

Abstract
Huntington disease (HD) is a neurodegenerative condition with prominent motor (including oculomotor), cognitive, and psychiatric effects. While neuropsychological deficits are present in HD, motor impairments may impact performance on neuropsychological measures, especially those requiring a speeded response, as has been demonstrated in multiple sclerosis and schizophrenia. The current study is the first to explore associations between oculomotor functions and neuropsychological performance in HD. Participants with impaired oculomotor functioning performed worse than those with normal oculomotor functioning on cognitive tasks requiring oculomotor involvement, particularly on psychomotor speed tasks, controlling for covariates. Consideration of oculomotor dysfunction on neuropsychological performance is critical, particularly for populations with motor deficits.
Keywords: Huntington disease, PREDICT-HD, oculomotor functioning, neuropsychology, processing speed
Huntington disease (HD) is an autosomal dominant progressive neurodegenerative disorder with a trinucleotide cytosine-adenine-guanine (CAG) expansion in the gene coding for huntingtin, found on chromosome 4 (The Huntington’s Disease Collaborative Research Group, 1993). Increased CAG repeat length is associated with earlier age of HD diagnosis (Duyao et al., 1993; Lee et al., 2012; Stine et al., 1993). HD onset typically occurs in adulthood, although persons with the gene expansions can be affected at any time, with documented onset ages between one and 80 years (Walker, 2007).
While HD patients can present with a range of cognitive, behavioral, and functional impairments, the presence of unequivocal motor signs such as chorea, dystonia, rigidity, bradykinesia, and gait instability is required for a clinical diagnosis of HD (Huntington Study Group, 1996). The main neuroanatomical feature of HD is atrophy of the caudate nucleus and putamen (Folstein, 1989; Gutekunst, Norflus, & Hersch, 2002; Rubinsztein, 2003; Schwarcz & Shoulson, 1987; Tobin, 1990; Vonsattel & DiFiglia, 1998). In genetically confirmed but non-clinically diagnosed patients, subtle motor abnormalities, including saccade initiation, was associated with smaller striatal volumes using volumetric MRI measurements (Biglan et al., 2009). However, motor abnormalities can manifest prior to clinical diagnosis. The stage known as prodromal HD is the period in which the patient may exhibit some signs, but not sufficient in severity or quantity to meet criteria for a formal clinical diagnosis (Biglan et al., 2009; Paulsen et al., 2006; Paulsen et al., 2008). Research has established that HD signs can precede formal motor diagnosis by as much as 15 years (Paulsen et al., 2006; Paulsen et al., 2008).
Chorea and oculomotor impairment are among the first signs that discriminate individuals with CAG expansions in the prodromal phase from non-gene-expanded individuals (Biglan et al., 2009; Paulsen, 2010). Oculomotor functions are classified into several different categories (Leigh & Zee, 1999) and typically include ocular pursuit and saccades, both of which are commonly affected in prodromal and early HD patients. Ocular pursuit and saccades involve bringing or holding images on the fovea and stabilizing one’s gaze (Leigh & Zee, 1999). Oculomotor impairments are found consistently in diagnosed HD patients (Blekher et al., 2004; Blekher et al., 2006; Dursun, Burke, Andrews, Mlynik-Szmid, & Reveley, 2000; Golding, Danchaivijitr, Hodgson, Tabrizi, & Kennard, 2006; Hicks, Robert, Golding, Tabrizi, & Kennard, 2008; Lasker & Zee, 1997; Tabrizi et al., 2009; Turner et al., 2011).
Cognitive deficits also are well-documented in prodromal and diagnosed HD patients (Paulsen, Smith, & Long, 2013) and in prodromal HD it may be a stronger indicator of future disease progression than motor signs. In a sample of prodromal HD participants, cognitive dysfunction accounted for a greater amount of variance (34.0%) relative to motor abnormalities (11.7%) when predicting a probability of diagnosis within 5 years (Stout et al., 2011). Cognitive impairments in prodromal HD participants include difficulties with psychomotor/processing speed, episodic memory, visuospatial processing, and executive functioning (Harrington et al., 2012; Paulsen et al., 2013; Stout et al., 2011). This pattern of deficits is similar to that observed in clinically diagnosed HD patients, where established deficits in attention, verbal fluency, processing speed, executive functioning, memory, and visuospatial abilities are well described (Beglinger, et al., 2005; Montoya et al., 2006; Paulsen & Conybeare, 2005; Stout & Johnson, 2005).
Assessment of many cognitive abilities relies on a motor response, including rapid written responses and eye movements. While intact motor abilities are required to complete neuropsychological tasks normally, these functions may not reflect the cognitive ability intended to be measured by the task. For example, tasks that assess processing speed tend also to include a motor component (e.g., visual scanning, providing rapid written or verbal responses). As such, motor deficits in persons with HD may interfere with performance on select cognitive measures. That is, in addition to assessing the primary domain of interest, these measures may unavoidably be capturing dysfunction in secondary domains, such as motor impairments.
While not thoroughly explored in HD patients, associations between oculomotor functions and neuropsychological performance have been documented in other clinical populations. For example, a sample of medicated schizophrenia patients, who are known to have documented oculomotor changes, showed significantly greater associations between oculomotor pursuit speed and most neuropsychological measures of processing speed, executive functioning, and motor speed relative to a matched sample of healthy controls (Radant, Claypoole, Wingerson, Cowley, & Roy-Byrne, 1997). Further, in patients with multiple sclerosis (MS), preliminary results show patients with slower eye movements on an eye tracking test demonstrated associations with neuropsychological tasks that included a visuomotor component (Glusman et al., 2013).
The current study is the first study to examine relationships between a global qualitative measure of oculomotor functioning and neuropsychological test performances. Specifically, we examined the impact of oculomotor abnormalities on neuropsychological performance in a sample of participants with prodromal HD, adjusting for other motor impairments and additional demographic covariates. We explored the effects of oculomotor functioning (ocular pursuit, saccade initiation, and saccade velocity) on neuropsychological measures in participants with prodromal HD and non-gene-expanded participants. We expected significant partial associations between oculomotor abilities and performance on timed neuropsychological tasks that involve speeded visual scanning, but modest or no relationships between oculomotor tasks and untimed cognitive tasks.
Method
Participants
Data in this study were collected from N = 1054 participants in the Neurobiological Predictors of Huntington’s Disease (PREDICT-HD) study (Paulsen et al., 2008). There were 821 gene-expanded participants and 233 non-gene-expanded at 32 worldwide sites. All participants had completed genetic testing for HD prior to (and independent from) study enrollment. Gene expansion status and CAG repeat length were confirmed at the initial study visit. Participants were considered to have prodromal HD if their CAG repeat expansion was equal to or greater than 36 and have a diagnostic confidence level of < 4 (Huntington Study Group, 1996). Those with repeats less than 36 served as gene mutation negative comparisons (controls). At study enrollment, participants were required to be 18 years of age or older and could not yet be diagnosed with manifest HD according to traditional motor criteria. Exclusion criteria included a history of a significant developmental cognitive disorder, other central nervous system disease or injury, evidence of an unstable medical or psychiatric illness (including substance abuse), a pacemaker or metallic implants, or having taken prescribed antipsychotic medication in the last six months or phenothiazine derivative antiemetic medication in the three months prior to enrollment. All participants provided informed consent (reviewed and approved by the Institutional Review Board at their respective sites) and were treated in accordance with the ethical standards of the American Psychological Association.
Progression groups.
It is necessary to index the stage of progression at study entry in order to make proper inferences. Progression groups were based on the CAG-Age Product (CAP) score (Zhang et al., 2011), which is computed as CAP = (Age at entry) × (CAG − 33.66). CAP is similar to the “disease burden” score of Penney, Vonsattel, MacDonald, Gusella, and Myers (1997) and reflects the cumulative toxicity of mutant huntingtin at the time of study entry. There are CAP cutoffs that can be used to form progression groups (Zhang et al., 2011). The Low group had CAP < 290, the Medium group had 290 ≤ CAP ≤ 368, and the High group had CAP > 368. Based on this stratification, the estimated time to diagnosis for each CAP group was > 12.78 years for the Low group, between 12.78 and 7.59 for the Medium group, and < 7.59 years for the High group.
Mean years in the study was 4.42 (SD = 2.79), with a range of 1 to 10. Furthermore, 9.59% of the sample had only one year in the study, 14.79% had two years, and 75.63% had three or more years. Table 1 shows demographic information by CAP group.
Table 1:
Demographic Information by CAP Group
| Control | Low | Medium | High | |
|---|---|---|---|---|
| N | 233 | 215 | 288 | 318 |
| Femalea | 148 (63.52%) | 146 (67.91%) | 188 (65.28%) | 187 (58.80%) |
| Age | 44.36 (11.41) | 34.98 (7.92) | 41.67 (9.56) | 44.93 (10.09) |
| Educ | 14.87 (2.56) | 14.57 (2.44) | 14.54 (2.61) | 14.33 (2.75) |
| Years in study | 4.39 (2.29) | 4.41 (2.68) | 4.64 (2.61) | 4.98 (2.52) |
| CAG | 20.27 (3.49) | 40.91 (1.62) | 42.02 (2.04) | 43.58 (2.74) |
| CAP | NA | 243.97 (34.55) | 330.50 (23.05) | 423.19 (51.25) |
Note. Educ = years of education
Percentage is based on group total.
Procedure
All participants underwent comprehensive baseline screening and annual visits at local study sites. Visits included blood draw, cognitive testing, a neurological evaluation, completion of psychiatric and psychological questionnaires, and brain MRI. All cognitive data was sent to a centralized location for quality control, including double or triple scoring of all protocols and double data entry to ensure accuracy.
Measures
The motor assessment portion of the Unified Huntington’s Disease Rating Scale (UHDRS) (Huntington Study Group, 1996) was rated by a trained motor examiner as part of a neurologic examination. The motor assessment portion consists of the subdomains of oculomotor examination (six items), chorea (seven items), dystonia (five items), bradykinesia (11 items), and rigidity (two items) (Marder et al., 2000). The primary UHDRS measures of interest for the current study are the oculomotor items of ocular pursuit (horizontal and vertical), saccade initiation (horizontal and vertical), and saccade velocity (horizontal and vertical). Ocular pursuit measures smooth eye movements while the participant pursues a stimulus. Saccade initiation examines the amount of time it takes a participant to initiate an eye movement to a designated stimulus. Saccade velocity indicates the strength and speed of a shifting gaze to a stimulus. Each task was rated for horizontal and vertical performance using a scale ranging from 0 (normal) to 4 (most severe impairment). The six items were summed to derive a total oculomotor score (possible range of 0 to 24; see Table 2 for distribution of oculomotor variable by group). Similar composites were computed for the other four motor domains of bradykinesia, rigidity, chorea, and dystonia (see Long et al., 2013, for additional details).
Table 2:
Demographics for oculomotor and non-oculomotor functioning by CAP group
| Control | Low | Medium | High | |
|---|---|---|---|---|
| Oculomotor functioning | 0.75 (1.43) | 1.07 (1.83) | 1.59 (2.32) | 2.96 (3.28) |
|
Non-oculomotor functioning |
1.89 (2.22) | 2.44 (3.39) | 4.03 (4.53) | 7.67 (7.53) |
A number of commonly used neuropsychological measures with and without visual motor or fine motor components were used in the current analyses. Tasks that involve timed visual scanning and a rapid motor response included Symbol Digit Modalities Test (total correct; Smith, 1991), a measure of processing speed and working memory measured through pairing symbols with digits; Trail Making Test A (time in seconds; Reitan, 1958), a measure of psychomotor speed in which participants connect circles containing numbers in ascending order; Stroop Color Naming, Word Reading, and Interference (total correct responses in 45 seconds; Stroop, 1935), measures of processing speed and executive functioning in which participants are asked to rapidly name colors, read words, and name color-words provided linearly and written in an incongruent color (e.g., the word “green” written in blue ink); and Buttons (the time between the release of one button and the depression of the next; Georgiou-Karistianis et al., 2014)1, an experimental computerized measure of cued movement sequencing.
Tasks with minimal motor speed demands included the Wechsler Abbreviated Scale of Intelligence (WASI) Matrix Reasoning (total correct; Wechsler, 1999), an untimed visual abstract reasoning task, and Hopkins Verbal Learning Test-Revised (HVLT) Delayed Recall (total words recalled after a delay; Brandt & Benedict, 2001), a measure of verbal list memory.
Statistical analysis.
Linear mixed effects regression (LMER; Verbeke & Molenberghs, 2013) was used to address the primary research question of whether oculomotor impairment predicted cognitive performance over time controlling for the four other domains of motor impairment (chorea, dystonia, bradykinesia, rigidity), baseline progression (as index by CAP groups), and the demographic variables of gender, years of education, and age at study entry. The time metric for the LMER analysis was duration, defined as the number of years in the study since entry. CAP group membership was represented by dummy codes (1 = in-group, 0 = otherwise), and each appeared in the model as a main effect and as an interaction term with duration (i.e., the product of a dummy code and years in study). The main effects represent overall CAP group level differences and the interactions represent CAP group differences in linear change over time. All motor impairment variables were time-varying predictors specified as main effects (overall level differences) and time interactions (slope differences). Based on previous research (see e.g., Paulsen et al., 2013), only the main effects of the demographic variables were specified. Individual variability and dependency due to repeated measures were modeled by random intercepts and slopes in all models. Maximum likelihood (ML) was used for estimation, which yields unbiased parameter estimates with missing data under the widely applicable assumption of an ignorable missing data mechanism (Little & Rubin, 2002). Additional details of the LMER models are presented in the appendix.
Eight outcome variables were examined separately: Symbol Digit Modalities Test (SDMT), Trail Making Test A (time to completion), Buttons Condition 1 Mean Movement Time, Stroop Color Naming Total, Stroop Word Reading Total, Stroop Interference Total, WASI Matrix Reasoning, and HVLT Delayed Recall. For each outcome, the following LMER models were fit: (1) a model with no oculomotor effects, (2) a model with main effects for oculomotor functioning, and (3) a model with main effects and time interactions (slope differences) for oculomotor functioning. All models were adjusted for non-oculomotor functioning (chorea, dystonia, bradykinesia, rigidity), CAP group, and demographic variables.
Models were compared in pairs (Model 1 vs. Model 2, Model 2 vs. Model 3) using a one degree of freedom (df) likelihood ratio test (LRT). The null hypothesis evaluated by the LRT is that the simpler model (e.g., Model 1) has an equal statistical fit to the more complex model (e.g., Model 2). Rejection of the null hypothesis indicates the more complex model should be adopted. Thus, the first LRT tested for the need of the oculomotor main effect, and the second LRT tested for the need of the oculomotor by time interaction.
Results
Results of the LMER model comparisons are shown in Table 3, along with the number of participants (N) and the number of time points (N*; i.e., all repeated measures among all participants). Missing data varied by variable due to timing of administration and other site and protocol perturbations.
Table 3:
Linear mixed model regression results by cognitive variable
| LRT |
||||
|---|---|---|---|---|
| Variable | N | N* | 1 vs 2 (Main Effect) |
2 vs 3 (Inter- action) |
| Symbol Digit Total | 1050 | 4114 | 12.13*** | 0.77 |
| Trail Making Test A | 1047 | 2601 | 21.11*** | 15.85*** |
| Buttons | 1041 | 2560 | 38.82*** | 0.88 |
| Stroop Color Reading | 1050 | 4104 | 18.1*** | 2.26 |
| Stroop Word Reading | 1050 | 4113 | 16.7*** | 1.99 |
| Stroop Interference | 1050 | 4107 | 18.64*** | 0.89 |
| WASI Matrix Reasoning | 880 | 1412 | 0.50 | 1.94 |
| HVLT Delayed Recall | 1049 | 2092 | 12.13*** | 2.41 |
Note. N = sample size;
= number of data points; LRT = likelihood ratio test (each test based on df = 1);
p < .001; Symbol Digit Total = Total of Symbol Digit Modalities Test; WASI = Wechsler Abbreviated Scale of Intelligence; HVLT = Hopkins Verbal Learning Test-Revised.
Table 2 shows that all outcomes except WASI Matrix Reasoning had a statistically significant main effect for oculomotor impairments (second to last column). The main effect was such that higher oculomotor scores were associated with worse performance. The last column of Table 3 shows that the oculomotor by time interaction was significant for Trail Making Test A (X2(1) = 15.85, p < .001). The significant interaction for Trail Making Test A indicates the rate of change over time was conditional on oculomotor impairments, controlling for the other variables. The nature of the effect is illustrated in Figure 1 that shows fitted curves based on Model 3 paneled by CAP group. The graph depicts fitted curves for a zero oculomotor score, representing no impairment (“none”), and a score of 3, presenting moderate impairment (“moderate”); the dichotomy was used only for graphing purposes with the continuous distribution used in the analysis (see above). These values were chosen to be the 50th and 75th percentile, respectively, of the empirical oculomotor distribution (note that 50% of the sample observations represented “normal”). In each panel, the blue line indicates normal oculomotor functioning (“none”), and the red line indicates moderately impaired functioning (note the continuous variable was used for the analysis and the binary grouping is used only for graphing). As the figure shows, there was an expected CAP group effect, such that time to completion increased as number of visits in the individual progression groups increased. The oculomotor effect is seen in each panel with the moderately impaired curve having a greater slope than the normal curve. Thus, impaired oculomotor functioning was associated with a faster rate of deterioration, controlling for non-oculomotor functioning and the covariates (see above). Detailed results not shown indicate that the rate of change for the unimpaired in the high CAP group was a decrease of 0.08 seconds per year on Trail Making Test A, whereas the impaired had an increase of 0.53 seconds per year.
Figure 1.
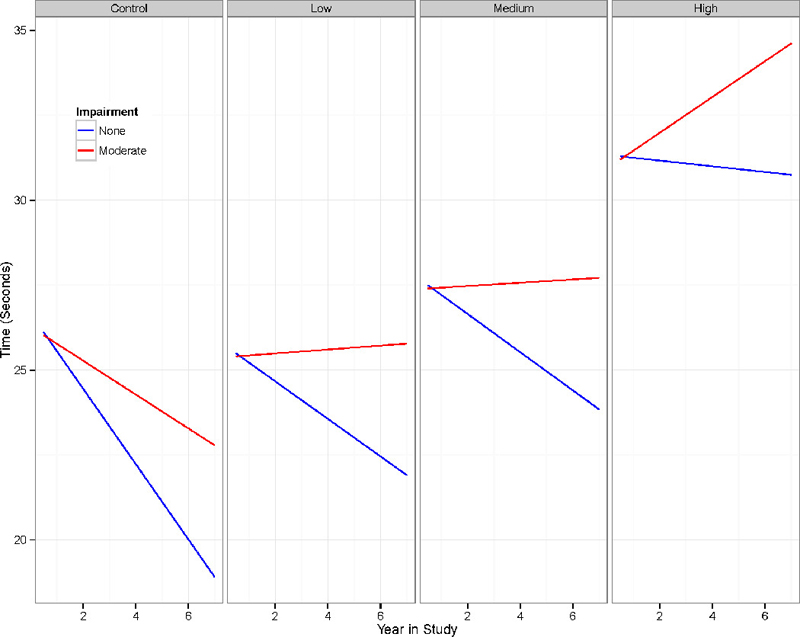
Fitted Trail Making Test A curves for two hypothetical impairment groups paneled by progression (one gene-negative and three gene-positive).
Discussion
The current study sought to examine the impact of oculomotor abnormalities on neuropsychological performance in a sample of individuals with prodromal HD. We found that on a test requiring quick visual scanning (Trail Making A), disease progression was related to oculomotor functioning. That is, oculomotor predicted both overall level (main effect) and change over time (interaction), such that greater oculomotor signs were associated with worse performance on Trail Making Test A (Figure 1). The association of greater oculomotor impairments with deterioration in Trail Making Test A performance over time occurred for all progression groups (Low, Medium, and High CAP groups). Notably, our finding was significant despite controlling for severity of non-oculomotor dysfunction.
Further, we found expected associations between oculomotor dysfunction and cognitive deficits on tasks that required rapid oculomotor activity. That is, the main effects indicate that for all the variables except WASI Matrix Reasoning, worse cognitive functioning was associated with greater oculomotor impairments. This is consistent with expectations as most neuropsychological tasks that showed an association with oculomotor functioning required oculomotor involvement/speeded responses.
Also consistent with expectations, oculomotor performance was not associated with a visual abstract reasoning task that requires no rapid oculomotor response (WASI Matrix Reasoning). This absence of associations between oculomotor effects and an untimed visual reasoning task provides some support for the sensitivity of our study. That is, effects were found on the expected tasks that required oculomotor activity, and a lack of an association was found on the aforementioned task that did not require a rapid oculomotor response.
We also had an unexpected finding of oculomotor effects on a verbal learning task (HVLT delayed recall). Associations between cognitive performance and oculomotor functioning may reflect overall motor dysfunction or disease burden rather than oculomotor abilities per se. However, the absence of an association between untimed visual reasoning and oculomotor performance, as well as the fact that we controlled for disease burden and non-oculomotor function, argues against these generalized motor effects (although the reason for this association with the verbal memory task remains unclear).
As previously discussed, there was a statistically significant decline in Trail Making Test A performance over time as oculomotor impairments worsened. That is, in Figure 1, it is evident that those without oculomotor impairment completed the Trail Making Test A faster after each evaluation (lower completion times), whereas those with impairment did not improve their completion times after each evaluation. While the size of the Trail Making Test A decline may appear minimal in regard to clinical significance, it may have important research implications. Though 0.81 seconds per year may not appear to be a very strong effect in some contexts, some clinical trials in HD rely on cognitive outcomes (e.g., Kieburtz et al., 2010), and even very small changes may suggest important treatment effects, particularly if the treatment group has an increasing slope (i.e., improved performance) while the untreated group has decreasing slope. This may be especially true in trials involving degenerative conditions like HD, where the ability to measure stability or improvement in a condition that progresses over time can be particularly challenging. As such, any degree of change (or lack thereof) that is not accurately accounted for by the formal intervention can result in erroneous conclusions about the utility of the intervention.
The current study has limitations. The gene negative controls also included participants with oculomotor impairment of unclear etiologies and gene positive participants also had a wide range of oculomotor performance. Thus, while our controls are confirmed to be negative for HD and did not meet exclusion criteria of other central nervous system disease or injury, we could not rule out the possibility of some of our controls having other early neurologic conditions that had not been diagnosed (though this suggests that our results are conservative). In addition, 75% of participants with abnormal impairments had a total oculomotor score of 5 or less, indicating the overall dysfunction of our sample was somewhat mild. Therefore, our results may not generalize to a more severely oculomotor impaired sample. Further, the sample is comprised of participants with gene positive prodromal HD and gene negative controls, all individuals who agreed to participate in this longitudinal study (though the current sample represents the largest group of prodromal HD participants ever studied). There is some evidence that HD patients who choose to undergo genetic testing may have better coping strategies than those who do not (Codori, Hanson, & Brandt, 1994). Therefore, we are uncertain if our findings would generalize to HD patients who would not elect to undergo genetic testing, nor opt to participate in studies of this kind. Our results revealed an association between oculomotor function and a verbal memory task, a finding which was not expected (though consistent with our hypothesis, we found no association between oculomotor function and non-verbal reasoning). While we did control for disease burden in our analyses, this raises some question as to whether our results in part are capturing overall disease burden. Finally, the longitudinal nature of the study also raises concern about practice effects on all cognitive tasks, which may have minimized the measurable impact of cognitive change over time. Future studies may consider also looking at other motor effects on cognitive dysfunction such as dysarthria, which was not conducted in the current study given the low variability of this motor sign in our sample.
The current study was the first to explore associations between oculomotor functions and cognitive performance in HD. However, effects of oculomotor performance on cognitive tasks have been documented in schizophrenia patients, in which associations between oculomotor functioning and gross motor speed, visual scanning, and executive functioning have been observed (Radant et al., 1997), and MS patients in which associations were found between oculomotor functioning and neuropsychological tasks that included a visuomotor component (Glusman et al., 2013). In our study, increases in oculomotor impairment was associated with a faster decline on a cognitive task requiring rapid visual scanning, and we found an association between oculomotor impairment and neuropsychological tasks that require a rapid visual response. The clinical implications of this decline depend on the extent of oculomotor impairment and the length of time over which individuals are examined. Researchers conducting clinical trials and other research studies, particularly in samples with concern about oculomotor involvement, should be aware of oculomotor effects when measuring cognitive performance over time.
Acknowledgments
We thank the PREDICT-HD sites, the study participants, the National Research Roster for Huntington Disease Patients and Families, the Huntington’s Disease Society of America and the Huntington Study Group.
This work was supported by the National Institutes for Health, National Institute of Neurological Disorders and Stroke under Grant 5R01NS040068 awarded to Jane Paulsen; CHDI Foundation, Inc. under Grants A6266 and A2015 awarded to Jane Paulsen; and Cognitive and Functional Brain Changes in Preclinical Huntington’s Disease (HD) under Grant R01NS054893 awarded to Jane Paulsen.
Appendix
Linear mixed effects regression (LMER)
Suppose yij is the cognitive variable score for the ith participant (i = 1, …, N) at the jth time point (j = 1, …, ni). Then the LMER model for the analysis can be written in matrix notation as
| , | (A1) |
where yi is the ni × 1 vector of cognitive variable scores over time; is the ni × 2 duration design matrix, with the first column a vector of 1s and the second column a vector of duration values indicating the year in the study at which the measurement was taken (i.e., yearij); is the ni × 6 CAP design matrix containing time-invariant dummy variables for the CAP groups (1 = in the group, 0 = otherwise) and the product of the dummy variables and duration (CAP by time interaction); is the ni × 8 motor design matrix containing the main effect of each of the four motor domains other than oculomotor (bradykinesia, rigidity, dystonia, chorea), and their product with duration; is the ni × 3 demographics design matrix with column values of gender, years of education, and age at entry; is the ni × 2 oculomotor design matrix with the first column being the main effect of oculomotor and the second column being the product with duration, with a row of the relevant matrix product being. The fixed effects vectors are, the random effects vector is ai, and the random error vector is. We make the typical assumptions,. Equation (A1) is Model 3 discussed in the text (see Table 3), Model 2 omits leaving the oculomotor main effect, and Model 1 omits all oculomotor effects. Thus, the likelihood ratio test (LRT) of Model 1 vs Model 2 is a test of H0: η1 = 0 (i.e., no oculomotor main effect), and the LRT of Model 2 vs Model 3 is a test of H0: η2 = 0 (i.e., no oculomotor by time effect).
PREDICT-HD Investigators, Coordinators, Motor Raters, Cognitive Raters
Isabella De Soriano, Courtney Shadrick, and Amanda Miller (University of Iowa, Iowa City, Iowa, USA);
Edmond Chiu, Joy Preston, Anita Goh, Stephanie Antonopoulos, and Samantha Loi (St. Vincent’s Hospital, The University of Melbourne, Kew, Victoria, Australia);
Phyllis Chua, and Angela Komiti (The University of Melbourne, Royal Melbourne Hospital, Melbourne, Victoria, Australia);
Lynn Raymond, Joji Decolongon, Mannie Fan, and Allison Coleman (University of British Columbia, Vancouver, British Columbia, Canada);
Christopher A. Ross, Mark Varvaris, Maryjane Ong, and Nadine Yoritomo (Johns Hopkins University, Baltimore, Maryland, USA);
William M. Mallonee and Greg Suter (Hereditary Neurological Disease Centre, Wichita, Kansas, USA);
Ali Samii, Emily P. Freney, and Alma Macaraeg (University of Washington and VA Puget Sound Health Care System, Seattle, Washington, USA);
Randi Jones, Cathy Wood-Siverio, and Stewart A. Factor (Emory University School of Medicine, Atlanta, Georgia, USA);
Roger A. Barker, Sarah Mason, and Natalie Valle Guzman (John van Geest Centre for Brain Repair, Cambridge, UK);
Elizabeth McCusker, Jane Griffith, Clement Loy, Jillian McMillan, and David Gunn (Westmead Hospital, Sydney, New South Wales, Australia);
Michael Orth, Sigurd Süβmuth, Katrin Barth, Sonja Trautmann, Daniela Schwenk, and Carolin Eschenbach (University of Ulm, Ulm, Germany);
Kimberly Quaid, Melissa Wesson, and Joanne Wojcieszek (Indiana University School of Medicine, Indianapolis, IN, USA);
Mark Guttman, Alanna Sheinberg, Albie Law, and Irita Karmalkar (Centre for Addiction and Mental Health, University of Toronto, Markham, Ontario, Canada);
Susan Perlman and Brian Clemente (UCLA Medical Center, Los Angeles, California, USA);
Michael D. Geschwind, Sharon Sha, Joseph Winer, and Gabriela Satris (University of California, San Francisco, California, USA);
Tom Warner and Maggie Burrows (National Hospital for Neurology and Neurosurgery, London, UK);
Anne Rosser, Kathy Price, and Sarah Hunt (Cardiff University, Cardiff, Wales, UK);
Frederick Marshall, Amy Chesire, Mary Wodarski, and Charlyne Hickey (University of Rochester, Rochester, New York, USA);
Peter Panegyres, Joseph Lee, Maria Tedesco, and Brenton Maxwell (Neurosciences Unit, Graylands, Selby-Lemnos & Special Care Health Services, Perth, Western Australia, Australia);
Joel Perlmutter, Stacey Barton, and Shineeka Smith (Washington University, St. Louis, Missouri, USA);
Zosia Miedzybrodzka, Daniela Rae, Vivien Vaughan, and Mariella D’Alessandro (Clinical Genetics Centre, Aberdeen, Scotland, UK);
David Craufurd, Judith Bek, and Elizabeth Howard (University of Manchester, Manchester, UK);
Pietro Mazzoni, Karen Marder, and Paula Wasserman (Columbia University Medical Center, New York, New York, USA);
Rajeev Kumar, Diane Erickson, Christina Reeves, and Breanna Nickels (Colorado Neurological Institute, Englewood, Colorado, USA);
Vicki Wheelock, Lisa Kjer, Amanda Martin, and Sarah Farias (University of California, Davis, Sacramento, California, USA);
Wayne Martin, Oksana Suchowersky, Pamela King, Marguerite Wieler, and Satwinder Sran (University of Alberta, Edmonton, Alberta, Canada);
Anwar Ahmed, Stephen Rao, Christine Reece, Alex Bura, and Lyla Mourany (Cleveland Clinic Foundation, Cleveland, Ohio, USA);
Executive Committee
Principal Investigator Jane S. Paulsen, Jeffrey D. Long, Hans J. Johnson, Thomas Brashers-Krug, Phil Danzer, Amanda Miller, H. Jeremy Bockholt, and Kelsey Montross.
Scientific Consultants
Deborah Harrington (University of California, San Diego); Holly Westervelt (Rhode Island Hospital/Alpert Medical School of Brown University); Elizabeth Aylward (Seattle Children’s Research Institute); Stephen Rao (Cleveland Clinic); David J. Moser, Janet Williams, Nancy Downing, Vincent A. Magnotta, Hans J. Johnson, Thomas Brashers-Krug, Jatin Vaidya, Daniel O’Leary, and Eun Young Kim (University of Iowa).
Core Sections
Biostatistics.
Jeffrey D. Long, Ji-In Kim, Spencer Lourens (University of Iowa); Ying Zhang and Wenjing Lu (University of Indiana).
Ethics.
Cheryl Erwin (Texas Tech University Health Sciences Center); Thomas Brashers-Krug, Janet Williams (University of Iowa); and Martha Nance (University of Minnesota).
Biomedical Informatics.
H. Jeremy Bockholt, Jason Evans, and Roland Zschiegner (University of Iowa).
Footnotes
Disclosure Statement: Dr. Janessa Carvalho has no interests to declare. Dr. Jeffrey Long has a consulting agreement with NeuroPhage, LLC. Dr. Megan Smith has no interests to declare. Dr. Jared Bruce has no interests to declare. Mr. James Mills has no interests to declare. Dr. Ji-In Kim has no interests to declare. Dr. Jane Paulsen has served on an advisory board for Lundbeck, LLC and has a consulting agreement with ProPhase, LLC.
On the Buttons task, a touch screen computer monitor displayed a series of “buttons” arranged in ten vertical pairs, with the pairs arranged in a line across the screen, along with additional buttons at the left and right of the rows to indicate the start and finish positions. The buttons were blue when depressed and turned white when pressed. Each participant was instructed to touch the white button in each pair as quickly and as accurately as possible. When each button was touched a sound was produced to indicate the correct response. The participant continued to touch each illuminated white button down the sequence until the last column was depressed. The computer recorded the time for which each button was held down and the time between the release of one button and the depression of the next.
Contributor Information
Janessa O. Carvalho, Department of Psychiatry and Human Behavior, Alpert Medical School of Brown University, Department of Psychiatry, Rhode Island Hospital, Providence, RI, 131 Summer Street, Bridgewater, MA 02325, janessacarvalho@gmail.com, 508-531-1975 (tel)
Jeffrey D. Long, Department of Psychiatry, Carver College of Medicine, The University of Iowa, Department of Biostatistics, The University of Iowa, 500 Newton Road, 1-328 MEB, Iowa City, IA 52242-1000, jeffrey-long@uiowa.edu, 319-335-8524 (tel)
Holly J. Westervelt, Department of Neurology, Vanderbilt University Medical Center, Vanderbilt Health, One Hundred Oaks, 719 Thompson Lane, Suite 24100, Nashville, TN 37204, holly.westervelt@vanderbilt.edu, 615-936-0060 (tel)
Megan M. Smith, Department of Neuropsychology, VA Maryland Healthcare System, 209 W. Fayette Street, Annex Room 519, Baltimore, MD 21201, megan.smith8@va.gov, 410-637-1389 (tel)
Jared M. Bruce, Department of Psychology, University of Missouri-Kansas City, 4825 Troost Building Suite 11-G, Kansas City, MO 64110, brucejm@umkc.edu, 816-235-5429 (tel)
Ji-In Kim, Department of Psychiatry, Carver College of Medicine, The University of Iowa, 500 Newton Road, 1-317 MEB, Iowa City, IA, 52242-1000, ji-in-kim@uiowa.edu, 319-335-7727 (tel).
James A. Mills, Department of Psychiatry, Carver College of Medicine, The University of Iowa, 500 Newton Road, 1-323 MEB, Iowa City, IA 52242-1000, jim-mills@uiowa.edu, 319-384-4517 (tel)
Jane S. Paulsen, Departments of Psychiatry and Neurology, Carver College of Medicine, The University of Iowa, Department of Psychology, The University of Iowa, 500 Newton Road, 1-305 MEB, Iowa City, IA 52242-1000, predict-publications@uiowa.edu, 319-353-4551 (tel)
References
- Beglinger LJ, Nopoulos PC, Jorge RE, Langbehn DR, Mikos AE, Moser DJ, … Paulsen JS (2005). White matter volume and cognitive dysfunction in early Huntington’s disease. Cognitive & Behavioral Neurology, 18, 102–107. doi: 00146965-200506000-00004 [DOI] [PubMed] [Google Scholar]
- Biglan KM, Ross CA, Langbehn DR, Aylward EH, Stout JC, Queller S, … and the PREDICT-HD Investigators of the Huntington Study Group (2009). Motor abnormalities in premanifest persons with Huntington’s disease: The PREDICT-HD study. Movement Disorders, 24, 1763–1772. doi: 10.1002/mds.22601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blekher T, Johnson SA, Marshall J, White K, Hui S, Weaver M, … Foroud T (2006). Saccades in presymptomatic and early stages of Huntington disease. Neurology, 67, 394–399. [DOI] [PubMed] [Google Scholar]
- Blekher TM, Yee RD, Kirkwood SC, Hake AM, Stout JC, Weaver MR, & Foroud TM (2004). Oculomotor control in asymptomatic and recently diagnosed individuals with the genetic marker for Huntington’s disease. Vision Research, 44, 2729–2736. [DOI] [PubMed] [Google Scholar]
- Brandt J, & Benedict RHB (2001). Hopkins Verbal Learning Test-Revised. Psychological Assessment Resources: Lutz. [Google Scholar]
- Codori A, Hanson R, & Brandt J (1994). Self-selection in predictive testing for Huntington’s disease. American Journal of Medical Genetics, 54, 167–173. doi: 10.1002/ajmg.1320540303 [DOI] [PubMed] [Google Scholar]
- Duff K, Paulsen J, Mills J, Beglinger LJ, Moser DJ, Smith MM, … Harrington DL (2010). Mild cognitive impairment in pre-diagnosed Huntington disease. Neurology, 75, 500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dursun SM, Burke JG, Andrews H, Mlynik-Szmid A, & Reveley MA (2000). The effects of antipsychotic medication on saccadic eye movement abnormalities in Huntington’s disease. Progress in Neuro-Psychopharmacology and Biological Psychiatry, 24, 889–896. doi: S0278-5846(00)00116-0 [DOI] [PubMed] [Google Scholar]
- Duyao M, Ambrose C, Myers R, Novelletto A, Persichetti F, Frontali M … MacDonald M (1993). Trinocleotide repeat length instability and age of onset in Huntington’s disease. Natural Genetics, 4, 387–392. [DOI] [PubMed] [Google Scholar]
- Folstein SE (1989). Huntington’s Disease. Baltimore, MD: Johns Hopkins University Press. [Google Scholar]
- Glusman M, Roberg B, Ness A, Thelen J, Feaster T, & Bruce J (2013). Speeded eye tracking is associated with performance on cognitive tests that require visual scanning in multiple sclerosis. Poster session presented at that annual meeting of the International Neuropsychological Society, Honolulu, HI. [Google Scholar]
- Georgiou-Karistianis N, Long JD, Lourens SG, Stout JC, Mills JA, Paulsen JS (2014). Movement sequencing in Huntington’s disease. World Journal of Biological Psychiatry, 15, 459–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golding CV, Danchaivijitr C, Hodgson TL, Tabrizi SJ, & Kennard C (2006). Identification of an oculomotor biomarker of prodromal Huntington disease. Neurology, 8, 485–487. [DOI] [PubMed] [Google Scholar]
- Gutekunst C, Norflus F, Hersch S (2002). The neuropathology of Huntington’s disease. In Bates G, Harper P, Jones L, (Eds.), Huntington’s disease (251–275). New York: Oxford University Press. [Google Scholar]
- Harrington DL, Smith MM, Zhang Y, Carlozzi NE, Paulsen JS, and the PREDICT-HD Investigators of the Huntington Study Group (2012). Cognitive domains that predict time to diagnosis in prodromal Huntington disease. Journal of Neurology, Neurosurgery, and Psychiatry, 83, 612–619. doi: 10.1136/jnnp-2011-301732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry JD, Crawford JR, & Phillips LH (2005). A meta-analytic review of verbal fluency deficits in Huntington’s disease. Neuropsychology, 19, 243–252. [DOI] [PubMed] [Google Scholar]
- Hicks SL, Robert MPA, Golding CVP, Tabrizi SJ, & Kennard C (2008). Oculomotor deficits indicate the progression of Huntington’s Disease. Progress in Brain Research, 171, 555–558. [DOI] [PubMed] [Google Scholar]
- Huntington Study Group (1996). Unified Huntington’s Disease Rating Scale: Reliability and consistency. Movement Disorders, 11, 136–142. [DOI] [PubMed] [Google Scholar]
- Kieburtz K, McDermott M, Voss TS, Corey-Bloom J, Deuel BA … The Dimebon in Subjects With Huntington Disease (DIMOND) Investigators of the Huntington Study Group (2010). A randomized, placebo-controlled trial of latrepirdine in Huntington disease. Archives of Neurology, 67, 154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasker AG & Zee DS (1997). Ocular motor abnormalities in Huntington’s disease. Vision Research, 34, 3639–3645. [DOI] [PubMed] [Google Scholar]
- Lee J-M, Ramos EM, Lee J-H, Gillis T, Mysore JS, Hayden MR, … Gusella JF (2012). CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashion. Neurology, 78, 690–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh RJ & Zee DS (1999). The neurology of eye movements (3rd ed.). New York: Oxford University Press. [Google Scholar]
- Long JD, Paulsen JS, Marder K, Zhang Y, Kim JI, Mills JA, Researchers of the PREDICT-HD Huntington’s Study Group. Tracking motor impairments in the progression of Huntington’s disease. Movement Disorders, 29, 311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marder K, Zhao H, Myers RH, Cudkowicz M, Kayson E, Kieburtz K, … and the Huntington Study Group. (2000). Rate of functional decline in Huntington’s disease. Neurology, 54, 452–458. [DOI] [PubMed] [Google Scholar]
- Montoya A, Pelletier M, Menear M, Duplessis E, Richer F, & Lepage M (2006). Episodic memory impairment in Huntington’s disease: A meta-analysis. Neuropsychologia, 44, 1984–1994. [DOI] [PubMed] [Google Scholar]
- The Huntington’s Disease Collaborative Research Group (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell, 72, 971–983. [DOI] [PubMed] [Google Scholar]
- O’Donnell BF, Wilt MA, Hake AM, Stout JC, Kirkwood SC, & Foroud T (2003). Visual function in Huntington’s disease patients and presymptomatic gene carriers. Movement Disorders, 18, 1027–1034. [DOI] [PubMed] [Google Scholar]
- O’Rourke JJF, Beglinger LJ, Smith MM, Mills J, Moser DJ, Rowe KC, … and the PREDICT-HD Investigators of the Huntington Study Group (2011). The trail making test in prodromal Huntington disease: Contributions of disease progression to test performance. Journal of Clinical and Experimental Neuropsychology, 33, 567–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen JS (2010). Early detection of Huntington’s disease. Future Neurology, 5, 85–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen JS & Conybeare RA (2005). Cognitive changes in Huntington’s disease. Advances in Neurology, 96, 209–225. [PubMed] [Google Scholar]
- Paulsen JS, Hayden M, Stout JC, Langbehn DR, Aylward E, Ross CA, … Penziner E (2006). Preparing for preventive clinical trials. The Predict-HD study. Archives of Neurology, 63, 883–890. [DOI] [PubMed] [Google Scholar]
- Paulsen JS, Langbehn DR, Stout JC, Aylward E, Ross CA, Nance M, … Hayden M (2008). Detection of Huntington’s disease decades before diagnosis: the Predict-HD study. Journal of Neurology, Neurosurgery, and Psychiatry, 79, 874–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen JS, Smith MM, & Long JD (2013). Cognitive decline in prodromal Huntington disease: Implications for clinical trials. Journal of Neurology, Neurosurgery & Psychiatry, 84, 1233–1239. doi: 10.1136/jnnp-2013-305114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penney JB, Vonsattel JP, MacDonald ME, Gusella JF, & Myers RH (1997). CAG repeat number governs the development rate of pathology in Huntington’s disease. Annals of Neurology, 41, 689–692. [DOI] [PubMed] [Google Scholar]
- Radant AD, Claypoole K, Wingerson DK, Cowley DS, & Roy-Byrne PP (1997). Relationships between neuropsychological and oculomotor measures in schizophrenia patients and normal controls. Biological Psychiatry, 42, 797–805. [DOI] [PubMed] [Google Scholar]
- Reitan RM (1958). Validity of the Trail Making Test as an indicator of organic brain damage. Perceptual and Motor Skills, 8, 271–276. [Google Scholar]
- Rubinsztein DC (2003). Molecular biology of Huntington’s disease (HD) and HD-like disorders. In Pulst S (Ed.), Genetics of movement disorders (365–377). California: Academic Press. [Google Scholar]
- Schwarcz R & Shoulson I (1987). Excitotoxins and Huntington’s disease. In Coyle JT (Ed.), Animal models of dementia: A synaptic Neurochemical Perspective (39–68). New York: Alan R. Liss. [Google Scholar]
- Smith A (1991). Symbol Digits Modalities Test. Western Psychological Services: Los Angeles, CA. [Google Scholar]
- Stine OC, Pleasant N, Franz ML, Abbott MH, Folstein SE, & Ross CA (1993). Correlation between the onset age of Huntington’s disease and length of the trinucleotide repeat in IT-15. Human Molecular Genetics, 2, 1547–1549. [DOI] [PubMed] [Google Scholar]
- Stout JC, Paulsen JS, Queller S, Solomon AC, Whitlock KB, Campbell JC, … Aylward EH (2011). Neurocognitive signs in prodromal Huntington disease. Neuropsychology, 25, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroop JR (1935). Studies of interference in serial verbal reactions. Journal of Experimental Psychology: General, 18, 643–662. [Google Scholar]
- Tabrizi SJ, Langbehn DR, Leavitt BR, Roos RAC, Durr A, Craufurd D, … and the TRACK-HD investigators (2009). Biological and clinical manifestations of Huntington’s disease in the longitudinal TRACK-HD study: Cross-sectional analysis of baseline data. The Lancet Neurology, 8, 791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin AJ (1990). Genetic disorders: Huntington’s disease. In Pearlman AL & Collins RC (Eds.), Neurobiology of disease. New York: Oxford University Press. [Google Scholar]
- Turner TH, Goldstein J, Hamilton JM, Jacobson M, Pirogovsky E, Peavy G, & Corey-Bloom J (2011). Behavioral measures of saccade latency and inhibition in manifest and premanifest Huntington’s disease. Journal of Motor Behavior, 43, 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbeke G & Molenberghs G (2013). The gradient function as an exploratory goodness-of-fit assessment of the random-effects distribution in mixed models. Biostatistics, 14, 477–490. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP & DiFiglia M (1998). Huntington disease. Journal of Neuropatholgy and Experimental Neurology, 57, 369–84. [DOI] [PubMed] [Google Scholar]
- Wechsler D (1999). Wechsler Abbreviated Scale of Intelligence. The Psychological Corporation: San Antonio, TX. [Google Scholar]
- Zhang Y, Long JD, Mills JA, Warner JH, Lu W, Paulsen JS, & the PREDICT-HD Investigators and Coordinators of the Huntington Study Group (2011). Indexing disease progression at study entry with individuals at-risk for Huntington disease. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics, 156, 751–763. [DOI] [PMC free article] [PubMed] [Google Scholar]