

Abstract
Uterine leiomyosarcoma (LMS) is a rare but deadly disease. Due to poor understanding of the molecular and genetic causes of the disease, the diagnosis of LMS has been based primarily on histology. Nuclear atypia is one of hallmarks in LMS, however, it also occurs in 2 clinically benign variants, including smooth muscle tumors with fumarate hydratase alteration (SMT‐FH) and leiomyoma with bizarre nuclei (LM‐BN). In addition to nuclear atypia, many well recognized biomarkers used for LMS are also frequently overexpressed in LM‐BN, and the histogenesis and molecular natures for LM‐BN and LMS remain largely unknown. To characterize the molecular profiling of LMS, SMT‐FH, and LM‐BN, we performed integrated comprehensive genomic profiling including whole‐genome sequencing (WGS) and RNA sequencing and genomic microarray analyses to assess genome‐wide copy number alterations (CNAs) and immunohistochemistry (IHC) in all 3 tumor types. We found that both LM‐BN and LMS showed genomic instability and harbored extensive CNAs throughout the whole genome. By contrast, the SMT‐FH presented its characteristic 1q43‐44 deletions in all cases tested, with minimal CNAs in the rest of genomic regions. Further analyses revealed that LMS and LM‐BN groups showed similar patterns of CNAs that are tended to cluster together and separated from the SMT‐FH group. The integrated molecular profiling enabled the detection of novel and traditional biomarkers and showed excellent discrimination between LM‐BN and LMS. Our study suggests that LM‐BN, despite having similar nuclear atypia to SMT‐FH, showed similar genomic instability but distinct genomic alterations with its malignant counterpart of LMS. The integrated molecular profiling is of clinical importance in characterizing these rare uterine smooth muscle tumors.
Keywords: copy number alteration, fumarate hydratase, gene expression, leiomyoma with bizarre nuclei, leiomyosarcoma, nuclear atypia
Leiomyosarcoma and 2 benign mimics (leiomyoma with FH alteration [SMT‐FH] and leiomyoma with bizarre nuclei [LM‐BN]) have characteristic nuclear atypia and often cause diagnostic challenge. LM‐BN and LMS are highly genomically unstable and harbor extensive CNAs in the genome, whereas SMT‐FH shows simple 1q43‐44 deletions. Integrated molecular profiling enabled the detection of novel and traditional biomarkers and showed excellent discrimination between LM‐BN and LMS.

1. INTRODUCTION
Uterine smooth muscle tumors (USMTs) show a broad range of histologic variants. Most such tumors are benign and are readily diagnosed and managed clinically. However, a small fraction of USMTs present with marked nuclear atypia, and these tumors can generally be divided into 3 tumor types, including smooth muscle tumors with fumarate hydratase alteration (SMT‐FH), leiomyoma with bizarre nuclei (LM‐BN), and leiomyosarcoma (LMS). LMS is a rare malignancy that is identified in 1 in 200‐800 hysterectomy specimens for smooth muscle tumors. 1 LMS is a highly aggressive malignancy, characterized by early metastasis, high rates of recurrence, and poor prognosis. 2 The diagnosis of LMS is primarily based on the histologic triad of nuclear atypia, high mitotic index, and tumor necrosis. 3 However, clinical manifestations of these tumors cannot always be accurately predicted by histology alone, 4 and a subset—particularly of those with severe nuclear atypia—may be misinterpreted. 5 , 6
LM‐BN is a rare variant of USMT, previously called “atypical leiomyoma” or “symplastic leiomyoma.” It demonstrates its cytological atypia with large and bizarre nuclei, 7 therefore the World Health Organization (WHO 2014) recommended using the term “leiomyoma with bizarre nuclei,” LM‐BN can show focal or diffuse nuclear atypia, with or without increased mitoses (generally up to 7‐8 mitoses/10 high power fields). Based on several large case series, LM‐BN tends to have a benign clinical course 7 with occasional recurrence but no identified fatalities. 5 A small fraction of LM‐BN has also been shown to harbor MED12 mutations, 7 suggesting a molecular relationship to the usual‐type leiomyomas.
SMT‐FH is another variant of USMT with marked nuclear atypia. 8 The nuclear atypia seen in SMT‐FH is characterized by mostly round/oval large and multinucleated nuclei with prominent nucleoli and perinucleolar halos. 5 , 9 , 10 , 11 Such nuclear features, along with other histologic features, can be seen in most but not all SMT‐FH. 7 SMT‐FH is caused by biallelic inactivation of fumarate hydratase (FH), which can be detected by the absence of FH staining using immunohistochemistry. 8 , 11 SMT‐FH can be induced by either germline or somatic FH alterations, but the exact molecular mechanism for the latter remains to be determined. 11
In contrast with SMT‐FH, the histogenesis and causes of LMS and LM‐BN remain largely unknown. Genomic studies demonstrate that both müllerian and non‐müllerian LMS show genomic instability, presenting with complex genomic alterations and inactivation of RB and TP53. 1 , 12 , 13 , 14 Cytogenetic studies have shown that LMS are genetically complex, often exhibiting chaotic complex karyotypes, and no typical pathognomonic chromosomal aberrations. 1 , 15 , 16 , 17 Prior studies by our group and others have suggested that LM‐BN may harbor some molecular changes commonly seen in LMS, 1 , 13 , 18 raising the question of whether these 2 tumor types may share a common pathogenesis or represent different stages of tumor progression, at least in some cases. Furthermore, due to its non‐negligible risk for recurrence and unclear relation to LMS, clinical management of patients with LM‐BN remains controversial, especially for those who want to preserve fertility. Further study comparing global genomic changes seen in LMS and LM‐BN may provide additional insight into the molecular relationship of these tumor types, with implications for tumorigenesis.
Our overall goal in this study was to examine the genome‐wide alterations including copy number alterations (CNAs), mutational profiling, and gene expression signatures in LMS, LM‐BN, and SMT‐FH, by using integrated comprehensive molecular profiling. In particularly, a clear separation of LM‐BN from SMT‐FH based on FH gene status will further justify the separation of SMT‐FH as a distinct molecular and diagnostic entity. Furthermore, we aimed to better understand the genomic alterations in LM‐BN and LMS to help elucidate their molecular relationship and identify potential biomarkers that could be useful clinically to help resolve diagnostically challenging cases.
2. MATERIALS AND METHODS
2.1. Case collection and clinical information
Cases were identified from our institutional laboratory information system database from 1994 to 2018. The candidate cases were reviewed and cases with typical histology that met the diagnostic criteria of SMT‐FH, LM‐BN or LMS were selected for this study (Figure S1). In total, 88 cases were included in this study: 28 cases of SMT‐FH, 25 of LM‐BN, and 35 of LMS. All tumors were evaluated by histology and immunohistochemistry by excluding other uterine mesenchymal tumors, including perivascular epithelioid cell tumor (PEComa), inflammatory myofibroblastic tumor, and endometrial stromal tumors. All tumors were included for biomarker expression analysis. Among them, 5 SMT‐FH, 13 LM‐BN, and 10 LMS cases and 15 normal myometrium controls were subjected to whole‐genome sequencing (WGS) and analyzed. In addition, 13 SMT‐FH and 8 myometrial controls were selected for Affymetrix OncoScan® copy number alterations (CNAs) analysis. Each patient’s clinical characteristics and patient’s demographic data were recorded, including the patient’s age, tumor size, surgical procedure, and original pathologic diagnosis (Tables 1 and S1). Clinical follow‐up was assessed both as recurrence and overall survival following the time of diagnosis. Recurrence was defined as histopathologically proven recurrence recorded in our institutional database, with patients censored for recurrence outcomes at the time of the last specimen in the database. Survival analyses, including assessment of the probability of recurrence‐free survival and overall survival at specific time points, were performed using the Kaplan‐Meier method. The study was approved by our Institutional Review Board (IRB#STU00205311).
TABLE 1.
Clinical and histological feature of SMT‐FH, LM‐BN, and LMS
| Type | SMT‐FH | LM‐BN | LMS | P‐value | |
|---|---|---|---|---|---|
| No. cases (n) | 28 | 25 | 35 | ||
| Age (y) | Mean ± SEM | 40.97 ± 1.56 | 42.88 ± 2.01 | 55.62 ± 2.26 | <.001 |
| Tumor size (cm) | Mean ± SEM | 9.74 ± 0.84 | 6.28 ± 0.97 | 10.64 ± 1.25 | .0182 |
| Surgery type [n (%)] | Hysterectomy | 13 (46.4) | 21 (84.0) | 33 (94.3) | |
| Tumorectomy | 15 (53.6) | 4 (16.0) | 2 (5.7) | <.001 | |
| Severe nuclear atypia | Focal/multifocal | 78.6% | 60.7% | 0.0% | |
| Diffuse | 21.4% | 39.3% | 100% | <.001 | |
| Mitosis/10 HPF | <1 | 25.0% | 24.0% | 0.0% | |
| 1‐5 | 71.4% | 60.0% | 5.7% | ||
| 6‐10 | 3.6% | 16.0% | 5.7% | ||
| >10 | 0.0% | 0.0% | 88.6% | <.001 | |
| Tumor necrosis | Absence | 96.4% | 100% | 5.7% | |
| Focal | 3.6% | 8.6% | |||
| Extensive | 58.7% | <.001 | |||
| Follow‐up (mo) | Median (IQR) | 85.5 (28.8, 204.3) | 41.5 (12.8, 134.3) | 55.2 (13.7, 120.2) | |
| Overall survival | 1 y | 100% | 100% | 76% (64%, 91%) | |
| (95% CI) | 2 y | 100% | 100% | 68% (55%, 85%) | |
| 5 y | 100% | 100% | 50% (36%, 69%) |
2.2. Genomic DNA extraction
DNA was isolated from formalin‐fixed paraffin‐embedded (FFPE) tumor tissue, including 13 SMT‐FH, 13 LM‐BN, 10 LMS, and 15 myometrial tissue samples. For each specimen, 12 FFPE sections (5 µm thickness) were cut from a single block. Two of the 12 sections for each specimen were stained with hematoxylin and eosin (H&E) to confirm the correct tumor types and serve as a guide for macrodissection of the remaining 10 unstained sections, which were subsequently collected for DNA extraction. Total genomic DNA was isolated using the ZYMO RESEARCH Quick‐DNA™ FFPE Kit (ZYMO RESEARCH, D3067, USA) and following the manufacturer's instructions. DNA was quantitated using a Qubit™ fluorometer (Life Technologies) and DNA quality was assessed by evaluating the sample's A260/A280 ratio and its integrity by 1% agarose gel electrophoresis (Figure S2A).
2.3. Library preparation and whole‐genome sequencing
A DNA library and WGS were prepared and performed using the BGISEQ‐500 platform (BGI). In brief, genomic DNA was fragmented using Covaris technology to produce a fragment size between 150 bp and 250 bp. End repair of DNA fragments was performed, and an “A” base was added at the 3′‐end of each strand. Adapters were ligated to both ends of the end‐repaired/dA‐tailed DNA fragments, then amplified by ligation‐mediated PCR (LM‐PCR), followed by single‐strand separation and cyclization. Rolling circle amplification (RCA) was performed to produce DNA nanoballs (DNBs). Each resulting qualified captured library was loaded onto the BGISEQ‐500 platform and high‐throughput sequencing was performed to ensure that each sample met the average sequencing coverage requirement. Raw data were filtered by removing sequences using the following rules: (a) reads from the adapter; (b) a low‐quality base ratio (base quality less than or equal to 5); (c) an unknown base (“N” base) ratio of more than 10%. After data cleaning, sequence data from each sample were mapped to the human reference genome (GRCh37/HG19) using Burrows‐Wheeler Aligner (BWA) software and a data analysis quality control (QC)system.
Bioinformatics data analysis was performed using Basepair software (https://www.basepairtech.com/), and raw sequencing data from the BGISEQ machine. In brief, data were analyzed as follows: reads from cleaned sequence data were mapped to the hg19 (UCSC) and DNA sequences from normal myometrium using BWA (v.0.7.5) with parameters “mem –t 8 ‐P –M.” Generated files were sorted, and PCR duplicates were removed using Picard (v.1.105) (http://broadinstitute.github.io/picard). Subsequently, the BAM files were indexed using SAMtools (v.0.1.19). The WGS had a quality score of 20 (Q20), and 95%‐96% of reads were successfully aligned (Figure S2).
2.4. Chromosomal microarray analysis
Chromosomal microarray analysis (CMA) was performed in 13 SMT‐FH tumors on Affymetrix OncoScan® CNA arrays (Thermo Fisher Scientific). The data were analyzed using Affymetrix Chromosome Analysis Suite (ChAS) software and reviewed independently by 2 cytogeneticists. CNAs, including copy number gains or losses, and copy neutral loss of heterozygosity (CN‐LOH), were manually reviewed.
2.5. Copy number alteration calling by whole‐genome sequencing
The CNAs from read‐depth data obtained from WGS were called using the Genome Analysis Toolkit 4.0 software (https://gatk.broadinstitute.org/hc/en‐us). The coverage of 10‐kb windows with sufficient mapping quality and read density was recorded and subsequently corrected for GC content and replication timing. In brief, the coverage values of tumors were normalized by myometrial controls and log‐transformed to yield log‐ratios. GISTIC 2.0 analysis was performed to identify significant foci of somatic CNAs. Significance of CNAs identified in specific chromosome regions was assessed through use of random permutation tests which simulate the null distribution of CNAs across the genome, 19 as implemented by the GISTIC 2.0 analysis. The generated P‐values were corrected for multiple comparisons using the Benjamini‐Hochberg false discovery rate (FDR) procedure 20 to obtain q‐values. The following criteria were used to determine significance of whole‐gene gain or loss events: fold change > 2.0 (gain) or <−2.0 (loss), and a q‐value < 0.25 (implying a 25% FDR).
2.6. Principal component analysis
Principal component analysis (PCA) of these based on significant focal CNA regions that were identified for LM‐BN and LMS using the above GISTIC 2.0 analysis was also performed using the online tool ClustVis 21 (https://biit.cs.ut.ee/clustvis/) with unit variance scaling applied to the row and singular value decomposition (SVD) imputation for principal components (PC). The first 2 PCs were considered sufficient to explain the observed variation and were visualized as a 2D scatter plot.
2.7. Gene enrichment and molecular pathway analysis
For gene enrichment analyses, we performed Gene Ontology (GO) analysis (GO annotations: biological process, molecular function, cellular compartment, and protein domain) using the STRING (v.11.0) database to confirm enrichment results with topological features from gene interaction networks. The route of gene cluster and related functions was determined using Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis (http://www.genome.jp/). A P‐value < .05 was considered to be statistically significant.
2.8. RNA sequencing
RNA‐seq was performed by Genewiz (South Plainfield). Total RNA was extracted from FFPE tumor tissue (n = 3) and myometrial tissue (n = 3) using the QIAGEN RNeasy FFPE kit (Qiagen). RNA was quantified using a Qubit 2.0 fluorometer and RNA integrity was checked using the Agilent TapeStation 4200 system (Agilent Technologies). RNA sequencing libraries were prepared using the NEBNext Ultra RNA Library Prep Kit for Illumina and following the manufacturer's instructions (NEB). Briefly, samples underwent ribosomal RNA depletion, stranded RNA library preparation, and multiplexing and cluster generation, followed by the Illumina HiSeq (Illumina) system at 2 × 150 base pairs for paired‐end sequencing in a high output mode. The clean reads were mapped to the Homo sapiens GRCh37 reference genome available on ENSEMBL using STAR aligner v.2.5.2b. Feature “Counts” within the Subread package v.1.5.2 was applied to calculate unique gene hit counts. After extraction of gene hit counts, the gene hit counts table was used for downstream differential expression analysis. DESeq2 was used to identify differential expression analysis between groups. Differentially expressed genes were identified using a threshold adjusted P‐value < .05 and absolute log2 fold change > 1. RNA sequence data were submitted to the NCBI Sequence Read Archive (SRA accession number: PRJNA649753).
2.9. Tissue microarrays
FFPE tissue blocks containing SMT‐FH, LM‐BN, LMS and myometrium controls were selected for each case, and 2 mm tissue cores were taken to create a tissue microarray (TMA). The TMA was arranged to allow a comfortable distance of 4 μm between samples. The first and last slides of each TMA were stained with H&E to conduct quality assurance by confirming the presence of the correct tumor types.
2.10. Immunohistochemistry
Immunohistochemical (IHC) analysis was performed on biomarkers selected based on the results of the above genomic analyses, including AGR2, ATAD3, DUSP2, MT1G, NR4A2, FH, PTBP1, p16, PGR, and SIK1. The antibody information and working conditions are summarized in Table S2. IHC was performed on a Ventana Nexus automated system, as described previously. 22 H&E and IHC slides were scanned using a slide scanner (Hamamatsu, pathology core facility, NU) and a selected tumor area of equal size for each core (c. 20% of a well stained area of tumor core with 0.8 mm diameter circle or square area) was quantified based on the integrated density of biomarker staining using ImageJ software. 23 In brief, the IHC stained TMA were analyzed using ImageJ IHC profiler (https://sourceforge.net/projects/ihcprofiler). Once the “Nuclear Stained Image” or “Cytoplasmic Stained Image” was selected for specific biomarkers, the threshold was adjusted in accordance with the software instructions, and quantitative scores were calculated.
2.11. Statistical analysis
Student t test and ANOVA were performed using GraphPad Prism software v.8. Histologic features (nonparametric) were analyzed using Pearson chi‐square, Fisher exact, and Kruskal‐Wallis tests, as appropriate. Age range, tumor size, and other parametric factors were analyzed by ANOVA, with a P‐value < .05 considered statistically significant. The selected biomarkers for IHC analysis were assessed on their ability to discriminate between LM‐BN and LMS when used in combination, combining the biomarkers that showed a significant difference in expression between LMS and LM‐BN. A logistic regression model (the “full” model) was developed using the integrated density of brown staining for all included biomarkers as the predictor variables and LMS as the binary outcome variable. This model was restricted only to the 25 LM‐BN cases and 35 LMS cases. Once the “full” model was developed, we determined the optimal combination of biomarkers for distinguishing between LMS and LM‐BN using the Least Absolute Shrinkage and Selection Operator (LASSO) algorithm, 24 with the lambda parameter automatically selected to minimize the cross‐validation error (lambda = 0.1108). A receiver operating characteristic (ROC) curve for discriminating LMS from LM‐BN was constructed using the linear predictors from this model, with leave‐one‐out cross‐validation used to estimate out‐of‐sample performance and prevent model overfitting. Overall discrimination was assessed via the area under the ROC curve (AUC), with confidence intervals estimated via the DeLong method. Logistic regression and AUC analyses were performed using R v.4.0.2 (https://www.R‐project.org), with the “pROC” package, 25 and the LASSO algorithm was performed using the “glmnet” package. 26
3. RESULTS
3.1. Histopathologic features
The purpose of this study was to investigate and compare the global genomic alterations in 3 different USMTs, including SMT‐FH, LM‐BN, and LMS. In particularly, we wanted to evaluate differences and common changes associated with these 3 different tumor types with similar nuclear pleomorphism/atypia. To this end, all selected cases were subjected to further histologic evaluation, in addition to the original clinical diagnosis. In brief, all tumor types showed marked nuclear atypia in focal, multifocal, or diffuse patterns, characterized by large, pleomorphic, and hyperchromatic nuclei. Most SMT‐FH could be readily diagnosed based on specific nuclear and histologic features (Figure 1A), and some cases showed overlapping nuclear features with LM‐BN (Figure S1). Therefore, all selected cases were subjected to immunostaining for FH. Cases with both typical nuclear features and immunonegativity for FH were classified as SMT‐FH, and they were all sporadic cases. All LM‐BN and LMS were positive for FH (Figure 1B). Close examination of the morphologic characteristics of the nuclear atypia in the 3 tumor types showed that: (a) 28 cases of SMT‐FH had large round and oval nuclei with hyperchromatic and coarse chromatin, distinct nuclear membranes, prominent nucleoli and some perinucleolar halos; (b) 25 cases of LM‐BN were recognized by large spindled and/or rounded nuclei with nuclear membrane irregularities and dark chromatin, commonly described bizarre nuclei. Nineteen cases had mitoses up to 4/10 high power fields, and 6 cases had mitoses of 5‐7/10 high power fields (Table S1). 5 , 7 , 27 The latter would be defined as STUMP in accordance with the new WHO tumor classification (2020); and (c) 35 cases of conventional LMS contained a broad range of nuclear atypia, including large nuclear size with pleomorphism, hyperchromasia, nuclear membrane irregularity, and prominent nucleoli (Figures 1A and S1). The general clinical and pathologic characteristics of these 3 tumor types are summarized in Table 1 and Table S1.
FIGURE 1.
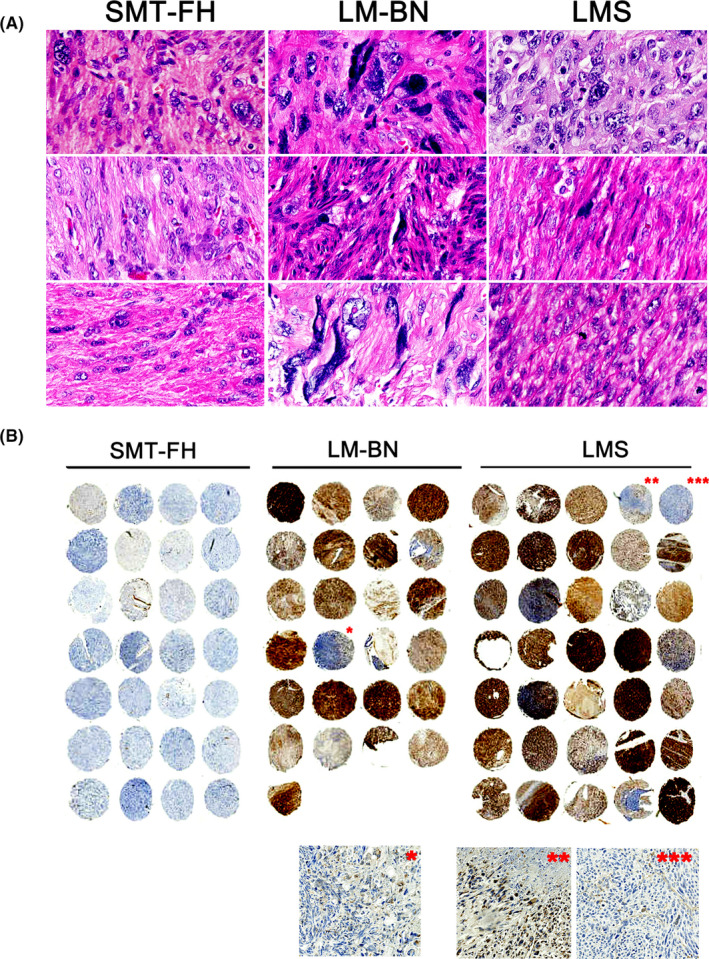
Histology of SMT‐FH, LM‐BN, and LMS. A, Photomicrographs of histologic and cytological features in each of 3 examples of SMT‐FH, LM‐BN, and LMS. B, Immunostaining for FH in tissue microarray of 28 cases of SMT‐FH, 25 cases of LM‐BN and 35 cases of LMS. *–***: enlarged inserts for detailed immunostaining for FH
3.2. Genome‐wide copy number alterations analysis
To explore the genome‐wide copy number changes, WGS on the BGISEQ‐500 platform was performed in 5 SMT‐FH, 13 LM‐BN, 10 LMS and 15 myometria (normal controls). The clean number of base pairs sequenced ranged from 10.5 to 11.7 billion in tumor samples, and 10.6 billion in corresponding myometrium samples. After WGS, tumor data were normalized to their corresponding myometrial sequences, CNAs were analyzed by GATK4 at a resolution of 10 kb (Figure S3). Overall, CNAs varied widely among 3 different tumor types, with an overall low level of CNAs seen in SMT‐FH, an intermediate level seen in LM‐BN, and a high level seen in LMS (Figures 2A and S4). Copy number losses were much more common than copy number gains in all tumor types.
FIGURE 2.
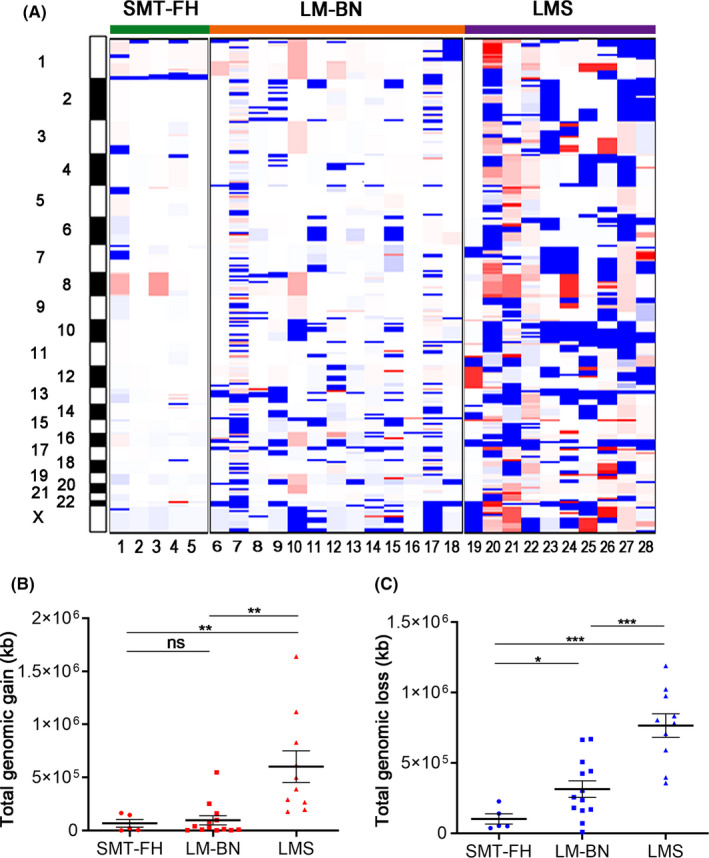
Genomic copy number alterations (CNAs) analysis of SMT‐FH, LM‐BN, and LMS. A, Copy number gains (red) and losses (blue) were displayed for 5 SMT‐FH, 13 LM‐BN and 10 LMS (x‐axis: tumor type; y‐axis: chromosomal location). B and C, Quantitation of the total base length of gains (B) and losses (C) by tumor types were shown. Error bars represent the standard error of the mean. *P < .05; **P < .005; ***P < .001; n.s., not significant
The total lengths (Mb) of the genome with copy number gains in SMT‐FH and LM‐BN were very low and no difference between the 2 tumor types was found. The length of copy number gains in LMS was significantly higher than that of SMT‐FH and LM‐BN (Figure 2B). The total length of copy number losses was significantly higher in LM‐BN and LMS than that of SMT‐FH (Figure 2C). Our data indicated that the SMT‐FH is a tumor type with less genomic complexity showing overall lower CNAs across the entire genome. The LM‐BN showed a median complexity level with a higher frequent focal CNAs, however its total basepair length of CNAs was much lower than that of LMS (Figure 2A) and LMS has the highest genomic complexity. No difference in CNAs was found between LM‐BN with <5 and >4 mitoses/10 high power fields (data not shown).
3.3. 1q43‐44 deletion in SMT‐FH
CNA analysis in 5 SMT‐FH showed loss of chromosome 1q43‐44, where the FH gene is located (Figure 3A). This finding prompted us to use a different platform for CNA analysis in more tumor samples. The original 5 cases and an additional 8 randomly selected SMT‐FH tumors were examined using Affymetrix OncoScan® CNA arrays. As shown in Figure 3B, loss of 1q43‐44 was observed in all 13 cases analyzed by GISTIC 2.0 software. Further analysis revealed that 53.8% (7/13) of SMT‐FH showed loss of both copies of 1q43‐44, indicating the homozygous deletions of the FH gene, and 46.2% (6/13) showed loss of a single copy (Figure 3C) indicating the heterozygous deletion of the FH gene. Of those tumors with only a single copy loss of FH, only 2 cases showed missense mutations of FH gene (Figure 3C). A plot of CNA frequency of chromosome 1 in 3 tumor types is summarized in Figure 3D. Our findings suggested that sporadic SMT‐FH may be frequently associated with deletion of FH genomic regions, and that point mutations account for a small fraction of somatic SMT‐FH, as we reported previously. 11 Genomic alterations in other chromosome regions were much less common in SMT‐FH (see below), suggesting a simple FH‐driven tumorigenesis process in this tumor type. Interestingly, the other significant CNA regions observed in SMT‐FH, although much less common than the loss of 1q43‐44, were also observed in LM‐BN (as described in later sections and in Figure S6).
FIGURE 3.
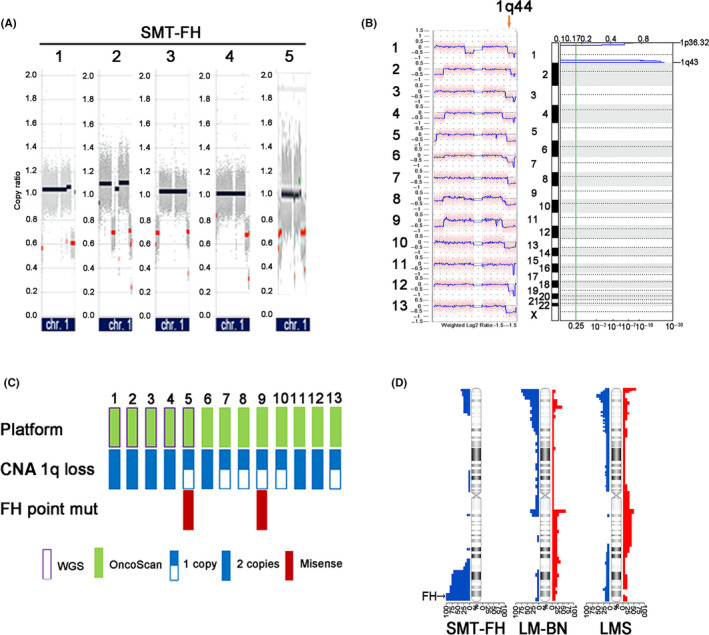
Genomic alterations in SMT‐FH. A, CNA plots of chr1 in 5 SMT‐FH analyzed by WGS. Red dots indicate the regions of copy number loss. B, CNA plots of chr1 as analyzed by Affymetrix OncoScan® microarrays in 13 SMT‐FH (left panel) and significant copy number losses analyzed by GISTIC v.2.0. (right panel). C, Integrated representation of copy number losses on chromosome 1 in 13 SMT‐FH analyzed on 2 platforms and FH mutation analysis. D, CNA plot on chromosome 1 in SMT‐FH, LM‐BN, and LMS tumors. Blue color (left) indicated loss and red gain (right)
3.4. CNAs comparison between LM‐BN and LMS
As most SMT‐FH showed very simple CNAs, as illustrated previously, that were completely different from other 2 tumor types, we were interested in comparing CNAs between LM‐BN and LMS. Both LM‐BN and LMS showed a high frequency of CNAs, including mostly copy number losses (Figure 2A,B). The GISTIC 2.0 analysis of LM‐BN and LMS identified 43 regions of CNA that were present at a higher frequency than would be expected due to chance (q < 0.25), including 9 copy number gains and 34 copy number losses (Figure 4A and Table S3). In all 43 significant peaks, the frequent changes occurring in ≥30% of both LM‐BN and LMS were segmental gains of chromosomes 1q21.1 and 15q14, and losses on 1p36.32, 2p25.1, 2q37.3, 5q35.3, 7q36.3, 9q34.3, 10q26.3, 11p15.5, 13q14.2, 14q11.2, 16p13.3, 16q21, 17p13.1, 19p13.3, 19q13.43, 22q13.2, Xp22.31 and Xq28. Of note, 3 copy number losses were presented in >50% of LMS and LM‐BN, including 9q34.3 (61.5% for LM‐BN and 50% for LMS), 13q14.2 (53.8% for LM‐BN and 90% for LMS) and 22q13.2 (61.5% for LM‐BN and 60% for LMS) (Figure 4A and Table S3). Further analysis of 43 significant CNA regions by Venn diagram revealed that 37 were shared between LM‐BN and LMS, and 6 were present in LM‐BN only (Figure 4B). Interestingly 3 of these 6 copy number losses in LM‐BN showed high frequency, including 8q24.3 (53.8%), 15q11.2 (69.2%), and 20q13.33 (53.8%) (Figure 4B and Table S3).
FIGURE 4.
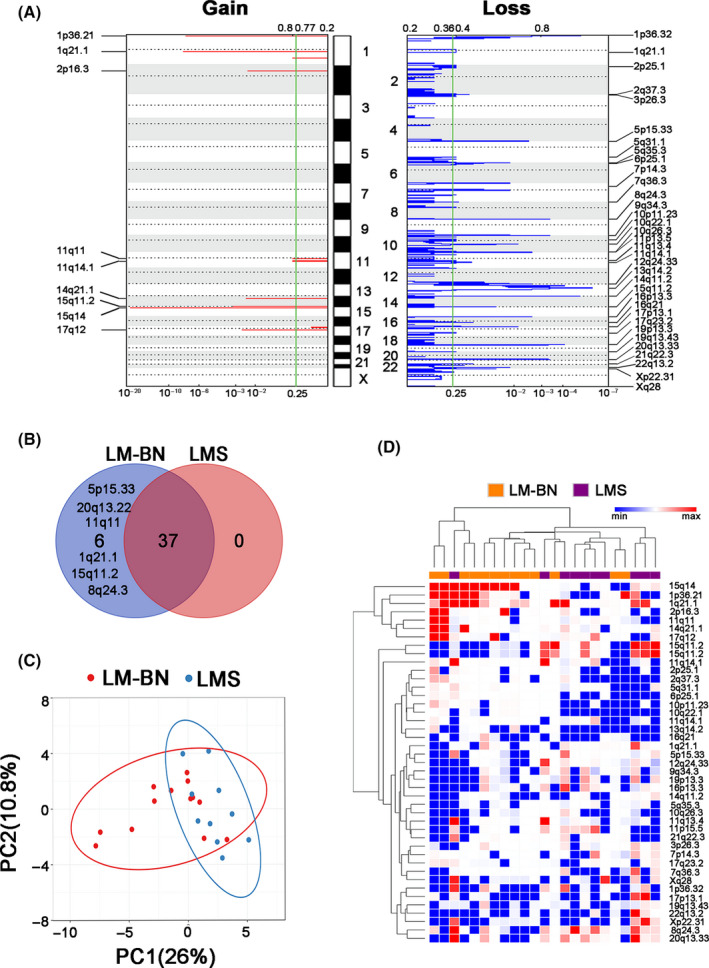
CNA analysis in LM‐BN and LMS. A, GISTIC analysis of significant copy number gain (left) and loss (right) in chromosomal regions in both LM‐BN and LMS (x‐axis: q‐values). B, Venn diagram showing the distribution of significant focal CNAs in LM‐BN, LMS and both. C, Scatter plot of principal component analysis (PCA) of LM‐BN (n = 13) and LMS (n = 10) samples. Each sample is represented as a colored point (red, LM‐BN; blue, LMS), and the axes reflect the first 2 PCs. D, Heatmap dendrogram of significant focal CNAs (row, gain red; loss blue) for each analyzed sample (column) of LM‐BN (orange) and LMS (purple). Dendrogram arm length is inversely proportional to the relatedness of the involved cluster
As both LM‐BN and LMS tumor types share many similar CNAs throughout the genome, a PCA was performed to determine whether CNAs data as a whole could be used to separate LM‐BN and LMS into distinct entities. PCA on the 43 significant CNA regions showed the 2 principal components, which explained 36.8% of the overall variance, shown in Figure 4C. These results also displayed some separation of LM‐BN and LMS into 2 related clusters, although significant overlap remained, with 69.6% of cases falling within both clusters. Unsupervised hierarchical clustering analysis of the same 43 CNA regions (Figure 4D) showed similar findings of a significant amount of overlap but distinction between the clusters. Overall, our findings indicated that although both LM‐BN and LMS show higher genomic instability, with many similar CNA changes, however the CNA patterns in these 2 tumor types are distinguishable.
3.5. CNAs associated candidate genes in LM‐BN
To evaluate whether CNAs in specific genes may be associated with tumor type, GISTIC2.0 analysis with a cut‐off of q < 0.25 was performed. Thirty‐three regions showed a high level of regional CNAs in LM‐BN, 26 losses and 7 gains (Figure 5A). The most frequent copy number gains (>40% of cases) were gains of 1p36.21, 1q21.1 and 15q14, and frequent copy number losses (>40% of cases) were observed in 1p36.33, 5p15.33, 8q24.3, 9q34.3, 13q14.3, 14q11.2, 15q11.2, 16p13.3, 17p13.2, 19p13.3, 20q13.33, 22q13.33, and Xp21.2 (Figure 5A and Table S4). These regions harbored many important candidate oncogenes and tumor suppressor genes, which could potentially be related to LM‐BN tumorigenesis. Among more than 700 genes found in these regions, there were 39 cancer‐associated genes that were frequently lost in LM‐BN (Table S4). Enrichment analysis (in the GO annotation and STRING databases) revealed a significantly predicted interaction network (P = 3.6 × 10−4). Among these 39 genes, 17 of them were centered and linked by NOTCH1 (Figure 5B), suggesting an important role of NOTCH1 in LM‐BN. In addition, gains of the PRAMEF family of genes (1p36.21), losses of the ATAD3 gene cluster (1p36.33), TERT (5p15.33), RB1 (13q14.3), TSC2 (16p13.3), and PTPB1 (19p13.3) were of greatest interest.
FIGURE 5.
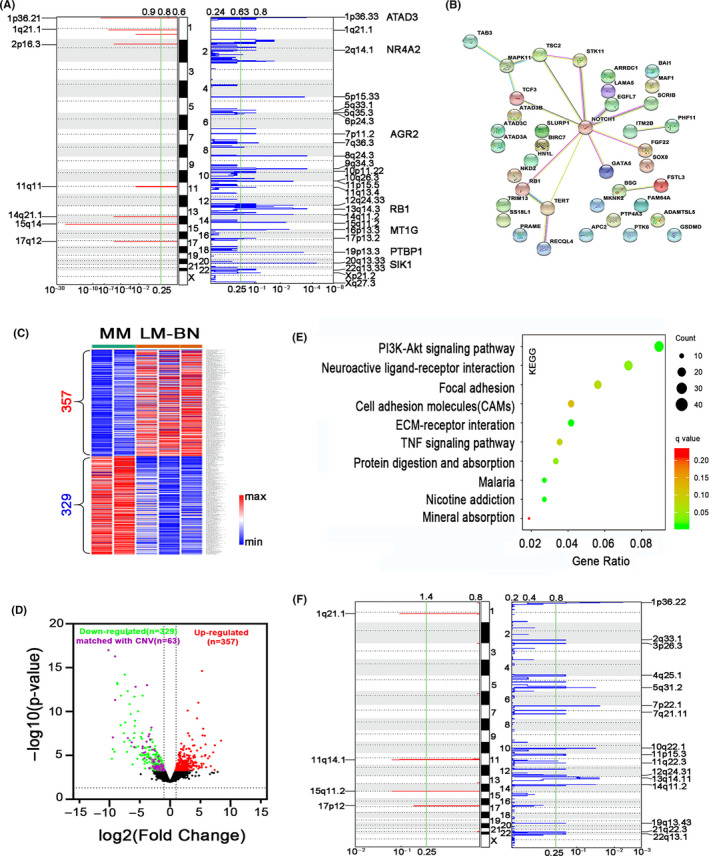
Gene alteration analysis in LM‐BN. A, GISTIC analysis of significant copy number gain (left) and loss (right) in chromosomal regions in LM‐BN (x‐axis: q‐values). B, Network graph showing topological relationship of 39 cancer‐associated genes centered on NOTCH1 from common CNA regions detected by GISTIC 2.0 analysis in LM‐BN. C, Heatmap illustrates 686 dysregulated genes in LM‐BN by RNA‐seq analysis, compared with myometrial controls (MM). D, Volcano plot showing the distribution of downregulated (green) and upregulated (red) genes in LM‐BN. Purple dots indicate downregulated genes in the regions of common copy number loss. E, Altered pathways by enrichment analysis in GO annotation (KEGG) for CNA (729 genes) and RNA‐seq (686 genes) in LM‐BN. F, GISTIC analysis of significant copy number gain (left) and loss (right) in chromosomal regions in LMS (x‐axis: q‐values)
To evaluate whether CNAs in LM‐BN affect the target gene expression, RNA‐seq analysis was performed in LM‐BN and matched normal myometrium controls. Overall, 357 and 329 genes were significantly upregulated or downregulated compared with normal myometrium (P‐value < .05 and fold change > 1.8; Figure 5C). Volcano plots visualized the significantly differently expressed significant genes in LM‐BN (Figure 5D) and 63 downregulated genes shown by RNA‐seq were present as copy number losses in LM‐BN and LMS (Figure 5A,D). These 63 downregulated genes were involved in protein binding, extracellular exosome, negative regulation of apoptosis, and positive regulation of smooth muscle cell proliferation as detected by GO analysis (Table S4). Copy number gains were less common in LM‐BN, very few upregulated genes found by RNA‐seq were matched to copy number gains (data not shown). When combining the candidate genes identified in CNAs (729 genes) and RNA‐seq (686 genes), the PI3K‐Akt signaling pathway ranked top (Figure 5E). Collectively, all data suggested that NOTCH1, extracellular exosome, smooth muscle proliferation, and AKT pathways might be important in LM‐BN pathogenesis.
As with LM‐BN, copy number losses accounted for the majority of CNAs found in LMS (Figure 5F). The most frequent copy number losses (≥40% of cases) were losses in 1p36.22, 4q35.1, 7p22.1, 7q21.11, 10q22.1, 11p15.3, 11q22.3, 13q14.11, 14q11.2, 21q22.3, and 22q13.1, and the most frequent copy number gains were gains in 1q21.1, 15q11.2, and 17p12 (Figure 5F and Table S4). Loss of ENO1 (1p36.22, 70%); SMAD1 (4q35.1, 40%); RB1 (13q14.11, 80% of cases); PGR (11q22.1, 40% of cases); SIRT4 (12q24.31, 30%); CCAR1, FASTK and PTEN (10q22, 90% of cases); and SMARCB1 and CHEK2 (22q13.1, 60%), as well as gain of PAK1 (11q14.1, 30%) were significantly common in LMS (Figure 5F and Table S4). Except for RB1, all were highly altered in LMS, but not in LM‐BN or SMT‐FH.
3.6. Selected biomarker expression analysis in 3 tumor types
Copy number loss of 1q43‐44 (Figure 3B) was responsible for loss of FH expression by IHC (Figure 1) in most sporadic SMT‐FH. We aimed to explore candidate biomarkers in genomic regions that showed differential CNAs between LM‐BN and LMS. Copy number loss of the 1p36.33 region containing the ATAD3 gene cluster was found in 76.9% of LM‐BN, but was found much less frequently in the other 2 tumor types (Figure 5A). Immunostaining revealed significantly lower ATAD3 expression in LM‐BN compared with either LMS or SMT‐FH (Figure 6A). Loss of the 19p13.3 genomic region containing the PTBP1 gene was frequently seen in LMS, and immunostaining showed a significantly reduced PTBP1 expression in LMS compared with LM‐BN and SMT‐FH (Figure 6A). Copy number loss for RB1 was seen in 80% of LMS and 30% LM‐BN (Figure 5A). P16 is an RB1 downstream effector. As shown in Figure 6A, strong and diffuse immunoreactivity for p16 was found in most LMS and some LM‐BN, but very few were found in SMT‐FH. Lastly, over 40% of LMS had loss of the PGR genomic region. PGR expression was absent in almost all LMS, but PGR was highly expressed in the other 2 tumor types (Figure 6A). The preliminary results suggested that CNAs in LM‐BN and LMS could be further explored for new biomarkers for pathogenesis of these 2 tumor types. Many cancer‐associated genes were found to have copy number loss and were downregulated in RNA‐seq in LM‐BN (Figure 5). We selected 5 such markers, including ARG2, DUSP2, MT1G, NR4A2, and SIK1, for immunohistochemistry, as they were present in regions of copy number loss and downregulated in RNA‐seq. All markers were downregulated in LM‐BN and LMS (Figure 6A), and 8 markers showed a significant difference in expression levels between LM‐BN and LMS, including AGR2, ATAD3, DUSP2, NR4A2, PR, p16, PTBP1, and SIK1. When these markers were combined via logistic regression, these 8 IHC markers showed excellent discrimination in our cohort, with an AUC of 0.99 (0.97, 1.00). When the LASSO algorithm was applied to this model, the optimal combination of markers for discriminating between LMS and LM‐BN included 6 IHC markers: AGR2, ATAD3, DUSP2, NR4A2, p16, and PTBP1. The optimal model also showed excellent discrimination in our cohort, with an AUC of 0.98 (0.96, 1.00). As shown in Figure 6B, the performance of this optimal model was robust when out‐of‐sample performance was estimated by leave‐one‐out cross‐validation, with an AUC of 0.89 (95% confidence interval (CI) 0.78, 1.00). Immunostaining for pAKT also showed significant upregulation in both LM‐BN and LMS (Figure S6B), indicating highly activated AKT signaling in these 2 tumor types.
FIGURE 6.
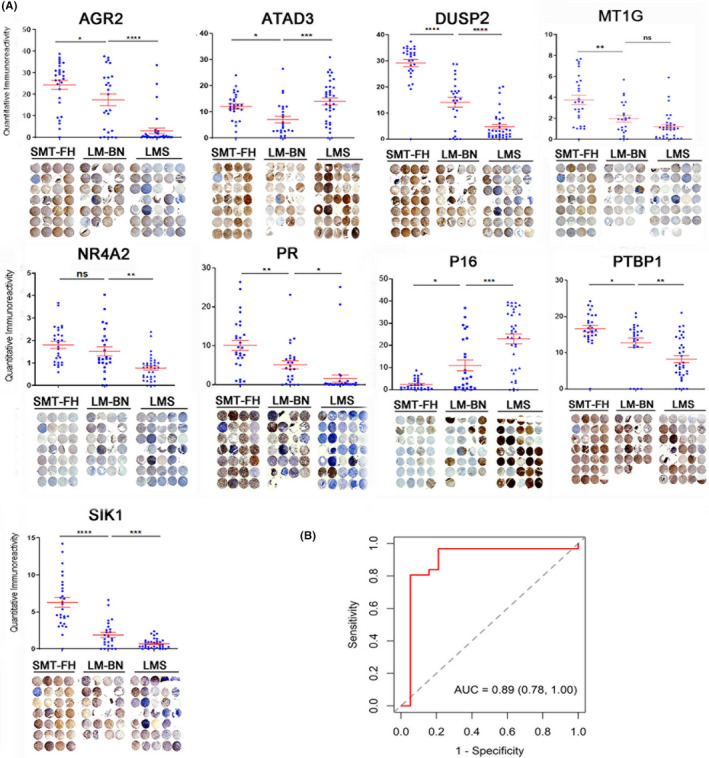
Immunohistochemistry analysis of the selected biomarkers in SMT‐FH, LM‐BN, and LMS. A, Immunohistochemical analysis of 9 biomarkers in 3 different tumors. Dot plot (upper) summarizes the quantitative scores of immunointensity using ImageJ software (see Materials and Methods). Red bars indicated mean and standard errors. Each dot represents one sample. *P < .05; **P < .005; ***P < .001; n.s., not significant. Tissue microarray (bottom) illustrates the actual immunostain (brown color) with counterstain of hematoxylin (blue color) in 3 tumor types. Cores that did not have sufficient tissue for an IHC score are marked with a red cross. B, ROC curve for optimal combination of IHC biomarkers. A receiver operating characteristic (ROC) curve for discriminating LMS from LM‐BN was constructed using the linear predictors from a logistic regression model and combining all 8 significant IHC biomarkers. The optimal combination of 6 biomarkers was selected using the LASSO algorithm. Leave‐one‐out cross‐validation was used on this optimal model to estimate out‐of‐sample performance and prevent overfitting
4. DISCUSSION
Although there have been many recent technical advances describing the genomic landscape of LMS, its underlying causes and pathogenesis remain to be established. Although the current classification of LMS and its mimics, including LM‐BN and other atypical smooth muscle tumors (ASMT), continue to rely on morphologic criteria, 3 diagnostic challenges remain due to: (i) interobserver variability on key histologic criteria; (ii) a lack of reliable clinical ancillary testing; and (iii) shared expression of highly relevant biomarkers. 1 This raises the question of whether LMS arises primarily as a de novo process or as a progression from previously existing ASMT or LM‐BN. As LMS is characterized by genomic instability—as evidenced by pervasive, seemingly random karyotypic abnormalities, especially for copy number changes 14 , 28 —a comparison of the genome‐wide CNAs between LMS and LM‐BN may provide insight into the tumorigenesis of these 2 tumor types. Furthermore, separating SMT‐FH from LM‐BN based on morphologic and molecular analysis would be expected to provide more homogenous tumor types and, thus, cleaner, more informative genomic data for LM‐BN than has been studied previously. 7 , 10 , 18 To our knowledge, this is the first comprehensive, high‐resolution molecular profiling in the 3 different tumor types of SMT‐FH, LM‐BN, and LMS to date.
LM‐BN were originally designated as “atypical leiomyomas with a low risk of recurrence,” 3 and several recent large case series studies have suggested that, unlike LMS, LM‐BN behave in a benign fashion with no contribution to patient mortality. 5 , 7 , 27 Both LMS and LM‐BN present morphologically with marked nuclear atypia and, although the latter typically shows nuclear atypia with a more degenerative appearance, clear distinction of the 2 tumor types based on morphologic characteristics of the nuclear atypia is not easy in all cases. 29 Of note, a case that showed progression from LM‐BN to LMS was reported recently. 10 Our group also showed previously that LM‐BN has a similar frequency of mutations with LMS in select genes, including TP53, MED12, and PTEN(1). Little information is known, however, about the genome‐wide genomic alterations seen in LM‐BN and how they compare with LMS. After histologic and IHC evaluation to exclude SMT‐FH, 13 LM‐BN samples were subjected to whole‐genome copy number analysis. Widespread genomic CNAs were observed, involving nearly all chromosomes (Figure 2), indicating chromosomal instability in LM‐BN. In particularly, LM‐BN showed frequent copy number losses in chromosome arms 16q, 17p, 17q, 19q, 22q, Xp, and Xq, as well as frequent copy number gains in chromosome arms 1q, 16p, and 19p, similar to previously published data (Figure S5). 18 The frequently observed CNAs in focal genomic regions included 7 peaks showing gains and 26 showing losses in LM‐BN (Figure 5 and Table S4).
LMS is characterized by genomic instability and frequent CNAs. 12 , 13 , 14 , 30 , 31 Similar to previous results (Figure S5), we observed multifocal, random, high‐frequency CNAs, involving nearly all chromosomes (Figure 2), with the most frequent losses seen in 10q22.1, 13q14.11, 1p36.22, 2q33.1, 22q13.1, and 11p15.3, and the most frequent gains seen in 15q11.2, 17p12, and 1q21.1. LMS tended to have large regions of CNAs, indicating that large‐scale genomic structural alterations appear to play an important role in LMS (Figure 2). This study displays a high frequency of CNAs that are present in both LM‐BN and LMS, with 37‐shared CNA peaks identified, including 8 gains and 29 losses (Figure 4B). Both LM‐BN and LMS also show large numbers of CNAs per tumor sample, particularly with copy number losses. PCA of the significant CNA foci demonstrated substantial overlap between LMS and LM‐BN (Figure 4C). Similarly, an unsupervised hierarchical cluster analysis of the same data showed the 2 tumor types branching together on dendrograms, indicating that these histologically similar entities show overlapping genomic copy number alteration, consistent with a recent study by Lehtonen and colleagues. 18
We also identified many CNAs that were commonly seen in LM‐BN, but not in LMS, such as losses of 1q21.1, 5p13.33, 11q11, 8q24.3, 15q11.2, and 20q13.33 (Figure 4 and Table S3). These CNA differences may provide a molecular basis for differentiating LM‐BN from LMS. Loss of 1p36.33 is frequently seen in LM‐BN (76.9% of cases), but is much less commonly seen in SMT‐FH and LMS (Figure 5 and Table S4). Interestingly, leiomyoma with loss of 1p, specifically loss of 1p36, was previously reported to be associated with cellular smooth muscle tumor with nuclear atypia 32 and such tumors had a gene expression profile that was more similar to LMS than to the usual‐type leiomyoma or myometrium. 32 Therefore, frequent losses of 1p36.33 in this study may be closely related to LM‐BN. 1p36.33 mainly includes cellular senescence, PI3K‐Akt, and NOD‐like receptor signaling pathways and the role of 1p36.33 loss in LM‐BN deserves further investigation. 14 , 33 The ATAD3 gene family, mapped to 1p36.33, includes ATAD3A, ATAD3B, and ATAD3C. Immunoreactivity for ATAD3 is lower to absent in LM‐BN compared with LMS (Figure 6), supporting the CNAs. ATAD3 is a tumor suppressor and a mitochondrial membrane ATPase. It is a target gene of c‐MYC, 34 and copy number gains or amplifications of ATAD3 are frequently associated with multiple tumor types probably by increasing resistance to apoptosis 35 , 36 , 37 , 38 and mitochondrial DNA integrity. 39 Our findings suggested that ATAD3 could be potentially used as a biomarker to distinguish LM‐BN and LMS. The role of ATAD3 alterations in tumorigenesis of LM‐BN warrants further investigation. AGR2 (p53 inhibitor), MT1G (promoting cell differentiation), DUSP2 (cellular proliferation and differentiation), NR4A2 (a nuclear transcription regulator), and SIK1(regulator of smooth muscle cells) are all tumor suppressor genes and are differentially expressed among the 3 tumor types (Figure 6). Due to the characteristic and differential expression patterns of these novel biomarkers as shown by immunohistochemistry, they have the potential to be diagnostically useful when used in combination to distinguish LM‐BN and LMS (Figure 6B). However, assessing the true clinical utility of these IHC biomarkers, as used by pathologists in clinical practice, requires assessment of whether manual scoring of the biomarkers would perform as well as the automated scoring utilized in our analysis; this remains an important question for future research studies.
SMT‐FH can be readily diagnosed by its histology in conjunction with IHC or CNA analysis. The significant finding was the homozygous or heterozygous loss of chromosome 1q43‐44 in 100% (13/13) of SMT‐FH, but not in LM‐BN and LMS. This region contains FH, which is the primary driver gene for SMT‐FH development. FH is one of the key enzymes in the Krebs tricarboxylic acid cycle and catalyzes the conversion of fumarate to malate. 40 Sporadic SMT‐FH is mostly caused by somatic FH alteration, 41 , 42 , 43 , 44 and about one‐quarter of them are caused by FH gene mutations. 11 , 45 Genomic deletion of FH in tumors has been detected by FISH 46 and CGH. 18 A recent study 18 reported copy number loss at the FH locus in 30.8% of LM‐BN, however this study did not separate SMT‐FH from LM‐BN. Our findings suggested that sporadic SMT‐FH is mainly associated with deletion of the FH genomic region and that point mutations account for a relatively small fraction of SMT‐FH. In addition to the 1q43‐44 deletion, SMT‐FH presents with a much lower global frequency of CNAs than LM‐BN and LMS.
In summary, to our knowledge, this is the first comprehensive analysis of genome‐wide CNAs in 3 distinct uterine smooth muscle tumor types with marked nuclear atypia. SMT‐FH showed characteristic genomic alterations leading to the loss of FH. Both LM‐BN and LMS showed a high level of chromosomal instability and significant overlap in recurrent loci of copy number losses, suggesting that these 2 tumor types may be closely related and have overlapping pathogenesis. Finally, this study suggests that ATAD3, and several other novel biomarkers, may be potentially useful surrogate markers to help distinguish LM‐BN and LMS. Future studies will focus on the mechanisms behind the observed genomic instability in LM‐BN and LMS.
DISCLOSURE
Authors have nothing to disclose.
Supporting information
Fig S1‐S6
Table S1‐S5
ACKNOWLEDGMENTS
We thank the technical support of Northwestern Pathology Core Facility and Sequence Core. We also thank Dr. Mandina Sukhanova from clinical cytogenetic laboratory at Northwestern University for her technical support. This work is partially supported by Friend of Prentice. Part of this work was presented at the 109th United States and Canadian Academy of Pathology (USCAP) annual meeting in Los Angeles, CA, USA.
Gao T, Finkelman BS, Ban Y, et al. Integrated histologic and molecular analysis of uterine leiomyosarcoma and 2 benign variants with nuclear atypia. Cancer Sci. 2021;112:2046–2059. 10.1111/cas.14775
Contributor Information
Chunfang Ha, Email: hachunfang@gmail.com.
Jian‐Jun Wei, Email: jianjun-wei@northwestern.edu.
REFERENCES
- 1. Zhang Q, Ubago J, Li LI, et al. Molecular analyses of 6 different types of uterine smooth muscle tumors: emphasis in atypical leiomyoma. Cancer. 2014;120(20):3165‐3177. [DOI] [PubMed] [Google Scholar]
- 2. Mas A, Alonso R, Garrido‐Gómez T, et al. The differential diagnoses of uterine leiomyomas and leiomyosarcomas using DNA and RNA sequencing. Am J Obstet Gynecol. 2019;221(4):320.e1‐320.e23. [DOI] [PubMed] [Google Scholar]
- 3. Bell SW, Kempson RL, Hendrickson MR. Problematic uterine smooth muscle neoplasms. A clinicopathologic study of 213 cases. Am J Surg Pathol. 1994;18(6):535‐558. [PubMed] [Google Scholar]
- 4. Mills AM, Ly A, Balzer BL, et al. Cell cycle regulatory markers in uterine atypical leiomyoma and leiomyosarcoma: immunohistochemical study of 68 cases with clinical follow‐up. Am J Surg Pathol. 2013;37(5):634‐642. [DOI] [PubMed] [Google Scholar]
- 5. Croce S, Young RH, Oliva E. Uterine leiomyomas with bizarre nuclei: a clinicopathologic study of 59 cases. Am J Surg Pathol. 2014;38(10):1330‐1339. [DOI] [PubMed] [Google Scholar]
- 6. Abeler VM, Røyne O, Thoresen S, Danielsen HE, Nesland JM, Kristensen GB. Uterine sarcomas in Norway. A histopathological and prognostic survey of a total population. Histopathology. 2009;54(3):355‐364.from 1970 to 2000 including 419 patients. [DOI] [PubMed] [Google Scholar]
- 7. Ubago JM, Zhang Q, Kim JJ, Kong B, Wei JJ. Two subtypes of atypical leiomyoma: clinical, histologic, and molecular analysis. Am J Surg Pathol. 2016;40(7):923‐933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Reyes C, Karamurzin Y, Frizzell N, et al. Uterine smooth muscle tumors with features suggesting fumarate hydratase aberration: detailed morphologic analysis and correlation with S‐(2‐succino)‐cysteine immunohistochemistry. Mod Pathol. 2014;27(7):1020‐1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bennett JA, Weigelt B, Chiang S, et al. Leiomyoma with bizarre nuclei: a morphological, immunohistochemical and molecular analysis of 31 cases. Mod Pathol. 2017;30(10):1476‐1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gregova M, Hojny J, Nemejcova K, et al. Leiomyoma with bizarre nuclei: a study of 108 cases focusing on clinicopathological features, morphology, and fumarate hydratase alterations. Pathol Oncol Res. 2019;26(3):1527‐1537. [DOI] [PubMed] [Google Scholar]
- 11. Zhang Q, Poropatich K, Ubago J, et al. Fumarate hydratase mutations and alterations in leiomyoma with bizarre nuclei. Int J Gynecol Pathol. 2018;37(5):421‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cuppens T, Moisse M, Depreeuw J, et al. Integrated genome analysis of uterine leiomyosarcoma to identify novel driver genes and targetable pathways. Int J Cancer. 2018;142(6):1230‐1243. [DOI] [PubMed] [Google Scholar]
- 13. Chudasama P, Mughal SS, Sanders MA, et al. Integrative genomic and transcriptomic analysis of leiomyosarcoma. Nat Commun. 2018;9(1):1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Abeshouse A, Adebamowo C, Adebamowo SN, et al. Comprehensive and integrated genomic characterization of adult soft tissue sarcomas. Cell. 2017;171(4):950‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Raish M, Khurshid M, Ansari MA, et al. Analysis of molecular cytogenetic alterations in uterine leiomyosarcoma by array‐based comparative genomic hybridization. J Cancer Res Clin Oncol. 2012;138(7):1173‐1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hu J, Khanna V, Jones M, Surti U. Genomic alterations in uterine leiomyosarcomas: potential markers for clinical diagnosis and prognosis. Genes Chromosomes Cancer. 2001;31(2):117‐124. [DOI] [PubMed] [Google Scholar]
- 17. Levy B, Mukherjee T, Hirschhorn K. Molecular cytogenetic analysis of uterine leiomyoma and leiomyosarcoma by comparative genomic hybridization. Cancer Genet Cytogenet. 2000;121(1):1‐8. [DOI] [PubMed] [Google Scholar]
- 18. Liegl‐Atzwanger B, Heitzer E, Flicker K, et al. Exploring chromosomal abnormalities and genetic changes in uterine smooth muscle tumors. Mod Pathol. 2016;29(10):1262‐1277. [DOI] [PubMed] [Google Scholar]
- 19. Mermel CH, Schumacher SE, Hill B, et al. 0 facilitates sensitive and confident localization of the targets of focal somatic copy‐number alteration in human cancers. Genome Biol. 2011;12(4):R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dugo M, Devecchi A, De Cecco L, et al. Focal recurrent copy number alterations characterize disease relapse in high grade serous ovarian cancer patients with good clinical prognosis: a pilot study. Genes. 2019;10(9):678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Metsalu T, Vilo J. ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015;43(W1):W566‐W570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bertsch E, Qiang W, Zhang Q, et al. MED12 and HMGA2 mutations: 2 independent genetic events in uterine leiomyoma and leiomyosarcoma. Mod Pathol. 2014;27(8):1144‐1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Varghese F, Bukhari AB, Malhotra R, De A. IHC Profiler: an open source plugin for the quantitative evaluation and automated scoring of immunohistochemistry images of human tissue samples. PLoS One. 2014;9(5):e96801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tibshirani R. Regression shrinkage and selection via the lasso. J R Stat Soc. 1996;58:267‐288. Series B. [Google Scholar]
- 25. Robin X, Turck N, Hainard A, et al. pROC: an open‐source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics. 2011;12:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw. 2010;33(1):1‐22. [PMC free article] [PubMed] [Google Scholar]
- 27. Ly A, Mills AM, McKenney JK, et al. Atypical leiomyomas of the uterus: a clinicopathologic study of 51 cases. Am J Surg Pathol. 2013;37(5):643‐649. [DOI] [PubMed] [Google Scholar]
- 28. Salawu A, Ul‐Hassan A, Hammond D, Fernando M, Reed M, Sisley K. High quality genomic copy number data from archival formalin‐fixed paraffin‐embedded leiomyosarcoma: optimisation of universal linkage system labelling. PLoS One. 2012;7(11):e50415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kefeli M, Caliskan S, Kurtoglu E, Yildiz L, Kokcu A. Leiomyoma with bizarre nuclei: clinical and pathologic features of 30 patients. Int J Gynecol Pathol. 2018;37(4):379‐387. [DOI] [PubMed] [Google Scholar]
- 30. Hemming ML, Klega K, Rhoades J, et al. Detection of circulating tumor DNA in patients with leiomyosarcoma with progressive disease. JCO Precis Oncol. 2019;3:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Silveira SM, Villacis RAR, Marchi FA, et al. Genomic signatures predict poor outcome in undifferentiated pleomorphic sarcomas and leiomyosarcomas. PLoS One. 2013;8(6):e67643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Christacos NC, Quade BJ, Cin PD, Morton CC. Uterine leiomyomata with deletions of Ip represent a distinct cytogenetic subgroup associated with unusual histologic features. Genes Chromosom Cancer. 2006;45(3):304‐312. [DOI] [PubMed] [Google Scholar]
- 33. Gibault L, Ferreira C, Pérot G, et al. From PTEN loss of expression to RICTOR role in smooth muscle differentiation: complex involvement of the mTOR pathway in leiomyosarcomas and pleomorphic sarcomas. Mod Pathol. 2012;25(2):197‐211. [DOI] [PubMed] [Google Scholar]
- 34. Zeller KI, Jegga AG, Aronow BJ, O'Donnell KA, Dang CV. An integrated database of genes responsive to the Myc oncogenic transcription factor: identification of direct genomic targets. Genome Biol. 2003;4(10):R69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hubstenberger A, Labourdette G, Baudier J, Rousseau D. ATAD 3A and ATAD 3B are distal 1p‐located genes differentially expressed in human glioma cell lines and present in vitro anti‐oncogenic and chemoresistant properties. Exp Cell Res. 2008;314(15):2870‐2883. [DOI] [PubMed] [Google Scholar]
- 36. Schaffrik M, Mack B, Matthias C, Rauch J, Gires O. Molecular characterization of the tumor‐associated antigen AAA‐TOB3. Cell Mol Life Sci. 2006;63(18):2162‐2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fang H‐Y, Chang C‐L, Hsu S‐H, et al. ATPase family AAA domain‐containing 3A is a novel anti‐apoptotic factor in lung adenocarcinoma cells. J Cell Sci. 2010;123(7):1171‐1180. [DOI] [PubMed] [Google Scholar]
- 38. Chen T‐C, Hung Y‐C, Lin T‐Y, et al. Human papillomavirus infection and expression of ATPase family AAA domain containing 3A, a novel anti‐autophagy factor, in uterine cervical cancer. Int J Mol Med. 2011;28(5):689‐696. [DOI] [PubMed] [Google Scholar]
- 39. He J, Mao C‐C, Reyes A, et al. The AAA+ protein ATAD3 has displacement loop binding properties and is involved in mitochondrial nucleoid organization. J Cell Biol. 2007;176(2):141‐146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Eng C, Kiuru M, Fernandez MJ, Aaltonen LA. A role for mitochondrial enzymes in inherited neoplasia and beyond. Nat Rev Cancer. 2003;3(3):193‐202. [DOI] [PubMed] [Google Scholar]
- 41. Joseph NM, Solomon DA, Frizzell N, Rabban JT, Zaloudek C, Garg K. Morphology and immunohistochemistry for 2SC and FH aid in detection of fumarate hydratase gene aberrations in uterine leiomyomas from young patients. Am J Surg Pathol. 2015;39(11):1529‐1539. [DOI] [PubMed] [Google Scholar]
- 42. Harrison WJ, Andrici J, Maclean F, et al. Fumarate hydratase–deficient uterine leiomyomas occur in both the syndromic and sporadic settings. Am J Surg Pathol. 2016;40(5):599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Barker KT, Spendlove HE, Banu NS, et al. No evidence for epigenetic inactivation of fumarate hydratase in leiomyomas and leiomyosarcomas. Cancer Lett. 2006;235(1):136‐140. [DOI] [PubMed] [Google Scholar]
- 44. Barker KT, Bevan S, Wang R, et al. Low frequency of somatic mutations in the FH/multiple cutaneous leiomyomatosis gene in sporadic leiomyosarcomas and uterine leiomyomas. Br J Cancer. 2002;87(4):446‐448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bayley J‐P, Launonen V, Tomlinson IP. The FH mutation database: an online database of fumarate hydratase mutations involved in the MCUL (HLRCC) tumor syndrome and congenital fumarase deficiency. BMC Med Genet. 2008;9(1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gross KL, Panhuysen CIM, Kleinman MS, et al. Involvement of fumarate hydratase in nonsyndromic uterine leiomyomas: genetic linkage analysis and FISH studies. Genes Chromosomes Cancer. 2004;41(3):183‐190. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S6
Table S1‐S5