

Abstract
Lowe Syndrome (LS) is a lethal genetic disorder caused by mutations in the OCRL1 gene which encodes the lipid 5′ phosphatase Ocrl1. Patients exhibit a characteristic triad of symptoms including eye, brain and kidney abnormalities with renal failure as the most common cause of premature death. Over 200 OCRL1 mutations have been identified in LS, but their specific impact on cellular processes is unknown. Despite observations of heterogeneity in patient symptom severity, there is little understanding of the correlation between genotype and its impact on phenotype.
Here, we show that different mutations had diverse effects on protein localization and on triggering LS cellular phenotypes. In addition, some mutations affecting specific domains imparted unique characteristics to the resulting mutated protein. We also propose that certain mutations conformationally affect the 5′-phosphatase domain of the protein, resulting in loss of enzymatic activity and causing common and specific phenotypes (a conformational disease scenario).
This study is the first to show the differential effect of patient 5′-phosphatase mutations on cellular phenotypes and introduces a conformational disease component in LS. This work provides a framework that explains symptom heterogeneity and can help stratify patients as well as to produce a more accurate prognosis depending on the nature and location of the mutation within the OCRL1 gene.
Introduction
Lowe Syndrome (LS) or Oculo-Cerebro-Renal syndrome of Lowe (OCRL) (OMIM#30900) is an X-linked genetic disorder caused by mutations in the OCRL1 gene (1). Affected children are born with bilateral cataracts, and they present neurological abnormalities and renal symptoms shortly after birth (2,3). Progressive renal dysfunction leads to end-stage renal disease and premature death (2). Unfortunately, currently there are no LS-specific therapeutics (4).
The gene product of OCRL1 is the inositol 5′ phosphatase Ocrl1 (EC 3.1.3.36) which has specificity for the signaling lipid phosphatidyl inositol 4,5-bisphophate, PI(4,5)P2 (5,6). Ocrl1 localizes at the trans-Golgi network (TGN) (7), endosomes (8) and transiently at the plasma membrane (9). In addition to its phosphatase domain, Ocrl1 displays N-terminal PH (10) and C-terminal ASH-RhoGAP (8,11) domains through which it interacts with several signaling and trafficking proteins, thereby participating in several basic cellular processes including cell migration, cell spreading, actin remodeling, ciliogenesis, vesicle trafficking, cytokinesis and phagocytosis (12–22). Further, we previously established that Ocrl1 participates in cellular processes through determinants spatially segregated within the protein. Specifically, the N-terminus is required for membrane remodeling (12) and the C-terminus for primary cilia assembly (15), while a functional Ocrl1 5′-phosphatase domain is required for both processes to proceed normally (12,15).
There are over 200 unique LS-causing mutations identified in OCRL1 (4,23) with many affecting different domains of Ocrl1 and a characteristic broad symptom variability among patients. However, little is known about the correlation between the genotype of patients and their cellular phenotypes.
Here we show that depending on the domain they affect, OCRL1 mutations have a differential impact on cell spreading and ciliogenesis, thereby suggesting cellular basis for LS patient symptom heterogeneity. In addition, specific mutants possess unique characteristics in terms of localization and protein stability. Importantly, some patient variants bearing a mutated phosphatase domain induced fragmentation of the Golgi apparatus. This phenotype was not induced by any other patient mutation tried, and although novel for LS, it has been previously observed in diseases affecting the nervous system (24–26). Therefore, and given that LS has neurological manifestations, we speculate that this defect may play a role in disease pathogenesis.
In addition, molecular dynamics analysis of Ocrl1 patient variants with residue changes at non-catalytic sites within the phosphatase domain predicted the existence of conformational changes affecting the active site. Further, experimental results showed impairment of 5′-phosphatase activity for these mutants as well as the presence of cellular phenotypes in cells expressing these variants. These results explain how mutations affecting non-catalytic residues cause LS, but also imply that some OCRL1 mutants lead to a conformational/protein misfolding disease scenario.
We believe that this study will provide the framework that contributes to explain symptom heterogeneity and will allow LS patient stratification as well as aid in creating tailored therapeutic strategies that would consider the nature and location of mutation in the gene as well as its effect on the biochemical activity of Ocrl1.
Results
Segregation of OCRL1 missense patient mutations according to the Ocrl1 domain they affect
This study is focused on understanding the consequences of OCRL1 missense mutations on several characteristics (stability, localization and ability to sustain specific cellular functions) of the encoded 5′-phosphatase Ocrl1.
Since 90% of missense mutations found in LS patients locate within OCRL1’s exons encoding for the phosphatase and ASH (ASPM-SPD-2-Hydin)-RhoGAP (RhoGTPase Activating Protein) domains (4,23,27–30), we selected patient missense mutations located within exons 9–23 for this study (Table 1 and Fig. 1A).
Table 1.
OCRL1 patient mutants used in this study
| Mutation | Exon | Domain affected |
|---|---|---|
| S256N | 9 | 5′ phosphatase |
| D451G | 13 | |
| V508D | 15 | |
| H524R | 15 | |
| V577E | 17 | ASH-RhoGAP |
| F668V | 18 | |
| I768N | 21 |
Figure 1.
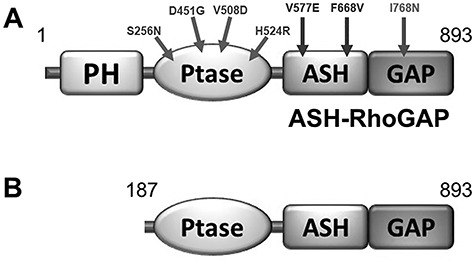
Ocrl1 variants used in this study. A: Residue change due to LS patient missense mutations are mapped on the Ocrl1 molecule. B: Representation of a proposed Ocrl1 variant resulting from alternative initiation at Met187. PH: Pleckstrin homology; Ptase: Inositol phosphatase domain; ASH: ASPM-SPD- 2-Hydin; RhoGAP: RhoGTPase Activating Protein.
There are nearly 80 unique missense, LS-causing mutations which lead to amino acid changes affecting Ocrl1’s phosphatase domain. While many of these replacements map to conserved regions important for catalysis and substrate recognition, other mutations affected residues not directly involved in substrate binding and processing (Supplementary Material, Fig. S1). Therefore, for this project we selected Ocrl1 patient variants to represent both categories; i.e. H524R (critical catalytic residue replaced), S256N, D451G and V508D (affecting non-catalytic residues) (Supplementary Material, Fig. S1). In addition, there are nearly 20 unique missense mutations resulting in changes in the amino acids encoding the C-terminal ASH-RhoGAP domain of Ocrl1. From these, here we selected variants V577E, F668V and I768N for functional studies (Table 1 and Fig. 1A).
Although a few Ocrl1 variants have been found to display amino acid changes in the PH domain, there are no reported LS-causing missense mutations in exons 2–7 that we know of. Nevertheless, several nonsense, frameshift mutations and deletions have been found within these 5′ exons of the OCRL1 gene. Strikingly, these mutations often cause a milder condition known as Dent-2 disease (27). Interestingly, Shrimpton et al. (31) proposed that in these cases an alternative initiation codon (corresponding to Ocrl1’s Met187) is used to translate an N-terminal truncation (lacking the PH domain) that would retain substantial functionality. We recreated such Ocrl1187–901 (ΔPH) product for characterization in this study (Fig. 1B).
Ocrl1 patient variants differentially affect cell spreading and ciliogenesis in human kidney epithelial cells
Since renal failure is the main cause of death among LS patients, in this study we mostly used human embryonic kidney 293 T epithelial and proximal tubule HK2 cells lacking OCRL1 (HEK293T KO and HK2 KO, respectively—see Materials and Methods). We also focused on cell spreading and ciliogenesis as cellular readouts as they are likely to impact organogenesis and renal function.
Cell spreading: We transfected different GFP-tagged Ocrl1 mutated variants or Ocrl1WT in HEK293T KO cells and performed standard spreading assays as described before (12) and in Materials and Methods. Briefly, 18 h after transfection, cells were lifted and allowed to attach and spread for 30 min on fibronectin-coated coverslips. Next, cells were fixed with 4% formaldehyde, and stained with rhodamine-phalloidin to label the actin cytoskeleton. Random fields containing transfected cells were imaged and the spreading area was measured using the magic wand tracing tool from ImageJ, fraction of cells versus cell area histograms were constructed, and statistical analysis was performed using the Kolmogorov–Smirnov test as described in Materials and Methods.
In agreement with the hypothesis that Ocrl1’s N-terminus region is required for membrane remodeling (12), Ocrl1ΔPH-expressing cells showed a significant spreading defect detected as a shift of their histogram toward smaller areas as compared to the one corresponding to Ocrl1WT (Fig. 2A).
Figure 3.
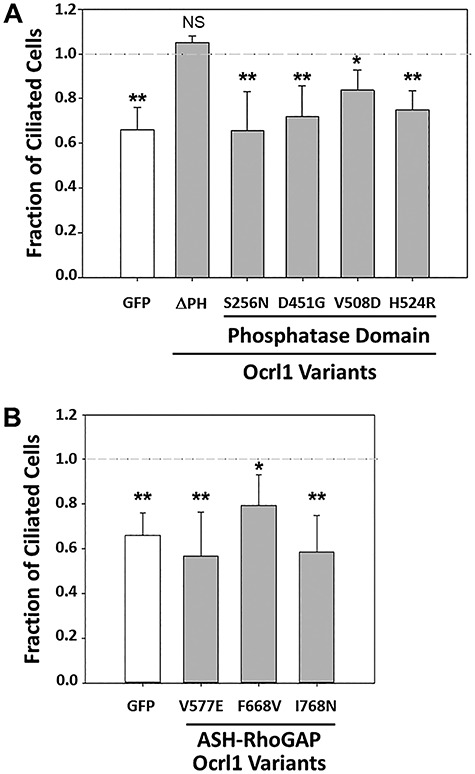
Ocrl1 patient variants differentially affect ciliogenesis. HEK293T KO cells were transfected with different Ocrl1WT, Ocrl1ΔPH and Ocrl1 phosphatase domain mutants (A) or ASH-RhoGAP domain mutants (B). Ciliogenesis assays were performed as described in Materials and Methods. A total of 20 random fields with at least 50 cells were imaged and the fraction of transfected cells with cilia was calculated. This number was normalized to the fraction of Ocrl1WT-expressing cells forming cilia. Each experiment was repeated at least thrice (n = 120–150 cells). Statistically significance of the mean difference with respect to Ocrl1WT was A: **P < 0.05, *P < 0.1 (Bonferroni corrected for 6 comparisons), N.S = not significant by Student’s t-test; B: **P < 0.05, *P < 0.1 (Bonferroni corrected for four comparisons) by the Student’s t-test. The horizontal reference line represents the normalized fraction of Ocrl1WT expressing cells making cilia.
Figure 2.
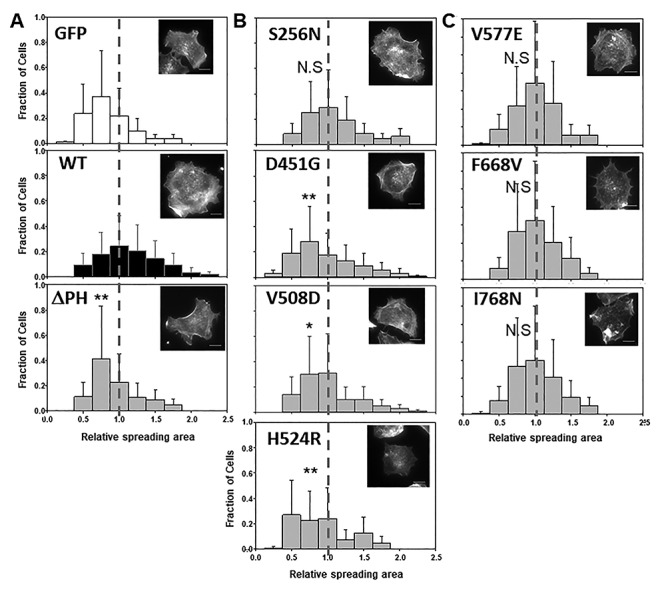
Ocrl1 patient variants differentially affect cell spreading. HEK293T KO cells were transfected with Ocrl1WT, Ocrl1ΔPH (A); phosphatase domain mutants (B); or ASH-RhoGAP domain mutants (C) and allowed to attach and spread on fibronectin-coated surfaces (See Materials and Methods). Median spreading areas of cells expressing mutants were normalized with respect to Ocrl1WT. Histograms were constructed using data from three independent experiments, with a total n = 120–150 cells each. Example of rhodamine-phalloidin stained cells representative of the high-frequency groups within each histogram. Scale bar: 10 μm. Statistically significance of the mean difference with respect to Ocrl1WT was A: **P < 0.05 by KS test; B: **P < 0.05, *P < 0.1 (Bonferroni corrected by 4 comparisons), N.S = not significant by KS test; C: N.S = not significant. The vertical reference line highlights WT-normalized median position.
We found that cells expressing most Ocrl1 patient variants bearing a mutated phosphatase domain also displayed significant smaller spreading areas compared to those expressing Ocrl1WT (Fig. 2B). However, Ocrl1S256N did not induce a significant spreading defect in cells (Fig. 2B). Mutated patient variants within this category induced different degree of severity in the cell spreading phenotype; e.g. while Ocrl1D451G and Ocrl1H524R showed a more severe spreading phenotype, Ocrl1V508D was modestly affected and as indicated above, Ocrl1S256N produced no spreading phenotype (Fig. 2B).
Although cells expressing ASH-RhoGAP mutants Ocrl1V577E, Ocrl1F668V and Ocrl1I768N were somewhat prone to show smaller cell spreading areas, the differences were not statistically significant as compared to WT (Fig. 2C). These results were in agreement with previous observations that indicated that the C-terminal region of Ocrl1 is not critical for membrane remodeling (12).
Ciliogenesis: We also tested the impact of Ocrl1 patient mutated variants on primary cilia (PC) assembly, to such effect HEK293T KO cells were seeded at ~30–50% confluency, transfected with plasmids for expression of Ocrl1WT/patient variants and ciliogenesis assays were performed as described previously (15) and in Materials and Methods. Briefly, 18 h after transfection, cells were serum-starved for 24 h by maintaining them in 0.1% fetal bovine serum (FBS) media to induce ciliogenesis. After that, cells were fixed with 4% formaldehyde, followed by indirect immunofluorescence using antibodies against acetylated tubulin (to label primary cilia) and pericentrin-2 (to label centriole at the base of cilia). Fields were randomly imaged and the fraction of Ocrl1 mutant transfected cells forming cilia was determined and compared to the fraction of ciliated Ocrl1WT-expressing cells (See Materials and Methods).
Interestingly, Ocrl1ΔPH was unaffected for ciliogenesis (Fig. 3A), likely due to the presence of functional phosphatase and ASH-RhoGAP domains which were previously shown by our lab to be required for PC assembly (15). These results also indicated that the N-terminal truncated Ocrl1 variant starting at M187 is partially functional; i.e. despite lacking the ability to facilitate cell spreading, it was capable of sustaining ciliogenesis (Figs 2A and 3A).
Figure 4.
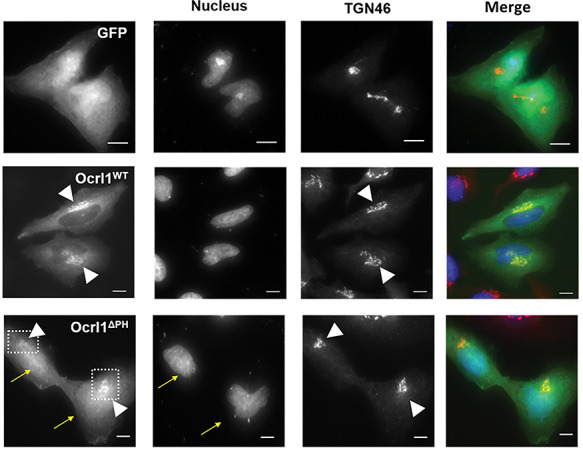
Truncation of Ocrl1 PH domain results in nuclear mislocalization of variant. HK2 KO cells transiently expressing GFP, Ocrl1WT or Ocrl1ΔPH and immunostained for TGN (see Materials and Methods). The arrows indicate Ocrl1ΔPH enrichment in the nuclear compartment; the arrowheads point to TGN and TGN-colocalizing Ocrl1 within the boxed area. Scale bar: 10 μm.
All cells expressing phosphatase mutants were affected for ciliogenesis in varying degrees (Fig. 3A). Similar to their effect on cell spreading, the severity of the ciliogenesis phenotype varied among different patient variants. While cells expressing Ocrl1H524R, Ocrl1S256N and Ocrl1D451G were clearly impaired for PC assembly, expression of Ocrl1V508D caused a significant but milder phenotype. In contrast to its lack of impact on cell spreading (Fig. 2B), Ocrl1S256N could not sustain ciliogenesis in HEK293T KO cells (Fig. 3A).
In agreement with previous findings, indicating that Ocrl1’s C-terminus was important for cilia assembly, cells expressing the ASH-RhoGAP V577E, F668V and I768N mutated variants were affected for ciliogenesis in different degree as compared to Ocrl1WT (with Ocrl1F668V showing a modest defect–Fig. 3B).
Cellular phenotypes and protein localization abnormalities associated with specific Ocrl1 patient mutated variants
In addition to well-established LS cellular phenotypes such as defects in membrane remodeling (e.g. cell spreading) and ciliogenesis, certain patient mutated variants exhibited the following specific phenotypes and/or localization abnormalities: (a) Ocrl1ΔPH: deficient localization; (b) Ocrl1 phosphatase mutants: induction of Golgi apparatus fragmentation and disperse punctate pattern with poor TGN co-localization. (c) Ocrl1 variants bearing mutated ASH-RhoGAP domain: mislocalization to centriolar structures (some of these variants also exhibited cytosolic aggregates).
Deficient localization of Ocrl1ΔPH
For this and other microscopy-based analysis (Figs 4–6) we used human kidney HK2 OCRL1 K.O. (HK2 KO) cells. HK2 cells are larger in size and possess a flatter morphology than HEK293T, making them more suitable to microscopy analysis (in addition, all phenotypes described before for each Ocrl1 mutated variant were also observed in HK2 KO cells). Using these cells, we observed that similar to Ocrl1WT, GFP-Ocrl1ΔPH localized to the TGN (Fig. 4: arrowheads, second and third image row; see also Supplementary Material, Fig. S2), but surprisingly, it also showed an increased cytosolic and nuclear localization (Fig. 4, arrows in bottom panels). Since it is well known that GFP displays nonspecific affinity for the nucleus (e.g.Fig. 4, top left panel), we speculate that lack of the PH domain weakens Ocrl1’s ability to localize, causing a fraction of the protein pool to remain in the cytosol and susceptible to be dragged into the nucleus by fusion to GFP. Therefore, this aberrant mislocalization of the Ocrl1ΔPH variant to the nucleus suggests that the N-terminus somehow contributes to maintain proper Ocrl1’s intracellular localization.
Figure 5.
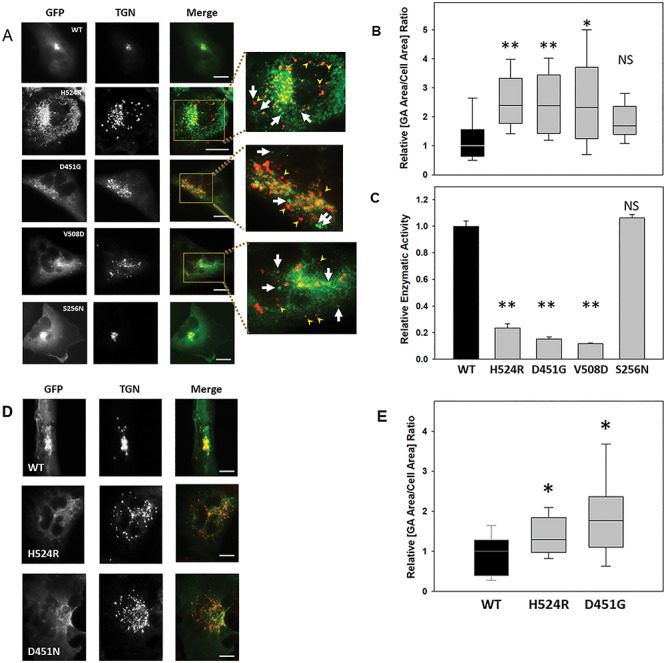
Phosphatase domain mutants produce Golgi apparatus fragmentation. A: HK2 KO cells transiently transfected with GFP-tagged Ocrl1WT, Ocrl1H524R, Ocrl1D451G, Ocrl1V508D or Ocrl1S256N were immunostained for TGN. The highlighted region in merged images corresponds to the TGN area which was enlarged 3X (inset images) to better visualize Golgi apparatus fragmentation. The arrowheads indicate fragmented TGN puncta that lack Ocrl1. The arrows indicate dispersed mutant Ocrl1 lacking TGN colocalization. Scale bar: 10 μm. B-C: The ability of the indicated Ocrl1 patient variants (see horizontal axis at bottom of panel C) to induce GA fragmentation (B) and to catalyze PI(4,5)P2 hydrolysis (C) was tested. B: At least 40 cells transfected with the indicated variants (see horizontal axis at bottom of panel C) were imaged randomly per experiment and the area occupied by TGN was quantified relative to the total cell area (See Materials and Methods). Each experiment was repeated at least thrice with total n = 120–150 cells/group. Statistically significance of the mean difference with respect to Ocrl1WT was **P < 0.05, *P < 0.1 (Bonferroni corrected for four comparisons), NS = No significant by the Wilcoxon test. C: Bacterially expressed and purified GST-Ocrl11–563; WT and the different phosphatase mutants were assayed for enzymatic activity in vitro using the malachite green method as described in Materials and Methods. Statistically significance of the mean difference with respect to Ocrl1WT was **P < 0.05 (Bonferroni corrected for four comparisons), NS = No significant by the t-test. D: HK2 KO cells stably expressing GFP-tagged Ocrl1WT, Ocrl1H524R or Ocrl1D451G (see Materials and Methods) were prepared and immunostained with an anti-TGN46 antibody. Scale bar: 10 μm. E: Golgi apparatus fragmentation determined as a function of total cell area. All cells stably expressing Ocrl1 variants were imaged and TGN area was quantified relative to the total cell area (see Materials and Methods). Statistically significance of the mean difference with respect to Ocrl1WT was *P < 0.1 (Bonferroni corrected for two comparisons) by the Wilcoxon test.
Figure 7.
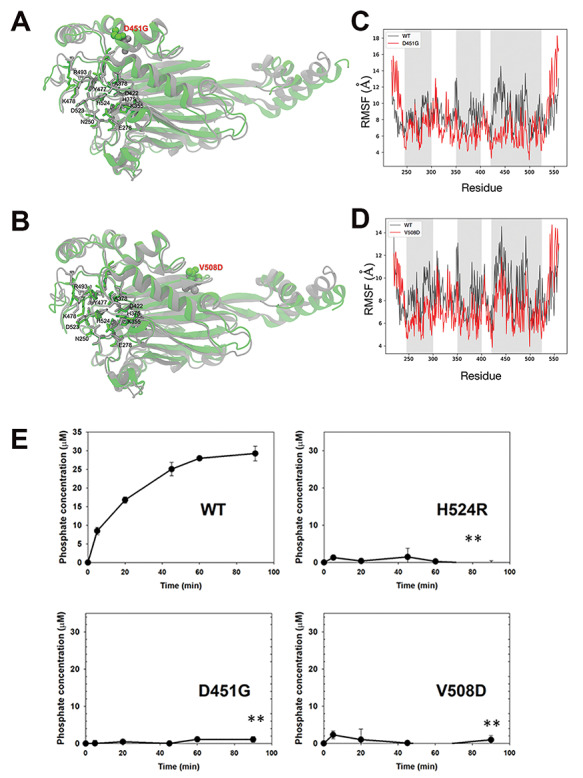
Molecular dynamics prediction of the effect of D451G/V508D mutations on Ocrl1 phosphatase domain structure. A-B: Conformational change in WT (gray) and mutant (green). The residues in catalytic site are represented by sticks, while point mutated variant D451G (A) or V508D (B) is shown by a space-filled model. C-D: Root mean square fluctuation (RMSF), comparison between WT and mutant D451G (C) or V508D (D) indicated that the flexibility of regions within the active site is affected (gray zones). E: Ocrl1 phosphatase domain mutants are impaired for 5′ phosphatase activity. Enzymatic activity was measured as described in Materials and Methods and Figure 5C. Experiments were done at constant enzyme and substrate concentrations while varying the incubation times. All experiments were done in triplicates and repeated at least thrice. Statistically significance of the mean difference with respect to Ocrl1WT at the latest time point was **P < 0.05 (Bonferroni corrected for three comparisons) by student t-test.
Induction of Golgi apparatus (GA) fragmentation
We had previously observed that overexpression of the patient’s phosphatase-dead Ocrl1H524R variant led to fragmentation of the Golgi complex in HeLa cells (12). It should be highlighted that GA fragmentation has been associated with neurological disorders including Alzheimer’s, Parkinson’s and Huntington’s diseases, as well as amyotrophic lateral sclerosis, Angelman syndrome, spinal muscular atrophy and epilepsy (24–26,32,33). Given the presence of a neurological component in LS (2,4), we speculated that this cellular phenotype is relevant to this disease pathogenesis. In addition, GA-related secretion defects have also been observed in polycystic kidney disease (34–36).
Since previous results (12) suggested that lack of phosphatase activity is important for this phenotype to manifest, we tested the ability of Ocrl1 patient variants bearing mutated 5′-phosphatase domains to induce GA fragmentation (measured as the ratio between the area occupied by the GA and the whole cell area, see Materials and Methods—Fig. 5A and B) in HK2 KO. While HK2 KO cells expressing Ocrl1WT showed a continuous and compact Golgi complex, as seen by labeling the trans-Golgi network (using an anti-TGN46 antibody) and the cis-Golgi network (using an anti-GM130 antibody, Supplementary Material, Fig. S3), cells expressing patient variants H524R, D451G and V508D displayed a discontinuous and fragmented Golgi complex that occupied a larger area than the one covered by the GA in cells expressing Ocrl1WT (Fig. 5A, B and Supplemental Material, Fig. S3). Interestingly, we also observed that these Ocrl1 mutated variants displayed a dispersed punctate pattern themselves with poor colocalization between the mutated Ocrl1 variant and TGN46 (Fig. 5A, insets; see also Supplementary Material, Fig. S2) and lacked phosphatase activity (Fig. 5C).
Figure 6.
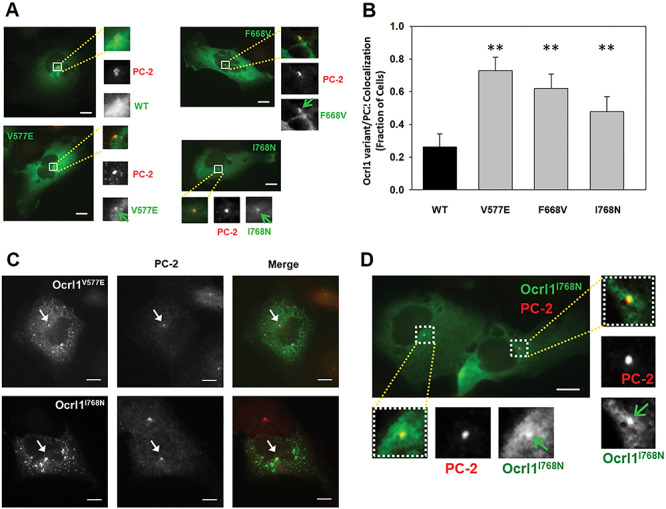
ASH-RhoGAP domain mutants exhibit enrichment or accumulation at the centriole under steady state conditions. A: HK2 KO cells transiently expressing Ocrl1WT or ASH-RhoGAP mutants and maintained in serum-supplemented complete media were immunostained for the centriole marker PC-2 (see Materials and Methods). The arrows indicate Ocrl1 at PC-2 labeled structures. B: Transfected cells were randomly imaged from at least 25 fields containing at least 40 cells. Cells with Ocrl1 colocalization to PC-2 were scored and fraction of cells exhibiting colocalization in a field was determined. Statistically significance of the mean difference with respect to Ocrl1WT was **P < 0.05 (Bonferroni corrected for three comparisons) by the t-test. C: HK2 KO cells transiently expressing Ocrl1V577E or Ocrl1I768N exhibiting protein aggregation. The arrows point to GFP-Ocrl1 colocalization with PC-2. D: HK2 KO cells stably expressing Ocrl1I768N were immunostained for PC-2 (centriole marker) (see Materials and Methods). The highlighted region in merged images corresponds to the perinuclear region which was scaled to 3X (inset images) to better visualize Ocrl1 and PC-2 colocalization. The arrows indicate Ocrl1 at PC-2 labeled structures. Scale bar: 10 μm.
Importantly, the magnitude of the GA fragmentation was independent of the amount of GFP-tagged Ocrl1 mutated variant expressed (measured as total cell-associated green fluorescence intensity, Supplementary Material, Fig. S4). Along the same lines, this phenotype was also observed in HK2 KO cells stably expressing the 5′-phosphatase affected Ocrl1 variants H524R and D451G (Fig. 5D and E). Similar results were observed using HeLa and HEK293T cells (Data not shown).
In contrast, cells expressing Ocrl1S256N did not show substantial GA fragmentation (Fig. 5A and B). Importantly, in vitro malachite green 5′ phosphatase assays using GST-Ocrl1S256N (see Materials and methods) revealed that this variant exhibited substantial 5′ phosphatase activity (Fig. 5C).
ASH-RhoGAP mutated patient variants show mislocalization to centriolar structures
Expression of GFP-tagged ASH-RhoGAP mutants Ocrl1V577E, Ocrl1F668V and Ocrl1I768N in HK2 KO cells under steady state conditions and in presence of serum led to the formation of a GFP-positive perinuclear puncta (Fig. 6A).
A similar localization was previously observed for Ocrl1WT, but only under ciliogenesis induction conditions (i.e. serum-starvation) (15). Specifically, we have shown that during ciliogenesis, Ocrl1WT localize transiently at the base of the cilium (where the axoneme-linked centrioles are), presumably for trafficking of ciliary cargo to the PC (15). Therefore, although steady state experiments (like the ones shown inFig. 6A) are not done under serum-starvation conditions, we wondered if the punctate structure observed in cells expressing ASH-RhoGAP mutated Ocrl1 would also correspond to the centrioles. Indeed, we found that GFP-tagged Ocrl1V577E, Ocrl1F668V and Ocrl1I768N perinuclear punctate structures colocalized with pericentrin-2 (PC-2: a centriole marker, Fig. 6A and B). In contrast, under the same conditions (using serum-supplemented media; e.g.Fig. 6A), we could not observe substantial localization of Ocrl1WT at the centriole.
In addition, ASH-RhoGAP mutated Ocrl1V577E, Ocrl1F668V and Ocrl1I768N patient variants had an overall diffuse cytosolic appearance in cells and lacked typical enrichment at the Golgi apparatus. We also analyzed centriolar localization of the ASH-RhoGAP mutated proteins as a function of their levels of expression. Specifically, we segregated transfected cells according to their total Ocrl1 variant content (i.e. total fluorescence intensity) into ‘low’ (up to 5.8 × 106 fluorescence arbitrary units, corresponding to >80% of cells) and ‘medium-high’ (above 5.8 × 106 fluorescence arbitrary units) groups covering the entire cell population (Supplementary Material, Fig. S5A and SB). Independently of the total fluorescence intensity, GFP-tagged Ocrl1 patient variants Ocrl1V577E, Ocrl1F668V and Ocrl1I768N colocalized with the centriole (Supplementary Material, Fig. S5A). Further, at higher levels of expression, Ocrl1 mutated proteins Ocrl1V577E and Ocrl1I768N showed evidence of protein aggregation (Fig. 6C and Supplementary Material, Fig. S5B). Although this may not occur at physiological levels of expression, it suggested a destabilizing effect on Ocrl1 of these ASH-RhoGAP mutations.
Importantly, and in contrast to Ocrl1WT, low copy number, stably transfected Ocrl1I768N cells also showed centriolar localization lacking perinuclear enrichment (Fig. 6D). In agreement with our observations on low-expressing transient transfectants, stably expressing cells did not exhibit protein aggregates (Fig. 6D, Supplementary Material, Fig. S4B).
Selected OCRL1 mutations affecting phosphatase domain likely cause a conformational change in the catalytic domain of the mutated protein
Although some of the tested Ocrl1 patient variants exhibited changes in phosphatase domain’s residues not directly involved in binding/processing of substrate (Supplementary Material, Fig. S1), they produced cellular defects linked to lack of enzymatic activity.
The phosphatase domain contains six conserved motifs essential for 5′ phosphatase activity (37–40), as well as other conserved residues involved in substrate binding and lipid chain interactions (39,41) (Supplementary Material, Fig. S1). For example, the mutation H524R affects a critical residue that interacts with the scissile 5′ phosphate (39,41) (Supplementary Material, Fig. S1), and has been demonstrated to abolish phosphatase activity (28). In contrast, D451G and V508D affect residues that are found outside the conserved catalytic motifs within the phosphatase domain (Supplementary Material, Fig. S1), and yet were affecting cellular functions of Ocrl1 (Figs 2, 3, 5; Supplementary Material, Figs S3, S4). We hypothesized that these mutations affect the conformation of the catalytic domain, impairing its phosphatase activity.
We used molecular dynamics (MD) to model residue changes D451G and V508D in the phosphatase domain of Ocrl1 (PDB ID: 4CMN). Since the 4CMN structural data did not include substrate molecule coordinates, we simulated a PI(4,5)P2-containing membrane and Ocrl1 system (see details in Materials and Methods). The Ocrl1WT protein-membrane system was equilibrated using MD to obtain a starting model of enzyme—substrate complex (Supplementary Material, Fig. S5).
Our results predict that although the changed amino acids were physically distant from the enzyme’s active site, they induce conformation changes affecting the position of critical catalytic residues (Fig. 7A and B). Further, we calculated per residue root mean square fluctuation (RMSF) to identify changes in the flexibility of the phosphatase domain of Ocrl1. Our results indicate that the D451G and V508D point mutations affected the flexibility of various regions of the catalytic site (Fig. 7C and D).
Although results from Figure 5C already showed that mutations D451G and V508D affected Ocrl1 phosphatase activity, we performed a time-course analysis of the catalytic function of these variants using in vitro malachite green phosphatase assays (Materials and Methods) and bacterially purified GST-fusions of Ocrl11–563 (i.e. truncations containing the PH-phosphatase domain) from patient variants (D451G, V508D and H524R) or WT.
As expected, Ocrl1H524R lacked phosphatase activity (Fig. 7E). Importantly, and in agreement with our MD results, although the D451G and V508D amino-acid changes do not directly affect the catalytic site of Ocrl1, they led to 5′-phosphatase domain inactivation (Fig. 7E).
Discussion
Complementing the pioneering works of different authors (12–16,28,31,42–49) this study contributes to better understand LS as a complex disease. Here we highlight the heterogenous nature of this condition by focusing on the impact of different OCRL1 missense mutations on typical cellular phenotypes while reporting previously unnoticed LS abnormalities triggered by specific patient variants. Indeed, to the best of our knowledge, this is the first systematic analysis of the impact on specific phenotypes of a group of mutations encoding for residue changes in all regions of Ocrl1.
On the one hand, data presented in this study support the hypothesis that some Ocrl1’s functional roles are segregated within the protein primary structure (12,15,46). Specifically, that integrity of the N-terminus is mostly required for membrane remodeling functions (e.g. cell spreading (12)), while Ocrl1’s C-terminal region is more relevant to ciliogenesis (15). As indicated before, the central phosphatase domain was required for both functions (12,15).
On the other hand, this study highlights specific phenotypes associated with certain mutations.
A predicted Ocrl1 patient variant that lacks its N-terminal PH domain (Ocrl1∆PH), but bears all other domains/regions should be able to interact with Rab GTPases and in consequence should have proper localization to the Golgi complex (50–52). Indeed, similar to Ocrl1WT this variant was enriched in the TGN; however, we also observed Ocrl1∆PH cytosolic/nuclear mislocalization. These observations led us to hypothesize that absence of the PH domain causes the loss of previously unnoticed localization determinants that allowed the GFP-tag to aberrantly drag/mislocalize the fusion protein to the nucleus. Nuclear localization is typical of free GFP (see Fig. 4 top panel), but it is not observed in GFP-tagged Ocrl1WT where the protein’s strong localization determinants overpower the tag tendency to accumulate in the nucleus. Since Ocrl1ΔPH lacks the N-terminal region that contains binding sites for the endocytic machinery, one could speculate of a possible role for proteins involved in endocytosis in sustaining Ocrl1’s proper localization. Alternatively, this may be revealing the presence of yet to be identified Ocrl1-interaction partners in the N-terminus that play a critical role in maintaining its cellular localization. Further investigations will attempt to shed light into the mechanism by which the PH domain contributes to Ocrl1 proper localization. This variant produced cell spreading abnormalities in agreement with the previously proposed segregation of Ocrl1’s functions across the protein domains (12,15).
Localization was also affected for Ocrl1 patient variants bearing amino acid changes within the ASH-RhoGAP domains. Specifically, we found that these mutants were predominantly cytosolic diffuse and lacking the Ocrl1WT-like enrichment at the GA. For these variants we also observed aggregates in a dose-dependent manner. Indeed, mutations affecting this domain are predicted to be destabilizing in nature, resulting in aggregated and/or degraded Ocrl1 (43,53).
The presence of the ASH domain in other proteins is associated with localization at the centriole and cilia (11). Indeed, it is within this region that Ocrl1contains Rab/Rab8-binding sites (50,51) that during starvation (ciliogenesis-stimulating conditions) contribute to localization at the base of and support the assembly of the primary cilia (15). However, Ocrl1 patient variants with their ASH-RhoGAP domains mutated localized at the centriole even under steady state conditions (which was not the case for Ocrl1WT). Importantly, this was observed even in a Rab-binding mutant Ocrl1F668V, suggesting that this phenomenon is likely related to unique characteristics that these mutants display.
Although these mutants produced aggregates, it is unclear if this phenomenon occurs in patients as it was most noticeable at higher levels of expression. Interestingly, in these cases we observed aggregates colocalizing with centrioles (Fig. 6D) and it is well known that subunits and substrates of the proteosome are enriched at these structures (54). Given the proposed unstable nature of ASH-RhoGAP mutants, it is tempting to speculate that this enrichment is due to unstable proteins being targeted for degradation by the centriole-associated proteasome known as the ‘aggresome’ (55–57). In fact, many other aggregated proteins implicated in diseases have been observed to be enriched in these compartments as well (55,57).
Several patient variants with mutated phosphatase domains (affecting residues with either catalytic or non-catalytic relevance) displayed a dispersed punctate pattern and induced Golgi apparatus fragmentation. Further, these Ocrl1 puncta colocalized poorly with the GA. It should be noted that Golgi complex fragmentation has been observed in neurological diseases (24–26), and therefore might be relevant for the neurological component of LS.
GA fragmentation was only dependent on lack of Ocrl1 catalytic activity; therefore, it is possible that PI(4,5)P2 accumulation affects GA integrity. Interestingly, the Nussbaum lab previously reported that Ocrl1 interacts with the GA tethering protein Golgin-84 (58), and other studies have demonstrated that trafficking defects of the latter led to GA fragmentation (59–61). It is possible that, in the absence of Ocrl1 catalytic activity, Golgin-84 is not properly localized and causes the GA phenotype.
In addition, these mutants produced substantial defects in cell spreading and ciliogenesis further supporting the idea that loss of enzymatic function affects both cellular processes. Interestingly, the LS patient variant Ocrl1S256N, despite having a mutation in the phosphatase domain, had a substantial phosphatase activity and in consequence did not induce GA fragmentation or displayed a dispersed punctate pattern (Fig. 5A, B, D). However, while cell spreading of cells expressing this variant was normal, Ocrl1S256N could not sustain normal ciliogenesis (Figs 2B and 3A, respectively). Therefore, the Ocrl1S256N variant may cause LS by an almost exclusively ciliogenesis-dependent mechanism and is the focus of current investigations.
We humbly believe that one of the most interesting hypotheses emerging from this work is that LS has a conformational disease component; i.e. that some patients would express Ocrl1 variants which are conformationally affected.
In fact, out of the 80 unique missense mutations within the phosphatase domain of Ocrl1, ~50% of the mutations (including D451G and V508D) are affecting residues not directly involved in catalysis and yet produce LS. In other words, we speculate that some patients express Ocrl1 mutated proteins with intact binding/catalytic sequences, but likely locked in a conformation unable to process substrate. Indeed, the lack of phosphatase activity of some of these mutants and molecular dynamics analysis supported our rationale (Fig. 7).
Therefore, we speculate that for patients expressing these specific variants, LS has a component of a ‘conformational or misfolded protein disease’. A conformational disease is one where mutations in the gene lead to loss of function in the resultant mutant protein, either by affecting synthesis, transport stability, protein folding or its enzymatic activity (62). These abnormalities cause the accumulation of the non-native conformation, rendering loss in function (62,63). This possibility is the focus of current and intense investigation.
Overall, this study, along with others, suggests the existence of several levels of complexity/variability contributing to make LS a disease with a broad spectrum of symptoms and phenotype severity among patients.
At tissue-cellular level, the severity or manifestations of Ocrl1 functional deficiency would depend on the levels of expression of the Ocrl1’s paralog Inpp5b in different cell types/tissues/organs (48). However, as is to be expected for different gene products, even in the presence of Inpp5b, Ocrl1-deficiency triggers Ocrl1-specific phenotypes (e.g. cell spreading and migration; (12)). Nevertheless, we speculate that different cell types/tissues/organs would show various levels of severity of these specific phenotypes also depending on factors like composition and compliance of the extracellular matrix. As a result of this composite of factors different organs would be affected in different manner by OCRL1 mutations.
The next level of variability affecting patients comes from the nature of the specific LS-causing OCRL1 mutation: missense, nonsense or deletion/insertion mutations (with the latter in either coding or noncoding regions of the gene) and their possible outcomes (e.g. presence of a mutated protein or absence of gene product, splicing defects and changes in expression levels of Ocrl1). This study focused on different missense mutations affecting different Ocrl1 domains or producing a ΔPH-truncated variant (31) and their effect on cellular phenotypes.
Our results support the idea of spatial segregation of functions for the Ocrl1 molecule: the N-terminus required for membrane remodeling, the C-terminus for ciliogenesis and the central phosphatase domain for both processes. Therefore, OCRL1 mutations affecting these different regions have different impact on cellular phenotypes (Fig. 8 and Supplementary Material, Table S3), adding another source of variability to LS patient’s spectrum of phenotypes and symptoms. Further, for different LS-causing mutations, the severity of the corresponding phenotype was not homogeneous even when affecting the same domain (Fig. 8 and Supplementary Material, Table S3). In addition, mutations led to domain-specific abnormalities such as mislocalization or Golgi apparatus fragmentation (Fig. 8 and Supplementary Material, Table S3).
Figure 8.
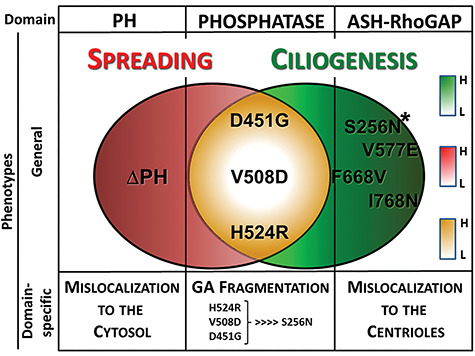
Different missense mutations effect on LS phenotypes. Specific mutations have differential effects on general LS phenotypes such as defects on cell spreading (red) and ciliogenesis (green). Different mutations associated with specific amino acid changes (or alternative protein initiation, e.g. ΔPH) at specific Ocrl1 domains are indicated. The Venn diagrams show patient variants inducing only cell spreading defects (ΔPH), only ciliogenesis abnormalities (e.g. ASH-RhoGAP mutated variants) or both (phosphatase mutated proteins, in yellow). Differential phenotype severity is represented by color tone (boxes on the right), the darker the tone the more severe the phenotype, while white indicates the mildest phenotype. Phenotypes associated with changes in specific Ocrl1 domains are indicated. *Patient variant inducing severe (but only) ciliogenesis phenotypes. Note that in the case of GFP-fusions mislocalization to the cytosol leads to GFP-mediated retention in the nucleus.
This study provides evidence for heterogeneity in LS cellular phenotypes indicating that the impact of OCRL1 mutations is a complex composite of all the above factors. In addition, genetic modifiers as well as environmental factors may play a role in how these defects manifest as patient symptoms and should be the focus of further studies.
Materials and Methods
Reagents and constructs
All reagents were procured from Fisher Scientific (Fairlawn, NJ) or Sigma Aldrich (St. Louis, MO) unless stated otherwise. Antibodies used in this study are listed in Supplementary Material, Table S1. Site-directed mutagenesis was performed using Quikchange Lightning mutagenesis kit (Agilent Technologies) and pEGFP-c1 hsOCRL1 (wild-type, isoform b) as template to create the various OCRL1 constructs used in this study. Plasmid constructs used in this study are listed in Supplementary Material, Table S2.
Cells and culture conditions and transfections
Normal human proximal tubule epithelial (HK2) and human embryonic kidney epithelial 293 T (HEK293T) cells were purchased from ATCC and cultured in DMEM, Streptomycin/Penicillin, 2 mM L-Glutamine and 10% FBS. Cells were maintained at 37°C in a 5% CO2 incubator. OCRL1−/− (OCRL KO) HK2 and HEK293T cells were prepared by GenScript Inc. Piscataway, NJ, USA (64) and maintained under identical culture conditions. Absence of Ocrl1 as well LS-specific phenotypes including cell spreading and ciliogenesis defects were previously validated in the KO cell lines (46,64). Plasmids encoding different Ocrl1 mutants were transfected using Fugene 6 reagent (Promega) according to manufacturer’s instructions.
Preparation of stable cell lines
HK2 (and HK2 KO) cells have been immortalized using a plasmid encoding SV40 large T antigen than confers it resistance to G418. Therefore, to prepare stable transfectants cells were co-transfected with plasmid for expression of GFP-Ocrl1WT/Mutants along with pTre2Hyg-6XHis vector (in a 4:1 ratio). Stably transfected clones were selected using 100ug/ml Hygromycin. Antibiotic-containing media was replaced every 48 h. Within 2–3 weeks, GFP-positive colonies were identified and isolated using sterile cloning discs. Although proliferation, cytokinesis and survival were affected, these clones were subcultured over several passages and expression of Ocrl1WT/Mutant was confirmed by microscopic analysis.
Cell spreading assays
Cells were transfected as described above and grown in complete media up to 18 h. Cell confluency was maintained at ~50% to ensure single-cell suspensions were obtained prior to spreading assays. 18 h post transfection, the cells were lifted with 20 mM EDTA (in 1X PBS), pelleted at 100 × g for 5 min and resuspended in complete media. Cell suspensions were then set in a rotator for 1 h before seeding them on 10ug/ml fibronectin-coated coverslips for 30 min, undisturbed, to allow attachment and spreading. At 30 min, coverslips were gently washed using 1X PBS and fixed in 4% formaldehyde for 10 min at room temperature. Cells were stained with rhodamine-phalloidin (used at 1:200) and imaged by epifluorescence microscopy. At least 40 cells were analyzed per experiment. The magic wand tool in ImageJ software was used to trace the cell boundaries and determine cell areas and perimeter.
In every experiment, spreading area of cells expressing Ocrl1 mutants was normalized to the median area calculated in Ocrl1WT-expressing cells for the same experiment. Histograms were constructed from the normalized spreading area values and Kolmogorov–Smirnov (KS) test was performed to determine statistical significance.
Ciliogenesis assays
Cells were seeded (in complete media) on glass cover slips coated with poly-D-lysine and transfected as described above. Confluency of cells was maintained in such a way that it did not exceed 50% at the time of ciliogenesis. 18 h after transfection, the media was replaced by 0.1% serum DMEM (starvation media) for another 24 h to induce ciliogenesis. Cells were washed with 1X PBS, fixed in 4% formaldehyde–PBS for 10 min. Indirect immunofluorescence was performed using antibodies against acetylated tubulin antibody (to label cilium) and pericentrin-2 (PC-2; to label centriole) (refer to Table 1). A total of 20 random fields, comprising of at least 50 cells, were imaged for every experiment and repeated at least thrice.
From all the transfected cells imaged, the fraction of Ocrl1 mutant-transfected cells forming a cilium was calculated. This was normalized to the fraction of cilia produced by Ocrl1WT-expressing cells from the same experiment. The upper limit of fraction of ciliated cells was 1 (represented by HEK293T KO cells expressing Ocrl1WT) and the lower limit of fraction of ciliated cell was 0.7 (HEK293T KO cells expressing Ocrl1GFP). Though we observed variability between experiments, individual experiments were statistically significant.
Indirect immunofluorescence and fluorescence microscopy
In all immunofluorescence procedures, antibodies were diluted in DMEM media containing 10% FBS (blocking agent) and 0.1% saponin (permeabilizing agent). Primary antibodies were incubated for 1 h at room temperature, washed two times with PBS. Fluorescent molecule-conjugated secondary antibodies were incubated with cells for 45 min in the dark, washed two times with PBS. Cells were then stained using DAPI to label the nucleus and mounted on pre-cleaned glass slides using Aqua-PolyMount reagent (Polysciences). Following indirect immunofluorescence, coverslips were imaged with constant fluorescence exposure times using a 40X objective on Zeiss Axiovert inverted microscope. Random fields were imaged to cover the entire coverslip. Exposure times were maintained consistent for all independent experiments.
Pericentrin-2 colocalization experiments: given the binary nature of the result (the Ocrl1 variant is either present or absent at the centrioles), the error of the fraction of cells showing colocalization was estimated using the Poisson counting standard error.
Protein purification
The PH and phosphatase domain of wild-type hsOcrl1 (1–563 amino acids) was cloned in a pGEX-4 T1 plasmid that contains an N-terminus glutathione S-transferase (GST) tag. Using site-directed mutagenesis, missense mutations H524R, D451G and V508D were introduced. Plasmids were transformed in Rosetta (DE3) competent cells. Bacterial cultures were grown overnight at 37°C, in LB medium supplemented with 2.5% glucose, 1X ampicillin and 1X chloramphenicol. The following day, cultures were expanded in super broth media containing 1X ampicillin and 1X chloramphenicol for 3 h at 37°C. Then, cultures were supplemented with 5% glycerol and 0.1 mM IPTG and incubated for 5 h at 30°C.
Cells were harvested by centrifugation (3000 × g, 10 min), and pellets were stored at −80°C until use. Cells were lysed in lysis buffer containing 200 mM Tris pH 7.4, 10% glycerol, 0.1% Tween 20, complete EDTA-free protease inhibitor, 1 mg/ml lysozyme was added to resuspend prepared cell pellets. Cells were disrupted by sonication at 50% power for three sets of 33 pulses with 30 s breaks in between pulses. Cell debris was removed via centrifugation at 21 500 × g, 30 min, 4°C. The supernatant was transferred to tubes containing glutathione resin (Pierce) and incubated at room temperature on a shaker (12 rpm) for 2 h. Beads were washed four times with lysis buffer (w/o protease inhibitor and lysozyme). Then, 100 mM glutathione (pH 8.0) was added to the tubes containing supernatant and incubated at room temperature on a shaker (12 rpm) for 2 h to elute the protein. Supernatant (after centrifugation at 1000 × g for 2 min) was loaded onto desalting columns (Thermo Scientific, Zeba, 89 891) and centrifuged (1000 × g, 2 min, acceleration: 5) and purified protein was obtained. Protein concentration was estimated using NanoDrop 1000 (ThermoFisher) and was used immediately for malachite green phosphatase assays.
Malachite green phosphatase assays
For 5′ phosphatase activity assays, the malachite green phosphate assay kit (Sigma-Aldrich, MAK307) was used. Briefly, in a 384-well plate, 10 μl of 2 μM of purified Ocrl1 (wildtype and mutant) was incubated with drug or vehicle for 1 h at room temperature, to obtain a final enzyme concentration of 1uM. After treatment, 10 μl of 50 μM PI(4,5)P2 diC8 (Echelon Biosciences, P-4508) was added to wells and incubated for 5 min at room temperature in the same buffer as the purified phosphatases, containing 200 mM Tris, pH 7.4. To stop enzyme reaction, 20 μl of 0.25X malachite green reagent was added to the reaction wells. After 20 min of color development, absorbance was measured at 620 nm. A standard phosphate curve was prepared (as per manufacturer’s instructions) in the enzyme buffer solution to determine the amount of free phosphate released by the enzyme variants tested. Experiments were repeated at least thrice and each condition was tested in triplicates. Student’s t-test was used to determine statistical significance.
MD simulation
For modeling a bound complex for Ocrl-1 (PDB: 4CMN) (39) and PI(4,5)P2, the protein is constructed with explicit lipid bilayer with membrane composition 90% phosphatidylcholine (PC), 5% phosphatidylserine (PS) and 5% PI(4,5)P2 (65) using CHARMM-GUI membrane builder (66,67) (Supplementary Material, Fig. S6A).
The protein and membrane systems are then solvated with TIP3P water and neutralized with 200 mM MgCl2. MD simulations are performed for 100 ns to obtain an equilibrated model for WT Ocrl-1 (enzyme) and PI(4,5)P2 (substrate) complex. Supplementary Material, Fig. S6B shows the trajectory for the head group atoms of the PI(4,5)P2 molecule that interacts with the residues in catalytic site of Ocrl1WT, starting from the membrane plane and diffusing inwards deeper into the catalytic site.
Next, the enzyme-substrate complex is used, and point mutations for D451G and V508D are performed using the MUTATOR plugin (https://www.ks.uiuc.edu/Research/vmd/plugins/mutator/) in VMD 1.9.3 (68). Independent MD simulations, each 100 ns long (in addition to equilibration), are performed for WT and mutant (D451G and V508D) systems.
All the MD simulations were performed with NAMD 2.13 (69) using CHARMM36m force field for lipid/protein (70) and a timestep of 2 fs. Long-range electrostatic interactions were evaluated with particle mesh Ewald (PME) (71) and periodic boundary conditions were used throughout the simulations. Nonbonded forces were calculated with a cutoff of 12 Å and switching distance of 10 Å. During the simulation, temperature (T = 310°K) and pressure (P = 1 atm) (NPT ensemble) were maintained by Nosé–Hoover Langevin piston method (72). For analysis we calculate, per residue RMSF using the software VMD 1.9.3.
Statistical analysis
Statistical significance of differences between spreading-distribution histograms were analyzed using the Kolmogorov–Smirnov (KS) test. The student’s t-test was used to evaluate the significance of differences of normally distributed (e.g. ciliogenesis experiments and Poisson-normalized colocalization data) samples, while the Wilcoxon’s test was employed when samples were non-normally distributed (quantification of Golgi apparatus fragmentation). For all comparisons involving Ocrl1WT and Ocrl1 mutants, the Bonferroni’s correction for multiple comparisons was performed whenever applicable [αC = p/n; n being the number of comparisons].
After carefully analyzing each data set distribution the most appropriate representation of data in each case was adopted. These representations included histograms and box plots as they allow to thoroughly examine the data distribution (73). When the data presented a normal distribution, a bar graph with standard deviations was used to represent the data.
Supplementary Material
Acknowledgements
We are indebted to Drs. Donna Fekete, Don Ready and Phil Low (Purdue University) for stimulating discussions. We also thank members of the Aguilar lab for discussions and critical reading of the manuscript.
Conflict of Interest statement. None.
Contributor Information
Swetha Ramadesikan, Department of Biological Sciences, Purdue University, West Lafayette, IN 47907, USA.
Lisette Skiba, Department of Biological Sciences, Purdue University, West Lafayette, IN 47907, USA.
Jennifer Lee, Department of Biological Sciences, Purdue University, West Lafayette, IN 47907, USA.
Kayalvizhi Madhivanan, Department of Biological Sciences, Purdue University, West Lafayette, IN 47907, USA.
Daipayan Sarkar, Department of Biological Sciences, Purdue University, West Lafayette, IN 47907, USA.
Agustina De La Fuente, Department of Biological Sciences, Purdue University, West Lafayette, IN 47907, USA.
Claudia B Hanna, Department of Biological Sciences, Purdue University, West Lafayette, IN 47907, USA.
Genki Terashi, Department of Biological Sciences, Purdue University, West Lafayette, IN 47907, USA.
Tony Hazbun, Department of Medicinal Chemistry and Molecular Pharmacology, Purdue University, West Lafayette, IN 47907, USA.
Daisuke Kihara, Department of Biological Sciences, Purdue University, West Lafayette, IN 47907, USA; Department of Computer Science, Purdue University, West Lafayette, IN 47907, USA.
R Claudio Aguilar, Department of Biological Sciences, Purdue University, West Lafayette, IN 47907, USA.
Funding
This work was supported by the National Institutes of Health [1R01DK109398-01 to R.C.A.]; the Clinical Translational Science Institute [CTSI 106564/8000063783 PDT Award to R.C.A.]; and the Lowe Syndrome Association to R.C.A. D.K. acknowledges partial support by the National Institutes of Health (R01GM123055) and the National Science Foundation (DMS1614777, CMMI1825941, MCB1925643, DBI2003635).
References
- 1. Attree, O., Olivos, I.M., Okabe, I., Bailey, L.C., Nelson, D.L., Lewis, R.A., McInnes, R.R. and Nussbaum, R.L. (1992) The Lowe's oculocerebrorenal syndrome gene encodes a protein highly homologous to inositol polyphosphate-5-phosphatase. Nature, 358, 239–242. [DOI] [PubMed] [Google Scholar]
- 2. Loi, M. (2006) Lowe syndrome. Orphanet J. Rare Dis., 1, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lowe, C.U., Terrey, M. and MacLachlan, E.A. (1952) Organic-aciduria, decreased renal ammonia production, hydrophthalmos, and mental retardation; a clinical entity. A.M.A. Am. J. Dis. Child., 83, 164–184. [DOI] [PubMed] [Google Scholar]
- 4. Bökenkamp, A. and Ludwig, M. (2016) The oculocerebrorenal syndrome of Lowe: an update. Pediatr. Nephrol., 31, 2201–2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang, X., Jefferson, A.B., Auethavekiat, V. and Majerus, P.W. (1995) The protein deficient in Lowe syndrome is a phosphatidylinositol-4,5-bisphosphate 5-phosphatase. Proc. Natl. Acad. Sci. U. S. A., 92, 4853–4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Suchy, S.F., Olivos-Glander, I.M. and Nussabaum, R.L. (1995) Lowe syndrome, a deficiency of phosphatidylinositol 4,5-bisphosphate 5-phosphatase in the Golgi apparatus. Hum. Mol. Genet., 4, 2245–2250. [DOI] [PubMed] [Google Scholar]
- 7. Dressman, M.A., Olivos-Glander, I.M., Nussbaum, R.L. and Suchy, S.F. (2000) Ocrl1, a PtdIns(4,5)P(2) 5-phosphatase, is localized to the trans-Golgi network of fibroblasts and epithelial cells. J. Histochem. Cytochem., 48, 179–190. [DOI] [PubMed] [Google Scholar]
- 8. Erdmann, K.S., Mao, Y., McCrea, H.J., Zoncu, R., Lee, S., Paradise, S., Modregger, J., Biemesderfer, D., Toomre, D. and De Camillil, P. (2007) A role of the Lowe syndrome protein OCRL in early steps of the endocytic pathway. Dev. Cell, 13, 377–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Faucherre, A., Desbois, P., Nagano, F., Satre, V., Lunardi, J., Gacon, G. and Dorseuil, O. (2005) Lowe syndrome protein Ocrl1 is translocated to membrane ruffles upon Rac GTPase activation: a new perspective on Lowe syndrome pathophysiology. Hum. Mol. Genet., 14, 1441–1448. [DOI] [PubMed] [Google Scholar]
- 10. Mao, Y., Balkin, D.M., Zoncu, R., Erdmann, K.S., Tomasini, L., Hu, F., Jin, M.M., Hodsdon, M.E. and De Camilli, P. (2009) A PH domain within OCRL bridges clathrin-mediated membrane trafficking to phosphoinositide metabolism. EMBO J., 28, 1831–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ponting, C.P. (2006) A novel domain suggests a ciliary function for ASPM, a brain size determining gene. Bioinformatics, 22, 1031–1035. [DOI] [PubMed] [Google Scholar]
- 12. Coon, B.G., Mukherjee, D., Hanna, C.B., Riese, D.J., II, Lowe, M. and Aguilar, R.C. (2009) Lowe syndrome patient fibroblasts display Ocrl1-specific cell migration defects that cannot be rescued by the homologous Inpp5b phosphatase. Hum. Mol. Genet., 18, 4478–4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Suchy, S.F. and Nussbaum, R.L. (2002) The deficiency of PIP2 5-phosphatase in Lowe syndrome affects actin polymerization. Am. J. Hum. Genet., 71, 1420–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.. Vicinanza, M., Di Campli, A., Polishchuk, E., Santoro, M., Di Tullio, G., Godi, A., Levtchenko, E., De Leo, M.G., Polishchuk, R., Sandoval, L.. et al. (2011) OCRL controls trafficking through early endosomes via PtdIns4,5P(2)-dependent regulation of endosomal actin. EMBO J., 30, 4970–4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Coon, B.G., Hernandez, V., Madhivanan, K., Mukherjee, D., Hanna, C.B., Barinaga-Rementeria Ramirez, I., Lowe, M., Beales, P.L. and Aguilar, R.C. (2012) The Lowe syndrome protein OCRL1 is involved in primary cilia assembly. Hum. Mol. Genet., 21, 1835–1847. [DOI] [PubMed] [Google Scholar]
- 16. Choudhury, R., Diao, A.P., Zhang, F., Eisenberg, E., Saint-Pol, A., Williams, C., Konstantakopoulos, A., Lucocq, J., Johannes, L., Rabouille, C.et al. (2005) Lowe syndrome protein OCRL1 interacts with clathrin and regulates protein trafficking between endosomes and the trans-Golgi network. Mol. Biol. Cell, 16, 3467–3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van Rahden, V.A., Brand, K., Najm, J., Heeren, J., Pfeffer, S.R., Braulke, T. and Kutsche, K. (2012) The 5-phosphatase OCRL mediates retrograde transport of the mannose 6-phosphate receptor by regulating a Rac1-cofilin signalling module. Hum. Mol. Genet., 21, 5019–5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dambournet, D., Machicoane, M., Chesneau, L., Sachse, M., Rocancourt, M., El Marjou, A., Formstecher, E., Salomon, R., Goud, B. and Echard, A. (2011) Rab35 GTPase and OCRL phosphatase remodel lipids and F-actin for successful cytokinesis. Nat. Cell Biol., 13, 981–U245. [DOI] [PubMed] [Google Scholar]
- 19. El Kadhi, K.B., Roubinet, C., Solinet, S., Emery, G. and Carreno, S. (2011) The inositol 5-phosphatase dOCRL controls PI(4,5)P2 homeostasis and is necessary for cytokinesis. Curr. Biol., 21, 1074–1079. [DOI] [PubMed] [Google Scholar]
- 20. Noakes, C.J., Lee, G. and Lowe, M. (2011) The PH domain proteins IPIP27A and B link OCRL1 to receptor recycling in the endocytic pathway. Mol. Biol. Cell, 22, 606–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bohdanowicz, M., Balkin, D.M., De Camilli, P. and Grinstein, S. (2012) Recruitment of OCRL and Inpp5B to phagosomes by Rab5 and APPL1 depletes phosphoinositides and attenuates Akt signaling. Mol. Biol. Cell, 23, 176–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Marion, S., Mazzolini, J., Herit, F., Bourdoncle, P., Kambou-Pene, N., Hailfinger, S., Sachse, M., Ruland, J., Benmerah, A., Echard, A.et al. (2012) The NF-kappa B Signaling protein Bcl10 regulates actin dynamics by controlling AP1 and OCRL-bearing vesicles. Dev. Cell, 23, 954–967. [DOI] [PubMed] [Google Scholar]
- 23. Hichri, H., Rendu, J., Monnier, N., Coutton, C., Dorseuil, O., Poussou, R.V., Baujat, G., Blanchard, A., Nobili, F., Ranchin, B.et al. (2011) From Lowe syndrome to dent disease: correlations between mutations of the OCRL1 gene and clinical and biochemical phenotypes. Hum. Mutat., 32, 379–388. [DOI] [PubMed] [Google Scholar]
- 24. Bexiga, M.G. and Simpson, J.C. (2013) Human diseases associated with form and function of the Golgi complex. Int. J. Mol. Sci., 14, 18670–18681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ayala, I. and Colanzi, A. (2017) Alterations of Golgi organization in Alzheimer's disease: a cause or a consequence? Tissue Cell, 49, 133–140. [DOI] [PubMed] [Google Scholar]
- 26. Martínez-Menárguez, J., Tomás, M., Martínez-Martínez, N. and Martínez-Alonso, E. (2019) Golgi fragmentation in neurodegenerative diseases: is there a common cause? Cell, 8, 748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Utsch, B., Bökenkamp, A., Benz, M.R., Besbas, N., Dötsch, J., Franke, I., Fründ, S., Gok, F., Hoppe, B., Karle, S.et al. (2006) Novel OCRL1 mutations in patients with the phenotype of dent disease. Am. J. Kidney Dis., 48, 942.e941–942.e914. [DOI] [PubMed] [Google Scholar]
- 28. Lin, T., Orrison, B.M., Leahey, A.M., Suchy, S.F., Bernard, D.J., Lewis, R.A. and Nussbaum, R.L. (1997) Spectrum of mutations in the OCRL1 gene in the Lowe oculocerebrorenal syndrome. Am. J. Hum. Genet., 60, 1384–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lin, T., Orrison, B.M., Suchy, S.F., Lewis, R.A. and Nussbaum, R.L. (1998) Mutations are not uniformly distributed throughout the OCRL1 gene in Lowe syndrome patients. Mol. Genet. Metab., 64, 58–61. [DOI] [PubMed] [Google Scholar]
- 30. Lichter-Konecki, U., Farber, L.W., Cronin, J.S., Suchy, S.F. and Nussbaum, R.L. (2006) The effect of missense mutations in the RhoGAP-homology domain on ocrl1 function. Mol. Genet. Metab., 89, 121–128. [DOI] [PubMed] [Google Scholar]
- 31. Shrimpton, A.E., Hoopes, R.R., Jr., Knohl, S.J., Hueber, P., Reed, A.A.C., Christie, P.T., Igarashi, T., Lee, P., Lehman, A., White, C.et al. (2009) OCRL1 mutations in dent 2 patients suggest a mechanism for phenotypic variability. Nephron Physiol., 112, 27–36. [DOI] [PubMed] [Google Scholar]
- 32. Fan, J., Hu, Z., Zeng, L., Lu, W., Tang, X., Zhang, J. and Li, T. (2008) Golgi apparatus and neurodegenerative diseases. Int. J. Dev. Neurosci., 26, 523–534. [DOI] [PubMed] [Google Scholar]
- 33. Caracci, M.O., Fuentealba, L.M. and Marzolo, M.P. (2019) Golgi complex dynamics and its implication in prevalent neurological disorders. Front. Cell Dev. Biol., 7, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jin, H., Carone, F.A., Nakamura, S., Liu, Z.Z. and Kanwar, Y.S. (1992) Altered synthesis and intracellular transport of proteoglycans by cyst-derived cells from human polycystic kidneys. J. Am. Soc. Nephrol., 2, 1726–1733. [DOI] [PubMed] [Google Scholar]
- 35. Charron, A.J., Bacallao, R.L. and Wandinger-Ness, A. (2000) ADPKD: a human disease altering Golgi function and basolateral exocytosis in renal epithelia. Traffic, 1, 675–686. [DOI] [PubMed] [Google Scholar]
- 36. Le Corre, S., Eyre, D. and Drummond, I.A. (2014) Modulation of the secretory pathway rescues zebrafish polycystic kidney disease pathology. J. Am. Soc. Nephrol., 25, 1749–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Whisstock, J.C., Romero, S., Gurung, R., Nandurkar, H., Ooms, L.M., Bottomley, S.P. and Mitchell, C.A. (2000) The inositol polyphosphate 5-phosphatases and the apurinic/apyrimidinic base excision repair endonucleases share a common mechanism for catalysis. J. Biol. Chem., 275, 37055–37061. [DOI] [PubMed] [Google Scholar]
- 38. Tsujishita, Y., Guo, S.L., Stolz, L.E., York, J.D. and Hurley, J.H. (2001) Specificity determinants in phosphoinositide dephosphorylation: crystal structure of an archetypal inositol polyphosphate 5-phosphatase. Cell, 105, 379–389. [DOI] [PubMed] [Google Scholar]
- 39. Tresaugues, L., Silvander, C., Flodin, S., Welin, M., Nyman, T., Graslund, S., Hammarstrom, M., Berglund, H. and Nordlund, P. (2014) Structure. Elsevier Ltd, United States, Vol. 22, pp. 744–755. [DOI] [PubMed] [Google Scholar]
- 40. Jefferson, A.B. and Majerus, P.W. (1996) Mutation of the conserved domains of two inositol polyphosphate 5-phosphatases. Biochemistry, 35, 7890–7894. [DOI] [PubMed] [Google Scholar]
- 41. Mills, S.J., Silvander, C., Cozier, G., Trésaugues, L., Nordlund, P. and Potter, B.V. (2016) Crystal structures of type-II inositol polyphosphate 5-phosphatase INPP5B with synthetic inositol polyphosphate surrogates reveal new mechanistic insights for the inositol 5-phosphatase family. Biochemistry, 55, 1384–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pirruccello, M. and De Camilli, P. (2012) Inositol 5-phosphatases: insights from the Lowe syndrome protein OCRL. Trends Biochem. Sci., 37, 134–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pirruccello, M., Swan, L.E., Folta-Stogniew, E. and De Camilli, P. (2011) Recognition of the F&H motif by the Lowe syndrome protein OCRL. Nat. Struct. Mol. Biol., 18, 789–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rbaibi, Y., Cui, S., Mo, D., Carattino, M., Rohatgi, R., Satlin, L.M., Szalinski, C.M., Swanhart, L.M., Folsch, H., Hukriede, N.A.et al. (2012) OCRL1 modulates cilia length in renal epithelial cells. Traffic, 13, 1295–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Luo, N., West, C.C., Murga-Zamalloa, C.A., Sun, L., Anderson, R.M., Wells, C.D., Weinreb, R.N., Travers, J.B., Khanna, H. and Sun, Y. (2012) OCRL localizes to the primary cilium: a new role for cilia in Lowe syndrome. Hum. Mol. Genet., 21, 3333–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Madhivanan, K., Ramadesikan, S., Hsieh, W.C., Aguilar, M.C., Hanna, C.B., Bacallao, R.L. and Aguilar, R.C. (2020) Lowe syndrome patient cells display mTOR- and RhoGTPase-dependent phenotypes alleviated by rapamycin and statins. Hum. Mol. Genet., 29, 1700–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bothwell, S.P., Farber, L.W., Hoagland, A. and Nussbaum, R.L. (2010) Species-specific difference in expression and splice-site choice in Inpp5b, an inositol polyphosphate 5-phosphatase paralogous to the enzyme deficient in Lowe syndrome. Mamm. Genome, 21, 458–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Janne, P.A., Suchy, S.F., Bernard, D., MacDonald, M., Crawley, J., Grinberg, A., Wynshaw-Boris, A., Westphal, H. and Nussbaum, R.L. (1998) Functional overlap between murine Inpp5b and Ocrl1 may explain why deficiency of the murine ortholog for OCRL1 does not cause Lowe syndrome in mice. J. Clin. Investig., 101, 2042–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Leahey, A.M., Charnas, L.R. and Nussbaum, R.L. (1993) Nonsense mutations in the OCRL-1 gene in patients with the oculocerebrorenal syndrome of Lowe. Hum. Mol. Genet., 2, 461–463. [DOI] [PubMed] [Google Scholar]
- 50. Hyvola, N., Diao, A., McKenzie, E., Skippen, A., Cockcroft, S. and Lowe, M. (2006) Membrane targeting and activation of the Lowe syndrome protein OCRL1 by Rab GTPases. EMBO J., 25, 3750–3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hagemann, N., Hou, X., Goody, R.S., Itzen, A. and Erdmann, K.S. (2012) Crystal structure of the Rab binding domain of OCRL1 in complex with Rab8 and functional implications of the OCRL1/Rab8 module for Lowe syndrome. Small GTPases, 3, 107–110. [DOI] [PubMed] [Google Scholar]
- 52. Hou, X., Hagemann, N., Schoebel, S., Blankenfeldt, W., Goody, R.S., Erdmann, K.S. and Itzen, A. (2011) A structural basis for Lowe syndrome caused by mutations in the Rab-binding domain of OCRL1. EMBO J., 30, 1659–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. McCrea, H.J., Paradise, S., Tomasini, L., Addis, M., Melis, M.A., De Matteis, M.A. and De Camilli, P. (2008) All known patient mutations in the ASH-RhoGAP domains of OCRL affect targeting and APPL1 binding. Biochem. Biophys. Res. Commun., 369, 493–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vora, S.M. and Phillips, B.T. (2016) The benefits of local depletion: the centrosome as a scaffold for ubiquitin-proteasome-mediated degradation. Cell Cycle, 15, 2124–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Park, J., Park, Y., Ryu, I., Choi, M.H., Lee, H.J., Oh, N., Kim, K., Kim, K.M., Choe, J., Lee, C.et al. (2017) Misfolded polypeptides are selectively recognized and transported toward aggresomes by a CED complex. Nat. Commun., 8, 15730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Olzmann, J.A., Li, L. and Chin, L.S. (2008) Aggresome formation and neurodegenerative diseases: therapeutic implications. Curr. Med. Chem., 15, 47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Johnston, J.A., Ward, C.L. and Kopito, R.R. (1998) Aggresomes: a cellular response to misfolded proteins. J. Cell Biol., 143, 1883–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bascom, R.A., Srinivasan, S. and Nussbaum, R.L. (1999) Identification and characterization of golgin-84, a novel Golgi integral membrane protein with a cytoplasmic coiled-coil domain. J. Biol. Chem., 274, 2953–2962. [DOI] [PubMed] [Google Scholar]
- 59. Jiang, Q., Wang, L., Guan, Y., Xu, H., Niu, Y., Han, L., Wei, Y.P., Lin, L., Chu, J., Wang, Q.et al. (2014) Golgin-84-associated Golgi fragmentation triggers tau hyperphosphorylation by activation of cyclin-dependent kinase-5 and extracellular signal-regulated kinase. Neurobiol. Aging, 35, 1352–1363. [DOI] [PubMed] [Google Scholar]
- 60. Munro, S. (2011) The golgin coiled-coil proteins of the Golgi apparatus. Cold Spring Harb. Perspect. Biol., 3, a005256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sohda, M., Misumi, Y., Yamamoto, A., Nakamura, N., Ogata, S., Sakisaka, S., Hirose, S., Ikehara, Y. and Oda, K. (2010) Interaction of Golgin-84 with the COG complex mediates the intra-Golgi retrograde transport. Traffic, 11, 1552–1566. [DOI] [PubMed] [Google Scholar]
- 62. Kopito, R.R. and Ron, D. (2000) Conformational disease. Nat. Cell Biol., 2, E207–E209. [DOI] [PubMed] [Google Scholar]
- 63. Kopito, R.R. (2000) Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol., 10, 524–530. [DOI] [PubMed] [Google Scholar]
- 64. Hsieh, W.C., Ramadesikan, S., Fekete, D. and Aguilar, R.C. (2018) Kidney-differentiated cells derived from Lowe syndrome patient's iPSCs show ciliogenesis defects and Six2 retention at the Golgi complex. PLoS One, 13, e0192635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Le Coq, J., Camacho-Artacho, M., Velázquez, J.V., Santiveri, C.M., Gallego, L.H., Campos-Olivas, R., Dölker, N. and Lietha, D. (2017) Structural basis for interdomain communication in SHIP2 providing high phosphatase activity. Elife, 6, e26640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jo, S., Kim, T. and Im, W. (2007) Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS One, 2, e880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lee, J., Patel, D.S., Ståhle, J., Park, S.J., Kern, N.R., Kim, S., Cheng, X., Valvano, M.A., Holst, O., Knirel, Y.A.et al. (2019) CHARMM-GUI membrane builder for complex biological membrane simulations with glycolipids and Lipoglycans. J. Chem. Theory Comput., 15, 775–786. [DOI] [PubMed] [Google Scholar]
- 68. Humphrey, W., Dalke, A. and Schulten, K. (1996) VMD: visual molecular dynamics. J. Mol. Graph., 14, 27–38. [DOI] [PubMed] [Google Scholar]
- 69. Phillips, J.C., Hardy, D.J., Maia, J.D.C., Stone, J.E., Ribeiro, J.V., Bernardi, R.C., Buch, R., Fiorin, G., Hénin, J., Jiang, W.et al. (2020) Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys., 153, 044130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Best, R.B., Zhu, X., Shim, J., Lopes, P.E., Mittal, J., Feig, M. and Mackerell, A.D. (2012) Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. J. Chem. Theory Comput., 8, 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Essmann, U., Perera, L., Berkowitz, M.L., Darden, T., Lee, H. and Pedersen, L.G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys., 103, 8577–8593. [Google Scholar]
- 72. Martyna, G.J., Tobias, D.J. and Klein, M.L. (1994) Constant pressure molecular dynamics algorithms. J. Chem. Phys., 101, 4177–4189. [Google Scholar]
- 73. Weissgerber, T.L., Milic, N.M., Winham, S.J. and Garovic, V.D. (2015) Beyond bar and line graphs: time for a new data presentation paradigm. PLoS Biol., 13, e1002128. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.