MOF depletion promotes the cell proliferation, migration, and invasion of HCC
We therefore turned to examine the biological function of MOF in hepatocellular carcinoma cell lines, HCCLM3, and Huh7 cells with stable knockdown of MOF were established for the series of experiments. First, colony formation experiments were performed, and the results showed that MOF knockdown promoted the colony forming in HCCLM3 and Huh7 cells (Figures 5A,B, S5A,B). Next, MTS experiments were also performed to detect cell viability, and the results showed that MOF knockdown significantly promoted the cell viability of HCCLM3 and Huh7 with or without E2 (Figure 5C,D). To clarify the role of ERα in MOF inhibiting HCC, we launched experiments to interfere with ERα expression. First, siRNA interference technology was used to knock down ERα, and we found that the inhibitory effect of MOF on HCCLM3 cells disappeared through the cell growth experiment (Figure 5E). In addition, we also used fulvestrant (ICI 182780), a potent estrogen receptor antagonist, to degrade ERα protein expression. Cell growth experiments confirmed that, as the concentration of fulvestrant gradually increased, the growth inhibitory effect of MOF on HCCLM3 cells gradually disappeared (Figure 5F). In addition, cell migration and invasion experiments were also performed in Huh7 cells, and the results showed that MOF knockdown promoted cell migration (Figure 5G) and invasion (Figure 5H) ability. At the same time, scratch experiments were also performed in HCCLM3 cells, and the results also showed that MOF knockdown promoted cell migration ability (Figure 5I). In addition, considering the important role of ERα in lipid metabolism, we tried to verify whether MOF also affects lipid metabolism. Lipid droplet formation experiments were performed, and the results showed that MOF knockdown promoted lipid droplet formation in HCCLM3 cells (Figure S5C,D). The above results indicated that MOF knockdown promotes cell proliferation, migration, and invasion of HCC cells, at least partly dependent on ERα.
FIGURE 5.
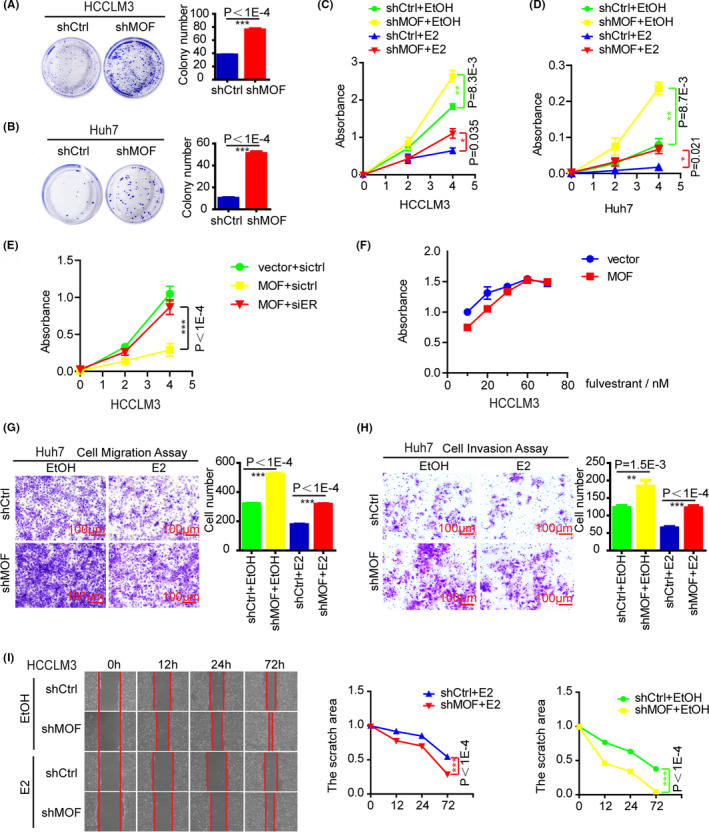
MOF depletion promotes cell proliferation, migration, and invasion in HCC cells. A, B, MOF depletion promoted colony formation. C, D, MOF depletion promotes cell viability. E, F, MOF inhibits HCC cell growth, partly dependent on ERα. MOF was stably overexpressed by viral incubation. G, MOF depletion promotes cell migration. H, MOF depletion promotes cell invasion. I, MOF depletion promotes cell migration. In A‐I, a two‐tailed Student t test was used for statistical analysis, and error bars represent mean ± SD (*P < .05, **P < .01, ***P < .001)
3.6. MOF depletion promotes subcutaneous tumor‐bearing growth in mice
To further investigate the biological function of MOF in animals, we performed a tumor formation experiment of subcutaneously implanted cells in BALB/c Nude mice. After a month of experiments, the results revealed that MOF knockdown significantly promoted the growth of subcutaneous tumors in mice (Figure 6A), including tumor volume (Figure 6B) and weight (Figure 6C). It was shown that MOF significantly inhibits the growth of subcutaneous tumors in mice. In addition, we examined the expression of MOF, SHP, and SMAD7 in mouse tumors. Western blot experiments showed that MOF knockdown significantly inhibited the protein expression of SHP and SMAD7 (Figure 6D,E). Overall, our data demonstrated that MOF knockdown promoted the cell growth of HCC in vivo.
FIGURE 6.
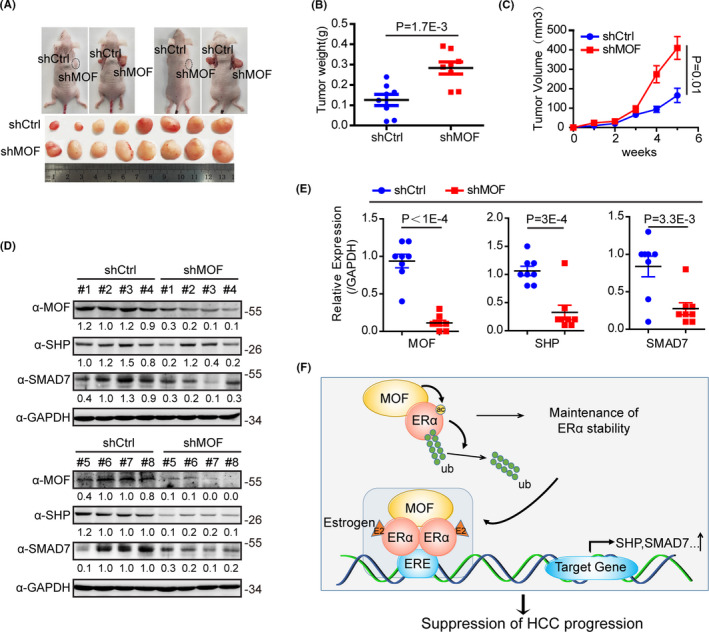
MOF depletion promotes subcutaneous tumor‐bearing growth in mice. A, Photograph showing the xenograft tumor in male SCID mice with shctrl (left) and shMOF (right) HCCLM3 cells. Tumors were dissected from mice at the 35th d post injection. B, The tumor weights were measured. C, The tumor volume was measured on the indicated weeks. D, The tumor protein expressions were analyzed by western blot with the indicated antibodies. E, Using the gray‐scale expression of MOF, SHP and SMAD7 in panel (D) and GAPDH as a reference. F, We propose a model to show that MOF promotes ERα activity and inhibits hepatocellular carcinoma. MOF interacts with ERα and acetylates ERα, thereby inhibiting ERα polyubiquitination to maintain ERα stability. At the same time, MOF as a co‐activator of ERα activates the endogenous ERα target genes, including SHP and SMAD7
4. DISCUSSION
HCC is one of the severe carcinomas with poor prognosis. The ERα signaling pathway plays an important role in inhibiting the occurrence and development of HCC. Therefore, to well understand the modulation of ERα signaling pathway would provide the potential target for HCC therapy. However, the regulation of ERα action in HCC is still elusive. In this study, we provided evidence to show that MOF as a key member of MYST family interacts with ERα and acetylates ERα, thereby inhibiting ERα polyubiquitination to maintain ERα stability. Moreover, our data demonstrated that MOF as a co‐activator of ERα activates the endogenous ERα target genes, including SHP and SMAD7. Functionally, the depletion of MOF promotes cell proliferation and migration in HCC cells (Figure 6F).
Having established in this study that low expression of MOF was significantly associated with poor overall survival in HCC, and that the lower expression of MOF is positively correlated with that of ERα in clinical HCC samples (Figure 1). Our data further provided the evidence that MOF is involved in maintaining ERα stability (Figure 2). MOF as a member of the MYST family has been shown to acetylate some non‐histone proteins, such as P53, FASN, LSD1, NoRC, and IRF3, to exert its multiple biological functions on tumor progression. 18 , 19 , 20 , 21 , 22 We therefore turned to examine whether MOF participates in acetylation of ERα, our results showed that MOF triggers the acetylation of ERα in HCC cell lines. In addition, our data demonstrated that acetylation activity of MOF is required for the inhibition of the polyubiquitination of ERα to maintain ERα stability (Figure 3). It has been mentioned that a series of E3 ubiquitin ligases, including murine double minute 2 (MDM2) and CHIP, trigger ERα ubiquitination to promote ubiquitin‐mediated ERα proteolysis. 30 , 31 Here, our study provides a new mechanism that MOF is involved in acetylation of ERα, thereby inhibiting the ubiquitination of ERα to stabilize ERα protein in HCC. Our results suggest that ERα acetylation may crosstalk with its deubiquitination to participate in the stability of ERα, although it is still unknown how ERα acetylation mediated by MOF correlates with ERα ubiquitination. The influence of MOF‐mediated ERα acetylation on the association between ERα and E3 ligase/deubiquitinase would be clarified in the future.
It has been reported that MOF acting as a histone H4K16 acetylase is involved in upregulating the transcriptional activation of nuclear factor‐κB and androgen receptor in prostate cancer. 32 , 33 G9a has been identified as an ERα co‐activator associated with the PHF20/MOF complex in breast cancer. 34 In this study, our results demonstrated that MOF identified as a co‐activator upregulates ERα‐mediated transactivation in HCC cells in the presence or absence of estrogen (Figure 4A‐C). Interestingly, we observed that ERα is distributed in the nucleus independently on treatment of estrogen in HCC cells, suggesting that the characteristics of ERα in HCC should be quite different from that in breast cancer. Modulation of ERα action might be a more effective strategy compared with the regulation of hormone levels for HCC therapy. Our data also showed that MOF is involved in upregulation of transcription of the endogenous ERα target genes. ChIP assay analyses further provided the evidence that MOF or ERα is recruited to cis‐regulatory elements of ERα target gene SMAD7 and SHP (Figure 4). Our results indicated that MOF acts as an ERα co‐activator and participates in enhancement of the transcription of ERα target genes in HCC cell lines.
Human MOF is a member of the MYST family of HATs. Histone deacetylase (HDAC) and HAT activity antagonize each other to balance intracellular acetylation status. The combination of monomethyl PHF20L1 reader and methylated pRb mediates higher levels of control by recruiting MOF acetyltransferase complexes to E2F target genes. 35 At present, various small molecule inhibitors such as A‐485, 36 B026, 37 CPI‐076, CPI‐090, 38 and compound 1r 39 have been mentioned as regulators for HAT. However, there are relatively few studies on activators, and our data provide a possibility to develop specific MOF activators or modulator to inhibit HCC progress.
In summary, we demonstrated that MOF as a crucial histone acetylase participates in the acetylation of ERα, thereby inhibiting the ubiquitination of ERα to stabilize ERα protein in HCC. MOF upregulates ERα‐induced transactivation. MOF depletion promotes cell proliferation, migration, and invasion in HCC cells. Our findings describe new regulatory mechanism between MOF and ERα, and therefore implicate the function of MOF on suppression of HCC progression. Therefore, we propose that the MOF‐ERα pathway provides effective strategies for the treatment of HCC.
DISCLOSURE
The authors declare no conflict of interest.
AUTHOR CONTRIBUTION
Shan Wei, Ning Sun, Huijuan Song, and Chunyu Wang designed the study and collected related patient information. Shan Wei, Wei Liu, and Yi Wu performed the experiments. Shengli Wang, Renlong Zou, Lin, Kai Zeng, Baosheng Zhou, Manlin Wang, Ruina Luan, and Fan Yang analyzed the data. Shan Wei and Yue Zhao wrote the manuscript. All authors read and approved the final manuscript.
Supporting information
Supplementary Material
Figs S1‐S5
Tables S1, S2
ACKNOWLEDGMENTS
We thank Dr. Yunlong Huo and Dr. Hongyan Zhang for excellent technique assistance. This study was supported by the 973 Program Grant from the Ministry of Science and Technology of China [2013CB945201]; National Natural Science Foundation of China [31871286 for Yue Zhao, 81872015 for Chunyu Wang, 31701102 for Shengli Wang, 81702800 for Renlong Zou]; Ministry of Education fund innovation team [IRT 13101]; Foundation for Special Professor of Liaoning Province for Yue Zhao (the 5th batch).
Wei S, Liu W, Sun N, et al. MOF upregulates the estrogen receptor α signaling pathway by its acetylase activity in hepatocellular carcinoma. Cancer Sci. 2021;112:1865–1877. 10.1111/cas.14836
REFERENCES
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394‐424. [DOI] [PubMed] [Google Scholar]
- 2. Shimizu I. Impact of oestrogens on the progression of liver disease. Liver Int. 2003;23:63‐69. [DOI] [PubMed] [Google Scholar]
- 3. Yu MW, Chang HC, Chang SC, et al. Role of reproductive factors in hepatocellular carcinoma: impact on hepatitis B‐ and C‐related risk. Hepatology. 2003;38:1393‐1400. [DOI] [PubMed] [Google Scholar]
- 4. El‐Serag HB, Mason AC, Key C. Trends in survival of patients with hepatocellular carcinoma between 1977 and 1996 in the United States. Hepatology. 2001;33:62‐65. [DOI] [PubMed] [Google Scholar]
- 5. Abou‐Alfa GK, Shi Q, Knox JJ, et al. Assessment of treatment with sorafenib plus doxorubicin vs sorafenib alone in patients with advanced hepatocellular carcinoma: phase 3 CALGB 80802 randomized clinical trial. JAMA Oncol. 2019;5:1582–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Reig M, da Fonseca LG, Faivre S. New trials and results in systemic treatment of HCC. J Hepatol. 2018;69:525‐533. [DOI] [PubMed] [Google Scholar]
- 7. Zhao YC, Xu LW, Ding S, et al. Nuclear receptor retinoid‐related orphan receptor alpha deficiency exacerbates high‐fat diet‐induced cardiac dysfunction despite improving metabolic abnormality. Biochim Biophys Acta Mol Basis Dis. 2017;1863:1991‐2000. [DOI] [PubMed] [Google Scholar]
- 8. Metzger E, Wissmann M, Yin N, et al. LSD1 demethylates repressive histone marks to promote androgen‐receptor‐dependent transcription. Nature. 2005;437:436‐439. [DOI] [PubMed] [Google Scholar]
- 9. Sheng ML, Xu GL, Zhang CH, et al. Aberrant estrogen receptor alpha expression correlates with hepatocellular carcinoma metastasis and its mechanisms. Hepatogastroenterology. 2014;61:146‐150. [PubMed] [Google Scholar]
- 10. Tu CC, Kumar VB, Day CH, et al. Estrogen receptor alpha (ESR1) over‐expression mediated apoptosis in Hep3B cells by binding with SP1 proteins. J Mol Endocrinol. 2013;51:203‐212. [DOI] [PubMed] [Google Scholar]
- 11. Deng L, Yang H, Tang J, et al. Inhibition of MTA1 by ERalpha contributes to protection hepatocellular carcinoma from tumor proliferation and metastasis. J Exp Clin Cancer Res. 2015;34:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang X, Lu Y, Wang E, et al. Hepatic estrogen receptor alpha improves hepatosteatosis through upregulation of small heterodimer partner. J Hepatol. 2015;63:183‐190. [DOI] [PubMed] [Google Scholar]
- 13. Lai K, Harnish DC, Evans MJ. Estrogen receptor alpha regulates expression of the orphan receptor small heterodimer partner. J Biol Chem. 2003;278:36418‐36429. [DOI] [PubMed] [Google Scholar]
- 14. Wu H, Yao S, Zhang S, et al. Elevated expression of Erbin destabilizes ERalpha protein and promotes tumorigenesis in hepatocellular carcinoma. J Hepatol. 2017;66:1193‐1204. [DOI] [PubMed] [Google Scholar]
- 15. Rea S, Xouri G, Akhtar A. Males absent on the first (MOF): from flies to humans. Oncogene. 2007;26:5385‐5394. [DOI] [PubMed] [Google Scholar]
- 16. Akhtar A, Zink D, Becker PB. Chromodomains are protein‐RNA interaction modules. Nature. 2000;407:405‐409. [DOI] [PubMed] [Google Scholar]
- 17. McCullough CE, Song S, Shin MH, Johnson FB, Marmorstein R. Structural and functional role of acetyltransferase hMOF K274 autoacetylation. J Biol Chem. 2016;291:18190‐18198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sykes SM, Mellert HS, Holbert MA, et al. Acetylation of the p53 DNA‐binding domain regulates apoptosis induction. Mol Cell. 2006;24:841‐851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lin HP, Cheng ZL, He RY, et al. Destabilization of fatty acid synthase by acetylation inhibits de novo lipogenesis and tumor cell growth. Cancer Res. 2016;76:6924‐6936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Luo H, Shenoy AK, Li X, et al. MOF acetylates the histone demethylase LSD1 to suppress epithelial‐to‐mesenchymal transition. Cell Rep. 2016;15:2665‐2678. [DOI] [PubMed] [Google Scholar]
- 21. Zhou Y, Schmitz KM, Mayer C, Yuan X, Akhtar A, Grummt I. Reversible acetylation of the chromatin remodelling complex NoRC is required for non‐coding RNA‐dependent silencing. Nat Cell Biol. 2009;11:1010‐1016. [DOI] [PubMed] [Google Scholar]
- 22. Huai W, Liu X, Wang C, et al. KAT8 selectively inhibits antiviral immunity by acetylating IRF3. J Exp Med. 2019;216:772‐785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang Y, Zhang R, Wu D, et al. Epigenetic change in kidney tumor: downregulation of histone acetyltransferase MYST1 in human renal cell carcinoma. J Exp Clin Cancer Res. 2013;32:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cao L, Zhu L, Yang J, et al. Correlation of low expression of hMOF with clinicopathological features of colorectal carcinoma, gastric cancer and renal cell carcinoma. Int J Oncol. 2014;44:1207‐1214. [DOI] [PubMed] [Google Scholar]
- 25. Zhang J, Liu H, Pan H, et al. The histone acetyltransferase hMOF suppresses hepatocellular carcinoma growth. Biochem Biophys Res Commun. 2014;452:575‐580. [DOI] [PubMed] [Google Scholar]
- 26. Remacle AG, Golubkov VS, Shiryaev SA, et al. Novel MT1‐MMP small‐molecule inhibitors based on insights into hemopexin domain function in tumor growth. Cancer Res. 2012;72:2339‐2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Le Romancer M, Poulard C, Cohen P, Sentis S, Renoir JM, Corbo L. Cracking the estrogen receptor's posttranslational code in breast tumors. Endocr Rev. 2011;32:597‐622. [DOI] [PubMed] [Google Scholar]
- 28. Ziemin‐van der Poel S, McCabe NR, Gill HJ, et al. Identification of a gene, MLL, that spans the breakpoint in 11q23 translocations associated with human leukemias. Proc Natl Acad Sci USA. 1991;88:10735‐10739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gao WW, Xiao RQ, Zhang WJ, et al. JMJD6 licenses ERalpha‐dependent enhancer and coding gene activation by modulating the recruitment of the CARM1/MED12 co‐activator complex. Mol Cell. 2018;70:340‐357 e348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Duong V, Boulle N, Daujat S, et al. Differential regulation of estrogen receptor alpha turnover and transactivation by Mdm2 and stress‐inducing agents. Cancer Res. 2007;67:5513‐5521. [DOI] [PubMed] [Google Scholar]
- 31. Fan M, Park A, Nephew KP. CHIP (carboxyl terminus of Hsc70‐interacting protein) promotes basal and geldanamycin‐induced degradation of estrogen receptor‐alpha. Mol Endocrinol. 2005;19:2901‐2914. [DOI] [PubMed] [Google Scholar]
- 32. Kim JY, Yu J, Abdulkadir SA, Chakravarti D. KAT8 regulates androgen signaling in prostate cancer cells. Mol Endocrinol. 2016;30:925‐936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jaganathan A, Chaurasia P, Xiao GQ, et al. Coactivator MYST1 regulates nuclear factor‐kappaB and androgen receptor functions during proliferation of prostate cancer cells. Mol Endocrinol. 2014;28:872‐885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang X, Peng D, Xi Y, et al. G9a‐mediated methylation of ERalpha links the PHF20/MOF histone acetyltransferase complex to hormonal gene expression. Nat Commun. 2016;7:10810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Carr SM, Munro S, Sagum CA, Fedorov O, Bedford MT, La Thangue NB. Tudor‐domain protein PHF20L1 reads lysine methylated retinoblastoma tumour suppressor protein. Cell Death Differ. 2017;24:2139‐2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lasko LM, Jakob CG, Edalji RP, et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage‐specific tumours. Nature. 2017;550:128‐132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ma H, Chang H, Yang W, Lu Y, Hu J, Jin S. A novel IFNalpha‐induced long noncoding RNA negatively regulates immunosuppression by interrupting H3K27 acetylation in head and neck squamous cell carcinoma. Mol Cancer. 2020;19:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gardberg AS, Huhn AJ, Cummings R, et al. Make the right measurement: discovery of an allosteric inhibition site for p300‐HAT. Struct Dyn. 2019;6:054702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu R, Zhang Z, Yang H, et al. Design, synthesis, and biological evaluation of a new class of histone acetyltransferase p300 inhibitors. Eur J Med Chem. 2019;180:171‐190. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Figs S1‐S5
Tables S1, S2