

Abstract
Intracranial mesenchymal tumors with FET‐CREB fusions are a recently described group of neoplasms in children and young adults characterized by fusion of a FET family gene (usually EWSR1, but rarely FUS) to a CREB family transcription factor (ATF1, CREB1, or CREM), and have been variously termed intracranial angiomatoid fibrous histiocytoma or intracranial myxoid mesenchymal tumor. The clinical outcomes, histologic features, and genomic landscape are not well defined. Here, we studied 20 patients with intracranial mesenchymal tumors proven to harbor FET‐CREB fusion by next‐generation sequencing (NGS). The 16 female and four male patients had a median age of 14 years (range 4–70). Tumors were uniformly extra‐axial or intraventricular and located at the cerebral convexities (n = 7), falx (2), lateral ventricles (4), tentorium (2), cerebellopontine angle (4), and spinal cord (1). NGS demonstrated that eight tumors harbored EWSR1‐ATF1 fusion, seven had EWSR1‐CREB1, four had EWSR1‐CREM, and one had FUS‐CREM. Tumors were uniformly well circumscribed and typically contrast enhancing with solid and cystic growth. Tumors with EWSR1‐CREB1 fusions more often featured stellate/spindle cell morphology, mucin‐rich stroma, and hemangioma‐like vasculature compared to tumors with EWSR1‐ATF1 fusions that most often featured sheets of epithelioid cells with mucin‐poor collagenous stroma. These tumors demonstrated polyphenotypic immunoprofiles with frequent positivity for desmin, EMA, CD99, MUC4, and synaptophysin, but absence of SSTR2A, myogenin, and HMB45 expression. There was a propensity for local recurrence with a median progression‐free survival of 12 months and a median overall survival of greater than 60 months, with three patients succumbing to disease (all with EWSR1‐ATF1 fusions). In combination with prior case series, this study provides further insight into intracranial mesenchymal tumors with FET‐CREB fusion, which represent a distinct group of CNS tumors encompassing both intracranial myxoid mesenchymal tumor and angiomatoid fibrous histiocytoma‐like neoplasms.
Keywords: angiomatoid fibrous histiocytoma (AFH), brain tumor, CREB, EWSR1, intracranial myxoid mesenchymal tumor, molecular neuropathology, sarcoma
Intracranial mesenchymal tumors with FET‐CREB fusion comprise a group of neoplasms that have been variably described as intracranial angiomatoid fibrous histiocytoma and intracranial myxoid mesenchymal tumor. Our investigation reveals that these tumors occur in a wide age range with a female predominance, usually have extra‐axial or intraventricular location, typically have solid and cystic growth pattern with enhancement after contrast administration, and demonstrate a propensity for local recurrence and occasionally dissemination or metastasis leading to mortality. These tumors demonstrate a broad range of histologic features, with those harboring CREB1 or CREM as the fusion partner enriched for stellate/spindle cell morphology and mucin‐rich stroma, versus those with ATF1 as the fusion partner enriched for epithelioid/rhabdoid morphology and mucin‐poor stroma.

1. INTRODUCTION
Intracranial mesenchymal tumors with FET‐CREB fusions comprise a rare group of neoplasms of the central nervous system (CNS) that have been variably described as intracranial angiomatoid fibrous histiocytoma (AFH), and more recently as intracranial myxoid mesenchymal tumor (IMMT) (1, 2, 3, 4). Despite their disparate names, intracranial AFH and IMMT are both molecularly defined by in‐frame gene fusions of the FET family of RNA‐binding proteins (EWSR1 or FUS) to the CREB (cyclic AMP response element‐binding protein) family of transcription factors, which includes ATF1, CREB1, and CREM. Notably, identical FET‐CREB fusions are recurrently found in extracranial AFH, clear cell sarcoma of soft tissue, clear cell sarcoma of the gastrointestinal tract, primary pulmonary myxoid sarcoma, hyalinizing clear cell carcinoma of the salivary gland, and a subset of malignant mesotheliomas lacking BAP1 and NF2 alterations (5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15). However, the exact relation of these intracranial mesenchymal tumors with FET‐CREB fusions to extracranial AFH and other FET‐CREB fusion‐driven neoplasms remains unknown.
AFH has been characterized in the soft tissue as an uncommon neoplasm of intermediate malignancy comprising 0.3% of all soft tissue tumors, most frequently occurring in the subcutaneous tissue of the extremities in children and young adults, but occasionally can be located within the viscera or retroperitoneum (5, 14, 16, 17). They display a characteristic histomorphology with four defining characteristics, although not all are required for diagnosis: solid nodules of epithelioid or spindled cells with a syncytial growth pattern, pseudoangiomatous spaces, a fibrous pseudocapsule, and robust pericapsular lymphoplasmacytic inflammatory infiltrates with germinal center formation. AFH is well‐described to display a spectrum of variant histomorphologies, such as prominent myxoid stroma prompting some to describe a myxoid variant of AFH (15, 18, 19, 20, 21). In one series, three AFH with myxoid features occurring in the extremities (two cases) or lung (one case) harbored EWSR1‐CREM fusions (15).
Several intracranial mesenchymal tumors histologically resembling AFH or the myxoid variant of AFH and harboring FET‐CREB fusions have now been reported (1, 2, 3, 4, 33). Kao et al first reported in 2017 four children and young adults with intracranial myxoid mesenchymal neoplasms with EWSR1‐CREB1, EWSR1‐CREM, or EWSR1‐ATF1 fusions (3). Shortly thereafter, Bale et al reported three additional pediatric patients with tumors composed of similar histologic features and EWSR1‐CREB1 or EWSR1‐CREM fusions (1). The authors postulated that these tumors may represent a myxoid variant of AFH occurring intracranially, or alternatively a novel intracranial tumor entity (IMMT). The reported cases of IMMT had mucin‐rich stroma, often lacked some of the characteristic histologic features of AFH, and were enriched for CREM fusions that are rare in AFH of extracranial soft tissue. Despite some morphologic similarities, they also have a spectrum of morphologic appearances that partially overlap with those of conventional AFH, including pseudo‐encapsulation, nodular proliferations of epithelioid cells, and prominent subcapsular lymphoplasmacytic aggregates with hemosiderin deposition (1, 3). The immunoprofiles of both intracranial AFH‐like tumors and IMMT are similar to conventional AFH of extracranial soft tissue (1, 2, 3). Tumor cells are consistently positive for desmin (at least focally), but are negative for skeletal muscle markers including myogenin and MyoD1, as well as smooth muscle markers including smooth muscle actin, caldesmon, and calponin. There is also positivity for CD99 and EMA in the majority of tumors, while they are consistently negative for melanocytic markers (S100, HMB45) and glial markers (GFAP and OLIG2). CD68 and CD163 have been positive in both neoplastic cells and tumor‐associated macrophages. However, the clinical features, patient outcomes, and optimal treatment strategies for these tumors remain largely undefined.
Therefore, to further investigate the clinical, radiologic, and pathologic features of intracranial mesenchymal tumors with FET‐CREB fusions, we have comprehensively studied a cohort of 20 patients and reviewed the previously reported cases of these tumors in the published literature.
2. MATERIALS AND METHODS
2.1. Study population and tumor specimens
The study cohort consisted of 20 patients who underwent surgical resection of a primary intracranial neoplasm that was identified to harbor a gene fusion of EWSR1 or the related FUS together with a CREB family member (ATF1, CREB1, or CREM). Pathologic review of all tumor specimens was performed by a group of expert neuropathologists (E.A.S., A.P., and D.A.S.).
2.2. Immunohistochemistry
Immunohistochemistry was performed on whole formalin‐fixed, paraffin‐embedded tissue sections using the following antibodies: desmin (Cell Marque, clone D33, undiluted, ER1 antigen retrieval), epithelial membrane antigen (EMA, Leica, clone GP1.4, undiluted, ER1 antigen retrieval), CD99 (Signet, clone CD99, 1:400 dilution, ER1 antigen retrieval), S100 (DAKO, polyclonal, 1:2,000 dilution, no antigen retrieval), MUC4 (Millipore, clone 8G7, 1:500 dilution, ER1 antigen retrieval), pan‐neurotrophic receptor tyrosine kinase protein family (pan‐NTRK, Abcam, clone EPR‐17341, 1:100 dilution, ER2 antigen retrieval), somatostatin receptor 2A (SSTR2A, Abcam, clone UMB1, 1:2,000 dilution, ER2 antigen retrieval), OLIG2 (Immuno Bio Labs, polyclonal, 1:200 dilution, ER1 antigen retrieval), glial fibrillary acidic protein (GFAP, DAKO, polyclonal, 1:3,000 dilution, no antigen retrieval), synaptophysin (Cell Marque, polyclonal, 1:100 dilution, ER2 antigen retrieval), CD68 (Leica, clone 514H12, undiluted, ER2 antigen retrieval), cytokeratin AE1/AE3 (Dako, clone AE1/AE3, 1:100 dilution, ER1 antigen retrieval), cytokeratin CAM5.2 (Becton Dickinson, clone CAM5.2, 1:100 dilution, ER1 antigen retrieval), HMB45 (Dako, clone HMB45, undiluted, CC1 antigen retrieval), myogenin (Cell Marque, clone F5D, undiluted, CC1 antigen retrieval), and Ki‐67 (Dako, clone Mib1, 1:50 dilution, ER2 antigen retrieval). Immunostaining for desmin, EMA, CD99, S100, MUC4, pan‐NTRK, SSTR2A, OLIG2, GFAP, synaptophysin, CD68, cytokeratin AE1/AE3, cytokeratin CAM5.2, and Ki‐67 was performed in a Leica Bond‐Max automated stainer. Immunostaining for HMB45 and myogenin was performed in a Ventana BenchMark Ultra‐automated stainer. Diaminobenzidine was used as the chromogen, followed by hematoxylin counterstain.
2.3. Targeted next‐generation DNA sequencing analysis
Tumor tissue was selectively scraped from unstained slides or punched from formalin‐fixed, paraffin‐embedded (FFPE) blocks using 2.0 mm disposable biopsy punches (Integra Miltex Instruments) to enrich for as high tumor content as possible. Genomic DNA was extracted from this macrodissected tumor tissue using the QIAamp DNA FFPE Tissue Kit (Qiagen). Tumor tissue from both the initial surgical resection and a subsequent recurrence was sequenced for one patient (ATF1 #2). Capture‐based next‐generation DNA sequencing was performed using an assay that targets all coding exons of 479 cancer‐related genes, select introns and upstream regulatory regions of 47 genes to enable detection of structural variants including gene fusions, and DNA segments at regular intervals along each chromosome to enable genome‐wide copy number and zygosity analysis, with a total sequencing footprint of 2.8 Mb (UCSF500 Cancer Panel) (34). Multiplex library preparation was performed using the KAPA Hyper Prep Kit (Roche) according to the manufacturer's specifications. Hybrid capture of pooled libraries was performed using a custom oligonucleotide library (NimbleGen SeqCap EZ Choice). Captured libraries were sequenced as paired‐end 100 bp reads on an Illumina HiSeq 2500 instrument. Sequence reads were mapped to the reference human genome build GRCh37 (hg19) using the Burrows–Wheeler aligner (BWA). Recalibration and deduplication of reads was performed using the Genome Analysis Toolkit (GATK). Coverage and sequencing statistics were determined using Picard CalculateHsMetrics and Picard CollectInsertSizeMetrics. Single nucleotide variant and small insertion/deletion mutation calling was performed with FreeBayes, Unified Genotyper, and Pindel. Large insertion/deletion and structural alteration calling was performed with Delly. Variant annotation was performed with ANNOVAR. Single nucleotide variants, insertions/deletions, and structural variants were visualized and verified using Integrative Genome Viewer. Genome‐wide copy number and zygosity analysis was performed by CNVkit and visualized using Nexus Copy Number (Biodiscovery).
2.4. Reverse‐transcription PCR
Total RNA was extracted from formalin‐fixed paraffin‐embedded tissue using the RNeasy FFPE Isolation Kit (Qiagen). Reverse transcriptase‐polymerase chain reaction (RT‐PCR) was performed using SuperScript III One‐Step RT‐PCR System (Life Technologies) and the following primers: FUS, 5′‐TGGACAGCAGAACCAGTACA‐3′ and CREM, 5′‐CAGTAGGAGCTCGGATCTGG‐3′. RT‐PCR products were resolved by agarose gel by electrophoresis and Sanger sequenced using BigDye terminator chemistry (Applied Biosystems) following standard techniques.
2.5. Kaplan–Meier survival plots and statistical analyses
Clinical outcomes were studied by Kaplan–Meier analysis using GraphPad Prism. The Kaplan–Meier survival analysis stratified by extent of resection includes only the 17 patients from this cohort with known extent of resection. Additional Kaplan–Meier survival analyses include the 20 patients from this cohort combined with the 18 prior cases of intracranial mesenchymal tumors with FET‐CREB fusions reported in the literature. p values were calculated by Log‐rank (Mantel‐Cox) test. Statistical comparison of histologic features was performed by Mann–Whitney unpaired two‐tailed t test using GraphPad Prism.
3. RESULTS
3.1. Clinical features
The 16 female and four male patients had a median age of 14 years (range 4–70 years) (Table 1). Two of the patients had a known history of prior neoplasia. Patient CREB1 #3 had a history of pre‐B acute lymphoblastic leukemia at 10 years of age (7 years prior to the diagnosis of intracranial mesenchymal tumor with EWSR1‐CREB1 fusion), for which he received treatment with prednisone and methotrexate but no radiation therapy. Patient CREB1 #7 had a history of high‐risk adrenal neuroblastoma for which he received local radiation treatment and systemic chemotherapy, but the posterior fossa where the intracranial mesenchymal tumor with EWSR1‐CREB1 fusion developed 5 years later was outside the radiation treatment field. None of the patients had a known tumor predisposition syndrome or other known predisposing factors to neoplasia, and none of the patients had evidence of extracranial neoplasms at time of diagnosis to suggest the possibility of metastatic disease. Presenting symptoms were variable and most frequently included severe headaches or nausea, and also occasionally seizures, tinnitus, or diplopia (Table S1).
TABLE 1.
Clinical data for the 20 patients with intracranial mesenchymal tumors with FET‐CREB fusions
| Patient ID | Age | Sex | Fusion | Tumor location | Extent of resection | Initial adjuvant therapy | Disease progression/recurrence | Clinical status at last follow up | Length of follow‐up |
|---|---|---|---|---|---|---|---|---|---|
| ATF1 #1 | 12 | M | EWSR1‐ATF1 | Cerebral convexity (parietal) | Subtotal | XRT (59.4 Gy) | Local recurrence x 2 (6 months; 10 months) | Alive with progressive disease | 24 months |
| ATF1 #2 | 9 | F | EWSR1‐ATF1 | Cerebral convexity (frontal) | Subtotal | None | Local recurrence (3 months) | Died of disease | 63 months |
| ATF1 #3 | 24 | F | EWSR1‐ATF1 | Cerebral convexity (occipital) | Unknown | Unknown | Unknown | Unknown | Unknown |
| ATF1 #4 | 13 | F | EWSR1‐ATF1 | Cerebral convexity (frontal) | Gross total | None | None | Alive, disease‐free | 24 months |
| ATF1 #5 | 34 | F | EWSR1‐ATF1 | Tentorium | Subtotal | None | Local recurrence (54 months) | Alive, disease‐free | 81 months |
| ATF1 #6 | 17 | F | EWSR1‐ATF1 | CP angle | Subtotal | XRT (59.4 Gy); ifosfamide, vincristine, adriamycin | Metastasis to thoracic lymph nodes and vertebrae (12 months) | Died of disease | 27 months |
| ATF1 #7 | 70 | M | EWSR1‐ATF1 | CP angle with spinal dissemination | Subtotal | None | Continuous progression | Died of disease | 1 month |
| ATF1 #8 | 17 | F | EWSR1‐ATF1 | CP angle | Subtotal | None | None | Alive with stable disease | 13 months |
| CREB1 #1 | 14 | F | EWSR1‐CREB1 | Lateral ventricle | Gross total | None | None | Alive, disease‐free | 59 months |
| CREB1 #2 | 39 | F | EWSR1‐CREB1 | Lateral ventricle | Gross total | None | None | Alive, disease‐free | 6 months |
| CREB1 #3 | 10 | M | EWSR1‐CREB1 | Falx (parietal) | Gross total | None | Local recurrence (9 months) | Alive, disease‐free | 57 months |
| CREB1 #4 | 14 | F | EWSR1‐CREB1 | Lateral ventricle | Unknown | None | None | Alive, disease‐free | 47 months |
| CREB1 #5 | 25 | F | EWSR1‐CREB1 | CP angle | Gross total | None | Local recurrence (11 months) | Alive with stable disease | 30 months |
| CREB1 #6 | 14 | F | EWSR1‐CREB1 | Cerebral convexity (parietal) | Unknown | None | Local recurrence (49 months) | Alive with progressive disease | 57 months |
| CREB1 #7 | 12 | M | EWSR1‐CREB1 | Tentorium | Gross total | None | None | Alive, disease‐free | 46 months |
| CREM #1 | 15 | F | EWSR1‐CREM | Spinal cord (thoracic) | Subtotal | XRT (dose unknown) | Local recurrence (6 months) | Alive, disease‐free | 30 months |
| CREM #2 | 14 | F | EWSR1‐CREM | Lateral ventricle | Gross total | None | None | Alive, disease‐free | 38 months |
| CREM #3 | 5 | F | EWSR1‐CREM | Cerebral convexity (frontal) | Subtotal | ifosfamide, carboplatin, etoposide | Local recurrence x 2 (2 months; 6 months) | Alive with progressive disease | 11 months |
| CREM #4 | 30 | M | EWSR1‐CREM | Falx (frontal) | Subtotal | XRT (54 Gy) | None | Alive, disease‐free | 6 months |
| FUS #1 | 4 | F | FUS‐CREM | Cerebral convexity (occipital) | Gross total | None | Local recurrence x 2 (9 months; 13 months) | Alive, disease‐free | 36 months |
Patients CREB1 #4 and CREB1 #7 have been previously described in part as case 2 and case 1 from Bale et al Brain Pathology 2018.
3.2. Radiologic features
Tumors were exclusively extra‐axial in the meninges with compression of the subjacent brain parenchyma or intraventricular, with locations at the cerebral convexities (n = 7), falx (2), lateral ventricles (4), tentorium (2), cerebellopontine angle (4), and spinal cord (1). Radiologic characteristics include circumscribed and lobulated growth, often with both solid and cystic components, avid enhancement after contrast administration, intratumoral blood products, and substantial peritumoral edema (Figure 1). Some tumors showed a dural tail or bony involvement of the overlying skull mimicking meningioma. One patient (ATF1 #7) demonstrated a mass in the cerebellopontine (CP) angle with diffuse leptomeningeal dissemination along the spinal cord, whereas all other patients had localized disease without dissemination at time of presentation.
FIGURE 1.
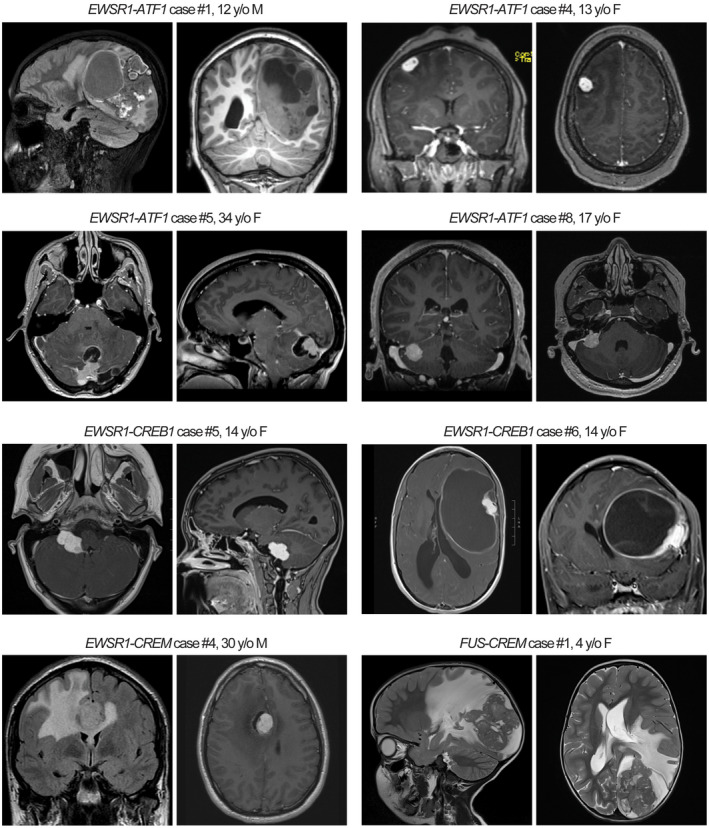
Radiologic features of intracranial mesenchymal tumors with FET‐CREB fusion. These tumors were typically contrast enhancing and often demonstrated both solid and cystic components with extensive peritumoral edema
3.3. Genomic features
Next‐generation sequencing (NGS) revealed that eight tumors harbored EWSR1‐ATF1 fusion, seven had EWSR1‐CREB1 fusion, four had EWSR1‐CREM fusion, and one had FUS‐CREM fusion. The fusions linked the 5′ coding exons of either EWSR1 or FUS to the 3′ coding exons of ATF1, CREB1, or CREM. The predicted fusion transcript most commonly linked exons 1–8 of EWSR1 to exons 5–7 of ATF1, 7–9 of CREB1, and 7–8 of CREM (Table S2). No likely pathogenic single nucleotide variants or indels were identified in any of the 20 tumors. Specifically, no TERT promoter hotspots mutations were present. No mutations or other alterations in genes known to be recurrently altered in meningiomas were identified (e.g., NF2, TRAF7, KLF4, AKT1, SMO, PIK3CA, SMARCB1, BAP1, and YAP1) (35, 36). All tumors lacked STAT6 rearrangements that are defining of solitary fibrous tumor/hemangiopericytoma, and lacked PAX3/7‐FOXO1 fusions that characterize most alveolar rhabdomyosarcomas (37, 38).
Many of the tumors demonstrated a diploid genome (n = 10), whereas the remaining demonstrated a paucity of chromosomal gains or losses (Table S3). All of the tumors with either EWSR1‐ATF1 or EWSR1‐CREB1 fusion appear to have resulted from balanced translocations with an absence of copy number breakpoints at chromosomes 22q12, 12q13, or 2q33 where these three genes are located (Figure 2). In contrast, all five tumors with either EWSR1‐CREM or FUS‐CREM fusion appear to have resulted from unbalanced translocations (Figure 2). Recurrent loss of distal 10p with a breakpoint occurring at the CREM locus at 10p11 was seen in all five tumors, accompanied by either loss of distal 22q with a breakpoint occurring at the EWSR1 locus at 22q12, or gain of distal 16p with a breakpoint at the FUS locus at 16p11.
FIGURE 2.
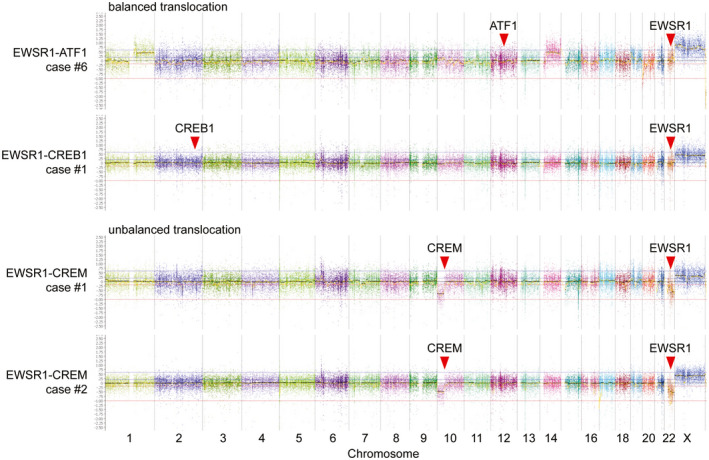
Chromosomal copy number analysis demonstrates that EWSR1‐ATF1 and EWSR1‐CREB1 fusions typically result from balanced translocations, whereas EWSR1‐CREM fusions typically result from unbalanced translocations
3.4. Histologic features
The intracranial mesenchymal tumors with FET‐CREB fusion in this series demonstrated a wide morphologic spectrum with several recurrent histologic features, some of which were observed among tumors with all fusion types and some of which were enriched in tumors with specific fusion types (Figures 3 and 4, and Table 2). A unifying feature of all tumors was a collagenous stroma with dense intercellular matrix, as highlighted by reticulin staining. A subset of tumors also demonstrated fibrous septa separating nodules of tumor cells. The background stroma ranged from mucin‐rich, to collagenous, to nearly absent. Tumor cell morphology included epithelioid/rhabdoid cells, stellate/spindle cells, and round cells (less frequent). In most cases, the stellate/spindle cell morphology occurred in those tumors with a mucin‐rich stroma, but occasional tumors were also seen with epithelioid morphology and a mucin‐rich stroma histologically mimicking chordoid meningioma (Figure 5). Mitotic activity was generally low, typically less than 5 mitoses per 1 mm2. Dense lymphoplasmacytic cuffing at the tumor periphery or along fibrous septa and evidence of prior intratumoral hemorrhage with hemosiderin or hematoidin were seen in the majority of cases. Hemangioma‐like collections of dilated thin‐walled vessels were a frequent finding, whereas staghorn‐like branching vessels characteristic of solitary fibrous tumor/hemangiopericytoma were infrequent. Blood‐filled pseudoangiomatous spaces characteristic of AFH occurring in extracranial soft tissue were not observed. Cellular whorls resembling those of meningioma were observed in a subset. Necrosis was not a common feature but was observed in two tumors. Most tumors demonstrated relatively monotonous round or ovoid nuclei, but two of the tumors (CREB1 #5 and FUS #1) demonstrated increased nuclear pleomorphism and cytologic atypia. Tumor CREB1 #5 with EWSR1‐CREB1 fusion was predominantly composed of stellate/spindle cells in a mucin‐rich stroma with abrupt transition to hypercellular foci of cytologically atypical and mitotically active cells lacking a myxoid stroma. Tumor FUS #1 with FUS‐CREM fusion was composed of dense sheets of epithelioid to rhabdoid cells with striking cytologic atypia and mitotic activity diffusely throughout the tumor (Figure 6).
FIGURE 3.
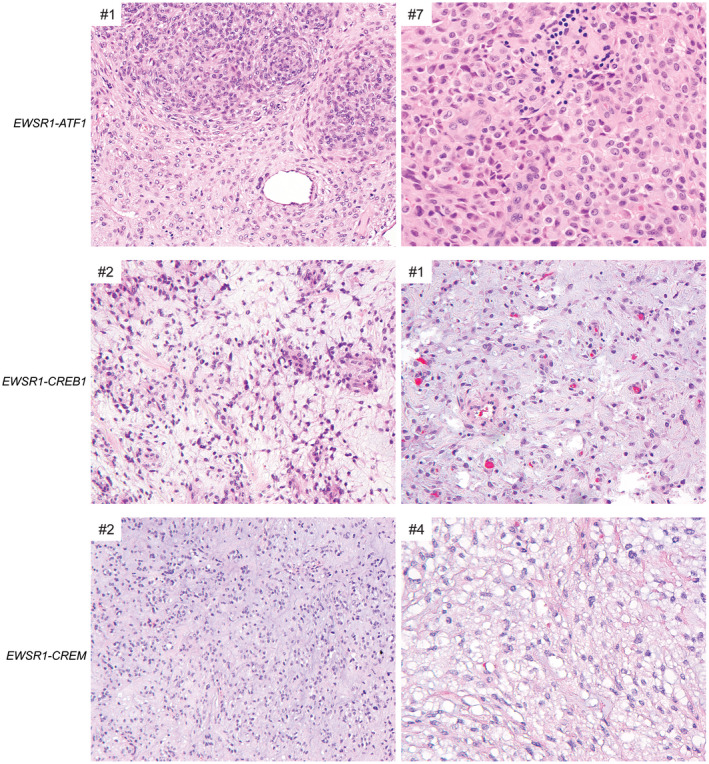
Histologic features of intracranial mesenchymal tumors with FET‐CREB fusion. Those tumors with EWSR1‐ATF1 fusion were most often composed of sheets of epithelioid to rhabdoid cells in a mucin‐poor collagenous stroma. Those tumors with EWSR1‐CREB1 or EWSR1‐CREM fusions were most often composed of cords and clusters of stellate to spindle cells in a mucin‐rich stroma
FIGURE 4.
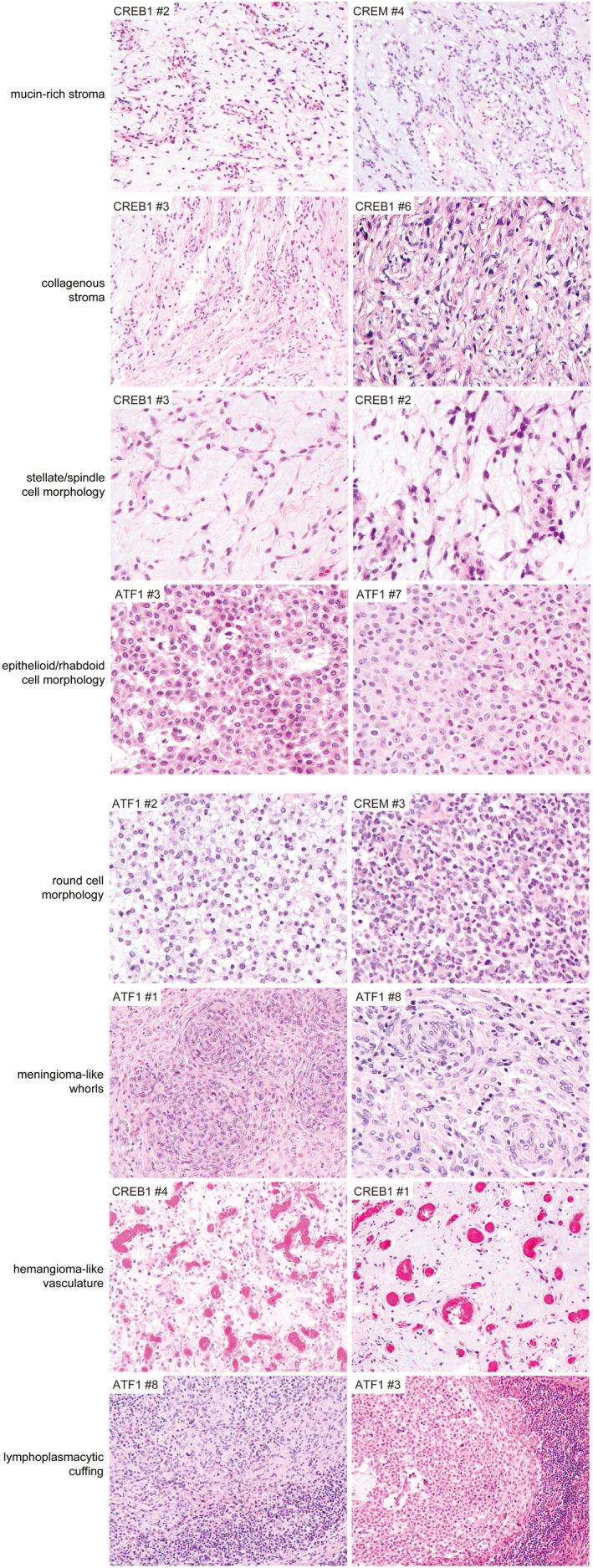
Specific histologic features recurrently observed in intracranial mesenchymal tumors with FET‐CREB fusion. Example illustrations are shown of the characteristic histologic features recurrently observed across all fusion types (e.g., collagenous stroma, lymphoplasmacytic cuffing) and those enriched in specific fusion types (e.g., epithelioid/rhabdoid cell morphology with EWSR1‐ATF1 fusion; mucin‐rich stroma, stellate/spindle cell morphology, and hemangioma‐like vasculature with EWSR1‐CREB1 fusion)
TABLE 2.
Histologic features of intracranial mesenchymal tumors with FET‐CREB fusions
| Histologic feature | All tumors | EWSR1‐ATF1 | EWSR1‐CREB1 | EWSR1‐CREM | FUS‐CREM | |
|---|---|---|---|---|---|---|
| Mucin‐rich stromaa | 12/20 (60%) | 3/8 (38%) | 7/7 (100%) | 2/4 (50%) | 0/1 (0%) | p = 0.03 |
| Collageneous stroma ‐ intercellular matrix | 20/20 (100%) | 8/8 (100%) | 7/7 (100%) | 4/4(100%) | 1/1 (100%) | |
| Collagenous stroma ‐ internodular septae | 14/20 (70%) | 4/8 (50%) | 6/7 (86%) | 3/4 (75%) | 1/1 (100%) | |
| Epithelioid/rhabdoid morphologya | 10/20 (50%) | 7/8 (88%) | 1/7 (14%) | 1/4 (25%) | 1/1 (100%) | p = 0.01 |
| Stellate/spindle cell morphologya | 10/20 (50%) | 1/8 (13%) | 7/7 (100%) | 2/4 (50%) | 0/1 (0%) | p = 0.001 |
| Round cell morphology | 2/20 (10%) | 1/8 (13%) | 0/7 (0%) | 1/4 (25%) | 0/1 (0%) | |
| Hemangioma‐like vasculaturea | 10/20 (50%) | 2/8 (25%) | 6/7 (86%) | 2/4 (50%) | 0/1 (0%) | p = 0.04 |
| Staghorn/HPC‐like vasculature | 4/20 (20%) | 1/8 (13%) | 1/7 (14%) | 2/4 (50%) | 0/1 (0%) | |
| Pseudoangiomatoid vasculature | 0/20 (0%) | 0/8 (0%) | 0/7 (0%) | 0/4 (0%) | 0/1 (0%) | |
| Dense lymphoplasmacytic cuffing | 12/20 (60%) | 4/8 (50%) | 6/7 (86%) | 1/4 (25%) | 1/1 (100%) | |
| Hemosiderin/hematoidin | 13/20 (65%) | 3/8 (38%) | 6/7 (86%) | 3/4 (75%) | 1/1 (100%) | |
| Meningioma‐like whorls | 4/20 (20%) | 3/8 (38%) | 0/7 (0%) | 1/4 (25%) | 0/1 (0%) | |
| Amianthoid fibers | 2/20 (10%) | 1/8 (13%) | 1/7 (14%) | 0/4 (0%) | 0/1 (0%) | |
| Necrosis | 2/20 (10%) | 1/8 (13%) | 0/7 (0%) | 1/4 (25%) | 0/1 (0%) |
Denotes histologic features which are (bold values) statistically significantly different (p < 0.05) when comparing tumors with EWSR1‐ATF1 fusion versus EWSR1‐CREB1 fusion via unpaired, non‐parametric t‐test (Mann‐Whitney).
FIGURE 5.
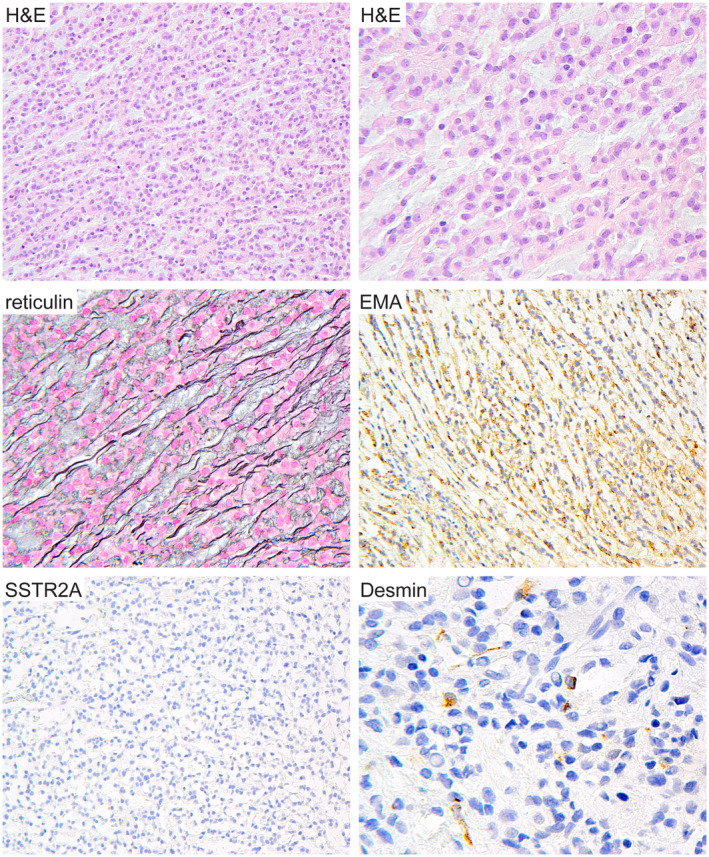
Intracranial mesenchymal tumor with FET‐CREB fusion can histologically mimic chordoid meningioma. Patient ATF1 #2 is a 9‐year‐old female who presented with an extra‐axial mass along the cerebral convexity of the left frontal lobe. Surgical resection demonstrated a neoplasm composed of cords and clusters of epithelioid cells in a mucin‐rich stroma closely resembling chordoid meningioma. However, the tumor was negative for SSTR2A expression and contained small foci of tumor cells with desmin expression. Genomic interrogation revealed an in‐frame EWSR1‐ATF1 fusion and absence of mutations involving NF2 and other genes characteristic of meningioma
FIGURE 6.
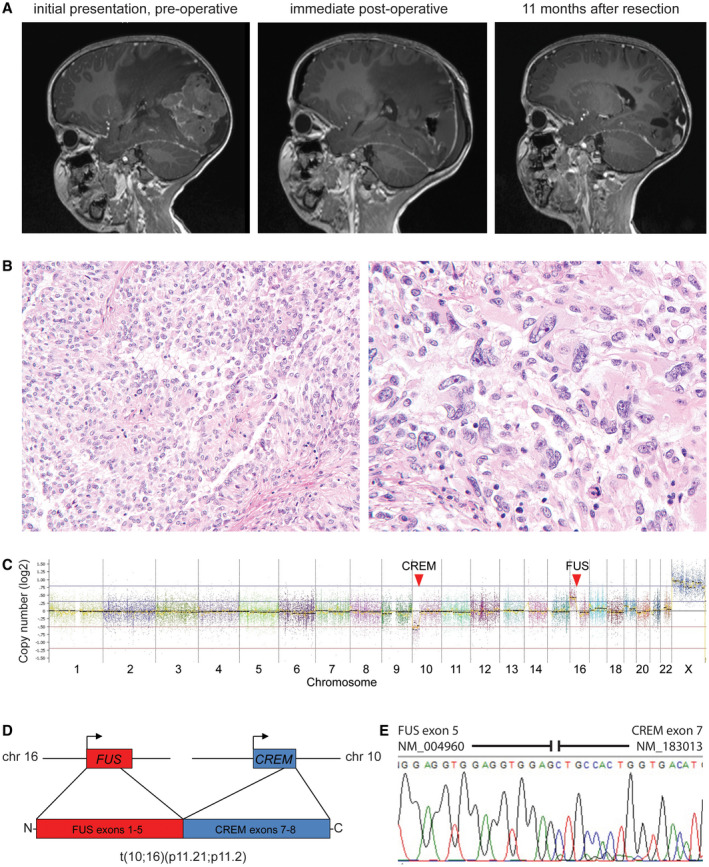
Intracranial mesenchymal tumor with FUS‐CREM fusion. A, A 4‐year‐old girl presented with fevers and headaches and was found to have a large circumscribed and heterogeneously‐enhancing mass in the left occipital region of the brain with significant peritumoral edema. After gross total resection, local tumor recurrence was seen on follow‐up imaging as multiple‐enhancing nodules at the periphery of the prior resection cavity. B, Histologic sections demonstrated a highly cellular neoplasm with both sheet‐like and papillary growth patterns composed of epithelioid to rhabdoid tumor cells with prominent nuclear pleomorphism and atypia. C, Chromosomal copy number plot demonstrates that the FUS‐CREM fusion was the result of an unbalanced translocation between chromosome 10p11.21 and chromosome 16p11.2. D, Schematic of the FUS‐CREM gene fusion. E, Sanger chromatogram following reverse‐transcription PCR of the FUS‐CREM fusion transcript composed of exons 1–5 of FUS linked with exons 7–8 of CREM
When comparing histologic features versus specific fusion type, four histologic features were significantly enriched in specific fusion types (Table 2). Epithelioid or rhabdoid cell morphology was more often observed in tumors with EWSR1‐ATF1 fusion versus EWSR1‐CREB1 fusion (p = 0.01). Conversely, mucin‐rich stroma, stellate/spindle cell morphology, and hemangioma‐like vasculature were more likely to be observed in tumors with EWSR1‐CREB1 fusion versus EWSR1‐ATF1 fusion (p = 0.03, 0.001, and 0.04, respectively).
3.5. Immunohistochemical features
All evaluated tumors in this cohort were positive for desmin expression, which ranged from diffuse strong labeling to limited focal clusters of tumor cells (Table 3 and Figure 7). The majority of cases were also positive for EMA and CD99 in a membranous pattern. Variable S100, synaptophysin, and CD68 immunostaining was also observed in a subset of tumors. Most tumors demonstrated no immunoreactivity for cytokeratins AE1/AE3 and CAM5.2, with only one tumor demonstrating focal labeling for AE1/AE3. MUC4 immunostaining was observed in five of seven evaluated tumors, which ranged from diffuse strong labeling to focal clusters of positive cells. Pan‐NTRK immunohistochemistry demonstrated positivity in only one of five evaluated tumors. All evaluated tumors were negative for expression of glial fibrillary acidic protein (GFAP), myogenin, somatostatin receptor 2A (SSTR2A), and HMB45. All tumors tested (n = 5) had intact/retained expression of INI‐1 (also referred to BAF47 or SMARCB1). Reticulin staining demonstrated abundant intercellular basement membrane deposition in all evaluated tumors (n = 12). The Ki‐67 labeling index was generally low (less than 5%, seven of the 13 evaluated tumors), but occasionally was elevated up to 15–20% (six of the 13 evaluated tumors). There was no significant association of specific CREB fusion partner with distinct immunophenotype or Ki‐67 labeling index observed.
TABLE 3.
Immunohistochemical features of intracranial mesenchymal tumors with FET‐CREB fusions
| All tumors | EWSR1‐ATF1 | EWSR1‐CREB1 | EWSR1‐CREM | FUS‐CREM | |
|---|---|---|---|---|---|
| Desmin | 16/16 (100%) | 7/7 (100%) | 6/6 (100%) | 2/2 (100%) | 1/1 (100%) |
| EMA | 13/14 (93%) | 5/5 (100%) | 5/6 (83%) | 2/2 (100%) | 1/1 (100%) |
| CD99 | 10/11 (91%) | 4/5 (80%) | 4/4 (100%) | 1/1 (100%) | 1/1 (100%) |
| S100 | 5/12 (42%) | 3/6 (50%) | 2/5 (40%) | ND | 0/1 (0%) |
| CD68 | 5/6 (100%) | 1/1 (100%) | 2/3 (100%) | 1/1 (100%) | 1/1 (100%) |
| MUC4 | 5/7 (71%) | 3/5 (60%) | 1/1 (100%) | 1/1 (100%) | ND |
| pan‐NTRK | 1/5 (20%) | 0/3 (0%) | 1/1 (100%) | 0/1 (0%) | ND |
| Synaptophysin | 5/8 (63%) | 1/2 (50%) | 2/4 (50%) | 1/1 (100%) | 1/1 (100%) |
| GFAP | 0/9 (0%) | 0/1 (0%) | 0/6 (0%) | 0/1 (0%) | 0/1 (0%) |
| Myogenin | 0/6 (0%) | ND | 0/4 (0%) | 0/1 (0%) | 0/1 (0%) |
| SSTR2a | 0/8 (0%) | 0/4 (0%) | 0/3 (0%) | ND | 0/1 (0%) |
| HMB45 | 0/6 (0%) | 0/3 (0%) | 0/2 (0%) | ND | 0/1 (0%) |
| Cytokeratin AE1/AE3 | 1/7 (17%) | 1/2 (50%) | 0/4 (0%) | 0/1 (0%) | ND |
| Cytokeratin CAM5.2 | 0/9 (0%) | 0/2 (0%) | 0/5 (0%) | 0/1 (0%) | 0/1 (0%) |
FIGURE 7.
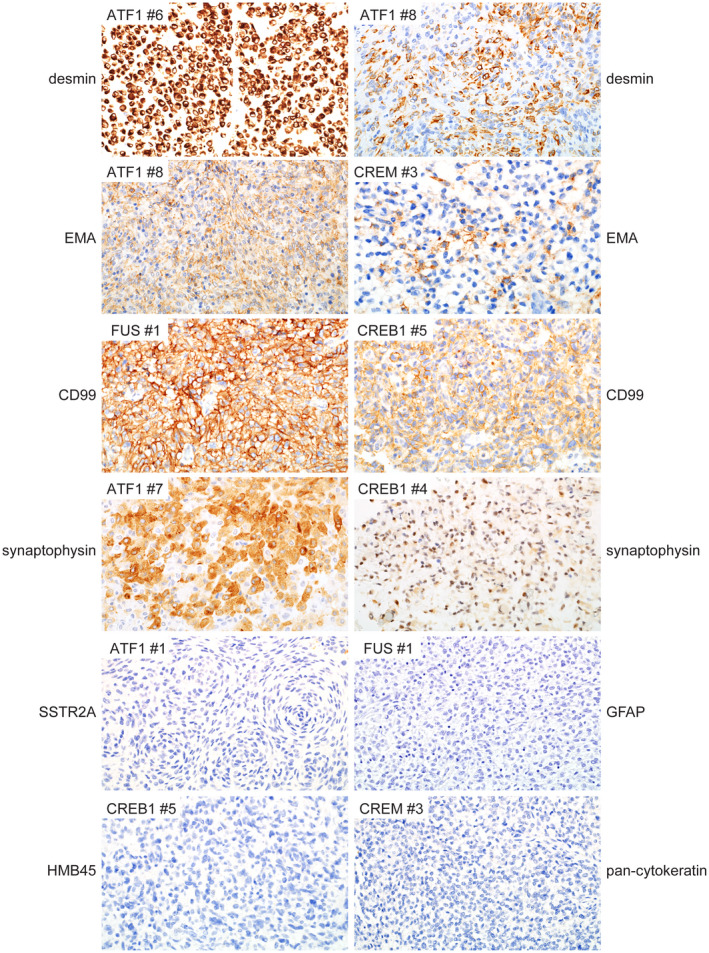
Immunohistochemical features of intracranial mesenchymal tumors with FET‐CREB fusion. These tumors were uniformly positive for desmin expression, which ranged from diffuse strong labeling to cases with focal clusters of positive cells only. Most tumors were also positive for EMA and CD99 expression in a membranous distribution. A subset of tumors demonstrated positivity for synaptophysin that ranged from diffuse strong to more focal and weak staining. These tumors were uniformly negative for SSTR2A, GFAP, and HMB45. Most tumors were negative for cytokeratin expression, with only a single tumor that demonstrated focal positivity for cytokeratin AE1/AE3 (not shown)
3.6. Clinical outcomes
The complete clinical data including extent of resection, treatment regimen, and outcome data from the 20 patients are presented in Table 1 and Table S1. Nine patients initially had a gross total resection, whereas only a subtotal resection was achieved in eight patients. Extent of resection was unknown in the remaining three patients. None of the patients who had gross total resection were treated with adjuvant therapy, whereas five of the eight patients who had subtotal resection were treated with adjuvant radiation (n = 3), adjuvant chemotherapy (n = 1), or both radiation and chemotherapy (n = 1). The chemotherapy regimens were both sarcoma‐based using ifosfamide together with vincristine and doxorubicin or carboplatin and etoposide. Among the 19 patients with available clinical follow‐up, 11 patients (58%) experienced tumor recurrence/progression and three patients (16%) died of disease, all of whom had tumors with EWSR1‐ATF1 fusion that were subtotally resected. Kaplan–Meier analysis of overall survival and progression‐free survival stratified by extent of resection revealed that subtotal resection was associated with increased risk of death and tumor recurrence, although neither was statistically significant (Figure 8). Seven of the nine patients (78%) with subtotal resection experienced local recurrence/progression within 12 months, and one patient (ATF1 #6) developed metastatic disease to the thoracic spine and lymph nodes. Kaplan–Meier analysis of overall survival and progression‐free survival stratified by mucin‐rich versus mucin‐poor stroma revealed a possible trend toward improved outcomes for those tumors with mucin‐rich stroma, although this was not statistically significant (Figure 9). Among the 12 cases with available clinical follow‐up data that were evaluated for Ki‐67 labeling index, the subset of patients with elevated tumor proliferative indices (greater than 5%) had increased frequency of recurrence (five of six patients [83%]), whereas the subset of patients with low tumor proliferative indices (less than 5%) had lower frequency of recurrence (three of six patients [50%]).
FIGURE 8.
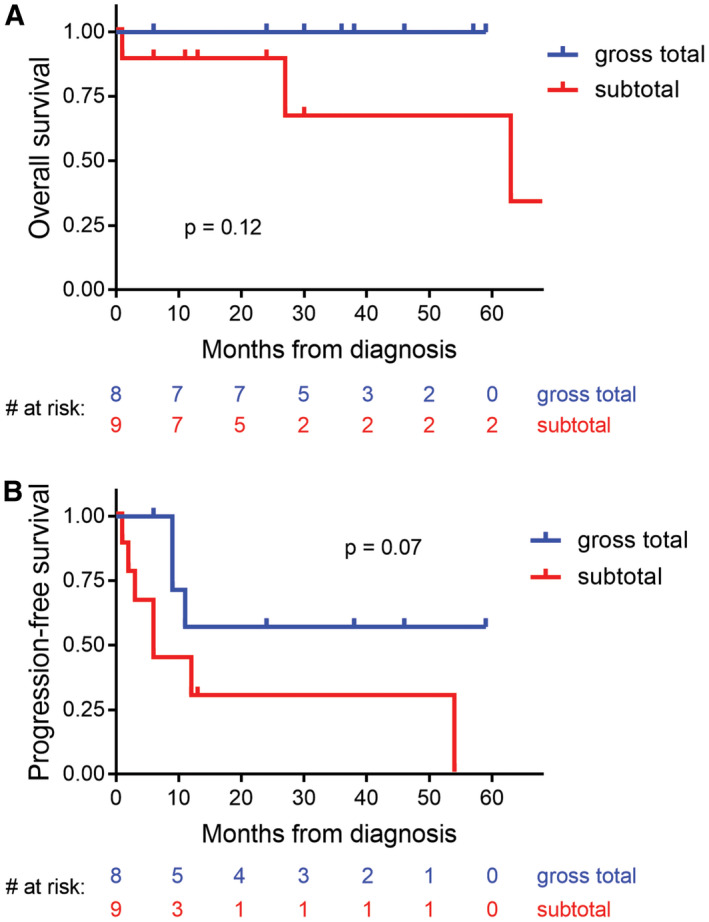
Clinical outcomes for patients with intracranial mesenchymal tumors with FET‐CREB fusion based on extent of surgical resection. A, Kaplan–Meier plot of the overall survival for the 17 patients from this study with known extent of resection. B, Kaplan–Meier plot of progression‐free survival for the 17 patients from this study with known extent of resection. Compared to gross total resection, subtotal resection was associated with increased risk of death and tumor recurrence (although neither was statistically significant)
FIGURE 9.
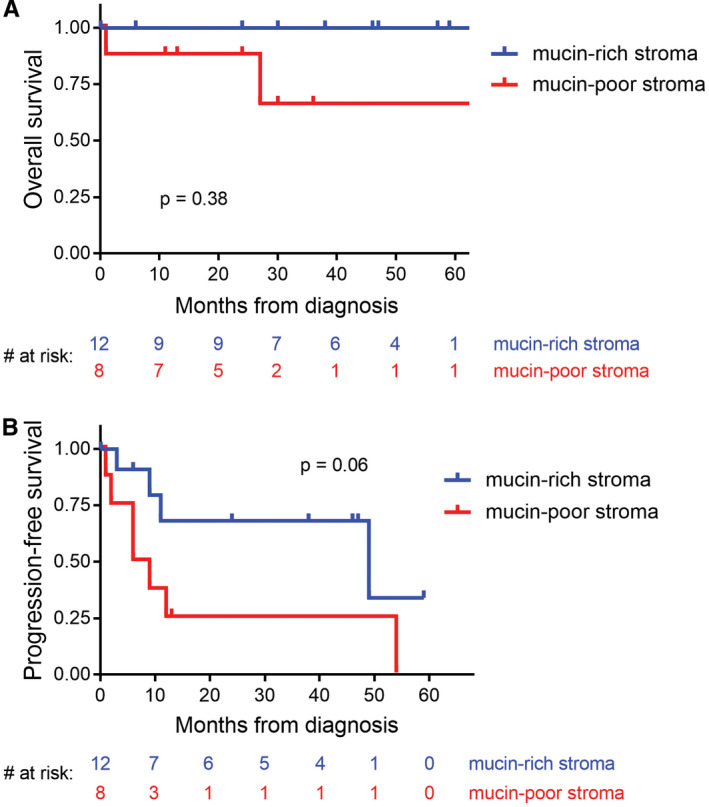
Clinical outcomes for patients with intracranial mesenchymal tumors with FET‐CREB fusion based on mucin content of the stroma. A and B, Kaplan–Meier plot of overall survival (A) and progression‐free survival (B) for the 20 patients from this study stratified by mucin‐rich versus mucin‐poor stroma
In order to further assess the clinical outcomes for these tumors, we next curated clinical data from all reported cases of intracranial mesenchymal tumors with confirmed FET‐CREB fusions published in the scientific literature to date (Table 4). Patient age, sex, tumor location, and survival data were analyzed from the 20 patients in our cohort together with these 18 previously reported patients (Figure 10). The median patient age was 17 years (range 4–70 years) at time of initial diagnosis. These 38 patients included 12 males and 26 females. Tumors were located at the cerebral convexities (n = 14), falx (6), lateral ventricle (6), third ventricle (1), skull base (1), CP angle (5), tentorium (3), and spinal cord (1). No significant association of fusion type with patient age or tumor location was apparent (Figure 10). Kaplan–Meier analysis for the 38 patients revealed a median overall survival of greater than 60 months with 91% survival rate at 5 years (Figure 11A), and a median progression‐free survival of 28 months (Figure 11B). Kaplan–Meier analysis of overall survival or progression‐free survival stratified by fusion type did not identify a statistically significant difference in outcomes (Figure 11C,D). However, three patients with tumors containing EWSR1‐ATF1 fusion succumbed to disease, while all patients with EWSR1‐CREB1 or EWSR1‐CREM fusion remained alive at time of last clinical follow‐up.
TABLE 4.
Clinicopathologic features of previously reported intracranial mesenchymal tumors with FET‐CREB fusions
| Age | Sex | Location | Fusion | Diagnosis | Adjuvant XRT or chemotherapy | Outcome | Length of follow‐up | Reference |
|---|---|---|---|---|---|---|---|---|
| 25 | M | Occipital | EWSR1‐ATF1 | AFH | Unknown | Unknown | None | Dunham 2008 |
| 15 | F | Not specified | EWSR1‐CREM | IMMT | None | Alive, disease‐free | 17 months | Kao 2017 |
| 23 | F | Occipital | EWSR1‐CREB1 | IMMT | Unknown | Unknown | None | Kao 2017 |
| 20 | M | Frontal | EWSR1‐CREB1 | IMMT | Unknown | Unknown | None | Kao 2017 |
| 12 | M | Frontal | EWSR1‐ATF1 | IMMT | Unknown | Unknown | None | Kao 2017 |
| 19 | M | CP angle | EWSR1‐CREM | AFH | XRT +chemo | Alive w/ local recurrence at 10 years | 120 months | Gareton 2018 |
| 17 | F | Frontal | EWSR1‐ATF1 | IMMT | None | Alive w/ local recurrences at 20 months and 7 years | 90 months | Sciot 2018 |
| 18 | M | Frontal | EWSR1‐CREM | IMMT | None | Alive, disease‐free | 12 months | Bale 2018 |
| 17 | M | CP angle | EWSR1‐CREB1 | IMMT | None | Alive w/ local recurrences at 8 months and 20 months | 36 months | Velz 2018 |
| 13 | F | Frontal | EWSR1‐ATF1 | AFH | None | Alive w/ local recurrence at 5 years | 72 months | Konstantinidis 2019 |
| 12 | F | Frontal | EWSR1‐CREM | AFH | None | Alive w/ local recurrence at 28 months | 28 months | Konstantinidis 2019 |
| 58 | F | Parietal | EWSR1‐CREB1 | myxoid AFH | None | Alive w/ residual disease | 3 months | Ghanbari 2019 |
| 9 | M | Frontal | EWSR1‐CREM | IMMT | None | Alive w/ local recurrence at 6 months | 6 months | White 2019 |
| 53 | F | Third ventricle | EWSR1‐CREB1 | IMMT | XRT | Alive, disease‐free | 3 months | Komastu 2020 |
| 67 | M | Temporal | EWSR1‐ATF1 | IMMT | Unknown | Unknown | None | Ballester 2020 |
| 50 | F | Frontal | EWSR1‐ATF1 | AFH | Unknown | Unknown | None | Vizcaino 2020 |
| 48 | F | Lateral ventricle | EWSR1‐ATF1 | IMMT | None | Alive w/ local recurrence at 4 months | 17 months | Ward 2020 |
| 58 | F | Lateral ventricle | EWSR1‐CREB1 | IMMT | None | Alive, disease‐free | 6 months | Aguiar 2020 |
FIGURE 10.
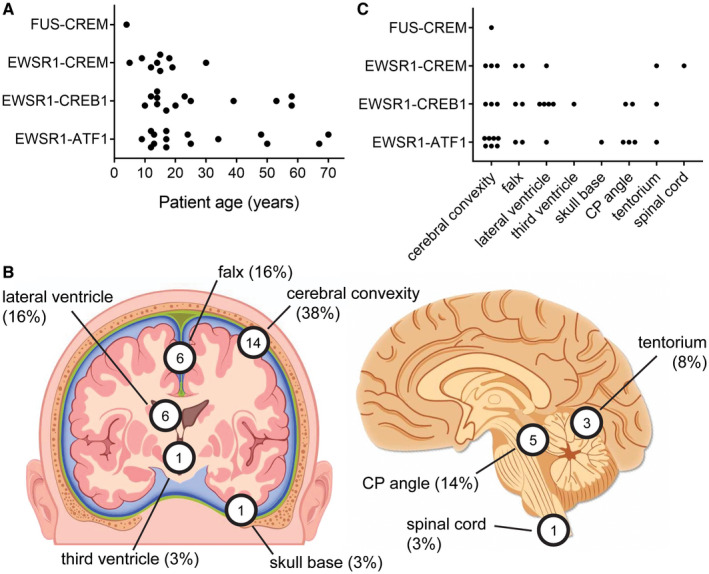
Anatomic location and age at diagnosis for intracranial mesenchymal tumors with FET‐CREB fusion from this study combined with all previously reported cases in the literature (see Table 4). A, Plot of patient age at diagnosis versus gene fusion. B, Diagram of tumor locations. C. Plot of tumor location versus gene fusion
FIGURE 11.
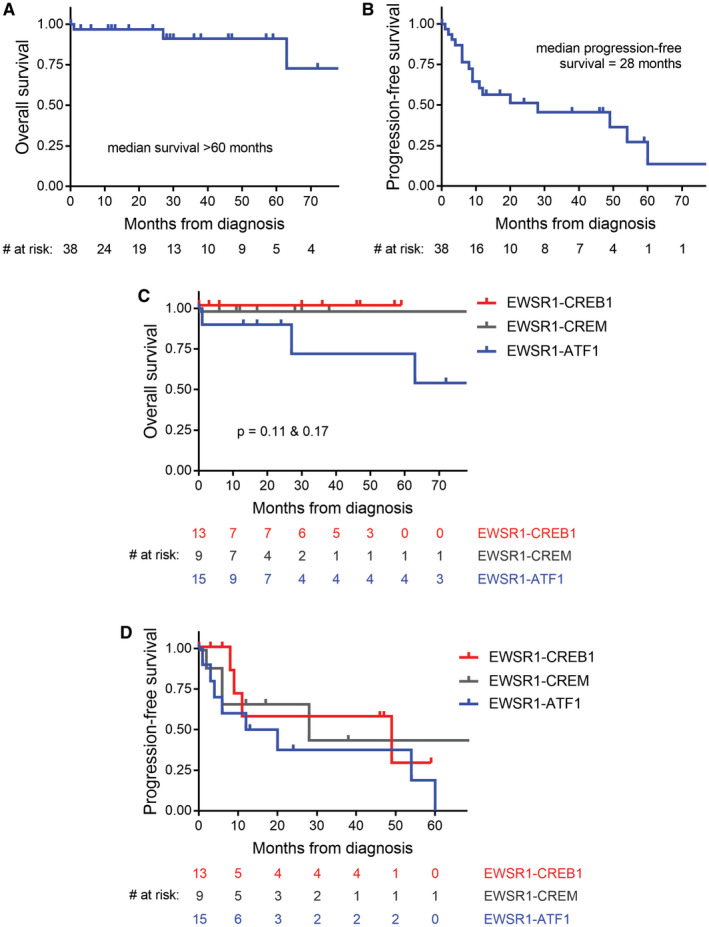
Clinical outcomes for patients with intracranial mesenchymal tumors with FET‐CREB fusion from this study combined with all previously reported cases in the literature (see Table 4). A, Kaplan–Meier plot of overall survival demonstrates a median overall survival >60 months. B, Kaplan–Meier plot of progression‐free survival demonstrates that a majority of patients experience disease recurrence with a median interval of 28 months. C, Kaplan–Meier plot of overall survival stratified by fusion type. While no statistically significant difference in overall survival based on fusion type is observed, three patients with tumors containing EWSR1‐ATF1 fusion succumbed to disease, while all patients with EWSR1‐CREB1 or EWSR1‐CREM fusion remained alive at time of last clinical follow‐up. D, Kaplan–Meier plot of progression‐free survival stratified by fusion type. No significant difference was observed based on fusion type (median recurrence‐free survival: EWSR1‐ATF1, 16 months; EWRS1‐CREB1, 49 months; EWSR1‐CREM, 28 months)
4. DISCUSSION
Here, we have studied a cohort of 20 patients with intracranial mesenchymal tumors harboring FET‐CREB fusions, and comprehensively characterized their radiologic, molecular, and clinicopathologic features. Additionally, we have compiled and reviewed the 18 previously reported cases of intracranial tumors demonstrated to have EWSR1 or FUS fused to a CREB family gene. Our findings demonstrate that these tumors occur with a female predominance (12 males/26 females) in a wide age range (4–70 years old), but most often occur in the second and third decades of life (median age of 17 years). They are predominantly extra‐axial or intraventricular tumors which can arise throughout the neuroaxis, including the falx, tentorium, cerebral convexities, skull base, spinal cord, lateral ventricle, and third ventricle. They are typically contrast‐enhancing masses, well circumscribed with solid and cystic growth patterns, and often have pronounced peritumoral edema. Of the 38 total tumors, 15 had EWSR1‐ATF1 fusion, 13 EWSR1‐CREB1, 9 EWSR1‐CREM, and 1 FUS‐CREM. Beyond the oncogenic fusions, no additional pathogenic mutations, amplifications, or deletions were identified in our tumor cohort by a broad NGS panel of approximately 500 cancer‐associated genes. Most of these tumors demonstrated a diploid or near‐diploid genome with a paucity of chromosomal gains and losses. While all tumors with EWSR1‐ATF1 and EWSR1‐CREB1 fusions were the result of balanced translocations, all tumors with EWSR1‐CREM or FUS‐CREM fusions demonstrated intrachromosomal copy number breakpoints occurring at the CREM and EWSR1 or FUS loci, indicative of unbalanced translocation as the mechanism of gene fusion. The reason for this divergence in unbalanced versus balanced translocations for tumors with CREM as the fusion partner versus those with ATF1 or CREB1 is uncertain.
Histologically, intracranial mesenchymal tumors with FET‐CREB fusions demonstrate a range of morphologic features that encompass both those previously described in AFH and IMMT. We note that tumors with CREB1 or CREM as the fusion partners are enriched for those with stellate/spindle cell morphology and prominent myxoid stroma, and those with ATF1 are enriched for those with epithelioid morphology with mucin‐poor stroma. However, this was not always the case, as we encountered tumors with EWSR1‐CREB1 fusion and epithelioid morphology with mucin‐poor stroma, as well as tumors with EWSR1‐ATF1 fusion and stellate/spindle cell morphology with mucin‐rich stroma. Additionally, a subset of tumors contained both of these morphologic patterns within the same resection specimen. No immunophenotypic differences were noted when stratifying tumors by fusion type or morphologic pattern.
In terms of clinical outcomes, these tumors demonstrate a propensity for local recurrence and occasionally dissemination or metastasis leading to mortality. The available data suggest improved outcomes for patients that are able to undergo gross total resection. The growth rates have been variable, and the interval to disease recurrence has been up to 10 years. While optimal treatment strategy remains to be defined, adjuvant radiation may be a consideration given the propensity for local recurrence, especially for incompletely resected tumors. Histologic features or other criteria to predict disease course were not identified by this study, although the three patients who died of disease all had EWSR1‐ATF1 fusions. Larger patient cohorts are needed to define prognostic criteria for these neoplasms.
These intracranial mesenchymal tumors with FET‐CREB fusion can both radiographically and histologically mimic a number of other intracranial tumor entities. The extra‐axial location and dural tail seen radiologically in a subset of tumors can result in a preoperative impression of meningioma or solitary fibrous tumor/hemangiopericytoma. Microscopically, these tumors can feature syncytial growth and meningioma‐like whorls that closely resemble meningioma. Additionally, those with rhabdoid cytology, or with cords of epithelioid cells in a mucin‐rich stroma, could be misinterpreted as rhabdoid or chordoid variants of meningioma (Figure 4). The frequent expression of EMA in these tumors can further confound the differential diagnosis of meningioma. Fortunately, these tumors are negative for SSTR2A and positive for desmin, which can be used to distinguish them immunohistochemically. Furthermore, FET‐CREB fusions are specific to these tumors and are not found in meningiomas, and all the 19 tumors analyzed by next‐generation sequencing in this study lacked meningioma‐associated mutations in NF2, TRAF7, KLF4, SMO, AKT1, PIK3CA, and BAP1. The extra‐axial location and prominent collagenous stroma can also raise the diagnostic possibility of solitary fibrous tumor/hemangiopericytoma. However, intracranial mesenchymal tumors with FET‐CREB fusions lack nuclear STAT6 immunopositivity and the characteristic NAB2‐STAT6 fusion. They are desmin positive, but lack expression of skeletal muscle markers including myogenin and MyoD1 that allows distinction from rhabdomyosarcoma. They uniformly have intact expression of INI1 and lack SMARCB1 mutations or deletions, which distinguishes them from atypical teratoid/rhabdoid tumor and proximal‐type epithelioid sarcoma. A subset of these tumors demonstrates MUC4 immunopositivity, a protein marker previously thought to be highly specific to low‐grade fibromyxoid sarcoma and sclerosing epithelioid fibrosarcoma, tumor entities which are defined by fusion of FUS (or rarely EWSR1) to either CREB3L1 or CREB3L2. The subset of tumors with round cell morphology, collagenous stroma, and polyphenotypic differentiation by immunohistochemistry with the combination of desmin, EMA, and synaptophysin positivity overlaps with desmoplastic small round cell tumor (DSRCT), which do rarely occur intracranially (39). Fluorescence in situ hybridization showing EWSR1 break‐apart does not allow distinction between DSRCT with EWSR1‐WT1 fusions and those intracranial mesenchymal tumors with EWSR1 fusions to CREB family members. Therefore, the distinction from DSRCT requires additional testing to determine the specific fusion partner. The possibility of a brain metastasis from a primary extracranial tumor should be considered as part of the workup of these patients, although all of these tumors described to date have appeared to be primary intracranial neoplasms. Noteworthy is that these intracranial mesenchymal tumors with FET‐CREB fusion are negative for expression of melanocytic markers such as HMB45 and MITF that is characteristic of clear cell sarcoma of soft tissue, and are almost always positive for desmin expression which is absent in both clear cell sarcoma of soft tissue and primary pulmonary myxoid sarcoma (11, 13).
Because of the overlapping histologic, immunophenotypic, and molecular features of IMMT and intracranial AFH‐like neoplasms, there has been ongoing uncertainty as to whether these tumors represent two distinct entities, versus a single entity defined by intracranial location and FET‐CREB fusions (1, 3). The overlapping histologic spectrum seen across these tumors with different fusion types (and even within the same tumor) suggests that intracranial AFH and IMMT may likely represent morphologic variants of a single biologic tumor entity. In support of this, the clinical outcome data did not show significant differences in overall or progression‐free survival when stratified by fusion type or mucin‐rich versus mucin‐poor stroma.
In summary, we have characterized a group of intracranial tumors with FET‐CREB fusions, which to date have been categorized as either intracranial AFH or IMMT. Further investigation including epigenetic profiling is needed to define the true nature of these unique intracranial neoplasms and their exact relationship to extracranial tumors with similar fusions. Future studies are also needed to explore the relationships between patient outcomes and additional clinicopathologic variables such tumor location, extent of resection, fusion type, and morphologic features.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
AUTHOR CONTRIBUTIONS
E.A.S. performed the DNA extractions. E.A.S. and D.A.S. performed the targeted next‐generation sequencing analysis. E.A.S., A.P., and D.A.S. performed pathologic assessment. J.E.V. performed radiologic assessment. E.A.S., J.C., S.A., J.M.E., D.J.C., W.W., M.M., N.U., E.C.B., M.W., M.P., R.O., A.O., A.G., B.K.D., M.B.L., C.R., M.S.B., S.C., A.R., B.R., S.P.F., J.C.L., J.W.H., S.J.C., A.E.H., M.P., T.T., A.W.B., F.R., D.W.E., A.P., and D.A.S. provided clinical care and contributed to the cohort. E.A.S. and D.A.S. conceptualized the study, reviewed all data, prepared the figures, and wrote the manuscript. All authors critically reviewed the manuscript and approved its submission.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
This study was approved by the Committee on Human Research of the University of California, San Francisco, with a waiver of patient consent.
Supporting information
ACKNOWLEDGMENTS
We thank the staff of the UCSF Clinical Cancer Genomics Laboratory for assistance with genetic profiling. This study was supported by the NIH Director's Early Independence Award from the Office of the Director, National Institutes of Health (DP5 OD021403) to D.A.S.
DATA AVAILABILITY STATEMENT
Scanned image files of H&E stained sections from 19 of the tumors in this cohort are available for downloading and viewing at the following link: https://figshare.com/projects/Intracranial_mesenchymal_tumors_with_EWS‐CREB_fusion/88661. Structural variant and chromosomal copy number data are available in the online supplementary material. Raw sequencing data files are available from the authors upon request.
REFERENCES
- 1.Bale TA, Oviedo A, Kozakewich H, Giannini C, Davineni PK, Ligon K, et al. Intracranial myxoid mesenchymal tumors with EWSR1‐CREB family gene fusions: myxoid variant of angiomatoid fibrous histiocytoma or novel entity? Brain Pathol. 2018;28:183–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dunham C, Hussong J, Seiff M, Pfeifer J, Perry A. Primary intracerebral angiomatoid fibrous histiocytoma: report of a case with a t(12;22)(q13;q12) causing type 1 fusion of the EWS and ATF‐1 genes. Am J Surg Pathol. 2008;32:478–84. [DOI] [PubMed] [Google Scholar]
- 3.Kao YC, Sung YS, Zhang L, Chen CL, Vaiyapuri S, Rosenblum MK, et al. EWSR1 fusions with CREB family transcription factors define a novel myxoid mesenchymal tumor with predilection for intracranial location. Am J Surg Pathol. 2017;41:482–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Konstantinidis A, Cheesman E, O'Sullivan J, Pavaine J, Avula S, Pizer B, et al. Intracranial angiomatoid fibrous histiocytoma with EWSR1‐CREB family fusions: a report of two pediatric cases. World Neurosurg. 2019;126:113–9. [DOI] [PubMed] [Google Scholar]
- 5.Antonescu CR, Dal Cin P, Nafa K, Teot LA, Surti U, Fletcher CD, et al. EWSR1‐CREB1 is the predominant gene fusion in angiomatoid fibrous histiocytoma. Genes Chromosomes Cancer. 2007;46:1051–60. [DOI] [PubMed] [Google Scholar]
- 6.Antonescu CR, Nafa K, Segal NH, Dal Cin P, Ladanyi M. EWS‐CREB1: a recurrent variant fusion in clear cell sarcoma—association with gastrointestinal location and absence of melanocytic differentiation. Clin Cancer Res. 2006;12:5356–62. [DOI] [PubMed] [Google Scholar]
- 7.Antonescu CR, Katabi N, Zhang L, Sung YS, Seethala RR, Jordan RC, et al. EWSR1‐ATF1 fusion is a novel and consistent finding in hyalinizing clear‐cell carcinoma of salivary gland. Genes Chromosomes Cancer. 2011;50:559–70. [DOI] [PubMed] [Google Scholar]
- 8.Antonescu CR, Tschernyavsky SJ, Woodruff JM, Jungbluth AA, Brennan MF, Ladanyi M. Molecular diagnosis of clear cell sarcoma: detection of EWS‐ATF1 and MITF‐M transcripts and histopathological and ultrastructural analysis of 12 cases. J Mol Diagn. 2002;4:44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Argani P, Harvey I, Nielsen GP, Takano A, Suurmeijer AJH, Voltaggio L, et al. EWSR1/FUS–CREB fusions define a distinctive malignant epithelioid neoplasm with predilection for mesothelial‐lined cavities. Mod Pathol. 2020;33(11):2233–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Desmeules P, Joubert P, Zhang L, Al‐Ahmadie HA, Fletcher CD, Vakiani E, et al. A subset of malignant mesotheliomas in young adults are associated with recurrent EWSR1/FUS‐ATF1 fusions. Am J Surg Pathol. 2017;41:980–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferrari A, Casanova M, Bisogno G, Mattke A, Meazza C, Gandola L, et al. Clear cell sarcoma of tendons and aponeuroses in pediatric patients: a report from the Italian and German Soft Tissue Sarcoma Cooperative Group. Cancer. 2002;94:3269–76. [DOI] [PubMed] [Google Scholar]
- 12.Skálová A, Weinreb I, Hyrcza M, Simpson RH, Laco J, Agaimy A, et al. Clear cell myoepithelial carcinoma of salivary glands showing EWSR1 rearrangement: molecular analysis of 94 salivary gland carcinomas with prominent clear cell component. Am J Surg Pathol. 2015;39:338–48. [DOI] [PubMed] [Google Scholar]
- 13.Thway K, Nicholson AG, Lawson K, Gonzalez D, Rice A, Balzer B, et al. Primary pulmonary myxoid sarcoma with EWSR1‐CREB1 fusion: a new tumor entity. Am J Surg Pathol. 2011;35:1722–32. [DOI] [PubMed] [Google Scholar]
- 14.Waters BL, Panagopoulos I, Allen EF. Genetic characterization of angiomatoid fibrous histiocytoma identifies fusion of the FUS and ATF‐1 genes induced by a chromosomal translocation involving bands 12q13 and 16p11. Cancer Genet Cytogenet. 2000;121:109–16. [DOI] [PubMed] [Google Scholar]
- 15.Yoshida A, Wakai S, Ryo E, Miyata K, Miyazawa M, Yoshida KI, et al. Expanding the phenotypic spectrum of mesenchymal tumors harboring the EWSR1‐CREM fusion. Am J Surg Pathol. 2019;43:1622–30. [DOI] [PubMed] [Google Scholar]
- 16.Thway K, Fisher C. Tumors with EWSR1‐CREB1 and EWSR1‐ATF1 fusions: the current status. Am J Surg Pathol. 2012;36:e1–e11. [DOI] [PubMed] [Google Scholar]
- 17.Thway K, Fisher C. Angiomatoid fibrous histiocytoma: the current status of pathology and genetics. Arch Pathol Lab Med. 2015;139:674–82. [DOI] [PubMed] [Google Scholar]
- 18.Bohman SL, Goldblum JR, Rubin BP, Tanas MR, Billings SD. Angiomatoid fibrous histiocytoma: an expansion of the clinical and histological spectrum. Pathology. 2014;46:199–204. [DOI] [PubMed] [Google Scholar]
- 19.Chen G, Folpe AL, Colby TV, Sittampalam K, Patey M, Chen MG, et al. Angiomatoid fibrous histiocytoma: unusual sites and unusual morphology. Mod Pathol. 2011;24:1560–70. [DOI] [PubMed] [Google Scholar]
- 20.Kao YC, Lan J, Tai HC, Li CF, Liu KW, Tsai JW, et al. Angiomatoid fibrous histiocytoma: clinicopathological and molecular characterisation with emphasis on variant histomorphology. J Clin Pathol. 2014;67:210–5. [DOI] [PubMed] [Google Scholar]
- 21.Shi H, Li H, Zhen T, Zhang F, Dong Y, Zhang W, et al. Clinicopathological features of angiomatoid fibrous histiocytoma: a series of 21 cases with variant morphology. Int J Clin Exp Pathol. 2015;8:772–8. [PMC free article] [PubMed] [Google Scholar]
- 22.Aizpurua M, Zebian B, Minichini V, Sanghvi H, Dudau C, Fisher C, et al. A case of childhood intracerebral angiomatoid fibrous histiocytoma radiologically mimicking infection and with unusual immunopositivity for placental alkaline phosphatase. Clin Neuropathol. 2019;38:245–8. [DOI] [PubMed] [Google Scholar]
- 23.Alshareef MA, Almadidy Z, Baker T, Perry A, Welsh CT, Vandergrift WA 3rd. Intracranial angiomatoid fibrous histiocytoma: case report and literature review. World Neurosurg. 2016;96:403–9. [DOI] [PubMed] [Google Scholar]
- 24.Ballester LY, Meis JM, Lazar AJ, Prabhu SS, Hoang KB, Leeds NE, et al. Intracranial myxoid mesenchymal tumor with EWSR1‐ATF1 fusion. J Neuropathol Exp Neurol. 2020;79:347–51. [DOI] [PubMed] [Google Scholar]
- 25.Gareton A, Pierron G, Mokhtari K, Tran S, Tauziède‐Espariat A, Pallud J, et al. ESWR1‐CREM fusion in an intracranial myxoid angiomatoid fibrous histiocytoma‐like tumor: a case report and literature review. J Neuropathol Exp Neurol. 2018;77:537–41. [DOI] [PubMed] [Google Scholar]
- 26.Ghanbari N, Lam A, Wycoco V, Lee G. Intracranial myxoid variant of angiomatoid fibrous histiocytoma: a case report and literature review. Cureus. 2019;11:e4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Komatsu M, Yoshida A, Tanaka K, Matsuo K, Sasayama T, Kojita Y, et al. Intracranial myxoid mesenchymal tumor with EWSR1‐CREB1 gene fusion: a case report and literature review. Brain Tumor Pathol. 2020;37:76–80. [DOI] [PubMed] [Google Scholar]
- 28.Sciot R, Jacobs S, Calenbergh FV, Demaerel P, Wozniak A, Debiec‐Rychter M. Primary myxoid mesenchymal tumour with intracranial location: report of a case with a EWSR1‐ATF1 fusion. Histopathology. 2018;72:880–3. [DOI] [PubMed] [Google Scholar]
- 29.Spatz M, Nussbaum ES, Lyons L, Greenberg S, Kallmes KM, Nussbaum LA. Primary intracranial angiomatoid fibrous histiocytoma: a case report and literature review. Br J Neurosurg. 2018;1–3. [DOI] [PubMed] [Google Scholar]
- 30.Valente Aguiar P, Pinheiro J, Lima J, Vaz R, Linhares P. Myxoid mesenchymal intraventricular brain tumour with EWSR1‐CREB1 gene fusion in an adult woman. Virchows Arch. 2020. 10.1007/s00428-020-02885-7. [DOI] [PubMed] [Google Scholar]
- 31.Vizcaino MA.Case 10 – Angiomatoid fibrous histiocytoma with rhabdoid features. Diagnostic Slide Session of the 2020 Annual Meeting of the American Association of Neuropathologists.
- 32.Ward B, Wang C, Macaulay RJB, Liu JKC. Adult intracranial myxoid mesenchymal tumor with EWSR1‐ATF1 gene fusion. World Neurosurg. 2020;143:91–6. [DOI] [PubMed] [Google Scholar]
- 33.White MD, McDowell MM, Pearce TM, Bukowinski AJ, Greene S. Intracranial myxoid mesenchymal tumor with rare EWSR1‐CREM translocation. Pediatr Neurosurg. 2019;54:347–53. [DOI] [PubMed] [Google Scholar]
- 34.Kline CN, Joseph NM, Grenert JP, van Ziffle J, Talevich E, Onodera C, et al. Targeted next‐generation sequencing of pediatric neuro‐oncology patients improves diagnosis, identifies pathogenic germline mutations, and directs targeted therapy. Neuro Oncol. 2017;19:699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sievers P, Chiang J, Schrimpf D, Stichel D, Paramasivam N, Sill M, et al. YAP1‐fusions in pediatric NF2‐wildtype meningioma. Acta Neuropathol. 2020;139:215–8. [DOI] [PubMed] [Google Scholar]
- 36.Yuzawa S, Nishihara H, Tanaka S. Genetic landscape of meningioma. Brain Tumor Pathol. 2016;33:237–47. [DOI] [PubMed] [Google Scholar]
- 37.Schweizer L, Koelsche C, Sahm F, Piro RM, Capper D, Reuss DE, et al. Meningeal hemangiopericytoma and solitary fibrous tumors carry the NAB2‐STAT6 fusion and can be diagnosed by nuclear expression of STAT6 protein. Acta Neuropathol. 2013;125:651–8. [DOI] [PubMed] [Google Scholar]
- 38.Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion‐positive and fusion‐negative tumors. Cancer Discov. 2014;4:216–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee JC, Villanueva‐Meyer JE, Ferris SP, Cham EM, Zucker J, Cooney T, et al. Clinicopathologic and molecular features of intracranial desmoplastic small round cell tumors. Brain Pathol. 2020;30:213–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Scanned image files of H&E stained sections from 19 of the tumors in this cohort are available for downloading and viewing at the following link: https://figshare.com/projects/Intracranial_mesenchymal_tumors_with_EWS‐CREB_fusion/88661. Structural variant and chromosomal copy number data are available in the online supplementary material. Raw sequencing data files are available from the authors upon request.