

Abstract
Lipotoxicity was recently reported in several forms of kidney disease, including focal segmental glomerulosclerosis (FSGS). Susceptibility to FSGS in African Americans is associated with the presence of genetic variants of the Apolipoprotein L1 gene (APOL1) named G1 and G2. If and how endogenous APOL1 may alter mitochondrial function by the modifying cellular lipid metabolism is unknown. Using transgenic mice expressing the APOL1 variants (G0, G1 or G2) under endogenous promoter, we show that APOL1 risk variant expression in transgenic mice does not impair kidney function at baseline. However, APOL1 G1 expression worsens proteinuria and kidney function in mice characterized by the podocyte inducible expression of nuclear factor of activated T-cells (NFAT), which we have found to cause FSGS. APOL1 G1 expression in this FSGS-model also results in increased triglyceride and cholesterol ester contents in kidney cortices, where lipid accumulation correlated with loss of renal function. In vitro, we show that the expression of endogenous APOL1 G1/G2 in human urinary podocytes is associated with increased cellular triglyceride content and is accompanied by mitochondrial dysfunction in the presence of compensatory oxidative phosphorylation (OXPHOS) complexes elevation. Our findings indicate that APOL1 risk variant expression increases the susceptibility to lipid-dependent podocyte injury, ultimately leading to mitochondrial dysfunction.
Introduction
Focal segmental glomerulosclerosis (FSGS) is the most common primary glomerular disorder causing chronic kidney disease (CKD) (1). Genetic variants of the Apolipoprotein L gene 1 (APOL1), G1 (S342G and I384M) and G2 (del388N389Y) have been shown to be associated with higher rates of end-stage renal disease and progression of CKD in African American patients (2). The expression of APOL1 variants was originally found to protect against African sleeping sickness caused by the Trypanosoma brucei (3). The APOL1 gene is only present in humans and some higher primates (4,5). While the presence of one APOL1 risk variant (RV) is associated with protection from trypanosomiasis (6–8), possession of two RVs was shown to be associated with an increased risk for FSGS and HIV-associated nephropathy (HIVAN) (9). APOL1 RVs have the highest allelic frequency in Western sub-Saharan Africa, occurring in 24% (G2) to 40% (G1) of the population (7,10). Approximately 13% of African Americans carry two RVs and are at high risk for CKD (11). However, most high-risk individuals do not develop kidney diseases in the absence of a ‘second hit’, including TNF, lipopolysaccharide and interferon (12–17). APOL1 is expressed in the kidney, where it is localized in glomeruli, cortical tubules and vascular endothelium. However, within normal glomeruli, APOL1 is localized exclusively in podocytes (12).
Associated with apolipoprotein A-I, APOL1 is an abundant secreted protein that circulates as an integral component of high-density lipoprotein (HDL) in the blood (18). We recently reported that APOL1 RV expression promotes cholesterol accumulation, using tissues and macrophages from APOL1 transgenic mice, as well as APOL1 overexpressing HeLa cells (19). In APOL1 transfected human podocytes and HEK293 cells, the translocation of APOL1 RVs from lipid droplets (LDs) to the endoplasmic reticulum (ER) was found to be associated with cytotoxicity (20). Additionally, overexpression of APOL1 G1 and G2 variants in HEK293 cells was shown to contribute to mitochondrial dysfunction and ATP depletion (21–24). Of note, to date APOL1 function was mostly assessed in model systems expressing tagged and overexpressed APOL1 protein rather than by studying APOL1 protein function at expression levels similar to endogenous levels.
Activation of calcineurin/nuclear factor of activated T-cells (NFAT) pathway in podocytes is thought to be a potential mediator of FSGS and results in proteinuria and glomerulosclerosis in mice (25,26). We previously demonstrated that podocyte-specific NFAT activation causes lipid-dependent albuminuria and an FSGS-like phenotype using Podocin-rtTA; NFATc1nuc mice (27). This new mouse model is a highly translational model of FSGS, as it mimics disease progression in humans and it is driven by a pathway responsive to calcineurin inhibitors, which are currently used to treat patients with steroid-resistant FSGS and nephrotic syndrome (28–30).
In the present study, we sought to determine if APOL1 RV expression contributes to lipid-mediated podocyte injury using two model systems where APOL1 is expressed under the endogenous APOL1 promoter. First, we investigated the renal phenotype of APOL1 RV expression in bacterial artificial chromosome (BAC) transgenic mice. Second, we used human urinary podocyte cell lines carrying either APOL1 G0/G0 or G1/G2 alleles derived from patients with FSGS. We demonstrate that APOL1 RV expression in APOL1 BAC transgenic mice does not impair kidney function at baseline. However, APOL1 G1 RV expression renders mice susceptible to renal disease in Podocin-rtTA; NFATc1nuc mice, a mouse model resembling clinical FSGS with progressive renal failure (27). Furthermore, we show that APOL1 G1-associated renal failure in this mouse model correlates with increased triglyceride and cholesterol ester contents in kidney cortices. Finally, we demonstrate that APOL1 RV expression in podocytes is associated with increased cellular triglyceride content and is accompanied by mitochondrial dysfunction in the presence of compensatory oxidative phosphorylation (OXPHOS) complexes elevation. Our findings shed new light on the role of endogenous APOL1 RV expression in lipid-induced mitochondrial dysfunction and podocyte injury.
Results
Transgenic APOL1 RV expression in mice does not impair kidney function at baseline
We investigated whether APOL1 variant expression contributes to kidney injury and found that APOL1 BAC transgenic mice do not develop proteinuria at 7 months of age (Fig. 1A). We found that glomerular expression of APOL1 mRNA is similar among G0 and G1 transgenic mice but is significantly lower in G2 transgenic mice (Supplementary Material, Fig. S1). Therefore, in order to study the specific effect of the presence of the risk allele irrespectively of APOL1 expression, we excluded G2 mice from further studies. Transmission electron microscopy (TEM) of G0 and G1 transgenic mice revealed intact podocyte foot process in kidney cortices in G1 transgenic mice (Fig. 1B).
Figure 1 .
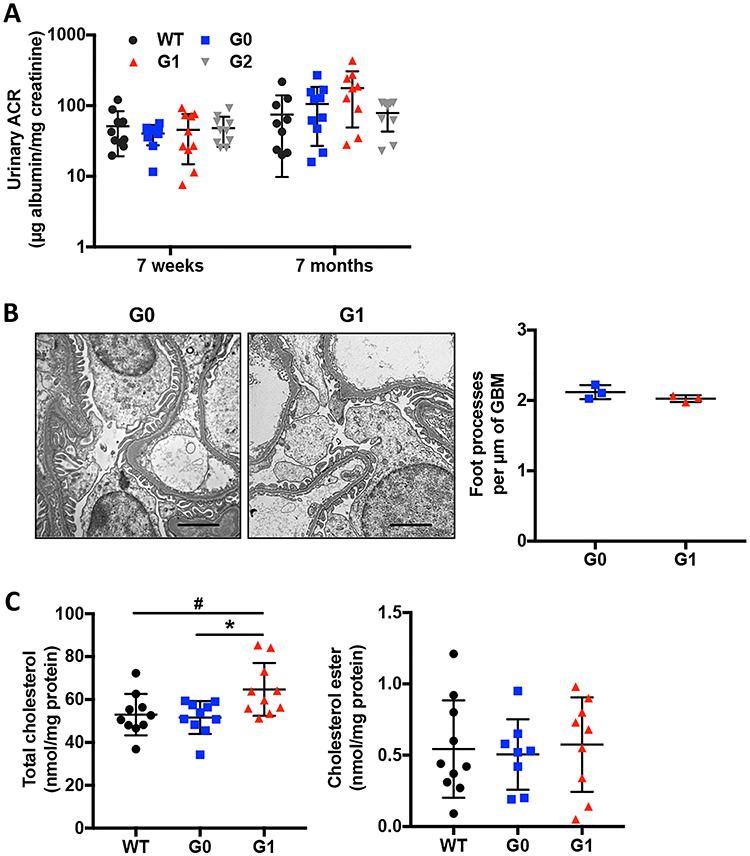
Transgenic APOL1 risk variant expression in mice does not impair kidney function at baseline. (A) Urinary albumin-to-creatinine ratio in 7-week-old and 7-month-old APOL1 BAC transgenic mice (n = 9–10). (B) Representative TEM images to identify podocyte foot process in APOL1 BAC transgenic mice carrying G0 and G1 alleles (scale bars: 2 μm) and scatter plot quantification of podocyte foot process per μm of GBM (n = 3). (C) Total cholesterol and cholesterol ester contents in kidney cortices of APOL1 BAC transgenic mice, normalized to protein concentrations (n = 8–10). The error bars represent mean ± SD of biologically independent experiments. One-way ANOVA followed by Tukey’s test (A); Two-tailed Student’s t-test (B); One-way ANOVA followed by Dunnett’s test (C). *P < 0.05. #WT compared with G1, *G0 compared with G1.
We next analyzed the lipid content of kidney cortices of APOL1 transgenic mice. Consistent with our prior observation that APOL1 may affect cholesterol metabolism (19), we demonstrated that the total cholesterol (TC) of kidney cortices of G1 transgenic mice is significantly higher when compared to G0, while cholesterol ester (CE) content remained unchanged (Fig. 1C). These results indicate that APOL1 RV expression under the endogenous APOL1 promoter is sufficient to drive lipid accumulation but does not cause proteinuria in mice, similar to what we described in mice with the podocyte-specific deletion of ATP-binding cassette transporter A1 (ABCA1) (31).
APOL1 G1 risk allele expression renders mice more susceptible to renal disease in a mouse model of lipotoxicity-driven FSGS
To investigate if APOL1 RV expression renders mice more susceptible to FSGS, we bred APOL1 transgenic mice to Podocin-rtTA; NFATc1nuc, a model of FSGS with progressive renal failure that we have developed in the laboratory (27). These triple transgenic (TT) mice (APOL1;Podocin-rtTA;NFATc1nuc) (Fig. 2A) were compared to double transgenic (DT) littermate (WT;Podocin-rtTA;NFATc1nuc) controls. All non-induced animals did not develop proteinuria (Supplementary Material, Fig. S2A and B). We found that doxycycline-induced G1 TT mice are characterized by more severe proteinuria post-induction (Fig. 2B). Increased proteinuria levels were accompanied by reduced gain in body weight (Supplementary Material, Fig. S3A) and increased blood urea nitrogen (BUN) levels (Fig. 2C).
Figure 2 .
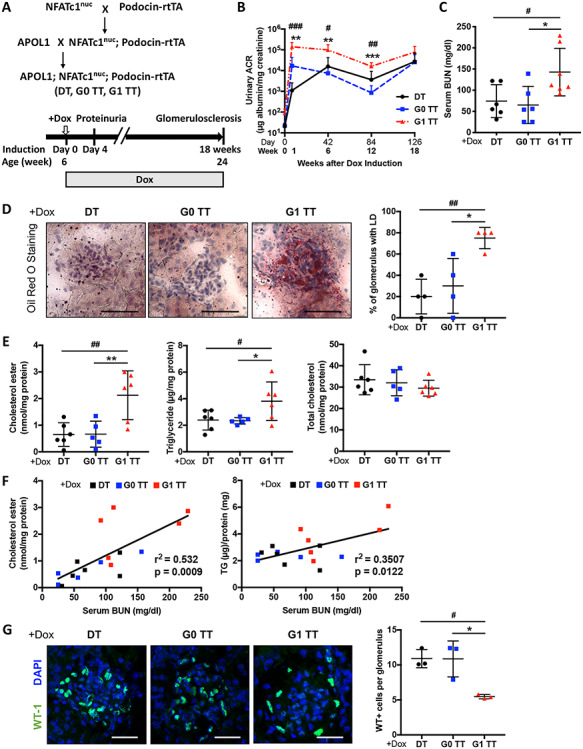
Podocyte-specific NFAT activation in mice carrying the APOL1 G1 allele (G1 TT) causes increased proteinuria, serum BUN and renal lipid accumulation. (A) Experimental design to generate wildtype; Podocin-rtTA; NFATc1nuc mice (DT), APOL1 G0; Podocin-rtTA; NFATc1nuc (G0 TT) and APOL1 G1; Podocin-rtTA; NFATc1nuc (G1 TT) transgenic mice (upper panel). Doxycycline induction was initiated at 6 weeks of age and was continued for 18 weeks. Mice were sacrificed at 24 weeks of age (lower panel). (B) Urinary albumin-to-creatinine ratio in DT, G0 TT mice and G1 TT mice. Urines were collected before doxycycline induction and 1, 6, 12, 18 weeks after induction (n = 6). (C) Scatter plots of BUN level of TT mice fed doxycycline chow for 18 weeks (n = 6–7). (D) Representative Oil Red-O (ORO) images of stained kidney cortices sections from TT mice fed doxycycline chow for 18 weeks (scale bars: 50 μm) and quantification of the number of glomeruli with lipid droplets (LD) in ORO stained slides from four mice per group (n = 4). (E) Scatter plots of cholesterol ester (ce), triglyceride (TG) and total cholesterol (TC) contents in kidney cortices of TT mice fed doxycycline chow for 18 weeks. Values are normalized to protein concentrations (n = 5–6). (F) Correlation analyses between the ce content of kidney cortices and BUN and between the TG content of kidney cortices and BUN (n = 17). (G) Representative images of kidney cortices of TT mice fed doxycycline chow for 18 weeks stained with WT1 (green) to detect podocytes and DAPI (blue) to reveal nuclei (scale bars: 25 μm) and quantification of the average number of WT1-positive podocytes per glomerulus in WT1 stained slides from three mice per group (n = 3). The error bars represent mean ± SD of biologically independent experiments. One-way ANOVA followed by Dunnett’s test (B–E and G); Pearson’s correlation coefficient (F). *P (#P) < 0.05, **P (##P) < 0.01, ***P (###P) < 0.001. #DT compared with G1 TT, *G0 TT compared with G1 TT.
Consistent with what we observed in APOL1 transgenic mice (Fig. 1C), we found an increased number of LD positive glomeruli in doxycycline-induced G1 TT mice as determined by Oil Red-O staining (Fig. 2D). We then extracted total lipids from kidney cortices to investigate lipid composition, and found increased ce and triglyceride contents in doxycycline-induced G1 TT mice. However, the TC content was similar in all three groups (Fig. 2E), suggesting that the presence of APOL1 risk alleles may differentially affect lipid metabolism in health and disease, and that accumulation of lipid species other than TC may characterize experimental models of glomerular diseases. In fact, we found a positive correlation of the ce content in kidney cortices of doxycycline-induced TT mice with serum BUN, similar to what we previously reported in podocyte-specific Abca1-deficient diabetic mice (31). Additionally, the triglyceride content in kidney cortices also positively correlated with BUN (Fig. 2F). These finding suggest that APOL1 G1 RV expression induces proteinuria and glomerular accumulation of ce and triglyceride, the latter of which correlates with loss of renal function.
We furthermore investigated if glomerular lipid accumulation in the context of FSGS is associated with podocyte loss and found that doxycycline-induced G1 TT mice have significantly decreased podocyte numbers per glomerulus (Fig. 2G) as indicated by significantly decreased Wilms tumor 1 (WT1)-positive cells per glomerulus. Glomeruli of doxycycline-induced G1 TT mice also exhibited significant mesangial matrix expansion (Fig. 3A) as determined by PAS staining, significantly increased glomerular and tubulointerstitial fibrosis as determined by Picrosirius red staining (Fig. 3B) and significantly increased podocyte foot process effacement as determined by TEM (Fig. 3C). Interestingly, in addition to increased foot process effacement, we observed mitochondrial vacuolization in doxycycline-induced G1 TT mice (Fig. 3C), suggesting a potential link between APOL1 G1 expression, lipid metabolism and mitochondrial structure in experimental FSGS.
Figure 3 .
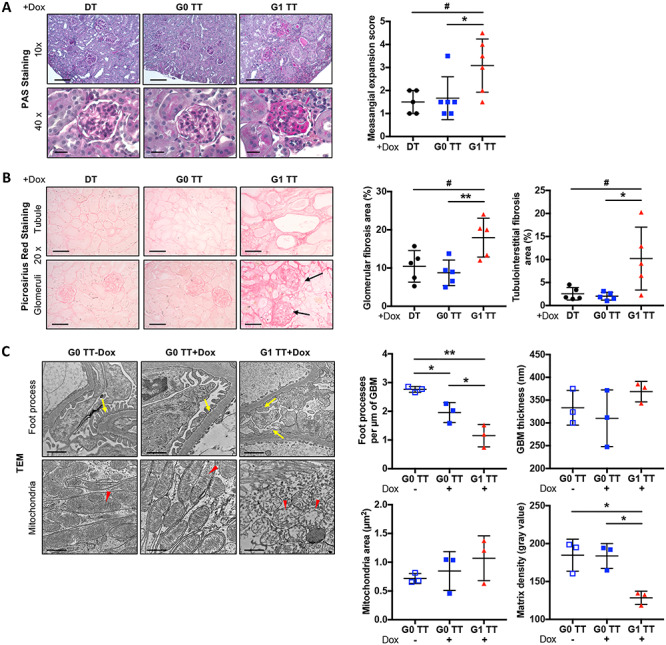
Podocyte-specific NFAT activation in mice carrying the APOL1 G1 allele (TT) is associated with increased glomerulosclerosis, fibrosis, foot process effacement and mitochondrial vacuolization. (A) Representative images of PAS staining of kidney cortices sections from DT, G0 TT and G1 TT mice fed doxycycline chow for 18 weeks (left panel, scale bars: 10×: 100 μm; 40×: 20 μm). Scatter plots of the mesangial expansion score as determined by analysis of PAS stained slides (right panel, n = 5–6). (B) Representative Picrosirius red staining of kidney cortices sections from DT, G0 TT and G1 TT mice fed doxycycline chow for 18 weeks (left panel, scale bars: 50 μm). Scatter plots of the quantification of fibrosis in glomeruli and tubules (right panel, n = 5). (C) Representative TEM images to identify podocyte foot process in TT mice fed doxycycline chow for 18 weeks. The arrows point to podocyte foot processes; arrow heads point to mitochondria (left panel, scale bars: 1 μm). Scatter plot quantification of podocyte foot process per μm of GBM and GBM thickness (upper-right panel, n = 3). Scatter plot quantification of mitochondrial size and matrix density of mitochondria (lower-right panel, n = 3). The error bars represent mean ± SD of biologically independent experiments. One-way ANOVA followed by Dunnett’s test (A and B); Two-tailed Student’s t-test (C). *P < 0.05, **P < 0.01.
The effect on lipid content in kidney cortices was not completely reflected by the effect on circulating lipids. While serum cholesterol levels in G1 TT mice were higher when compared to DT and G0 TT mice, no significant differences were detected in serum triglyceride content (Supplementary Material, Fig. S3B and C). Additionally, we observed the colocalization of podocyte marker Synaptopodin (SYNPO) and LD-associated protein Perilipin 2 (PLIN2) in kidney cortices of doxycycline-induced G1 TT, indicating that LDs harbor in podocytes (Supplementary Material, Fig. S4).
APOL1 G1/G2 podocytes from patients with FSGS are characterized by increased triglyceride content and altered mitochondrial structure
To investigate the mechanism by which APOL1 contributes to podocyte injury in FSGS, we utilized urinary podocyte cell lines established from patients affected by FSGS and carrying either G0/G0 or G1/G2 alleles. The expression of podocyte-specific markers Nephrin and Synaptopodin was confirmed (Supplementary Material, Fig. S5A). We also extracted total lipids to measure the lipid content in these podocytes but did not find significant differences in TC and ce contents between G0/G0 and G1/G2 podocytes (Fig. 4A). However, consistent with the experimental mouse model of FSGS, we observed an increased triglyceride content in G1/G2 podocytes (Fig. 4B). We also found decreased hexosyl ceramide, monosialodihexosylganglioside (GM3), lysophosphatidylglycerol (LPG) and sphingomyelin contents in G1/G2 podocytes (Supplementary Material, Table S3), indicating that expression of APOL1 risk alleles may alter membrane lipids composition as suggested by overexpression study (20).
Figure 4 .
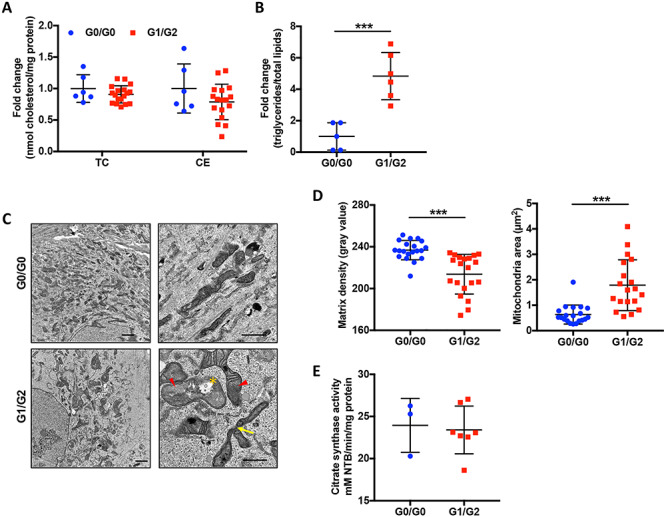
Human urinary podocytes carrying APOL1 G1/G2 risk alleles are characterized by increased triglyceride content and changes in mitochondrial morphology. (A and B) Scatter plot quantification of total cholesterol (TC), cholesterol ester (ce) (A) and triglyceride (TG) (B) content in G0/G0 and G1/G2 expressing podocytes (n = 6 & 18). (C) Representative TEM images of G0/G0 and G1/G2 expressing podocytes. The arrow heads point to mitochondria with loss of cristae membranes and matrix swelling; the asterisks indicate rupture of outer mitochondrial membrane and the arrows point to hyperbranched mitochondria. Scale bar: 2 μm (left), 1 μm (right). (D) Scatter plots of matrix density of mitochondria and mitochondrial size quantified from at least five different images using ImageJ (n = 20). (E) Scatter plot quantification of citrate synthase activity determined by the rate of absorbance gain at 412 nm normalized to protein concentration (n = 3 & 7). The error bars represent mean ± SD of biologically independent experiments. Two-tailed Student’s t-test. ***P < 0.001.
Podocytes are high energy demanding cells during differentiation (32). We performed TEM analysis of podocytes and found that mitochondria of G0/G0 podocytes are characterized by the presence of dark matrix, indicating normal cristae structure. However, in G1/G2 podocytes, we detected reduced mitochondrial matrix density, while the mitochondrial area was found to be increased and hyperbranched mitochondria were observed (Fig. 4C and D). Interestingly, the activity of the Krebs cycle enzyme citrate synthase, which is commonly used as marker of mitochondrial content, was not affected in G1/G2 podocytes (Fig. 4E). Lastly, the changes in mitochondrial morphology in G1/G2 podocytes were accompanied by a significant increase in the mRNA levels of mitochondrial fission and fusion proteins FIS1 and MFN1 (Supplementary Material, Fig. S5B and C). These data suggest that G1/G2 expression may be associated with mitochondrial structural alterations similar to one observed under stress conditions (33,34).
Mitochondrial dysfunction in the presence of increased OXPHOS complexes occurs in G1/G2 expressing podocytes
The trypanolytic activity of APOL1 is associated with the depolarization of the mitochondrial membrane of Trypanosoma brucei (35). The structural abnormalities found in mitochondria of podocytes led us to hypothesize that energy production could be compromised in G1/G2 podocytes. Initially, we assessed endogenous respiration in intact cells and observed a significant decrease in KCN-sensitive oxygen consumption rate (OCR) of G1/G2 podocytes compared to G0/G0 podocytes (Fig. 5A). Next, we measured substrate-driven OCR in digitonin-permeabilized cells. Both state 3 and state 4 respirations were significantly reduced in G1/G2 podocytes (Fig. 5B). Moreover, titration of the uncoupler carbonyl cyanide m-chlorophenylhydrazone (CCCP) was carried out to progressively decrease MMP and achieve maximal OCR. We found that G0/G0 and G1/G2 podocytes respond differently to CCCP (Fig. 5C), which maybe a consequence of differences in the mitochondrial membrane permeability. Meanwhile, maximal respiratory capacity, respiratory control (RC) ratio and spare capacity were not altered in G1/G2 cells (Fig. 5B).
Figure 5 .
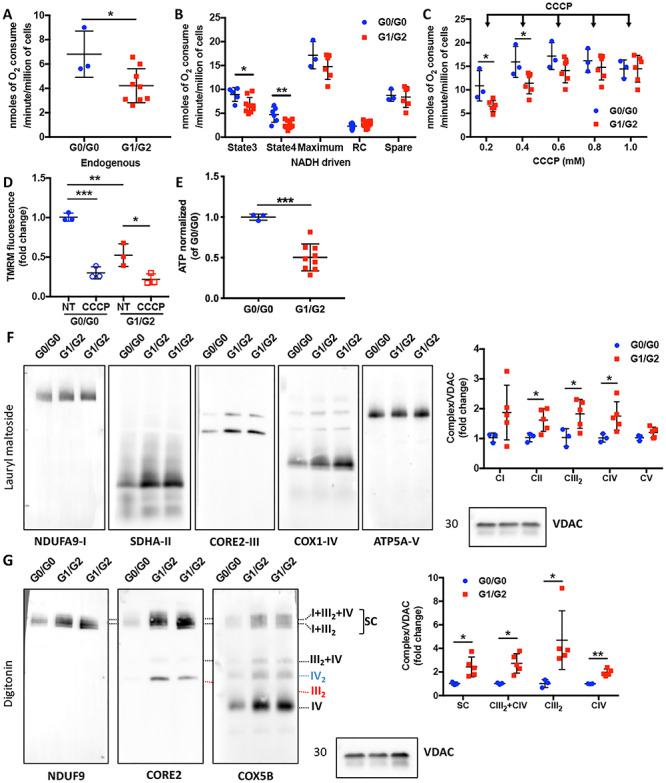
Podocytes carrying APOL1 G1/G2 risk alleles are characterized by mitochondrial dysfunction in association with increased OXPHOS complexes. (A and B) Scatter plot quantification of endogenous (A) and substrate-driven (B) oxygen consumption rates (OCR, nmol of oxygen consumed per minute per million of cells) in podocytes carrying G0/G0 and G1/G2 alleles (n = 3–9). (C) Scatter plot quantification of oxygen consumption rates during carbonyl cyanide m-chlorophenylhydrazone (CCCP) titrations. The arrows indicate sequential additions of CCCP (1 μl incremental addition of 0.1 M CCCP) (n = 3 & 6). (D) Scatter plot quantification of mitochondrial membrane potential in untreated (NT) and uncoupler agent (CCCP) treated podocytes carrying G0/G0 and G1/G2 (n = 3). (E) ATP content was determined in podocytes carrying G0/G0 and G1/G2 alleles using a luminescence assay and normalized to cell number (n = 3 & 9). (F) Representative Western blot images of BN-PAGE analysis using lysates obtained by lauryl maltoside extraction and probed for NDUFA9 (Complex I: CI), SDHA (CII), CORE2 (CIII), COX1 (CIV), ATP5A (CV) and VDAC was detected in SDS-PAGE as a loading control for the detection of steady state levels of individual complexes (left panel). Signals from the BN-PAGE analysis of lauryl maltoside extracts were quantified by densitometry and normalized to VDAC (right panel, n = 3 & 5). (G) Representative Western blot images of BN-PAGE analysis using lysates obtained by digitonin extraction and probed for CORE2, NDUFA9, COX5B and VDAC was detected in SDS-PAGE as a loading control to determine the OXPHOS complex distribution at the mitochondrial membrane (left panel). Signals from the BN-PAGE experiments of digitonin extracts were quantified by densitometry and normalized to VDAC (right panel, n = 3 & 5). The error bars represent mean ± SD of biologically independent experiments. Two-tailed Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001.
Reduced electron transfer is normally associated with decreased mitochondrial membrane potential (MMP). To measure MMP podocytes were incubated with tetramethylrhodamine, methyl ester (TMRM). As expected, G0/G0 podocytes retained TMRM fluorescence, while it was reduced in G1/G2 podocytes, indicating a decrease in MMP (Fig. 5D), which could ultimately lead to a defect in mitochondrial ATP generation. Indeed, cellular ATP levels were reduced in G1/G2 podocytes (Fig. 5E). These results suggest that endogenous G1/G2 RV expression in podocytes is associated with mitochondrial dysfunction and ATP depletion, similar to overexpression of G1 and G2 in HEK293 cells (21,22,24).
We next assessed whether there are alterations in mitochondrial OXPHOS complexes. Increased individual OXPHOS complexes (Fig. 5F) and supercomplexes (Fig. 5G) were observed in G1/G2 expressing podocytes, suggesting that G1/G2 RV expression may impair OXPHOS function and an increase in OXPHOS biogenesis could represent a compensatory mechanism in an attempt to preserve mitochondrial ATP production. However, elevated OXPHOS complex steady-state levels were not sufficient to fully restore mitochondrial bioenergetic capacity.
To validate that lipotoxicity mediates mitochondrial dysfunction in G1/G2 podocytes, cells were treated with lipid-modifying agents, including a liver X receptor (LXR) agonist (36), and an ABCA1 inducer (31). Mitochondrial membrane potential in G1/G2 podocytes was restored to levels similar to those in vehicle-treated G0/G0 cells, as indicated by an increase in the aggregate/monomer fluorescence ratio (Fig. 6A). Additionally, ATP production in G1/G2 podocytes was significantly improved (Fig. 6B), though to a level that was still lower than in vehicle-treated G0/G0 podocytes. These data suggest that reducing the lipid content in podocytes ameliorates APOL1-mediated mitochondrial dysfunction and may represent a new therapeutic strategy to treat APOL1 nephropathy.
Figure 6 .
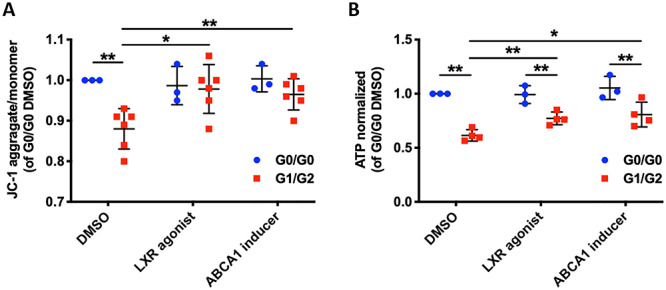
Lipid-modifying agents improve mitochondrial membrane potential and ATP production in G1/G2 expressing podocytes. (A) Scatter plot of mitochondrial membrane potential determined by JC-1 probe comparing G0/G0 and G1/G2 expressing podocytes treated with lipid-modifying agents (n = 3 & 6). (B) Scatter plot of ATP content normalized to cell number comparing G0/G0 and G1/G2 expressing podocytes treated with lipid-modifying agents (n = 3 & 4). The error bars represent mean ± SD of biologically independent experiments. Two-tailed Student’s t-test. *P < 0.05, **P < 0.01.
APOL1 RV binds to mitochondrial specific cardiolipin with higher affinity
To determine the specific role of APOL1 in mitochondrial function of podocytes, we first co-stained the cells with MitoTracker and APOL1 antibody. We confirmed that in podocytes both APOL1 wildtype and variants localized to mitochondria (Fig. 7A).
Figure 7 .
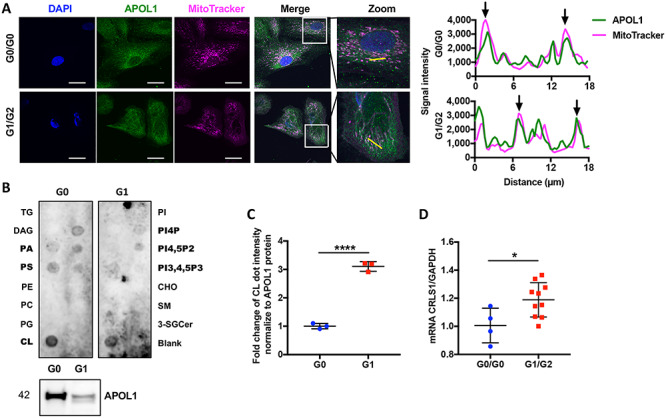
APOL1 is localized in mitochondria of urinary podocytes and binds with mitochondria-specific phospholipid cardiolipin. (A) Representative confocal images of differentiated podocytes stained with DAPI (blue), APOL1 (green) and MitoTracker (magenta). White color indicates overlapping pixels. Zoom: Regional magnification of the box area with the yellow line indicating the scanned segment (left panel, scale bar: 50 μm). Line scans of APOL1 and MitoTracker from left panel. The arrows represent colocalization (right panel). (B) APOL1 G0-6xHis and APOL1 G1-6xHis protein isolated from HeLa cells were overlaid in a lipid strip (upper panel). Purified APOL1-6xHis was confirmed by Western blot analysis (lower panel). (C) Scatter plots of the densitometric measurement of binding of Cardiolipin (CL) with APOL1 G0 and G1, normalized to 6xHis tagged APOL1 expression (n = 3). (D) Scatter plots of CRLS1 mRNA expression in G0/G0 and G1/G2 expressing podocytes (n = 4 & 10). The error bars represent mean ± SD of biologically independent experiments. Two-tailed Student’s t-test. *P < 0.05, ****P < 0.0001.
To further study other factors that may contribute to mitochondrial dysfunction, the interaction of APOL1 with mitochondrial lipids was investigated. We therefore purified APOL1-6xHis protein using HeLa cells stably infected with lentivirus carrying the APOL1 G0, G1 under the CMV promoter (19). A protein-lipid overlay assay was performed. We found an interaction of APOL1 with mitochondrial specific cardiolipin, as well as lipid precursors phosphatidic acid (PA), phosphatidylserine (PS) and the phosphorylated derivatives of phosphatidylinositol (PI4P, PI4,5P2, PI3,4,5P3) (Fig. 7B). Additionally, we found that the affinity of APOL1 G1 to cardiolipin is significantly higher than APOL1 G0 to cardiolipin, when normalized to 6xHis tagged APOL1 expression (Fig. 7C). We next examined mRNA expression of Cardiolipin Synthase 1 (CRLS1), the enzyme that catalyzes the synthesis of cardiolipin and found CRLS1 expression to be significantly increased in G1/G2 podocytes (Fig. 7D). Since cardiolipin is an important constituent of mitochondrial inner membrane and plays a key role in OXPHOS biogenesis and function (37), increased CRLS1 mRNA expression is consistent with overexpressed OXPHOS complexes in G1/G2 podocytes. APOL1 binding to cardiolipin may interfere with cardiolipin function and lead to compensatory CRLS1 overexpression.
Discussion
With this study, we aimed at investigating the role of endogenous APOL1 RV expression in cellular lipid metabolism and mitochondrial function utilizing clinically relevant experimental models of FSGS. We focused on podocytes, as podocytes are highly susceptible to injury in the prevalent cohort of African American patients who carries APOL1 RVs (2,38–41). We first generated human APOL1 BAC transgenic mice expressing either the G0 allele or the risk alleles G1 and G2 under the endogenous APOL1 promoter. We demonstrate that the expression of endogenous levels of APOL1 is not associated with proteinuria and renal injury, distinct from mice with podocyte-specific overexpression (42) or inducible expression of APOL1 RV (43). Similarly, mice expressing the APOL1 G2 under the Nephrin promoter also did not develop proteinuria (44). Taken together, these studies suggest that podocyte APOL1 RV expression per se may not cause injury (Fig. 1). These data are furthermore consistent with clinical evidence indicating that carrying two APOL1 RVs are not sufficient to develop renal diseases in humans, thus opening up the possibility that a ‘second hit’ is necessary to cause podocyte injury (45). To test this hypothesis, we utilized mice with podocyte-specific expression of NFATc1nuc, a mouse model for FSGS that we have demonstrated to mimic progressive FSGS in patients and that we have demonstrated to be linked to lipotoxic podocyte injury (27). We utilized Podocin-rtTA; NFATc1nuc mice to generate mice that simultaneously express different APOL1 alleles (TT). We fed TT mice with low dose doxycycline which mildly induces proteinuria. We demonstrate that by low dose doxycycline induction, TT with APOL1 G1 expression showed increased susceptibility to podocytes injury thus worsening the FSGS-like phenotype of mice (Fig. 2). Furthermore, we found that APOL1 G1 TT mice is associated with the accumulation of ce and triglycerides in kidney cortices, suggesting that APOL1 G1 expression might contribute to lipotoxicity-induced renal injury, similarly to what we previously described in other mouse models of renal disease (19,27,31,46). Interestingly, TEM analysis of kidney cortex sections of doxycycline-induced G1 TT mice revealed mitochondrial vacuolization in addition to foot process effacement (Fig. 3), suggesting a possible role for APOL1 in lipid-induced mitochondrial dysfunction in podocytes.
To further investigate the role of APOL1 RV expression in lipid-induced mitochondrial dysfunction, we analyzed human urinary podocytes carrying either APOL1 G0/G0 or G1/G2 alleles (Fig. 4). In-depth analysis of the mitochondrial phenotype allowed us to demonstrate that G1/G2 podocytes are characterized by mitochondrial vacuolization and coexistence of hyperfusion and fragmentation of mitochondria as detected by TEM. Fusion and fission are opposing reactions that determine the shape and function of mitochondria (47). Under stress and starvation, mitochondria undergo hyperfusion in response to stress. Accumulation and prolonged exposure to cellular stress can ultimately lead to mitochondrial fragmentation activated by calcineurin (33,48). Both FIS1 and MFN1 mRNA levels are increased in G1/G2 podocytes further supporting the idea that fusion and fission of mitochondria occurs at the same time and that these cells are exposed to metabolic stress.
We also observed incomplete energy production in combination with increased expression of OXPHOS complexes (Fig. 5). We showed that G1/G2 podocytes have impaired mitochondria function, including reduced ATP generation, reduced state 3 and state 4 respiration and reduced MMP, consistent with previous studies in APOL1 overexpressing HEK293 cells (21–24). Additionally, CCCP titration revealed differences in mitochondrial membrane permeability in G0/G0 and G1/G2 podocytes. These findings led us to examine how APOL1 RVs drive mitochondrial dysfunction. APOL1 was previously localized by other groups in various organelles including mitochondria, ER, endosomes, autophagosomes and LD (20–22,42). We were able to confirm the localization of APOL1 to mitochondria (Fig. 7). Using protein-lipid overlay assay, we showed an interaction of APOL1 with cardiolipin, similar to what was described in previous studies (49,50). Additionally, we found that the affinity of APOL1 G1 to cardiolipin is higher when compared to APOL1 G0, suggesting that APOL1 G1 binding to cardiolipin might interfere with the proper function of cardiolipin in OXPHOS complexes. This model is also supported by the increased and likely compensatory expression of CRLS1, the catalytic enzyme responsible for increased cardiolipin production. Located in the inner mitochondrial membrane, cardiolipin binds to complexes I, III, IV and V with high affinity (37). It is possible that the increased expression of OXPHOS complexes and CL production occur in an attempt of the cells to compensate for the deficient energy production. It is noteworthy that cardiolipin released from disrupted mitochondria can interact with NLRP3 inflammasome, resulting in caspase-1 activation (51). Interestingly, it was reported that the expression of the APOL1 RVs causes inflammatory-mediated podocyte death and glomerular scarring (42). Therefore, our findings may reveal a novel mechanism connecting APOL1-mediated mitochondrial dysfunction with inflammatory pathways.
Overexpression of APOL1 RVs in human podocytes was previously shown to induce cytotoxicity (20,52). Therefore, the human urinary podocyte cell lines established by us will enable us to study mechanisms of APOL1 RV expression under endogenous conditions. We previously demonstrated glomerular cholesterol accumulation in FSGS mouse model (27,46). To investigate the role of lipid-mediated podocyte toxicity of APOL1 RV expression, we determined the lipid content of podocytes and in renal cortices of APOL1 transgenic mice. We found increases in the triglyceride content in both, G1/G2 podocytes as well as in doxycycline-induced G1 TT mice, although we did not detect differences in the TC and ce contents between G0/G0 and G1/G2 podocytes. It is possible that the fact that cells were isolated from patients with established FSGS masked a difference in TC and ce contents, as we have shown increased cholesterol content in podocytes in several other models of FSGS unrelated to APOL1 (27,46). In this respect, it would be great to study G0/G0 and G1/G2 podocytes isolated from healthy individuals; however, we were unable to do so as an insufficient number of urinary cells can be retrieved from healthy donors (data not shown). Others demonstrated that APOL1 is expressed in LD (20), which are organelles primarily composed of triglycerides and ce. Therefore, it is possible that APOL1-dependent podocyte cytotoxicity could be mediated by triglycerides. Finally, we found decreased GM3 levels in G1/G2 compared to G0/G0 podocytes using mass spectrometry (Supplementary Material, Table S3). GM3 is a glycosphingolipid that is highly enriched in lipid rafts on the podocyte surface where it interacts with receptors of vascular endothelial growth factor (VEGFR), thus promoting actin organization (53). A reduction of GM3 may cause reorganization of the podocyte actin cytoskeleton and foot process effacement. Taken together, the use of human urinary podocytes to study the role of APOL1 RVs in podocyte health and disease offers a unique perspective to investigate lipid profile and mitochondrial function in a more clinical-relevant setting.
This study has several limitations. First, this study did not discern possible differences in APOL1 G1 and G2 function as we did not analyze APOL1 G2 mice due to low expression of the transgene in G2 mice. Second, the podocyte-specific NFAT-inducible model as an experimental model of progressive FSGS is rather new and may not represent all forms of human FSGS. Third, the use of mouse models for the study of APOL1 function in vivo is not ideal, as the APOL1 gene is not conserved in mice, therefore rendering this system partially artificial. However, the fact that APOL1 is only expressed in higher primates renders any in vivo investigation of APOL1 function difficult. Last, immortalized urinary podocyte cell lines were derived from a limited number of patients with established FSGS, where a second hit may have influenced the presented findings. Therefore, a direct comparison of our findings to G0/G0 podocytes established from healthy individuals would add further value. However, the establishment of cell lines from healthy individuals is difficult as healthy subjects lose only few podocytes in urine. Though our in vitro and in vivo data obtained in this study are consistent, a validation of these results in another system in the future is warranted.
In summary, we established a mouse model to study APOL1 RV associated susceptibility to NFAT-mediated FSGS. We provide for the first-time evidence that APOL1 G1 induced glomerular lipid accumulation correlates with loss of renal function. Our results also confirm in clinically relevant experimental models that APOL1 G1/G2 RVs is associated with mitochondrial dysfunction and that APOL1 RV expression interacts with cardiolipin thus interfering with the proper function of cardiolipin. We also describe for the first time that APOL1 G1/G2 RVs related mitochondrial dysfunction is linked to podocyte lipid accumulation and compensatory OXPHOS complexes elevation. Further understanding of this mechanism will enable the identification of new pathway-relevant targets and personalized therapeutic strategies aimed at the treatment of the large number of patients carrying APOL1 RVs.
Materials and Methods
Study Approval
All studies involving mice were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Miami. The University of Miami (UM) has an Animal Welfare Assurance on file with the Office of Laboratory Animal Welfare, NIH (A-3224-01, effective November 24, 2015). Additionally, UM is registered with the US Department of Agriculture Animal and Plant Health Inspection Service, effective December 2014, registration 58-R-007. As of October 22, 2013, the Council on Accreditation of the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC International) has continued UM’s full accreditation.
Animal Studies
Human BAC transgenic mice (129S1) that express different APOL1 genetic variants (G0, G1, G2) under the endogenous APOL1 promoter were used for in vivo studies (19). Mice were made by pronuclear injection of an engineered linearized human BAC construct (~47.2 kb) that also included a portion of the APOL2 and the MYH9 genes assuring that all relevant regulatory regions were included in the transgene. Inducible constitutively active NFATc1 transgenic mice (NFATc1nuc) were a gift from Dr Gerald R. Crabtree (B6BAF1/J) and were bred to Podocin-reverse tetracycline-controlled transactivator, Podocin-rt-TA mice (FVB/N-Tg(NPHS2-rtTA2*M2)1Jbk/J, stock number: 008202, Jackson Laboratory) to generate podocyte-specific tetracycline-inducible constitutively active NFATc1 (Podocin-rtTA;NFATc1nuc) mice, a mouse model of FSGS previously characterized by us (27). The latter mice were further bred to APOL1 mice to generate triple transgenic mice (TT). The following three groups of mice were analyzed: WT, Podocin-rtTA; NFATc1nuc mice (DT), APOL1 G0;Podocin-rtTA;NFATc1nuc (G0 TT) and APOL1 G1;Podocin-rtTA;NFATc1nuc (G1 TT). To account for background variations of the three background strains, only F1 animals were used in the experiments and their DT littermates were served as controls. Mice of both genders were used. Transgene expression of podocyte-specific NFATc1nuc was induced in 6 weeks old DT and TT mice with 200 ppm doxycycline added to the food pellets. All induced mice were sacrificed 18 weeks after the induction of transgene expression.
Phenotypic analysis of mice
Urinary albumin-to-creatinine ratios
Morning spot urine samples were collected at different time points (APOL1 BAC transgenic mice: 7 weeks old and 7 months old, at the time of sacrifice; TT/DT + Dox: before doxycycline induction and 1, 6, 12, 18 weeks after induction; TT/DT-Dox: 24 weeks of age, at the time of sacrifice). Urinary albumin-to-creatinine ratios (ACR) were determined using the Mouse Albumin ELISA Quantitation Set (Bethyl Laboratories) and Creatinine LiquiColor (Stanbio). Albuminuria values are expressed as microgram albumin per milligram creatinine.
Serology
Blood samples were collected at the time of sacrifice. Serum BUN, cholesterol and TG were analyzed in the Comparative Laboratory Core Facility at the University of Miami.
Molecular analysis of glomeruli
Glomeruli were isolated by sieving technique and mRNA was extracted using the RNeasy Mini Kit (Qiagen). Glomerular expression of APOL1 mRNA was analyzed in APOL1 BAC transgenic mice by quantitative real-time polymerase chain reaction (PCR) as described below.
Oil Red-O staining
4 μm kidney cortices optimal cutting temperature (OCT) sections were incubated with 100 μl freshly prepared Oil Red-O solution (Electron Microscopy Science) for 15 min and counterstained with Hematoxylin Harris solution for 5 min (VWR, 10143-606) to detect lipid deposition. Images were examined under a light microscope (Olympus BX 41, Tokyo, Japan), and quantified by the percentage of LD positive glomeruli.
Podocyte loss
To measure podocyte number per glomerulus, glomerular sections embedded in OCT were stained with a Wilms tumor 1 (WT1) antibody (Santa Cruz Biotechnology, sc-192, rabbit, 1:300), followed by a secondary antibody (Invitrogen, A-11008, 1:500) and Mounting Medium with DAPI (Vectorlabs, H-1200). Images were acquired by confocal microscopy using a Leica SP5 inverted microscope with the 40× wet objective. 5–10 glomeruli per mouse were quantified.
Kidney histology analysis
Perfused kidneys were fixed in 10% formalin and paraffin embedded, and then cut in 4 μm thick sections. Periodic acid-Schiff (PAS) staining was performed to investigate mesangial expansion following a standard protocol. Mesangial expansion was visualized under a light microscope (Olympus BX 41, Tokyo, Japan) and 20 glomeruli per kidney cortex section were scored by semiquantitative analysis (scale 0–5), performed by two blinded investigators. Picrosirius red staining was performed to measure fibrosis. Paraffin-embedded sections were deparaffinized with xylene and a graded alcohol series. Sections were rinsed, stained for 1 h with Picrosirius red in saturated aqueous picric acid. Sections were examined under a light microscope (Olympus BX 41, Tokyo, Japan), followed by analysis with Image J.
Podocyte cell culture
Podocyte cell lines were established from FSGS patients’ urine following a method previously described (54). These cell lines were kindly provided by Dr Jeffrey B. Kopp from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). Cell lines carrying either APOL1 G0/G0 or G1/G2 alleles were cultured at 33°C in RPMI culture medium containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (complete growth medium), supplemented with insulin-transferrin-selenium (ITS, Corning), then thermo-shifted at 37°C for 14 days to achieve terminal differentiation (55). All cell culture dishes were coated with 0.1 mg/ml type I collagen. For experiments with pharmacological compounds, differentiated podocyte cell lines were serum-restricted overnight. Cells were treated for 24 h with LXR agonist (Roche, 1 μM) (36) and ABCA1 inducer (Roche, 5 μM) (31).
Quantitative real-time PCR
RNA was extracted from differentiated podocytes or mouse glomeruli using the RNeasy Mini Kit (Qiagen). Reverse transcription was performed using QuantiTect Reverse Transcription Kit (Qiagen). Quantitative real-time PCR was carried out using the StepOnePlus system (Applied biosystems) with PerfeCTa SYBR Green FastMix (Quanta). Relative quantification was determined as 2−ΔΔCt. The list of primers used can be found in Supplementary Material, Table S1.
Western blot analysis
Cells lysates were prepared using 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonic (CHAPS) acid buffer. Protein concentration was measured with the bicinchoninic acid (BCA) reagent (Thermo Scientific). 20–30 μg of protein extract was loaded onto 4 to 20% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) gels (Bio-Rad) and transferred to Immobilon-P PVDF membranes (Bio-Rad). Western blot analysis was performed using a standard protocol and the following primary antibodies: APOL1 (Sigma-Aldrich, HPA018885, rabbit, 1:500), NEPHRIN (Santa Cruz Biotechnology, sc-28 192, rabbit, 1:1000), SYNAPTOPODIN (Santa Cruz Biotechnology, sc-21 537, goat, 1:1000), GAPDH (Sigma-Aldrich, CB1001, mouse, 1:10000); or secondary antibodies: anti-mouse IgG HRP (Promega, W402B, 1:10000), anti-rabbit IgG HRP (Promega, W401B, 1:10000) or anti-rabbit IgG HRP (Promega, V805A, 1:10000). Signal was detected with Radiance ECL (Azure) using Azure c600 Imaging System. The list of all antibodies can be found in Supplementary Material, Table S2.
Lipid extraction
Tissues/cells were homogenized in a buffer containing 50 mM pH 7.4 potassium phosphate and cOmplete Protease Inhibitor Cocktail tablet (Roche, 1 pill in 10 ml buffer) by sonication for 20s, twice, on ice. Total lipids were extracted from homogenates using hexane:isopropanol (3:2) and placed in a mixer (1000 rpm) for 30 min. The mixed homogenate was then spun at top speed, lipids contained in the supernatants were collected, and pellets were disrupted by two sequential lipid extractions. Total lipids were then pooled and dried using a speed vacuum at 37°C and reconstituted with 100 μl isopropanol:NP-40 (9:1). Proteins were extracted from the pellets using 8 M Urea, 0.1% SDS, 0.1 M NaOH. Extracted lipids were used for determining total cholesterol (TC), cholesterol ester (ce) and triglyceride contents, and normalized to protein concentrations.
Triglyceride assay
The TG content was determined using Triglyceride Colorimetric Assay Kit (Cayman) following manufacturer’s protocol. A total of 10 μl standards and lipid samples from above-mentioned extraction were added into a 96-well plate. The reaction was initiated by adding 150 μl enzyme buffer to each well. Plates were incubated at room temperature for 15 min and absorbance at 540 nm was measured using a SpectraMax M5 plate reader (Molecular Devices).
Cholesterol assay
Cholesterol assays were performed using the Amplex Red Cholesterol Assay Kit (ThermoFisher Scientific) following the manufacturer’s instructions with some modifications. To measure TC, endogenous peroxides (60 μl 1:100 diluted samples, 24 μl 100 U/ml catalase, 36 μl 1xAssay buffer) were removed using catalase. The reaction was incubated for 30 min at 37°C and 50 μl of the reaction was transferred and incubated with 50 μl 2x TC reagent (2 U/ml cholesterol oxidase, 1 U/ml cholesterol esterase, 2 U/ml horseradish peroxidase, 75 μM Amplex Red) at 37°C for 30 min in a duplicate format. Fluorescence was read at 530/580 nm. ce assays were performed using direct enzymatic method as previously described with modifications (56). A total of 25 μl lipid extracts were mixed with 150 μl free cholesterol decomposing reagent (100 μl 45 U/ml catalase and 50 μl 3.5 U/ml cholesterol oxidase in 45 U/ml catalase solution). The reaction was mixed and incubated overnight at 37°C. From this reaction, 75 μl were transferred and incubated with 25 μl 4x cholesterol ester reagent (2 U/ml cholesterol oxidase, 4 U/ml cholesterol esterase, 24 U/ml horseradish peroxidase, 300 μM Amplex Red) at 37°C for 30 min in a duplicate format. Fluorescence was read at 530/580 nm. SpectraMax M5 plate reader (Molecular Devices) was used.
Transmission electron microscopy
Kidneys from three mice per group were harvested at the time of sacrifice and transferred into a solution of 0.1 M phosphate buffer (pH 7.4) containing 4% paraformaldehyde, 1% gluteraldehyde. Podocytes were seeded in a 6-well plate with 80% confluency for differentiation. Differentiated cells were washed with 0.1 M Na-cacodylate, and fixed with 2% Paraformaldehyde, 2.5% Glutaraldehyde in 0.1 M Sodium Cacodylate Buffer (pH 7.4). Kidney cortices and podocytes were processed for ultrastructural examination at the Electron Microscopy & Histology Core Facility at the Weill Cornell Medical College. Images were taken by a digital electron microscope (JEOL USA JEM-1400) by one blinded reviewer. Mitochondrial area, matrix density, glomerular basement membrane (GBM) thickness and foot process effacement were quantified using Image J by two blinded investigators. Five representative images were obtained from each group and 20 representative mitochondria were selected for analysis. Foot process effacement was determined as podocyte foot process numbers per μm of GBM in 10 glomerular loops of three mice per group. GBM thickness was measured in 20 different points of three mice per group.
ATP determination assay
The ATP determination assay was performed using the ATP Determination Kit (Molecular Probes) following manufacturer’s protocol. The production of cellular ATP was quantified using the bioluminescence assay in which luciferase converts D-luciferin in the presence of ATP. The production of light is proportional to the amount of ATP in cells. Data were normalized to cell numbers.
OCR assays
OCR was measured using an oxygen electrode in a micro water-jacketed, magnetically stirred chamber set at 37°C (Hansatech) following a method previously described (57). A total of 1 × 106 differentiated podocytes were collected and then resuspended in 500 μl 37°C permeabilized-cell respiration buffer (PRB) containing 0.3 M mannitol, 10 mM KCl, 5 mM MgCl2, 0.5 mM EDTA, 0.5 mM EGTA, 10 mM KH3PO4 pH 7.4 and 1 mg/ml BSA. Resuspened cells were immediately placed into the chamber to measure endogenous respiration. For substrate-driven respiration, cells were suspended in 500 μl PRB supplemented with 10 U/ml hexokinase and 2 mM ADP. After being placed in the chamber, cells were permeabilized with digitonin (3.4 μg/106 cells). Complex I substrates (10 mM glutamate plus 10 mM malate) were then added to the chamber using a Hamilton microsyringe, and the OCR under phosphorylating condition (state 3, coupled) was measured. Oligomycin (1 μM final concentration) was successively added to inhibit ATP synthesis, and non-phosphorylating condition (state 4, uncoupled) was measured. Mitochondrial respiration was uncoupled by 1 μl incremental addition of 0.1 M carbonyl cyanide m-chlorophenylhydrazone (CCCP) to reach maximal oxygen consumption. The highest value observed during CCCP titrations was considered as the maximal OCR. Respiration was inhibited by addition of potassium cyanide (KCN, 0.48 mM final concentration), which is a specific CIV inhibitor. Cell respiration was recorded as nmol O2 consumed for 1 min and normalized to cell numbers. Respiratory control (RC) ratio (state 3/state 4) and spare capacity (maximal-state 3) were calculated.
Mitochondrial membrane potential
MMP was determined by MitoProbe™ TMRM Assay Kit (Invitrogen) according to manufacturer’s protocol. Briefly, 1 × 106 differentiated podocytes were resuspended in 1 ml phosphate-buffered saline (PBS). For the control samples, 2 μl of 50 mM of CCCP was added to induce MMP depolarization. Experimental samples were incubated with 20 nM tetramethylrhodamine, methyl ester (TMRM) for 30 min at 37°C, 5% CO2. Data were acquired on a BD LSR Fortessa flow cytometer (BD Biosciences) with 561 nm excitation and the PE-Texas red filter. For experiments with lipid-modifying compounds, mitochondrial membrane potential was determined using the JC-1 probe (Invitrogen) in a 96-well plate. 10 μg/ml of JC-1 was added to differentiated podocytes for 30 min at 37°C. The JC-1 fluorescence was detected at both 535/595 and 485/535 nm on a SpectraMax M5 plate reader (Molecular Devices) in order to quantify the aggregate versus the monomer signal, respectively. Membrane potential was calculated as the aggregate/monomer ratio.
BN-PAGE analysis of OXPHOS complexes
Blue native-polyacrylamide gel electrophoresis (BN-PAGE) was performed as previously described (31,57). A total of 2 × 106 differentiated podocytes were collected and resuspended in 0.2 ml PBS with 70 μl 8 mg/ml digitonin. Permeabilized podocytes were pelleted and resuspended in 100 μl buffer containing 1.5 M aminocaproic acid, 50 mM Bis-Tris pH 7.0 and proteins were extracted in the presence of either lauryl maltoside (to analyze individual OXPHOS complexes) or digitonin (to analyze OXPHOS complex organization into supercomplexes) at the 1:4 protein-detergent ratios. Native PAGE Novex® 3%–12% Bis-Tris Protein Gels (Life Technologies) were loaded with 150 μg of total cell extracts. After electrophoresis, proteins were transferred to PDVF membranes and were probed with specific antibodies for OXPHOS subunits. The following primary antibodies were used: ATP5A (Abcam, ab14748, 1:500), CORE2 (Abcam, ab14745, 1:500), COX1 (Abcam, ab14705, 1:500), COX5b (Santa Cruz Biotechnology, sc-374 417, 1:500), NDUFA9 (Abcam, ab14713, 1:500), SDHA (Proteintech, 14 865-1-AP, 1:500) and VDAC1 (Abcam, ab14734, 1:1000) was detected in SDS-PAGE as a loading control.
Citrate synthase assay
Citrate synthase assay was performed by measuring the reduction of DTNB (5,5′-dithiobis-(2-nitrobenzoic acid)) as previously described (57). Differentiated podocytes were harvested and permeabilized by mechanical breakage (four freeze–thaw cycles), followed by resuspension in a buffer containing 225 mM mannitol, 75 mM sucrose, 10 mM Tris pH 7.2, 0.1 mM EDTA. A total of 10 μl of cell homogenate was added to 10 mM Tris–HCl, pH 7.5 buffer containing 0.2% Triton X-100, 0.1 mM DTNB, 0.2 mM acetyl-CoA. Baseline absorbance was measured at 412 nm. 0.5 mM oxalacetic acid was added to initiate the reaction, and the increase in absorbance at 412 nm was recorded for 1 min. The linear rate of increase was used to calculate citrate synthase activity as mM of NTB (nitro-thiobenzoate, ε = 13.6 mM−1 cm−1) product generated per minute and normalized to protein concentration of the homogenate.
Confocal immunofluorescence microscop
Podocytes were plated on collagen coated coverslips. After 14 days of differentiation, cells were incubated for 40 min in medium containing 250 nM MitoTracker Red (Invitrogen), washed with PBS three times and fixed in 4% PFA/Sucrose, followed by permeabilization with 0.3% Triton in PBS for 15 min. Coverslips were blocked in Universal blocker for 1 h, and incubated with APOL1 antibody (Proteintech, 11 486-2-AP, rabbit, 1:50) for 2 h at room temperature and rinsed three times with PBS. Secondary antibody (Invitrogen, A-11008, 1:500) was applied at room temperature for 1 h and washed with PBS three times. Coverslips were mounted with Mounting Medium with DAPI (Vectorlabs, H-1200). To measure the colocalization of podocyte marker SYNPO and LD-associated protein PLIN2, glomerular sections embedded in OCT were stained with primary antibodies SYNAPTOPODIN (Santa Cruz Biotechnology, sc-21 537, goat, 1:300) and PLIN2 (Progen, GP40, guinea pig, 1:100), followed by secondary antibodies (Invitrogen, A-11055 and A-11075, 1:500) and Mounting Medium with DAPI (Vectorlabs, H-1200). Images were acquired using Olympus IX81 confocal Microscope coupled with a 60× or 40× oil immersion objective lens and colocalization was analyzed using Fiji/Image J.
Protein-lipid overlay assay
HeLa cells stably infected with lentivirus carrying the 6xHis-tagged APOL1 G0 were cultured in DMEM containing 10% FBS and 1% penicillin/streptomycin at 37°C (19). Polyhistidine-tagged proteins were purified using MagneHis Protein Purification System (Promega). Binding of APOL1-6xHis protein to lipid strips (Echelon Biosciences) was performed following manufacturer’s protocol. The strips were blocked in 3% fatty acid free BSA in TBST at room temperature for 1 h, followed by incubation with 5 μg 6xHis APOL1 protein for 1 h at room temperature. Strips were washed with TBST 10 min for three times. Strips were then probed with APOL1 antibody, secondary antibody and detected by ECL.
Mass spectrometry analysis
Total lipid was extracted from podocytes using chloroform: methanol (2:1). Mass spectrometry was performed in the Miami Integrative Metabolomics Research Center (MIMRC) of the University of Miami as previously described (31). The list of lipids species assessed via mass spectrometry can be found in Supplementary Material, Table S3.
Statistics
All values are presented as mean ± SD. Statistical analysis was performed using Prism GraphPad 7 software. Animals were grouped unblinded, but randomized, and investigators were blinded for the quantification. When comparing between two groups, a two-tailed Student’s t-test was performed. Otherwise results were analyzed using one-way analysis of variance (ANOVA) followed by Tukey’s or Dunnett’s test. A P-value less than 0.05 was considered statistically significant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Supplementary Material
Acknowledgements
A.F. and S.M. are supported by National Institutes of Health grants R01DK117599, R01DK104753 and R01CA227493. A.F. is also supported by Miami Clinical Translational Science Institute (U54DK083912, UM1DK100846, U01DK116101 and UL1TR000460). F.F. is supported by Department of Defense (DoD) Discovery Award (PR180598). M.G. is supported by a predoctoral fellowship of the American Heart Association (18PRE34030042). We would like to thank the Katz family for supporting this study, and the Ophthalmology mass spectrometry facility, vision core grant EY14801 and Miami Integrative Metabolomics Research Center (MIMRC) for mass spectrometry analysis.
Conflict of Interest statement: A.F. and S.M. are inventors on pending or issued patents (PCT/US11/56272, PCT/US12/62594, PCT/US2019/041730, PCT/US2019/032215, PCT/US13/36484 and PCT 62/674897) aimed at diagnosing or treating proteinuric kidney diseases. They stand to gain royalties from the future commercialization of these patents. A.F. is Vice-President of L&F Health LLC and is a consultant for ZyVersa Therapeutics, Inc. ZyVersa Therapeutics, Inc has licensed worldwide rights to develop and commercialize hydroxypropyl-beta-cyclodextrin from L&F Research for the treatment of kidney disease. A.F. is the founder of LipoNexT LLC. S.M. is a consultant for Kintai Therapeutics, Inc and holds equity interest in L&F Research. A.F. and S.M. are supported by Hoffman-La Roche and by Boehringer Ingelheim.
Contributor Information
Mengyuan Ge, Katz Family Division of Nephrology and Hypertension, Department of Medicine, University of Miami Miller School of Medicine, Miami, Florida 33136, USA; Peggy and Harold Katz Family Drug Discovery Center, University of Miami Miller School of Medicine, Miami, Florida 33136, USA.
Judith Molina, Katz Family Division of Nephrology and Hypertension, Department of Medicine, University of Miami Miller School of Medicine, Miami, Florida 33136, USA; Peggy and Harold Katz Family Drug Discovery Center, University of Miami Miller School of Medicine, Miami, Florida 33136, USA.
G Michelle Ducasa, Katz Family Division of Nephrology and Hypertension, Department of Medicine, University of Miami Miller School of Medicine, Miami, Florida 33136, USA; Peggy and Harold Katz Family Drug Discovery Center, University of Miami Miller School of Medicine, Miami, Florida 33136, USA.
Shamroop K Mallela, Katz Family Division of Nephrology and Hypertension, Department of Medicine, University of Miami Miller School of Medicine, Miami, Florida 33136, USA; Peggy and Harold Katz Family Drug Discovery Center, University of Miami Miller School of Medicine, Miami, Florida 33136, USA.
Javier Varona Santos, Katz Family Division of Nephrology and Hypertension, Department of Medicine, University of Miami Miller School of Medicine, Miami, Florida 33136, USA; Peggy and Harold Katz Family Drug Discovery Center, University of Miami Miller School of Medicine, Miami, Florida 33136, USA.
Alla Mitrofanova, Katz Family Division of Nephrology and Hypertension, Department of Medicine, University of Miami Miller School of Medicine, Miami, Florida 33136, USA; Peggy and Harold Katz Family Drug Discovery Center, University of Miami Miller School of Medicine, Miami, Florida 33136, USA; Department of Surgery, University of Miami Miller School of Medicine, Miami, Florida 33136, USA.
Jin-Ju Kim, Katz Family Division of Nephrology and Hypertension, Department of Medicine, University of Miami Miller School of Medicine, Miami, Florida 33136, USA; Peggy and Harold Katz Family Drug Discovery Center, University of Miami Miller School of Medicine, Miami, Florida 33136, USA.
Xiaochen Liu, Katz Family Division of Nephrology and Hypertension, Department of Medicine, University of Miami Miller School of Medicine, Miami, Florida 33136, USA; Peggy and Harold Katz Family Drug Discovery Center, University of Miami Miller School of Medicine, Miami, Florida 33136, USA.
Alexis Sloan, Katz Family Division of Nephrology and Hypertension, Department of Medicine, University of Miami Miller School of Medicine, Miami, Florida 33136, USA; Peggy and Harold Katz Family Drug Discovery Center, University of Miami Miller School of Medicine, Miami, Florida 33136, USA.
Armando J Mendez, Diabetes Research Institute, University of Miami Miller School of Medicine, Miami, Florida 33136, USA.
Santanu Banerjee, Department of Surgery, University of Miami Miller School of Medicine, Miami, Florida 33136, USA.
Shaoyi Liu, Social Profit Network Research Lab, Alexandria Launch Labs, New York, New York 10016, USA.
Hazel H Szeto, Social Profit Network Research Lab, Alexandria Launch Labs, New York, New York 10016, USA.
Myung K Shin, Merck & Company, Inc., Kennilworth, New Jersey 07033, USA.
Maarten Hoek, Merck & Company, Inc., Kennilworth, New Jersey 07033, USA.
Jeffrey B Kopp, Kidney Disease Section, NIDDK, NIH, Bethesda, Maryland 20892, USA.
Flavia Fontanesi, Department of Biochemistry and Molecular Biology, University of Miami Miller School of Medicine, Miami, Florida 33136, USA.
Sandra Merscher, Katz Family Division of Nephrology and Hypertension, Department of Medicine, University of Miami Miller School of Medicine, Miami, Florida 33136, USA; Peggy and Harold Katz Family Drug Discovery Center, University of Miami Miller School of Medicine, Miami, Florida 33136, USA.
Alessia Fornoni, Katz Family Division of Nephrology and Hypertension, Department of Medicine, University of Miami Miller School of Medicine, Miami, Florida 33136, USA; Peggy and Harold Katz Family Drug Discovery Center, University of Miami Miller School of Medicine, Miami, Florida 33136, USA.
Author contributions
M.G. conceived the project, performed the in vitro and in vivo experiments, analyzed the data and wrote the manuscript. J.M., M.D., A.M., J.J.K., X.L. and A.S. assisted with some of the in vitro and in vivo experiments. J.V.S. and A.J.M. designed experiments related to cholesterol determination. S.K.M. and S.B. helped with the mass spectrometry analysis of lipids. H.H.S. and S.L. performed TEM experiments. J.K. established and provided the human urinary podocyte cell lines. M.K.S. and M.H. generated and provided the APOL1 BAC mice. F.F., S.M. and A.F. conceived the project, designed and supervised the study, analyzed the data and edited the manuscript. A.F. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
References
- 1. D'Agati, V.D., Kaskel, F.J. and Falk, R.J. (2011) Focal segmental glomerulosclerosis. N. Engl. J. Med., 365, 2398–2411. [DOI] [PubMed] [Google Scholar]
- 2. Genovese, G., Friedman, D.J., Ross, M.D., Lecordier, L., Uzureau, P., Freedman, B.I., Bowden, D.W., Langefeld, C.D., Oleksyk, T.K., Uscinski Knob, A.L.et al. (2010) Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science, 329, 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Thomson, R., Genovese, G., Canon, C., Kovacsics, D., Higgins, M.K., Carrington, M., Winkler, C.A., Kopp, J., Rotimi, C., Adeyemo, A.et al. (2014) Evolution of the primate trypanolytic factor APOL1. Proc. Natl. Acad. Sci. U. S. A., 111, E2130–E2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lugli, E.B., Pouliot, M., Portela Mdel, P., Loomis, M.R. and Raper, J. (2004) Characterization of primate trypanosome lytic factors. Mol. Biochem. Parasitol., 138, 9–20. [DOI] [PubMed] [Google Scholar]
- 5. Smith, E.E. and Malik, H.S. (2009) The apolipoprotein L family of programmed cell death and immunity genes rapidly evolved in primates at discrete sites of host-pathogen interactions. Genome Res., 19, 850–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Freedman, B.I., Limou, S., Ma, L. and Kopp, J.B. (2018) APOL1-associated nephropathy: a key contributor to racial disparities in CKD. Am. J. Kidney Dis., 72, S8–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cooper, A., Ilboudo, H., Alibu, V.P., Ravel, S., Enyaru, J., Weir, W., Noyes, H., Capewell, P., Camara, M., Milet, J.et al. (2017) APOL1 renal risk variants have contrasting resistance and susceptibility associations with African trypanosomiasis. elife, 6, e25461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kamoto, K., Noyes, H., Nambala, P., Senga, E., Musaya, J., Kumwenda, B., Bucheton, B., Macleod, A., Cooper, A., Clucas, C.et al. (2019) Association of APOL1 renal disease risk alleles with Trypanosoma brucei rhodesiense infection outcomes in the northern part of Malawi. PLoS Negl. Trop. Dis., 13, e0007603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kopp, J.B., Nelson, G.W., Sampath, K., Johnson, R.C., Genovese, G., An, P., Friedman, D., Briggs, W., Dart, R., Korbet, S.et al. (2011) APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J. Am. Soc. Nephrol., 22, 2129–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Limou, S., Nelson, G.W., Kopp, J.B. and Winkler, C.A. (2014) APOL1 kidney risk alleles: population genetics and disease associations. Adv. Chronic Kidney Dis., 21, 426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dummer, P.D., Limou, S., Rosenberg, A.Z., Heymann, J., Nelson, G., Winkler, C.A. and Kopp, J.B. (2015) APOL1 kidney disease risk variants: An evolving landscape. Semin. Nephrol., 35, 222–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Madhavan, S.M., O'Toole, J.F., Konieczkowski, M., Ganesan, S., Bruggeman, L.A. and Sedor, J.R. (2011) APOL1 localization in normal kidney and nondiabetic kidney disease. J. Am. Soc. Nephrol., 22, 2119–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhaorigetu, S., Wan, G., Kaini, R., Jiang, Z. and Hu, C.A. (2008) ApoL1, a BH3-only lipid-binding protein, induces autophagic cell death. Autophagy, 4, 1079–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Horrevoets, A.J., Fontijn, R.D., vanZonneveld, A.J., deVries, C.J., tenCate, J.W. and Pannekoek, H. (1999) Vascular endothelial genes that are responsive to tumor necrosis factor-alpha in vitro are expressed in atherosclerotic lesions, including inhibitor of apoptosis protein-1, stannin, and two novel genes. Blood, 93, 3418–3431. [PubMed] [Google Scholar]
- 15. Monajemi, H., Fontijn, R.D., Pannekoek, H. and Horrevoets, A.J. (2002) The apolipoprotein L gene cluster has emerged recently in evolution and is expressed in human vascular tissue. Genomics, 79, 539–546. [DOI] [PubMed] [Google Scholar]
- 16. Aghajan, M., Booten, S.L., Althage, M., Hart, C.E., Ericsson, A., Maxvall, I., Ochaba, J., Menschik-Lundin, A., Hartleib, J., Kuntz, S.et al. (2019) Antisense oligonucleotide treatment ameliorates IFN-gamma-induced proteinuria in APOL1-transgenic mice. JCI Insight, 4, e126124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nichols, B., Jog, P., Lee, J.H., Blackler, D., Wilmot, M., D'Agati, V., Markowitz, G., Kopp, J.B., Alper, S.L., Pollak, M.R.et al. (2015) Innate immunity pathways regulate the nephropathy gene apolipoprotein L1. Kidney Int., 87, 332–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Duchateau, P.N., Pullinger, C.R., Orellana, R.E., Kunitake, S.T., Naya-Vigne, J., O'Connor, P.M., Malloy, M.J. and Kane, J.P. (1997) Apolipoprotein L, a new human high density lipoprotein apolipoprotein expressed by the pancreas. Identification, cloning, characterization, and plasma distribution of apolipoprotein L. J. Biol. Chem., 272, 25576–25582. [DOI] [PubMed] [Google Scholar]
- 19. Ryu, J.H., Ge, M., Merscher, S., Rosenberg, A.Z., Desante, M., Roshanravan, H., Okamoto, K., Shin, M.K., Hoek, M., Fornoni, A.et al. (2019) APOL1 renal risk variants promote cholesterol accumulation in tissues and cultured macrophages from APOL1 transgenic mice. PLoS One, 14, e0211559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chun, J., Zhang, J.Y., Wilkins, M.S., Subramanian, B., Riella, C., Magraner, J.M., Alper, S.L., Friedman, D.J. and Pollak, M.R. (2019) Recruitment of APOL1 kidney disease risk variants to lipid droplets attenuates cell toxicity. Proc. Natl. Acad. Sci. U. S. A., 116, 3712–3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ma, L., Chou, J.W., Snipes, J.A., Bharadwaj, M.S., Craddock, A.L., Cheng, D., Weckerle, A., Petrovic, S., Hicks, P.J., Hemal, A.K.et al. (2017) APOL1 renal-risk variants induce mitochondrial dysfunction. J. Am. Soc. Nephrol., 28, 1093–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Granado, D., Muller, D., Krausel, V., Kruzel-Davila, E., Schuberth, C., Eschborn, M., Wedlich-Soldner, R., Skorecki, K., Pavenstadt, H., Michgehl, U.et al. (2017) Intracellular APOL1 risk variants cause cytotoxicity accompanied by energy depletion. J. Am. Soc. Nephrol., 28, 3227–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ma, L., Ainsworth, H.C., Snipes, J.A., Murea, M., Choi, Y.A., Langefeld, C.D., Parks, J.S., Bharadwaj, M.S., Chou, J.W., Hemal, A.K.et al. (2020) APOL1 kidney-risk variants induce mitochondrial fission. Kidney Int. Rep., 5, 891–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shah, S.S., Lannon, H., Dias, L., Zhang, J.Y., Alper, S.L., Pollak, M.R. and Friedman, D.J. (2019) APOL1 kidney risk variants induce cell death via mitochondrial translocation and opening of the mitochondrial permeability transition pore. J. Am. Soc. Nephrol., 30, 2355–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schlondorff, J., Del Camino, D., Carrasquillo, R., Lacey, V. and Pollak, M.R. (2009) TRPC6 mutations associated with focal segmental glomerulosclerosis cause constitutive activation of NFAT-dependent transcription. Am. J. Physiol. Cell Physiol., 296, C558–C569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang, Y., Jarad, G., Tripathi, P., Pan, M., Cunningham, J., Martin, D.R., Liapis, H., Miner, J.H. and Chen, F. (2010) Activation of NFAT signaling in podocytes causes glomerulosclerosis. J. Am. Soc. Nephrol., 21, 1657–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pedigo, C.E., Ducasa, G.M., Leclercq, F., Sloan, A., Mitrofanova, A., Hashmi, T., Molina-David, J., Ge, M., Lassenius, M.I., Forsblom, C.et al. (2016) Local TNF causes NFATc1-dependent cholesterol-mediated podocyte injury. J. Clin. Invest., 126, 3336–3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Campbell, K.N. and Tumlin, J.A. (2018) Protecting podocytes: a key target for therapy of focal segmental glomerulosclerosis. Am. J. Nephrol., 47, 14–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rao, A. (2009) Signaling to gene expression: calcium, calcineurin and NFAT. Nat. Immunol., 10, 3–5. [DOI] [PubMed] [Google Scholar]
- 30. Meyrier, A. (2005) Treatment of focal segmental glomerulosclerosis. Expert. Opin. Pharmacother., 6, 1539–1549. [DOI] [PubMed] [Google Scholar]
- 31. Ducasa, G.M., Mitrofanova, A., Mallela, S.K., Liu, X., Molina, J., Sloan, A., Pedigo, C.E., Ge, M., Santos, J.V., Hernandez, Y.et al. (2019) ATP-binding cassette A1 deficiency causes cardiolipin-driven mitochondrial dysfunction in podocytes. J. Clin. Invest., 129, 3387–3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Imasawa, T., Obre, E., Bellance, N., Lavie, J., Imasawa, T., Rigothier, C., Delmas, Y., Combe, C., Lacombe, D., Benard, G.et al. (2017) High glucose repatterns human podocyte energy metabolism during differentiation and diabetic nephropathy. FASEB J., 31, 294–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shutt, T.E. and McBride, H.M. (2013) Staying cool in difficult times: mitochondrial dynamics, quality control and the stress response. Biochim. Biophys. Acta, 1833, 417–424. [DOI] [PubMed] [Google Scholar]
- 34. Vincent, A.E., Ng, Y.S., White, K., Davey, T., Mannella, C., Falkous, G., Feeney, C., Schaefer, A.M., McFarland, R., Gorman, G.S.et al. (2016) The Spectrum of mitochondrial ultrastructural defects in mitochondrial myopathy. Sci. Rep., 6, 30610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vanwalleghem, G., Fontaine, F., Lecordier, L., Tebabi, P., Klewe, K., Nolan, D.P., Yamaryo-Botte, Y., Botte, C., Kremer, A., Burkard, G.S.et al. (2015) Coupling of lysosomal and mitochondrial membrane permeabilization in trypanolysis by APOL1. Nat. Commun., 6, 8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Panday, N., Benz, J., Blum-Kaelin, D., Bourgeaux, V., Dehmlow, H., Hartman, P., Kuhn, B., Ratni, H., Warot, X. and Wright, M.B. (2006) Synthesis and evaluation of anilinohexafluoroisopropanols as activators/modulators of LXRalpha and beta. Bioorg. Med. Chem. Lett., 16, 5231–5237. [DOI] [PubMed] [Google Scholar]
- 37. Paradies, G., Paradies, V., De Benedictis, V., Ruggiero, F.M. and Petrosillo, G. (2014) Functional role of cardiolipin in mitochondrial bioenergetics. Biochim. Biophys. Acta, 1837, 408–417. [DOI] [PubMed] [Google Scholar]
- 38. Blazer, A., Wang, B., Simpson, D., Kirchhoff, T., Heffron, S., Clancy, R.M., Heguy, A., Ray, K., Snuderl, M. and Buyon, J.P. (2017) Apolipoprotein L1 risk variants associate with prevalent atherosclerotic disease in African American systemic lupus erythematosus patients. PLoS One, 12, e0182483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen, T.K., Appel, L.J., Grams, M.E., Tin, A., Choi, M.J., Lipkowitz, M.S., Winkler, C.A. and Estrella, M.M. (2017) APOL1 risk variants and cardiovascular disease: results from the AASK (African American study of kidney disease and hypertension). Arterioscler. Thromb. Vasc. Biol., 37, 1765–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Parsa, A., Kao, W.H., Xie, D., Astor, B.C., Li, M., Hsu, C.Y., Feldman, H.I., Parekh, R.S., Kusek, J.W., Greene, T.H.et al. (2013) APOL1 risk variants, race, and progression of chronic kidney disease. N. Engl. J. Med., 369, 2183–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ng, D.K., Robertson, C.C., Woroniecki, R.P., Limou, S., Gillies, C.E., Reidy, K.J., Winkler, C.A., Hingorani, S., Gibson, K.L., Hjorten, R.et al. (2017) APOL1-associated glomerular disease among African-American children: a collaboration of the chronic kidney disease in children (CKiD) and nephrotic syndrome study network (NEPTUNE) cohorts. Nephrol. Dial. Transplant., 32, 983–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Beckerman, P., Bi-Karchin, J., Park, A.S., Qiu, C., Dummer, P.D., Soomro, I., Boustany-Kari, C.M., Pullen, S.S., Miner, J.H., Hu, C.A.et al. (2017) Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat. Med., 23, 429–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kumar, V., Paliwal, N., Ayasolla, K., Vashistha, H., Jha, A., Chandel, N., Chowdhary, S., Saleem, M.A., Malhotra, A., Chander, P.N.et al. (2019) Disruption of APOL1-miR193a Axis induces disorganization of podocyte actin cytoskeleton. Sci. Rep., 9, 3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bruggeman, L.A., Wu, Z., Luo, L., Madhavan, S.M., Konieczkowski, M., Drawz, P.E., Thomas, D.B., Barisoni, L., Sedor, J.R. and O'Toole, J.F. (2016) APOL1-G0 or APOL1-G2 transgenic models develop preeclampsia but not kidney disease. J. Am. Soc. Nephrol., 27, 3600–3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Freedman, B.I., Langefeld, C.D., Turner, J., Nunez, M., High, K.P., Spainhour, M., Hicks, P.J., Bowden, D.W., Reeves-Daniel, A.M., Murea, M.et al. (2012) Association of APOL1 variants with mild kidney disease in the first-degree relatives of African American patients with non-diabetic end-stage renal disease. Kidney Int., 82, 805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mitrofanova, A., Molina, J., Varona Santos, J., Guzman, J., Morales, X.A., Ducasa, G.M., Bryn, J., Sloan, A., Volosenco, I., Kim, J.J.et al. (2018) Hydroxypropyl-beta-cyclodextrin protects from kidney disease in experimental Alport syndrome and focal segmental glomerulosclerosis. Kidney Int., 94, 1151–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Youle, R.J. and van derBliek, A.M. (2012) Mitochondrial fission, fusion, and stress. Science, 337, 1062–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cribbs, J.T. and Strack, S. (2007) Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep., 8, 939–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wan, G., Zhaorigetu, S., Liu, Z., Kaini, R., Jiang, Z. and Hu, C.A. (2008) Apolipoprotein L1, a novel Bcl-2 homology domain 3-only lipid-binding protein, induces autophagic cell death. J. Biol. Chem., 283, 21540–21549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Uzureau, S., Lecordier, L., Uzureau, P., Hennig, D., Graversen, J.H., Homble, F., Mfutu, P.E., Oliveira Arcolino, F., Ramos, A.R., La Rovere, R.M.et al. (2020) APOL1 C-terminal variants may trigger kidney disease through interference with APOL3 control of Actomyosin. Cell Rep., 30, 3821–3836e3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Iyer, S.S., He, Q., Janczy, J.R., Elliott, E.I., Zhong, Z., Olivier, A.K., Sadler, J.J., Knepper-Adrian, V., Han, R., Qiao, L.et al. (2013) Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity, 39, 311–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lan, X., Jhaveri, A., Cheng, K., Wen, H., Saleem, M.A., Mathieson, P.W., Mikulak, J., Aviram, S., Malhotra, A., Skorecki, K.et al. (2014) APOL1 risk variants enhance podocyte necrosis through compromising lysosomal membrane permeability. Am. J. Physiol. Renal Physiol., 307, F326–F336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jin, J., Sison, K., Li, C., Tian, R., Wnuk, M., Sung, H.K., Jeansson, M., Zhang, C., Tucholska, M., Jones, N.et al. (2012) Soluble FLT1 binds lipid microdomains in podocytes to control cell morphology and glomerular barrier function. Cell, 151, 384–399. [DOI] [PubMed] [Google Scholar]
- 54. Sakairi, T., Abe, Y., Kajiyama, H., Bartlett, L.D., Howard, L.V., Jat, P.S. and Kopp, J.B. (2010) Conditionally immortalized human podocyte cell lines established from urine. Am. J. Physiol. Renal Physiol., 298, F557–F567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shankland, S.J., Pippin, J.W., Reiser, J. and Mundel, P. (2007) Podocytes in culture: past, present, and future. Kidney Int., 72, 26–36. [DOI] [PubMed] [Google Scholar]
- 56. Mizoguchi, T., Edano, T. and Koshi, T. (2004) A method of direct measurement for the enzymatic determination of cholesteryl esters. J. Lipid Res., 45, 396–401. [DOI] [PubMed] [Google Scholar]
- 57. Barrientos, A., Fontanesi, F. and Diaz, F. (2009) Evaluation of the mitochondrial respiratory chain and oxidative phosphorylation system using polarography and spectrophotometric enzyme assays. Curr. Protoc. Hum. Genet., Chapter 19, Unit19 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.