

Abstract
Exercise can alter the skeletal muscle DNA methylome, yet little is known about the role of the DNA methylation machinery in exercise capacity. Here, we show that DNMT3A expression in oxidative red muscle increases greatly following a bout of endurance exercise. Muscle‐specific Dnmt3a knockout mice have reduced tolerance to endurance exercise, accompanied by reduction in oxidative capacity and mitochondrial respiration. Moreover, Dnmt3a‐deficient muscle overproduces reactive oxygen species (ROS), the major contributors to muscle dysfunction. Mechanistically, we show that DNMT3A suppresses the Aldh1l1 transcription by binding to its promoter region, altering its epigenetic profile. Forced expression of ALDH1L1 elevates NADPH levels, which results in overproduction of ROS by the action of NADPH oxidase complex, ultimately resulting in mitochondrial defects in myotubes. Thus, inhibition of ALDH1L1 pathway can rescue oxidative stress and mitochondrial dysfunction from Dnmt3a deficiency in myotubes. Finally, we show that in vivo knockdown of Aldh1l1 largely rescues exercise intolerance in Dnmt3a‐deficient mice. Together, we establish that DNMT3A in skeletal muscle plays a pivotal role in endurance exercise by controlling intracellular oxidative stress.
Keywords: DNA methylation, exercise, oxidative stress
Subject Categories: Metabolism, Musculoskeletal System
Skeletal muscle‐expressed DNA methyltransferase 3 maintains mitochondrial function and oxidative capacity by suppressing Aldh1l1 transcription.

Introduction
Endurance exercise, an aerobic exercise, is generally characterized by high‐frequency, long duration, and low power output activity, such as marathon running and swimming. Endurance exercise exerts many positive effects on health, prevents disease, and even acts as therapeutics for a wide range of non‐communicable diseases (Hawley et al, 2014; Baskin et al, 2015). Despite these benefits of exercise, there is very limited understanding in the regulatory factors that affect endurance exercise.
Mitochondria are the organelles where oxidation meets phosphorylation to generate ATP for contracting muscles (Huertas et al, 2019). In response to endurance exercise, skeletal muscle increases energy production through aerobic metabolism through involvement of enhancing mitochondrial oxidative capacity (Clausen & Trap‐Jensen, 1970). It has been suggested that the degree of mitochondrial health and adaptations can be dependent on ROS levels. Thus, a moderate increase in skeletal muscle ROS production in the acute phase of exercise is thought to activate signaling pathways that lead to cellular adaptation, thereby protecting against future stress (He et al, 2016). On the other hand, excessive ROS can oxidatively damage macromolecules, such as DNA, lipids, and proteins, as well as modify cellular redox status and cellular functions. Consequently, ROS elevation is also associated with pathophysiological states of muscle and contractile dysfunction (Gomes et al, 2012; He et al, 2016). Mitochondria make a large contribution to ROS production at rest, but not during muscle contraction (Powers et al, 2011). The majority of ROS produced during contraction arise from non‐mitochondrial sources, such as NADPH oxidase (NOX), located in the microtubules (Cross & Segal, 2004; Panday et al, 2015; Di Meo et al, 2016). The redox‐mediated crosstalk between NOX and mitochondria exacerbates ROS production and disrupts redox homeostasis. For example, NOX‐derived ROS promote the opening of mitochondrial ATP‐sensitive K+ channels (Daiber, 2010; Dan Dunn et al, 2015; Daiber et al, 2017). The resultant potassium influx into the matrix lowers the mitochondrial membrane potential, which causes mitochondrial swelling, opening of permeability transition pores, and elevated ROS production (Daiber, 2010; Dan Dunn et al, 2015; Daiber et al, 2017). In addition, NOX‐derived ROS causes leakage of Ca2+ from the sarcoplasmic reticulum or entry of extracellular Ca2+, resulting in mitochondrial Ca2+ overload and mitochondrial ROS emission, which ultimately results in muscle fatigue and dysfunction (Görlach et al, 2015; Steinbacher & Eckl, 2015; He et al, 2016).
DNA methylation, a reversible epigenetic mark that usually occurs on a cytosine residue followed by a guanine (CpG), is mediated by a member of the DNA methyltransferase (DNMT) family (Jaenisch & Bird, 2003). Methylation prevents the binding of transcriptional machinery that requires interaction with cytosine, usually resulting in transcriptional silencing (Bird & Wolffe, 1999). Exercise significantly alters the DNA methylation profile of skeletal muscle (Barrès et al, 2012; Nitert et al, 2012; Brown, 2015; Kanzleiter et al, 2015; Li et al, 2017; Valenzuela et al, 2017; Seaborne et al, 2018; Song et al, 2019; Widmann et al, 2019). Acute and chronic forms of exercise induce both hyper‐ and hypo‐CpG methylation of target loci (Barrès et al, 2012; Nitert et al, 2012; Brown, 2015; Kanzleiter et al, 2015; Li et al, 2017; Valenzuela et al, 2017; Seaborne et al, 2018; Song et al, 2019; Widmann et al, 2019), and some of these modifications are inversely correlated with gene expression (Barrès et al, 2012; Nitert et al, 2012; Brown, 2015). For example, a single bout of aerobic endurance exercise in human subjects transiently induces hypomethylation at the promoter region of important mitochondria‐related transcripts (e.g., PPARGC1A, PDK4, TFAM, and PPARD), followed by an increase in their expression (Barrès et al, 2012). Moderate‐intensity exercise in humans has been reported to result in hypermethylation of FABP3 and COX4L1, which is inversely associated with their gene expression (Seaborne et al, 2018). As such, despite the obvious link between altered DNA methylation and exercise‐associated gene expression, the underlying function of DNMTs in exercise performance remains unclear.
Here, we report that skeletal muscle DNMT3A is a critical epigenetic modulator of endurance exercise. Muscle‐specific Dnmt3a‐deficient mice greatly reduced the exercise capacity accompanied by increased signs of myopathy. Remarkably, knockout (KO) muscles, especially soleus and gastrocnemius (GA) muscles, exhibited a dramatic reduction in oxidative capacity and mitochondrial dysfunction accompanied by an increase in ROS during exercise. Our transcriptomic analysis identifies Aldh1l1 as a key direct target of repression by DNMT3A in soleus muscle and GA muscles. Overexpression of ALDH1L1 was sufficient to recapitulate Dnmt3a KO‐mediated mitochondrial dysfunction and oxidative stress by promoting accumulation of NADPH and thereby increasing NOX activity. Conversely, Aldh1l1 KO or pharmacological inhibition of NOX rescued mitochondrial decline and oxidative stress caused by Dnmt3a deficiency. Lastly, we demonstrate that resolving oxidative stress with an antioxidant and Aldh1l1 knockdown largely rescues exercise incapacity in Dnmt3a KO mice. Together, our results provide novel insights into the epigenetic regulation of the muscle response to exercise and reveal a surprising molecular target that is important for sustaining endurance exercise.
Results
DNMT3A level in the soleus muscle increases after endurance exercise
DNMT1 is the major enzyme involved in maintenance of the DNA methylation pattern following DNA replication, whereas DNMT3A and DNMT3B are primarily responsible for de novo DNA methylation (Jaenisch & Bird, 2003). Hence, we postulated that de novo DNMTs might be more important for adaptive responses to environmental changes. To begin to characterize the role of de novo DNMTs in endurance exercise, we examined their expression patterns in soleus, extensor digitorum longus (EDL), and GA muscles, which are red, white, and mixed muscles, respectively, at rest and after a bout of endurance exercise, in C57BL/6J wild‐type mice. We also measured the mRNA expression of PPARγ‐coactivator 1α (Ppargc1a), which is known to be induced by exercise in skeletal muscle (Barrès et al, 2012; Gouspillou et al, 2014). Ppargc1a mRNA levels increased after 50 min of treadmill running, especially in the soleus and GA (Fig EV1A). Strikingly, Dnmt3a mRNA levels also increased by ~ 3‐fold after exercise in soleus, but not in EDL (Fig EV1B–D). By contrast, exercise caused no significant changes in Dnmt3b transcript levels in any muscle types (Fig EV1B–D). The increase in the level of DNMT3A after a bout of exercise was confirmed by Western blotting (Fig EV1E–F).
Figure EV1. Dnmt3a levels are upregulated in soleus and GA muscles after a bout of endurance exercise.
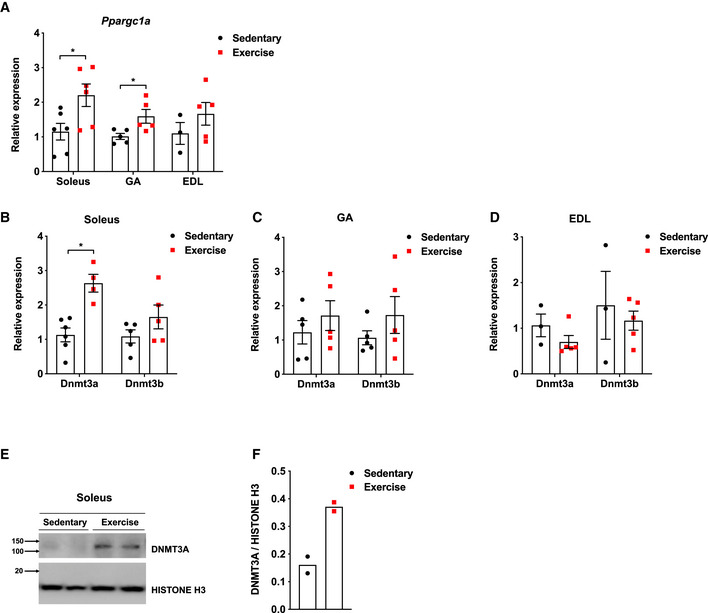
-
APpargc1a was measured in various muscle types from C57BL/6J mice at rest and after a bout of low‐intensity exercise (n = 6 Soleus, n = 5 GA, n = 3 EDL Sedentary and n = 5 EDL exercise, means ± SEM, *P < 0.05, two‐tailed Student's t‐test).
-
B–DTranscript levels of genes encoding de novo DNMTs were measured in soleus (B), GA (C), and EDL (D) from C57BL/6J mice at rest and after a bout of low‐intensity exercise (B: n = 6 Sedentary and n = 4 Exercise for Dnmt3a measurement and n = 5 for Dnmt3b measurement, C: n = 5, D: n = 3 Sedentary and n = 5 Exercise. means ± SEM, *P < 0.05, two‐tailed Student's t‐test).
-
E, F(E) Soleus DNMT3A protein level in the nuclear fraction from (A) was measured by immunoblotting and (F) normalizing to Histone H3 using ImageJ.
Source data are available online for this figure.
Muscle‐specific Dnmt3a ablation decreases the capacity for endurance exercise
Endurance exercise has been shown to lead to the greater relative increase in contractile activity in the soleus and red portion of the GA muscle relative to white muscles, such as EDL (Laughlin et al, 2006; Haizlip et al, 2015). The increase of DNMT3A expression in soleus led us to hypothesize that DNMT3A plays an important role in endurance exercise. To test this, we generated muscle‐specific Dnmt3A knockout mice (MCK‐Dnmt3a KO; Fig EV2) by using the well‐characterized muscle creatine kinase (MCK)‐Cre, which excises floxed alleles in muscle fibers but not satellite cells starting at embryonic day 17 (Brüning et al, 1998). MCK‐Dnmt3a KO mice were viable, exhibited normal growth and fertility, and displayed no significant differences in body weight, body composition, energy balance, or muscle mass, relative to wild‐type littermates harboring two floxed alleles without Cre (WT; Appendix Fig S1).
Figure EV2. Establishment of muscle‐specific knockout of Dnmt3a using MCK‐Cre.
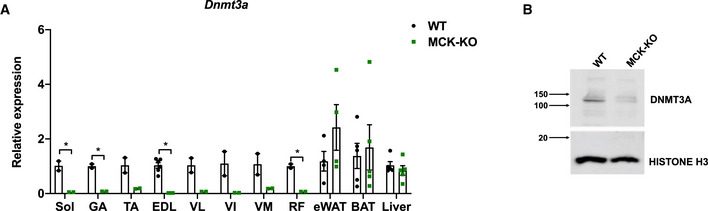
- Dnmt3a mRNA expression measured in various tissues from WT and MCK‐KO by qPCR analysis (n = 2 for soleus, GA, TA, VL, VI, VM, and RF n = 5 for EDL, BAT, Liver, n = 4 for eWAT, means ± SEM, *P < 0.05, two‐tailed Student's t‐test). eWAT (epididymal white adipose tissue), Quad (quadriceps), Sol (soleus), TA (tibialis anterior), VL (vastus lateralis), VI (vastus intermedius), VM (vastus medialis), RF (rectus femoris).
- DNMT3A protein expression was assessed by immunoblotting using nuclear extract from WT and MCK‐KO soleus muscle.
Source data are available online for this figure.
To assess tolerance to endurance exercise, we employed two different regimens: (i) a low‐intensity regimen (Fig 1A) that tested the ability to run steadily at relatively low speed (12 m/min) for an initial 40 min, followed by a gradual increase in speed until exhaustion (DeBalsi et al, 2014), and (ii) a high‐intensity regimen (Fig 1D) that rapidly increased the running speed (6 m/min and increased by 2 m/min every 5 min) to a maximal pace of 20 m/min, which persisted until exhaustion (DeBalsi et al, 2014). During low‐intensity exercise, oxidative muscle fibers predominantly rely on fatty acid oxidation for their ATP production. On the other hand, high‐intensity exercise raises the ATP utilization rate and induces a metabolic switch from fatty acid to glucose oxidation (DeBalsi et al, 2014). We tested the exercise performance of the MCK‐Dnmt3a KO mice at 8 weeks of age under these two regimens. Remarkably, the running capacity of the MCK‐Dnmt3a KO mice was greatly impaired: Both distance and duration were reduced by 30–40% under both the low‐ and high‐intensity regimens (Fig 1B, C, E and F). To test whether the exercise capacity was affected at a younger age, we put WT and KO mice at 5 weeks of age into the low‐intensity regimen and found that the KO still showed reduced exercise capacity compared to WT mice (Appendix Fig S2). Next, we tested whether the KO mice showed reduced trainability. We put the cohort of mice on the training regimen, which consisted of a single bout of running 5 days/week for 4 weeks. Although the KO mice improved their running capacity after the exercise training, the KO mice still ran less time and distance compared to WT mice (Fig EV3A and B). Overall, there was a trend toward that KO muscles overproduce ROS compared to WT muscle after the exercise training (Fig EV3C–E).
Figure 1. MCK‐Dnmt3a KO mice display a reduced tolerance to endurance exercise.
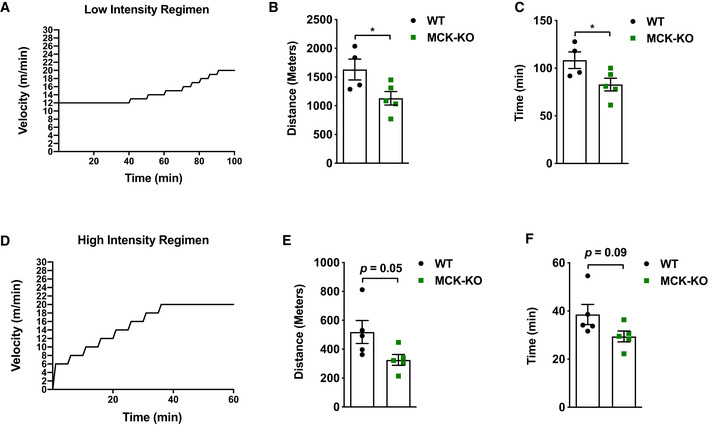
-
ASchematic of low‐intensity exercise regimen.
-
B, CExercise capacity of MCK‐KO and WT mice from the low‐intensity regimen was conducted. (n = 4 WT, n = 5 KO mice, mean ± SEM, *P < 0.05, two‐tailed Student's t‐test).
-
DSchematic of high‐intensity exercise regimen.
-
E, FExercise capacity of MCK‐KO and WT mice from the high‐intensity regimen (n = 5 mice, means ± SEM, two‐tailed Student's t‐test).
Source data are available online for this figure.
Figure EV3. Response to an exercise training of MCK‐Dnmt3a KO mice.
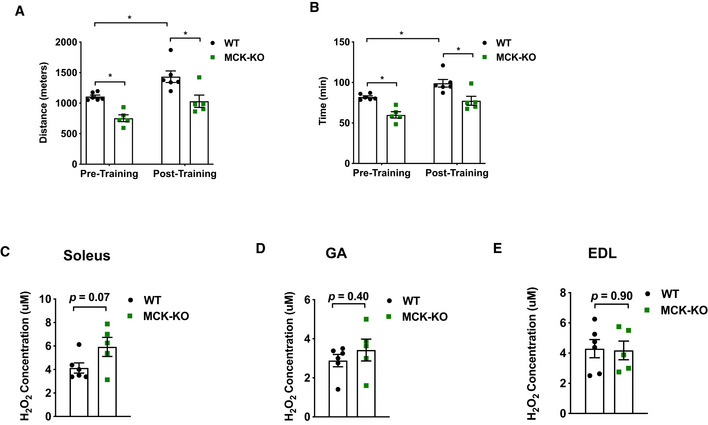
-
A, BExercise capacity of MCK‐KO and WT from the low‐intensity regimen accomplished during initial (pretraining) and final exercise testing (post‐training) following 4 weeks of treadmill training (n = 6 WT, n = 5 MCK‐KO, means ± SEM, *P < 0.05, two‐tailed Student's t‐test and two‐way ANOVA followed by Bonferroni post hoc testing).
-
C–EHydrogen peroxide (H2O2) levels were measured in WT and MCK‐KO after 4 weeks of treadmill training in soleus (C), GA (D) and EDL (E). (n = 6 WT, n = 5 MCK‐KO means ± SEM, two‐tailed Student's t‐test).
Source data are available online for this figure.
Next, we investigated whether reduced exercise tolerance of MCK‐Dnmt3a KO mice accompanies other morphological and biochemical changes as indications of muscle fatigue. First, nuclear dislocation is a hallmark of dysfunctional muscle with or without degeneration/regeneration (Roman & Gomes, 2018). Concordant with impaired exercise capacity, the KO muscles displayed an increased frequency of muscle damage, evidenced by an increased number of dislocated nuclei in the soleus (2.4% vs 4.8% in WT vs KO) and GA 2.3 vs 3.5% muscles from 8‐week‐old mice (Fig 2A–D) after a single bout of low‐intensity exercise for the same duration. Similarly, 5‐week‐old KO mice showed a trend toward more centro‐nucleated soleus and GA muscle fibers after exercise (Appendix Fig S3). To ask whether these defects exist at rest, we performed analogous studies and found that KO soleus and GA muscles tend to have increased numbers of centro‐nucleated muscle fibers at rest (Appendix Fig S4). Second, we measured ROS levels, as high levels of ROS are associated with contractile dysfunction and muscle fatigue (Powers et al, 2011). Remarkably, we found that ROS levels greatly increased in red soleus and mixed GA muscles, but not in white EDL, at both 5 and 8 weeks of age (Appendix Fig S5, Fig 2E–G). Lastly, we also measured blood lactate levels, which essentially serve as an indirect marker for biochemical events such as fatigue within exercising muscle (Finsterer, 2012). As expected, blood lactate levels were also increased in exercised KO mice (Fig 2H). Together, these data indicate that Dnmt3a KO muscles display increased signs of muscle fatigue which was associated with reduced exercise capacity.
Figure 2. MCK‐Dnmt3a KO mice display increased muscle damage following exercise.
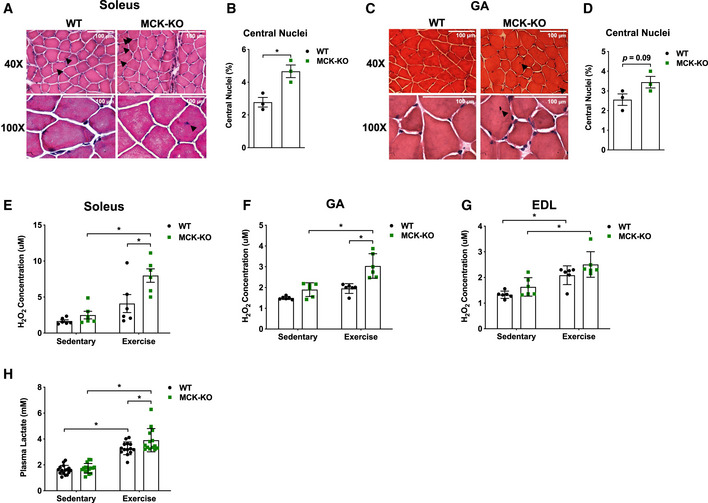
-
AThe H&E staining of MCK‐KO and WT soleus muscle after a bout of low‐intensity treadmill running (top 40×, bottom 100× magnifications). Black arrow indicates centralized nuclei.
-
BThe percentage of myofibers with centralized nuclei was determined by manual counting 300 myofibers in 20× magnification. (n = 3, means ± SEM, *P < 0.05, two‐tailed Student's t‐test).
-
C, DThe analogous set of data is shown with WT and MCK‐KO GA muscles. (n = 3, means ± SEM, two‐tailed Student's t‐test. GA, gastrocnemius).
-
E–GHydrogen peroxide (H2O2) levels were measured in WT and MCK‐KO soleus (E), GA (F), and EDL (G) at rest and after a bout of exercise (n = 6 per group, means ± SEM, *P < 0.05, two‐tailed Student's t‐test and two‐way ANOVA followed by Bonferroni post hoc testing).
-
HSerum levels of lactate were measured before and after a bout of low‐intense exercise for 50 min in WT and MCK‐KO mice (n = 16 for sedentary and n = 14 for exercise groups, means ± SEM, *P < 0.05, two‐tailed Student's t‐test and two‐way ANOVA followed by Bonferroni post hoc testing).
Source data are available online for this figure.
Dnmt3a‐KO muscle has reduced oxidative capacity and diminished mitochondrial respiration
High power of oxidative capacity and mitochondrial function of skeletal muscle is critical for supporting endurance exercise. Succinate dehydrogenase (SDH), located in the inner membrane of the mitochondrion, is responsible for oxidizing succinate to fumarate in the citric acid cycle (Old & Johnson, 1989). Hence, we performed SDH staining to distinguish between oxidative and less‐oxidative muscles. Remarkably, KO soleus muscles displayed ~ 20% less SDH activity relative to WT tissues even at the sedentary (Fig 3A and B) and that this difference became exacerbated after exercise showing a 50% decline in KO tissues from 8‐week‐old mice (Fig 3C and D). KO GA muscles showed a similar pattern of SDH activity at rest and following exercise (Fig 3E–H). By contrast, EDL muscles did not show marked differences between genotypes at both conditions (Fig 3I–L). Similar to other histological parameters, we also found that 5‐week‐old KO mice exhibited a trend toward reduced SDH activity in soleus and GA, but not in EDL, muscles compared to WT tissues (Fig EV4). We then assessed whether such differences of SDH activity is related to mitochondrial respiration rates and noted that KO soleus muscles exhibit a reduced oxygen consumption rate in both sedentary and exercise conditions (Fig 3M and N). We were also able to confirm that myotube‐autonomous effect of DNMT3A loss of function in oxygen consumption rates (Fig 3O–P). Lastly, we performed H&E staining and SDH staining using successive tissue sections to examine whether damaged myofibers with centro‐nucleation overlap with reduced SDH activity. We found that nuclear dislocation and reduced SDH activity do not necessarily overlap each other (Appendix Fig S6).
Figure 3. Dnmt3a‐KO soleus muscle displays a decreased oxidative capacity with a reduced mitochondrial respiration.
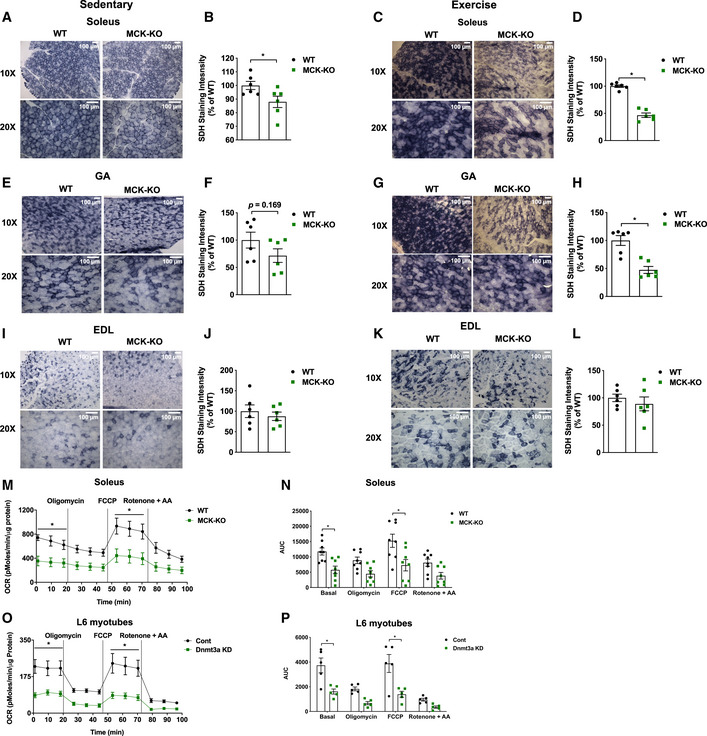
-
A–DSuccinate dehydrogenase staining was performed in WT and MCK‐KO soleus at sedentary (A, B) and after a bout of low‐intensity exercise for 50 min (C, D; 10×, 20× magnifications), and the staining intensity was quantified using ImageJ (n = 6, means ± SEM, *P < 0.05, two‐tailed Student's t‐test).
-
E–HThe analogous set of data is shown with WT and MCK‐KO GA muscles. (n = 6, means ± SEM, *P < 0.05, two‐tailed Student's t‐test).
-
I–LThe analogous set of data is shown with WT and MCK‐KO EDL muscles. (n = 6, means ± SEM, two‐tailed Student's t‐test. EDL, extensor digitorum longus).
-
M, NMitochondrial respiration was measured in WT and MCK‐KO soleus tissue after a bout of low‐intensity exercise for 50 min under basal conditions and in response to 4 mM oligomycin (complex V inhibitor), 4 mM FCCP (uncoupler), or 4 mM each of rotenone and antimycin A (complex I inhibitor; n = 8, means ± SEM, *P < 0.05, two‐tailed Student's t‐test and two‐way ANOVA followed by Bonferroni post hoc testing). AUC: Area under the curve.
-
O, PMitochondrial respiration was measured in Dnmt3a knocked down L6 myotubes which were transduced with lentiviral under basal conditions and in response to 4 mM oligomycin (complex V inhibitor), 4 mM FCCP (uncoupler), or 4 mM rotenone and antimycin A (complex I inhibitor; n = 5, means ± SEM, *P < 0.05, two‐tailed Student's t‐test and two‐way ANOVA followed by Bonferroni post hoc testing). AUC: Area under the curve.
Source data are available online for this figure.
Figure EV4. Dnmt3a‐KO soleus and GA muscles display a decreased trend in oxidative capacity at 5 weeks of age following exercise.
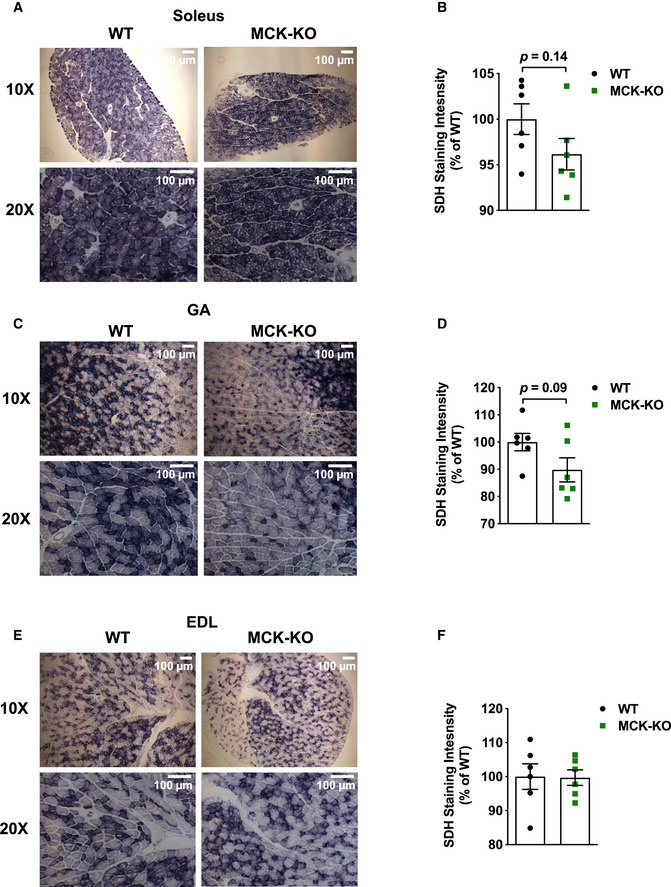
-
A–FSuccinate dehydrogenase staining and quantification was performed in WT and MCK‐KO after a bout of low‐intensity exercise for 50 min in soleus (A, B) GA (C, D) and EDL (E, F; 10×, 20× magnifications), and the staining intensity was quantified using ImageJ (n = 6, means ± SEM, two‐tailed Student's t‐test).
Source data are available online for this figure.
Skeletal muscle depots are composed of heterogeneous populations of muscle fibers, which are categorized largely as slow‐twitch (type I) and fast‐twitch (type II) based upon biophysical property of contractility (Talbot & Maves, 2016). Slow‐twitch fibers are dense in mitochondria to allow high oxidative capacity and sustain long‐term energy demands (Bourdeau Julien et al, 2018). By contrast, fast‐twitch fibers are subdivided into fast‐oxidative (type IIA) or fast‐glycolytic (type IIB/X), which correlate with their mitochondrial density (Bourdeau Julien et al, 2018). The soleus muscle is rich in type I and some IIA myosin heavy chains (MHCs), whereas muscles like EDL are enriched in the faster MHC IIB fibers that are for the fast‐twitch property (Arany et al, 2007). Previous studies suggest that shifting muscle fiber composition is engaged with altered exercise capacity and muscle dysfunction (Handschin et al, 2007). To ask if that is the case with MCK‐Dnmt3a KO mice, we assessed muscle fiber distribution by immunostaining of fiber‐specific MHC and found no obvious differences in muscle fiber composition between WT and KO soleus, GA and EDL muscles (Fig EV5A–F). In agreement, no discernible change in the expression of muscle fiber type‐specific MHC isoforms between WT and KO soleus muscles (Fig EV5G). Collectively, our data suggest that DNMT3A is required for the full oxidative capacity of skeletal muscle and is not associated with fiber type determination.
Figure EV5. Distribution of MHC isoforms is unchanged in Dnmt3a KO muscles.
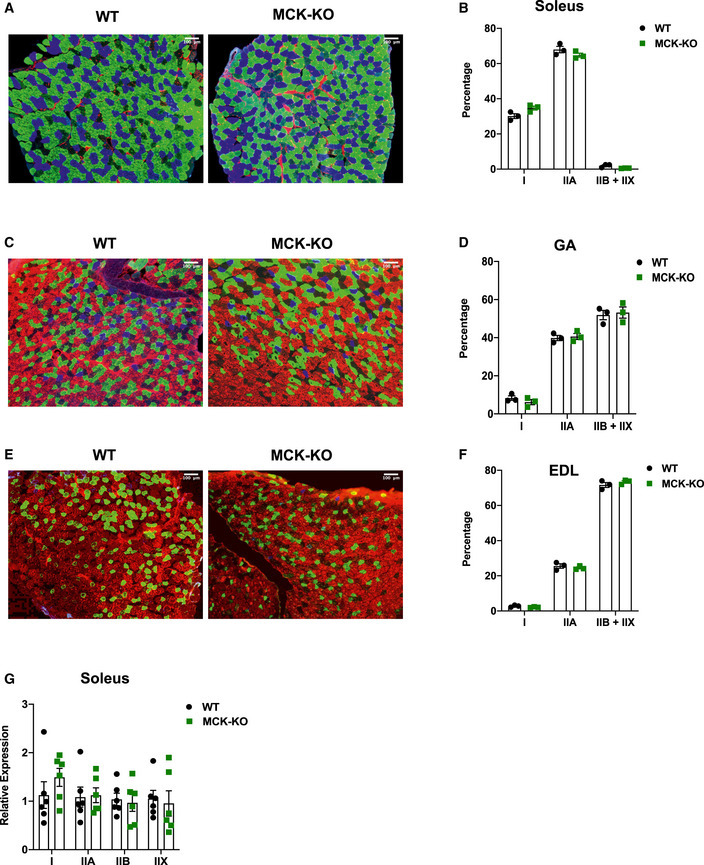
-
A–FCross sections and quantification of WT and MCK‐KO soleus (A, B), GA (C, D), and EDL (E, F) stained with antibodies that are MHC fiber‐specific (type I [blue], IIA [green], IIB [red], and IIX [unstained]). (n = 3, data are expressed as means ± SEM, two‐tailed Student's t‐test).
-
GThe mRNA expression of muscle subtype‐specific MHC isoforms was measured in WT and MCK‐KO soleus muscles (n = 6, *P < 0.05, means ± SEM, two‐tailed Student's t‐test).
Source data are available online for this figure.
Gene profiling identifies muscle‐specific DNMT3A target genes
We and others have shown that DNMT3A regulates biological processes by regulating non‐overlapping sets of cell type‐specific target genes (Nguyen et al, 2007; Challen et al, 2011; Nishikawa et al, 2015). To elucidate the underlying molecular basis by which DNMT3A regulates exercise capacity, we performed RNA‐Seq on WT and KO soleus muscle at rest and after exercise. The transcriptome profiles detected that 23 genes were upregulated, and three genes were downregulated in Dnmt3a‐deficient soleus muscle at rest, while 18 genes were up and 17 genes were downregulated in the exercise condition (Fig 4A and B). While several of the upregulated genes in the KO overlapped conditions, none of the downregulated genes overlapped (Fig. 4A and B, Appendix Fig S7A and B). Since whole muscle contains multiple cell types, such as fibroblasts, endothelial cells, and satellite cells, we extracted myofibers by collagenase treatment from WT and KO mice at rest and measured DNMT3A target genes that are commonly upregulated in the KO at rest and after exercise. We found that most of the genes were upregulated in KO myofibers (Appendix Fig S7C).
Figure 4. Transcriptome analysis identifies Aldh1l1 as a key target gene of DNMT3A in the soleus muscle.
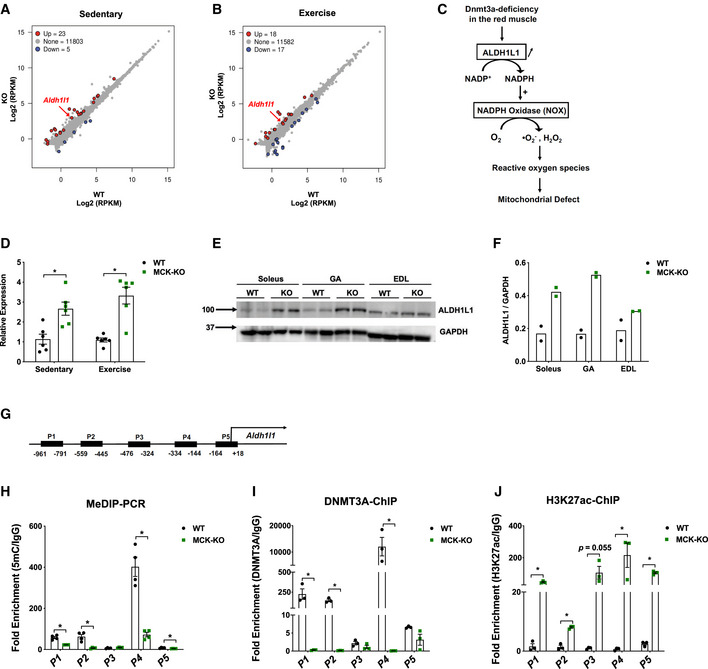
-
A, B(A) RNA‐Seq was performed in WT and MCK‐KO soleus muscle at rest. The scatter plots show differentially expressed genes in MCK‐KO soleus muscle at sedentary (A) and after a bout of low‐intensity exercise for 50 min (B; FDR < 0.05, P < 0.05).
-
CProposed model for ROS regulation during loss of Dnmt3a. Loss of DNMT3A increases ALDH1L1 expression, thus leading to the increase in NADPH levels and increased activity of NADPH oxidase, leading to increased ROS levels. The increased oxidative stress contributes to mitochondrial dysfunction and muscle fatigue.
-
DqPCR validation of Aldh1l1 at sedentary and after exercise for 50 min (n = 6, means ± SEM, *P < 0.05, two‐tailed Student's t‐test).
-
E, FALDH1L1 protein expression and the quantification in WT vs MCK‐KO soleus, GA, and EDL muscles at sedentary and normalizing to GAPDH using ImageJ.
-
GThe map of CpG‐rich promoter regions of Aldh1l1 and MeDIP and ChIP primers (P1‐P5) that cover the CpG‐rich regions. The numbers correspond to the position from the transcriptional start site of Aldh1l1.
-
HMeDIP‐qPCR was performed in WT and MCK‐KO soleus muscle to assess differential methylation using primer sets from (G; n = 4, means ± SEM, *P < 0.05, two‐tailed Student's t‐test).
-
IDNMT3A ChIP‐PCR was conducted in WT and MCK‐KO soleus muscles using primer sets from (G; n = 3, means ± SEM, *P < 0.05, two‐tailed Student's t‐test).
-
JH3K27ac ChIP‐PCR was conducted in WT and MCK‐KO soleus muscles using primer sets from (G; n = 3, means ± SEM, *P < 0.05, two‐tailed Student's t‐test).
Source data are available online for this figure.
Because a relatively small number of genes were differentially regulated in the KO tissue, we did not identify any specific biological pathways enriched among the DNMT3A target genes. However, genes involved in the detoxification of reactive oxygen species (e.g., Gpx1 and Gpx3) were upregulated in KO muscle, reflecting the increased levels of oxidative stress in KO muscles (Appendix Fig S7A). Our search to identify the targets responsible for the increase in ROS production led us to investigate Aldh1l1, which encodes aldehyde dehydrogenase 1 family member L1 (ALDH1L1), a cytosolic enzyme involved in folate and one‐carbon metabolism. Specifically, ALDH1L1 oxidizes 10‐formyltetrahydrofolate to tetrahydrofolate, simultaneously producing NADPH as a byproduct (Krupenko, 2009) (Fig 4C). Notably in this regard, NADPH plays a dual role in the regulation of oxidative stress. On the one hand, it is a reducing agent for glutathione, thioredoxins, peroxiredoxins, and glutathione peroxidases, which neutralize ROS (Fernandez‐Marcos & Nóbrega‐Pereira, 2016). On the other hand, it contributes to ROS generation, through the activity of the NADPH oxidase complex (NOX; Fig 4C) located within the sarcoplasmic reticulum, transverse tubules, and sarcolemma in skeletal muscle fibers (Ferreira & Laitano, 2016). We hypothesized that ALDH1L1‐mediated NADPH production feeds into NOX, thereby increasing intracellular ROS in Dnmt3a KO muscle.
First, we examined the regulation of Aldh1l1 by DNMT3A. We measured the levels of Aldh1l1 mRNA and ALDH1L1 protein to be elevated in KO muscle tissues (Fig 4D–F). Notably, ALDH1L1 expression was elevated only in the red soleus and mixed GA muscles, but not in the white EDL muscle (Fig 4E and F), suggesting that muscle depot‐selective regulation of Aldh1l1 by DNMT3A. To determine whether Aldh1l1 is indeed a direct target of DNMT3A, we performed methylated DNA immunoprecipitation (MeDIP)‐qPCR analysis of the CpG‐rich Aldh1l1 promoter regions (Fig 4G). In KO soleus muscle, DNA methylation was greatly reduced in four of the five regions we examined, including the CpG island (P4; Fig 4H). In vivo ChIP assay confirmed strong enrichment of DNMT3A at those differentially methylated regions (Fig 4I). In fact, the DNA methylation and histone regulation machineries often engage in crosstalk (Du et al, 2015). Therefore, we also conducted a ChIP assay for H3K27ac, a histone modification marker of active promoters and enhancers, and detected strong signals at Aldh1l1 promoter regions in KO soleus tissues (Fig 4J). Collectively, these data demonstrate that DNMT3A directly regulates expression of Aldh1l1 by modifying the epigenetic profile at its regulatory regions.
ALDH1L1 drives the increase in ROS and mitochondrial dysfunction in KO soleus muscle
To determine whether elevated ALDH1L1 expression is responsible for NADPH‐dependent generation of ROS, we compared NADPH levels between WT and KO muscle. Indeed, NADPH levels were significantly increased in KO muscle (Fig 5A). To assess whether this in turn results in increased flux into NOX (Reusch & Burger, 1974; Hidalgo et al, 2006), indeed, we detected elevated NOX level in KO tissues (Fig 5B). Next, to evaluate the functional significance of ALDH1L1, we performed an ALDH1L1 gain‐of‐function study in L6 rat myotubes in the presence and absence of apocynin, a specific NOX inhibitor (Petrônio et al, 2013). Overexpression of ALDH1L1 had no effect on myogenesis (Appendix Fig S8), but remarkably, it was sufficient to recapitulate the redox changes in Dnmt3a‐KO muscles, including the increases in the levels of NADPH (Fig 5C) and ROS (Fig 5D and E). More strikingly, ALDH1L1‐overexpressing myotubes exhibited a reduced oxygen consumption relative to controls (Figs 5F and G). Importantly, all of these changes associated with ALDH1L1 overexpression were largely reversed by treatment with apocynin (Fig 5C–G). To obtain evidence that ALDH1L1 is required for the phenotype of the Dnmt3a loss‐of‐function model, we knocked out Aldh1l1 in Dnmt3a knockdown L6 myotubes, which resulted in dramatic rescue of the oxidative stress that is mediated by Dnmt3a deficiency (Fig 5H–K).
Figure 5. ALDH1L1 contributes to the oxidative stress and mitochondrial defect in loss of Dnmt3a .
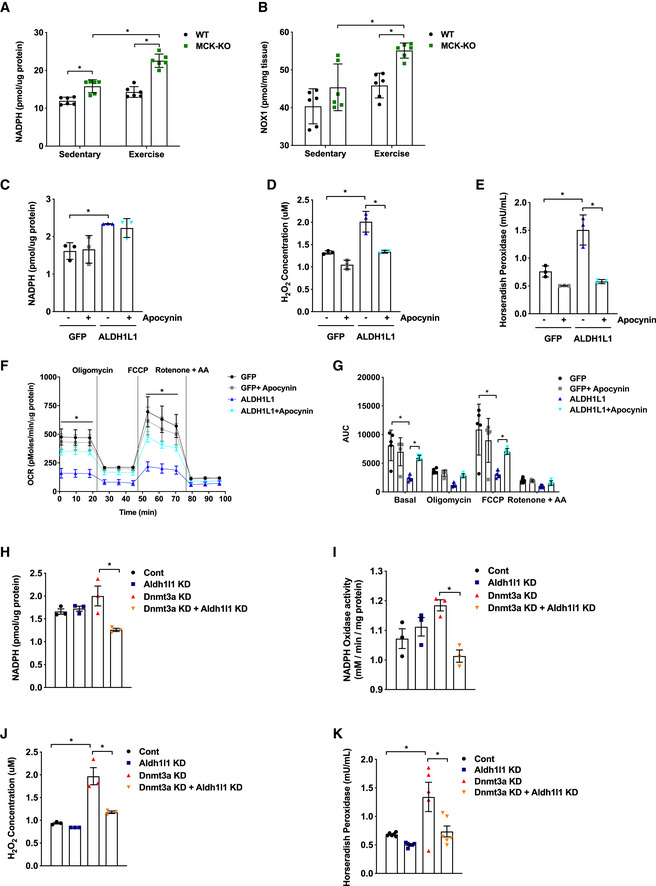
-
A, BNADPH levels (A) and NOX (B) level was measured in WT and MCK‐KO muscles at rest and after a bout of low‐intensity exercise for 50 min (n = 6, means ± SEM, *P < 0.05, two‐tailed Student's t‐test and two‐way ANOVA followed by Bonferroni post hoc testing).
-
C–GL6 myotubes were transduced with lentiviral expression plasmids for Flag‐ALDH1L1 and GFP. NADPH levels (C), H2O2 levels (D, E), Mitochondrial respiration (F, G) were measured from these cells in the presence and absence of NADPH oxidase inhibitor apocynin (n = 3 per group, for (C–E) and n = 5 for (F, G), means ± SEM, *P < 0.05, two‐tailed Student's t‐test and two‐way ANOVA followed by Bonferroni post hoc testing). AUC: Area under the curve.
-
H–KSingle and double knockdowns of Dnmt3a and Aldh1l1 in L6 myotubes were achieved by lentiviral transduction. NADPH levels (H) and NOX activity (I) were measured in single and double knockdowns of Dnmt3a and Aldh1l1 in L6 myotubes. (n = 3, means ± SEM, *P < 0.05, two‐tailed Student's t‐test and two‐way ANOVA followed by Bonferroni post hoc testing). (J, K) H2O2 levels were measured in single and double knockdowns of Dnmt3a and Aldh1l1 in L6 myotubes. (J: n = 3 and K: n = 6 for Control, Aldh1l1 KD, Aldh1l1 KD + Dnmt3a KD, n = 5 for Dnmt3a KD, means ± SEM, *P < 0.05, two‐tailed Student's t‐test and two‐way ANOVA followed by Bonferroni post hoc testing).
Source data are available online for this figure.
Resolving oxidative stress and muscle‐targeted Aldh1l1 silencing in MCK‐Dnmt3a KO mice partially rescues exercise intolerance
Since we hypothesized that excessive ROS production is a main driver of muscle dysfunction and exercise incapacity during loss of Dnmt3a, we sought to determine whether an antioxidant could resolve the issues. Remarkably, a single i.p. injection of N‐acetylcysteine (NAC) rescued exercise capacity in KO mice by 43% but had no significant effect on WT's exercise performance (Fig 6A and B). Consistent with this data, we confirmed that NAC reduced ROS levels in Dnmt3a KD myotubes (Fig 6C) and significantly improved mitochondrial respiration (Fig 6D and E). Lastly, we sought to determine whether decreasing ALDH1L1 levels can rescue the exercise intolerance detected in Dnmt3a KO mice. To this end, we chose to use in vivo transfection method which allows us to specifically transfect the nuclei of terminally differentiated adult muscle fibers, but not the nuclei of satellite cells or connective tissue cells (Welle, 2009). We delivered gRNA against gAldh1l1 or control to the GA and soleus muscles in the KO mice, and we achieved ~ 50% knockdown of Aldh1l1 mRNA and protein in soleus and GA, but without a major change in EDL (Fig 7A–C). Remarkably, Dnmt3a KO mice that were delivered with Aldh1l1 knockdown gRNA partially restored the ability of treadmill running by 47 and 38% by time and distance, respectively, compared to the KO mice transfected with control gRNA (Fig 7D and E). Together, our results suggest that ALDH1L1 plays a critical role in producing muscle dysfunction and exercise intolerance arising from Dnmt3a deficiency.
Figure 6. NAC treatment partially rescues reduced oxidative capacity in Dnmt3a KD myotubes and exercise intolerance in MCK‐Dnmt3a KO mice.
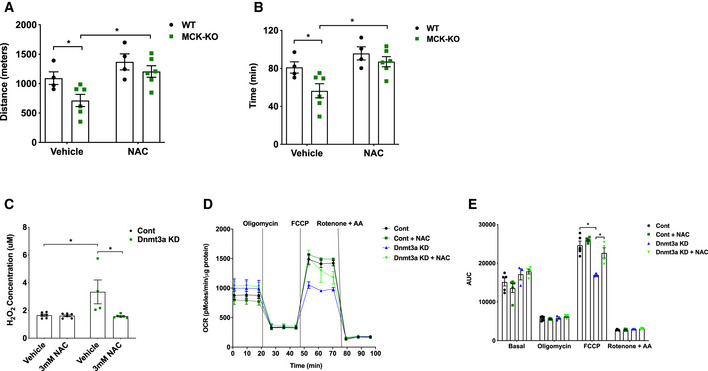
-
A, BExercise capacity of MCK‐KO and WT mice treated with PBS (vehicle) or NAC (200 mg/kg, i.p). (n = 4 WT and n = 6 MCK‐KO, mean ± SEM, *P < 0.05, two‐tailed Student's t‐test and two‐way ANOVA).
-
C–E(C) Hydrogen peroxide (H2O2) levels (n = 6 for Control groups and Dnmt3a KD with NAC treatment group n = 4 Dnmt3a KD, means ± SEM, *P < 0.05, two‐tailed Student's t‐test and two‐way ANOVA followed by Bonferroni post hoc testing). (D, E) mitochondrial respiration in Dnmt3a knockdown and control L6 myotubes treated with NAC (3 mM) or vehicle treatment for 24 h (n = 5 Control groups n = 3 Dnmt3a KD, n = 4 Dnmt3a KD with NAC treatment groups. means ± SEM, *P < 0.05, two‐tailed Student's t‐test and two‐way ANOVA followed by Bonferroni post hoc testing). AUC: Area under the curve.
Source data are available online for this figure.
Figure 7. ALDH1L1 knockdown in vivo partially rescues exercise intolerance in MCK‐Dnmt3a KO mice.
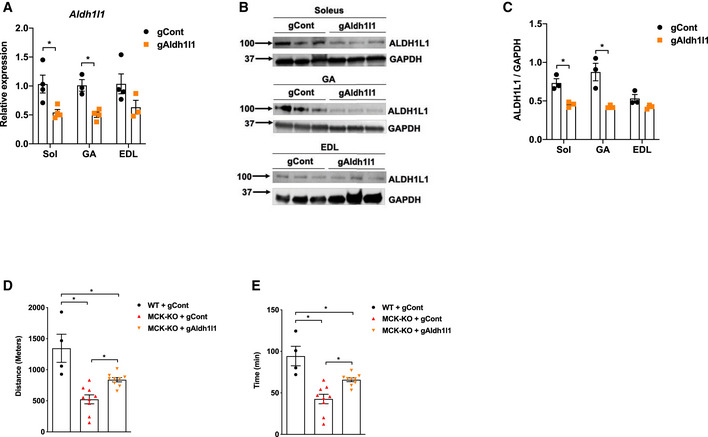
-
AAldh1l1 mRNA expression in various muscles in MCK‐KO mice that were transfected with gRNA DNAs against Aldh1l1 or control. (n = 4 for Soleus, n = 3 GA gCont and n = 4 GA gAldh1l1, n = 4 EDL gCont and n = 3 EDL gAldh1l1, means ± SEM, *P < 0.05, two‐tailed Student's t‐test).
-
B, CALDH1L1 protein expression and the quantification in MCK‐KO muscles that were transfected with gCont vs gAldh1l1 using ImageJ. (n = 3, means ± SEM, *P < 0.05, two‐tailed Student's t‐test).
-
D, EExercise capacity of WT and MCK‐KO with or without transfection of gCont vs gAldh1l1 under the low‐intensity regimen (n = 4 WT + gCont, n = 9 MCK‐KO + gCont, and MCK‐KO + gAldh1l1, *P < 0.05, means ± SEM, two‐tailed Student's t‐test and one‐way ANOVA).
Source data are available online for this figure.
Discussion
Exercise significantly alters the DNA methylation profile of skeletal muscle (Barrès et al, 2012; Nitert et al, 2012; Brown, 2015; Kanzleiter et al, 2015; Li et al, 2017; Valenzuela et al, 2017; Seaborne et al, 2018; Song et al, 2019; Widmann et al, 2019); however, it has not been studied whether DNA methylation machinery has functional roles in exercise capacity. Here, we demonstrate that skeletal muscle DNMT3A plays an essential role for the full capacity to perform endurance exercise. Our findings indicate that Dnmt3a deficiency in skeletal muscle leads to oxidative stress and muscle fatigue and consequently a dramatic reduction of exercise capacity. Surprisingly, our studies find that skeletal muscle DNMT3A is necessary to maintain mitochondrial function and oxidative capacity, which are critical to support endurance exercise. Our mechanistic studies reveal that ALDH1L1 is a key downstream effector of DNMT3A loss of function as evidenced by that knockdown of ALDH1L1 that can largely rescue defects from Dnmt3a deficiency in vitro and in vivo, while overexpression of ALDH1L1 recapitulates them. These findings demonstrate a surprising role for DNMT3A as an epigenetic modulator of endurance exercise by controlling intracellular oxidative stress and reveal ALDH1L1 as a key mediator.
Studies have reported that aerobic exercise changes DNA methylation profile. For example, a human study has shown that a single bout of aerobic exercise transiently induces promoter DNA hypomethylation of promoter regions of important mitochondria‐related genes (e.g., PPARGC1A, PDK4, TFAM, and PPARD), followed by their induction (Barrès et al, 2012). On the basis of this human study, our results showing the requirement of DNMT3A in endurance exercise as counterintuitive. However, it should be noted that exercise affects DNA methylation profile in either direction depending on loci. As an example, another human study reported that moderate‐intensity exercise results in hypermethylation of FABP3 and COX4L1, which negatively correlated with their expression (Lane et al, 2015; Seaborne et al, 2018). By the same token, genome‐wide studies have reported that both acute and chronic exercise interventions produce profound changes in CpG methylation (Barrès et al, 2012; Nitert et al, 2012; Brown, 2015; Kanzleiter et al, 2015; Li et al, 2017; Valenzuela et al, 2017; Seaborne et al, 2018; Song et al, 2019; Widmann et al, 2019). We did not observe that Dnmt3a deficiency in soleus alters the expression of the genes that were shown to be differentially methylated in association with exercise.
Mitochondria in skeletal muscle are highly dynamic organelles that exhibit remarkable plasticity, adapting their content, structure, and metabolism in response to a variety of physiological and pathophysiological stresses including exercise, disuse, and aging (Gouspillou & Hepple, 2016; Hood et al, 2019). Exercise training increases mitochondrial biogenesis to satisfy elevated energy requirements by increasing oxidative capacity to ensure optimal ATP supply; this has the consequence of favoring lipid metabolism (Boushel et al, 2014; Kuzmiak‐Glancy & Willis, 2014). Thus, exercise represents a viable therapy, with the potential to reverse the impairment of mitochondrial function associated with diseases such as type 2 diabetes, and aging‐related sarcopenia (Kim et al, 2017; Yoo et al, 2018; Picca et al, 2019). In this regard, a key link between exercise and control of mitochondrial biology was revealed by the observation that PGC‐1α expression is transiently induced in skeletal muscle following an acute bout of exercise (Safdar et al, 2011). Since that discovery, a great deal of research effort has been devoted to elucidating the role of PGC1A in skeletal muscle mitochondrial biology and exercise. For example, transgenic expression of PGC1A increases mitochondrial content and function and the abundance of oxidative type I muscle fibers, while decreasing muscle fatigue. However, loss of PGC1A has only a mild effect on exercise capacity and does not alter fiber type composition in muscle (Zechner et al, 2010) or affect training‐induced increase in the expression of genes involved in oxidative phosphorylation (Rowe et al, 2012). This suggests that PGC1A is sufficient, but not necessary, to mediate metabolic adaptations in response to exercise. We believe that the role of DNMT3A in the regulation of mitochondrial biology is likely to be PGC1A‐independent as PGC1A mRNA and protein levels are not altered in Dnmt3a KO muscles.
Overproduction of ROS induced by unaccustomed, exhaustive exercise training or other stresses can lead to oxidative stress‐related tissue damage and reduced contractility (Tidball & Wehling‐Henricks, 2007; Bowen et al, 2015), involving impaired cellular function, macromolecule damage, and apoptosis (Simioni et al, 2018). Mitochondria are highly susceptible to chronic high levels of ROS‐mediated damage, and a heterogeneous class of human diseases, such as aging, cancer, neurodegenerative disorders, and diabetes, have been linked to mitochondrial defects and oxidative stress (Lejay et al, 2014; Di Meo et al, 2017; Simioni et al, 2018). Despite the clinical significance, the molecular mechanisms involved in mitochondrial dysfunction and increased ROS production are not well understood. Recent studies have proposed a link between epigenetic factors and ROS‐mediated adaptation in skeletal muscle (Radak et al, 2011, 2013; Rasmussen et al, 2014; Dimauro et al, 2020). Our results suggest DNMT3A is a critical epigenetic modulator of ROS and thereby helps to prevent oxidative stress‐mediated myopathy. NADPH oxidases are major contributors to ROS production in skeletal muscle (Sakellariou et al, 2013; Henríquez‐Olguin et al, 2019). It has been shown that physical stretching can increase the activity of NADPH oxidase, especially NOX2, causing production of ROS in microtubule‐dependent processes (Prosser et al, 2011). While that study described mechanotransduction‐dependent activation of NADPH in cardiac muscles, here we identified ALDH1L1‐dependent activation of NADPH oxidase as a contributor to ROS overproduction, especially in oxidative muscles, during loss of Dnmt3a. Our finding that inhibiting NAnDPH oxidase rescued both oxidative stress and mitochondrial dysfunction raises the possibility of repurposing these inhibitors to improve exercise trainability. Further understanding the epigenetic and molecular basis of DNMT3A to moderate ROS will help us address several critical health issues that are derived from exercise‐induced high levels of ROS in the pathogenic processes of relevant human diseases.
ALDH1L1 is a folate‐metabolizing enzyme that controls the overall flux of one‐carbon groups in folate‐dependent biosynthetic pathways, with simultaneous production of NADPH from NADP. Differential methylation of ALDH1L1 has been reported with an implication in tumor development and progression in several cancer models (Oleinik et al, 2011; Kang et al, 2016; Krupenko & Krupenko, 2019). Other than that, little has been known about physiological functions of ALDH1L1 in skeletal muscle biology. Here, we outline that ALDH1L1, whose transcriptional level is epigenetically regulated by DNMT3A especially in soleus and GA muscles, plays a determining role in Dnmt3a deficiency‐induced oxidative stress and exercise intolerance.
Overall, we highlight the surprising role of DNMT3A in endurance exercise and skeletal muscle mitochondrial biology. Mechanistically, we reveal that ALDH1L1 serves as a novel molecular link that contributes to oxidative stress and mitochondrial dysfunction following the loss of Dnmt3a in red muscle. This is of great importance from the standpoint of exercise physiology, as physical activity is strongly encouraged as a key strategy for preventing and treating a wide range of human diseases. Understanding the epigenetic and molecular basis of exercise tolerance will help us to address critical health issues that arise from reduced ability to perform exercise.
Materials and Methods
Animals
Animal Care Mice were maintained under a 12‐h light/12‐h dark cycle at constant temperature (23°C) with free access to food and water. All mice were extensively back‐crossed onto a C57BL/6J background. All animal work was approved by UC Berkeley ACUC. In vivo assays were done with 5‐ to 20‐week‐old littermate male mice.
Measurement of exercise capacity
All mice were acclimated to the treadmill prior to the exercise test session. For each session, food was removed 2 h before exercise. Acclimation began at a low speed of 5–8 m/min for a total of 10 min on Day 1 and was increased to 5–10 m/min for a total of 10 min on Day 2. The experiments were performed on Day 3. For the low‐intensity treadmill test, the treadmill began at a rate of 12 m/min for 40 min. After 40 min, the treadmill speed was increased at a rate of 1 m/min every 10 min for a total of 30 min and then increased at the rate of 1 m/min every 5 min until the mice were exhausted. The high‐intensity treadmill test was conducted on the same open‐field six‐lane treadmill set at a 10% incline. Following a 5‐min 0 m/min acclimation period, the speed was raised to 6 m/min and increased by 2 m/min every 5 min to a maximal pace of 20 m/min until exhaustion. Mice were considered exhausted when they were unable to respond to continued prodding with a soft brush (Kong et al, 2018). For chronic exercise training, the treadmill‐acclimatized mice were subjected to the low‐intensity exhaustion test to record the time to exhaustion and were returned back to the home cage to rest for a week. For exercise training, these mice performed a single bout of running 5 days/week for 4 weeks. Mice ran 12 m/min for 30 min per day with a 5° incline for the first week of exercise training, followed by an increase to 40 min per day in the second and third weeks and 50 min per day in the last week. Mice were subjected to the low‐intensity exhaustion test to record the time to exhaustion after completion of exercise training. For the rescue experiment with NAC (Sigma, #A7250), mice were intraperitoneally injected with 200 mg/kg 4 h prior to the exercise capacity test.
RNA‐Seq library generation and analysis
RNA samples were extracted using the RNeasy Mini kit (Qiagen, 74104), and the quality of total RNA was assessed by the 2100 Bioanalyzer (Agilent) and agarose gel electrophoresis. Libraries were prepared using the BGI Library Preparation Kit, and sequencing was performed on the BGISEQ (BGI, China). RNA‐Seq reads were aligned to the UCSC mm10 genome using HISAT2 (Hierarchical Indexing for Spliced Alignment of Transcripts (Kim et al, 2015), and mapping was done using Bowtie2 (Kim et al, 2015). Differentially regulated genes were calculated using DEseq2 (Love et al, 2014).
Measurement of NADPH
The NADPH measurement was performed on GA/Soleus or L6 myotube extracts using the NADP/NADPH assay kit (Cat#K347, BioVision) according to the manufacturer's instructions. Briefly, ~ 20 mg tissue samples or 4 × 106 cells were extracted in 400 µl of the given extraction buffer, and 50 µl was processed following instructions of the kit. OD450 measurements were made on a plate reader (SpectraMAX i3 Plate reader) at 25°C, and the data were calculated using a standard curve.
Measurement of NADPH oxidase activity
NOX activity was measured by accessing oxidation of NADPH through Continuous Spectrophotometric Rate Determination (Reusch & Burger, 1974; Hidalgo et al, 2006). Briefly, cells were extracted in 200 µl of potassium phosphate buffer. The cells were first homogenized and then sonicated. The homogenate was centrifuged at top speed for 10 min, and the supernatant was used for reading. Oxidation of NADPH was monitored at 340 nm on the SpectraMAX i3 Plate Reader at 30°C (Reusch & Burger, 1974; Hidalgo et al, 2006).
Measurement of NADPH oxidase level
The NOX1 measurement was performed using the Abbexa assay kit (Cat#, abx255148, Abbexa) according to the manufacturer's instructions. Briefly, 20 mg of tissues were extracted in 500 µl of phosphate buffer saline. The tissues were first homogenized and then sonicated. The homogenate was centrifuged at 5,000 g for 5 min, and the supernatant was used for read at OD450 on the SpectraMAX i3 Plate Reader at 25°C. The data was calculated based upon the standard curve.
MeDIP‐qPCR
Genomic DNA was sheared using a Covaris S220 sonicator to an average of 200–600 bp. 600 ng of denatured DNA was incubated with 2 mg of 5‐methylcytosine (5‐mC) monoclonal antibody ([33D3] Diagenode, Cat No # C15200081) in IP buffer (0.1% SDS, 1 Triton X‐100, 2 mM EDTA, 20 mM Tris–HCl pH 8.1, 150 mM NaCl) for 1 h at 4°C on a rotating wheel. Antibody‐bound DNA was collected with 20 ml of protein A/G PLUS‐Agarose (#sc‐2003) for 1 h at 4°C on a rotating wheel and successively washed three times with IP buffer (0.1% SDS, 1 Triton X‐100, 2 mM EDTA, 20 mM Tris–HCl pH 8.1, 150 mM NaCl). DNA was recovered in 100 ml of digestion buffer (50 mM Tris pH 8.0, 0.5% SDS, 35 mg proteinase K) and incubated overnight at 65°C. Recovered DNA was used for qPCR analysis. Primers for MeDIP‐qPCR studies are listed in Appendix Table S1. All data were normalized to input.
ChIP‐qPCR
Soleus muscles were homogenized in dounce homogenizer using Nuclei Preparation Buffer (10 mM HEPES (pH 7.5), 10 mM KCl, 1.5 mM MgCl2, 0.1% NP40) and crosslinked with 1% formaldehyde for 10 min, then neutralized with glycine and rinsed with cold phosphate‐buffered saline. After nuclei isolation, samples were sonicated using an S220 Covaris to generate DNA fragments of ∼ 200–500 bp. Inputs were taken from cleared lysates, and the rest were rotated O/N at 4°C with DNMT3A, H3K27ac, and IgG antibodies for immunoprecipitation. An aliquot of 20 μl of protein A/G PLUS‐Agarose (#sc‐2003) were added per IP and rotated 1 h at 4°C. Beads were successively washed in low‐salt RIPA buffer (20 mM Tris–HCl [pH 8.0], 1 mM EDTA, 1% Triton x‐100, 0.1% SDS,140 mM NaCl, 0.1% Na deoxycholate), high‐salt RIPA buffer (20 mM Tris–HCl [pH 8.0], 1 mM EDTA, 1% Triton x‐100, 0.1% SDS, 500 mM NaCl, 0.1% Na deoxycholate), LiCl buffer (250 mM LiCl, 0.5% NP40, 0.5% Na deoxycholate, 1 mM EDTA, 10 mM Tris–HCl [pH 8.0]) and TE buffer (10 mM Tris–HCl [pH 8.0] and 1 mM EDTA). Each reaction was then incubated in digestion buffer (50 mM Tris–HCl [pH 8.0], 1 mM EDTA, 100 mM NaCl, 0.5% SDS, proteinase K) for a minimum of 4 h at 65°C to reverse cross‐links. DNA was recovered using a phenol‐chloroform extraction. Recovered DNA was used for qPCR analysis. Primers for CHIP‐qPCR studies are listed in Appendix Table S1. All data were normalized to input.
Cell culture
L6 rat myoblasts (UCSF Cell Culture Facility Core) were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. Culture conditions were maintained in a humidified incubator under an atmosphere of 5% CO2 at 37°C. Differentiation was carried out in DMEM supplemented with 2% horse serum. To generate lentivirus particles, lentiviral constructs were co‐transfected with pMD2.G‐ and psPAX2‐expressing plasmids into 293T cells. After 48 h, the virus‐containing supernatant was collected, filtered through 0.45‐mm filters, and added to mature L6 myotubes for 48 h along with 8 mg/ml polybrene. Transduction efficiency was determined by comparing to cells transduced in parallel with a GFP‐expressing lentivirus.
In vivo electroporation
Mice were anesthetized by an IP injection of 91 mg/kg ketamine and 9.1 mg/kg xylazine, after which hindlimbs were shaved, and the GA muscles were injected with 30 μl hyaluronidase solution (which was prepared by resuspending bovine placental hyaluronidase [Sigma] in sterile injectable 0.9% NaCl at a concentration of 0.4 U/μl). Mice were anesthetized 2 h later, and the GAs were injected with 180 μg plasmid DNA in sterile saline. After injection of plasmid DNA, the hind limbs were placed between two‐paddle electrodes and subjected to 10 pulses (20 ms) of 175 V/cm (with 480‐ms intervals between pulses) using an ECM‐830 electroporator (BTX Harvard Apparatus, Holliston, MA).
ROS measurement
Accumulation of hydrogen peroxide (H2O2) and horseradish peroxidase was measured using OxiSelect™ Hydrogen Peroxide/Peroxidase Assay Kit (Cell Biolabs, Inc., San Diego, CA), a sensitive quantitative fluorometric assay for hydrogen peroxide or peroxidase activity levels. To investigate H2O2 accumulation, the muscle tissues or L6 myotubes were homogenized in 1× assay buffer provided from the kit followed by centrifuging to remove debris. These lysates were then assayed according to the manufacturer's procedure.
Mitochondrial respiration
For tissue respiration, mice were allowed to run for 50 min on the low‐intensity regime. Following this, WT and MCK‐Dnmt3a KO soleus tissues were isolated and seeded in XF24 plates (catalog #101122‐100, Seahorse Bioscience). For cellular respiration, lentivirally transduced L6 were plated on XF24 Cell Culture Microplates catalog #100777‐004, Seahorse Bioscience). Measurement of intact tissue and cellular respiration was performed using the Seahorse XF24 analyzer (Seahorse Bioscience). Oxygen consumption rates (OCRs; picomoles of O2 per minute) were measured under basal conditions after three consecutive injections of the following: (i) oligomycin (ATP synthase inhibitor; 4 μM); (ii) the electron transport chain accelerator ionophore FCCP (4 μM; FCCP treatment gives the maximal OCR capacity of the cells); and (iii) the electron transport chain inhibitors Rotenone (4 μM) and Antimycin A (4 μM).
Statistical analysis
Data are presented as means ± SEM and individual data points are plotted. Sample size was determined by our experience with inherent variability. No statistical method was used to predetermine sample size. Statistical analyses and the number of samples (n) were described in detail for each figure panel. Statistical analyses and the number of samples (n) are described in detail for each figure panel. Two‐tailed unpaired Student's t‐test was used for the comparison between two groups. One‐way analysis of variance (ANOVA) or two‐way ANOVA followed by the Bonferroni's test was used for the multiple comparisons. Statistical analyses were performed using excel and GraphPad Prism. All reported P values were two‐sided and differences were considered significant at P < 0.05.
Author contributions
SK supervised experiments and wrote the manuscript. SDV drafted the result, method, and legend sections. Experiments were carried out by SDV, DY, JK, HHP, BCJ, TT and SK. HL analyzed RNA‐Seq. SME and CMA contributed to mouse histology and in vivo transfection assays.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View/Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We thank Dr. Hei Sook Sul, Dr. Jen‐Chywan Wally Wang, and Dr. Anders Näär for helpful conversations about the manuscript. We thank Dr. Boris Rubinsky and Dr. Chenang Lyu for providing us with ECM‐830 electroporator equipment. We are very grateful to Dr. Brian Black and Emily Wilson for technical help with histology, Sarah Fung, Lilian Kim, Anna Pi, Sarah, and Sarah Ampalloor, Mrinalini Jain, Pouya Amin, and Michelle Tampa for their technical supports. Work was funded by AHA Award # 19POST34380834 to DY and R01 DK116008 to SK.
The EMBO Journal (2021) 40: e106491.
Data availability
RNA‐Seq data can be found GSE159105 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE159105).
References
- Arany Z, Lebrasseur N, Morris C, Smith E, Yang W, Ma Y, Chin S, Spiegelman BM (2007) The transcriptional coactivator PGC‐1beta drives the formation of oxidative type IIX fibers in skeletal muscle. Cell Metab 5: 35–46 [DOI] [PubMed] [Google Scholar]
- Barrès R, Yan J, Egan B, Treebak JT, Rasmussen M, Fritz T, Caidahl K, Krook A, O'Gorman DJ, Zierath JR (2012) Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab 15: 405–411 [DOI] [PubMed] [Google Scholar]
- Baskin KK, Winders BR, Olson EN (2015) Muscle as a “mediator” of systemic metabolism. Cell Metab 21: 237–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird AP, Wolffe AP (1999) Methylation‐induced repression–belts, braces, and chromatin. Cell 99: 451–454 [DOI] [PubMed] [Google Scholar]
- Bourdeau Julien I, Sephton CF, Dutchak PA (2018) Metabolic networks influencing skeletal muscle fiber composition. Front Cell Dev Biol 6: 125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boushel R, Lundby C, Qvortrup K, Sahlin K (2014) Mitochondrial plasticity with exercise training and extreme environments. Exerc Sport Sci Rev 42: 169–174 [DOI] [PubMed] [Google Scholar]
- Bowen TS, Schuler G, Adams V (2015) Skeletal muscle wasting in cachexia and sarcopenia: molecular pathophysiology and impact of exercise training. J Cachexia Sarcopenia Muscle 6: 197–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown WM (2015) Exercise‐associated DNA methylation change in skeletal muscle and the importance of imprinted genes: a bioinformatics meta‐analysis. Br J Sports Med 49: 1567–1578 [DOI] [PubMed] [Google Scholar]
- Brüning JC, Michael MD, Winnay JN, Hayashi T, Hörsch D, Accili D, Goodyear LJ, Kahn CR (1998) A muscle‐specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol Cell 2: 559–569 [DOI] [PubMed] [Google Scholar]
- Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, Bock C, Vasanthakumar A, Gu H, Xi Y et al (2011) Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet 44: 23–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen JP, Trap‐Jensen J (1970) Effects of training on the distribution of cardiac output in patients with coronary artery disease. Circulation 42: 611–624 [DOI] [PubMed] [Google Scholar]
- Cross AR, Segal AW (2004) The NADPH oxidase of professional phagocytes–prototype of the NOX electron transport chain systems. Biochim Biophys Acta 1657: 1–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daiber A (2010) Redox signaling (cross‐talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim Biophys Acta 1797: 897–906 [DOI] [PubMed] [Google Scholar]
- Daiber A, Di Lisa F, Oelze M, Kröller‐Schön S, Steven S, Schulz E, Münzel T (2017) Crosstalk of mitochondria with NADPH oxidase via reactive oxygen and nitrogen species signalling and its role for vascular function. Br J Pharmacol 174: 1670–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan Dunn J, Alvarez LA, Zhang X, Soldati T (2015) Reactive oxygen species and mitochondria: a nexus of cellular homeostasis. Redox Biol 6: 472–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBalsi KL, Wong KE, Koves TR, Slentz DH, Seiler SE, Wittmann AH, Ilkayeva OR, Stevens RD, Perry CGR, Lark DS et al (2014) Targeted metabolomics connects thioredoxin‐interacting protein (TXNIP) to mitochondrial fuel selection and regulation of specific oxidoreductase enzymes in skeletal muscle. J Biol Chem 289: 8106–8120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Meo S, Reed TT, Venditti P, Victor VM (2016) Role of ROS and RNS sources in physiological and pathological conditions. Oxid Med Cell Longev 2016: 1245049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Meo S, Iossa S, Venditti P (2017) Skeletal muscle insulin resistance: role of mitochondria and other ROS sources. J Endocrinol 233: R15–R42 [DOI] [PubMed] [Google Scholar]
- Dimauro I, Paronetto MP, Caporossi D (2020) Exercise, redox homeostasis and the epigenetic landscape. Redox Biol 35: 101477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Johnson LM, Jacobsen SE, Patel DJ (2015) DNA methylation pathways and their crosstalk with histone methylation. Nat Rev Mol Cell Biol 16: 519–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez‐Marcos PJ, Nóbrega‐Pereira S (2016) NADPH: new oxygen for the ROS theory of aging. Oncotarget 7: 50814–50815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira LF, Laitano O (2016) Regulation of NADPH oxidases in skeletal muscle. Free Radic Biol Med 98: 18–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finsterer J (2012) Biomarkers of peripheral muscle fatigue during exercise. BMC Musculoskelet Disord 13: 218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes EC, Silva AN, de Oliveira MR (2012) Oxidants, antioxidants, and the beneficial roles of exercise‐induced production of reactive species. Oxid Med Cell Longev 2012: 756132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Görlach A, Bertram K, Hudecova S, Krizanova O (2015) Calcium and ROS: a mutual interplay. Redox Biol 6: 260–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouspillou G, Sgarioto N, Norris B, Barbat‐Artigas S, Aubertin‐Leheudre M, Morais JA, Burelle Y, Taivassalo T, Hepple RT (2014) The relationship between muscle fiber type‐specific PGC‐1α content and mitochondrial content varies between rodent models and humans. PLoS One 9: e103044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouspillou G, Hepple RT (2016) Editorial: mitochondria in skeletal muscle health, aging and diseases. Front Physiol 7: 446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haizlip KM, Harrison BC, Leinwand LA (2015) Sex‐based differences in skeletal muscle kinetics and fiber‐type composition. Physiology (Bethesda) 30: 30–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handschin C, Chin S, Li P, Liu F, Maratos‐Flier E, Lebrasseur NK, Yan Z, Spiegelman BM (2007) Skeletal muscle fiber‐type switching, exercise intolerance, and myopathy in PGC‐1alpha muscle‐specific knock‐out animals. J Biol Chem 282: 30014–30021 [DOI] [PubMed] [Google Scholar]
- Hawley JA, Hargreaves M, Joyner MJ, Zierath JR (2014) Integrative biology of exercise. Cell 159: 738–749 [DOI] [PubMed] [Google Scholar]
- He F, Li J, Liu Z, Chuang C‐C, Yang W, Zuo L (2016) Redox mechanism of reactive oxygen species in exercise. Front Physiol 7: 486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henríquez‐Olguin C, Knudsen JR, Raun SH, Li Z, Dalbram E, Treebak JT, Sylow L, Holmdahl R, Richter EA, Jaimovich E et al (2019) Cytosolic ROS production by NADPH oxidase 2 regulates muscle glucose uptake during exercise. Nat Commun 10: 4623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo C, Sánchez G, Barrientos G, Aracena‐Parks P (2006) A transverse tubule NADPH oxidase activity stimulates calcium release from isolated triads via ryanodine receptor type 1 S ‐glutathionylation. J Biol Chem 281: 26473–26482 [DOI] [PubMed] [Google Scholar]
- Hood DA, Memme JM, Oliveira AN, Triolo M (2019) Maintenance of skeletal muscle mitochondria in health, exercise, and aging. Annu Rev Physiol 81: 19–41 [DOI] [PubMed] [Google Scholar]
- Huertas JR, Casuso RA, Agustín PH, Cogliati S (2019) Stay fit, stay young: mitochondria in movement: the role of exercise in the new mitochondrial paradigm. Oxid Med Cell Longev 2019: 7058350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenisch R, Bird A (2003) Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 33(Suppl): 245–254 [DOI] [PubMed] [Google Scholar]
- Kang JH, Lee S‐H, Hong D, Lee J‐S, Ahn H‐S, Ahn J‐H, Seong TW, Lee C‐H, Jang H, Hong KM et al (2016) Aldehyde dehydrogenase is used by cancer cells for energy metabolism. Exp Mol Med 48: e272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanzleiter T, Jähnert M, Schulze G, Selbig J, Hallahan N, Schwenk RW, Schürmann A (2015) Exercise training alters DNA methylation patterns in genes related to muscle growth and differentiation in mice. Am J Physiol Endocrinol Metab 308: E912–E920 [DOI] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12: 357–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Triolo M, Hood DA (2017) Impact of aging and exercise on mitochondrial quality control in skeletal muscle. Oxid Med Cell Longev 2017: 3165396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong X, Yao T, Zhou P, Yao T, Kazak L, Tenen D, Lyubetskaya A, Dawes BA, Tsai L, Kahn BB, Spiegelman BM, Liu T & Rosen ED, (2018) Brown adipose tissue controls skeletal muscle function via the secretion of myostatin. Cell Metab. 28: 631–643.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupenko SA (2009) FDH: an aldehyde dehydrogenase fusion enzyme in folate metabolism. Chem Biol Interact 178: 84–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupenko SA, Krupenko NI (2019) Loss of ALDH1L1 folate enzyme confers a selective metabolic advantage for tumor progression. Chem Biol Interact 302: 149–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmiak‐Glancy S, Willis WT (2014) Skeletal muscle fuel selection occurs at the mitochondrial level. J Exp Biol 217: 1993–2003 [DOI] [PubMed] [Google Scholar]
- Lane SC, Camera DM, Lassiter DG, Areta JL, Bird SR, Yeo WK, Jeacocke NA, Krook A, Zierath JR, Burke LM et al (2015) Effects of sleeping with reduced carbohydrate availability on acute training responses. J Appl Physiol 119: 643–655 [DOI] [PubMed] [Google Scholar]
- Laughlin MH, Cook JD, Tremble R, Ingram D, Colleran PN, Turk JR (2006) Exercise training produces nonuniform increases in arteriolar density of rat soleus and gastrocnemius muscle. Microcirculation 13: 175–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lejay A, Meyer A, Schlagowski A‐I, Charles A‐L, Singh F, Bouitbir J, Pottecher J, Chakfé N, Zoll J, Geny B (2014) Mitochondria: mitochondrial participation in ischemia‐reperfusion injury in skeletal muscle. Int J Biochem Cell Biol 50: 101–105 [DOI] [PubMed] [Google Scholar]
- Li F, Xiao H, Zhou F, Hu Z, Yang B (2017) Study of HSPB6: insights into the properties of the multifunctional protective agent. Cell Physiol Biochem 44: 314–332 [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen S, Meletis K, Fu D, Jhaveri S, Jaenisch R (2007) Ablation of de novo DNA methyltransferase Dnmt3a in the nervous system leads to neuromuscular defects and shortened lifespan. Dev Dyn 236: 1663–1676 [DOI] [PubMed] [Google Scholar]
- Nishikawa K, Iwamoto Y, Kobayashi Y, Katsuoka F, Kawaguchi S, Tsujita T, Nakamura T, Kato S, Yamamoto M, Takayanagi H et al (2015) DNA methyltransferase 3a regulates osteoclast differentiation by coupling to an S‐adenosylmethionine‐producing metabolic pathway. Nat Med 21: 281–287 [DOI] [PubMed] [Google Scholar]
- Nitert MD, Dayeh T, Volkov P, Elgzyri T, Hall E, Nilsson E, Yang BT, Lang S, Parikh H, Wessman Y et al (2012) Impact of an exercise intervention on DNA methylation in skeletal muscle from first‐degree relatives of patients with type 2 diabetes. Diabetes 61: 3322–3332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Old SL, Johnson MA (1989) Methods of microphotometric assay of succinate dehydrogenase and cytochrome c oxidase activities for use on human skeletal muscle. Histochem J 21: 545–555 [DOI] [PubMed] [Google Scholar]
- Oleinik NV, Krupenko NI, Krupenko SA (2011) Epigenetic silencing of ALDH1L1, a metabolic regulator of cellular proliferation, in cancers. Genes Cancer 2: 130–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panday A, Sahoo MK, Osorio D, Batra S (2015) NADPH oxidases: an overview from structure to innate immunity‐associated pathologies. Cell Mol Immunol 12: 5–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrônio MS, Zeraik ML, da Fonseca LM, Ximenes VF (2013) Apocynin: chemical and biophysical properties of a NADPH oxidase inhibitor. Molecules 18: 2821–2839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picca A, Calvani R, Leeuwenburgh C, Coelho‐Junior HJ, Bernabei R, Landi F, Marzetti E (2019) Targeting mitochondrial quality control for treating sarcopenia: lessons from physical exercise. Expert Opin Ther Targets 23: 153–160 [DOI] [PubMed] [Google Scholar]
- Powers SK, Ji LL, Kavazis AN, Jackson MJ (2011) Reactive oxygen species: impact on skeletal muscle. Compr Physiol 1: 941–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser BL, Ward CW, Lederer WJ (2011) X‐ROS signaling: rapid mechano‐chemo transduction in heart. Science 333: 1440–1445 [DOI] [PubMed] [Google Scholar]
- Radak Z, Bori Z, Koltai E, Fatouros IG, Jamurtas AZ, Douroudos II, Terzis G, Nikolaidis MG, Chatzinikolaou A, Sovatzidis A et al (2011) Age‐dependent changes in 8‐oxoguanine‐DNA glycosylase activity are modulated by adaptive responses to physical exercise in human skeletal muscle. Free Radic Biol Med 51: 417–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radak Z, Zhao Z, Koltai E, Ohno H, Atalay M (2013) Oxygen consumption and usage during physical exercise: the balance between oxidative stress and ROS‐dependent adaptive signaling. Antioxid Redox Signal 18: 1208–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen M, Zierath JR, Barrès R (2014) Dynamic epigenetic responses to muscle contraction. Drug Discov Today 19: 1010–1014 [DOI] [PubMed] [Google Scholar]
- Reusch VM, Burger MM (1974) Distribution of marker enzymes between mesosomal and protoplast membranes. J Biol Chem 249: 5337–5345 [PubMed] [Google Scholar]
- Roman W, Gomes ER (2018) Nuclear positioning in skeletal muscle. Semin Cell Dev Biol 82: 51–56 [DOI] [PubMed] [Google Scholar]
- Rowe GC, El‐Khoury R, Patten IS, Rustin P, Arany Z (2012) PGC‐1α is dispensable for exercise‐induced mitochondrial biogenesis in skeletal muscle. PLoS One 7: e41817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safdar A, Little JP, Stokl AJ, Hettinga BP, Akhtar M, Tarnopolsky MA (2011) Exercise increases mitochondrial PGC‐1alpha content and promotes nuclear‐mitochondrial cross‐talk to coordinate mitochondrial biogenesis. J Biol Chem 286: 10605–10617 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Sakellariou GK, Vasilaki A, Palomero J, Kayani A, Zibrik L, McArdle A, Jackson MJ (2013) Studies of mitochondrial and nonmitochondrial sources implicate nicotinamide adenine dinucleotide phosphate oxidase(s) in the increased skeletal muscle superoxide generation that occurs during contractile activity. Antioxid Redox Signal 18: 603–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaborne RA, Strauss J, Cocks M, Shepherd S, O'Brien TD, van Someren KA, Bell PG, Murgatroyd C, Morton JP, Stewart CE et al (2018) Methylome of human skeletal muscle after acute & chronic resistance exercise training, detraining & retraining. Sci Data 5: 180213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simioni C, Zauli G, Martelli AM, Vitale M, Sacchetti G, Gonelli A, Neri LM (2018) Oxidative stress: role of physical exercise and antioxidant nutraceuticals in adulthood and aging. Oncotarget 9: 17181–17198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S, Wen Y, Tong H, Loro E, Gong Y, Liu J, Hong S, Li L, Khurana TS, Chu M et al (2019) The HDAC3 enzymatic activity regulates skeletal muscle fuel metabolism. J Mol Cell Biol 11: 133–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbacher P, Eckl P (2015) Impact of oxidative stress on exercising skeletal muscle. Biomolecules 5: 356–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot J, Maves L (2016) Skeletal muscle fiber type: using insights from muscle developmental biology to dissect targets for susceptibility and resistance to muscle disease. Wiley Interdiscip Rev Dev Biol 5: 518–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidball JG, Wehling‐Henricks M (2007) The role of free radicals in the pathophysiology of muscular dystrophy. J Appl Physiol 102: 1677–1686 [DOI] [PubMed] [Google Scholar]
- Valenzuela N, Soibam B, Li L, Wang J, Byers LA, Liu Y, Schwartz RJ, Stewart MD (2017) HIRA deficiency in muscle fibers causes hypertrophy and susceptibility to oxidative stress. J Cell Sci 130: 2551–2563 [DOI] [PubMed] [Google Scholar]
- Welle SL (2009) Myostatin and muscle fiber size. Focus on “Smad2 and 3 transcription factors control muscle mass in adulthood” and “Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size”. Am J Physiol Cell Physiol 296: C1245–C1247 [DOI] [PubMed] [Google Scholar]
- Widmann M, Nieß AM, Munz B (2019) Physical exercise and epigenetic modifications in skeletal muscle. Sports Med 49: 509–523 [DOI] [PubMed] [Google Scholar]
- Yoo S‐Z, No M‐H, Heo J‐W, Park D‐H, Kang J‐H, Kim SH, Kwak H‐B (2018) Role of exercise in age‐related sarcopenia. J Exerc Rehabil 14: 551–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zechner C, Lai L, Zechner JF, Geng T, Yan Z, Rumsey JW, Collia D, Chen Z, Wozniak DF, Leone TC et al (2010) Total skeletal muscle PGC‐1 deficiency uncouples mitochondrial derangements from fiber type determination and insulin sensitivity. Cell Metab 12: 633–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View/Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Data Availability Statement
RNA‐Seq data can be found GSE159105 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE159105).