

Abstract
Preparation of samples for nuclear magnetic resonance (NMR) characterization of larger proteins requires enrichment with less abundant, NMR-active, isotopes such as 13C and 15N. This is routine for proteins that can be expressed in bacterial culture where low-cost isotopically enriched metabolic substrates can be used. However, it can be expensive for glycosylated proteins expressed in mammalian culture where more costly isotopically enriched amino acids are usually used. We describe a simple, relatively inexpensive procedure in which standard commercial media is supplemented with 13C-enriched glucose to achieve labeling of all glycans plus all alanines of the N-terminal domain of the highly glycosylated protein, CEACAM1. We demonstrate an ability to detect partially occupied N-glycan sites, sites less susceptible to processing by an endoglycosidase, and some unexpected truncation of the amino acid sequence. The labeling of both the protein (through alanines) and the glycans in a single culture requiring no additional technical expertise past standard mammalian expression requirements is anticipated to have several applications, including structural and functional screening of the many glycosylated proteins important to human health.
Keywords: CEACAM1, glycoprotein, metabolic labeling, NMR, top–down MS
Introduction
Glycans are thought to decorate more than half of all mammalian proteins, making the biology of glycoproteins of particular concern to human health (Apweiler et al. 1999). The glycans on these proteins play important roles in protein folding (Varki and Gagneux 2015; Xu et al. 2018), in modulating protein interactions (Sheikh et al. 2015) and in propagating cell signals (Takeuchi and Haltiwanger 2014). They also provide sites for initial attachment of invading pathogens (Lu et al. 2015). While significant progress has been made in the development of methodology for the study of glycoproteins, structural and functional characterization remains difficult. Nuclear magnetic resonance (NMR) offers some advantages in that it can be applied in a native aqueous environment, can return information on both structural and dynamic aspects and can target particular parts of complex systems, including glycans. Targeting is typically done through labeling with isotopes that have useful magnetic properties (13C, 15N). When proteins can be expressed in Escherichia coli, this is straightforward; inexpensive substrates like15N-ammonium sulfate and 13C-glucose are commonly used to produce proteins uniformly labeled with high percentages of 13C and 15N.
Expressing uniformly labeled proteins in cells that are capable of producing native mammalian glycosylation proves expensive, because the isotopically labeled amino acids usually used as substrates are costly. There are ongoing efforts to develop less expensive media, primarily with uniform labeling of all amino acids in mind (Skelton et al. 2010; Sastry et al. 2011); progress in this area has been reviewed recently (Yanaka et al. 2018). However, labeling with a single or select set of amino acids (sparse labeling, sometimes called selective labeling; Kato et al. 1991) is far less expensive, and it returns adequate structural information for many applications (Prestegard et al. 2014; Minato et al. 2016). Among the applications are screens for ligand binding and assessment of interdomain geometry in multidomain proteins or protein–protein geometry in multiprotein complexes (Gao et al. 2016; Minato et al. 2016).
Here, we focus on achieving sparse labeling of glycans as well as a single amino acid by the simple addition of 13C-enriched glucose to a commonly used mammalian cell expression medium. Labeling of glycans with 13C-labeled sugars is not new; in fact there is an early demonstration (Yamaguchi et al. 1998), and since that time substantial effort has gone into minimizing metabolism of labeled sugars and using a select subset of sugars to improve sensitivity and facilitate resonance assignment (Kato et al. 2010). The labeling of the glycans by the simple addition of 13C-enriched glucose also continues to be practiced (Subedi et al. 2017). Here we show that, in addition to labeling ring structures in glucose and other sugars produced by epimerization (mannose and galactose), certain methyl groups are efficiently labeled, namely acetyl groups in acetylated sugars and alanines in the protein itself. The simple addition of 13C-glucose to a standard mammalian cell culture medium thus provides, in one preparation, simplified spectra that allow simultaneous monitoring of both the protein and attached glycans.
13C-labeling of methyl groups is particularly advantageous for NMR observation, because of the enhanced signal intensity resulting from three equivalent protons and altered relaxation properties that lead to very sharp resonances (Ollerenshaw et al. 2003). This has led to the widespread use of ILV labeling (methyl labeling in isoleucine, leucine and valine amino acids using media supplemented with labeled α-keto precursors) (Goto et al. 1999; Kerfah et al. 2015). There have been extensions to methyl labeling of alanine (Ayala et al. 2009) and stereo array isotope labeling of these groups (Kainosho et al. 2006). While full advantage requires perdeuteration of nonmethyl protons, some advantage will extend to 13C alanine and N-acetyl methyls labeled by addition of 13C-glucose to standard culture media. While the distribution of labeled sites will be sparse, and direct structural information locally concentrated, the range can be extended using long-range perturbations by paramagnetic moieties that produce relaxation enhancements (Clore 2015), pseudo contact shifts (Nitsche and Otting 2017) or residual dipolar couplings (Prestegard et al. 2004; Bax and Grishaev 2005).
The 13C-labeling of alanine and acetyl methyls is not surprising. For acetyl groups, the production of pyruvate by glycolysis of glucose followed by a single decarboxylation step leads to isotopically labeled acetyls of acetyl-CoA; this in turn adds to an amino sugar to produce a methyl-labeled N-acetylated sugar (Brockhausen et al. 2016). For alanine, pyruvate is transaminated to yield labeled alanine (Franke et al. 2018; McNeill et al. 2020). What is unknown is the level to which N-acetyl groups of acetylated sugars and alanines are labeled under conditions of simple glucose addition to a mammalian cell culture medium and whether that level will be adequate for routine NMR observation.
We choose to investigate the above issues using the 12.5 kDa N-terminal Ig-like V-type (IgV) domain of CEACAM1, which contains three N-X-S/T N-glycosylation sequons. Expression in MGAT1-deficient HEK293 cells produces high levels of Man5GlcNAc2N-glycan structures at these sites. The limited number of glycans and the well-resolved crosspeaks seen in heteronuclear single quantum coherence (HSQC) spectra for anomeric 1H–13C pairs and N-acetyl methyl 1H–13C pairs allow a qualitative analysis of site occupancy and the level of isotopic labeling in the sugar rings and acetyl methyls. When combined with additional mass spectrometry data, a more quantitative assessment of occupancy and incorporation results. We had previously produced the samples of 13C-methyl alanine-labeled CEACAM1-IgV by supplementing media with isotopically labeled alanine and knew the expected seven alanine methyl resonances to be well resolved and fall in spectral regions relatively unobstructed by natural abundance background. In this new work, 13C-glucose addition produced easily observed alanine methyl resonances, allowing analysis of labeling levels. However, observation of an extra alanine resonance suggested some heterogeneity in the sample. Mass spectrometry data showed this to be the result of unanticipated protein proteolysis. In short, we show that metabolic labeling by the simple addition of 13C-glucose to expression media can provide simultaneous monitoring of protein and glycan properties when conducting NMR studies of glycoproteins.
Results
Figure 1 presents sections of a 600 MHz 1H–13C HSQC spectrum of a 470 μM sample of CEACAM1-IgV. The sample was produced by expression in 1 L of defined commercial medium to which uniformly labeled 13C-labeled glucose had been added in an amount that equals the amount of unlabeled glucose originally present. The expression vector, which includes a His-tagged GFP moiety and TEV cleavage site ahead of the CEACAM1-IgV coding region, along with purification procedures has been described in previous publications (Subedi et al. 2015; Zhuo et al. 2016; Moremen et al. 2018).
Fig. 1.
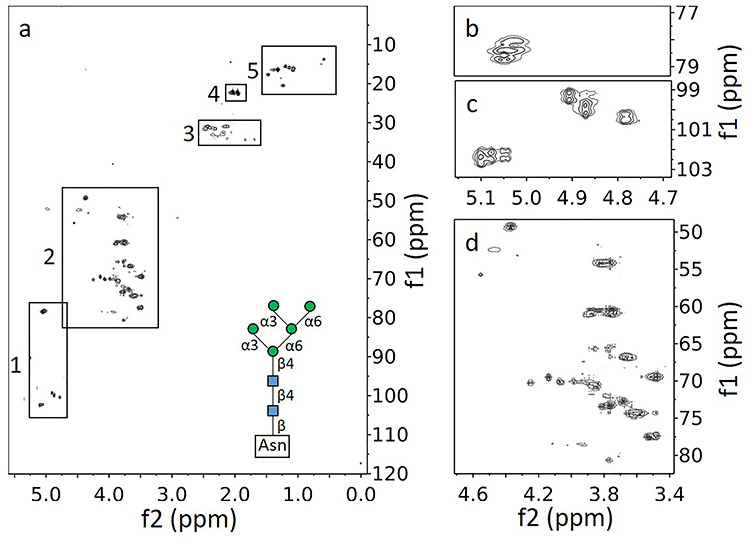
1H–13C HSQC of CEACAM1-IgV labeled metabolically through supplementation with u-13C-glucose. (A) Full spectrum with regions marked by boxes as well as a diagram of Man5GlcNAc2. (B) Close-up of GlcNAcs attached to Asn. (C) Close-up of anomeric region for the O-linked GlcNAcs and mannose residues. (D) Close-up focusing on the region of the spectrum associated with the sugar carbons 2–6.
Figure 1A shows a full spectrum with clusters of peaks (crosspeak multiplets due to scalar coupling) in five distinct regions. Region 1 is centered on 5.0 ppm for 1H and 90.4 ppm for 13C. Based on expected chemical shifts, peaks in this region clearly arise from protons directly attached to anomeric carbons of glycans. Region 2, centered on 3.7 and 65 ppm, arises primarily from the remaining proton–carbon pairs in pyranose sugar rings. Peaks in region 3, centered on 2.1 and 31 ppm, are decidedly weaker than peaks from other regions. Chemical shifts are consistent with methylene peaks from amino acids such as glutamate and glutamine, suggesting that under our protein expression conditions there is metabolic labeling of some amino acids. Both glutamate and glutamine are abundant in CEACAM1-IgV (6.2 and 8.9%, respectively) and both are produced from α-ketoglutarate, an early intermediate of the citric acid cycle which utilizes pyruvate, the end product of glycolysis. So, some labeling is expected, but it is clearly at a much lower level than that of the glycans. Region 4, centered on 2.0 and 22.3 ppm, has a set of very intense peaks. The chemical shifts of these peaks are consistent with N-acetyl methyl groups. Region 5, centered on 1.3 and 17 ppm, again has a set of fairly intense peaks. The proton shifts are consistent with amino acid side-chain methyl groups, but further downfield than one would expect for longer chain amino acids such as valine, leucine and isoleucine. They clearly belong to alanine methyls. All of the peaks in these regions must result from metabolic labeling that begins with u-13C-glucose. Signals from amino acids having natural abundance 13C or near natural abundance 13C are minimal, as can be seen in expansions of the aromatic region and region 3, respectively, of the HSQC plotted at a much lower threshold (Supplementary Figure S1).
Figure 1B and C shows a two-part expansion of the anomeric region (region 1). In Figure 1B, we can clearly identify two crosspeak clusters, each with four peaks due to scalar coupling in both the proton and carbon dimensions; these belong to GlcNAcs that are β-linked to the nitrogen of Asn side chains. Both the large proton–proton coupling and their unique chemical shifts (~78.5 and ~5.05 ppm) support this assignment. The cluster expected for the third Asn-linked GlcNAc overlaps one of the other clusters. The pattern of connectivities from these crosspeaks to other ring protons, as established by HSQC-total correlated spectroscopy (TOCSY) and 13C–13C correlation spectroscopy (COSY) spectra (Supplementary Figures S2 and S3), adds support to this conclusion. Figure 1C shows two anomeric crosspeak clusters near 100 and 4.9 ppm that have large coupling constants in the proton dimension consistent with β1-4-O-linked GlcNAcs. The third expected peak cannot be seen in Figure 1 as it is considerably weaker and lies largely under the water peak. The number of β-O-linked GlcNAc residues is consistent with all three consensus sites, N104, N111 and N115 (based on UniProt P13688 numbering) having some level of glycosylation. The variation in intensity most likely indicates that one of the sites is under occupied. Additional anomeric peaks with small, unresolved, proton couplings (Supplementary Figure S2) belong to mannose residues of Man5GlcNAc2 type glycans. There are some assignments of Man5GlcNAc2 resonances in the literature suggesting that the two resolved clusters with splitting only in the carbon dimension near 102 and 5.0 ppm belong to the α1,3-Man residues linked to the core β-1,4-Man and those near 100 and 5.1 ppm belong to the α1,6-Man residues. These are remote from the protein attachment sites, and crosspeaks from the different attachment sites are likely to be degenerated. Anomeric crosspeaks from β-O-linked mannoses fall close to the water resonance in the proton dimension and are often obscured. However, one can be seen in a plot at a lower threshold. Figure 1D shows an expansion of region 2 with crosspeaks for the remaining glycan 1H–13C pairs. Many of these can be connected to anomeric peaks using COSY and TOCSY experiments. All anomeric peaks, as well as many additional peaks resolved in TOCSY and COSY spectra, have been assigned to a residue type and pyranose ring position; these assignments are reported in Table I.
Table I.
Chemical shifts of types of resonances of glycans attached to native CEACAM1-IgV
| Attached to: | Name | H1, C1 | H2, C2 | H3, C3 | H4, C4 | H5, C5 | H6, C6 |
|---|---|---|---|---|---|---|---|
| Asn | β-GlcNAc | 5.06, 78.61 | 3.82, 54.19 | 3.62, 74.35 | 3.49, 77.42 | 3.48, 69.46 | 3.76, 61.11 |
| β-GlcNAc | 5.03, 78.25 | 3.77, 54.19 | ND | 3.53, 77.53 | 3.52, 69.48 | 3.75, 60.51 | |
| 1-4 GlcNAc | β-GlcNAc | 4.79, 100.42 | ND | ND | ND | ND | ND |
| β-GlcNAc | 5.04, 102.29 | ND | ND | ND | ND | ND | |
| β-GlcNAc | 4.71, 95.09 | 3.74, 56.32 | 71.97 | ND | ND | ND | |
| 1-4 GlcNAc | β-Man | 4.91, 99.40 | 3.89, 69.92 | ND | ND | ND | ND |
| β-Man | 4.87, 100.02 | 4.14, 69.66 | 78.48 | ND | ND | ND | |
| α-Man | 5.10, 102.6 | 4.08, 70.24 | ND | ND | ND | ND | |
| α-Man | 5.08, 102.36 | 4.07, 70.16 | ND | ND | ND | ND | |
| α-Man | 5.05, 102.27 | 4.06, 69.99 | ND | ND | ND | ND | |
| Unspecified/overlap | α-Man | ND | ND | 3.85, 70.59 | 3.68, 66.78 | 3.66, 72.84 | 3.74, 60.44 |
ND, not determined. Italics represents shifts of clusters representing multiple nuclei.
Region 4 is particularly interesting, in which the peaks are quite intense and well resolved. Expansions of this region are shown in Figure 2, one before treatment with Endoglycosidase-F1 (Endo-F) (2a), which cleaves between the two core GlcNAc residues at each glycosylation site, and one after treatment (2b). The chemical shifts are consistent with N-acetyl methyl groups. Note that all peaks occur in pairs separated in the carbon dimension by about 50 Hz. This is typical of scalar coupling to a single 13C-enriched carbonyl carbon. An 1H–13C heteronuclear multiple bond correlation (HMBC) spectrum (not depicted) also shows connectivities of carbonyls to the methyl protons. Before Endo-F treatment (Figure 2A) at least four 13C-coupled pairs can be counted. The expectation for two GlcNAcs (one N-linked and one O-linked) at each of the three N-glycosylation sites in CEACAM1-IgV would be six, but spectral overlap and variation in intensity due to differences in site occupancy or mobility can easily account for the lack of resolved peaks.
Fig. 2.
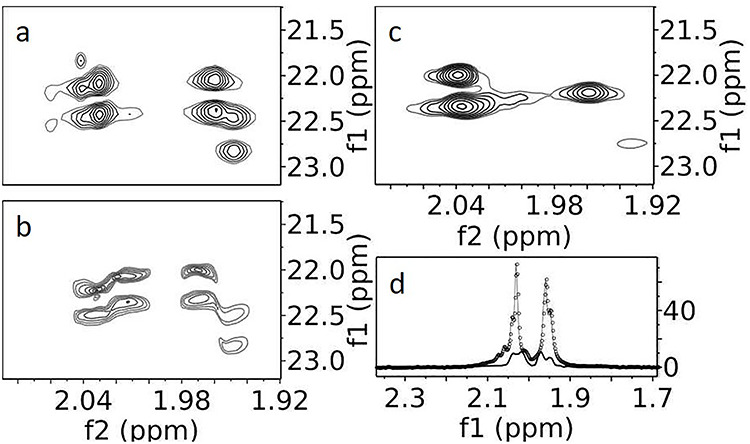
1H–13C HSQCs of acetyl methyl region of CEACAM1-IgV. (A) Supplemented with u-13C-glucose and grown in HEK293S (GnTI-) cells leading to a dominant Man5GlcNAc2 glycan population. (B) Similarly prepared CEACAM1-IgV with an additional Endo-F treatment to trim the glycans to a single GlcNAc. (C) Analogous to A, but a CEACAM1-IgV S96C mutant collected with a constant time HSQC. (D) Projections of a slice of A (−o-) and B (−), normalized to the noise.
The spectrum is simplified after Endo-F treatment (Figure 2B). There are now two intense doublets, one moderately intense doublet (2.04 ppm in the proton dimension) and one weak doublet (1.95 ppm in the proton dimension). The most intense doublets in the Endo-F-treated sample do not actually line up with the most intense doublets in the untreated sample; these were partially obscured in Figure 2A by overlap with the more intense O-GlcNAc signals. The difference in N-linked and O-linked intensities becomes clear by comparing projections of Figure 2A (dotted line) and Figure 2B (solid line) along their vertical 13C axes (Figure 2D). Therefore, the most intense doublets in the untreated sample come from the second GlcNAcs in the Man5GlcNAc2 structure (the O-GlcNAcs), which are further from the protein surface and are more mobile. The two intense doublets in the treated sample clearly belong to N-GlcNAcs at highly occupied sites; a third lower intensity doublet is expected for the lower occupancy site of the WT CEACAM1-IgV sample, but a second low-intensity doublet is unexpected. This additional doublet appears to result from incomplete Endo-F cleavage at one of the sites. In fact, examination of the anomeric region of an HSQC spectrum for the Endo-F-treated sample, similar to that shown in Figure 1B for the untreated sample, shows residual mannose and O-linked GlcNAc crosspeaks; the single well-resolved O-linked GlcNAc cluster integrates to ~25% of that seen in the Man5GlcNAc2 spectrum.
In principle, the N-acetyl region could be greatly simplified by the use of a constant time HSQC (CT-HSQC) in which 13C–13C couplings in the indirect (13C) dimension are removed. In Figure 2C, we show an enlargement from a CT-HSQC run on a very similar protein construct, which had not been Endo-F processed but had a much higher and more uniform level of glycosylation (a S96C mutant with 90% glycosylation at all three sites). There are now clearly just three intense noncoupled crosspeaks, two close to one another at 2.04 ppm and one shifted more upfield in the proton dimension (1.96 ppm). Unfortunately, the use of a constant time experiment causes loss of sensitivity, selectively for resonances broadened due to lack of internal motion; the three crosspeaks clearly belong to the three more mobile O-linked GlcNAcs. Identifying these allows a more definitive assignment of the two weaker doublets seen in Figure 2A. One of the two crosspeaks at 2.04 ppm corresponds to the intense doublet at 2.03 in Figure 2A, and the one at 1.96 corresponds to the intense doublet at 1.96 ppm. The weak doublet near 2.04 in Figure 2A, therefore, comes from an O-GlcNAc at an under glycosylated site in the WT sample as it resides at the chemical shift of the second intense peak at 2.04 ppm in the spectrum of the mutant sample. The weak doublet at 1.95 ppm in Figure 2A then comes from the third N-GlcNAc, and the one remaining peak in Figure 2B must be from an O-GlcNAc that is under processed by Endo-F, possibly the same site that is under occupied in WT CEACAM1-IgV.
The fact that we can analyze the N-acetyl region in such detail comes from the generally high-peak intensity and resolution in this region. The overall high intensity of peaks is partly due to the three equivalent protons on acetyl methyls and partly due to a high level of labeling. A high level of labeling is not unexpected, given the single metabolic step from pyruvate to acetyl-CoA. Isotopomer analysis of oxonium ions, from released GlcNAcs in MS2 spectra, shows isotopic labeling to be 42 ± 12% in the acetyl group. This is actually very near the level of labeling observed for carbons in all sugar rings (43 ± 14%). This, in turn, is near the maximum labeling expected due to the 50:50 mix of labeled and unlabeled glucose in our expression medium.
Figure 3A and B shows expansions of region 5 at different thresholds. The intense crosspeaks in Figure 3A belong to alanines and the weak peaks that appear at lower threshold levels in Figure 3B belong to natural abundance methyl groups of other amino acids. The alanine peaks are centered about the average chemical shifts of alanine methyl protons and carbons at 1.35 and 19.06 ppm, respectively, as reported in the Biological Magnetic Resonance Data Bank (Ulrich et al. 2008). There are eight intense peaks, one more than expected for the seven alanine residues in our construct. These intense peaks are unlikely to be from any other methyl containing amino acid, as those are all essential amino acids and would be unlikely to be synthesized from 13C-labeled glucose. Sample heterogeneity is a more likely cause. Some of the alanine methyl crosspeaks had been previously assigned using data from a uniformly labeled nonglycosylated sample expressed in E. coli (Zhuo et al. 2016); assignments have been transferred as shown in Figure 3A. In addition, we have assigned the two very narrow peaks to A34 (UniProt numbering), a residue just four amino acids from the N-terminus of our CEACAM1-IgV construct and one that was absent in E. coli construct. The four N-terminal amino acids are the glycine left following TEV cleavage and three linker amino acids (SGG) that were inserted to make the TEV cleavage site more accessible. These residues are likely to be disordered and can contribute to the enhanced mobility and narrow resonances for A34. All alanine peaks are intense, again partly due to the three equivalent protons on a methyl group and partly due to efficient labeling via the one-step conversion of pyruvate to alanine by alanine transaminase. The issue of the extra peak for A34 will be addressed in mass spectrometry data presented below.
Fig. 3.
1H–13C HSQC spectra of the methyl region of CEACAM1-IgV. (A) WT CEACAM1 labeled using u-13C-glucose; plotted at a high threshold. (B) Same as A, but plotted at a threshold where natural abundance methyls are visible. (C) A constant time HSQC of S96C mutant labeled with u-13C-glucose and dimethyl-13C-valine.
Many of the peaks seen when spectra are plotted at a lower contour (Figure 3B) come from natural abundance valine methyls. The constant time experiment mentioned in our discussion of acetyl methyls (Figure 2C) actually provides a means of assigning these peaks, as the sample for the constant time experiment was labeled using both 13C-glucose and dimethyl-13C-valine. A section of the constant time experiment showing alanine and valine methyl crosspeaks is shown in Figure 3C. The valine peaks are more intense than the alanine peaks, as expected given the upwards of 96% labeling (mass spectrometry [MS] confirmation) that results from using a combination of valine dropout media and addition of dimethyl-13C-valine. Moreover, these peaks are of opposite sign (red vs. blue). The sign change comes from that fact that the valine methyls have only a very small two-bond 13C–13C coupling. With the constant time interval selected for our experiment (13.3 ms), alanine peaks with a large one-bond coupling evolve to have the opposite phase of the valine peaks with a very small two-bond coupling. We can now use the natural abundance valine peaks in Figure 3B as a reference in estimating the % alanine labeling. Intensities are in general affected by internal motion, but both alanine and valine methyls are close to the backbone and, except for those in mobile segments at the protein termini, are likely to be quite rigid. By excluding the sharpest ~ 25% of crosspeaks for each amino acid type to avoid the effect of backbone flexibility, we can compare intensities. We find the alanine methyls to be isotopically labeled to 23 ± 3%. This is high given the maximum of 50% labeling expected from the glucose in the medium, and the additional natural abundance alanine available from the GlutaMAX (L-Ala-L-Gln) that was used to provide a controlled level of glutamine in the medium.
The suggested partial occupancy of one or more of the putative N-glycosylation sites in CEACAM1-IgV is easily confirmed using electrospray mass spectrometry of the intact protein. Ideally, this would be applied to the actual samples used in the NMR analysis. However, the presence of 13C at variable percentages of enrichment in both glycans and amino acids can complicate analysis. Hence, a sample with only alanine carbons enriched with 13C was treated with Endo-F. As shown in Figure 4, two series of peaks, each with peaks differing in mass by that expected for the presence of one, two or three GlcNAc residues, were observed. No mass peaks were observed for a completely nonglycosylated species. Assuming equal ionization tendencies for these species, the intensities do not fit a pattern of partial, but equal, occupancy of all three sites. They are consistent with two high occupancy sites and one lower occupancy site.
Fig. 4.
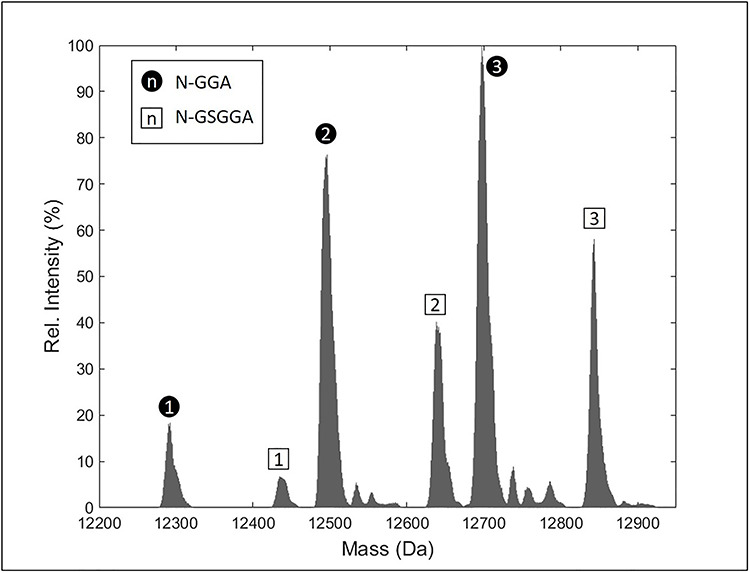
Deconvoluted mass spectrum of u-13C-Ala-labeled CEACAM1-IgV, Endo-F treated. Peaks labeled with black circles have an amino acid sequence beginning with GGA, while the peaks labeled with squares begin with GSGGA. In each case, the number within the label indicates the number of GlcNAc modifications.
The presence of two series of peaks in Figure 4 suggests additional sample heterogeneity. The separation in mass does not correspond to any deletion or addition of a glycan residue. Instead, the lighter of the two series, which is 145 Da less than expected according to the construct design, corresponds to the mass of glycine plus serine, the two residues expected at the N-terminus of our TEV-cleaved construct. If these residues were absent, the alanine closest to the N-terminus would be preceded by just a GG sequence and not the GSGG sequence expected for TEV cleavage. This change in termination could easily account for the shifted extra alanine methyl peak in our NMR spectra. There appear to be no reports of TEV cleavage at a noncanonical site (Kapust et al. 2002); however, it is interesting that cleavage between S and G would mimic the hydrophilic neutral (Q), followed by G, found in the canonical ENLYFQ/G TEV recognition site. It is also possible that the alternate truncation is the product of other contaminating proteases during purification.
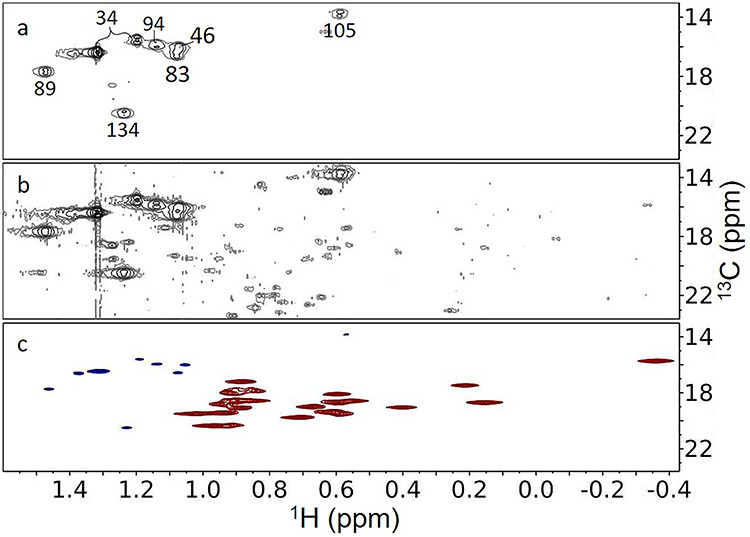
To identify the site with low N-glycosylation occupancy and determine the relative occupancy more accurately, we employed top–down electron capture dissociation (ECD) tandem mass spectrometry (MS2) on an Endo-F-treated wild-type sample having no isotopic labeling. Representative results are shown in Figure 5 for the protein isomer showing glycosylation at all three consensus sites. The term “top–down” means that no previous proteolytic digestion step is used and that the intact protein is subjected to fragmentation and MS2 analysis (Kelleher 2004). This is especially convenient for a high purity protein where the chromatographic separation typically used in a bottom-up analysis is not necessary. The fragmentation method, ECD, produces widespread cleavage of N–Cα bonds along the polypeptide backbone while preserving labile post-translational modifications such as glycosylation (Zubarev et al. 2000), thus allowing site-specific assessment of GlcNAc occupancy. As indicated in Figure 5, fragment ions were produced between each of the glycosylation sites confirming their localization and allowing assessment of their relative occupancy. By comparing the top–down fragmentation data for the mono-, di- and triglycoforms, it is observed that N115 is clearly the low occupancy site, with relatively high levels of occupancy at the other two sites, more specifically 75, 78 and 51% for the N104, N111 and N115 sites.
Fig. 5.
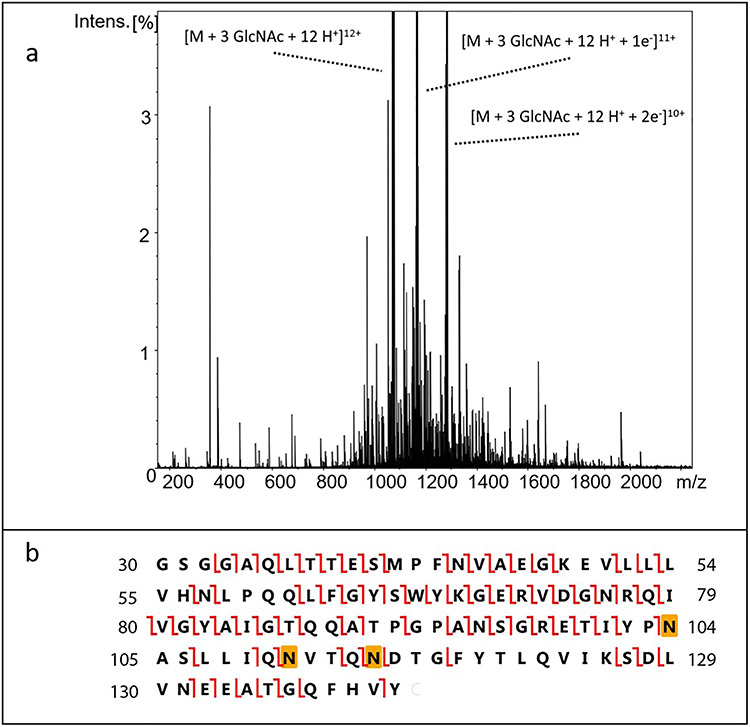
Top–down ECD MS2 determination of glycan occupancy. (A) Example of the ECD MS2 spectrum of the proteoform having three GlcNAcs. The vertical scale was chosen to emphasize the large number of low-intensity fragment ions. Three intense peaks corresponding to the precursor ion and two charge-reduced ions are observed. (B) Construct sequence showing observed c and z fragment ions. The highlighted Asn residues indicate the glycosylation sites.
Discussion
With the increased interest in the characterization of glycoproteins, whether it be for biophysical studies (Sakae et al. 2017; Arda and Jimenez-Barbero 2018; Flugge and Peters 2019) or characterization of the biologics that now dominate products of the pharmaceutical industry (Anish et al. 2014; Yang et al. 2015; O'Flaherty et al. 2018), it is clearly important to have methodology that can rapidly and effectively assess protein structure and the nature of glycosylation. Mass spectrometry provides rapid and effective characterization of glycoforms and peptide sequence, but it is much less effective at assessing preservation of protein tertiary structure, characterizing interactions between glycans and the protein to which they are attached, or characterizing interactions between a specific glycoprotein and its ligands. NMR is better suited to this type of structural and functional characterization, but applications are inhibited by the need to isotopically labeled, particularly when expression in eukaryotic hosts is required. Here we have illustrated the successful labeling of both protein and glycans in a single inexpensive culture by the simple addition of 13C-labeled glucose to an existing, chemically defined expression medium at a level that doubles the glucose content. Under these expression conditions, sugar residues of the Man5GlcNAc2 glycans produced in HEK293S (GnTI-) cells are labeled to near the maximum level expected based on the ratio of labeled glucose added to unlabeled glucose originally present in the medium (for GlcNAc, 43 ± 14% compared to the expected 50%). Similar levels are expected for both galactose and sialic acids found on glycans of proteins expressed in wild-type cells, as they are produced in single enzyme steps from glucose and mannose, respectively. N-acetyl methyls are also labeled to a similar percentage (42 ± 12%); alanine methyls of the protein are labeled to a useful level (23 ± 3%) despite dilution by natural abundance alanine inherent in the medium. Eliminating unlabeled glucose from the expression medium and replacing it with 99% u-13C-labeled glucose would raise these percentages by nearly a factor of two. However, effective labeling by simple addition to existing media is sufficient for screening structure changes and protein interactions and may extend NMR observation to a range of proteins expressed in eukaryotic cell media for other applications.
Isotope labeling of single amino acid types (sparse labeling), and even uniform isotope labeling of carbohydrates at less than 90%, does impose limits on the type of NMR experiments that can be performed. For proteins, the normal triple resonance experiments used for resonance assignment become inefficient. However, we and other groups have been working on methods to replace this approach (Prestegard et al. 2014; Kerfah et al. 2015; Kainosho et al. 2018; Yanaka et al. 2018). For glycans, even labeling at 50%, compared to 90%, results in nearly a 4-fold loss of sensitivity when HMBC experiments are used as a method to establish trans-glycosidic connectivities. Using NOE (nuclear Overhauser effect) experiments as an alternative method may provide higher sensitivity, but analysis suffers from ambiguities due to sampling of multiple conformations and from poor proton resolution. Even without assignment, properly processed glycan crosspeaks can allow a qualitative assessment of glycoform composition and differential site occupancy for multiply glycosylated proteins. Appearance of shifted peaks and additional peaks for either glycan or protein components can signal conformation changes or heterogeneities in sample preparation.
When assignments can be made, chemical shift perturbations can be indicators of site-specific interaction. Other methods based on distance constraints on sparsely labeled sites using paramagnetic tags provide an alternative strategy for more complete structure determination (Yagi et al. 2013). These structural methods can also be extended to association of glycan resonances with particular glycosylation sites and glycan conformational analysis (Kato and Yamaguchi 2015; Gao et al. 2016).
Methyl groups found on alanine, valine, leucine, isoleucine or methionine prove particularly useful in screening for protein stability and interactions (Tugarinov and Kay 2003; Flugge and Peters 2018). The crosspeak positions in HSQC or heteronuclear multiple quantum coherence spectra vary significantly with perturbations to both secondary and tertiary structures. Positions are also perturbed on binding of ligands or other proteins. The fact that the alanine crosspeaks we detect in proteins expressed in 13C-glucose-containing cultures are well separated from crosspeaks coming from dimethyl-13C-valine supplemented cultures means that simultaneous labeling of valines, alanine and glycans is an option, giving a more complete mapping of structure preservation and glycoprotein interaction.
For the current application, the methods described have proven particularly valuable in uncovering heterogeneities in samples previously assumed to be homogeneous. The observation of an extra alanine crosspeak in the spectrum of our CEACAM1-IgV construct led to the mass spectrometric identification of a species missing two residues at the N-terminus and association of the extra peak with A34 in the truncated product. The fraction of this species was large and unexpected. Whether this is due to some proteolytic activity in our expression medium or some lack of specificity in the TEV enzyme will require further investigation. The incomplete occupancy of N-glycosylation sites was also suggested by the intensity variation in anomeric and N-acetyl crosspeaks for the N-glycans. Top–down mass spectrometry then identified the low occupancy site as N115. Partial occupancy of this site may be associated with the central residue of the sequon, an aspartic acid. The presence of aspartic acid has been suggested to decrease glycosylation at the preceding asparagine (Mellquist et al. 1998). Enhanced occupancy of the N104 and N111 sites may be associated with the central residue as well. The presence of alanine and valine, as found in these sites, has been suggested to promote glycosylation (Petrescu et al. 2004). Also, N115 is the last position in a sequence of two closely spaced N-glycan sequons. Under occupancy at this position may also be a result of a local depletion of glycan donors on glycosylation of the upstream glycosylation site. Further investigation of the origin of these variations is clearly justified.
The origin of a fourth N-acetyl methyl peak in our spectra of Endo-F-processed protein appears to be the result of incomplete removal of Man5GlcNAc2 groups by this enzyme. This could be associated with some steric hindrance to approach of the Endo-F enzyme at the site. These heterogeneities may have some consequences. For example, our previous observation of inhibition of CEACAM1-IgV dimer formation by the presence of even minimal glycosylation has been difficult to reproduce (Zhuo et al. 2016). This may have been a consequence of more extensive proteolysis, abnormal glycosylation of a particular sample or chemical modification on long-term storage.
For future applications, close monitoring of glycosylation patterns and protein integrity will be very important. The sparse labeling strategy described here can play a role. While overall costs are less than is typical of uniform labeling with isotope enriched amino acids, cost remains a consideration. In the present work, we grew 1 L cultures, something required for the production of the >10 mg of protein typically used in complete NMR structural characterization of proteins. Adding 5 g of uniformly 13C-labeled glucose to this culture cost nearly $1000. However, for screening applications that use methyl groups (N-acetyl or Alanine methyls), <250 μg of 25% labeled protein proves adequate for 1 h HSQC screening applications using modern cryogenic, small volume, probes. This would reduce isotope costs to ~$25 per sample. It may also be possible to use media with lower levels of glucose. Common media, such as Basal Medium Eagle or Minimum Essential Medium Eagle contain as low as 1 g/L glucose, an amount approaching normal serum levels (Weil et al. 2009), and a chemically defined medium lacking all natural abundance glucose could be used.
There is obviously more work to do to optimize the above strategy to the protein production strategies of individual labs. However, the simplicity of adding 13C-glucose to standard culture media used for the expression of mammalian proteins has a great deal of appeal. It can certainly provide an assessment of the structural and glycosylation homogeneity of a preparation. It may also provide useful information comparing structural characteristics of closely related proteins, such as those differing due to a mutation, differing due to glycoform or differing due to culture conditions. In these cases, small cultures can be grown in parallel with 13C-glucose additions to each. Spectra can then be compared to detect structural perturbations of the protein or variation in glycoform production.
Materials and methods
Cell growth
13C-labeled versions of the CEACAM1-IgV domain were prepared by overexpression in HEK293S (GnTI-) (MGAT1 knockout) cells (ATCC), which produce proteins primarily labeled with Man5GlcNAc2 glycans, using an expression vector and purification procedures previously described for preparation of unlabeled and labeled samples (Zhuo et al. 2016; Moremen et al. 2018). Briefly, a construct containing a secretion signal sequence, 6xHis-tag, AviTag, GFP, a TEV protease cleavage site and CEACAM1-IgV domain in the pGEn2 vector was transfected into HEK293S (GnTI-) cells (Reeves et al. 2002; Subedi et al. 2015). The transcript for the CEACAM1-IgV domain comprised residues 34–141 (UniProt P13688) and used codon optimization for mammalian expression. It was also preceded by codons for a short linker (SGG), which was added to minimize steric interactions between the GFP and CEACAM1-IgV and provide greater accessibility for TEV protease cleavage. For 13C-labeling of glycans, cells were maintained in shake flask cultures using 1 L FreeStyle 293 expression media at 37°C in a humidified CO2 platform shaker. U-13C-glucose (5 g/L, Cambridge Isotope Labs, Tewksbury, MA) was added to the cells 24 h after transfection and the culture was maintained at 37 °C for 5 days. The protein was harvested by removing the cells via centrifugation and recovering the media, which contained the expressed protein. The protein was collected using a Ni2+-NTA superflow column (Qiagen, Germantown, MD) and eluted using 25 mM HEPES, 300 mM NaCl, 300 mM imidazole, pH 7.0. All buffer components used for this study were purchased from Sigma-Aldrich. The eluted fusion protein was concentrated to 1 mg/mL and mixed with recombinant TEV protease at ratio of 1:10 relative to GFP-CEACAM1-IgV and incubated at 4°C for 24 h. The sample was applied to a second Ni2+-NTA column and the flow-through, containing cleaved CEACAM1-IgV, was collected. The cleavage event should leave a four amino acid (GSGG) scar. The purified protein was run over a Superdex-75 (GE Healthcare Life Sciences, Chicago, IL) column and exchanged into a buffer containing 50 mM sodium phosphate, 100 mM NaCl, 1 mM TCEP, pH 6.5. The yield was ~11 mg. All NMR experiments presented here employed a further buffer exchange into the D2O version of the same buffer, supplemented with 5 mM NaN3 and 50 μM DSS (as an NMR internal reference).
A sample of CEACAM1-IgV was also labeled with u-13C-Ala (Cambridge Isotope Labs) to verify the chemical shifts of alanine and compare the labeling levels using MS. This was prepared as above with the following modifications. A custom dropout FreeStyle Expression medium without GlutaMAX (L-Ala-L-Gln) was supplemented with 100 mg/L u-13C-Ala and unlabeled Gln. After the protein was collected from the media using Ni2+-NTA resin, the protein was treated with both TEV protease and Endo-F, which cleaves between the first and second GlcNAc of Man5GlcNAc2, generating a protein that contains a single GlcNAc at the glycosylation site, but is otherwise structurally identical (based on NMR) to versions with more complete glycosylation. Mass spectrometry was used to verify enrichment of alanine as well as determine if there was any other unexpected enrichment (Ala ~ 40%, other labeling minimal). For analysis of site occupancy by mass spectrometry, a similar procedure was followed except cultures of ~100 mL were used and no labeled glucose or isotopically enriched amino was added.
In addition, a sample of a CEACAM1-IgV mutant (S96C) was metabolically labeled with dimethyl-13C-Val (Cambridge Isotope Labs) and u-13C-glucose to identify valine resonances. Labeling was achieved using a culture medium depleted in a subset of essential amino acids deemed useful for NMR experiments (Val, Phe, Tyr, and Lys). Dimethyl-13C-Val (150 mg/L, CIL) and u-13C-glucose were added to the media along with unlabeled forms of Phe, Tyr and Lys. The media also contained the normal concentration of GlutaMAX found in the FreeStyle Expression medium formulation. The S96C mutation resulted in a protein in which the major species was glycosylated at all three sites (MS verification). All samples were quantified using a Nanodrop 2000c spectrophotometer using an extinction coefficient of 14,400 M−1 cm−1.
Nuclear magnetic resonance
All experiments were carried out on 600 MHz Bruker AVANCE NEO spectrometers with TCI or TXO (13C-optimized) cryoprobes using 5 mm samples at 298 K. All samples were in 50 mM sodium phosphate buffer, pH 6.5, with 100 mM NaCl and 5 mM NaN3. The 1H–13C HSQCs utilized the Bruker pulse sequence hsqcetgpsisp2, which uses gradient pulses as well as sensitivity enhancement to reduce artifacts and optimize signals. The u-13C-glucose supplemented sample was 470 μM. Spectra were taken over a spectral width of 13 ppm in the proton dimension and 120 ppm in the carbon dimension with offsets of 4.7 and 60 ppm, respectively. The size of the fid was 6248 × 1024 and the relaxation delay was 1.5 s. For the sample made with both dimethyl-13C-valine labeling and supplemented with u-13C-glucose, the concentration was 420 μM and the fid size was 1024 × 140. The other parameters remained. All data were processed using a combination of TOPSPIN (version 4.0) and Mnova (V14). All NMR figures were made in Mnova.
Top–down mass spectrometry
For top–down site occupancy analysis, CEACAM1-IgV was prepared at a concentration of 20 μM in a solution of 49.5/49.5/1 (v/v/v) % water/methanol/formic acid and introduced into the mass spectrometer via direct infusion electrospray ionization at a flow rate of 2 μL/min. Mass spectrometry was performed using a 12T Bruker Solarix FT-ICR-MS. ECD MS2 spectra were collected using a 20 ms electron pulse, 1.0 V bias voltage and 15.0 V lens voltage. Peak picking and deconvolution were performed in Bruker DataAnalysis. Initial fragment assignments were determined using Protein Prospector MS-Product (prospector.ucsf.edu/prospector/mshome.htm) with a 50 ppm tolerance. A subsequent internal calibration was applied with a 2 ppm mass tolerance. All ions used for glycosylation site mapping were verified manually.
Glycopeptide analysis of isotope enrichment of sugars
For isotopic enrichment analysis of the CEACAM1-IgV glycans prepared with u-13C-glucose, 100 μg of the glycoprotein in 17 mM NH4HCO3 was carbamidomethylated with 30 mM iodoacetamide (IAA). Excess IAA was removed via centrifugal filtration (Amicon Ultra with a 10 kDa cut-off). The sample was digested with 5 μg of sequencing grade trypsin (Promega, Madison, WI) for 12 h at 37°C followed by digestion with 5 μg of elastase (Promega, Madison, WI) overnight at 37°C. The digest was analyzed on an Orbitrap Fusion Tribrid mass spectrometer equipped with a nanospray ion source and connected to a Dionex nano-LC system. A prepacked reverse phase column (C18 from Dionex) was used to separate peptides. Parent peaks for the peptides ETIYPN(HexNAc)AS and TQN(HexNAc)DTGFYT were subjected to higher-energy collisional dissociation and collision-induced dissociation MS2 to evaluate isotope peaks corresponding to oxonium ions from the HexNAc as either completely 12C-labeled (m/z 204.0874), 13C-acetate/12C-hexose (m/z 206.0930), 12C-acetate/13C-hexose (m/z 210.1061) or completely 13C-labeled (m/z 212.1027). Data analysis was performed using Data Xcalibur 3.0 and Byonic software as well as manual verification. Errors are standard deviations from four independent measures of isotope incorporation.
Supplementary Material
Acknowledgements
We thank Dr. John Glushka for his assistance in collecting NMR data and Dr. Parastoo Azadi and her staff for their assistance with MS-based isotopic analysis of sugars. Manuscript content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- CID
collision-induced dissociation
- COSY
correlation spectroscopy
- ECD
electron capture dissociation
- Endo-F
Endoglycosidase-F1
- HCD
higher-energy collisional dissociation
- HMBC
heteronuclear multiple bond correlation
- HMQC
heteronuclear multiple quantum coherence
- HSQC
heteronuclear single quantum coherence
- MS
mass spectrometry
- MS2
tandem mass spectrometry
- NMR
nuclear magnetic resonance
- TOCSY
total correlated spectroscopy
Supplementary data
Supplementary data for this article are available online at http://glycob.oxfordjournals.org/.
Funding
This work was supported by grants from the US National Institutes of Health (grant numbers R01GM033225, P41GM103390, T32GM107004, S10OD025118, S10OD021623, S10OD018530).
Conflict of interest statement
The authors declare no conflict of interest.
References
- Anish C, Schumann B, Pereira CL, Seeberger PH. 2014. Chemical biology approaches to designing defined carbohydrate vaccines. Chem Biol. 21:38–50. [DOI] [PubMed] [Google Scholar]
- Apweiler R, Hermjakob H, Sharon N. 1999. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim Biophys Acta. 1473:4–8. [DOI] [PubMed] [Google Scholar]
- Arda A, Jimenez-Barbero J. 2018. The recognition of glycans by protein receptors. Insights from NMR spectroscopy. Chem Commun. 54:4761–4769. [DOI] [PubMed] [Google Scholar]
- Ayala I, Sounier R, Use N, Gans P, Boisbouvier J. 2009. An efficient protocol for the complete incorporation of methyl-protonated alanine in perdeuterated protein. J Biomol NMR. 43:111–119. [DOI] [PubMed] [Google Scholar]
- Bax A, Grishaev A. 2005. Weak alignment NMR: a hawk-eyed view of biomolecular structure. Curr Opin Struct Biol. 15:563–570. [DOI] [PubMed] [Google Scholar]
- Brockhausen I, Nair DG, Chen M, Yang XJ, Allingham JS, Szarek WA, Anastassiades T. 2016. Human acetyl-CoA:glucosamine-6-phosphate N-acetyltransferase 1 has a relaxed donor specificity and transfers acyl groups up to four carbons in length. Biochem Cell Biol. 94:197–204. [DOI] [PubMed] [Google Scholar]
- Clore GM. 2015. Practical aspects of paramagnetic relaxation enhancement in biological macromolecules. In: Qin PZ, Warncke K, editors. Electron Paramagnetic Resonance Investigations of Biological Systems by Using Spin Labels, Spin Probes, and Intrinsic Metal Ions, Pt B.Elsevier Academic Press Inc, San Diego, CA USA p. 485–497. [DOI] [PMC free article] [PubMed]
- Flugge F, Peters T. 2018. Complete assignment of Ala, Ile, Leu, Met and Val methyl groups of human blood group A and B glycosyltransferases using lanthanide-induced pseudocontact shifts and methyl-methyl NOESY. J Biomol NMR. 70:245–259. [DOI] [PubMed] [Google Scholar]
- Flugge F, Peters T. 2019. Insights into allosteric control of human blood group A and B glycosyltransferases from dynamic NMR. ChemistryOpen. 8:760–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke B, Opitz C, Isogai S, Grahl A, Delgado L, Gossert AD, Grzesiek S. 2018. Production of isotope-labeled proteins in insect cells for NMR. J Biomol NMR. 71:173–184. [DOI] [PubMed] [Google Scholar]
- Gao Q, Chen CY, Zong C, Wang S, Ramiah A, Prabhakar P, Morris LC, Boons GJ, Moremen KW, Prestegard JH. 2016. Structural aspects of heparan sulfate binding to Robo1-Ig1-2. ACS Chem Biol. 11:3106–3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto NK, Gardner KH, Mueller GA, Willis RC, Kay LE. 1999. A robust and cost-effective method for the production of Val, Leu, Ile (delta 1) methyl-protonated N-15-, C-13-, H-2-labeled proteins. J Biomol NMR. 13:369–374. [DOI] [PubMed] [Google Scholar]
- Kainosho M, Miyanoiri Y, Terauchi T, Takeda M. 2018. Perspective: next generation isotope-aided methods for protein NMR spectroscopy. J Biomol NMR. 71:119–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kainosho M, Torizawa T, Iwashita Y, Terauchi T, Mei Ono A, Guntert P. 2006. Optimal isotope labelling for NMR protein structure determinations. Nature. 440:52–57. [DOI] [PubMed] [Google Scholar]
- Kapust RB, Tozser J, Copeland TD, Waugh DS. 2002. The P1' specificity of tobacco etch virus protease. Biochem Biophys Res Commun. 294:949–955. [DOI] [PubMed] [Google Scholar]
- Kato K, Matsunaga C, Igarashi T, Kim H, Odaka A, Shimada I, Arata Y. 1991. Complete assignment of the methionyl carbonyl carbon resonances in switch variant anti-dansyl antibodies labeled with [1-13C]methionine. Biochemistry. 30:270–278. [DOI] [PubMed] [Google Scholar]
- Kato K, Yamaguchi T. 2015. Paramagnetic NMR probes for characterization of the dynamic conformations and interactions of oligosaccharides. Glycoconj J. 32:505–513. [DOI] [PubMed] [Google Scholar]
- Kato K, Yamaguchi Y, Arata Y. 2010. Stable-isotope-assisted NMR approaches to glycoproteins using immunoglobulin G as a model system. Prog Nucl Magn Reson Spectrosc. 56:346–359. [DOI] [PubMed] [Google Scholar]
- Kelleher NL. 2004. Peer reviewed: top-down proteomics. Anal Chem. 76:196 A–203 A. [PubMed] [Google Scholar]
- Kerfah R, Plevin MJ, Sounier R, Gans P, Boisbouvier J. 2015. Methyl-specific isotopic labeling: a molecular tool box for solution NMR studies of large proteins. Curr Opin Struct Biol. 32:113–122. [DOI] [PubMed] [Google Scholar]
- Lu Q, Li S, Shao F. 2015. Sweet talk: protein glycosylation in bacterial interaction with the host. Trends Microbiol. 23:630–641. [DOI] [PubMed] [Google Scholar]
- McNeill AS, Dallas BH, Eiler JM, Bylaska EJ, Dixon DA. 2020. Reaction energetics and C-13 fractionation of alanine transamination in the aqueous and gas phases. J Phys Chem A. 124:2077–2089. [DOI] [PubMed] [Google Scholar]
- Mellquist JL, Kasturi L, Spitalnik SL, Shakin-Eshleman SH. 1998. The amino acid following an Asn-X-Ser/Thr sequon is an important determinant of N-linked core glycosylation efficiency. Biochemistry. 37:6833–6837. [DOI] [PubMed] [Google Scholar]
- Minato Y, Suzuki S, Hara T, Kofuku Y, Kasuya G, Fujiwara Y, Igarashi S, Suzuki E, Nureki O, Hattori Met al. 2016. Conductance of P2X4 purinergic receptor is determined by conformational equilibrium in the transmembrane region. Proc Natl Acad Sci U S A. 113:4741–4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moremen KW, Ramiah A, Stuart M, Steel J, Meng L, Forouhar F, Moniz HA, Gahlay G, Gao ZW, Chapla Det al. 2018. Expression system for structural and functional studies of human glycosylation enzymes. Nat Chem Biol. 14:156–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitsche C, Otting G. 2017. Pseudocontact shifts in biomolecular NMR using paramagnetic metal tags. Prog Nucl Magn Reson Spectrosc. 98-99:20–49. [DOI] [PubMed] [Google Scholar]
- O'Flaherty R, Trbojevic-Akmacic I, Greville G, Rudd PM, Lauc G. 2018. The sweet spot for biologics: recent advances in characterization of biotherapeutic glycoproteins. Expert Rev Proteomics. 15:13–29. [DOI] [PubMed] [Google Scholar]
- Ollerenshaw JE, Tugarinov V, Kay LE. 2003. Methyl TROSY: explanation and experimental verification. Magn Reson Chem. 41:843–852. [Google Scholar]
- Petrescu AJ, Milac AL, Petrescu SM, Dwek RA, Wormald MR. 2004. Statistical analysis of the protein environment of N-glycosylation sites: implications for occupancy, structure, and folding. Glycobiology. 14:103–114. [DOI] [PubMed] [Google Scholar]
- Prestegard JH, Agard DA, Moremen KW, Lavery LA, Morris LC, Pederson K. 2014. Sparse labeling of proteins: structural characterization from long range constraints. J Magn Reson. 241:32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prestegard JH, Bougault CM, Kishore AI. 2004. Residual dipolar couplings in structure determination of biomolecules. Chem Rev. 104:3519–3540. [DOI] [PubMed] [Google Scholar]
- Reeves PJ, Callewaert N, Contreras R, Khorana HG. 2002. Structure and function in rhodopsin: high-level expression of rhodopsin with restricted and homogeneous N-glycosylation by a tetracycline-inducible N-acetylglucosaminyltransferase I-negative HEK293S stable mammalian cell line. Proc Natl Acad Sci U S A. 99:13419–13424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakae Y, Satoh T, Yagi H, Yanaka S, Yamaguchi T, Isoda Y, Iida S, Okamoto Y, Kato K. 2017. Conformational effects of N-glycan core fucosylation of immunoglobulin G Fc region on its interaction with Fc gamma receptor IIIa. Sci Rep. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastry M, Xu L, Georgiev IS, Bewley CA, Nabel GJ, Kwong PD. 2011. Mammalian production of an isotopically enriched outer domain of the HIV-1 gp120 glycoprotein for NMR spectroscopy. J Biomol NMR. 50:197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh MO, Xu Y, Wel H, Walden P, Hartson SD, West CM. 2015. Glycosylation of Skp1 promotes formation of Skp1-cullin-1-F-box protein complexes in dictyostelium. Mol Cell Proteomics. 14:66–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skelton D, Goodyear A, Ni DQ, Walton WJ, Rolle M, Hare JT, Logan TM. 2010. Enhanced production and isotope enrichment of recombinant glycoproteins produced in cultured mammalian cells. J Biomol NMR. 48:93–102. [DOI] [PubMed] [Google Scholar]
- Subedi GP, Johnson RW, Moniz HA, Moremen KW, Barb A. 2015. High yield expression of recombinant human proteins with the transient transfection of HEK293 cells in suspension. J Vis Exp. 106:e53568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subedi GP, Falconer DJ, Barb AW. 2017. Carbohydrate-Polypeptide Contacts in the Antibody Receptor CD16A Identified through Solution NMR Spectroscopy. Biochemistry. 56:3174–3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi H, Haltiwanger RS. 2014. Significance of glycosylation in notch signaling. Biochem Biophys Res Commun. 453:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tugarinov V, Kay LE. 2003. Ile, Leu, and Val methyl assignments of the 723-residue malate synthase G using a new labeling strategy and novel NMR methods. J Am Chem Soc. 125:13868–13878. [DOI] [PubMed] [Google Scholar]
- Ulrich EL, Akutsu H, Doreleijers JF, Harano Y, Ioannidis YE, Lin J, Livny M, Mading S, Maziuk D, Miller Zet al. 2008. BioMagResBank. Nucleic Acids Res. 36:D402–D408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A, Gagneux P. 2015. Biological functions of glycans. In: Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, Darvill AG, Kinoshita T, Packer NH, Prestegard JHet al., editors. Essentials of Glycobiology. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press. p. 77–88. [PubMed] [Google Scholar]
- Weil BR, Abarbanell AM, Herrmann JL, Wang Y, Meldrum DR. 2009. High glucose concentration in cell culture medium does not acutely affect human mesenchymal stem cell growth factor production or proliferation. Am J Phys Regul Integr Comp Phys. 296:R1735–R1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XZ, Eletsky A, Sheikh MO, Prestegard JH, West CM. 2018. Glycosylation promotes the random coil to helix transition in a region of a Protist Skp1 associated with F-box binding. Biochemistry. 57:511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi H, Pilla KB, Maleckis A, Graham B, Huber T, Otting G. 2013. Three-dimensional protein fold determination from backbone amide pseudocontact shifts generated by lanthanide tags at multiple sites. Structure. 21:883–890. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Kato K, Shindo M, Aoki S, Furusho K, Koga K, Takahashi N, Arata Y, Shimada I. 1998. Dynamics of the carbohydrate chains attached to the Fc portion of immunoglobulin G as studied by NMR spectroscopy assisted by selective C-13 labeling of the glycans. J Biomol NMR. 12:385–394. [DOI] [PubMed] [Google Scholar]
- Yanaka S, Yagi H, Yogo R, Yagi-Utsumi M, Kato K. 2018. Stable isotope labeling approaches for NMR characterization of glycoproteins using eukaryotic expression systems. J Biomol NMR. 71:193–202. [DOI] [PubMed] [Google Scholar]
- Yang Z, Wang S, Halim A, Schulz MA, Frodin M, Rahman SH, Vester-Christensen MB, Behrens C, Kristensen C, Vakhrushev SYet al. 2015. Engineered CHO cells for production of diverse, homogeneous glycoproteins. Nat Biotechnol. 33:842–844. [DOI] [PubMed] [Google Scholar]
- Zhuo Y, Yang JY, Moremen KW, Prestegard JH. 2016. Glycosylation alters dimerization properties of a cell-surface signaling protein, carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1). J Biol Chem. 291:20085–20095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubarev RA, Horn DM, Fridriksson EK, Kelleher NL, Kruger NA, Lewis MA, Carpenter BK, McLafferty FW. 2000. Electron capture dissociation for structural characterization of multiply charged protein cations. Anal Chem. 72:563–573. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.