

Abstract
Besides insulin‐mediated transport of glucose into the cells, an important role is also played by the non‐insulin‐mediated transport. This latter process is called glucose effectiveness (acronym SG), which is estimated by modeling of glucose and insulin data after an intravenous glucose administration, and accounts for ≈70% of glucose disposal. This review summarizes studies on SG, mainly in humans and rodents with focus on results achieved in model experiments in mice. In humans, SG is reduced in type 2 diabetes, in obesity, in liver cirrhosis and in some elderly populations. In model experiments in mice, SG is independent from glucose levels, but increases when insulin secretion is stimulated, such as after administration of the incretin hormones, glucagon‐like peptide‐1 and glucose‐dependent insulinotropic polypeptide. SG is reduced in insulin resistance induced by high‐fat feeding and by exogenous administration of glucagon. Glucose‐dependent (insulin‐independent) glucose disposal is therefore important for glucose elimination, and it is also well regulated. It might be of pathophysiological relevance for the development of type 2 diabetes, in particular during insulin resistance, and might also be a target for glucose‐reducing therapy. Measuring SG is essentially important when carrying out metabolic studies to understand glucose homeostasis.
Keywords: Glucose disposal, Glucose effectiveness, Mathematical modeling
Besides insulin‐mediated transport of glucose into the cells, an important role is also played by the non‐insulin‐mediated transport which is called glucose effectiveness (acronym SG). This process accounts for ≈70% of glucose disposal, is reduced in type 2 diabetes and obesity, and increased by incretin hormones. This review summarizes studies on SG, mainly in humans and mice with focus on results achieved in model experiments in mice, and concludes that glucose effectiveness may be of pathophysiological relevance for development of type 2 diabetes, in particular during insulin resistance, and may also be a target for glucose‐reducing therapy.

INTRODUCTION
A major mechanism for glucose disappearance from the circulation is insulin‐mediated transport into the cells. However, as shown >80 years ago, there is also a non‐insulin‐dependent process that is mediated by glucose itself to enhance its uptake and metabolism 1 . This was confirmed >40 years ago, when the minimal modeling of glucose and insulin data from an intravenous glucose tolerance test (IVGTT) showed that non‐insulin‐mediated processes play a major role in glucose disappearance; these processes were described by the term “glucose effectiveness” 2 . The aim of the present review was to elucidate the relevance of glucose effectiveness for glucose disappearance under various physiological and pathophysiological conditions. Understanding the regulation of glucose effectiveness might also have potential therapeutic benefits for glucose‐lowering attempts in type 2 diabetes. We have therefore reviewed the clinical studies reporting glucose effectiveness as estimated from IVGTT, and we have also retrospectively analyzed changes of glucose effectiveness in multiple different conditions in mice, where a series of IVGTTs have been carried out under standardized conditions.
HISTORY AND DEFINITION
The history of glucose effectiveness goes back to the late 1970s, when Bergman et al. 2 formulated the equation system of the minimal model to describe glucose disappearance during an intravenous glucose administration in dogs. They then found that it was not possible to describe glucose disappearance only with the contribution of insulin 2 . Instead, a parameter describing the insulin‐independent mechanism was necessarily introduced. This parameter was termed “glucose effectiveness” (p1), although no specific discussion on p1 appeared in this first paper, which was focused on insulin sensitivity (SI). The existence of a non‐insulin‐dependent glucose disposal was also shown in the first study in humans with the minimal model, where again the parameter p1 was termed “glucose effectiveness” 3 . Similarly, in a study of glucose uptake in the absence of a sustained insulin response, it was observed that hyperglycemia increases glucose uptake, further suggesting an insulin‐independent glucose‐dependent glucose uptake in humans 4 .
Glucose effectiveness is today referred to as the ability of glucose per se to suppress endogenous glucose production and stimulate peripheral glucose uptake, as was elegantly shown in dogs by Ader et al. 5 The acronym for glucose effectiveness that we use today (SG) was first used in a human study in 1985, where it was stated that “SG (formerly p1) [is] the insulin‐independent fractional glucose disappearance” 6 . In the classic review by Bergman et al. 7 of the same year that canonized the minimal model approach as a reliable method to assess insulin sensitivity, parameter p1 was still used in the equations, but it was stated that p1 is SG, defined as a “measure of the effect of glucose to enhance its own disappearance [within the extracellular glucose pool] at basal insulin, independent of any increase in insulin”. In subsequent years, papers exploiting the minimal model have also reported glucose effectiveness, either as p1 8 , 9 or as SG 10 , 11 , 12 , or only mentioned SG without discussing it 13 . Glucose effectiveness has also been estimated with combined eu‐ and hyperglycemic clamp 14 , with similar conclusions as achieved by the minimal modeling approach, as recently was reviewed 15 .
GLUCOSE EFFECTIVENESS AND CLINICAL CONDITIONS
As glucose effectiveness is the “ability of glucose per se without any change in insulin to disappear from blood” 5 , 16 , it quantifies the fractional rate (min–1) of glucose utilization in the brain, central nervous system, red blood cells and other insulin‐independent tissues/organs, such as kidneys. Renal excretion of glucose, which is an insulin‐independent process, also contributes to SG. Glucose effectiveness is calculated from a minimal model of insulin and glucose data after an IVGTT 17 , 18 . The model assumes a first‐order non‐linear insulin‐controlled kinetic, and accounts for the effect of insulin and glucose itself on glucose disappearance after exogenous glucose injection. The model provides two parameters: SI, which is defined as the ability of insulin to enhance glucose disappearance and inhibit glucose production (i.e., insulin sensitivity), and SG, representing glucose disappearance from plasma without any change in dynamic insulin 3 , 17 , 18 . The mathematical procedure for minimal model parameters (thus SG) is explained in detail previous studies17,18, which show how SG is estimated through a series of mathematical steps when the model differential equations are applied to a set of IVGTT data.
Several early studies documented the large contribution of this insulin‐independent glucose disposal to overall glucose disposal in humans 19 , 20 , which was continuously appreciated 21 , 22 , even recently 15 , 23 . Several studies also examined SG in various clinical conditions. Table 1 summarizes many of these studies. Studies have thus shown that SG is reduced in obesity 24 , type 2 diabetes 9 , 25 , 26 , gestational diabetes 27 , liver cirrhosis 28 and USA older adults 29 , 30 , whereas SG is increased in growth hormone deficiency 31 and after administration of glucagon‐like peptide‐1 (GLP‐1) 32 , 33 , 34 . In contrast, SG is not changed in impaired glucose tolerance 20 or by treatment with thiazolidinediones 35 ; the GLP‐1 receptor agonist, liraglutide 36 ; or the dipeptidyl peptidase‐4 (DPP‐4) inhibitor, vildagliptin 37 , in type 2 diabetes patients; or after carbohydrate dieting in USA older adults 11 or in Italian older adults with a normal oral glucose test 12 . These studies have been undertaken mainly in white people, but the result that SG is reduced in type 2 diabetes has also been reported in Malaysian 38 , Japanese 39 and Chinese people 40 , whereas in contrast, similar SG in type 2 diabetes patients as controls has been reported in African Americans 41 and Ghanaians 42 . Therefore, different ethnic groups might show differences in the impact on type 2 diabetes by SG. However, in impaired glucose tolerance, SG was found to be lower than in controls in Japanese people 43 , but not reduced in white people 20 or in African Americans 41 . These differences are of interest on the background that type 2 diabetes in Asian people is primarily characterized by impaired insulin secretion rather than an interplay between insulin resistance and failed islet compensation 44 . The finding of reduced SG in individuals with impaired glucose tolerance among Japanese individuals 43 would suggest that reduced glucose effectiveness contributes to diabetes development in these patients, and this is supported by the results of a study showing reduced SG in the offspring of Japanese patients with type 2 diabetes even at normal glucose tolerance 45 . However, to study whether the contribution by SG to the development of type 2 diabetes is different in ethnic groups, direct comparisons need to be carried out in individual studies. One such study has compared SG in two different ethnic groups (Mexican Americans and non‐Hispanic white Americans) showing no difference 46 . However, more studies are required for examining SG in other ethnic groups.
Table 1.
Glucose effectiveness in various clinical studies
| Studies | Comparisons | SG (No. participants) | Reference |
|---|---|---|---|
| Obesity |
Lean Obese |
0.030 ± 0.003 (18) 0.016 ± 0.002 (18)* |
24 |
| Type 2 diabetes |
Type 2 diabetes Controls |
0.014 ± 0.002 0.024 ± 0.003* |
25 |
| Type 2 diabetes |
Type 2 diabetes Controls |
0.016 ± 0.009 (25) 0.023 ± 0.012 (130)* |
26 |
| Gestational diabetes |
GDM NGT |
0.022 ± 0.002 (10) 0.021 ± 0.003 (9) |
27 |
| Cirrhosis |
Cirrhosis Controls |
0.015 ± 0.002 (9) 0.024 ± 0.003 (6) |
28 |
| Aging |
Mean 65 years Mean 20 years |
0.017 ± 0.002 (20) 0.025 ± 0.002 (20) |
29 |
| Aging |
Young men (aged 18–36 years) Elderly men (65–82 years) |
0.029 ± 0.005 (8) 0.031 ± 0.004 (10) |
30 |
| GH administration in GH deficiency |
Controls GH deficiency GH administration in GH deficiency |
0.020 ± 0.003 (8) 0.010 ± 0.001 (8) * 0.015 ± 0.001 (8) * |
31 |
| GLP‐1 administration in healthy individuals |
Controls GLP‐1 |
0.018 ± 0.001 (6) 0.026 ± 0.003 (6) |
32 |
| GLP‐1 administration in healthy individuals |
Controls GLP‐1 |
0.018 ± 0.002 (17) 0.025 ± 0.002 (17) |
33 |
| GLP‐1 administration in healthy individuals |
Controls GLP‐17–36NH2 GLP‐17.37 GLP‐19–36NH2 |
0.018 ± 0.002 (10) 0.025 ± 0.003 (10) * 0.024 ± 0.002 (10) * 0.018 ± 0.002 (10) |
34 |
| Women with IGT |
IGT NGT |
0.019 ± 0.003 (10) 0.020 ± 0.003 (10) |
20 |
| Treatment with TZD of women at high risk for type 2 diabetes |
Women with recent GDM and IGT After 12 weeks TZD treatment |
0.014 ± 0.003 (14) 0.015 ± 0.004 (14) |
35 |
| Treatment with liraglutide in type 2 diabetes |
Placebo Liraglutide |
Change 0.0008 (–0.003, 0.006) Change 0.0016 (–0.0005, 0.006) |
36 |
| Treatment with vildagliptin in type 2 diabetes |
Placebo Vildagliptin |
0.018 ± 0.002 (14) 0.019 ± 0.002 (14) |
37 |
| Carbohydrate diet |
Young men (18–36 years) Elderly men (65–82 years) |
0.029 ± 0.005 (8) 0.027 ± 0.004 (10) |
11 |
| Type 2 diabetes in Malaysians |
Type 2 diabetes Controls |
0.012 ± 0.005 0.025 ± 0.001* |
38 |
| Type 2 diabetes in Japanese people |
Type 2 diabetes Controls (offspring) |
0.011 ± 0.003 (9) 0.024 ± 0.003 (11)* |
39 |
| Type 2 diabetes in Chinese people |
Insulin sensitive type 2 diabetes Insulin resistant type 2 diabetes |
0.013 ± 0.008 (71) 0.016 ± 0.009 (51)* |
40 |
| Type 2 diabetes and IGT in African Americans |
NGT IGT Type 2 diabetes |
0.029 ± 0.002 (101) 0.025 ± 0.002 (36) 0.024 ± 0.002 (17) |
41 |
| Type 2 diabetes in Ghanaians |
Type 2 diabetes Controls |
0.023 ± 0.005 (10) 0.027 ± 0.004 (15) |
42 |
| IGT in Japanese people |
NGT Insulin‐resistant IGT Insulin sensitive IGT |
0.023 ± 0.002 (15) 0.016 ± 0.002 (6)* 0.013 ± 0.002 (9)* |
43 |
| Offspring to Japanese patients with type 2 diabetes |
Offspring Controls |
0.016 ± 0.003 (10) 0.023±0.002 (10)* |
45 |
| Ethnic groups |
Mexican Americans Non‐Hispanic whites |
0.022 ± 0.002 (10) 0.026 ± 0.008 (11) |
46 |
Values are the mean ± standard error or median (95% confidence intervals). *Significant differences between the groups (P < 0.05). GDM, gestational diabetes mellitus; GH, growth hormone; GLP‐1, glucagon‐like peptide‐1; IGT, impaired glucose tolerance; NGT, normal glucose tolerance; TZD, thiazolidinedione.
APPROACH TO STUDY GLUCOSE EFFECTIVENESS IN MICE
To study the physiological and pathophysiological meaning of glucose effectiveness and its mechanism of action, in the 1990s we adapted the minimal model to standardized mouse experiments 18 , 47 . This allowed more detailed studies on physiology and regulation of SG, and the knowledge of SG has therefore been expanded during the past decades. When translated for studies that use the minimal model in mice, the following protocol has been used: after a 5‐h fast during the late morning hours, mice (most often from the NMRI or C57BL/6J strain) are anesthetized with an intraperitoneal injection of a fixed‐dose combination of fentanyl (0.02 mg/mouse)–fluanisone (0.5 mg/mouse) and midazolam (0.125 mg/ mouse). After 30 min, a blood sample (40 µL) is taken from the retrobulbar, intraorbital sinus capillary plexus in pipette tubes that have been pre‐rinsed in heparin solution (100 U/mL in 0.9% NaCl). Thereafter, mice are given an intravenous bolus dose of glucose (dissolved in saline) over a period of 3 s in a tail vein, and whole blood is sampled as aforementioned at 1, 5, 10, 20, 30 and 50 min after glucose injection. Glucose is detected in whole blood, and plasma is immediately separated after collection and stored at –20°C until analysis for insulin. Regarding the possible influence on the estimation of SG during IVGTT of the renal glucose excretion, it is worth noting that the peak glucose levels after the injection could be above the kidney glucose threshold. However, although it is known that the renal glucose threshold for mice is ≈22 mmol/L 48 , such high values are rarely seen after the standard glucose injection or are observed for only a very short period of time after the glucose challenge, and therefore, it is likely that the contribution of this process to SG is negligible.
With this technique, SG has been shown to be approximately 0.050 min–1 in normal mice, with a standard error of the mean of 0.006 47 . This is equivalent to a glucose disposal of 5% of the extracellular glucose pool per min by glucose‐dependent insulin‐independent mechanisms. This value is higher than the 0.021 min–1 reported in humans 22 and 0.028 min–1 in dogs 5 , but comparable to the reported values in rats, which range from ≈0.040 min–1 in obese Zucker rats to ≈0.053 min–1 in lean Zucker rats 49 , and ≈0.070 min–1 in Long‐Evans rats 50 and ≈0.090 min–1 in endurance‐trained animals 51 . Furthermore, several studies have been carried out to understand the factors that might regulate SG in mice, as it is summarized in Table 2. The incretin hormones, GLP‐1 and glucose‐dependent insulinotropic polypeptide (GIP), as well as exogenous administration of insulin, increase SG; whereas SG is not significantly affected by the neuropeptide, pituitary adenylate cyclase activating polypeptide, or in gastrin‐releasing peptide knockout mice, and reduced by glucagon, in GLP‐1 receptor knockout mice, in high‐fat fed and insulin‐resistant mice, as well as after inhibition of insulin secretion 47 , 52 , 53 , 54 , 55 , 56 , 57 . These data thus show that SG is a process exposed to a complex regulation, and suggest that SG might contribute to changes in glucose tolerance under a number of different conditions.
Table 2.
Glucose effectiveness in mouse experiments
| Studies | Comparisons | SG (No. animals) | Reference |
|---|---|---|---|
| GIP receptor knockout |
GIP receptor knockout Controls |
0.061 ± 0.004 (26) 0.057 ± 0.005 (30) |
52 |
| GLP‐1 receptor knockout |
GLP‐1 receptor knockout Controls |
0.027 ± 0.004 (17)* 0.044 ± 0.005 (17) |
52 |
| Incretin hormones |
GIP GLP‐1 Controls |
0.072 ± 0.004 (40)* 0.066 ± 0.005 (47)* 0.045 ± 0.003 (106) |
53 |
| GRP receptor knockout |
GRP receptor knockout Controls |
0.052 ± 0.007 (50) 0.038 ± 0.004 (50) |
54 |
| High‐fat feeding |
High‐fat feeding for 10 months Controls |
0.030 ± 0.004 (24)* 0.056 ± 0.006 (23) |
55 |
| Effect of insulin |
Insulin administration Blocking of insulin secretion Controls |
0.075 ± 0.004 (48)* 0.014 ± 0.002 (24)* 0.050 ± 0.002 (202) |
47 |
| PACAP‐27 |
PACAP‐27 Controls |
0.041 ± 0.005 (16) 0.040 ± 0.006 (16) |
56 |
| PACAP‐38 |
PACAP‐38 Controls |
0.057 ± 0.008 (24) 0.043 ± 0.006 (24) |
56 |
| Glucagon |
Glucagon (10 nmol/kg) Controls |
0.038 ± 0.004 (24) 0.058 ± 0.005 (135) |
57 |
| GLP‐1 |
GLP‐1 (3.0 nmol/kg) Controls |
0.066 ± 0.005 (47)* 0.045 ± 0.003 (106) |
53 |
| GIP |
GIP (3.0 nmol/kg) Controls |
0.072 ± 0.004 (40)* 0.045 ± 0.003 (106) |
53 |
*Significant differences between the groups (P < 0.05). GIP, glucose‐dependent insulinotropic polypeptide; GLP‐1, glucagon‐like peptide‐1; GRP, gastrin releasing peptide; PACAP, pituitary adenylate cyclase activating polypeptide.
CONTRIBUTION BY GLUCOSE EFFECTIVENESS TO GLUCOSE DISAPPEARANCE
The importance of SG on glucose tolerance was proposed in a study by Best et al. 19 from analyzing the linear regression between intravenous glucose elimination rate, KG, and SG. That study showed that insulin‐independent glucose uptake contributes by ≈72% to glucose disappearance, indicating that it is the major determinant of intravenous glucose tolerance. This evidence confirmed what was previously shown in dogs 5 , where it emerged that SG contributes by 70–80% to glucose disappearance, later further corroborated by Ader et al. 16 .
In normal mice, we reached a similar conclusion of a large contribution by glucose effectiveness to glucose disposal with a complex study exploiting IVGTT and glucose clamp 47 . We used sensitivity analysis, which provides estimates of changes of a dependent variable (KG) for a unit change of independent variables, and accurately describes in quantitative terms the relationships among those variables 47 , 58 . Some requirements, however, had to be fulfilled for a correct use of this method. First, we showed that SG is independent from both insulin and SI in the model. Also, we considered that the total contribution to the net glucose disappearance was ascribed to SG when insulin did not change. With these assumptions, we showed that insulin (through secretion and effect) contributes to glucose tolerance by 29 ± 6% in normal conditions (Figure 1). Therefore, we confirmed that insulin‐independent mechanisms; that is, SG, contributes by more than two‐thirds to glucose disappearance.
Figure 1.
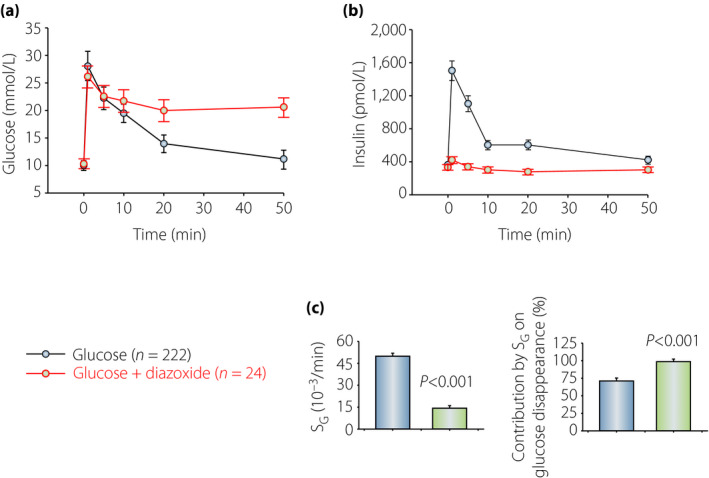
(a,b) Glucose and insulin concentrations before and after intravenous injection of glucose (1 g/kg) with or without diazoxide (25 mg/kg) in NMRI mice. (c) Glucose effectiveness (SG) and the relative contribution by SG on glucose disappearance in the two groups. The mean ± standard error of the mean is shown for glucose and insulin data, and for SG, and the mean ± standard deviation for the contribution. Data from experiments reported in Pacini et al.47 SG, glucose effectiveness.
We also studied SG when insulin secretion had been completely blocked and therefore no change in dynamic insulin is possible. This was achieved by the drug, diazoxide, which completely inhibits insulin secretion through a direct effect on the β‐cells 59 ; in mice, it was not possible to use somatostatin, as it never completely inhibited insulin secretion 47 . Diazoxide was administered subcutaneously to mice at the dose of 25 mg/kg 10 min before the intravenous administration of glucose 47 . This resulted in complete inhibition of insulin secretion, but yet an efficient glucose disappearance persisted. Figure 1 shows the results. It is seen that glucose disposal was impaired, but not absent, during diazoxide (in red), although the insulin response was totally inhibited (Figure 1b). SG contributed by approximately 75% to glucose disposal without diazoxide (Figure 1c). Therefore, elimination of insulin secretion during the intravenous glucose challenge resulted in impairment of glucose disposal by just ≈30%, which verified that insulin‐independent mechanisms are quantitatively more important than insulin‐dependent mechanisms for glucose disposal 47 , 58 .
RELATIONSHIP BETWEEN GLUCOSE EFFECTIVENESS AND INSULIN
SG is estimated as the insulin‐independent glucose disposal, and should therefore be independent from insulin. However, under certain conditions, there is a relationship between SG and the insulin secretory function. We verified this by showing that SG is reduced when insulin secretion is blocked by diazoxide 47 . This could suggest either that SG is overestimated by the minimal model (as SG during diazoxide should theoretically estimate the “true” SG) or that SG also requires insulin, even though its dynamics are not dependent on changes in insulin. However, when correlating SG with the area under the insulin curves (AUCinsulin) in studies with a wide span of insulin concentrations, no correlation was observed, except for extremely elevated values of peak insulin when SG was reduced, perhaps as a protection against hypoglycemia 47 . These results suggest that basal insulin and SG synergistically cooperate such that an increase in insulin during IVGTT is required for SG and, furthermore, that at extremely high insulin levels, SG is reduced.
RELATIONSHIP BETWEEN GLUCOSE EFFECTIVENESS AND GLUCOSE
To evaluate whether the estimation of SG is affected by the prevailing glycemia, we collected a series of IVGTT experiments carried out in 83 normal mice (glucose dose 0.35 g/kg)53. The total AUCglucose ranged from 380 to 880 mol/L·min in 50 min (averaging 555 ± 11 mol/L·min), and the mean peak (1 min) value of glucose was 17 ± 0.3 mmol/L. The mean SG was 0.045 ± 0.003 min–1, and did not correlate with either AUCglucose (R 2 = 0.0003; P > 0.5) or the peak glucose (R 2 = 10−5; P > 0.1). This shows that the estimation of SG is independent of glucose levels reached during the tests. This is also evident from a novel ad hoc series of experiments with IVGTT with two different doses of glucose in mice. Mice were anesthetized as explained above, and injected intravenously with glucose at either 0.35 g/kg (low dose; n = 17) or at 0.75 g/kg (high dose; n = 16), which yield extremely different glucose levels; samples were taken with the usual protocol, and SG estimated from glucose and insulin data. The results are reported in Figure 2. It is evident that the estimation of SG is independent of the glucose levels reached during the test: SG was 0.053 ± 0.003 min–1 at the glucose dose of 0.35 g/kg, and 0.057 ± 0.004 min–1 at 0.75 g/kg (not significantly different; P = 0.47). Hence, levels of circulating glucose do not affect the assessment of SG.
Figure 2(a,b).
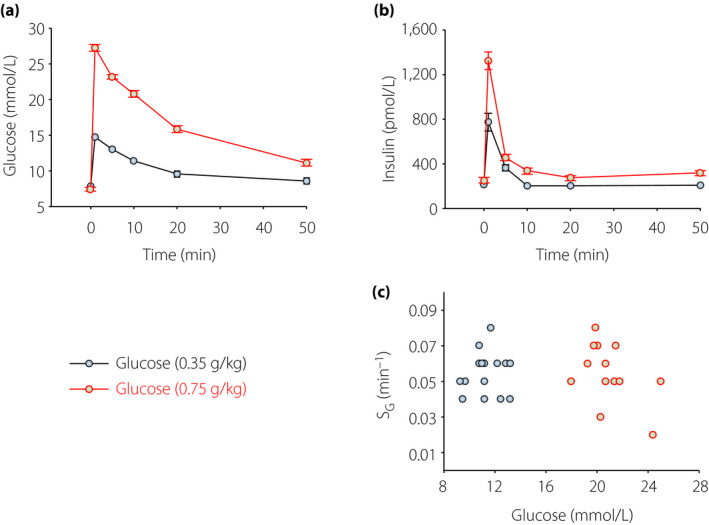
Glucose and insulin concentrations before and after intravenous administration of glucose at 0.35 g/kg (n = 17) or 0.75 g/kg (n = 16) in C57BL/6J mice. The mean ± standard error of the mean is shown. (c) Glucose effectiveness (SG) versus 1‐min glucose level after injection in individual mice in the two groups.
RELATIONSHIP BETWEEN GLUCOSE EFFECTIVENESS AND INSULIN RESISTANCE
Elevated insulin is a characteristic of insulin resistance. In humans, insulin resistance in obesity 24 , liver cirrhosis 28 and pregnancy with or without gestational diabetes 27 are associated with a 30–50% reduction in SG. Therefore, it has been of interest to deeply evaluate the role of SG in insulin resistance; that is, if either SG follows the pattern of insulin sensitivity or is increased in insulin resistance to augment glucose uptake. To study this, we used mice given a high‐fat diet for 10 months 55 . In this model, bodyweight is increased, along with a reduction in insulin sensitivity and an adaptive increase in insulin secretion; nevertheless, glucose disposal is reduced 60 . We carried out IVGTT at 1 week, and 1, 3 and 10 months after initiation of a high‐fat diet 55 . As expected, we found that bodyweight increased, SI was markedly reduced and insulin levels were compensatorily increased. Figure 3 shows the SG in these experiments. It is seen that SG was reduced by high‐fat feeding, and this effect was already evident after 1 week. The contribution of SG to glucose disappearance was reduced to approximately 40% at this time point. Interestingly, SG slightly improved after the first week of high‐fat feeding, although it was always lower than in mice fed a control diet. This was at variance with insulin sensitivity, which progressively deteriorated over time in mice fed a high‐fat diet. Increased SG over time in insulin resistance might therefore be a counterbalance of the elevated insulin resistance, but the main conclusion of this study is that insulin resistance is also associated with a reduced SG, which therefore might add to the glucose intolerance in this condition.
Figure 3.
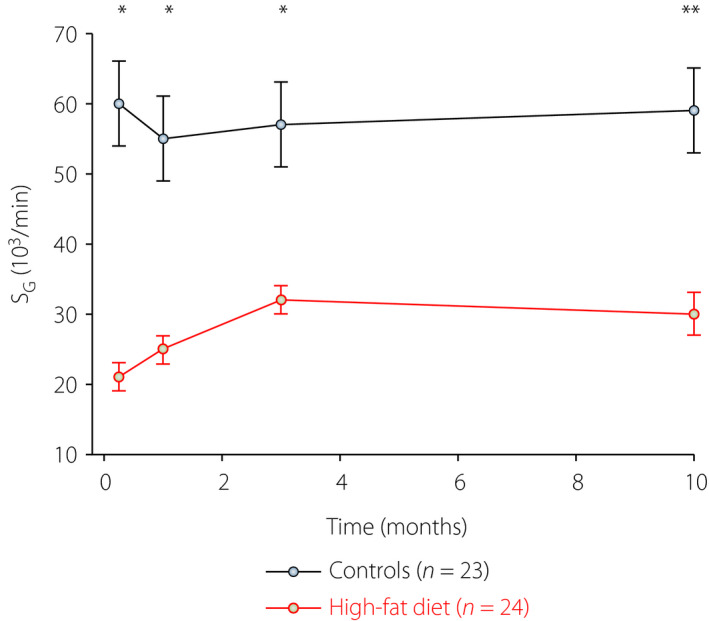
Glucose effectiveness (SG) in mice fed a control diet (11% fat; n = 23) or a high‐fat diet (58% fat, n = 24) for up to 10 months. The mean ± standard error of the mean is shown. Data from experiments reported by Ahrén et al.55 Asterisks indicate probability level of random difference between the groups, *P < 0.05, **P < 0.01.
GLUCOSE EFFECTIVENESS AND INCRETIN HORMONES
GLP‐1 and GIP are known to stimulate insulin secretion, and therefore enhance insulin levels 61 . This is a major effect behind the development of GLP‐1 receptor agonists 62 and DPP‐4 inhibitors 63 as glucose‐lowering therapy for type 2 diabetes. We carried out a study on the effects of GIP versus GLP‐1 in C57BL/6J mice 53 . We found that both incretin hormones augmented glucose‐stimulated insulin secretion in a dose‐dependent manner 53 . We found that both incretin hormones also increased SG 53 . Here, we have revisited those data and explored the SG results in relation to various administered dose of incretin hormone. Interestingly, as seen in Figure 4, GIP was more potent that GLP‐1 in augmenting SG, as a clear effect was observed by the dose of 0.03 nmol/kg, whereas the lowest effective dose of GLP‐1 was 10‐fold higher. In contrast, an earlier study in NMRI mice showed only modest changes in SG by increasing GLP‐1 doses 64 . In humans, it was also shown that GLP‐1 augments SG 32 , 33 , 34 . This suggests that increased SG, together with the classical incretin effect to stimulate insulin secretion, might be a mechanism to prevent hyperglycemia. This would also be supported by a finding that glucose effectiveness is increased during the early phase of an oral glucose tolerance test when the incretin effect is at its zenith 65 . We have also shown that SG is reduced in GLP‐1 receptor knockout mice, which further shows the impact of GLP‐1 on insulin‐independent glucose disappearance 52 . In contrast, SG is not significantly altered in GIP receptor knockout mice 52 .
Figure 4.
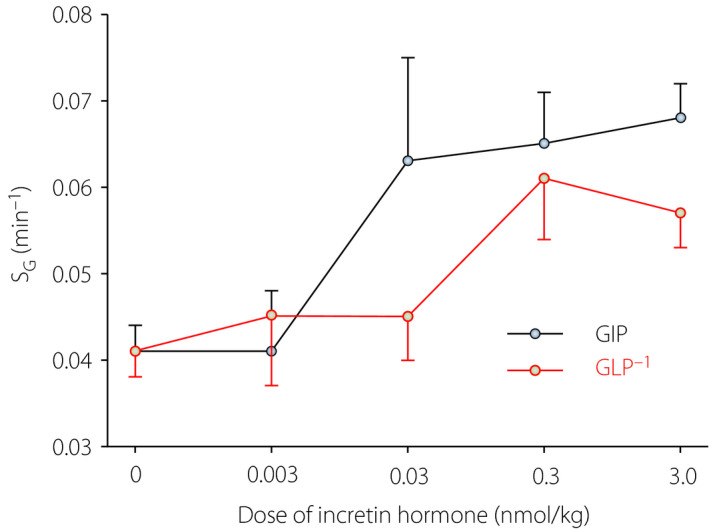
Glucose effectiveness (SG) after intravenous administration of glucose‐dependent insulinotropic polypeptide (GIP) or glucagon‐like peptide‐1 (GLP‐1) at different dose levels in an intravenous glucose tolerance test in C57BL/6J mice. The mean ± standard error of the mean is shown. There were 83 mice in the glucose‐only group (dose 0), and a total of 152 animals in the GLP‐1/GIP supplemented groups. Revisited data from results reported by Pacini et al.53
As incretin hormones increase circulating insulin after intravenous glucose, it is still not established whether the increase by GIP and GLP‐1 of SG is due either to the increasing insulin, regardless of the stimulus, or to a primary effect of incretins themselves. Evidence from other studies seem to support the first hypothesis, as other potent enhancers of glucose‐stimulated insulin secretion also similarly increase SG in mice, such as the neuropeptide, pituitary adenylate cyclase activating polypeptide 47 , 56 . However, as previously discussed, high insulin levels, if anything, reduce SG; thus, it is more likely that incretin hormones enhance SG through an extrapancreatic effect independently from their stimulation of insulin secretion. In support of this, we consider again the lack of association between SG and AUCinsulin after GLP‐1 and GIP 53 . Such an effect would be consistent with extrapancreatic actions of GIP 66 , 67 ; also, GLP‐1 has been shown to have extrapancreatic effects that might directly (through the liver) or indirectly (through neural effects) affect glucose disposal 68 , 69 , 70 , 71 .
An interesting consequence of the finding of enhanced SG by incretin hormones is that the proportion of the relative contribution of insulin‐dependent and non‐insulin‐dependent mechanisms to glucose disposal is increased, which was significant for GIP. Thus, a GIP‐induced increase in glucose disappearance was associated with a higher dependency on SG than after glucose alone and after glucose plus GLP‐1 53 . This suggests that GIP enhances the processes driving non‐insulin‐dependent glucose clearance, which, again, would fit with extrapancreatic actions of GIP.
GLP‐1 receptor agonists and DPP‐4 inhibitors are frequently used as antihyperglycemic therapy in type 2 diabetes patients 61 , 62 , 63 . Both these therapies work through GLP‐1 receptors, the GLP‐1 receptor agonists by achieving a pharmacological activation of the receptors, and DPP‐4 inhibitors by preventing the inactivation of endogenously produced GLP‐1, thereby increasing the GLP‐1 receptor activation by endogenous GLP‐1. It is therefore of interest to discuss whether the improved SG observed when GLP‐1 is administered to healthy volunteers 32 , 33 , 34 might contribute to the metabolic benefits of these therapies. One study explored this by comparing SG after 12 weeks of treatment with the GLP‐1 receptor agonist liraglutide in combination with metformin versus metformin alone in type 2 diabetes for 12 weeks using a cross‐over design 36 , and another study explored the effect of the DPP‐4 inhibitor, vildagliptin, versus a placebo during 10 days of treatment in type 2 diabetes patients 37 . It was found, however, that neither liraglutide nor vildagliptin did increase SG in these studies 36 , 37 . This would therefore suggest that although GLP‐1 is able to increase SG in healthy individuals, therapy with GLP‐1 receptor agonists or DPP‐4 inhibitors does not seem to increase the low SG associated with type 2 diabetes. This could be explained by the reduced SG in type 2 diabetes, which might be more difficult to increase than in healthy individuals, but it might also be due to a failure of GLP‐1 to continuously increase SG over a long period of time. Further studies are required to solve whether GLP‐1 receptor agonists and DPP‐4 inhibitors affect SG during prolonged treatment of type 2 diabetes.
GLUCOSE EFFECTIVENESS AND GLUCAGON
The decrease of glucose concentration during the IVGTT after the peak caused by the bolus glucose injection is mainly due to glucose uptake and inhibition of glucose production. It is known that glucagon is strictly related to endogenous (liver) glucose production; therefore, studying the effects of glucagon on SG could provide information on the probable actions that this pancreatic hormone exerts on glucose effectiveness and, consequently, hypothesize possible relationships between SG and glucose production.
To this aim, glucagon at different doses was added to the glucose bolus 57 . The results show (Table 1) that supplementing glucagon to glucose reduces SG by approximately 30% on average 57 . This indicates that glucagon diminishes glucose effectiveness, suggesting that SG reflects glucose production during hyperglycemia. As GLP‐1 increases SG, we carried out a series of experiments in mice where GLP‐1 was added to glucagon. This addition, however, did not modify SG compared with glucagon alone, indicating that GLP‐1 does not increase SG under conditions when glucagon levels are elevated. We conclude that glucagon is more potent as an inhibitor of SG than GLP‐1 as an enhancer.
POSSIBLE MECHANISMS OF GLUCOSE EFFECTIVENESS
Glucose per se is a fundamental substrate for liver metabolism 72 , and understanding the mechanisms of its regulation is paramount. Glucose effectiveness plays an essential role in this regulation; however, the molecular mechanisms underlying glucose effectiveness are not well defined yet. A study in individuals with hepatic cirrhosis showed that SG is reduced by 38%, which explained 65% of the glucose intolerance in these individuals 28 . This would be consistent with a hypothesis that SG is exerted in the liver, where SG would be linked to the stimulation of glucose uptake. However, as liver cells do not have the capacity to take up glucose, and there is no correlation between SG and liver enzymes in cirrhotic patients 28 , it is more likely that the reduction of SG in cirrhotic patients is a result of a reduced muscle mass, suggesting that SG is primarily exerted in the muscles 73 . Glucose transporters might be candidates for new studies; for instance, it is known that the glucose transporter 4 causes entry of glucose into muscular cells after its translocation to the membrane 74 . However, as the molecular bases for SG are still largely unknown, further studies on these topics are required.
RELEVANCE OF MONITORING INSULIN‐INDEPENDENT GLUCOSE DISPOSAL
As already seen, SG has been evaluated in several clinical conditions (Table 1), where it has been shown to vary, making it a relevant factor for the assessment of glucose tolerance and turnover of an individual. It is worth noting that the recent availability of sodium–glucose cotransporter 2 inhibitors as antidiabetic agents has offered a therapeutic approach acting directly on the kidneys without requiring insulin action 75 , 76 . In line with this, sodium–glucose cotransporter 2 inhibition has been shown to improve the reduced glucose effectiveness in the liver in diabetic Zucker fatty rats 77 . For this reason, glucose effectiveness might become a fundamental parameter for the evaluation of the influence of such compounds on glucose disposal. When the molecular mechanisms underlying SG are more established, there will also be a potential to target these mechanisms to increase SG in glucose‐lowering therapy of type 2 diabetes patients.
CONCLUSIONS
Glucose effectiveness describes the processes of insulin‐independent mechanisms of glucose disposal. It is estimated by modeling glucose and insulin data after an intravenous glucose administration, and it accounts for ≈70% of glucose disposal. It is reduced in type 2 diabetes 9 and obesity 24 , 78 , and experimental model studies in mice have characterized the regulation of glucose effectiveness with special emphases on the role of glucose, insulin and processes stimulating insulin secretion. It is essential, therefore, to evaluate this parameter any time a metabolic test is carried out, especially in large population studies 79 . Further studies are warranted to explore the regulation of glucose effectiveness, its molecular basis and the potential of targeting glucose effectiveness as a glucose‐lowering approach in type 2 diabetes patients.
DISCLOSURE
The authors declare no conflict of interest.
Acknowledgments
The expert technical assistance of Tina Ovlund in the reported experimental series is gratefully acknowledged. When the great majority of the described studies were carried out, GP was affiliated with the Metabolic Unit of the Institute of Biomedical Engineering (ISIB‐CNR), Padova, Italy.
J Diabetes Investig 2021; 12: 675–685
References
- 1. Soskin S, Levine R. A relationship between blood sugar level and the rate of sugar utilization, affecting the theories of diabetes. Am J Physiol 1937; 120: 761–770. [Google Scholar]
- 2. Bergman RN, Ider YZ, Bowden CR, et al. Quantitative estimation of insulin sensitivity. Am J Physiol 1979; 236: E667–E677. [DOI] [PubMed] [Google Scholar]
- 3. Bergman RN, Phillips LS, Cobelli C. Physiologic evaluation of factors controlling glucose tolerance in man: measurement of insulin sensitivity and beta‐cell glucose sensitivity from the response to intravenous glucose. J Clin Invest 1981; 68: 1456–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Best JD, Taborsky GJ Jr, Halter JB, et al. Glucose disposal is not proportional to plasma glucose level in man. Diabetes 1981; 34: 847–850. [DOI] [PubMed] [Google Scholar]
- 5. Ader M, Pacini G, Yang YJ, et al. Importance of glucose per se to intravenous glucose tolerance. Diabetes 1985; 34: 1092–1103. [DOI] [PubMed] [Google Scholar]
- 6. Chen M, Bergman RN, Pacini G, et al. Pathogenesis of age‐related glucose intolerance in man: insulin resistance and decreased beta‐cell function. J Clin Endocrinol Metab 1985; 60: 13–20. [DOI] [PubMed] [Google Scholar]
- 7. Bergman RN, Finegood DR, Ader M. Assessment of insulin sensitivity in vivo. Endocr Rev 1985; 6: 45–86. [DOI] [PubMed] [Google Scholar]
- 8. Beard JC, Bergman RN, Ward WK, et al. The insulin sensitivity index in nondiabetic man. Correlation between clamp‐derived and IVGTT‐derived values. Diabetes 1986; 35: 362–369. [DOI] [PubMed] [Google Scholar]
- 9. Welch S, Gebhart SS, Bergman RN, et al. Minimal model analysis of intravenous glucose tolerance test‐derived insulin sensitivity in diabetic subjects. J Clin Endocrinol Metab 1990; 71: 1508–1518. [DOI] [PubMed] [Google Scholar]
- 10. Yang YJ, Youn JH, Bergman RN. Modified protocols improve insulin sensitivity estimation using the minimal model. Am J Phsyiol 1987; 253: E595–E602. [DOI] [PubMed] [Google Scholar]
- 11. Chen M, Bergman RN, Porte D Jr. Insulin resistance and beta‐cell dysfunction in aging: The importance of dietary carbohydrate. J Clin Endocrinol Metab 1988; 67: 951–957. [DOI] [PubMed] [Google Scholar]
- 12. Pacini G, Valerio A, Beccaro F, et al. Insulin sensitivity and beta cell responsivity are not decreased in elderly subjects with normal OGTT. J Am Geriatr Soc 1988; 36: 317–323. [DOI] [PubMed] [Google Scholar]
- 13. Bergman RN, Prager R, Vølund A, et al. Equivalence of the insulin sensitivity index in man derived by the minimal model method and the euglycemic glucose clamp. J Clin Invest 1987; 79: 790–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Christopher MJ, Rantzau C, Ward GM, et al. Insulinopenia and hyperglycemia influence the in vivo partitioning of GE and SI. Am J Physiol Endocrinol Metab 1995; 268: E410–E421. [DOI] [PubMed] [Google Scholar]
- 15. Alford FP, Henriksen JE, Rantzau C, et al. Glucose effectiveness is a critical pathogenetic factor leading to glucose intolerance and type 2 diabetes: an ignored hypothesis. Diabetes Metab Res Rev 2018; 34: e2989. [DOI] [PubMed] [Google Scholar]
- 16. Ader M, Ni TC, Bergman RN. Glucose effectiveness assessed under steady state and dynamic conditions. J Clin Invest 1997; 99: 1187–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pacini G, Bergman RN. MINMOD: a computer program to calculate insulin sensitivity and pancreatic responsitivity from the frequently sampled intravenous glucose tolerance test. Comput Methods Progr Biomed 1986; 23: 113–122. [DOI] [PubMed] [Google Scholar]
- 18. Pacini G, Ahrén M, Ahrén B. Reappraisal of the intravenous glucose tolerance index for a simple assessment of insulin sensitivity in mice. Am J Physiol Regul Integr Comp Physiol 2009; 296: R1315–1324. [DOI] [PubMed] [Google Scholar]
- 19. Best JD, Kahn SE, Ader M, et al. Role of glucose effectiveness in the determination of glucose tolerance. Diabetes Care 1996; 19: 1018–1030. [DOI] [PubMed] [Google Scholar]
- 20. Ahrén B, Pacini G. Impaired adaptation of first‐phase insulin secretion in postmenopausal women with glucose intolerance. Am J Physiol 1997; 273: E701–707. [DOI] [PubMed] [Google Scholar]
- 21. Kahn SE, Prigeon RL, McCulloch DK, et al. The contribution of insulin‐dependent and insulin‐independent glucose uptake to intravenous glucose tolerance in healthy human subjects. Diabetes 1994; 43: 587–592. [DOI] [PubMed] [Google Scholar]
- 22. Bergman RN. Toward physiological understanding of glucose tolerance. Minimal‐model approach. Diabetes 1989; 38: 1512–1527. [DOI] [PubMed] [Google Scholar]
- 23. Dube S, Errazuriz‐Cruzat I, Basu A, et al. The forgotten role of glucose effectiveness in the regulation of glucose tolerance. Curr Diabet Rep 2015; 15: 31–36. [DOI] [PubMed] [Google Scholar]
- 24. Kautzky‐Willer A, Pacini G, Ludvik B, et al. B‐cell hypersecretion and not reduced hepatic insulin extraction is the main cause of hyperinsulinemia in obese nondiabetic subjects. Metabolism 1992; 41: 1304–1312. [DOI] [PubMed] [Google Scholar]
- 25. Ludvik B, Waldhäusl Prager R, Kautzky‐Willer A, et al. Mode of action of Ipomoea Batatas (Caiapo) in type 2 diabetic patients. Metabolism 2003; 52: 875–880. [DOI] [PubMed] [Google Scholar]
- 26. Martin BC, Warram JH, Krolewski AS, et al. Role of glucose and insulin resistance in development of type 2 diabetes mellitus: results of a 25‐year follow‐up study. Lancet 1992; 340: 925–929. [DOI] [PubMed] [Google Scholar]
- 27. Kautzky‐Willer A, Prager R, Waldhäusl W, et al. Pronounced insulin resistance and inadequate B‐cell secretion characterize lean gestational diabetes during and after pregnancy. Diabetes Care 1997; 20: 1717–1723. [DOI] [PubMed] [Google Scholar]
- 28. Marchesini G, Pacini G, Bianchi G, et al. Glucose disposal, beta‐cell secretion, and hepatic insulin extraction in cirrhosis: a minimal model assessment. Gastroenterology 1990; 99: 1715–1722. [DOI] [PubMed] [Google Scholar]
- 29. Ahrén B, Pacini G. Age‐related reduction in glucose elimination is accompanied by reduced glucose effectiveness and increased hepatic insulin extraction in man. J Clin Endocrinol Metab 1998; 83: 3350–3556. [DOI] [PubMed] [Google Scholar]
- 30. Chen M, Bergman RN, Pacini G, et al. Pathogenesis of age‐related glucose intolerance in man: insulin resistance and decreased beta‐cell function. J Clin Endocrinol Metab 1985; 60: 13–20. [DOI] [PubMed] [Google Scholar]
- 31. Riedl M, Ludvik B, Pacini G, et al. The increased insulin sensitivity in growth hormone‐deficient adults is reduced by growth hormone replacement therapy. Eur J Clin Invest 2000; 30: 771–778. [DOI] [PubMed] [Google Scholar]
- 32. D’Alessio DA, Kahn SE, Leusner CR, et al. Glucagon‐like peptide 1 enhances glucose tolerance both by stimulation of insulin release and by increasing insulin‐independent glucose disposal. J Clin Endocinol Metab 1994; 93: 2263–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. D’Alessio DA, Prigeon RL, Ensinck JW. Enteral enhancement of glucose disposition by both insulin‐dependent and insulin‐independent processes. A physiological role of glucagon‐like peptide 1. Diabetes 1995; 44: 1433–1437. [DOI] [PubMed] [Google Scholar]
- 34. Vahl TP, Paty BW, Fuller BD, et al. Effects of GLP‐1‐(7–36)NH2, GLP‐1‐(7–37), and GLP‐1‐(9–36)NH2 on intravenous glucose tolerance and glucose‐induced insulin secretion in healthy humans. J Clin Endocrinol Metab 2003; 88: 1772–1779. [DOI] [PubMed] [Google Scholar]
- 35. Buchanan TA, Xiang AH, Peters RK, et al. Response of pancreatic beta‐cells to improved insulin sensitivity in women at high risk for type 2 diabetes. Diabetes 2000; 49: 782–788. [DOI] [PubMed] [Google Scholar]
- 36. Anholm C, Kumarathurai P, Pedersen LR, et al. Liraglutide effects on beta‐cell, insulin sensitivity and glucose effectiveness in patients with stable coronary artery disease and newly diagnosed type 2 diabetes. Diabet Obes Metab 2017; 19: 850–857. [DOI] [PubMed] [Google Scholar]
- 37. Dalla Man C, Bock G, Giesler PD, et al. Dipeptidyl peptidase‐4 inhibition by vildagliptin and effect on insulin secretion and action in response to meal ingestion in type 2 diabetes. Diabetes Care 2009; 32: 14–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hor CP, Yeow TP, Lim SL, et al. Elevated dynamic insulin clearance characterizes obese young Asian with type 2 diabetes with reduced peripheral insulin. Front Endocrinol 2020; 67: 2057‐P. [Google Scholar]
- 39. Taniguchi A, Nakai Y, Fukushima M, et al. Pathogenic factors responsible for glucose intolerance in patients with NIDDM. Diabetes 1992; 41: 1540–1546. [DOI] [PubMed] [Google Scholar]
- 40. Lin JD. Metabolic syndrome in drug‐naïve Chinese patients with insulin‐sensitive and insulin‐resistant type 2 diabetes. Ann Saudi Med 2016; 36: 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Osei K, Gaillard T, Schuster DP. Pathogenetic mechanisms of impaired glucose tolerance and type II diabetes in African‐Americans. The significance of insulin secretion, insulin sensitivity, and glucose effectiveness. Diabetes Care 1997; 20: 396–404. [DOI] [PubMed] [Google Scholar]
- 42. Amoah AGB, Owusu SK, Schuster DP, et al. Pathogenic mechanism of type 2 diabetes in Ghanaians – the importance of beta cell secretion, insulin sensitivity and glucose effectiveness. S Afr Med J 2002; 92: 377–384. [PubMed] [Google Scholar]
- 43. Taniguchi A, Nakai Y, Doi K, et al. Glucose effectiveness in two subtypes within impaired glucose tolerance. A minimal model analysis. Diabetes 1994; 43: 1211–1217. [DOI] [PubMed] [Google Scholar]
- 44. Yabe D, Seino Y, Fukushima M, et al. ß cell dysfunction versus insulin resistance in the pathogenesis of type 2 diabetes I East Asians. Curr Diab Rep 2015; 15: 602–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Doi K, Taniguchi A, Nakai Y, et al. Decreased glucose effectiveness but not insulin resistance in glucose‐tolerant offspring of Japanese non‐insulin‐dependent diabetic patients: a minimal‐model analysis. Metabolism 1997; 46: 880–883. [DOI] [PubMed] [Google Scholar]
- 46. Haffner SM, Stern MP, Watanabe RM, et al. Relationship of insulin clearance and secretion to insulin sensitivity in non‐diabetic Mexican Americans. Eur J Clin Invest 1992; 22: 147–153. [DOI] [PubMed] [Google Scholar]
- 47. Pacini G, Thomaseth K, Ahrén B. Contribution to glucose tolerance of insulin‐independent vs. insulin‐dependent mechanisms in mice. Am J Physiol Endocrinol Metab 2001; 281: E693–703. [DOI] [PubMed] [Google Scholar]
- 48. Noonan WT, Banks RO. Renal function and glucose transport in male and female mice with diet‐induced type II diabetes mellitus. Proc Soc Exp Biol Med 2000; 225: 221–230. [DOI] [PubMed] [Google Scholar]
- 49. Ferrari B, Arnold M, Carr RD, et al. Subdiaphragmatic vagal deafferentation affects body weight gain and glucose metabolism in obese male Zucker (fa/fa) rats. Am J Physiol Regul Integr Comp Physiol 2005; 289: R1027–R1034. [DOI] [PubMed] [Google Scholar]
- 50. Rojas JM, Matsen ME, Mundlinger TO, et al. Glucose intolerance induced by blockade of central FGF receptors is linked to an acute stress response. Molecul Metab 2015; 4: 561–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tokuyama K, Suzuki M. Intravenous glucose‐tolerance test‐derived glucose effectiveness in endurance‐trained rats. Metabolism 1998; 47: 190–194. [DOI] [PubMed] [Google Scholar]
- 52. Tura A, Pacini G, Yamada Y, et al. Glucagon and insulin secretion, insulin clearance, and fasting glucose in GIP receptor and GLP‐1 receptor knockout mice. Am J Physiol Regul Integr Comp Physiol 2019; 316: R27–R37. [DOI] [PubMed] [Google Scholar]
- 53. Pacini G, Ahrén B. Glucagon‐like peptide‐1 and glucose‐dependent insulinotropic peptide: effects alone and in combination on insulin secretion and glucose disappearance in mice. Physiol Rep 2017; 5: e13280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Persson K, Pacini G, Sundler F, et al. Islet function phenotype in gastrin‐releasing peptide receptor gene‐deficient mice. Endocrinology 2002; 143: 3717–3726. [DOI] [PubMed] [Google Scholar]
- 55. Ahrén B, Pacini G. Insufficient islet compensation to insulin resistance vs. reduced glucose effectiveness in glucose‐intolerant mice. Am J Physiol Endocrinol Metab 2002; 283: E738–E744. [DOI] [PubMed] [Google Scholar]
- 56. Filipsson K, Pacini G, Scheurink AJW, et al. PACAP stimulates insulin secretion but inhibits insulin sensitivity in mice. Am J Physiol Endocrinol Metab 1998; 274: E834–E842. [DOI] [PubMed] [Google Scholar]
- 57. Pacini G, Ahrén B. Glucagon and GLP‐1 exhibit no synergistic enhancement of glucose‐stimulated insulin secretion in mice. Peptides 2015; 71: 66–71. [DOI] [PubMed] [Google Scholar]
- 58. Ahrén B, Pacini G. A novel approach to assess insulin sensitivity reveals no increased insulin sensitivity in mice with a dominant‐negative mutant hepatocyte nuclear factor‐1 alpha. Am J Physiol Regul Integr Comp Physiol 2006; 29: R131–R137. [DOI] [PubMed] [Google Scholar]
- 59. Henquin JC, Meissner HP. Opposite effects of tolbutamide and diazoxide on 86Rb fluxes and membrane potential in pancreatic B cells. Biochem Pharmacol 1982; 31: 1407–1415. [DOI] [PubMed] [Google Scholar]
- 60. Sörhede Winzell M, Ahrén B. The high‐fat fed mouse: a model for studying mechanisms and treatment of impaired glucose tolerance and type 2 diabetes. Diabetes 2004; 53(suppl 3): S215–S219. [DOI] [PubMed] [Google Scholar]
- 61. Nauck MA, Meier JJ. Incretin hormones: their role in health and disease. Diabet Obes Metab 2018; 20(suppl 1): 5–21. [DOI] [PubMed] [Google Scholar]
- 62. Ahrén B. Glucagon‐like peptide‐1 receptor agonists for type 2 diabetes: a rational drug development. J Diabet Invest 2019; 10: 196–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ahrén B. DPP‐4 inhibition and the path to clinical proof. Frontiers Endocrinol 2019; 10: 376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ahrén B, Pacini G. Dose‐related effects of GLP‐1 on insulin secretion, insulin sensitivity, and glucose effectiveness in mice. Am J Physiol 1999; 277: E996–E1004. [DOI] [PubMed] [Google Scholar]
- 65. Kahn SE, Ader M, Watanabe RM, et al. Role of glucose effectiveness in the determination of glucose tolerance. Diabetes Care 1996; 19: 1018–1030. [DOI] [PubMed] [Google Scholar]
- 66. Hansotia T, Maida A, Flock G, et al. Extrapancreatic incretin receptors modulate glucose homeostasis, body weight, and energy expenditure. J Clin Invest 2007; 117: 143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Seino Y, Yabe D. Glucose‐dependent insulinotropic polypeptide and glucagon‐like peptide‐1: incretin actions beyond the pancreas. J Diabetes Investig 2013; 4: 108–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tomas E, Stanojevic V, Habener JF. GLP‐1‐derived nonapeptide GLP‐1(28–36)amide targets to mitochondria and suppresses glucose production and oxidative stress ion isolated mouse hepatocytes. Regul Pept 2011; 167: 177–184. [DOI] [PubMed] [Google Scholar]
- 69. Kakei M, Yada T, Nakagawa A, et al. Glucagon‐like peptide‐1 evokes action potentials and increases cytosolic Ca2+ in rat nodose ganglion neurons. Autonomic Neurosci 2002; 102: 39–44. [DOI] [PubMed] [Google Scholar]
- 70. Krieger JP, Arnold M, Pettersen KG, et al. Knockdown of GLP‐1 receptors in vagal afferents affects normal food intake and glycemia. Diabetes 2016; 65: 34–43. [DOI] [PubMed] [Google Scholar]
- 71. Ahrén B. Sensory nerves contribute to insulin secretion by glucagon‐like peptide‐1 in mice. Am J Physiol Regul Integr Comp Physiol 2004; 286: R269–R272. [DOI] [PubMed] [Google Scholar]
- 72. Katz J, McGarry JD. The glucose paradox: is glucose a substrate for liver metabolism? J Clin Invest 1984; 74: 1901–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Baron AD, Brechtel G, Wallace P, et al. Rates and tissue sites of non‐insulin‐ and insulin‐mediated glucose uptake in humans. Am J Physiol Endocrinol Metab 1988; 255: E769–E774. [DOI] [PubMed] [Google Scholar]
- 74. Galante P, Mosthaf L, Kellerer M, et al. Acute hyperglycemia provides an insulin‐independent inducer for GLUT4 translocation in C2C12 myotubes and rat skeletal muscle. Diabetes 1995; 44: 646–651. [DOI] [PubMed] [Google Scholar]
- 75. Freeman JS. Review of insulin‐dependent and insulin‐independent agents for treating patients with type 2 diabetes mellitus and potential role for sodium‐glucose co‐transporter 2 inhibitors. Postgrad Med 2013; 125: 214–226. [DOI] [PubMed] [Google Scholar]
- 76. Seufert J. SGLT2 inhibitors—an insulin‐independent therapeutic approach for treatment of type 2 diabetes: focus on canagliflozin. Diabetes Metab Syndr Obes 2015; 8: 543–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. O’Brien TP, Jenkins EC, Estes SK, et al. Correcting postprandial hyperglycemia in Zucker diabetic fatty rats with an SGLT2 inhibitor restores glucose effectiveness in the liver and reduces insulin resistance in skeletal muscle. Diabetes 2017; 66: 1172–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Morettini M, Di Nardo F, Ingrillini L, et al. Glucose effectiveness and its components in relation to body mass index. Eur J Clin Invest 2019; 49: e13099. [DOI] [PubMed] [Google Scholar]
- 79. Lorenzo C, Wagenknecht LE, Karter AJ, et al. Cross‐sectional and longitudinal changes of glucose effectiveness in relation to glucose tolerance: the insulin resistance atherosclerosis study. Diabetes Care 2011; 34: 1959–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]