

Abstract
The glycosylation of proteins is typically considered as a stabilizing modification, including resistance to proteolysis. A class of peptidases, referred to as glycopeptidases or O-glycopeptidases, circumvent the protective effect of glycans against proteolysis by accommodating the glycans in their active sites as specific features of substrate recognition. IMPa from Pseudomonas aeruginosa is such an O-glycopeptidase that cleaves the peptide bond immediately preceding a site of O-glycosylation, and through this glycoprotein-degrading function contributes to the host-pathogen interaction. IMPa, however, is a relatively large multidomain protein and how its additional domains may contribute to its function remains unknown. Here, through the determination of a crystal structure of IMPa in complex with an O-glycopeptide, we reveal that the N-terminal domain of IMPa, which is classified in Pfam as IMPa_N_2, is a proline recognition domain that also shows the properties of recognizing an O-linked glycan on the serine/threonine residue following the proline. The proline is bound in the center of a bowl formed by four functionally conserved aromatic amino acid side chains while the glycan wraps around one of the tyrosine residues in the bowl to make classic aromatic ring-carbohydrate CH-π interactions. This structural evidence provides unprecedented insight into how the ancillary domains in glycoprotein-specific peptidases can noncatalytically recognize specific glycosylated motifs that are common in mucin and mucin-like molecules.
Keywords: IMPa, mucin, O-glycan, O-glycopeptidase, proline
Introduction
O-gycopeptidases are a growing class of enzyme that is able to hydrolyze the peptide backbone of O-glycosylated proteins, and do so in a manner that depends upon specific recognition of a glycan on the substrate. The MEROPS database classifies peptidases into families based on amino acid sequence relatedness. At present, O-glycopeptidases are found in families M26, M60, M66, M72, M88, M98 and S6 (Haurat et al., 2020; Nakjang et al., 2012; Noach et al., 2017; Shon et al., 2020; Yu et al., 2012). A common property of O-glycopeptidases is their presence in host-adapted microbes, such as bacteria that are commensals of gastrointestinal tract (e.g. Bacteroides and Akkermansia species) or that are notable pathogens (e.g. Pseudomonas aeruginosa, Streptococcus pneumoniae and Acinetobacter baumanii) (Haurat et al., 2020; Nakjang et al., 2012; Noach et al., 2017; Shon et al., 2020; Yu et al., 2012).
PA0572 from Pseudomonas aeruginosa was identified as a protease by its ability to cleave glycoproteins involved in leukocyte homing, thus compromising immune function and leading to the enzyme being called IMPa (immunomodulating protease of Pseudomonas aeruginosa) (Bardoel et al., 2012). Through its ability to cleave CD44, IMPa also inhibits phagocytosis (Tian et al., 2019). This secreted effector, therefore, is a potentially important factor in the host-pathogen interaction. The previously determined structures of IMPa shows that it adopts a fold comprising four domains (Figure 1A) (Noach et al., 2017). The N-terminal domain is classified in Pfam as “IMPa_N_2” (PF18650) and is a mixed α/β fold with a central β-sheet sandwiched by α-helices. This is followed by an “IMPa_helical” domain (PF18642) and an Ig-like domain that closely resembles “M60-like_N” domains (PF17291), which typically precede “Peptidase_M60” (PF13402) domains. The C-terminal domain of IMPa is a Peptidase_M60 domain that is classified into MEROPS family M88 and thereby contains the gluzincin active site motif typical of metallopeptidase clan MA (Rawlings et al., 2016). This latter domain is responsible for the hydrolysis of the peptide bond immediately N-terminal to a serine or threonine residue bearing an O-glycan (Figure 1B). This posttranslational modification is a required substrate recognition element for peptide hydrolysis by IMPa, and this enzyme appears able to recognize a variety of linear and branched O-glycans based on core 1, 2, 3, 4 and 6 structures (Noach et al., 2017).
Fig. 1.
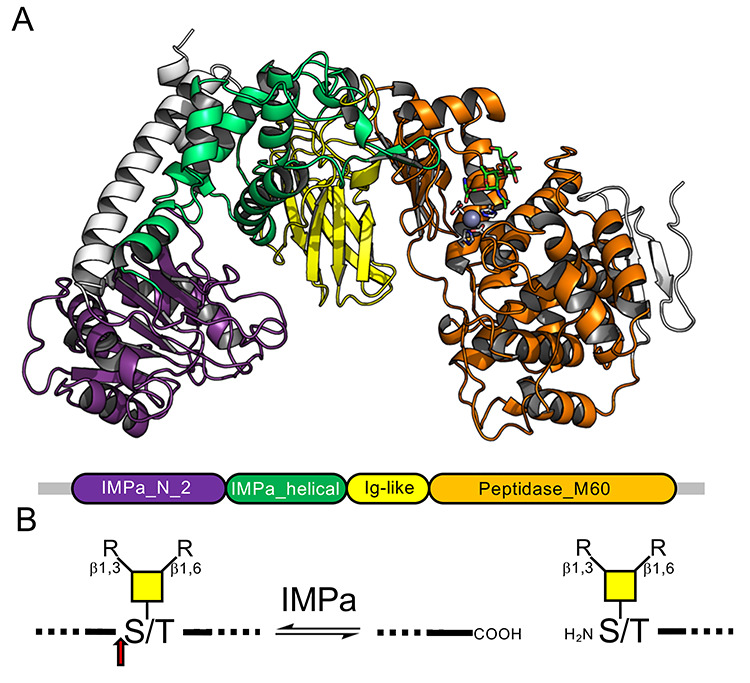
Domain structure and activity of IMPa. (A) Cartoon representation of IMPa (PDB ID 5KDW) colored according to the domains as defined by Pfam (shown beneath the structure). (B) Schematic representation of the IMPa O-glycopeptidase activity. The red arrow indicates the point of hydrolysis.
We have previously used the synthetic GAEAEAPS[TAg]AVPDAAG O-glycopeptide (referred to as F15-TAg where TAg is the core 1 Galβ1–3GalNAcα1- T-antigen) as a substrate for IMPa (Noach et al., 2017). In an effort to provide greater insight into how IMPa recognizes the glycan and peptide portion of its substrates, we pursued the crystallization of a catalytically inactivated IMPa E697Q mutant with F15-TAg. While this glycopeptide failed to occupy the active site of IMPa, it did bind to the IMPa_N_2 domain, providing unanticipated insight into the unique function of this domain. Here, therefore, we provide structural evidence for an unprecedented proline-specific glycopeptide binding domain that is associated with IMPa-like peptidases.
Materials and methods
Protein production
Using the previously described pET 22b construct encoding IMPa from Pseudomonas aeruginosa as a template, the In-Fusion HD Cloning Kit (Clontech) was used to introduce a glutamate 697 to glutamine mutation with previously described procedures and the forward and reverse oligonucleotide primers 5′-CAT CAG CTG GGC CAC AAC CTG CAA GT-3′ and 5′-TTG TGG CCC AGC TGA TGG CTT TCG CCC-3′, respectively (Noach et al., 2017). The resulting plasmid encoded an N-terminal pelB signal sequence fused to the mutant gene encoding IMPaE697Q followed by a C-terminal six-histidine tag. The DNA sequence fidelity was verified by bidirectional sequencing.
Escherichia coli strain BL21 (DE3) cells (Invitrogen) were transformed with pET 22b-IMPaE697Q and grown at 37°C in 2 L of sterile YT media supplemented with ampicillin until the culture reached an optical density of ~0.8 at 600 nm. Recombinant protein expression induced by the addition of isopropyl-β-D-1-thiogalacto-pyranoside (IPTG) to a final concentration of 0.5 mM after cooling the cultures to 16°C for 1 h. Cultures were kept overnight at 16°C with shaking. Cells were harvested by centrifugation at 5,000 x g for 10 min at 10°C and disrupted by lysozyme-chemical lysis. Cell-lysate was centrifuged at 15,000 × g for 30 min at 10°C and proteins were purified from the cleared cell-lysate by Ni2+-NTA immobilized metal affinity chromatography. Purified protein was concentrated using a stirred ultrafiltration unit (Amicon, Beverly, MA) with a 10 kDa molecular weight cutoff membrane (EMD Millipore, MA). The C-terminal six-histidine tag was cleaved from IMPa by bovine carboxypeptidase A, according to the manufacturer’s procedures, prior to size exclusion chromatography purification using a Sephacryl S-200 HR column (GE Health-care) in 200 mM Tris–HCl (pH 8.0) and 300 mM NaCl. The final purified protein was again concentrated in a stirred ultrafiltration cell. Protein concentration was determined by measuring the absorbance at 280 nm and using the calculated molar extinction coefficients of 174,680 cm−1 M−1.
IMPaE697Q (18 mg/mL) was preincubated with 2 mM F15-TAg [synthesized previously (Noach et al., 2017)] prior to crystallization in 20% polyethylene glycol 3350, 0.22 M NaH2PO4, 0.1 M HEPES (pH 7.5) at 18oC using hanging-drop vapor diffusion using a 1:1 protein to crystallization solution ratio. Crystals were cryoprotected in crystallization solution containing 25% ethylene glycol and flash cooled in liquid nitrogen. Diffraction data were collected at the Canadian Light Source (Saskatoon, Canada) beamline 08ID-1 (CMCF-ID) and processed with XDS and AIMLESS. Data collection and processing statistics are shown in Table I.
Table I.
Data collection and refinement statistics for the IMPaE697Q glycopeptide complex
| IMPa E657Q complex | |
|---|---|
| Data collection | |
| Beamline | CLS |
| Wavelength | 0.97949 Å |
| Space group | P21 |
| Cell dimensions | |
| a, b, c (Å) | 90.34, 156.50, 95.68 (β = 114.31) |
| Resolution (Å) | 30.00–2.45 (2.49–2.45) |
| R merge | 0.140 (0.686) |
| Rpim | 0.072 (0.375) |
| I/σI | 7.3 (2.0) |
| CC1/2 | 0.986 (0.698) |
| Completeness (%) | 99.6 (99.3) |
| Redundancy | 5.2 (4.8) |
| Total number of observations | 462786 (21261) |
| Total number unique | 88358 (4448) |
| Refinement | |
| Resolution (Å) | 2.45 |
| Number reflections (work/test) | 88301/4348 |
| Rwork/Rfree | 0.187/0.234 |
| Number of atoms | |
| Protein | 6777 (Monomer 1) 6790 (Monomer 2) |
| Ligand | 57 (Monomer 1) 48 (Monomer 2) |
| Water | 665 |
| B-factors | |
| Protein | 37.9 (Monomer 1) 39.7 (Monomer 2) |
| Ligand | 73.8 (Monomer 1) 84.9 (Monomer 2) |
| Water | 39.1 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.008 |
| Bond angles (°) | 0.901 |
| Ramachandran | |
| Preferred (%) | 97.0 |
| Allowed (%) | 2.9 |
| Disallowed (%) | 0.1 (1 residue) |
Values in parentheses are for highest-resolution shell.
The structure of IMPaE697Q in complex with F15-TAg was solved by molecular replacement using PHASER and native IMPa coordinates as a search model (PDB code 5KDW). The model was manually corrected with COOT (Emsley et al., 2010) followed by refinement with Phenix.refine (Liebschner et al., 2019). Five percent of the reflections were flagged as “free” and used to monitor the model building and refinement procedures (Brünger, 1992). Waters were added using FINDWATERS in COOT and inspected manually. All models were validated using MOLPROBITY (Chen et al., 2010). Model quality statistics are given in Table I. The coordinates and structure factors have been deposited with the PDB code 7JTV.
Results and discussion
The inactivated E697Q mutant of IMPa was cocrystallized with the F15-TAg substrate in a previously unobserved crystal form. Though the catalytic site architectures of the two IMPa E697Q molecules in the asymmetric unit did not show any overt structural changes that would prevent substrate binding, neither of the active sites contained electron density consistent with a bound peptide. However, electron density was found for a glycopeptide associated with the IMPa_N_2 domains in both IMPa molecules (Figure 2A and B, Supplementary Figure S1). For the two bound glycopeptides, only EAPS[TAg]A and EAPS[TAg] of the F15-TAg could be modeled with the remaining portions of the peptide too disordered to model (Figure 2B and Supplementary Figure S1). The poise of the glycopeptide was observed to be essentially identical in each of the two IMPa monomers in the asymmetric unit (Supplementary Figure S2).
Fig. 2.
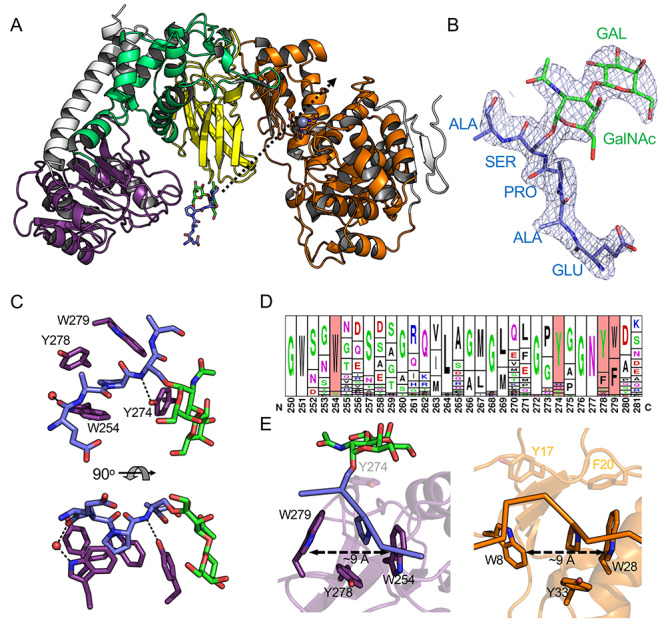
Structure of IMPa E697Q in complex with an O-glycopeptide. (A) Cartoon representation of the complex colored the same as Figure 1A. The glycopeptide bound to the IMPa_N_2 domain is shown as blue and green sticks. The trajectory of the glycopeptide backbone through the IMPa active site (shown by the zinc ion as a grey sphere) is indicated by a dashed arrow. (B) Representative electron density of one of the modeled glycopeptides (see also Supplementary Figure S1.). The 2Fo–Fc electron density map (1σ contour level) produced after refinement is shown as grey mesh. (C) Close-up of the proline binding site in the IMPa_N_2 domain. (D) Sequence logo in the frequency representation of a portion of the IMPa_N_2 domain with the aromatic residues involved in binding highlighted in red (Crooks, 2004). The alignment was generated using 44 nonredundant sequences identified by BLAST search (Altschul et al., 1997). (E) A close-up of the IMPa proline binding site in the IMPa_N_2 domain (purple with a blue and green glycopeptide ligand) compared with the proline binding site of a GYP domain (orange, PDB ID 1I2Z) reveals similarities in their organization. Peptides are shown in ribbon representation with the proline side chains as sticks.
The interaction of the glycopeptide with the IMPa_N_2 domain displays two key features. First, the side chain of the proline preceding the O-glycosylated serine residue sits in a bowl created by the aromatic side chains of two tryptophan residues and two tyrosine residues (Figure 2C). The dimensions of the bowl are roughly 9 Å by 9 Å, with each aromatic side-chain providing van der Waals interactions with the proline sidechain; however, W254 is positioned with the plane of its indole ring parallel to the proline ring such that it would appear to make significant CH-π interactions and perhaps be a driving feature of proline binding (Zondlo, 2013). The second notable feature of the interaction involves the glycan, which curls around the sidechain of Y274. The coplanar nature of the pyranose rings in the Galβ1–3GalNAc disaccharide, which is afforded by the β-1,3-glycosidic linkage, results in a relatively flat carbohydrate surface that lies parallel against the tyrosine sidechain, making a classic carbohydrate-aromatic amino acid sidechain interaction with the glycosidic bond roughly centered over the tyrosine side chain. Overall, the glycan lies along the exterior surface of the IMPa_N_2 domain in a manner that suggests a wide variety of the core O-glycan types could be accommodated (Supplementary Figure S3), which is consistent with the glycan-binding activity of the Peptidase_M60 domain of IMPa (Noach et al., 2017). Only a single direct hydrogen bond is made between the protein and ligand, and that is between the side-chain hydroxyl of Y274 and the backbone nitrogen of the glycosylated serine. A potential water-mediated hydrogen bond is made between the indole nitrogen of W254 and the backbone oxygen of the glutamic acid residue in the peptide. Notably, despite the presence of a second proline residue in the F15-Tag peptide (residue 11), there was no evidence of an alternate binding mode of the peptide suggesting that the presence of the glycan-modified serine following the proline drives selectivity for the observed manner of recognition.
Given this unexpected functional feature of the IMPa_N_2 domain, we probed its distribution in the sequence databases and the conservation of the aromatic bowl through BLAST searches. We used the 221 amino acids that structurally define the domain with specific criteria of 35–98% sequence identity (to exclude the large number of identical strain-specific P. aeruginosa sequences) and 75% sequence coverage. This yielded 44 nonredundant hits. Notably, all of these originate from IMPa-like sequences (> ~ 30% sequence identity over the full IMPa sequence), with only one protein having more than one IMPa_N_2 domain (WP_110616014.1 from Pseudomonas sp. OV647 has three at its N-terminus). When accounting for conservative amino acid substitutions, the aromatic bowl is completely conserved in ~75% of the sequences (Figure 2D). Y274, however, which primarily interacts with the glycan, is less well-conserved. When this is excluded, the other three positions (W254, Y278 and W279) are functionally conserved (i.e. with tyrosine, tryptophan, or phenylalanine) in over 90% of the sequences.
The well-conserved W254, Y278 and W279 trio of residues form a pocket that is structurally very similar to the central proline binding residues of GYF domains (Figure 2E) (Freund et al., 2002), despite the IMPa_N_2 domain sharing no fold-similarity to GYF domains, or any other proline recognition domains. The poise of proline in the GYF binding site, particularly the indole ring-proline ring interaction is also very similar, thus pointing to a conserved mode of proline recognition. This arrangement of proline binding residues is not conserved with other classes of proline recognition domains, which have extended binding sites to recognize peptide sequences containing multiple proline residues (Ball et al., 2005; Zarrinpar et al., 2003). Indeed, even GYF domains have additional binding subsites that enable recognition of extended peptide sequences (Ball et al., 2005; Freund et al., 2002; Zarrinpar et al., 2003). Thus, the proline binding site of the IMPa_N2_2 domain is unique, in particular, because of the function of Y274, which appears to be a specific adaptation to glycan recognition.
Overall, this provides compelling structural evidence that the N-terminal IMPa_N_2 domain of IMPa recognizes a proline-serine (or likely proline-threonine as well) motif where the serine (or threonine) bears an O-linked glycan. The conservation of the aromatic bowl enabling this recognition is largely conserved amongst IMPa_N_2 domains, which are only found in IMPa-like proteins, suggesting that recognition of this glycosylated motif is also conserved amongst most IMPa homologs. Regions of proteins that are dense with O-glycosylation are often rich in proline, threonine, and serine residues, as typified by the PTS (proline/serine/threonine) domains that are common to mucin proteins and other cell-surface adhesion molecules (Hansson, 2020; Van Klinken et al., 1995; Pinzón Martín et al., 2019). This suggests, therefore, that the IMPa_N_2 domain functions to target IMPa to proteins rich in O-glycosylated P-S/T motifs, which would assist in keeping IMPa in proximity to O-glycosylated proteins that could act as substrates, much as carbohydrate-binding modules in carbohydrate-active enzymes function (Boraston et al., 2004). Indeed, extrapolation of the pentapeptide bound to the IMPa_N_2 domain follows a trajectory that extends directly through the catalytic site of IMPa with the N- to C-terminus polarity consistent with the orientation of the substrate in the active site (Figure 2A and Figure 3) (Noach et al., 2017). The distance from the proline bound in the aromatic bowl to the catalytic machinery in the Peptidase_M60 domain is 32 Å, which remains consistent for the structures of IMPa that have now been determined from three different crystal forms, pointing to the relative rigidity of the enzyme structure. Heavily O-glycosylated mucin and mucin-like domains are described as having extended conformations (Hansson, 2020). Therefore, this distance of 32 Å would equate to a stretch of roughly 10–12 extended amino acids separating the IMPa_N_2 recognition site from the site of peptide hydrolysis in the catalytic domain, and lends to the potential for simultaneous recognition by both the Peptidase_M60 and IMPa_N_2 domains. This invokes the concept that the two-point recognition of substrate by the IMPa_N_2 domain and catalytic domain would provide the potential for avid recognition, thereby increasing affinity, while the selectivity of each domain for glycosylated motifs would generally enhance recognition of O-glycosylated substrates.
Fig. 3.
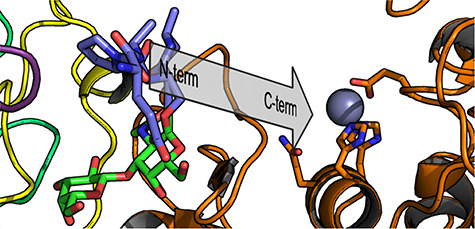
A perspective view of the glycopeptide backbone trajectory. The glycopeptide is shown as blue sticks (amino acid portion) and green sticks (carbohydrate portion). The trajectory of the peptide backbone extrapolates through to the active site, indicated by the zinc binding site (orange sticks for amino acids and grey sphere for the zinc ion). The N- to C-terminus polarity of the peptide is consistent with the orientation of the substrate as it binds in the active site. The distance from the proline residue in the glycopeptide ligand to the zinc ion is ~ 32 Å.
Supplementary Material
Acknowledgements
We thank the staff at the Canadian Light Source (CLS) where diffraction data were collected. The CLS is supported by the Natural Sciences and Engineering Research Council of Canada, the National Research Council of Canada, the Canadian Institutes of Health Research, the Province of Saskatchewan, Western Economic Diversification Canada, and the University of Saskatchewan.
Contributor Information
Ilit Noach, Biochemistry & Microbiology, University of Victoria, PO Box 3055 STN CSC, Victoria, BC V8W 3P6, Canada.
Alisdair B Boraston, Biochemistry & Microbiology, University of Victoria, PO Box 3055 STN CSC, Victoria, BC V8W 3P6, Canada.
Funding
This research was supported by an operating grant to ABB from the Canadian Institutes of Health Research (FRN 130305).
References
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball LJ, Kühne R, Schneider-Mergener J, Oschkinat H. 2005. Recognition of proline-rich motifs by protein-protein-interaction domains. Angew Chem Int Ed. 44:2852–2869. [DOI] [PubMed] [Google Scholar]
- Bardoel BW, Hartsink D, Vughs MM, Haas CJC, Strijp JAG, Kessel KPM. 2012. Identification of an immunomodulating metalloprotease of Pseudomonas aeruginosa (IMPa). Cell Microbiol. 14:902–913. [DOI] [PubMed] [Google Scholar]
- Boraston AB, Bolam DN, Gilbert HJ, Davies GJ. 2004. Carbohydrate-binding modules: Fine-tuning polysaccharide recognition. Biochem J. 382:769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger AT. 1992. Free R value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature. 355:472–475. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. 2010. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr Sect D Biol Crystallogr. 66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE. 2004. WebLogo: A sequence logo generator. Genome Res. 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr Sect D Biol Crystallogr. 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund C, Kühne R, Yang H, Park S, Reinherz EL, Wagner G. 2002. Dynamic interaction of CD2 with the GYF and the SH3 domain of compartmentalized effector molecules. EMBO J. 21:5985–5995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson GC. 2020. Mucins and the microbiome. Annu Rev Biochem. 89:769–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haurat MF, Scott NE, Di Venanzio G, Lopez J, Pluvinage B, Boraston AB, Ferracane MJ, Feldman MF. 2020. The glycoprotease CpaA secreted by medically relevant Acinetobacter species targets multiple O-linked host glycoproteins. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [DOI] [PMC free article] [PubMed]
- Van Klinken BJ, Dekker J, Buller HA, Einerhand AW. 1995. Mucin gene structure and expression: Protection vs. adhesion. Am J Physiol Liver Physiol. 269:G613–G627. [DOI] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Baker ML, Bunkóczi G, Chen VB, Croll TI, Hintze B, Hung LW, Jain S, McCoy AJet al. 2019. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr Sect D Struct Biol. 75:861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakjang S, Ndeh DA, Wipat A, Bolam DN, Hirt RP. 2012. A novel extracellular metallopeptidase domain shared by animal host-associated mutualistic and pathogenic microbes. (E. a. Permyakov, Ed.). PLoS One. 7:e30287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noach I, Ficko-Blean E, Pluvinage B, Stuart C, Jenkins ML, Brochu D, Buenbrazo N, Wakarchuk W, Burke JE, Gilbert Met al. 2017. Recognition of protein-linked glycans as a determinant of peptidase activity. Proc Natl Acad Sci. 114:E679–E688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinzón Martín S, Seeberger PH, Varón SD. 2019. Mucins and pathogenic mucin-like molecules are immunomodulators during infection and targets for diagnostics and vaccines. Front Chem. 7:710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlings ND, Barrett AJ, Finn R. 2016. Twenty years of the MEROPS database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 44:D343–D350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shon DJ, Malaker S, Pedram K, Yang E, Krishnan V, Dorigo O, Bertozzi C. 2020. An enzymatic toolkit for selective proteolysis, detection, and visualization of mucin-domain glycoproteins. Proc Natl Acad Sci. 117:21299–21307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Z, Cheng S, Xia B, Jin Y, Bai F, Cheng Z, Jin S, Liu X, Wu W. 2019. Pseudomonas aeruginosa ExsA regulates a metalloprotease, IMPa, that inhibits phagocytosis of macrophages. Infect Immun. 87:e00695–e00619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu ACY, Worrall LJ, Strynadka NCJ. 2012. Structural insight into the bacterial mucinase StcE essential to adhesion and immune evasion during enterohemorrhagic E. coli infection. Structure. 20:707–717. [DOI] [PubMed] [Google Scholar]
- Zarrinpar A, Bhattacharyya RP, Lim WA. 2003. The structure and function of proline recognition domains. Sci Signal. 179:re8. [DOI] [PubMed] [Google Scholar]
- Zondlo NJ. 2013. Aromatic–proline interactions: Electronically tunable ch/π interactions. Acc Chem Res. 46:1039–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.