

Abstract
Seven rhenium(I) complexes of the general formula fac-[Re(CO)3(NN)(OH2)]+ where NN = 2,2′-bipyridine (8), 4,4′-dimethyl-2,2′-bipyridine (9), 4,4′-dimethoxy-2,2′-bipyridine (10), dimethyl 2,2′-bipyridine-4,4′-dicarboxylate (11), 1,10-phenanthroline (12), 2,9-dimethyl-1,10-phenanthroline (13), or 4,7-diphenyl-1,10-phenanthroline (14), were synthesized and characterized by 1H NMR spectroscopy, IR spectroscopy, mass spectrometry, and X-ray crystallography. With the exception of 11, all complexes exhibited 50% growth inhibitory concentration (IC50) values that were less than 20 μM in HeLa cells, indicating that these compounds represent a new potential class of anticancer agents. Complexes 9, 10, and 13 were as effective in cisplatin-resistant cells as wild-type cells, signifying that they circumvent cisplatin resistance. The mechanism of action of the most potent complex, 13, was explored further by leveraging its intrinsic luminescence properties to determine its intracellular localization. These studies indicated that 13 induces cytoplasmic vacuolization that is lysosomal in nature. Additional in vitro assays indicated that 13 induces cell death without causing an increase in intracellular reactive oxygen species or depolarization of the mitochondrial membrane potential. Further studies revealed that the mode of cell death does not fall into one of the canonical categories such as apoptosis, necrosis, paraptosis, and autophagy, suggesting that a novel mode of action may be operative for this class of rhenium compounds. The in vivo biodistribution and metabolism of complex 13 and its 99mTc analogue 13* were also evaluated in naïve mice. Complexes 13 and 13* exhibited comparable biodistribution profiles with both hepatic and renal excretion. High-performance liquid chromatography inductively coupled plasma mass-spectrometry (HPLC-ICP-MS) analysis of mouse blood plasma and urine post administration showed considerable metabolic stability of 13, rendering this potent complex suitable for in vivo applications. These studies have shown the biological properties of this class of compounds and demonstrated their potential as promising theranostic anticancer agents that can circumvent cisplatin resistance.
Graphical abstract
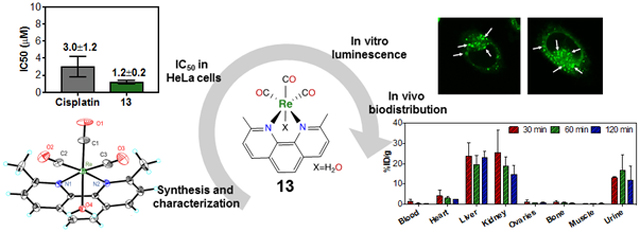
The potent anticancer activity of rhenium(I) tricarbonyl complexes circumventing Pt-resistance was investigated in a series of detailed biological studies.
Introduction
Cancer is a leading cause of death worldwide1 for which chemotherapy remains the most effective strategy for prolonging patient survival. Among the FDA-approved chemotherapeutic agents, the platinum-based drugs cisplatin, carboplatin, and oxaliplatin are especially common and effective, as they are used in approximately 50% of all chemotherapy regimens.2 These relatively simple coordination compounds induce their anticancer activity by forming covalent Pt-DNA crosslinks,3 which inhibit transcription and give rise to apoptotic cell death.4 Despite their widespread use, there are several limitations to the continued implementation of these platinum drugs. For example, they induce toxic side effects, which include nephrotoxicity, ototoxicity, and peripheral neuropathy.5 Additionally, after the first-line round of platinum chemotherapy, tumors often relapse in a platinum-resistant form, which signifies an extremely poor patient prognosis.6 Lastly, the platinum drugs are not amenable to detection by in vitro or in vivo imaging. The lack of spectroscopic handles for imaging these compounds in biological settings hinders the possibility of tracking tumor response in vivo and understanding the significance of intracellular localization in vitro.7
The exploration of alternative anticancer metal complexes that overcome the limitations associated with platinum drugs is an expanding field of research.8 These efforts have led to the development of titanium and ruthenium anticancer agents, some of which have progressed to Phase I and II clinical trials.8 These advances have provided an impetus for the continued investigation of the periodic table for the discovery of new anticancer drugs.
Anticancer applications of rhenium have only recently been explored. These studies have revealed rhenium(I) tricarbonyl complexes to be of particular interest.9–33 This class of compounds, most commonly utilized as CO2 reduction catalysts,34 possess several features that make them amenable for use as anticancer agents. For example, like the platinum-based drugs, they can bind covalently to DNA nucleobases.35–38 Furthermore, the ligand substitution kinetics for rhenium(I) tricarbonyl complexes are on the same order of magnitude as those for the platinum-based drugs.39 However, a key advantage of these compounds over conventional platinum anticancer agents is their rich spectroscopic properties that may be leveraged for imaging. The triplet-based luminescent emission of these rhenium(I) tricarbonyl complexes has been successfully used in cellular fluorescence microscopy imaging applications,40 and their distinct C≡O stretching frequency enables imaging by vibrational microscopy.41 Additionally, analogous 99mTc compounds can be synthesized and used for in vivo SPECT imaging applications.42
In this study, we report a systematic evaluation of a small library of these rhenium(I) tricarbonyl complexes as potential anticancer agents. We have found that these complexes are potent anticancer agents that induce cell death in a manner very different from that of cisplatin. We have also carried out in vivo studies that establish 99mTc analogues as suitable diagnostic companions for these agents. This study demonstrates that these compounds represent a promising novel class of anticancer agents worthy of continued investigation.
Results
Synthesis and Characterization.
The diimine rhenium(I) tricarbonyl complexes were synthesized via previously reported methods (Scheme 1).43,44 Treatment of Re(CO)5Cl with a diimine ligand in refluxing toluene affords the fac-[Re(CO)3(NN)Cl] compounds (1–7), where NN represents the diimine ligand. Because these compounds exhibit poor water solubility, the aqua complexes fac-[Re(CO)3(NN)(OH2)]+ (8–14) were prepared by the treatment of the chlorido complexes with AgOTf in acetone to remove the axial chloride ligand as insoluble AgCl. The resulting triflato complexes were then suspended in water to form the aqua complexes. The enhanced water solubility of the aqua complexes was verified by the fact that we could prepare them as millimolar aqueous solutions, an impossibility for most of the chlorido species.
Scheme 1.
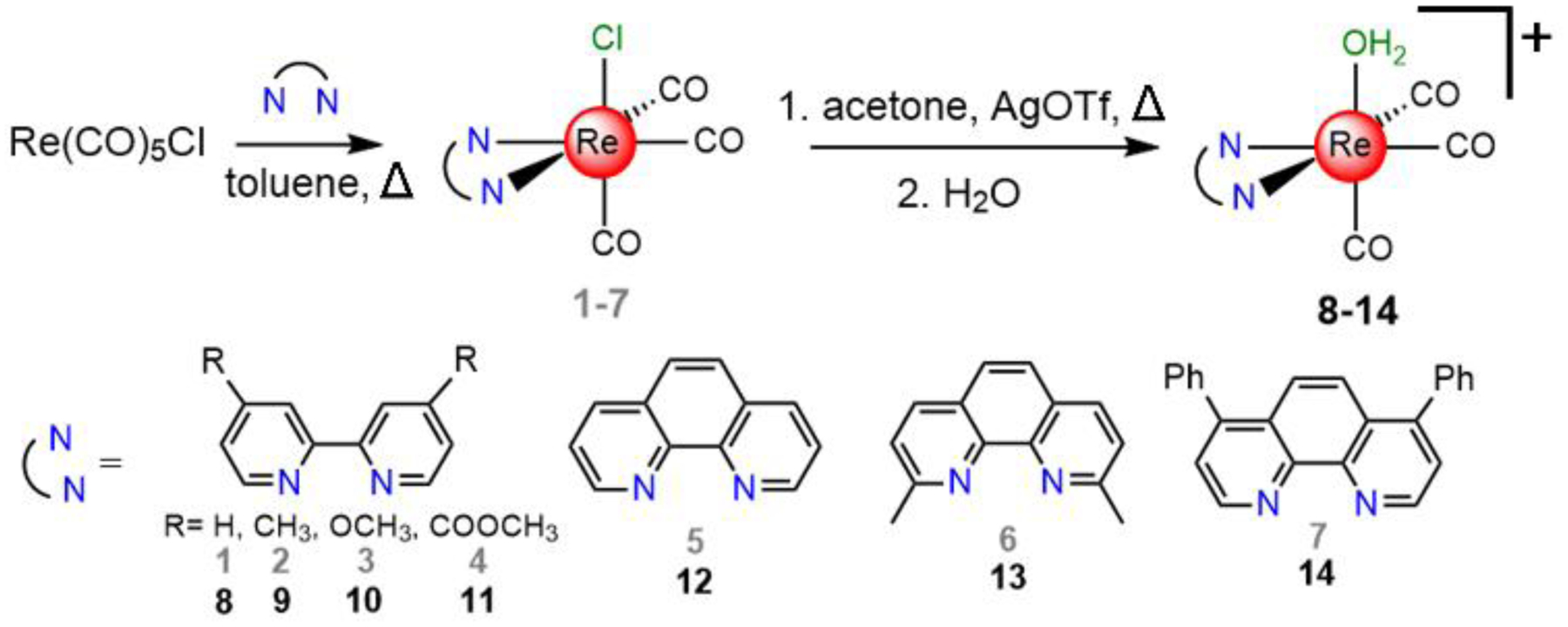
Synthesis of the rhenium complexes investigated in this study.
Complexes 8–14 were characterized by IR spectroscopy (Figure S1, Supporting Information), 1H NMR spectroscopy (Figures S2–S8), and electrospray ionization mass spectrometry (ESI-MS, Figures S9–S15). Their purity was verified to be greater than 95% by elemental analysis and HPLC. By elemental analysis, the samples analyzed either as the aqua-triflato salts or as the anhydrous triflato complexes, which may form under vacuum during sample drying. The relative lability of the axial ligand was evidenced by HPLC and NMR spectroscopy. For example, analysis of the chlorido complexes 1–7 by HPLC gives rise to a chromatogram containing two distinct peaks. A representative chromatogram for 5 is shown in the top panel of Figure 1a. By contrast, the chromatograms of the analogous aqua or triflato complexes 8–14 feature a single peak, matching the earlier peak of the chlorido complex, as shown for 12 in the bottom of Figure 1a. Hence, the more lipophilic peak of the chlorido complex is attributed to the intact chloride-bound compound in equilibrium with the aquated species, which has the same retention time as the cationic complex. The HPLC analysis of related rhenium and technetium chlorido complexes also shows two distinct peaks in the HPLC chromatogram, presumably arising from the same phenomenon described here.45,46 The 1H NMR spectra of the cationic complexes 8–14 also reveals a solution equilibrium. Acquisition of these spectra in MeOD-d4 show the presence of signals for two distinct complexes (Figure 1b). The addition of D2O to these samples leads to a coalescence of the signals. Therefore, we hypothesize that in MeOD-d4 solution these compounds comprise an equilibrium mixture of axial-bound MeOD-d4 and D2O complexes; the addition of D2O acts to drive the equilibrium exclusively to the aqua complex. As such, NMR spectra reported in the Experimental section for these complexes were acquired in a mixture of MeOD-d4 and D2O.
Figure 1.
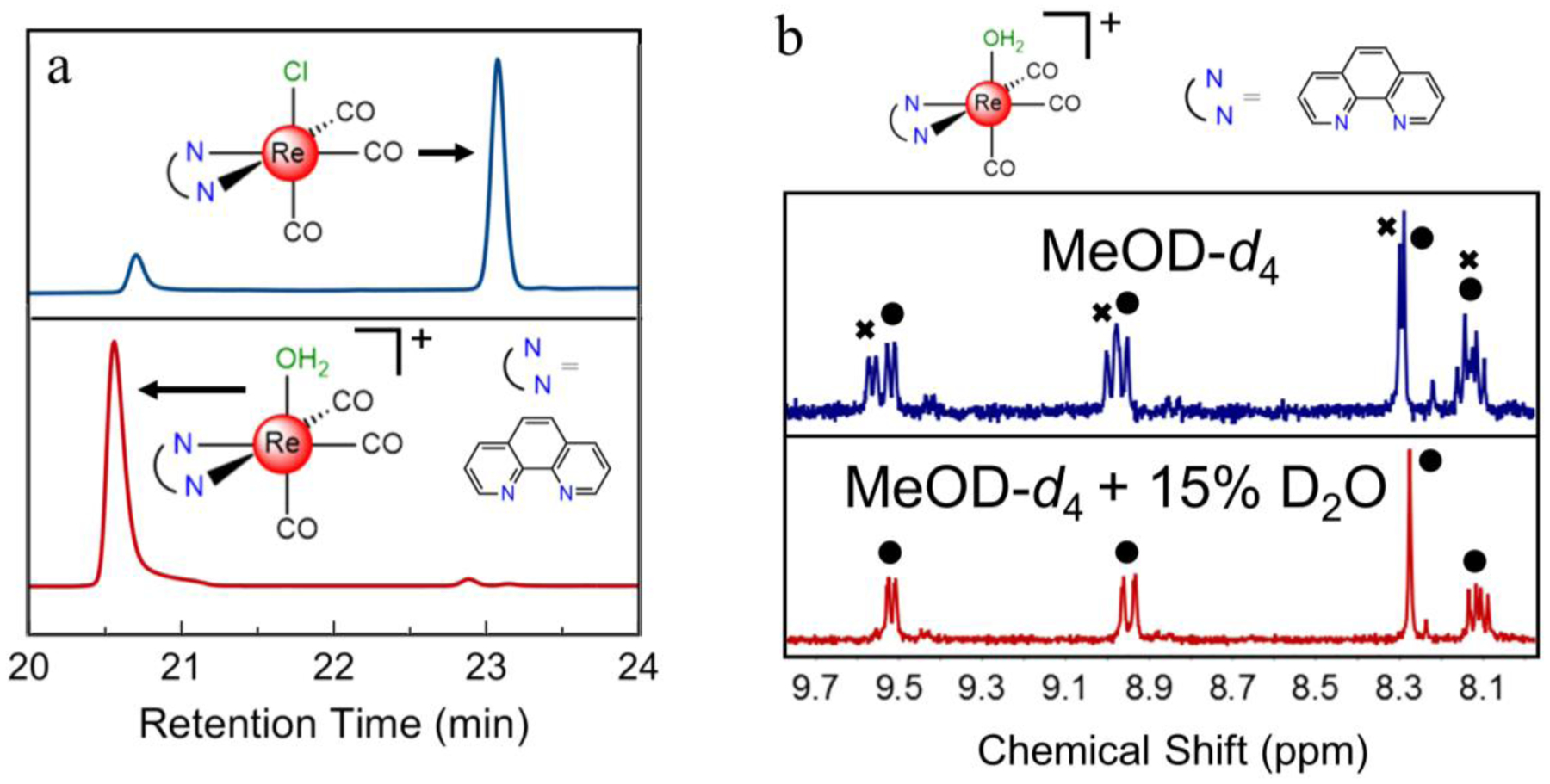
a) HPLC chromatogram of the phen chlorido species 5 (blue, top trace) and the corresponding aqua species 12 (red, bottom trace) using a methanol gradient elution and monitoring 260 nm. b) 1H NMR spectra of the phen aqua species 12 in MeOD-d4 (blue, top trace) and in MeOD-d4 with 15% D2O (red, bottom trace). The circles designate peaks due to the aqua complex and the x’s designate peaks due to the methanol adduct.
Single-Crystal X-ray Diffraction.
Single crystals of 11 and 13, as well as derivatives of 9 and 10 with the triflate ion substituted for a nitrate (9-NO3) and tetrafluoroborate (10-BF4) counterion, were obtained and analyzed by single-crystal X-ray diffraction to determine their molecular structures (Figure 2). Selected interatomic distances and angles are presented in Table 1. The diimine rhenium tricarbonyl core is maintained in all four structures, but each structure bears different axial ligands. The structures of compounds 9-NO3 and 11 reveal direct coordination of the nitrate and triflate counterions to the rhenium center, whereas 10-BF4 and 13 display coordination of the solvent (acetonitrile and water, respectively). The direct coordination of a nitrate counterion to related rhenium tricarbonyl diimine complexes is a rare occurrence, as only three such structures are reported;47,48 coordination of triflate and acetonitrile is a more common phenomenon.49–51 The nature of the axial ligand appears to have a minor influence on the overall interatomic distances of the complexes, as no statistically significant differences are observed.
Figure 2.
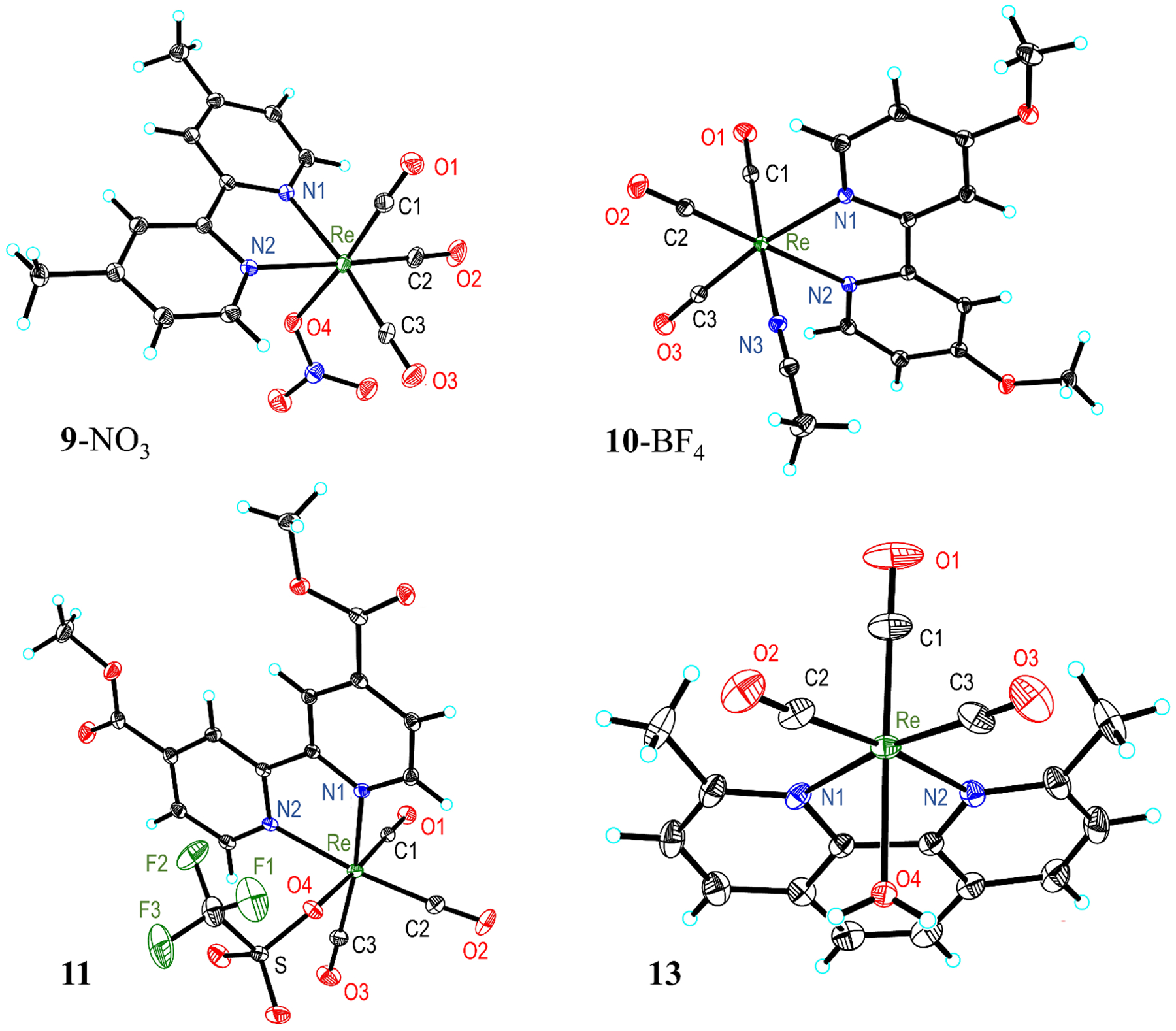
Crystal structures of 9-NO3, 10-BF4, 11, and 13. Outer-sphere solvent molecules and anions are omitted for clarity. Thermal ellipsoids are shown at the 50% probability level.
Table 1.
Selected interatomic distances (Å) and angles (°) from the crystal structures shown in Figure 2. Atom X represents the axial atom, either O4 or N3.
| Complex | ||||
|---|---|---|---|---|
| 9-NO3 | 10-BF4 | 11 | 13 | |
| Re–N1 | 2.178(4) | 2.182(2) | 2.180(3) | 2.210(3) |
| Re–N2 | 2.178(4) | 2.180(2) | 2.175(3) | 2.200(3) |
| Re–X | 2.154(4) | 2.149(2) | 2.191(2) | 2.196(2) |
| Re–C1 | 1.899(6) | 1.917(3) | 1.896(3) | 1.896(4) |
| Re–C2 | 1.915(6) | 1.927(3) | 1.921(4) | 1.929(4) |
| Re–C3 | 1.916(6) | 1.929(3) | 1.924(4) | 1.928(4) |
| C1–O1 | 1.163(7) | 1.147(3) | 1.149(4) | 1.147(5) |
| C2–O2 | 1.157(7) | 1.145(3) | 1.154(4) | 1.150(4) |
| C3–O3 | 1.152(7) | 1.144(3) | 1.150(4) | 1.148(5) |
| N1–Re–N2 | 74.60(15) | 74.42(7) | 75.68(9) | 76.18(9) |
| N1–Re–X | 87.73(14) | 82.77(8) | 79.09(9) | 77.99(9) |
| N1–Re–C1 | 93.68(19) | 94.18(9) | 92.69(12) | 98.36(15) |
| N1–Re–C2 | 99.00(2) | 99.72(11) | 98.09(13) | 100.72(13) |
| C2–Re–C3 | 89.0(2) | 87.77(13) | 89.02(16) | 82.84(17) |
Given the desired use of these complexes in aqueous solutions, the aqua structure of 13 is of particular interest. The distances and angles found in 13 are similar to those observed in the related 2,2′-bipyridine (bpy) and 1,10-phenanthroline (phen) aqua complexes.39,52–55 In platinum anticancer complexes with coordinated water ligands, the Pt–O distance typically ranges from 2.05–2.12 Å.56,57 The rhenium aqua compounds have longer Re–O distances of approximately 2.20 Å (2.196 Å in 13). This slightly longer distance may partly be a consequence of the trans π-accepting CO ligand and a different charge. However, the similarity of the bond lengths between the two types of metal complexes implies the feasibility of using rhenium in place of platinum for biological applications.
The four rhenium complexes adopt similar geometries with fairly consistent interatomic distances and angles. Notably, the C≡O bond lengths are approximately the same among all the complexes. This result is consistent with IR spectroscopy, which also shows that the energies of the C≡O vibrations are invariant between complexes. However, the Re–N distances and N1–Re–N2 bite angle are slightly larger in complex 13 compared to those in the bpy analogues; the Re–N distance is approximately 0.03 Å longer and the N1–Re–N2 angle is 1° wider. This difference is most likely due to the methyl groups on the 2,9-dimethyl-1,10-phenanthroline (dmphen) ligand that sterically crowd the rhenium center, giving rise to elongated interatomic distances. For example, the [Re(phen)(CO)3(H2O)]+ complex39 exhibits comparable Re–N distances to the bpy complexes because it does not contain any sterically repulsive methyl groups. By contrast, the crystal structure of [Re(dmphen)(CO)3Cl] has Re–N distances that are very close to those found in 13.58
Capacity Factor.
The activity of drug candidates may often be correlated to their lipophilicity. The lipophilicity of a class of compounds can be readily compared by determining their retention times on a reverse-phase HPLC column under the same isocratic elution conditions. The capacity factor k, given by the equation59 k = (tR−t0)/t0, where tR is the time at which the compound elutes and t0 is the dead time of the system, is a quantitative measure of retention on a RP-HPLC column that can be correlated directly with the lipophilicity of a compound. As an example, these values were used to determine water-octanol partition coefficients for a library of platinum anticancer agents.60
The capacity factors of the rhenium complexes and the isolated diimine ligands are given in Table 2. For these studies, an isocratic elution was used (40:60 MeCN:H2O, each containing 0.1% TFA). Because MeCN is an effective ligand for rhenium(I), two peaks in the chromatogram were observed, one corresponding to the aqua complex and the other to the MeCN adduct. The peak with the larger capacity factor is assigned to the MeCN adduct, which should be more lipophilic than the aqua complex. As anticipated, functionalization of the diimine gives rise to more lipophilic ligands. The rhenium complexes followed this same trend. Notably, the {Re(CO)3} core increases the lipophilicity of the complexes relative to the free ligands. The most lipophilic ligand, dpphen, gave rise to the most lipophilic rhenium complex 14, as evidenced by its capacity factor that exceeded 17.
Table 2.
Capacity factors of complexes 8–14 and their respective free ligands on a C18 column.
| Complex | Free Ligand Capacity Factor | Aqua Complex Capacity Factor | MeCN Complex Capacity Factor |
|---|---|---|---|
| 8 | 0.42 | 1.5 | 3.3 |
| 9 | 0.66 | 3.3 | 7.1 |
| 10 | 0.74 | 3.6 | 7.2 |
| 11 | 2.4 | 4.2 | 8.2 |
| 12 | 0.44 | 2.1 | 4.6 |
| 13 | 0.97 | 4.4 | 8.9 |
| 14 | >10 | >17 | >17 |
In Vitro Anticancer Activity.
The in vitro anticancer activities of cisplatin and complexes 8–14 were evaluated in HeLa cells by the MTT assay. The resulting 50% growth inhibitory concentration (IC50) values are displayed in Figure 3 and Table 3. Representative dose-response curves are shown in Figures S16–S21. All of the compounds except for 11 exhibited anticancer activity at concentrations under 20 μM. Notably, compounds 9 and 10 gave rise to IC50 values of less than 10 μM. The most potent compound screened was 13. Its low IC50 value (1.2 ± 0.2 μM) indicates that it is more active than the conventional metal-based anticancer drug cisplatin in HeLa cells (3.0 ± 1.2 μM).
Figure 3.
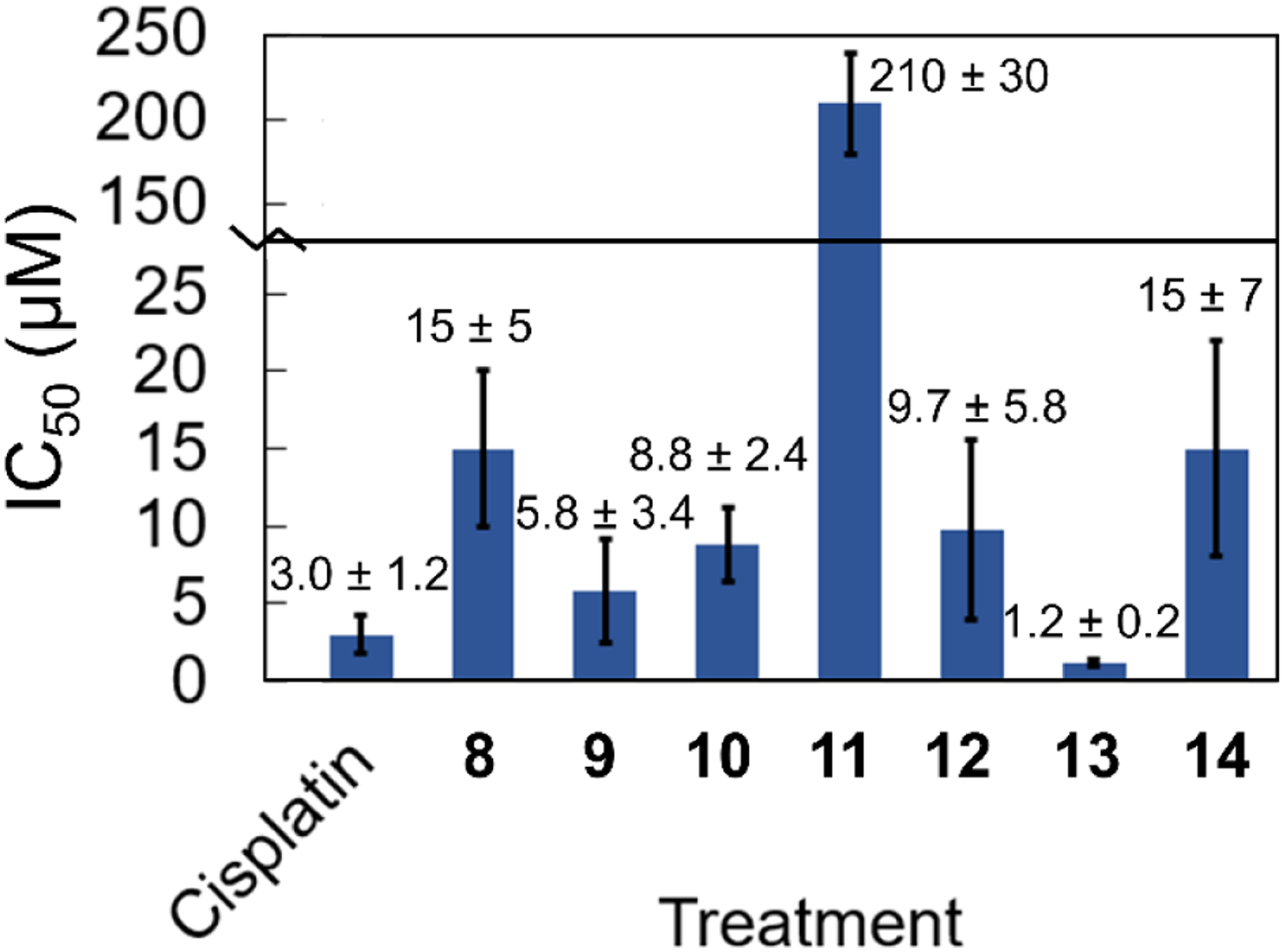
Cell viability data in HeLa cells. The error bars represent one standard deviation from three independent experiments.
Table 3.
Cell viability data in KB-3-1, KBCP20, A2780, A2780CP70, A549, A549 CisR, H460, H460 CisR, and MRC-5 cells. Error represents one standard deviation.
| IC50 (μM) or RFa of Complex | ||||
|---|---|---|---|---|
| Cell Line | Cisplatin | 9 | 10 | 13 |
| KB-3-1 | 1.0 ± 0.3 | 4.3 ± 1.6 | 0.77 ± 0.17 | 0.92 ± 0.20 |
| KBCP20 | 36 ± 7 | 5.3 ± 2.1 | 7.2 ± 1.2 | 1.6 ± 0.4 |
| RFa (KB-3-1) | 36 | 1.2 | 9.4 | 1.7 |
| A2780 | 0.23 ± 0.07 | 3.5 ± 2.8 | 2.2 ± 1.8 | 2.2 ± 0.2 |
| A2780CP70 | 8.2 ± 1.8 | 4.7 ± 1.4 | 2.8 ± 2.5 | 3.0 ± 0.7 |
| RFa (A2780) | 36 | 1.3 | 1.3 | 1.4 |
| A549 | 3.0 ± 1.8 | 5.2 ± 4.0 | 9.7 ± 4.1 | 6.7 ± 4.9 |
| A549 CisR | 12.4 ± 8.5 | 3.9 ± 4.6 | 5.7 ± 1.8 | 5.4 ± 1.8 |
| RFa (A549) | 4.1 | 0.8 | 0.6 | 0.8 |
| H460 | 0.75 ± 0.43 | 14 ± 1 | 9.0 ± 5.0 | 4.5 ± 0.7 |
| H460 CisR | 3.4 ± 1.6 | 21 ± 12 | 8.0 ± 2.1 | 5.3 ± 2.9 |
| RFa (H460) | 4.5 | 1.5 | 0.9 | 1.2 |
| MRC-5 | 0.43 ± 0.14 | 10.7 ± 0.5 | 6.0 ± 1.9 | 4.1 ± 0.9 |
RF is the resistance factor, which is the IC50 in the cisplatin-resistant cell line divided by the IC50 in the non-resistant matched cell line.
The most potent compounds 9, 10, and 13 were further investigated in wild-type and cisplatin-resistant matched cervical cancer cell lines KB-3-1 and KBCP20,61,62 ovarian cancer cell lines A2780 and A2780CP70,63 and lung cancer cell lines A549, A549 CisR, H460, and H460 CisR.64 In all of the cell lines, the rhenium complexes exhibited similar cytotoxic activity, characterized by IC50 values below 20 μM (Table 3). Notably, these rhenium complexes were nearly equally effective in the cisplatin-resistant cell lines. The resistance factors (RF), the ratio of the IC50 values in cisplatin-resistant and wild-type cells, ranged from 0.6 to 9.4. For comparison, the resistance factor determined for cisplatin was 36 for the A2780 and KB-3-1 cell lines. Complexes 9, 10, and 13 all exhibited lower resistance factors than cisplatin for the given matched cell lines, indicating that they can overcome cisplatin resistance mechanisms. These compounds were also tested in normal lung fibroblasts (MRC-5) as a representative model for non-cancerous cells (Table 3). The IC50 values of the rhenium complexes in these cells were about the same or slightly greater than those in the cancer cell lines. By contrast, cisplatin was more cytotoxic to the MRC-5 cells compared to the rhenium complexes.
Nucleobase and Amino Acid Binding.
The reactivity of 13, the most potent complex, with relevant biomolecules was probed using HPLC. This study was conducted to better understand the origin of in vitro anticancer activity of the rhenium complexes. The reaction of 9-ethylguanine, a small-molecule model for the most reactive nucleobase in RNA and DNA,65 was initially evaluated. The reaction of 13 with 9-ethylguanine in pH 7.3 MOPS buffer gave rise to a new lipophilic peak in the HPLC, corresponding to the covalent adduct (Figure 4). Additionally, complex 13 interacted appreciably with N-acetyl cysteine and N-acetyl histidine, models for amino acid residues on proteins. Qualitatively, the reaction of 13 was faster with 9-ethylguanine than either N-acetyl cysteine or N-acetyl histidine. The reactions of 13 with methionine, serine, and glycine were also investigated, but these studies revealed no significant interaction between the rhenium complex and the amino acids.
Figure 4.
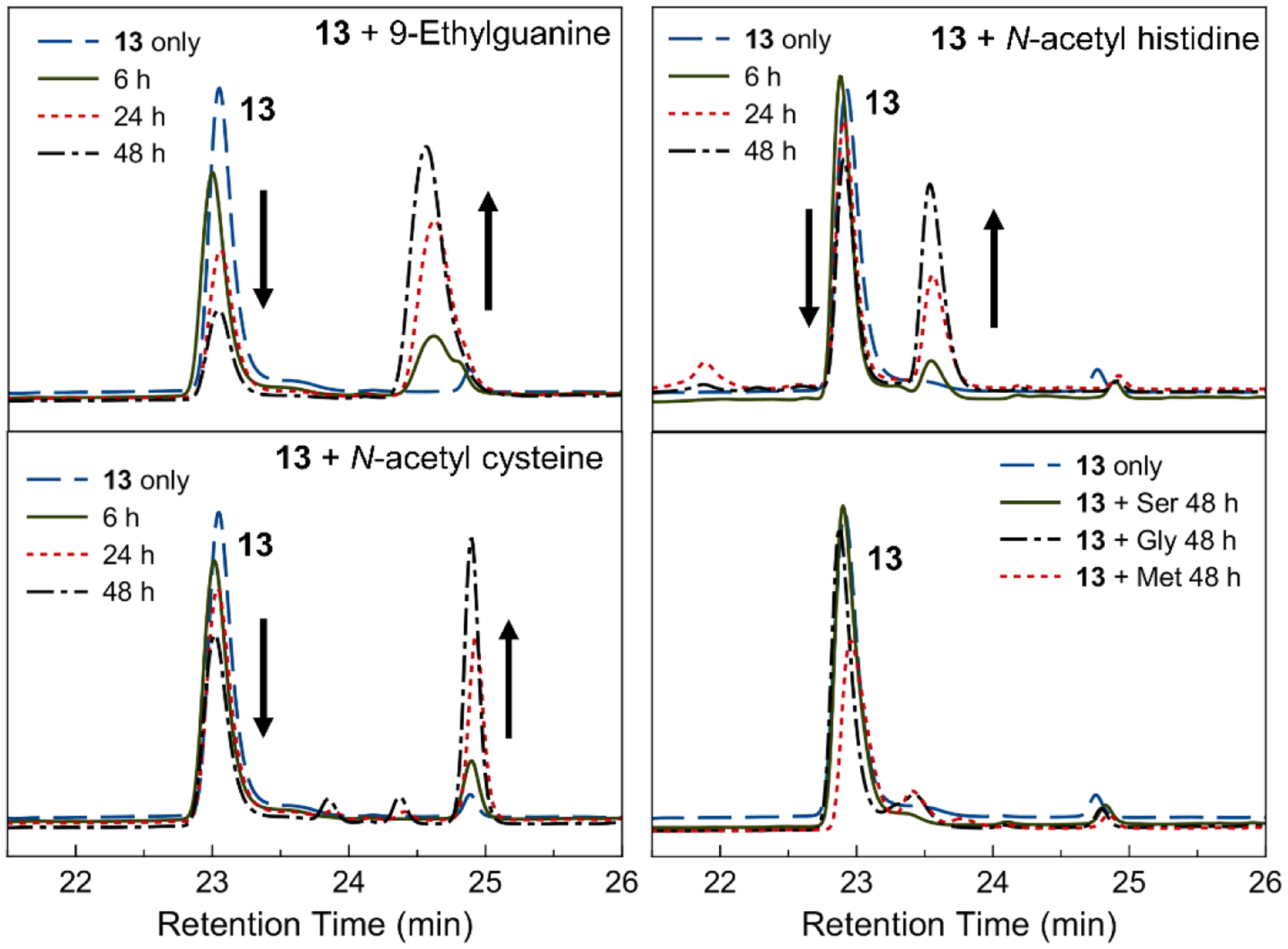
HPLC traces of the reaction of 13 with 9-ethylguanine, N-acetyl cysteine, N-acetyl histidine, or amino acids (serine, glycine, and methionine) for the indicated time monitored at 260 nm.
Fluorescence Microscopy.
Diimine rhenium(I) tricarbonyl complexes possess a luminescent triplet MLCT excited state that typically emits photons in the yellow region (560–590 nm) of the visible spectrum. These complexes have been successfully utilized for intracellular imaging applications27,66–69 via fluorescence microscopy. The ability to image the intracellular localization of the most potent rhenium complex 13 by confocal fluorescence microscopy was investigated. HeLa cells were treated with 13 and incubated for 4 or 24 h prior to imaging. The emission of 13 was detectable well above the background autofluorescence within the cells (Figure 5). The yellow emission of the rhenium was distributed throughout the cytosol. Notably, cytoplasmic vacuoles were observed, an apparent effect of the rhenium complex. The outer membranes of these vacuoles were brightly luminescent, indicating a large accumulation of the rhenium complexes.
Figure 5.
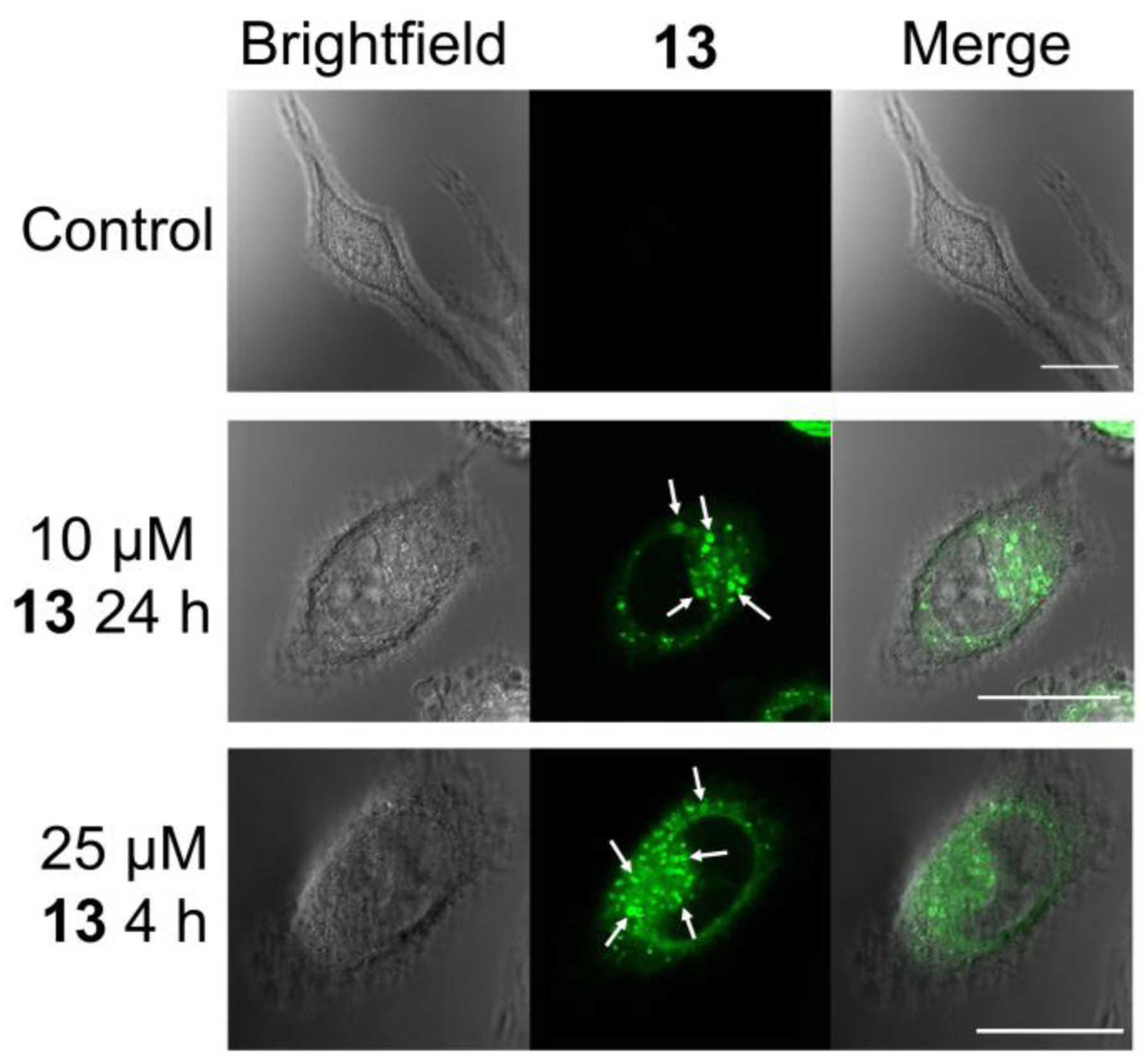
Brightfield and confocal fluorescent microscope images of control HeLa cells and HeLa cells treated with 13. Arrows point to vacuoles induced by 13 treatment. Scale bars = 20 μm.
To further explore the localization of the rhenium complexes, HeLa cells were treated with 13 and different organelle-localizing dyes or transfected to express organelle-specific proteins fused with a fluorescent protein (Figure 6 and Figures S22–S24). These co-localization studies readily reveal that 13 does not accumulate in the nucleus, mitochondria, or endoplasmic reticulum (Figure S23). In addition to its cytosolic distribution, the rhenium complex localizes to the large cytoplasmic vacuoles. The nature of these vacuoles was probed by transfecting the cells to express RFP-Rab5 and RFP-2×FYVE fusion proteins. Rab5 is a GTPase that localizes to the outer membrane of the early endosomes,70 and 2×FYVE is a tandem arrangement of a protein domain that binds to the lipid phosphatidylinositol 3-phosphate (PI3P),71 which is highly abundant in early endosomes and in the internal vesicles of multivesicular endosomes. The fluorescence microscopy images indicate that 13 co-localizes with RFP-Rab5, and partially with the RFP-2×FYVE conjugate (Figure 6). This observation suggests that 13 accumulates in some populations of endosomes and further implies that the cytoplasmic vacuoles are endosomal in origin. The lysosomal marker LysoTracker Red DND-99 was also employed. The fluorescent images indicate that the intracellular localization of 13 also correlates strongly with the lysosomes (Figure 6). This result suggests that the vacuoles also have lysosomal character and may be part of a compromised endosome-lysosome fusion process,72,73 or that 13 marks a broad population of endosomes and lysosomes. The cells were also transfected to express an RFP-LC3 fusion protein. LC3 is a protein that accumulates on autophagosomes, digestive double-membrane vacuoles that occur during the process of autophagy. Fluorescence microscopy images (Figure S24) indicate that the cytoplasmic vacuoles induced by 13 are not autophagosomes.74
Figure 6.
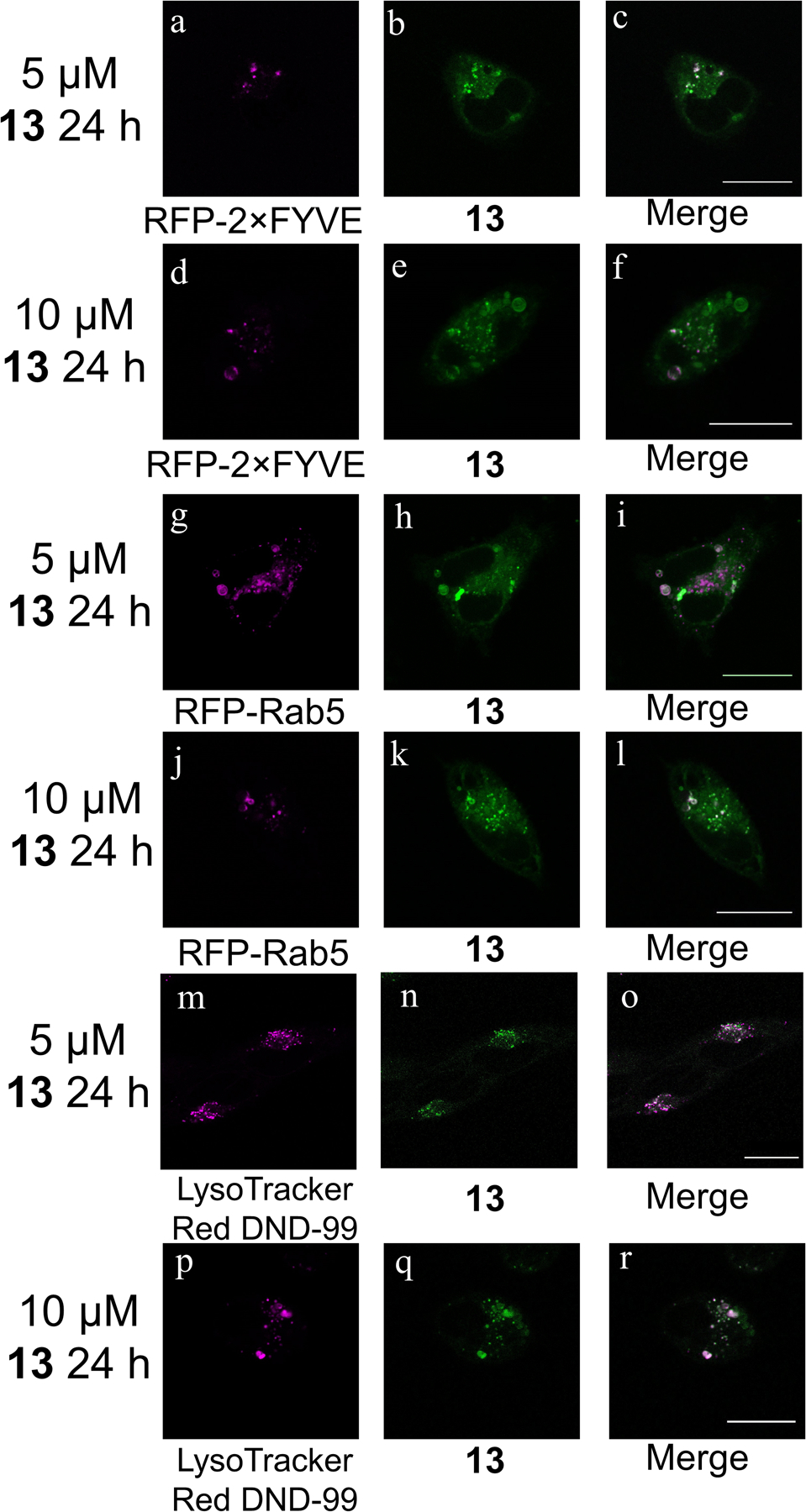
Confocal fluorescent microscope images of HeLa cells treated with 13 and transfected or stained with the indicated plasmid or dye. Scale bars = 20 μm.
Cell Cycle Analysis.
Anticancer agents often interfere with the cell cycle. The extent and nature of the cell cycle interruption may be indicative of the agent’s mechanism of action.75 The relative populations of cells in different phases of the cell cycle can be determined by fixing them, treating them with the fluorescent dye propidium iodide (PI), and then analyzing them with flow cytometry. Cells in the G2/M phase contain twice as much DNA as cells in the G1 phase. In the S phase, cells are actively replicating DNA.76 After binding DNA, cisplatin is known to stall cells in the S and G2/M phases.77 Accordingly, the treatment of HeLa cells with 5 μM cisplatin for either 24 or 48 h (Figure S25) gave results that are consistent with previous studies using this cell line.78
The effect of the rhenium complex on the cell cycle was probed by treating HeLa cells with 13 at 1, 5, or 10 μM for either 24 or 48 h (Figure S26). The most significant changes in the cell cycle population are visible at the 10 μM concentration level. After 24 h, 51.6% of the cells are in the G2/M phase, indicating that this compound stalls cells in these phases.
Annexin V/PI Assay.
The flipping of phosphatidylserine to the outer leaflet of the cell membrane is a hallmark feature of apoptotic cell death. The protein annexin V binds to extracellular phosphatidylserine with high affinity and specificity. The treatment of cells with annexin V conjugated to a fluorescent dye (annexin V-Alexa Fluor 488) enables the detection of apoptotic cells. This dye can be used in conjunction with PI to selectively label cells with compromised cell membranes, a feature of necrotic cell death.79
When HeLa cells were treated with 13 (5 μM), 19% of the total cell population was alive and possessed exposed phosphatidylserine after 24 h. When 13 was administered at a concentration of 10 μM, a greater proportion of cells label positive for PI, indicating that they are non-viable (Figure S27). However, the population of living cells positive for annexin did not increase. This annexin V assay was also conducted in the presence of the pan-caspase inhibitor Z-VAD-FMK. The inhibitor had no effect on the histogram of 13-treated cells, but was able to reduce the apoptotic population of cells treated with etoposide, a well characterized apoptosis-inducer.80
ROS Analysis.
Elevated levels of intracellular reactive oxygen species (ROS) often accompanies cell death.81 The amount of ROS present in cells treated with 13 were analyzed using 2′,7′-dichlorofluorescin diacetate (DCFDA) in conjunction with flow cytometry. Upon exposure to ROS, non-emissive DCFDA oxidizes to a brightly fluorescent product. The emission intensity in each cell, therefore, correlates with the amount of ROS present. Treatment of HeLa cells with 0.03% H2O2 gave rise to a greater than 30-fold increase in the intracellular ROS. Upon treating HeLa cells with 13, however, no significant increases (> 2-fold) in the intracellular ROS were observed (Figures S28–S29), indicating that this compound does not induce the formation of ROS.
JC-1 Assay.
The depolarization of the mitochondrial membrane potential (MMP) is an event that occurs early during the course of various cell death modes, such as apoptosis82 and paraptosis.83 This event leads to the release of cytochrome c and apoptosis-inducing factor from the mitochondria.82
HeLa cells were treated with 5 or 10 μM of 13 and the depolarization of the MMP was assessed by the JC-1 assay (Figures S30–S31). These results show that compound 13 does not induce depolarization of the MMP, as the percent of JC-1 aggregates is equivalent to that found in the untreated control. By contrast, carbonyl cyanide m-chlorophenyl hydrazine (CCCP), a known mitochondrial depolarizer, led to a substantial reduction in the number of JC-1 aggregates, owing to loss of the MMP.
Cell Viability in the Presence of Inhibitors.
To gain further insight into the mechanism of cell death induced by 13, its cytotoxicity in HeLa cells was evaluated in the presence of different chemical inhibitors that are well characterized in their ability to block chemical processes that mediate cell death. The IC50 value of 13 did not change in the presence of the autophagy inhibitor 3-methyladenine (Figure S32). This result indicates that 13 most likely does not induce cell death via autophagy. Treatment of the cells with cycloheximide, a protein synthesis inhibitor that prevents the cell death mode known as paraptosis,84,85 also failed to protect cells from the cytotoxic effects of 13 (Figure S32). Similarly, the addition of the necroptosis inhibitor necrostatin-1 did not significantly alter the dose-response curves (Figure S32). Because 13 induces cytoplasmic vacuolization that is endolysosomal in nature, the possibility of lysosomal protease-mediated cell death86 was investigated by using the protease inhibitor leupeptin.87 Likewise, leupeptin conferred no protective effects on the cells. Lastly, the influence of the pan-caspase inhibitor Z-VAD-FMK was investigated (Figure 7 and Figure S33). The IC50 value of cisplatin, which induces caspase-dependent apoptotic cell death,88 increased by a factor of four in the presence of Z-VAD-FMK. By contrast, Z-VAD-FMK had no protective effect on the IC50 value of 13, indicating that this compound gives rise to caspase-independent cell death. The small increase in potency of 13 in the presence of Z-VAD-FMK is consistent with similar observations for compounds that induce caspase-independent cell death.89,90
Figure 7.
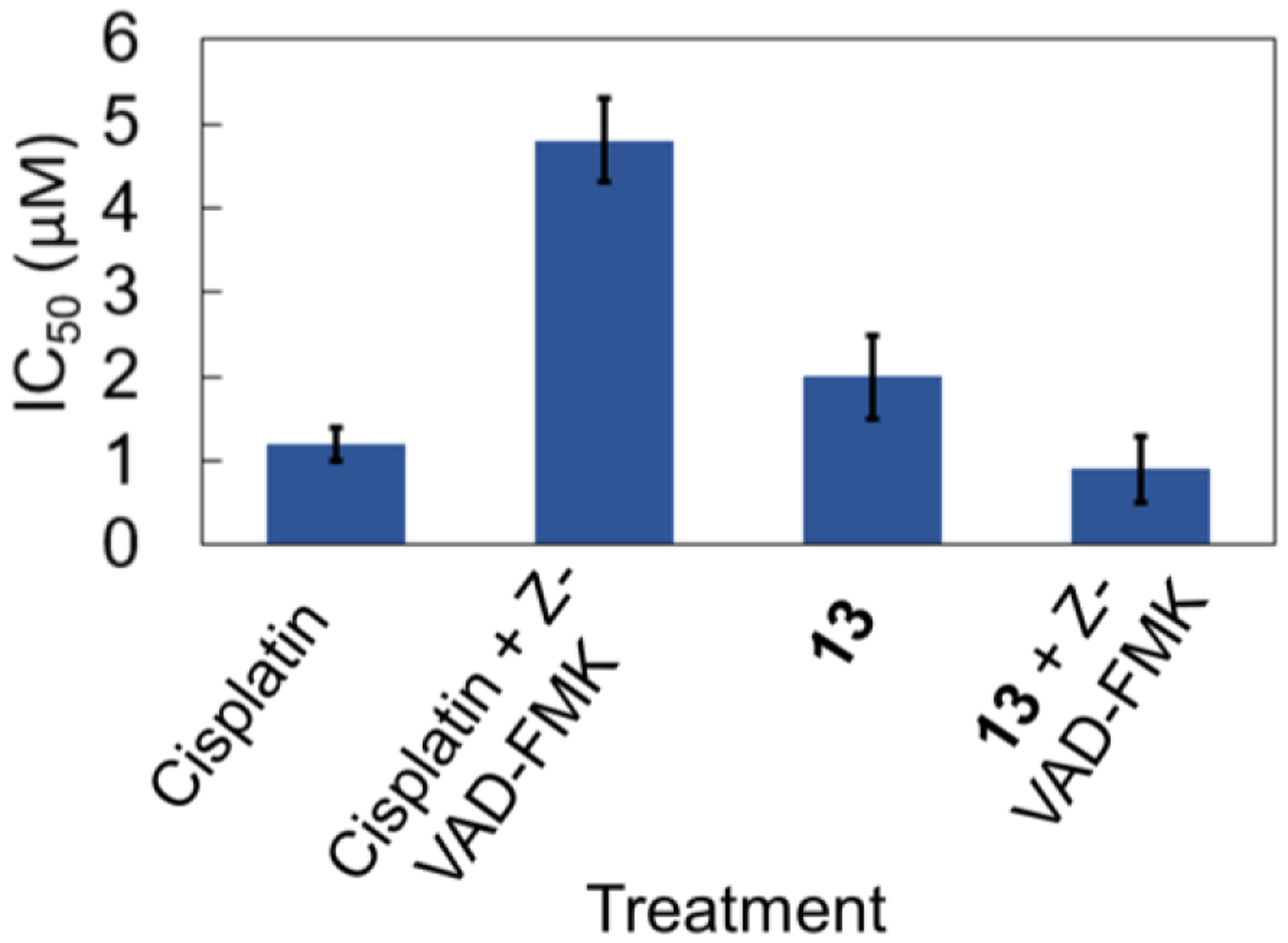
Cell viability in HeLa cells of cisplatin and 13 in the presence and absence of 15 μM of the caspase inhibitor Z-VAD-FMK. Error bars represent one standard deviation.
Western Blot Analysis of Protein Expression.
Different cell death pathways give rise to differential enhancement of specific protein expression levels, or may induce post-translational modification in these proteins. Poly (ADP-ribose) polymerase (PARP), for example, is cleaved by caspases during aptoposis.91 In addition, the protein LC3 is upregulated during the process of autophagy,92 and ERK is phosphorylated (p-ERK) in paraptosis.93,94 The expression levels of all of these proteins in HeLa cells were evaluated in the presence of complex 13 by Western blots (Figure S34). This compound did not significantly alter the expression levels of any of these proteins, providing evidence against these mechanisms of cell death. By contrast, cisplatin induced the increase of cleaved PARP levels in a manner that is consistent with the known apoptotic cell death mechanism of this drug.
Cellular Uptake Analysis with Flow Cytometry.
Because most cancer drug targets are intracellular, the cellular uptake of drug candidates may be a determining factor in their in vitro or in vivo activity. The cellular uptake of 13 was conveniently probed by flow cytometry, leveraging the luminescence properties of the complex for detection (Figures S35–S38).95 As the dose concentration of 13 is increased, the intensity of intracellular luminescence measured by flow cytometry also increased (Figure 8a). The cellular uptake scales linearly with the treated concentration up to 50 μM, at which point the uptake begins to level off. Cell uptake of 13 is inhibited when cells are treated at 4 °C, and it is reduced upon co-treatment with the endocytosis inhibitor chlorpromazine (Figure 8b). However, uptake is not reduced in the presence of the micropinocytosis inhibitor amiloride (Figure S38). These results implicate active transport of 13 via endocytosis.
Figure 8.
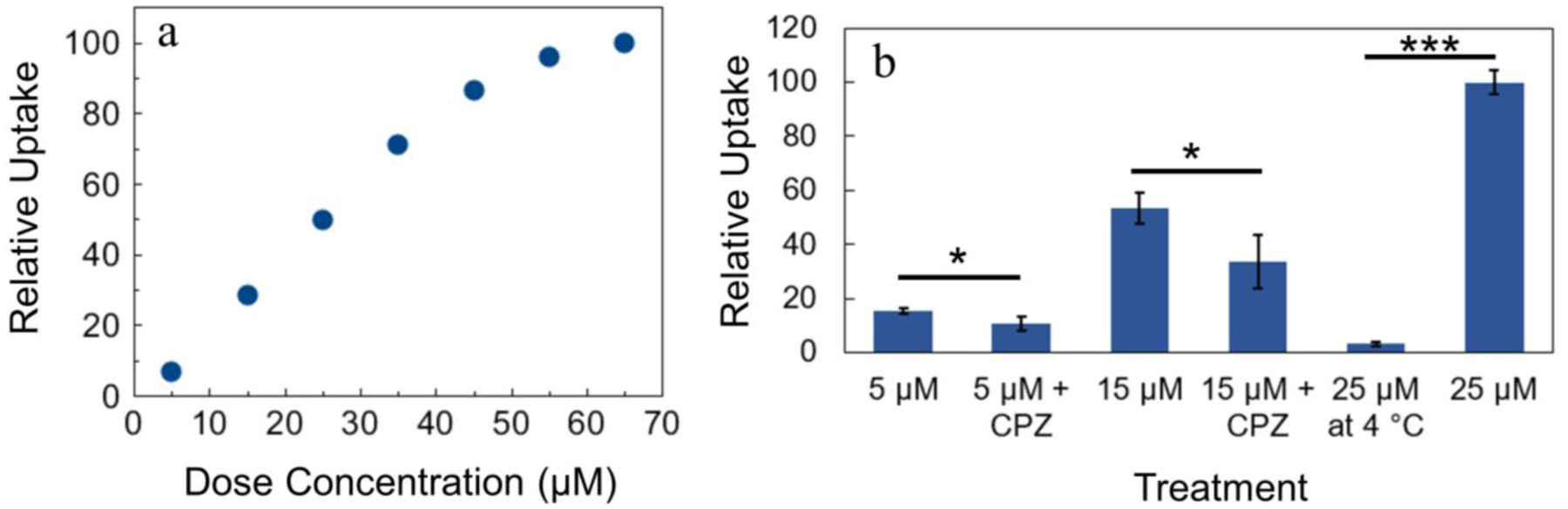
Relative uptake of 13 by investigating a) dose concentration and b) mechanism of uptake (all treatments lasted 4 h). For Student’s t-test analysis, p < 0.05 (*) or p < 0.001 (***). Error bars represent one standard deviation from three trials. CPZ represents 25 μM chlorpromazine. For the four total 5 μM and 15 μM treatments in b), propidium iodide was used to gate only live cells because dead cells may have different uptake, and chlorpromazine was somewhat toxic to the cells.
NCI-60 Screening.
To understand the activity of 13 in comparison to a wide range of validated anticancer drugs, compound 13 was submitted for analysis in the NCI-60 tumor cell panel screen.96 In this screening service, the compound is administered in a single-dose of 10 μM to a range of 60 different cancer cell lines. The relative cytotoxicity of a drug candidate in this diverse set of cell lines may reveal cancer types that are particularly susceptible to the tested compound. Additionally, the unique spectrum of activity of a given compound may be correlated with other drug candidates within the NCI database. The results of the NCI-60 single-dose screen are shown in Figure S39. They reveal that 13 is highly effective in all leukemia cell lines tested. This compound also exhibits potent activity in the lung cancer cell line NCI-H522, the melanoma cell line LOX IMVI, and the triple negative breast cancer cell line MDA-MB-468. The COMPARE algorithm, which quantitatively correlates activity spectra in the 60 cell lines of different drug candidates, was carried out for 13. These results are shown in Table 4. The similarity between different compounds is given by the Pearson correlation coefficient (PCC); values close to 1 indicate a high degree of similarity between drug candidates. The highest correlations for 13 are the natural products macbecin II (PCC 0.649) and rifamycin SV (PCC 0.625). Notably, the platinum(IV) drug candidate iproplatin97 is the only metal-containing compound to correlate with the spectrum of activity of 13.98
Table 4.
COMPARE analysis results for 13 based on the NCI-60 screening data.
| Pearson Correlation Coefficient (PCC) | Compound | NSC Number |
|---|---|---|
| 0.649 | macbecin II | S330500 |
| 0.625 | rifamycin SV | S133100 |
| 0.605 | L-cysteine analogue | S303861 |
| 0.585 | pibenzimol hydrochloride | S322921 |
| 0.572 | diglycoaldehyde | S118994 |
| 0.572 | actinomycin D | S3053 |
| 0.557 | CHIP (iproplatin) | S256927 |
| 0.557 | anguidine | S141537 |
| 0.550 | paclitaxel (Taxol) | S125973 |
| 0.541 | 5-azacytidine | S102816 |
Synthesis and In Vivo Evaluation of the 99mTc-Analogue of 13.
Tc is the lighter congener of Re, and exhibits similar chemistry. This similarity enables the use of 99mTc analogues of these rhenium anticancer agents as diagnostic partners for SPECT imaging or biodistribution studies. To assess in vivo behavior of 13, we synthesized the 99mTc analogue 13*. Compound 13* was prepared from the well-known precursor [99mTc(H2O)3(CO)3]+ and the dmphen ligand (Figure S40), and purified using preparative HPLC (Figure S41). After removal of the organic solvent, 13* was reconstituted and administered to naïve C57Bl6 mice via tail vein catheter simultaneously with a 0.10 μmol/kg dose of 13. Biodistribution was carried out at 30, 60 and 120 minutes post injection. Residual activity in select organs, tissues, and fluids (blood, heart, liver, kidney, ovaries, bone, muscle, urine) was quantified (Figure 9 and Table S1). We observed rapid renal and hepatic clearance of 13*. No significant non-specific uptake was observed in any organs studied, paving the way for future studies of the distribution of 13* in models of disease.
Figure 9.
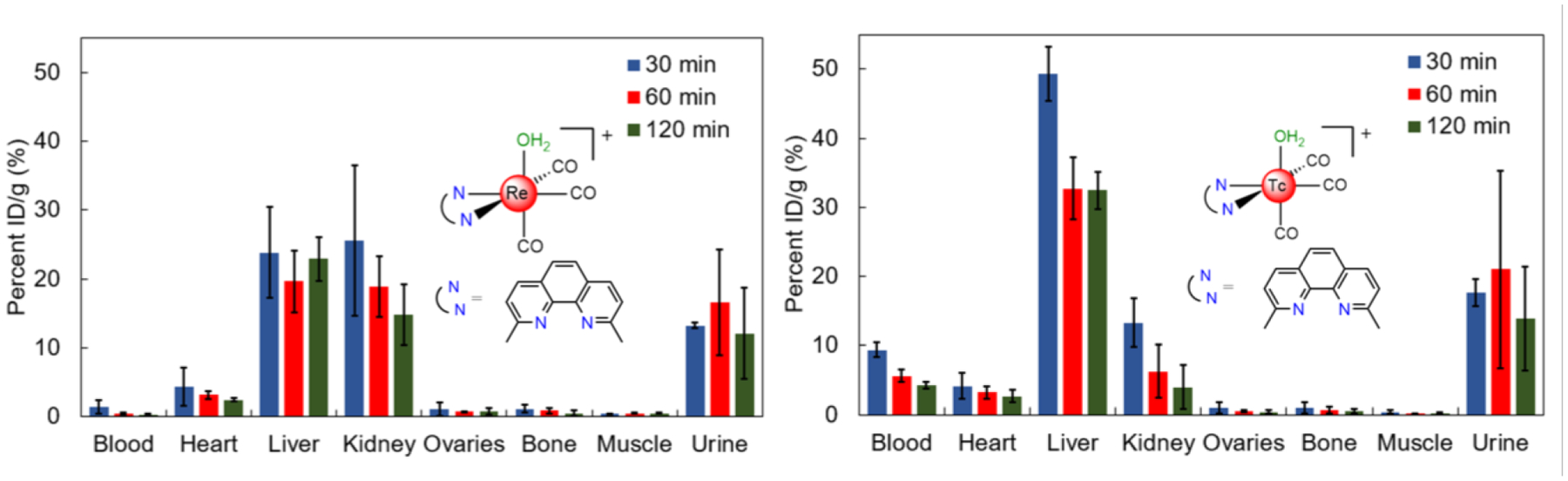
Biodistribution of the Re and 99mTc tricarbonyl complexes with the dmphen ligand using ICP-MS to detect rhenium and a gamma counter to detect technetium.
Biodistribution and Metabolite Analysis of 13.
We also assessed biodistribution and the metabolic profile of 13 in naïve C57Bl6 mice. After allowing for decay of 99mTc, the rhenium concentration in select organs, tissues, and fluids (blood, heart, liver, kidney, ovaries, bone, muscle, urine) was quantified using inductively coupled plasma–mass spectrometry (ICP-MS). Biodistribution of 13 revealed comparable behavior to 13* in most organs (Figure 9), suggesting the suitability of using the 99mTc analogue as a diagnostic partner. Notably, 13 exhibits higher uptake in the kidneys and accelerated blood clearance properties than 13*.
Additionally, fractions of blood plasma and urine from each time point were collected and subjected to analysis using HPLC-coupled ICP-MS detecting Re-species (Figure 10). Traces of samples collected at 30, 60 and 120 minutes, as well as reference traces of 13 with H2O as the axial ligand (aqua-) and 6 with Cl– as the axial ligand (chlorido-). Both blood plasma and urine analysis show similar trends. In vivo, most of intact 13 experiences an exchange of the axial aqua ligand to the chloride, as indicated by a shift in retention time from 13.8 to 15.6 minutes, with a small fraction of aqua complex detectable. Furthermore, two distinct metabolite peaks are observed: a hydrophilic species (3.3 min) and a more lipophilic species (13.6 min). At later time points, a relative increase of the hydrophilic species is observed, but both aqua and chlorido species of 13 can be detected in both blood plasma and urine at all time points. The presence of 13 or its chlorido form 6 at all time points suggests that this complex may reach tumor cells in vivo prior to decomposition.
Figure 10.
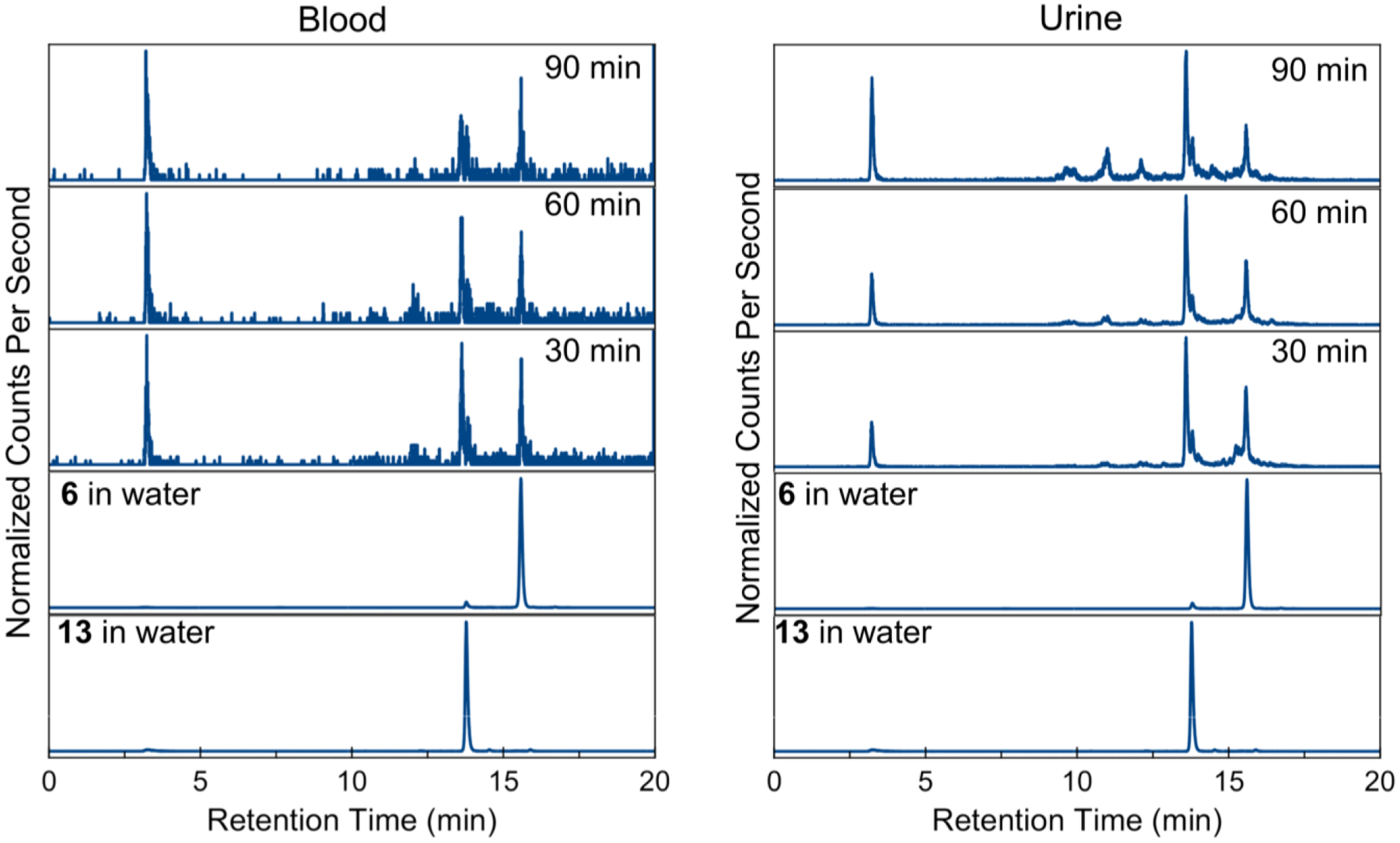
Normalized HPLC-ICP-MS traces of analysis of blood serum (left) and urine (right) of 13 in mice at the indicated time points.
Discussion
Although the platinum-based drugs have been a mainstay in first-line chemotherapy for decades, their toxic side effects and susceptibility to resistance remain significant challenges for their ongoing use in the clinical setting. These limitations have driven the search for alternative metal-based drugs, efforts that have led to the clinical trials of titanium, gallium, and ruthenium complexes.8 More recently, complexes of rhenium have emerged as alternatives for the traditional platinum-based drugs.99 For example, an increasing number of rhenium compounds with IC50 values under 50 μM in cancer cell lines have been discovered.99,100
In this study, we developed a small library of rhenium(I) tricarbonyl aqua complexes and evaluated their anticancer potential. Structural variety of these complexes was provided by seven different diimine ligands, each bearing functional groups with different electronic-withdrawing and lipophilic properties (Scheme 1). These compounds were characterized by standard techniques, including 1H NMR spectroscopy, mass spectrometry, and X-ray crystallography (Figure 2). Both 1H NMR spectroscopy and HPLC indicate that the axial aqua ligand is relatively labile, and subject to substitution with coordinating solvents, such as MeCN, or anions, such as chloride. The aqua ligands were employed to confer increased aqueous solubility to these complexes. However, the aqua ligands also potentially introduce another complication of acid-base chemistry via deprotonation of the coordinated water to form a hydroxide. The ligand substitution kinetics and therefore the biological activity of the aqua and hydroxido species are expected to be substantially different. For the related fac-[Re(CO)3(OH2)3]+ complex, the pKa value of the coordinated water is 7.5.101 Assuming that the rhenium(I) aqua complexes studied here have a similar pKa value, the complexes will exist in approximately a 50:50 mixture of the aqua and hydroxido forms at physiological pH. The studies in this manuscript therefore represent the composite effects of these two species under biological conditions.
These compounds were initially screened in HeLa cells (Figure 3). The IC50 values of the compounds span a wide range, depending on the nature of the coordinated diimine ligand. Some structure-activity relationships for this class of compounds can be discerned from this study. Namely, compound 11, which bears the diester-bpy ligand, is the least active. Thus, the presence of electron-withdrawing functional groups on the diimine ligand may act to reduce the biological activity of the complex. The lipophilicities of the complexes were determined using HPLC capacity factors to examine its relation to the biological activity as well. The capacity factor of 13 is the second largest among the seven compounds. The most lipophilic compound is 14, which bears the large 4,7-diphenyl-1,10-phenanthroline ligand. The cytotoxicity of 14, however, is substantially diminished relative to that of 13. Therefore, a direct correlation of lipophilicity with anticancer activity is not observed for this class of compounds.
The chlorido complexes bearing bpy (1), phen (5), and dmphen (6) diimine ligands were previously investigated for anticancer activity in PC-3 (prostate cancer), MCF-7 (breast cancer), and H522 (lung cancer) cell lines.58,102 Complex 6 was the most active, consistent with our studies of the aqua analogue 13. Notably, the use of the aqua complexes in this study enabled us to dissolve the compounds in pure water prior to dissolution in culture medium. By contrast, the chlorido analogues were diluted from DMSO stock solutions. Because the presence of DMSO may alter the biological activity of metal-based anticancer agents, caution should be taken when using this solvent for any new class of compounds.103,104
Based on the initial screening in HeLa cells, the most active compounds, 9, 10, and 13, were further evaluated in cisplatin-resistant cell lines. Because platinum resistance represents a significant problem in the clinic, the development of new metal-based drugs that are not cross-resistant to cisplatin is of significant importance. Cisplatin was 36 times less effective in resistant ovarian cancer (A2780CP70)63 and cervical cancer (KBCP20)61,62 cell lines compared to the parental wild-type cell lines (Table 3). Additionally, in the lung cancer cell lines, cisplatin was 4.1 times less effective in resistant A549 cells and 4.5 times less effective in resistant H460 cells.64 By contrast, the activity of the rhenium complexes in the cisplatin-resistant cell lines was always equivalent to that in the wild-type cells, with the exception of 10 in KBCP20 cells. These results indicate that this class of rhenium complexes can broadly circumvent platinum resistance mechanisms in a wide range of cancer types. Platinum resistance is multi-factorial, entailing decreased drug uptake, increased glutathione production, and increased DNA repair capacity.63,105,106 The origin of the lack of cross-resistance of the rhenium complexes with cisplatin is not clear, indicating that these rhenium complexes are operating by different mechanisms of action. Non-cancerous MRC-5 lung fibroblasts were used as a model for healthy cells. The cytotoxic activities of 9, 10, and 13 in this cell line were typically 2–4 fold lower than in HeLa, KB-3-1, and A2780 cells, but possessed similar toxicity in A549 and H460 cells. For comparison, cisplatin was about 2–7 times more cytotoxic in MRC-5 cells than in the HeLa, A549, and H460 cancer cell lines. This result suggests that this rhenium compound class may possess favorable therapeutic indices for further in vivo applications.
The interaction of the most potent complex, the dmphen aqua complex 13, with relevant biological nucleophiles, was explored to investigate potential biological targets. Metal complexes typically bind to guanine in RNA or DNA, or amino acids such as histidine and cysteine.107 By HPLC, 13 has high affinity for 9-ethylguanine, N-acetyl cysteine, and N-acetyl histidine, but does not show substantial binding to the other amino acids tested including methionine (Figure 4). Notably, 13 binds to 9-ethylguanine more rapidly than N-acetyl cysteine and N-acetyl histidine. Rhenium(I) tricarbonyl complexes, like 13, are known to interact with guanine38 in DNA35,36 and histidine residues in proteins;108–111 the interaction of such complexes with cysteine, however, is less documented.112 These studies suggest that cysteine residues may also be important intracellular targets.
Compound 13 was further investigated to probe its mechanism of action. Using microscopy, cell viability assays, flow cytometry assays, and Western blotting, it was concluded that 13 induces a non-canonical form of cell death. The details of this cell death and its failure to fit well-characterized cell death modes is explained in this section. First, confocal fluorescence microscopy was used to image the yellow 3MLCT luminescence of 13 directly in living HeLa cells (Figure 5). The fluorescence microscope images reveal that 13 induces cytoplasmic vacuolization and is localized diffusely throughout the cytosol and within the membranes of these vacuoles. Interestingly, no nuclear accumulation is seen despite the similarity of the rhenium complexes to platinum-based drugs in their ability to bind to 9-ethylguanine. Co-localization studies (Figure 6 and Figure S22) with LysoTracker Red DND-99 and the RFP-Rab5 fusion protein suggest that these vacuoles are endolysosomal in origin. The possibility of these vacuoles arising from the endoplasmic reticulum or from autophagosomes during autophagy was ruled out by colocalization studies employing an ER-localizing RFP-STIM1 fusion protein and an autophagosome-localizing RFP-LC3 marker (Figure S23 and S24). The yellow luminescence of 13 present in the vacuoles shows no overlap with either of these markers. Furthermore, microscopy images obtained of cells treated with 13 in the presence of the autophagy inhibitor 3-methyladenine still display the characteristic cytoplasmic vacuolization, further eliminating the possibility of these structures as autophagosomes (Figure S23). Like autophagy, paraptosis is a mode of cell death that proceeds in part via cytoplasmic vacuolization. The vacuoles formed during paraptosis are derived from the ER.84 Based on the negative result for colocalization with the RFP-STIM1 fusion protein, paraptosis as a mechanism of cell death may be ruled out as well. Additionally, treatment of the cells with the protein synthesis inhibitor cycloheximide or the Ca2+ channel inhibitor 2-aminoethoxydiphenyl borate, which are both known to inhibit paraptosis,84 failed to prevent formation of the vacuoles upon exposure to 13. These results further confirm that the cytoplasmic vacuolization does not arise from the induction of paraptosis.
The mechanism of cell death was also investigated by evaluating the cytotoxicity of 13 in the presence of various cell death inhibitors. Consistent with the imaging studies described above, 3-methyladenine and cycloheximide had no effect on the cytotoxicity of 13 (Figure S32) further ruling out autophagy and paraptosis as the mechanism of cell death. Because necroptosis, a regulated form of necrosis, was characterized as the cell death pathway induced by rhenium(V)-oxo compounds,88 the cytotoxicity of 13 was probed in the presence of the necroptosis inhibitor necrostatin-1. Necrostatin-1 had no effect on the cytotoxic activity of 13, suggesting that necroptosis is not operative. Because the vacuoles induced by 13 are endolysosomal in origin, the possibility of cell death induced by lysosomal proteases was investigated with the serine and cysteine protease inhibitor leupeptin. Again, no decrease in the cytotoxic effects of 13 was observed in the presence of this protease inhibitor. Caspases are proteases that regulate programmed cell death. Their activation is implicated in apoptosis, and their downregulation in cancer cells has been linked to drug resistance. The use of the pan-caspase inhibitor Z-VAD-FMK revealed that 13 retains its cytotoxicity when caspases are inhibited and therefore induces cell death in a caspase-independent manner (Figure 7).
Western blots were performed to evaluate protein expression levels that might be altered by different cell death modes. A Western blot for PARP and cleaved PARP in HeLa cells treated with 13 showed no significant alteration of the expression levels of these proteins, further ruling out apoptosis (Figure S34). Levels of LC3 were also unaffected by 13, indicating that autophagy was not operative. Western blots for ERK and p-ERK, proteins activated from ER stress related to paraptosis,94 showed no change in expression level either. These studies validate the novel mode of cell death induced by 13.
Further studies were carried out to investigate the potential role of ROS and depolarization of the MMP in mediating the cell death induced by 13. Compound 13 did not lead to an increase in intracellular ROS (Figure S28 and S29) nor did it depolarize the MMP (Figure S30 and S31). Compound 13 did give rise to flipping of phosphatidylserine to the outer membrane (Figure S27). Both paraptosis and necrosis give rise to an overproduction of ROS within the cell113–116 and apoptosis is known to depolarize the MMP.117,118 Thus, the cell death mechanism of 13 does not categorically fit within any of these descriptions. Although the flipping of phosphatidylserine is usually associated with apoptosis, alternative forms of cell death such as necrosis may also give rise to this phenomenon.119 Therefore, although 13 produces a small population of annexin positive living cells, it is not caused by apoptosis based on the other assays showing non-apoptotic characteristics of cell death. Additionally, the annexin/PI histogram of 13 is much different than that of etoposide, a known apoptosis-inducer.
Cell cycle analysis indicates that 13 arrests cells in the G2/M phases (Figure S26) implying that it may have antimetastatic effects. Anticancer drugs like celastrol93 and taxol120 also inhibit cells in these phases. By contrast, cisplatin, a DNA-binding agent, inhibits cells predominantly in the S-phase (Figure S25).
Thus far, there have been few studies that investigate the mechanism of cell death induced by potential rhenium anticancer agents. These investigations reveal a diverse range of pathways possible for these compounds. Bis(quinoline) rhenium(I) tricarbonyl complexes give rise to both apoptosis and necrosis.24,26 A diimine rhenium(I) tricarbonyl complex designed as a histone deacetylase inhibitor induces paraptosis.15 Rhenium N-heterocyclic carbene complexes induce caspase-independent cell death associated with cell cycle arrest, similar to 13.9 Higher oxidation state rhenium complexes have also been investigated, namely rhenium(IV) compounds bearing a chelating diimine and four chloride ligands that kill cells via apoptosis,121 and rhenium(V) oxo complexes that give rise to necroptosis, a regulated form of necrosis.88 Despite the array of investigations carried out in this work, the cell death mechanism for 13 remains uncertain. It is possible that 13 gives rise to an as-of-yet uncharacterized mode of cell death. The implications of this feature for the potential use of 13 as an anticancer drug are uncertain, but the fact that this compound is able to circumvent cisplatin resistance and kill cells independently of caspase function suggests its potential in anticancer therapy.
The cellular uptake of 13 was investigated using flow cytometry, capitalizing on its inherent luminescence properties. The cellular uptake of 13 exhibits saturation behavior at high concentrations and is substantially decreased at 4 °C (Figure 8). These results collectively indicate that 13 enters cells via active transport. This result is also consistent with the lack of correlation of the cytotoxicity with lipophilicity, as this correlation holds primarily for compounds that enter cells via passive diffusion. Treatment of cells with the clathrin-coated pit endocytosis inhibitor chlorpromazine122 decreased cell uptake, further suggesting that 13 is taken up via endocytosis. This result is significant because the confocal fluorescence microscope images indicate that 13 localizes to the enlarged endolysosomes. These enlarged vacuoles may therefore be a consequence of the endocytotic uptake of 13. The mechanism of cell uptake has been explored for several related rhenium complexes. Rhenium compounds decorated with fructose, for example, are taken up actively by a fructose transporter.28 Similarly, a glucose analogue enters the cells via the GLUT transporter.123 A hydroxamic acid-functionalized rhenium tricarbonyl complex is taken up via an active but non-endocytotic pathway.15 The results determined here for 13, which bears no additional targeting group, suggest that endocytotic uptake may be the default uptake pathway for such rhenium(I) tricarbonyl complexes.
The NCI-60 screening results used in conjunction with the COMPARE algorithm (Table 4) relates 13 to several organic natural products. The top correlations arise for macbecin II and rifamycin SV. The lack of any strong correlations with the FDA-approved platinum-based drugs confirm that 13 acts via a very different mechanism of action. Macbecin II is a well-characterized inhibitor of heat shock protein 90 (Hsp90),124 and rifamycin SV is an inhibitor of DNA-dependent RNA polymerase.125 Hsp90 is a highly abundant cytosolic protein that is involved in protein folding.126 It is overexpressed in leukemia and other cancer types127 and has recently arisen as a promising drug target.128 Notably, rifamycin SV is also known to possess Hsp90 inhibitory properties.129 Thus, the high correlation between macbecin II, rifamycin SV, and 13 suggests that Hsp90 could be a common molecular target.
To validate the potential of this compound for clinical use, the in vivo properties of 13 and its 99mTc analogue 13* were investigated to determine the metabolic outcomes of the complexes. When injected simultaneously in the same animal, both complexes exhibit similar profiles in biodistribution studies in mice (Figure 9). This result is somewhat surprising given the much faster ligand substitution kinetics of Tc(CO)3 complexes compared to Re(CO)3 complexes,130 but bodes well for the potential use of 99mTc analogues as diagnostic partners. Consistent with previous studies on 99mTc(CO)3 complexes,131,132 both compounds undergo renal and hepatobiliary modes of excretion, further evidenced by Re and 99mTc in the urine and liver. Metabolite analysis of 13 using LC-ICP-MS reveals the presence of four main Re-species at all time points analyzed (Figure 10); in vivo, most of 13 experiences exchange of the axial aqua ligand to chloride, as well as the conversion to two more hydrophilic metabolites of unknown nature. The presence of intact 13 in vivo at all time points suggests that this compound can access tumor sites prior to decomposition. Ongoing studies are aimed at evaluating the in vivo anticancer activity of this novel compound.
Conclusion
A systematic study on the anticancer potential of rhenium(I) tricarbonyl complexes was performed. These efforts revealed that this class of compounds exhibit in vitro anticancer activity. Compound 13 was discovered as a new lead candidate, as it is more potent than the established metal-based anticancer drug cisplatin. In addition, this complex overcomes cisplatin resistance and is trackable by luminescence imaging. Its framework also allows for the facile synthesis of the 99mTc analogue for diagnostic imaging, which has been used in this study to determine biodistribution, and will facilitate future in vivo studies. Mechanistic studies on 13 indicate that it induces caspase-independent cell death accompanied by cytoplasmic vacuolization. Categorization of the cell death in one of the canonical modes was unsuccessful, suggesting that 13 may be inducing cytotoxicity via a novel pathway. The molecular target of 13 and related rhenium(I) complexes also remains uncertain. Our current efforts are aimed towards identifying the target of 13 and pursuing the anticancer properties of these compounds within in vivo models.
Supplementary Material
Acknowledgements
This work was supported by Cornell University and by the Office of the Assistant Secretary of Defense for Health Affairs through the Ovarian Cancer Research Program under Award No. W81XWH-17-1-0097. B.L.M. acknowledges support for undergraduate summer research from the Gerald A. Hill and Kathleen Holmes Hill Fellowship at Cornell. J.M.B. acknowledges support from the National Institutes of Health (R00GM110121). E.B. acknowledges the NHLBI for support (K99HL125728). LCICP instrumentation at Massachusetts General Hospital was funded by NIH, the office of the director (S10OD010650). This work made use of the NMR facility at Cornell University, which is supported by the NSF under award number CHE-1531632. This work also made use of the flow cytometer and Zeiss LSM 880 microscope (NYSTEM CO29155 and NIH S10OD018516) at the Cornell University Biotechnology Resource Center (BRC). We also thank the Hu and Selvaraj labs at Cornell University for sharing DNA plasmids and reagents. Ms. Nicole Spiegelman is thanked for assistance with Western blot experiments.
Footnotes
Supporting Information. The Supporting Information is available free of charge on the ACS publications website at http://pubs.acs.org.
Experimental procedures, characterization data, cell viability curves, microscope images, flow cytometry plots and histograms, NCI-60 single-dose mean graph, and crystal data tables (PDF)
Crystallographic data (CIF).
References
- (1).Siegel RL; Miller KD; Jemal A CA Cancer J. Clin 2017, 67, 7–30. [DOI] [PubMed] [Google Scholar]
- (2).Galanski MA; Jakupec M; Keppler BK Curr. Med. Chem 2005, 12, 2075–2094. [DOI] [PubMed] [Google Scholar]
- (3).Jamieson ER; Lippard SJ Chem. Rev 1999, 99, 2467–2498. [DOI] [PubMed] [Google Scholar]
- (4).Todd RC; Lippard SJ Metallomics 2009, 1, 280–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Hartmann JT; Lipp H-P Expert Opin. Pharmacother 2003, 4, 889–901. [DOI] [PubMed] [Google Scholar]
- (6).Galluzzi L; Senovilla L; Vitale I; Michels J; Martins I; Kepp O; Castedo M; Kroemer G Oncogene 2012, 31, 1869–1883. [DOI] [PubMed] [Google Scholar]
- (7).Cheff DM; Hall MD J. Med. Chem 2017, 60, 4517–4532. [DOI] [PubMed] [Google Scholar]
- (8).Ott I; Gust R Arch. Pharm. Chem. Life Sci 2007, 340, 117–126. [DOI] [PubMed] [Google Scholar]
- (9).Simpson PV; Casari I; Paternoster S; Skelton BW; Falasca M; Massi M Chem. Eur. J 2017, 23, 6518–6521. [DOI] [PubMed] [Google Scholar]
- (10).Chakraborty I; Carrington SJ; Roseman G; Mascharak PK Inorg. Chem 2017, 56, 1534–1545. [DOI] [PubMed] [Google Scholar]
- (11).Carreño A; Aros AE; Otero C; Polanco R; Gacitúa M; Arratia-Pérez R; Fuentes JA New J. Chem 2017, 41, 2140–2147. [Google Scholar]
- (12).Raszeja LJ; Siegmund D; Cordes AL; Güldenhaupt J; Gerwert K; Hahn S; Metzler-Nolte N Chem. Commun 2017, 3, 905–908. [DOI] [PubMed] [Google Scholar]
- (13).Collery P; Santoni F; Ciccolini J; Tran TNN; Mohsen A; Desmaele D Anticancer Res. 2016, 36, 6051–6058. [DOI] [PubMed] [Google Scholar]
- (14).Kumar CA; Nagarajaprakash R; Victoria W; Veena V; Sakthivel N; Manimaran B Inorg. Chem. Commun 2016, 64, 39–44. [Google Scholar]
- (15).Ye R-R; Tan C-P; Lin Y-N; Ji L-N; Mao Z-W Chem. Commun 2015, 51, 8353–8356. [DOI] [PubMed] [Google Scholar]
- (16).Collery P; Mohsen A; Kermagoret A; Corre S; Bastian G; Tomas A; Wei M; Santoni F; Guerra N; Desmaële D; d’Angelo J Invest. New Drugs 2015, 33, 848–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Balakrishnan G; Rajendran T; Senthil K; Sathish M; Kamaraj V; Ganesan M Inorg. Chim. Acta 2015, 434, 51–59. [Google Scholar]
- (18).Medley J; Payne G; Banerjee HN; Giri D; Winstead A; Wachira JM; Krause JA; Shaw R; Pramanik SK; Mandal SK Mol. Cell. Biochem 2015, 398, 21–30. [DOI] [PubMed] [Google Scholar]
- (19).Clède S; Lambert F; Saint-Fort R; Plamont MA; Bertrand H; Vessières A; Policar C Chem. Eur. J 2014, 20, 8714–8722. [DOI] [PubMed] [Google Scholar]
- (20).Collery P; Bastian G; Santoni F; Mohsen A; Wei M; Collery T; Tomas A; Desmaele D; D’Angelo J Anticancer Res. 2014, 34, 1679–1690. [PubMed] [Google Scholar]
- (21).Langdon-Jones EE; Symonds NO; Yates SE; Hayes AJ; Lloyd D; Williams R; Coles SJ; Horton PN; Pope SJ A. Inorg. Chem 2014, 53, 3788–3797. [DOI] [PubMed] [Google Scholar]
- (22).Kaplanis M; Stamatakis G; Papakonstantinou VD; Paravatou-petsotas M; Demopoulos CA; Mitsopoulou CA J. Inorg. Biochem 2014, 135, 1–9. [DOI] [PubMed] [Google Scholar]
- (23).Parson C; Smith V; Krauss C; Banerjee HN; Reilly C; Krause JA; Wachira JM; Giri D; Winstead A; Mandal SK Br. J. Pharm. Res 2014, 4, 362–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Leonidova A; Pierroz V; Adams LA; Barlow N; Ferrari S; Graham B; Gasser G ACS Med. Chem. Lett 2014, 5, 809–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wähler K; Ludewig A; Szabo P; Harms K; Meggers E Eur. J. Inorg. Chem 2014, 2014, 807–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Kitanovic I; Can S; Alborzinia H; Kitanovic A; Pierroz V; Leonidova A; Pinto A; Spingler B; Ferrari S; Molteni R; Steffen A; Metzler-Nolte N; Wölfl S; Gasser G Chem. Eur. J 2014, 20, 2496–2507. [DOI] [PubMed] [Google Scholar]
- (27).Leonidova A; Pierroz V; Rubbiani R; Heier J; Ferrari S; Gasser G Dalton Trans. 2014, 43, 4287–4297. [DOI] [PubMed] [Google Scholar]
- (28).Yin Zhang K; Ka-Shun Tso K; Louie MW; Liu HW; Kam-Wing Lo K Organometallics 2013, 32, 5098–5102. [Google Scholar]
- (29).Redshaw C; Watkins S; Humphrey SM; Bulman Page PC; Ashby S; Chao Y; Herbert CJ; Mueller A RSC Adv. 2013, 3, 23963–23966. [Google Scholar]
- (30).Choi AW-T; Louie M-W; Li SP-Y; Liu H-W; Chan BT-N; Lam TC-Y; Lin AC-C; Cheng S-H; Lo KK-W Inorg. Chem 2012, 51, 13289–13302. [DOI] [PubMed] [Google Scholar]
- (31).Lo KK-W; Zhang KY; Li SP-Y Eur. J. Inorg. Chem 2011, 2011, 3551–3568. [Google Scholar]
- (32).Kermagoret A; Morgant G; d’Angelo J; Tomas A; Roussel P; Bastian G; Collery P; Desmaële D Polyhedron 2011, 30, 347–353. [Google Scholar]
- (33).Rajagopalan R; Grummon GD; Bugaj J; Hallemann LS; Webb EG; Marmion ME; Vanderheyden JL; Srinivasan A Bioconjugate Chem. 1997, 8, 407–415. [DOI] [PubMed] [Google Scholar]
- (34).Takeda H; Ishitani O Coord. Chem. Rev 2010, 254, 346–354. [Google Scholar]
- (35).Zobi F; Blacque O; Sigel RKO; Alberto R Inorg. Chem 2007, 46, 10458–10460. [DOI] [PubMed] [Google Scholar]
- (36).Zobi F; Spingler B; Alberto R ChemBioChem 2005, 6, 1397–1405. [DOI] [PubMed] [Google Scholar]
- (37).Zobi F; Spingler B; Fox T; Alberto R Inorg. Chem 2003, 42, 2818–2820. [DOI] [PubMed] [Google Scholar]
- (38).Oriskovich TA; White PS; Thorp HH Inorg. Chem 1995, 34, 1629–1631. [Google Scholar]
- (39).Salignac B; Grundler PV; Cayemittes S; Frey U; Scopelliti R; Merbach AE; Hedinger R; Hegetschweiler K; Alberto R; Prinz U; Raabe G; Kölle U; Hall S Inorg. Chem 2003, 42, 3516–3526. [DOI] [PubMed] [Google Scholar]
- (40).Lo KK-W Acc. Chem. Res 2015, 48, 2985–2995. [DOI] [PubMed] [Google Scholar]
- (41).Clède S; Policar C Chem. Eur. J 2015, 21, 942–958. [DOI] [PubMed] [Google Scholar]
- (42).Jürgens S; Herrmann WA; Kühn FE J. Organomet. Chem 2014, 751, 83–89. [Google Scholar]
- (43).Kurz P; Probst B; Spingler B; Alberto R Eur. J. Inorg. Chem 2006, 2006, 2966–2974. [Google Scholar]
- (44).Smieja JM; Kubiak CP Inorg. Chem 2010, 49, 9283–9289. [DOI] [PubMed] [Google Scholar]
- (45).Chan CY; Pellegrini PA; Greguric I; Barnard PJ Inorg. Chem 2014, 53, 10862–10873. [DOI] [PubMed] [Google Scholar]
- (46).Gottschaldt M; Koth D; Müller D; Klette I; Rau S; Görls H; Schäfer B; Baum R; Yano S Chem. Eur. J 2007, 13, 10273–10280. [DOI] [PubMed] [Google Scholar]
- (47).Domínguez SE; Alborés P; Fagalde F Polyhedron 2014, 67, 471–480. [Google Scholar]
- (48).Tzeng B; Chen B; Chen C; Chang Y; Tzeng W; Lin T; Lee G; Chou P; Fu Y; Chang AH Inorg. Chem 2011, 50, 5379–5388. [DOI] [PubMed] [Google Scholar]
- (49).Coe BJ; Foxon SP; Pilkington RA; Sánchez S; Whittaker D; Clays K; Van Steerteghem N; Brunschwig BS Organometallics 2016, 35, 3014–3024. [Google Scholar]
- (50).Chiarella GM; Cotton FA; Dalal NS; Murillo CA; Wang Z; Young MD Inorg. Chem 2012, 51, 5257–5263. [DOI] [PubMed] [Google Scholar]
- (51).Rodríguez MC; Bravo J; Freijanes E; Oñate E; Soledad G-F; Rodríguez-Seoane P Polyhedron 2004, 23, 1045–1053. [Google Scholar]
- (52).Mella P; Cabezas K; Cerda C; Cepeda M; Günther G; Pizarro NA; Vega A New J. Chem 2016, 40, 6451–6459. [Google Scholar]
- (53).Schutte M; Kemp G; Visser HG; Roodt A Inorg. Chem 2011, 50, 12486–12498. [DOI] [PubMed] [Google Scholar]
- (54).Rillema DP; Kirgan RA; Smucker B; Moore C Acta Crystallogr., Sect. E: Struct. Rep. Online 2007, 63, m1404–m1405. [Google Scholar]
- (55).Connick WB; Di Bilio AJ; Schaeffer WP; Gray HB Acta Crystallogr., Sect. C: Cryst. Struct. Commun 1999, 55, 913–916. [DOI] [PubMed] [Google Scholar]
- (56).Singh A; Anandhi U; Cinellu MA; Sharp PR Dalton Trans. 2008, 2314–2327. [DOI] [PubMed] [Google Scholar]
- (57).Hollis LS; Lippard SJ J. Am. Chem. Soc 1981, 103, 6761–6763. [Google Scholar]
- (58).Orsa DK; Nettles CR; Pramanik SK; Iwunze MO; Greco GE; Krause JA; Mandal SK In Handbook of Prostate Cancer Cell Research; Nova Science: Hauppauge, 2009; pp 323–362. [Google Scholar]
- (59).In OECD Guidelines for the Testing of Chemicals; OECD Publishing: Paris, 2004; pp 1–11. [Google Scholar]
- (60).Platts JA; Oldfield SP; Reif MM; Palmucci A; Gabano E; Osella DJ Inorg. Biochem 2006, 100, 1199–1207. [DOI] [PubMed] [Google Scholar]
- (61).Shen DW; Akiyama S; Schoenlein P; Pastan I; Gottesman MM Br. J. Cancer 1995, 71, 676–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Akiyama S; Fojo A; Hanover JA; Pastan I; Gottesman MM Somat. Cell Mol. Genet 1985, 11, 117–126. [DOI] [PubMed] [Google Scholar]
- (63).Godwin AK; Meister A; O’Dwyer PJ; Huang CS; Hamilton TC; Anderson ME Proc. Natl. Acad. Sci. U. S. A 1992, 89, 3070–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Barr MP; Gray SG; Hoffmann AC; Hilger RA; Thomale J; O’Flaherty JD; Fennell DA; Richard D; O’Leary JJ; O’Byrne KJ PLoS One 2013, 8, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Robertazzi A; Platts JA J. Biol. Inorg. Chem 2005, 10, 854–866. [DOI] [PubMed] [Google Scholar]
- (66).Thorp-Greenwood FL; Balasingham RG; Coogan MP J. Organomet. Chem 2012, 714, 12–21. [Google Scholar]
- (67).Fernandez-Moreira V; Thorp-Greenwood FL; Coogan MP Chem. Commun 2010, 46, 186–202. [DOI] [PubMed] [Google Scholar]
- (68).Lo KKW; Louie MW; Zhang KY Coord. Chem. Rev 2010, 254, 2603–2622. [Google Scholar]
- (69).Fernández-Moreira V; Thorp-Greenwood FL; Amoroso AJ; Cable J; Court JB; Gray V; Hayes AJ; Jenkins RL; Kariuki BM; Lloyd D; Millet CO; Williams CF; Coogan MP Org. Biomol. Chem 2010, 8, 3888–3901. [DOI] [PubMed] [Google Scholar]
- (70).Bucci C; Parton RG; Mather IH; Stunnenberg H; Simons K; Hoflack B; Zerial M Cell 1992, 70, 715–728. [DOI] [PubMed] [Google Scholar]
- (71).Gillooly DJ; Morrow IC; Lindsay M; Gould R; Bryant NJ; Gaullier JM; Parton RG; Stenmark H EMBO J 2000, 19, 4577–4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Luzio JP; Gray SR; Bright NA Biochem. Soc. Trans 2010, 38, 1413–1416. [DOI] [PubMed] [Google Scholar]
- (73).Luzio JP; Pryor PR; Bright NA Nat. Rev. Mol. Cell Biol 2007, 8, 622–632. [DOI] [PubMed] [Google Scholar]
- (74).Kimura S; Noda T; Yoshimori T Autophagy 2007, 3, 452–460. [DOI] [PubMed] [Google Scholar]
- (75).Senese S; Lo YC; Huang D; Zangle TA; Gholkar AA; Robert L; Homet B; Ribas A; Summers MK; Teitell MA; Damoiseaux R; Torres JZ Cell Death Dis 2014, 5, e1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Morgan D The Cell Cycle: Principles of Control; Lawrence E, Ed.; Sinauer Associates: London, 2007. [Google Scholar]
- (77).Zhang D; Piao H-L; Li Y-H; Qiu Q; Li D-J; Du M-R; Tsang BK Exp. Mol. Pathol 2016, 100, 506–513. [DOI] [PubMed] [Google Scholar]
- (78).Swift LH; Golsteyn RM Biol. Cell 2016, 108, 127–148. [DOI] [PubMed] [Google Scholar]
- (79).van Engeland M; Nieland L; Ramaekers F; Schutte B; Reutelingsperger C Cytometry 1998, 31, 1–9. [DOI] [PubMed] [Google Scholar]
- (80).Mizukami S; Kikuchi K; Higuchi T; Urano Y; Mashima T; Tsuruo T; Nagano T FEBS Lett. 1999, 453, 356–360. [DOI] [PubMed] [Google Scholar]
- (81).Zamzami N; Marchetti P; Castedo M; Decaudin D; Macho A; Hirsch T; Susin SA; Petit PX; Mignotte B; Kroemer GJ Exp. Med 1995, 182, 367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Kluck RM; Bossy-Wetzel E; Green DR; Newmeyer DD Science 1997, 275, 1132–1136. [DOI] [PubMed] [Google Scholar]
- (83).Maltese WA; Overmeyer JH Am. J. Pathol 2014, 184, 1630–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Yoon MJ; Lee AR; Jeong SA; Kim Y-S; Kim JY; Kwon Y-J; Choi KS Oncotarget 2014, 5, 6816–6831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Li B; Zhao J; Wang CZ; Searle J; He TC; Yuan CS; Du W Cancer Lett. 2011, 301, 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Guicciardi ME; Leist M; Gores GJ Oncogene 2004, 23, 2881–2890. [DOI] [PubMed] [Google Scholar]
- (87).Kieran D; Greensmith L Neuroscience 2004, 125, 427–439. [DOI] [PubMed] [Google Scholar]
- (88).Suntharalingam K; Awuah SG; Bruno PM; Johnstone TC; Wang F; Lin W; Zheng YR; Page JE; Hemann MT; Lippard SJ J. Am. Chem. Soc 2015, 137, 2967–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Flamme M; Cressey PB; Lu C; Bruno PM; Eskandari A; Hemann MT; Hogarth G; Suntharalingam K Chem. Eur. J 2017, 23, 9674–9682. [DOI] [PubMed] [Google Scholar]
- (90).Moriwaki K; Bertin J; Gough PJ; Orlowski GM; Chan FK M. Cell Death Dis 2015, 6, e1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Boulares AH; Yakovlev AG; Ivanova V; Stoica BA; Wang G; Iyer S; Smulson MJ Biol. Chem 1999, 274, 22932–22940. [DOI] [PubMed] [Google Scholar]
- (92).Tanida I; Ueno T; Kominami E In Autophagosome and Phagosome; Deretic V, Ed.; Humana Press: Totowa, NJ, 2008; pp 77–88. [Google Scholar]
- (93).Wang WB; Feng LX; Yue QX; Wu WY; Guan SH; Jiang BH; Yang M; Liu X; Guo DA J. Cell. Physiol 2012, 227, 2196–2206. [DOI] [PubMed] [Google Scholar]
- (94).Tripathy SK; De U; Dehury N; Laha P; Panda MK; Kim HS; Patra S Dalton Trans. 2016, 45, 15122–15136. [DOI] [PubMed] [Google Scholar]
- (95).Puckett CA; Ernst RJ; Barton JK Dalton Trans. 2010, 39, 1159–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Shoemaker RH Nat. Rev. Cancer 2006, 6, 813–823. [DOI] [PubMed] [Google Scholar]
- (97).Lira-Puerto V; Silva A; Morris M; Martinez R; Groshen S; Morales-Canfield F; Tenorio F; Muggia F Cancer Chemother. Pharmacol 1991, 28, 391–396. [DOI] [PubMed] [Google Scholar]
- (98).Huang R; Wallqvist A; Covell DG Biochem. Pharmacol 2005, 69, 1009–1039. [DOI] [PubMed] [Google Scholar]
- (99).Leonidova A; Gasser G ACS Chem. Biol 2014, 9, 2180–2193. [DOI] [PubMed] [Google Scholar]
- (100).Louie MW; Liu HW; Lam MHC; Lau TC; Lo KKW Organometallics 2009, 28, 4297–4307. [Google Scholar]
- (101).Egli A; Hegetschweiler K; Alberto R; Abram U; Schibli R; Hedinger R; Gramlich V; Kissner R; Schubiger PA Organometallics 1997, 16, 1833–1840. [Google Scholar]
- (102).Orsa DK; Haynes GK; Pramanik SK; Iwunze MO; Greco GE; Ho DM; Krause JA; Hill DA; Williams RJ; Mandal SK Inorg. Chem. Commun 2008, 11, 1054–1056. [Google Scholar]
- (103).Huang H; Humbert N; Bizet V; Patra M; Chao H; Mazet C; Gasser GJ Organomet. Chem 2016, 839, 15–18. [Google Scholar]
- (104).Hall MD; Telma KA; Chang KE; Lee TD; Madigan JP; Lloyd JR; Goldlust IS; Hoeschele JD; Gottesman MM Cancer Res. 2014, 74, 3913–3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Ferreira JA; Peixoto A; Neves M; Gaiteiro C; Reis CA; Assaraf YG; Santos LL Drug Resist. Updat 2016, 24, 34–54. [DOI] [PubMed] [Google Scholar]
- (106).Giaccone G Drugs 2000, 59, 9–17. [DOI] [PubMed] [Google Scholar]
- (107).Jovanović S; Petrović B; Bugarčić ŽD J. Coord. Chem 2010, 63, 2419–2430. [Google Scholar]
- (108).Zobi F; Spingler B Inorg. Chem 2012, 51, 1210–1212. [DOI] [PubMed] [Google Scholar]
- (109).Santoro G; Blacque O; Zobi F Metallomics 2012, 4, 253. [DOI] [PubMed] [Google Scholar]
- (110).Binkley SL; Leeper TC; Rowlett RS; Herrick RS; Ziegler CJ Metallomics 2011, 3, 909–916. [DOI] [PubMed] [Google Scholar]
- (111).Miller JE; Grǎdinaru C; Crane BR; Di Bilio AJ; Wehbi WA; Un S; Winkler JR; Gray HB J. Am. Chem. Soc 2003, 125, 14220–14221. [DOI] [PubMed] [Google Scholar]
- (112).Tavaré R; Williams J; Howland K; Blower PJ; Mullen GED J. Inorg. Biochem 2012, 114, 24–27. [DOI] [PubMed] [Google Scholar]
- (113).Ye R-R; Tan C-P; Chen M-H; Hao L; Ji L-N; Mao Z-W Chem. Eur. J 2016, 22, 7800–7809. [DOI] [PubMed] [Google Scholar]
- (114).Ghosh K; De S; Das S; Mukherjee S; Sengupta Bandyopadhyay S PLoS One 2016, 11, e0168488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Wang Y; Zhu X; Yang Z; Zhao X Biochem. Biophys. Res. Commun 2013, 430, 876–882. [DOI] [PubMed] [Google Scholar]
- (116).Xue X; Piao JH; Nakajima A; Sakon-Komazawa S; Kojima Y; Mori K; Yagita H; Okumura K; Harding H; Nakano HJ Biol. Chem 2005, 280, 33917–33925. [DOI] [PubMed] [Google Scholar]
- (117).Matsuyama S; Llopis J; Deveraux QL; Tsien RY; Reed JC Nat. Cell Biol 2000, 2, 318–325. [DOI] [PubMed] [Google Scholar]
- (118).Lemasters JJ; Nieminen A; Qian T; Trost LC; Elmore SP; Nishimura Y; Crowe RA; Cascio WE; Bradham CA; Brenner DA; Herman B Biochim. Biophys. Acta 1998, 1366, 177–196. [DOI] [PubMed] [Google Scholar]
- (119).Sawai H; Domae N Biochem. Biophys. Res. Commun 2011, 411, 569–573. [DOI] [PubMed] [Google Scholar]
- (120).Sun Q; Chen T; Wang XJ Cell. Physiol 2010, 222, 421–432. [DOI] [PubMed] [Google Scholar]
- (121).Martínez-Lillo J; Mastropietro TF; Lappano R; Madeo A; Alberto ME; Russo N; Maggiolini M; De Munno G Chem. Commun 2011, 47, 5283–5285. [DOI] [PubMed] [Google Scholar]
- (122).Pho MT; Ashok A; Atwood WJ J. Virol 2000, 74, 2288–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Louie MW; Liu HW; Lam MHC; Lam YW; Lo KK W. Chem. Eur. J 2011, 17, 8304–8308. [DOI] [PubMed] [Google Scholar]
- (124).Martin CJ; Gaisser S; Challis IR; Carletti I; Wilkinson B; Gregory M; Prodromou C; Roe SM; Pearl LH; Boyd SM; Zhang MJ Med. Chem 2008, 51, 2853–2857. [DOI] [PubMed] [Google Scholar]
- (125).Wehrli W; Knüsel F; Schmid K; Staehelin M Proc. Natl. Acad. Sci. U. S. A 1968, 61, 667–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Blagg BSJ; Kerr TD Med. Res. Rev 2006, 26, 310–338. [DOI] [PubMed] [Google Scholar]
- (127).Khajapeer KV; Baskaran R Leuk. Res. Treatment 2015, 2015, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Holzbeierlein JM; Windsperger A; Vielhauer G Curr. Oncol. Rep 2010, 12, 95–101. [DOI] [PubMed] [Google Scholar]
- (129).Davenport J; Balch M; Galam L; Girgis A; Hall J; Blagg B; Matts R Biology 2014, 3, 101–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Alberto R Eur. J. Inorg. Chem 2009, 2009, 21–31. [Google Scholar]
- (131).Yazdani A; Janzen N; Czorny S; Valliant JF Inorg. Chem 2017, 56, 2958–2965. [DOI] [PubMed] [Google Scholar]
- (132).Yazdani A; Janzen N; Banevicius L; Czorny S; Valliant JF Inorg. Chem 2015, 54, 1728–1736. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.