

Abstract
Gene editing by the CRISPR-Cas9 nuclease system technology can be considered among the most promising strategies to correct hereditary mutations in a variety of monogenic diseases. In this paper, we present for the first time the correction, by CRISPR-Cas9 gene editing, of the β039-thalassemia mutation, one of the most frequent in the Mediterranean area. The results obtained demonstrated the presence of normal β-globin genes after CRISPR-Cas9 correction of the β039-thalassemia mutation performed on erythroid precursor cells from homozygous β039-thalassemia patients. This was demonstrated by allele-specific PCR and sequencing. Accumulation of corrected β-globin mRNA and relevant “de novo” production of β-globin and adult hemoglobin (HbA) were found with high efficiency. The CRISPR-Cas9-forced HbA production levels were associated with a significant reduction of the excess of free α-globin chains. Genomic toxicity of the editing procedure (low indels and no off-targeting) was analyzed. The protocol might be the starting point for the development of an efficient editing of CD34+ cells derived from β039 patients and for the design of combined treatments using, together with the CRISPR-Cas9 editing of the β-globin gene, other therapeutic approaches, such as, for instance, induction of HbA and/or fetal hemoglobin (HbF) using chemical inducers.
Keywords: CRISPR-Cas9, genome editing, β039-thalassemia, erythroid precursors, β-globin, personalized therapy
Graphical abstract
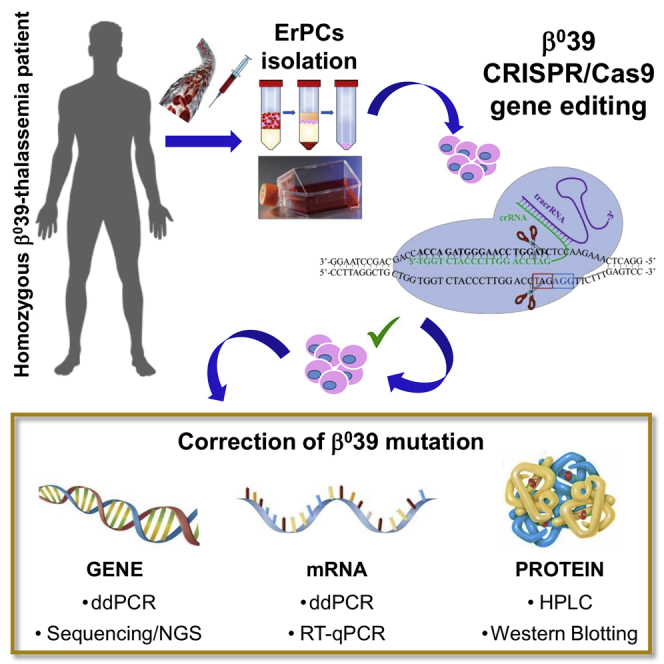
The “CRISPR-Cas9 gene editing” is among the most promising strategies to correct hereditary mutations. In this paper, we present the efficient correction of the nonsense β039 mutation, very frequent in β-thalassemia patients of the Mediterranean area. The protocol might be the starting point for innovative personalized therapeutic intervention in β-thalassemia.
Introduction
The β-thalassemias are a group of hereditary diseases caused by more than 300 mutations of the adult β-globin gene, leading to low or absent production of adult hemoglobin (HbA).1, 2, 3, 4 Together with sickle-cell disease (SCD), thalassemia syndromes are among the most impactful diseases in developing countries, in which the lack of genetic counseling and prenatal diagnosis have contributed to the maintenance of a very high frequency in the population.1,2 The management of β-thalassemia patients is mostly based on blood transfusion, chelation therapy, and alternatively, bone marrow transplantation.2
Recently, novel therapeutic options have been explored, such as gene therapy and fetal hemoglobin (HbF) induction.4,5 Even though these approaches are promising, they are, at present, still under deep experimental development and limited to a low number of clinical trials.4, 5, 6 With respect to gene therapy for β-thalassemia, significant progress is expected, also considering fundamental insights into globin switching and new technology developments, which might have a strong impact on novel gene-therapy approaches.4 A robust information is, however, available regarding the management of β-thalassemia, i.e., that patients exhibiting high levels of endogenous HbF might exhibit a milder clinical status, as in the case of hereditary persistence of HbF (HPFH).6, 7, 8, 9 In this context, one of the most exciting strategies recently proposed for hereditary diseases, including β-thalassemia, is genome editing, using a variety of strongly validated approaches targeting human hematopoietic cells.10 Among these strategies, the CRISPR and CRISPR-associated protein 9 (Cas9; CRISPR-Cas9) nuclease system is considered the most promising.10, 11, 12, 13
Although efficient gene editing has been reported for a variety of genetic diseases,14, 15, 16, 17 CRISPR-Cas9-based gene editing of the β-globin gene in β0-thalassemia has to be optimized to reach clinically relevant levels of HbA production. The protocol is based on the design of a CRISPR-Cas9 strategy to replace the mutated with normal gene sequences with the precise homology-directed repair (HDR) mechanism.4,5,12,13 To the best of our knowledge, no data are available in the literature on the CRISPR-Cas9-based gene correction of the β039-thalassemia mutation. Most of the gene-editing intervention for β-thalassemia is focusing on other mutations (e.g., IVS2-654 or IVS I-110)4,5,18, 19, 20, 21, 22 or on the reactivation of fetal globin gene expression by disrupting the γ-globin gene repressor BCL11A.4,5,23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33
An example is the paper by Antoniani et al.24 They designed a CRISPR-Cas9 strategy to disrupt a 13.6-kb genomic region encompassing the δ- and β-globin genes and a putative γ-δ intergenic HbF. Disruption of just the putative HbF silencer resulted in a mild increase in γ-globin expression, whereas deletion or inversion of a 13.6-kb region causes a robust reactivation of HbF synthesis in adult erythroblasts that is associated with epigenetic modifications and changes in chromatin contacts within the β-globin locus. In primary SCD patient-derived hematopoietic stem cell (HSC)/progenitor cell (HSPCs), targeting the 13.6-kb region results in a high proportion of γ-globin expression in erythroblasts, increased HbF synthesis, and amelioration of the sickling-cell phenotype. Overall, this study provides clues for a potential CRISPR-Cas9 genome-editing approach to the therapy of β-hemoglobinopathies.
A further example has been recently reported by Métais et al.32 They disrupted an HBG1/HBG2 gene promoter motif that is bound by the transcriptional repressor BCL11A. Electroporation of a Cas9 single-guide RNA (sgRNA) ribonucleoprotein (RNP) complex into normal and SCD donor CD34+ HSPCs resulted in high frequencies of on-target mutations and the induction of HbF to potentially therapeutic levels in erythroid progeny generated in vitro and in vivo after transplantation of HSPC mouse models.
Disruption of BCL11A in HSCs by using CRISPR-Cas9 protocols is, at present, under clinical validation and is expected to implement information deriving from similar zinc finger nuclease (ZFN)-based interventions (ClinicalTrials.gov: NCT03432364, NCT03653247, NCT03655678, and NCT03745287).34
This study aims to determine whether the CRISPR-Cas9 technology can be applied to correct the β0-thalassemia mutation in erythroid precursors isolated from patients affects by β039-thalassemia. This nonsense mutation causes the introduction of a stop codon with the production of a truncated, non-functional β-globin, leading to a β0-globin phenotype. Furthermore, the β039-globin mRNA is unstable, due to the well-known “nonsense-mediated decay” (NMD).1, 2, 3
Results
Experimental strategy for CRISPR-Cas9 correction of the thalassemia β039 mutation
Figure 1A shows the experimental strategy for the correction of the β039-thalassemia mutation in erythroid precursor cells (ErPCs) isolated from β039 homozygous patients. Cells are cultured for 7 days in culture medium in the absence of erythropoietin (EPO) (phase I). Then, cells are transferred to an EPO-dependent medium (phase II) to stimulate erythroid differentiation and hemoglobin production. After 3 days of phase II culture, the cells are electroporated, adding to the reaction mix all the components of the CRISPR-Cas9 system. After electroporation, the cells are maintained in phase II medium for a further 5 days and then analyzed to evaluate the correction of the β039-thalassemia mutation at the following levels: genomic (sequencing and droplet digital polymerase chain reaction [ddPCR]), transcriptomic (qRT-PCR and RT-ddPCR), and protein (western blotting and high-performance liquid chromatography [HPLC]).
Figure 1.

Experimental strategy
(A) Experimental strategy for the correction of the β039-thalassemia mutation in ErPCs isolated from β039 homozygous patients. After the first step of cell culture (phase I), the ErPCs are transferred in medium containing EPO (phase II). At day 3, cells are electroporated, and the CRISPR-Cas9 genomic editing was performed. After genomic editing, the cells have been maintained in phase II medium for a further 5 days and then collected and analyzed to evaluate the correction of the β039-thalassemia mutation at genomic, transcriptomic, and proteomic levels. (B) Scheme representing the CRISPR-Cas9 model system used for the β039 correction.
Figure 1B shows the CRISPR-Cas9-based design for the correction of the β039-thalassemia mutation. The CRISPR-Cas9 system binds, by CRISPR RNA (crRNA), on exon 2 of the β-globin gene, cutting the DNA near the protospacer adjacent motif (PAM; AGG, in blue) sequence causing the formation of a DSB (double-strand break) and inducing the cell to activate repair mechanisms, such as homologous recombination. The cells will use a single-strand (ss) donor DNA (ssDNA) as a template to repair the damage; this allows the insertion of the correct nucleotide sequence instead of the one containing the β039 stop mutation.
Culture and editing procedure using ErPCs isolated from a β039-thalassemia patient
The blood samples were collected from β039-thalassemia patients after informed consent. The ErPCs, obtained from 25 mL of peripheral blood, were cultured following the Fibach protocol7 and electroporated on the 3rd day of phase II culture in the presence of the CRISPR-Cas9 system and a ssDNA β-globin donor template (fwd [forward] donor; rev [reverse] donor). After the genomic-editing procedure, the cells were monitored, observed under the microscope, and counted at 3, 5, and 7 days after electroporation in order to monitor any possible difference between edited and not edited (C−) cells. The data reported in Figure S1 show, after the CRISPR-Cas9 editing, the maintenance of a good survival and growth capacity (in comparison to those of control cells) in cells from all of the patients analyzed, comparable with those of control cells. These data suggest that no clonal selection is occurring following the gene-editing protocol.
Efficiency of β039 gene editing: genomic analyses
Two complementary approaches have been followed to demonstrate the presence of the normal β-globin gene after CRISPR-Cas9 correction of the β039-thalassemia mutation performed on ErPCs from homozygous β039-thalassemia patients: (1) allele-specific PCR, performed using ddPCR and (2) direct gene sequencing (Sanger sequencing). Additional information about editing efficiency, obtained with whole genome sequencing (WGS) and amplicon next-generation sequencing (NGS), is presented and discussed in the relative sections.
Figures 2A and 2B show a representative analysis of gene correction with the CRISPR-Cas9 system performed on ErPC genomic DNA isolated from a β039-thalassemia patient (Th-ErPC-2) and analyzed by ddPCR assay. The correction data plotted are represented in the1-dimensional (1d) and 2-dimensional (2d) dot plots (Figure 2A). The blue dot plots, related to the recognition of the correct β-globin gene sequence, and the green dot plots, for the recognition of the β-globin gene sequence with the β039 mutation, have been done with two different probes (edited 6-carboxyfluorescein [FAM] and mutated hexachloro-6-carboxy fluorescein [HEX], respectively). The samples indicated as C−, gene editing (GE) Fwd, and GE Rev represent control untreated cells, cells treated with the CRISPR-Cas9 system with fwd donor DNA template, and cells treated with the CRISPR-Cas9 system with rev donor DNA template, respectively.
Figure 2.

Genomic analysis of the CRISPR-Cas9 system in ErPC cultures
(A–C) The data relating to a representative example of ddPCR analysis of a β039-thalassemia patient-derived ErPC (Th-ErPC-2) (A and B), whereas (C) shows the ddPCR average result of all ErPCs analyzed. (A) Representative results obtained after gene-correction treatment performed by the CRISPR-Cas9 system on a culture of ErPCs isolated from a β039-thalassemia patient and analyzed by ddPCR assay. C−, untreated cells; GE forward (Fwd), cells treated with the CRISPR-Cas9 system with fwd donor DNA; GE reverse (Rev), cells treated with the CRISPR-Cas9 system with rev donor DNA. Top and middle: 1d dot plot showing edited and mutated β-globin sequence. Colored dots reported in (A) represent the events (droplets) in function of the respective FAM or HEX fluorescence. (B) Fractional abundance related at the representative ddPCR example reported in (A). (C) Average fractional abundance calculated from the average concentrations of six ddPCR analyses performed on ErPC cultures isolated from different β039-thalassemia patients. (D) Electropherograms related to a small portion of the β-globin gene in which the β039 mutation is highlighted with the mutated nucleotide T (red) and the corrected nucleotide C (blue) and obtained by DNA sequencing from the edited ErPC cultures.
As clearly seen, the presence of amplified, edited β-globin gene sequences are absent in C− but present in both GE Fwd and GE Rev samples (Figure 2A, blue spots in the upper panel). As expected, mutated β-globin gene sequences are present in all the samples (Figure 2A, middle panel). In the bottom panel of Figure 2A, the 2d dot plot, which represents the specificity of the assay used for genomic analysis, is visualized. The correction level obtained is reported in the form of fractional abundance %, calculated as the edited/(edited + mutated) concentration. In Figures 2B and 2C, the fractional abundance, in purple, is shown. In Figure 2B, the data obtained from the same representative experiment shown in Figure 2A are reported, whereas in Figure 2C, the average concentrations of ddPCR analyses performed on six different ErPC cultures isolated from β039/β039-thalassemia patients are shown. The average fractional abundance was found to be 6.67 ± 1.00 in GE Fwd samples and 4.60 ± 1.01 in GE Rev samples.
A representative Sanger sequencing experiment reporting the electropherograms obtained by sequencing of the DNA extracted from the ErPC cultures treated with the CRISPR-Cas9 system, containing both potential edited cells and cells in which editing has not occurred, is shown in Figure 2D. In particular, only a small portion of the β-globin gene is reported, in which the β039 mutation is highlighted with the mutated nucleotide T (red) and the corrected nucleotide C (blue). Although the mutated nucleotide is still predominant even in the edited samples (GE Fwd and GE Rev), the peaks corresponding to the corrected sequence are increased in both GE Fwd and GE Rev samples. The proportion of the sequencing peaks corresponding to the corrected nucleotide (C) increased in both CRISPR-Cas9-edited ErPCs GE Fwd and GE Rev. The percentage of this increase was calculated as the area subtended by the peaks, and in particular, the % of C/C + T was found to range between 34.29% and 25.97% for fwd and rev samples, respectively.
In conclusion, the experiments performed using ddPCR and shown in Figures 2A−2C demonstrate that amplification of a normal corrected β-globin gene sequence was detectable only when CRISPR-Cas9-treated ErPCs were employed. No amplification was detectable using untreated β039/ β039 ErPCs. Analysis of the fractional abundance of the edited genomes suggests that at least 4.5% of the β-globin genes have been corrected by this approach. With the consideration of the differences between the two technologies employed (digital PCR and sequencing), we can anyway conclude that the obtained data display the same trend.
High expression of normal β-globin mRNA following CRISPR-Cas9 correction of the thalassemia β039 mutation
In order to evaluate the correction level of the β039-thalassemia mutation obtained from ErPCs treated with our the CRISPR-Cas9 system, we analyzed both mutated β039 as well as edited β-globin mRNAs using RT-PCR and RT-ddPCR approaches. Figures 3A and 3B show a representative example of accumulation of β-globin mRNA from a β039/β039 homozygous β-thalassemia patient (Th-ErPC-2) and analyzed by RT-ddPCR assay. The amplification events are represented in 1d (Figure 3A, upper and middle panels) and 2d dot plots (Figure 3A, bottom panel). The blue dot plots (correct β-globin mRNA) and the green dot plots (β-globin mRNA with the β039 mutations) have been obtained with two different and selective probes: edited FAM and mutated HEX, respectively. In Figure 3B, the data obtained from the representative experiment (Figure 3A) are reported in the form of fractional abundance %, calculated as an edited/(edited + mutated) concentration, whereas in Figure 2C, the average fractional abundance % of different mRNA samples isolated from six CRISPR-Cas9-treated β039/β039-thalassemia ErPC cultures derived from different β-thalassemia patients is shown. The average fractional abundance was found to be 48.28 ± 1.21 in GE Fwd samples and 50.50 ± 0.91 in GE Rev samples. The different fractional abundance found at the genomic (Figure 2C) and transcriptomic (Figure 3C) levels is expected, since it is known that the mutated β039-globin mRNA is far less stable than the normal β-globin mRNA. It is known indeed that the accumulated mutated β039-globin mRNA does not exceed 2%–5% with respect to the expected content values predicted on the basis of transcriptional efficiency.35
Figure 3.

Gene-editing correction by the CRISPR-Cas9 system evaluated in globin mRNAs
(A) Representative example of gene-correction treatment performed by the CRISPR-Cas9 system on Th-ErPC-2 and analyzed by RT-ddPCR assay. The treatment points shown in the figure are: C− (untreated); GE Fwd (cells treated with the CRISPR-Cas9 system with fwd donor DNA); GE Rev (cells treated with the CRISPR-Cas9 system with rev donor DNA). Top and middle: 1d dot plots obtained after analysis of edited β-globin mRNA (top) or non-corrected cultures; bottom: 2d dot plot showing edited and mutated β-globin mRNA. Colored dots reported in (A) represent the events (droplets) in function of the respective FAM or HEX fluorescence. (B) Fractional abundance (in purple) related at the representative example reported in (A) is shown. (C) The fractional abundance calculated on the average of six RT-ddPCR analyses performed on ErPC cultures isolated from different β039-thalassemia patients and represented by the histogram. (D) The histogram shows the relative content β039 non-treated (NT) values related to the α (blue)-, β (red)-, and γ (green)-globins obtained from the treated/NT ratio for each sample analyzed. All of the data were normalized using β-actin and GAPDH as housekeeping genes.
Accordingly, it was expected to find that the relatively slight increase of β-globin gene correction generated a sharp increase (at least 10-fold) when corrected β-globin transcripts were analyzed.
The relative content of globin mRNAs (α-, β-, and γ-globins) shown in Figure 3D is obtained analyzing, by qRT-PCR, the cDNA from ErPCs isolated from six different β-thalassemia patients treated with the CRISPR-Cas9 system. The histograms show the mean relative content values related to the α-globin (blue), β-globin (red), and γ-globin (green) mRNAs relative to C− untreated cells. The sharp increase in β-globin mRNA is well in agreement with the fractional abundance data shown in Figure 3C and demonstrated a high content of the edited β-globin mRNA in CRISPR-Cas9-treated ErPCs, which reached a level of total β-globin mRNA eight times higher than the level of C− samples. Further details of the qRT-PCR analyses are shown in Figure S2.
Western blotting and HPLC analyses demonstrate “de novo” production of β-globin and HbA following CRISPR-Cas9 correction of the β039-thalassemia mutation
We conclusively confirmed the correction of the β039-globin gene by performing two complementary approaches: western blotting (Figure 4) and HPLC (Figure 5) analyses.
Figure 4.

Evaluation of β- and α-globin proteins, analysis by western blotting
(A) The amount of β-globin protein (16 kDa) corrected and α-globin protein (15 kDa) was assessed by western blotting analysis by comparing it with the quantity of housekeeping β-actin (45 kDa) protein in the protein extracts obtained from a Th-ErPC-2 treated by the CRISPR-Cas9 system. The genome editing efficiency are reported in Table 2. (B) The quantification of the bands, carried out by means of a ChemiDoc densitometric analysis, reveals a significant increase in the quantity of β-globin protein (top panel) corrected for both donor templates using GE Fwd (p < 0.001) and GE Rev (p < 0.001). These results are reported as relative values calculated by normalizing the data with respect to the β-actin protein and as β-globin fold increase over C−. At the bottom of (B), the α-globin protein expression showed no difference reported either as relative value with respect to the β-actin protein or as fold increase with respect to the untreated sample (C−). The original uncut version of the gel is shown in Figure S3.
Figure 5.

Evaluation of adult hemoglobin (HbA) by HPLC analysis
(A−C) Chromatograms relating to the HPLC analysis conducted on the protein lysates of the ErPC cultures derived from three different β039-thalassemic patients (A, Th-ErPC-2; B, Th-ErPC-4; C, Th-ErPC-6). For each of them, the expression pattern of hemoglobins in the untreated cells and in the CRISPR-Cas9 system-treated cells was analyzed. (D) Percentages of HbA (left panel) and free α-globin chain (right panel) were represented as different colored spots for each sample analyzed. An inverse trend can be displayed for the two parameters analyzed. An opposite trend was recorded for hemoglobin HbA and free α chains for the untreated sample and for GE Fwd and GE Rev. The relationship between the trends of the two protein components was represented graphically and reported in the graph in (E). The genome editing efficiency is reported in Table 2.
Results obtained from western blotting evidenced a sharp increase of β-globin protein expression in CRISPR-Cas9-treated ErPCs. The band relative to the β-globin protein detected for the samples treated with the GE Fwd and GE Rev system is significantly more intense with respect to the untreated C−. On the other hand, no variation of β-actin protein was detected in all samples. The content of the α-globin was also analyzed and found not modified following treatment of the cells with the CRISPR-Cas9 system (Figure 4A). The relative levels of β-globin and α-globin proteins were calculated after normalization with the densitometric values obtained using an anti-β-actin antibody.
In particular, the level of β-globin protein in edited samples reaches up to a 7.1-fold increase (upper panels of Figure 4B) with respect to the non-edited control sample. On the contrary, the α-globin relative content did not exhibit any differences when edited samples are compared to untreated sample (C−) (Figure 4B, lower panels). The uncut versions of the gels are shown in Supplemental materials and methods (Figure S3).
The HPLC analyses performed on CRISPR-Cas9-edited ErPCs from three β039/β039-thalassemia patients are reported in Figure 5. The HPLC data, as clearly represented in chromatograms (Figures 5A−5C), indicate a de novo production of HbA in all edited samples analyzed (GE Fwd and GE Rev samples). The percentage of HbA, with respect to total hemoglobin content obtained after the CRISPR-Cas9 correction, was found to be between 17.04% and 28.75%.
This efficiency of HbA production is very high considering that only 4.6%–6.67% (genomic average fractional abundance; see Figure 2) of the β-globin genes have been corrected within the CRISPR-Cas9-treated cell populations. In order to understand what level of HbA was reached within the CRISPR-Cas9-corrected ErPCs, we developed an algorithm based on the expected assumption that (1) no increase in HbA is expected in ErPCs that were not corrected by the gene-editing treatment; (2) no major changes in the hemoglobin pattern occur in these cells (this hypothesis is supported by the concept that the pattern of control, untreated, electroporated cells is over-imposable to that of untreated controls, data no shown); (3) no major changes of expression of HbF and HbA2 occur in successfully CRISPR-Cas9-edited cells; and (4) the maximum correction was achieved in the edited cells (i.e., both the alleles were corrected). With the consideration of all of these hypotheses, the virtual HbA percentage increase within corrected ErPCs, as demonstrated by Figure 5, can be calculated applying the following algorithm:
where “HbAc” represents the area percentage obtained by HPLC analysis following the CRISPR-Cas9 correction, “HbAnc” represents the area percentage obtained by HPLC analysis in the sample not corrected (C− sample) with the CRISPR-Cas9 system, and “FAc” represents the fractional abundance value obtained in CRISPR-Cas9-corrected samples after ddPCR analysis of genomic DNA (see Figure 2).
The results obtained by applying this algorithm are shown in Table 1 and suggest a very high production of HbA (87.73% in Th-ErPC-2 GE Rev and Th-ErPC-4 GE Fwd; 94.08% in Th-ErPC-6 Rev) within the CRISPR-Cas9-corrected samples. These values are very close to the percentage of HbA that could be detected in ErPCs from healthy subjects (96% ± 3.2%, data derived from 8 independent cell cultures) (data not shown).
Table 1.
HbA percentage calculated by applying the algorithm that connects the genomic fraction abundance correction values and percentage areas obtained from HPLC analysis of the same ErPC cultures
| Sample | Virtual HbA% in corrected ErPCs |
|---|---|
| Th-ErPC-2 GE Fwd | 81.03 |
| Th-ErPC-2 GE Rev | 87.73 |
| Th-ErPC-4 GE Fwd | 87.73 |
| Th-ErPC-4 GE Rev | 87.61 |
| Th-ErPC-6 GE Fwd | 80.89 |
| Th-ErPC-6 GE Rev | 94.08 |
As far as possible clinically relevant effects, we found that in all of the CRISPR-Cas9-edited cultures, a strong reduction of the excess-free α-globin chains is present (Figure 5D). This should be considered an important effect since the excess of free α-globins is a major issue in pathological characteristics of β-thalassemia.1,36,37 The relationship between the increase of HbA and decrease of the excess of free α-globin chains is reported in Figure 5E, demonstrating high correlation levels (r2 = 0.85).
It is very important to underline that no modulation of the expression of the α-globin protein is induced with the CRISPR-Cas9 system (Figure 4) but rather, a reduction of the free α chains, as a part of them complexes with the correct beta chains to form the tetramer of the HbA.
Amplicon-based sequencing results
Bar-coded amplicons were sequenced on a NovaSeq 6000 platform 150 phycoerytrin (PE) mode. The obtained number of fragments was ∼400,000 for the 18 (nine in duplicate) samples sequenced. The calculation of the frequency of the edited base and the indels at the sites of interest showed an editing rate between 6.5% and 14.4% for the edited base (chr11:5,226,774), with deletion rate between 1.3% and 17.8%. Very low occurrence of insertions (lower than 0.1%) was detected (Table 2).
Table 2.
Editing results obtained by amplicon sequencing (replicates 1 and 2)
| Sample name | Replicate ID | Tot reads | Reads “T” allele | %AF T | Reads “C” allele | %AF C | Reads del | %AF del | Reads ins | %AF ins |
|---|---|---|---|---|---|---|---|---|---|---|
| Th-ErPC-2 C− | 1 | 458,006 | 456,607 | 99.7 | 725 | 0.2 | 19 | 0 | 0 | 0 |
| 1A | 218,508 | 217,278 | 99.4 | 318 | 0.1 | 6 | 0 | 0 | 0 | |
| Th-ErPC-2 GE Fwd | 2 | 340,907 | 24,731 | 7.3 | 38,805 | 11.4 | 53,527 | 15.7 | 622 | 0.2 |
| 2A | 228,217 | 162,305 | 71.1 | 24,163 | 10.6 | 40,733 | 17.8 | 464 | 0.2 | |
| Th-ErPC-2 GE Rev | 3 | 487,416 | 354,441 | 72.7 | 69,534 | 14.3 | 61,375 | 12.6 | 444 | 0.1 |
| 3A | 291,744 | 210,678 | 72.2 | 41,252 | 14.1 | 3,861 | 13.2 | 203 | 0.1 | |
| Th-ErPC-4 C− | 4 | 427,363 | 425,828 | 99.6 | 715 | 0.2 | 32 | 0.01 | 0 | 0 |
| 4A | 291,416 | 290,403 | 99.7 | 468 | 0.2 | 19 | 0.01 | 1 | 0 | |
| Th-ErPC-4 GE Fwd | 5 | 417,987 | 354,157 | 84.7 | 28,731 | 6.9 | 33,668 | 8.1 | 401 | 0.1 |
| 5A | 270,785 | 230,672 | 85.2 | 17,534 | 6.5 | 21,653 | 8.0 | 241 | 0.1 | |
| Th-ErPC-4 GE Rev | 6 | 397,882 | 335,472 | 84.3 | 29,596 | 7.4 | 31,592 | 7.9 | 53 | 0 |
| 6A | 276,418 | 233,346 | 84.4 | 20,950 | 7.6 | 21,124 | 7.6 | 52 | 0 | |
| Th-ErPC-6 C− | 7 | 461,288 | 459,674 | 99.7 | 893 | 0.2 | 23 | 0 | 0 | 0 |
| 7A | 262,450 | 261,364 | 99.6 | 557 | 0.2 | 53 | 0.02 | 0 | 0 | |
| Th-ErPC-6 GE Fwd | 8 | 377,829 | 31,396 | 8.3 | 26,798 | 7.1 | 35,671 | 9.4 | 538 | 0.1 |
| 8A | 262,572 | 218,433 | 83.2 | 23,086 | 8.8 | 19,472 | 7.4 | 1,206 | 0.5 | |
| Th-ErPC-6 GE Rev | 9 | 428,089 | 326,031 | 76.2 | 61,707 | 14.4 | 38,718 | 9.0 | 576 | 0.1 |
| 9A | 364,864 | 2,9097 | 80 | 35,703 | 9.8 | 36,575 | 10.0 | 589 | 0.2 |
The columns report the sample name, the replicate identification (ID), the total (Tot) number of reads containing the base position (chr11:5,226,774, corresponding to the β039 mutation), the number of reads containing the allele T (mutated), the allele frequency (AF) of T, the number of reads containing the allele C (not mutated), the AF of C, the number of reads containing the deletion (del), the AF of del, the number of reads containing the insertion (ins), and the AF of ins.
All samples showed an editing rate above the control samples, generated as expected background values, indicating that the editing worked on all samples analyzed. The two replicates showed high reproducibility of the allele frequencies (AFs) on the target site.
The data show that a consistent proportion of corrected sequences are present in all of the edited samples, in agreement with the digital PCR data shown in Figure 2, and demonstrate that both fwd and rev donors sustain gene correction. As far as indel effects, no significant insertions were found. On the contrary, deletions were detected to a proportion similar to the insertion of corrected sequences. A representative example among the analysis performed (Table 2) is shown in Figure 6 and further details in Figures S4A and S4B focusing, in particular, on indel effects. The efficacy of this editing protocol was confirmed in CD34+ purified hematopoietic cells. Representative examples are shown in Table S3, including data related to the percentage of correctly edited genomes (12.37% and 9.16% in the two CD34+ samples shown) and to the percentage of indels (17.64% and 6.72% deletions and 0.45% and 0.14% insertions, respectively). The full description of the efficacy of this protocol of CD34+-purified cells is the issue of a forthcoming study (unpublished data).
Figure 6.

Alignment of the fragments generated by the amplicon sequencing obtained from a sample edited with the β039 CRISPR-Cas9 system
The figure shows sections of some representative reads obtained following the sequencing of one of the two amplicons generated for Th-ErPC-2 GE Fwd (only one of the replicates is reported). In particular, the alignment relative to position chr11:5,226,774 (in which the β039-thalassemia mutation is present) is indicated. In addition to the four nucleotides reported each with a different color, deletions are visible, shown with a black line surmounted by the number of deleted nucleotides. The reads were aligned by placing the same unedited patient as the reference genome sequence.
WGS results
In order to confirm these data using the WGS approach and to extend the analysis on possible off-target effects of the gene-editing protocol, the study was focused on two randomly selected samples: Th-ErPC-2 and Th-ErPC-6 (C− and GE Fwd).
The whole genome libraries were sequenced on a NovaSeq 6000 platform using 150 PE mode. The obtained number of fragments was between 450,000 and 600,000 for the 4 samples sequenced (data not shown, C− and GE Fwd edited in both cases). The achieved insert length was ∼500 bp, as expected. Results of the alignment of reads showed that all samples obtained the expected mean coverage of ∼40×, with an average duplication rate of 8%. The genotypability reached 86.9% at a standard read depth of 3 (86.8% if considering 10 reads covering the site), and the fold 80 base penalty showed a value of ∼1.3, which indicates a high uniformity of coverage (fold 80 close to 1).
In the WGS study, the calculation of the frequency of the edited base and of the indels was supported by a coverage of 81−82× on Th-ErPC-2 GE Fwd and of 33−35× on Th-ErPC-6 GE Fwd on the sites of interest. As expected, the target site was edited in both samples. Moreover, no insertions were found in any of the sites of interest, whereas there was a low number of deletions on the target site, in agreement with data obtained with an amplicon sequencing approach.
As a key issue of the WGS approach, we further investigated the potential off-target sites of CRISPR-Cas9, as calculated by Cas-OFFinder, together with a manual inspection of all of the insertions identified in the VCF file of Mutect2. Both of the analyses reported no off-target mutations, indicating that the editing did not produce any off-target effects outside the region of interest. The data generated by Cas-OFFinder software and reporting the predicted off-target sites are shown in Figure 7.
Figure 7.

Potential off-target sites in the WGS data obtained by Cas-OFFinder analysis
The off-target sites identified by the tool were inspected in the VCF files using bedtools intersect (v. 2.29.2). The software was set up to allow a maximum of three mismatches with respect to the input query sequence, with default values for the size of the DNA and RNA bulge (both with bulge size = 0).
Moreover, we manually investigated supplementary alignments containing stretch homologs to the region of interest, identifying 35 for Th-ErPC-2 GE Fwd and 10 for Th-ErPC-6 GE Fwd supplementary alignments, not included in the potential off-target site found by Cas-OFFinder, supported only by one read, as reported in the Tables S1 and S2. In addition, by checking the primary alignments of these reads, none of the regions was present in Th-ErPC-2 GE Fwd and Th-ErPC-6 GE Fwd samples. These data support the concept of extremely low, occasional, and random alignments, with no accumulation on genomic DNA, strongly highlighting the conclusion that the editing approach did not produce noticeable off-target effects outside the target site (no off-target mutation and no insertion of donor-derived DNA sequences).
Discussion
Gene editing by the CRISPR-Cas9 technology can be considered among the most promising strategies to correct hereditary mutations in a variety of monogenetic diseases. For instance, CRISPR-Cas9 has been employed in cystic fibrosis,38,39 SCD,40,41 Huntington’s chorea disorder,42,43 Duchenne muscular dystrophy,44,45 hemophilia,46,47 and chronic granulomatous disease.48,49
As far as thalassemia, this approach has been the object of several studies,18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33 has been the main inventive part of several patents (for instance US patents US9822372, US10550372, and U8945839),50, 51, 52 and is considered in different clinical trials (ClinicalTrials.gov: NCT03728322, NCT03655678, and NCT04208529),34 demonstrating the translation of this research from the laboratory to the clinic. These studies are based on CTX001 (autologous CD34+ human HSPCs [hHSPCs] modified with CRISPR-Cas9 at the erythroid lineage-specific enhancer of the BCL11A gene), and recruited subjects will receive a single infusion of CTX001 through a central venous catheter. This is expected to cause an increase in the production of HbF in the patient’s red blood cells. The hypothesis sustaining this last development is that no major genome alterations occur in CRISPR-Cas9-edited cells.29, 30, 31, 32, 33, 34
In this paper, we present for the first time the correction, by CRISPR-Cas9 gene editing, of the β039-thalassemia mutation, one of the most frequent in the Mediterranean area.1,2 The protocol here described is expected to be of interest for all of the clinicians working on hematological diseases, such as β-thalassemia, in particular, for those working on β0-thalassemia. It should be underlined that researchers also working with SCD patients might be interested because, apart the gene editing of the SCD locus, several patients are compound subjects carrying the SCD locus together with a β0-39-thalassemia mutation.
In addition, this protocol might be of great interest for researchers involved in developing alternative approaches for gene editing. For instance, peptide nucleic acids (PNAs) were found to be useful reagents for non-enzymatic gene editing. By forming high-affinity heterotriplex structures within the genome, PNAs have been used to correct multiple human disease-relevant mutations, including β-thalassemia. In a model of β-thalassemia, treated animals demonstrated clinically relevant protein restoration and disease phenotype amelioration, suggesting a potential for curative therapeutic application of PNAs to monogenic disorders. In this context, our CRISPR-Cas9 protocols might be considered to test the efficacy of alternative strategies finalized to gene editing for thalassemia.53, 54, 55, 56
Moreover, the protocol might be the starting point to develop combined treatments using, together with the CRISPR-Cas9 editing of the β-globin gene, other therapeutic approaches, such as HbF induction using chemical inducers.57, 58, 59, 60, 61, 62, 63, 64 In this context, several clinical trials in β-thalassemia and/or SCD are ongoing using HbF inducers, such as ClinicalTrials.gov: NCT01245179 (based on the histone deacetylase [HDAC] inhibitor panobinostat), NCT00790127 (based on 2,2-dimethylbutyrate, HQK-1001),64 and NCT03877809 (based on the mTOR inhibitor sirolimus).60 Possible combined treatment of erythroid cells with HbF inducers and gene-editing protocols might stimulate the co-increase of both HbF and HbA. In this context, the possibility of inducing HbA by gene therapy and HbF by co-treatment with HbF inducers has been demonstrated in recent studies.4,65
Finally, the protocol here described might be of interest to researchers working on gene editing of coding BCL11A and other transcription repressors of the γ-globin genes. In the case of combined CRISPR-Cas9 treatments (using our protocol and elsewhere-described strategies for correcting the genes coding for the γ-globin gene repressor), it might be possible to obtain induction of both HbF and HbA.
Concerning the efficiency, a very important point of our protocol is that the CRISPR-Cas9-corrected cells exhibit a very high production of HbA, approaching the percentage of HbA that could be detected in ErPCs from healthy subjects. On the other hand, we should consider the heterogeneity of the CRISPR-Cas9-corrected populations. This is one of the limits of this and other CRISPR-Cas9-based protocols. However, in order to have an idea about the level of correction with respect to the maximum level of HbA that can be obtained in the overall population that underwent the gene-editing protocol, the following considerations can be done. When the information about (1) the HbA content in the heterogenous cell population containing corrected erythroid cells, (2) the correction efficiency, and (3) the proportion of the indel fraction observed is considered together, the amount of HbA expected is, in any case, lower than that exhibited by ErPCs from healthy subjects, mainly due to the presence of indels containing cells. Therefore, future experiments about functional rescue should be considered, as well as pharmacological induction of an increase of HbA (from the de novo-corrected β-globin mRNAs) and HbF (from endogenous γ-globin genes).
Despite these limitations, a key advantage of this protocol is that the CRISPR-Cas9-forced HbA production levels are associated with the reach of a clinically relevant goal, i.e., the reduction of the excess of α-globin chains. This is an important achievement, together with the very efficient increase of HbA. In this specific context, recent studies suggest that the reduction of the excess of free α-globin chains might be associated in β-thalassemia to activation of autophagy.66 Therefore, a combination of the CRISPR-Cas9 approach with inducers of autophagy might lead to full suppression of the α-globin chains’ excess.
In conclusion, the possible combination of our CRISPR-Cas9 protocols with other therapeutic strategies for β-thalassemia might lead to a further increase of HbA and HbA/HbF, in association with a full reduction of the excess α-globin chains, allowing the reach of clinically relevant endpoints.
A critical issue of our approach is that the percentage of gene-edited cells is low, with respect to the finding that a very high level of production of HbA is activated in CRISPR-Cas9-corrected cells (see Table 1). Both the ssODN donors (fwd and rev) were found to sustain the editing with similar efficiencies, and only minor differences were appreciated with the use of the different technological approaches employed. In addition, no apparent insertions were detected at the target site, where, on the contrary, several deletions were found. These expected results are shown by the representative example of Figure 6 and by the summary of the results obtained reported in Table 2. Although the deletions at the target sites do not represent a major functional issue, since both the β0-globin genes carry a β039 mutation, leading to a β0-globin phenotype and to the absence of HbA production, the overall percentage of CRISPR-Cas9-mediated changes never exceeded 30% (%AF “C” + %AF deletion [DEL] + %AF insertion [INS]) (see Table 2). This finding confirms that an important challenge of the CRISPR system is the delivery to the target cells.67, 68, 69, 70 Many delivery approaches are being tested both to eliminate the problem of the size of the transgenic DNA to be inserted and to identify a safer and more efficient delivery system than viral vectors. Some of them involve the use of microinjection, transduction induced by osmocytosis and propane-betaine, lipid-mediated transfection, or systems that provide for the insertion of the donor DNA template using gold nanoparticles, or cell-penetrating peptides, and still hydrodynamic delivery.71
Finally, no major off-targeting effects were detectable (see Figure 7; Tables S1 and S2). This is clearly a positive finding, fully in agreement with the results of preclinical experiments with CTX001, presented in December 2017 at the American Society of Hematology (ASH) Annual Meeting, confirming that CTX001 was able to efficiently edit the target gene causing an increase of HbF production, without off-target effects on HSCs, thereby appearing to be a safe potential treatment.72
Although the described gene-editing protocols might be considered a promising starting point on the road of the development of therapeutic approaches for β-thalassemia, we like to point out that the final demonstration of functional genetic modification of HSCs will require in vivo xenotransplantation experiments and in vivo model systems mimicking β-thalassemia.
Materials and methods
ErPC isolation and culture
The blood samples used to conduct our experiments were collected from patients with β039-thalassemia with informed consent. About 25 mL of peripheral blood was collected in tubes treated with the Vacutainer lithium heparin (LH) (BD Vacutainers; Becton Dickinson, UK). The separation and isolation of peripheral blood mononuclear cells (PBMCs) were obtained starting from a whole-blood sample by centrifugation of the Ficoll-Hypaque density gradient (Lympholyte-H Cell Separation Media; Cedarlane, Euroclone, Italy). The PBMC ring, obtained after the separation of the different blood components, was collected and washed with 1× DPBS W/O CA-MG (Dulbecco’s phosphate-buffered saline without calcium-magnesium; Gibco, Invitrogen, Life Technologies, Carlsbad, CA, USA). The cells obtained were maintained in culture with the Fibach protocol, characterized by the succession of two different culture media.73 During phase I, the culture medium contains an α-minimum essential medium (α-MEM; Sigma-Genosys, St. Louis, MO, USA), prepared from a powder and diluted with water; a penicillin-streptomycin (PEN-STREP) solution (PEN-STREP 10,000 U/mL; Lonza, Verviers, Belgium); 10% fetal bovine serum (FBS; Celbio, Milan, Italy); 10% conditioned medium (CM), obtained from cell cultures of bladder cancer cells (5,637); and 1 μg/mL of cyclosporin A (Sigma-Aldrich), prepared with cyclosporine absolute ethanol and diluted in 1× DPBS (Gibco), in the ratio 1:1. After 7 days in phase I culture, the non-adherent cells were taken, washed once with 1× DPBS (Gibco), and then grown in phase II medium. The composition of this second medium includes the following: α-MEM (Sigma-Genosys), 30% FBS (Celbio), 1% deionized bovine serum albumin (BSA; Sigma-Genosys), 10−5 M β-mercaptoethanol (Sigma-Genosys), L-glutamine 2 mM (Sigma-Genosys), dexamethasone 10−6 M (Sigma-Genosys), and 1 U/mL of recombinant human EPO (Tebu-bio, Magenta, Milan, Italy) and stem cell factor (SCF; Life Technologies, Carlsbad, CA, USA) at a final concentration of 10 ng/mL.
Treatment with CRISPR-Cas9 system: Cell electroporation
The hemoglobin subunit beta synthetic donor template used was purchased from Integrated DNA Technologies (IDT; Tema, Coralville, IA, USA) with chemically modified nucleotides at the three terminal nucleotides at both the 5′ and 3′ ends containing 2′-O-methyl-3′-phosphorothioate. The genomic sgRNA target sequence, with PAM underlined, is 5′-TGGTCTACCCTTGGACCTAGAGG-3′; the guide RNA (gRNA) complex begins by joining a trans-activating crRNA (tracrRNA; ATTO 550-labeled Alt-R CRISPR-Cas9 tracrRNA; IDT) and the Alt-R CRISPR-Cas9 crRNA (IDT) oligonucleotide in Thermoblock at 95°C for 5 min. Subsequently, Cas9 RNP was made by incubating gRNA and Cas9 at a molar ratio of 1:2 at 25°C for 10 min immediately before electroporation. The ErPCs were electroporated on the 3rd day of phase II in the presence of Cas9 RNP and 6 μM of β-globin PAM-modified donor template by using the Lonza Nucleofector IIb (program U-008). The cells and all components necessary for gene correction were resuspended and electroporated in Mirus Ingenio solution (Mirus Bio, Madison, WI, USA) in a final volume of 100 μL. After 7 days, the erythroid precursors were collected by extracting RNA and DNA, and approximately 4 × 106 cells were lysed to analyze the hemoglobins produced by HPLC.
Genomic DNA extraction
The DNA was extracted from 200−300 μL of whole blood using the QiAmp DNA Blood Mini Kit (QIAGEN, Hilden, Germany), according to the manufacturer’s protocol. The DNA obtained was visualized on a UV transilluminator after 0.8% agarose gel electrophoresis and quantified using the spectrophotometer SmartSpec Plus (Bio-Rad).
PCR and Sanger sequencing reaction
The β-globin gene was amplified starting from 300 ng of genomic DNA. Each reaction was carried out in a final volume of 100 μL in the presence of 1 × buffer (10 mM Tris-HCl, pH 8.8, 1.5 mM MgCl2, 50 mM KCl, 0.1% Triton X-100), 33 μM deoxyribonucleotide triphosphates (dNTPs), 0.25 μM fwd and rev primers (Table 3), 2 U of DyNAzyme II DNA Polymerase (Finnzymes, Espoo, Finland) or DNA polymerase DreamTaq 5 U/μL (MBI Fermentas, Burlington, ON, Canada), and ultra-pure water. Each reaction was subjected to an initial denaturation step of 2 min at 94°C. The 35 PCR cycles used were as follows: denaturation, 30 s at 94°C; annealing, 30 s at 65°C; and elongation, 1 min at 72°C. PCR products were analyzed by agarose gel electrophoresis to 1% before being purified for sequencing. The PCR products were visualized on a UV transilluminator after 1% agarose gel electrophoresis. The PCR products were purified with MicroClean (Microzones, Hayward Heath, West Sussex, UK) and were sequenced in both directions using the PCR primers fwd and rev (Table 3) and the BigDye Terminator v.1.1 Cycle Sequencing Kit (Life Technologies). The reaction products were purified from unincorporated dideoxynucleotides (ddNTPs) by using a 96-well MultiScreen (Millipore) plate containing Sephadex G-50 Superfine (Amersham Biosciences, UK). Sequencing was performed by BMR Genomics (Padua, Italy), while the obtained sequence data were analyzed by the Sequence Scanner, v.1.0 (Applied Biosystems, Life Technologies), software.
Table 3.
List of oligonucleotides used to conduct the different analyses and evaluate the correction degree obtained on the β-globin gene and mRNA with the CRISPR-Cas9 system
| Oligonucleotide and assay | Sequence | Application |
|---|---|---|
| α-globin forward primer | 5′-GGTCTTGGTGGTGGGGAAG-3′ | qRT-PCR |
| α-globin reverse primer | 5′-CGACAAGACCAACGTCAAGG-3′ | qRT-PCR |
| α-globin probe | 5′-/5HEX/ACATCCTCT/ZEN/CCAGGGCCTCCG/3IABkFQ/-3′ | qRT-PCR |
| β-globin forward primer | 5′-GGTGAATTCTTTGCCAAAGTGAT-3′ | qRT-PCR |
| β-globin reverse primer | 5′-GGGCACCTTTGCCACAC-3′ | qRT-PCR |
| β-globin probe | 5′-/5Cy5/ACGTTGCCCAGGAGCCTGAAG/3IAbRQSp/-3′ | qRT-PCR |
| γ-globin forward primer | 5′-TTCTTTGCCGAAATGGATTGC-3′ | qRT-PCR |
| γ-globin reverse primer | 5′-TGACAAGCTGCATGTGGATC-3′ | qRT-PCR |
| γ-globin probe | 5′-/56-FAM/TCACCAGCA/ZEN/CATTTCCCAGGAGC/3IABkFQ/-3′ | qRT-PCR |
| GAPDH forward primer | 5′-TGTAGTTGAGGTCAATGAAGGG-3′ | qRT-PCR |
| GAPDH reverse primer | 5′-ACATCGCTCAGACACCATG-3′ | qRT-PCR |
| GAPDH probe | 5′-/56-FAM/AAGGTCGGTCGGA/ZEN/GTCAACGGATTTGGTC/3IABkFQ/-3′ | qRT-PCR |
| β-actin forward primer | 5′-ACAGAGCCTCGCCTTTG-3′ | qRT-PCR |
| β-actin reverse primer | 5′-ACGATGGAGGGGAAGACG-3′ | qRT-PCR |
| β-actin probe | 5′-/5Cy5/CCTTGCACATGCCGGAGCC/3IAbRQSp/-3′ | qRT-PCR |
| β-globin forward primer | 5′-CACTGACTCTCTCTGCCTATTG-3′ | ddPCR β-globin gene |
| β-globin reverse primer | 5′-ACCTTAGGGTTGCCCATAAC-3′ | ddPCR β-globin gene |
| β-globin β039 probe (HEX) | 5′-/5HEX/TCTACCCTT/ZEN/GGACCTAGAGGTTCT/3IABkFQ/-3′ | ddPCR β-globin gene |
| β-globin edit probe (FAM) | 5′-/56-FAM/TCTACCCTT/ZEN/GGACCCAGAGATTCT/3IABkFQ/-3′ | ddPCR β-globin gene |
| β-globin forward primer | 5′-TGGATGAAGTTGGTGGTGAG-3′ | ddPCR β-globin mRNA |
| β-globin reverse primer | 5′-CCTTAGGGTTGCCCATAACA-3′ | ddPCR β-globin mRNA |
| β-globin β039 probe (HEX) | 5′-/5HEX/TCTACCCTT/ZEN/GGACCTAGAGGTTCTT/3IABkFQ/-3′ | ddPCR β-globin mRNA |
| β-globin edit probe (FAM) | 5′-/56-FAM/TCTACCCTT/ZEN/GGACCCAGAGGTTCTT/3IABkFQ/-3′ | ddPCR β-globin mRNA |
| BGF β-globin forward primer | 5′-GTGCCAGAAGAGCCAAGGACAGG-3′ | β-globin PCR and sequencing |
| T12R β-globin reverse primer | 5′-AGTTCTCAGGATCCACGTGCA-3′ | β-globin PCR and sequencing |
RNA isolation and reverse transcription (RT)
The total cellular RNA was extracted by TRI Reagent (Sigma-Aldrich). The isolated RNA was washed once with cold 75% ethanol, dried, and dissolved in diethylpyrocarbonate-treated water (WMBR [water molecular biology reagent] nuclease-free; Sigma-Aldrich) before use. For gene-expression analysis, 0.5 μg of total RNA was reverse transcribed by using the TaqMan Rev Transcription Reagents and Random Hexamer (Applied Biosystems, Life Technologies, Thermo Fisher Scientific).
Levels of α-, β-, and γ-globins were quantified by multiplex qPCR using primers and FAM, HEX, and Cy5/ZEN/IBFQ-labeled hydrolysis probes purchased as custom-designed PrimeTime qPCR Assays from IDT and reported in Table 3. The obtained data were normalized using β-actin and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as housekeeping genes.
ddPCR to evaluate genomic and transcriptomic β039-globin correction
The evaluation of the β-globin gene-correction levels in the position of the codon 39 was carried out by ddPCR. The ddPCR allows the absolute quantification of certain nucleic acids by dividing the analyte into multiple reactions divided as limit dilutions allowing us to also determine substrates or targets that are poorly expressed or present in very low concentrations. In these experiments, TaqMan probes marked with FAM and HEX fluorophores were used, designed specifically for the identification of the sequence containing the β039 mutation (HEX) in the β-globin gene and the corresponding corrected sequence (FAM). Both probes were used for genomic and transcriptional analysis and were designed using IDT tools, whereas specific pairs of primers have been designed for the selective amplification of DNA or transcript (in introns in the case of genomic analysis and in exons in the case of transcriptomic analysis). The predetermined quantity of DNA or cDNA obtained following extraction from ErPCs treated with the CRISPR-Cas9 system was added to the ddPCR reaction mix containing 2× ddPCR Supermix for probes (no dUTP) (Bio-Rad) and 20× TaqMan β039 Assay or β-Edited Assay (IDT) (Table 3). The ddPCR reaction mix was mixed with Automated Droplet Generation Oil for Probes (Bio-Rad), and droplet emulsion (water in oil) was automatically generated using Automated Droplet Generator (AutoDG) (Bio-Rad). The emulsion was amplified using the GeneAmp PCR System 9700 (Thermo Fisher Scientific) using the following thermal cycler condition at 95°C for 10 min, 94°C for 30 s, and 61°C for 1 min; repeated for 40 cycles; and then a final phase of inactivation of the enzyme DNA polymerase at 98°C for 10 min. The plate must be kept at 4°C for 1 h before reading in order to stabilize the analysis. Generated droplets were read using the QX200 Droplet Reader, and data analysis was performed with QuantaSoft v.1.7.4 (Bio-Rad).
Western blotting analysis
The accumulation of the β-globin protein (16 kDa) was assessed by western blotting. For whole-cell extract preparation, the cells were lysed through three freeze-thaw cycles in dry ice and quantified by BCA assay (Pierce BCA Protein Assay Kit; Thermo Fisher Scientific). For each sample, 20 μg of ErPCs extracts was loaded on a 16% acrylamide SDS-PAGE gel (40% acrylamide/bis solution; Bio-Rad). After separation by an electrophoretic run, the proteins were transferred onto nitrocellulose paper and incubated with different primary antibodies: anti-β-globin chain antibody (monoclonal antibody [mAb], sc-21757; Santa Cruz), anti-α-globin chain antibody (mAb, TA307691; OriGene), and anti-β-actin (13E5 rabbit mAb; Cell Signaling Technology), a constitutive protein (45 kDa) expressed similarly in all treated and untreated samples, used as housekeeping to normalize the quantification of the target protein.
HPLC analysis of hemoglobins
To evaluate the effectiveness of the genome-editing systems in ErPCs, an HPLC analysis was carried out on the protein extracts obtained at the end of the treatments in order to evaluate the occurred correction found by increasing the level of HbA produced. The ErPCs were centrifuged at 8,000 rpm for 8 min and washed with PBS. The pellet is then resuspended in a predefined volume of water for HPLC (Sigma-Aldrich, St. Louis, MO, USA), following 3 freeze/thaw cycles on dry ice in order to lyse the cells and obtain the protein extracts. Hemoglobin analysis is performed by loading the protein extracts into a PolyCAT-A cation exchange column and then eluting in a sodium-chlorine-BisTris- potassium cyanide (KCN) aqueous mobile phase using the HPLC Beckman Coulter instrument, System Gold 126 Solvent Module-166 Detector, which allows us to obtain a quantification of the hemoglobins present in the sample. The reading is performed at a wavelength of 415 nm, and a commercial solution of purified human HbA0 (Sigma-Aldrich) extracts has been used as standard. The values thus obtained are processed using “32 Karat software.”
WGS and amplicon sequencing
WGS experiments and bioinformatic analysis were performed at Genartis-Innovative Genomic Technologies Laboratories (Genartis srl, Verona, Italy; https://genartis.it/).
Genomic DNA quality control was performed using the 4150 TapeStation System (Agilent Technologies, Santa Clara, CA, USA) and quantified using the Qubit double-stranded DNA (dsDNA) High-Sensitivity (HS) Assay Kit (Thermo Fisher Scientific). Whole Genome libraries were prepared from 1 μg of genomic DNA. DNA shearing was performed with a Covaris S220 (Covaris) in order to obtain an insert size of around 500 bp. Libraries were obtained using a KAPA HyperPrep Kit and unique dual-indexed adapters (5 μL of a 15-μM stock), according to the supplier’s protocol (Roche, Basel, Switzerland), with minor modification in order to obtain the desired insert size. Specifically, the post-ligation products were cleaned up using 0.8 × Ampure XP beads followed by a Size Selection step using 0.7× of Ampure XP beads. The libraries were analyzed on the 4150 TapeStation System and quantified with the KAPA Library Quantification Kit for Illumina platforms, using QuantStudio3 Real-Time PCR Systems (Thermo Fisher Scientific). Libraries were pooled at equimolar concentrations and sequenced on the NovaSeq 6000 platform (Illumina, San Diego, CA, USA) using the 2 × 150-bp mode.
Amplicons from eighteen samples in duplicate were generated using fwd primer 5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGAAACTGGGCATGTGGAGAC-3′ and rev primer 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGGCACCGAGCACTTTCTT-3′ for the Nextera XT kit. PCR products were cleaned up using 0.9× Ampure XP beads. Dual indexes barcodes, provided by Nextera XT Index Kit Set B (Illumina, San Diego, CA, USA), were added to each amplicon by 8 PCR cycles. The final product was purified using 1.12× Ampure XP beads, and the average size was checked by the 4150 TapeStation System (average size 526–573 bp) and quantified using the Qubit dsDNA BR Assay Kit (Thermo Fisher Scientific). Amplicons were pooled at equimolar concentration, and a final quantification was done by real-time PCR, as mentioned above. Sequencing was performed on the NovaSeq 6000 platform (Illumina, San Diego, CA, USA) using the 2 × 150-bp mode.
Bioinformatics pipeline
The obtained fastq files were trimmed with the fastp tool (v.0.21.0), and reads were aligned against the hg38 reference genome using BWA-mem (v.0.7.17). After that, the generated BAM files were cleaned performing the soft clipping using bamUtils (v.1.0.14), followed by the removal of the duplicates with Picard (v.2.21.1) and the base recalibration using GATK BaseRecalibrator (v.4.1.9.0). For the amplicon-based samples, duplicate removal was not performed. GATK CallableLoci v.3.8.0.1 was used to calculate, for the genome and the target sequence for amplicons, the average coverage, the percentage of the genome and of the target covered by at least 5×, 10×, 20×, and the genotypability at minimum read depth of 3 (%PASS) and at a minimum read depth of 10 (%PASS RD ≥ 10). Fold80 penalty value (uniformity of coverage) was calculated using Picard CollectHSMetrics v.2.21.1 (http://broadinstitute.github.io/picard/). Then, a variant calling on WGS was performed using Mutect2 by GATK (v.4.1.9.0) applying the “tumor-normal” mode, considering as “tumor” the edited sample and as “normal” the wild-type, obtaining only the variants present in the edited cells. For the amplicon-based samples, variant calling was performed using VarScan (v.2.4.4.). The bam files were manually curated by removing short reads (≤60 bp) and supplementary alignments on the target region. The Python (v.2.7) pysamstats package was then used to calculate the frequencies of the edited base and of the indels located in the positions of interest (chr11:5,226,774 and chr11:5,226,769) for both the WGS samples and the ones processed with the amplicon protocol.74, 75, 76, 77, 78
CRISPR-Cas9 off-target site estimation
Cas-OFFinder was used to investigate the potential off-target sites in the WGS data.79 The software was set to allow a maximum of three mismatches with respect to the input query sequence, with the default values for DNA and RNA bulge size. The off-target sites identified by the tool were inspected in the VCF files using bedtools intersect (v.2.29.2).80
Moreover, the supplementary alignments in the target site were manually investigated to identify potential off-target sites not identified with Cas-OFFinder.
Off-target insertion of target sequence
To investigate the presence of the insertion of the target sequence (5′-ACCCTTAGGCTGCTGGTGGTCTACCCTTGGACCCAGAGATTCTTTGAGTCCTTTGGGGAT-3′) in the WGS samples, the VCF files obtained from variant calling were filtered by variant type retaining only the insertions. Manual inspection of the insertions was then performed.
Acknowledgments
The authors are grateful to Prof. Massimo Delledonne, Dott. Marzia Rossato, and their PhD researcher team (Genartis srl, Verona, Italy) for WGS and bioinformatic analysis support in the analyses of editing results, off-target sites, and indels estimation at the genomic level. This paper is dedicated to the memory of Elio Zago, past AVLT president, and Chiara Gemmo, a young investigator of our team. This study was sponsored by the Wellcome Trust (innovator award 208872/Z/17/Z) and AIFA (AIFA-2016-02364887). The research leading to these results has received funding also by the UE THALAMOSS Project (Thalassemia Modular Stratification System for Personalized Therapy of Beta-Thalassemia; no. 306201-FP7-HEALTH-2012-INNOVATION-1) and FIR and FAR funds from the University of Ferrara. This research was also supported by A.L.T. (Associazione per la lotta alla Talassemia) Rino Vullo–Ferrara, AVLT (Associazione Veneta per la Lotta alla Talassemia), and by the Interuniversity Consortium for the Biotechnology (CIB), Italy. L.C.C. was supported by a fellowship from “Tutti per Chiara Onlus.”
Author contributions
L.C.C., J.G., N.R., M.Z., and C.Z. conducted the experiments. A.F., L.C.C., and R.G. conceived and designed the experiments, analyzed the data, and drafted and wrote the paper. L.C.C. and C.Z. were in charge of blood collection from patients and ErPC cultures and did the HPLC experiments. L.C.C. and N.R. did the nucleofection with CRISPR-Cas9. L.C.C. and J.G. performed the ddPCR, RT-ddPCR, and qRT-PCR analyses and conducted the western blotting. All authors read and approved the final manuscript. All of the authors gave their consent to publish this study.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2021.03.025.
Contributor Information
Roberto Gambari, Email: gam@unife.it.
Alessia Finotti, Email: alessia.finotti@unife.it.
Supplemental information
References
- 1.Origa R. β-Thalassemia. Genet. Med. 2017;19:609–619. doi: 10.1038/gim.2016.173. [DOI] [PubMed] [Google Scholar]
- 2.Fucharoen S., Weatherall D.J. Progress Toward the Control and Management of the Thalassemias. Hematol. Oncol. Clin. North Am. 2016;30:359–371. doi: 10.1016/j.hoc.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 3.Quek L., Thein S.L. Molecular therapies in beta-thalassaemia. Br. J. Haematol. 2007;136:353–365. doi: 10.1111/j.1365-2141.2006.06408.x. [DOI] [PubMed] [Google Scholar]
- 4.Finotti A., Breda L., Lederer C.W., Bianchi N., Zuccato C., Kleanthous M., Rivella S., Gambari R. Recent trends in the gene therapy of β-thalassemia. J. Blood Med. 2015;6:69–85. doi: 10.2147/JBM.S46256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Finotti A., Gambari R. Recent trends for novel options in experimental biological therapy of β-thalassemia. Expert Opin. Biol. Ther. 2014;14:1443–1454. doi: 10.1517/14712598.2014.927434. [DOI] [PubMed] [Google Scholar]
- 6.Thein S.L. The emerging role of fetal hemoglobin induction in non-transfusion-dependent thalassemia. Blood Rev. 2012;26(Suppl 1):S35–S39. doi: 10.1016/S0268-960X(12)70011-5. [DOI] [PubMed] [Google Scholar]
- 7.Breveglieri G., Bianchi N., Cosenza L.C., Gamberini M.R., Chiavilli F., Zuccato C., Montagner G., Borgatti M., Lampronti I., Finotti A., Gambari R. An Aγ-globin G->A gene polymorphism associated with β039 thalassemia globin gene and high fetal hemoglobin production. BMC Med. Genet. 2017;18:93. doi: 10.1186/s12881-017-0450-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ye L., Wang J., Tan Y., Beyer A.I., Xie F., Muench M.O., Kan Y.W. Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: An approach for treating sickle cell disease and β-thalassemia. Proc. Natl. Acad. Sci. USA. 2016;113:10661–10665. doi: 10.1073/pnas.1612075113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Traxler E.A., Yao Y., Wang Y.D., Woodard K.J., Kurita R., Nakamura Y., Hughes J.R., Hardison R.C., Blobel G.A., Li C., Weiss M.J. A genome-editing strategy to treat β-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat. Med. 2016;22:987–990. doi: 10.1038/nm.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Magrin E., Miccio A., Cavazzana M. Lentiviral and genome-editing strategies for the treatment of β-hemoglobinopathies. Blood. 2019;134:1203–1213. doi: 10.1182/blood.2019000949. [DOI] [PubMed] [Google Scholar]
- 11.Hu X. CRISPR/Cas9 system and its applications in human hematopoietic cells. Blood Cells Mol. Dis. 2016;62:6–12. doi: 10.1016/j.bcmd.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 12.Dever D.P., Bak R.O., Reinisch A., Camarena J., Washington G., Nicolas C.E., Pavel-Dinu M., Saxena N., Wilkens A.B., Mantri S. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539:384–389. doi: 10.1038/nature20134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Finotti A., Borgatti M., Gambari R. Ground state naïve pluripotent stem cells and CRISPR/Cas9 gene correction for β-thalassemia. Stem Cell Investig. 2016;3:66. doi: 10.21037/sci.2016.09.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boulad F., Mansilla-Soto J., Cabriolu A., Rivière I., Sadelain M. Gene Therapy and Genome Editing. Hematol. Oncol. Clin. North Am. 2018;32:329–342. doi: 10.1016/j.hoc.2017.11.007. [DOI] [PubMed] [Google Scholar]
- 15.Lau C.H. Applications of CRISPR-Cas in Bioengineering, Biotechnology, and Translational Research. CRISPR J. 2018;1:379–404. doi: 10.1089/crispr.2018.0026. [DOI] [PubMed] [Google Scholar]
- 16.El-Kenawy A., Benarba B., Neves A.F., de Araujo T.G., Tan B.L., Gouri A. Gene surgery: Potential applications for human diseases. EXCLI J. 2019;18:908–930. doi: 10.17179/excli2019-1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Papasavva P., Kleanthous M., Lederer C.W. Rare Opportunities: CRISPR/Cas-Based Therapy Development for Rare Genetic Diseases. Mol. Diagn. Ther. 2019;23:201–222. doi: 10.1007/s40291-019-00392-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu P., Tong Y., Liu X.Z., Wang T.T., Cheng L., Wang B.Y., Lv X., Huang Y., Liu D.P. Both TALENs and CRISPR/Cas9 directly target the HBB IVS2-654 (C > T) mutation in β-thalassemia-derived iPSCs. Sci. Rep. 2015;5:12065. doi: 10.1038/srep12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Niu X., He W., Song B., Ou Z., Fan D., Chen Y., Fan Y., Sun X. Combining Single Strand Oligodeoxynucleotides and CRISPR/Cas9 to Correct Gene Mutations in β-Thalassemia-induced Pluripotent Stem Cells. J. Biol. Chem. 2016;291:16576–16585. doi: 10.1074/jbc.M116.719237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patsali P., Turchiano G., Papasavva P., Romito M., Loucari C.C., Stephanou C., Christou S., Sitarou M., Mussolino C., Cornu T.I. Correction of IVS I-110(G>A) β-thalassemia by CRISPR/Cas-and TALEN-mediated disruption of aberrant regulatory elements in human hematopoietic stem and progenitor cells. Haematologica. 2019;104:e497–e501. doi: 10.3324/haematol.2018.215178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patsali P., Mussolino C., Ladas P., Floga A., Kolnagou A., Christou S., Sitarou M., Antoniou M.N., Cathomen T., Lederer C.W., Kleanthous M. The Scope for Thalassemia Gene Therapy by Disruption of Aberrant Regulatory Elements. J. Clin. Med. 2019;8:E1959. doi: 10.3390/jcm8111959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiong Z., Xie Y., Yang Y., Xue Y., Wang D., Lin S., Chen D., Lu D., He L., Song B. Efficient gene correction of an aberrant splice site in β-thalassaemia iPSCs by CRISPR/Cas9 and single-strand oligodeoxynucleotides. J. Cell. Mol. Med. 2019;23:8046–8057. doi: 10.1111/jcmm.14669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shariati L., Khanahmad H., Salehi M., Hejazi Z., Rahimmanesh I., Tabatabaiefar M.A., Modarressi M.H. Genetic disruption of the KLF1 gene to overexpress the γ-globin gene using the CRISPR/Cas9 system. J. Gene Med. 2016;18:294–301. doi: 10.1002/jgm.2928. [DOI] [PubMed] [Google Scholar]
- 24.Antoniani C., Meneghini V., Lattanzi A., Felix T., Romano O., Magrin E., Weber L., Pavani G., El Hoss S., Kurita R. Induction of fetal hemoglobin synthesis by CRISPR/Cas9-mediated editing of the human β-globin locus. Blood. 2018;131:1960–1973. doi: 10.1182/blood-2017-10-811505. [DOI] [PubMed] [Google Scholar]
- 25.Liu N., Hargreaves V.V., Zhu Q., Kurland J.V., Hong J., Kim W., Sher F., Macias-Trevino C., Rogers J.M., Kurita R. Direct Promoter Repression by BCL11A Controls the Fetal to Adult Hemoglobin Switch. Cell. 2018;173:430–442.e17. doi: 10.1016/j.cell.2018.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martyn G.E., Wienert B., Yang L., Shah M., Norton L.J., Burdach J., Kurita R., Nakamura Y., Pearson R.C.M., Funnell A.P.W. Natural regulatory mutations elevate the fetal globin gene via disruption of BCL11A or ZBTB7A binding. Nat. Genet. 2018;50:498–503. doi: 10.1038/s41588-018-0085-0. [DOI] [PubMed] [Google Scholar]
- 27.Grevet J.D., Lan X., Hamagami N., Edwards C.R., Sankaranarayanan L., Ji X., Bhardwaj S.K., Face C.J., Posocco D.F., Abdulmalik O. Domain-focused CRISPR screen identifies HRI as a fetal hemoglobin regulator in human erythroid cells. Science. 2018;361:285–290. doi: 10.1126/science.aao0932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shariati L., Rohani F., Heidari Hafshejani N., Kouhpayeh S., Boshtam M., Mirian M., Rahimmanesh I., Hejazi Z., Modarres M., Pieper I.L., Khanahmad H. Disruption of SOX6 gene using CRISPR/Cas9 technology for gamma-globin reactivation: An approach towards gene therapy of β-thalassemia. J. Cell. Biochem. 2018;119:9357–9363. doi: 10.1002/jcb.27253. [DOI] [PubMed] [Google Scholar]
- 29.Chung J.E., Magis W., Vu J., Heo S.J., Wartiovaara K., Walters M.C., Kurita R., Nakamura Y., Boffelli D., Martin D.I.K. CRISPR-Cas9 interrogation of a putative fetal globin repressor in human erythroid cells. PLoS One. 2019;14:e0208237. doi: 10.1371/journal.pone.0208237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu Y., Zeng J., Roscoe B.P., Liu P., Yao Q., Lazzarotto C.R., Clement K., Cole M.A., Luk K., Baricordi C. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat. Med. 2019;25:776–783. doi: 10.1038/s41591-019-0401-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khosravi M.A., Abbasalipour M., Concordet J.P., Berg J.V., Zeinali S., Arashkia A., Azadmanesh K., Buch T., Karimipoor M. Targeted deletion of BCL11A gene by CRISPR-Cas9 system for fetal hemoglobin reactivation: A promising approach for gene therapy of beta thalassemia disease. Eur. J. Pharmacol. 2019;854:398–405. doi: 10.1016/j.ejphar.2019.04.042. [DOI] [PubMed] [Google Scholar]
- 32.Métais J.Y., Doerfler P.A., Mayuranathan T., Bauer D.E., Fowler S.C., Hsieh M.M., Katta V., Keriwala S., Lazzarotto C.R., Luk K. Genome editing of HBG1 and HBG2 to induce fetal hemoglobin. Blood Adv. 2019;3:3379–3392. doi: 10.1182/bloodadvances.2019000820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weber L., Frati G., Felix T., Hardouin G., Casini A., Wollenschlaeger C., Meneghini V., Masson C., De Cian A., Chalumeau A. Editing a γ-globin repressor binding site restores fetal hemoglobin synthesis and corrects the sickle cell disease phenotype. Sci. Adv. 2020;6:eaay9392. doi: 10.1126/sciadv.aay9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hirakawa M.P., Krishnakumar R., Timlin J.A., Carney J.P., Butler K.S. Gene editing and CRISPR in the clinic: current and future perspectives. Biosci. Rep. 2020;40 doi: 10.1042/BSR20200127. BSR20200127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peixeiro I., Silva A.L., Romão L. Control of human beta-globin mRNA stability and its impact on beta-thalassemia phenotype. Haematologica. 2011;96:905–913. doi: 10.3324/haematol.2010.039206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nienhuis A.W., Nathan D.G. Pathophysiology and Clinical Manifestations of the β-Thalassemias. Cold Spring Harb. Perspect. Med. 2012;2:a011726. doi: 10.1101/cshperspect.a011726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mettananda S., Gibbons R.J., Higgs D.R. α-Globin as a molecular target in the treatment of β-thalassemia. Blood. 2015;125:3694–3701. doi: 10.1182/blood-2015-03-633594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schwank G., Koo B.K., Sasselli V., Dekkers J.F., Heo I., Demircan T., Sasaki N., Boymans S., Cuppen E., van der Ent C.K. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell. 2013;13:653–658. doi: 10.1016/j.stem.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 39.Firth A.L., Menon T., Parker G.S., Qualls S.J., Lewis B.M., Ke E., Dargitz C.T., Wright R., Khanna A., Gage F.H., Verma I.M. Functional gene correction for cystic fibrosis in lung epithelial cells generated from patient iPSCs. Cell Rep. 2015;12:1385–1390. doi: 10.1016/j.celrep.2015.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoban M.D., Lumaquin D., Kuo C.Y., Romero Z., Long J., Ho M., Young C.S., Mojadidi M., Fitz-Gibbon S., Cooper A.R. CRISPR/Cas9-mediated correction of the sickle mutation in human CD34+ cells. Mol. Ther. 2016;24:1561–1569. doi: 10.1038/mt.2016.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park S.H., Lee C.M., Deshmukh H., Bao G. Therapeutic CRISPR/Cas9 genome editing for treating sickle cell disease. Blood. 2016;128:4703. [Google Scholar]
- 42.Shin J.W., Kim K.H., Chao M.J., Atwal R.S., Gillis T., MacDonald M.E., Gusella J.F., Lee J.M. Permanent inactivation of Huntington’s disease mutation by personalized allele-specific CRISPR/Cas9. Hum. Mol. Genet. 2016;25:4566–4576. doi: 10.1093/hmg/ddw286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Monteys A.M., Ebanks S.A., Keiser M.S., Davidson B.L. CRISPR/Cas9 editing of the mutant huntingtin allele in vitro and in vivo. Mol. Ther. 2017;25:12–23. doi: 10.1016/j.ymthe.2016.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ousterout D.G., Kabadi A.M., Thakore P.I., Majoros W.H., Reddy T.E., Gersbach C.A. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat. Commun. 2015;6:6244. doi: 10.1038/ncomms7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H.L., Fujimoto N., Sasakawa N., Shirai S., Ohkame T., Sakuma T., Tanaka M., Amano N., Watanabe A., Sakurai H. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Reports. 2015;4:143–154. doi: 10.1016/j.stemcr.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park C.Y., Kim D.H., Son J.S., Sung J.J., Lee J., Bae S., Kim J.H., Kim D.W., Kim J.S. Functional correction of large factor VIII gene chromosomal inversions in Hemophilia A patient-derived iPSCs using CRISPR-Cas9. Cell Stem Cell. 2015;17:213–220. doi: 10.1016/j.stem.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 47.Guan Y., Ma Y., Li Q., Sun Z., Ma L., Wu L., Wang L., Zeng L., Shao Y., Chen Y. CRISPR/Cas9-mediated somatic correction of a novel coagulator factor IX gene mutation ameliorates hemophilia in mouse. EMBO Mol. Med. 2016;8:477–488. doi: 10.15252/emmm.201506039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Flynn R., Grundmann A., Renz P., Hänseler W., James W.S., Cowley S.A., Moore M.D. CRISPR-mediated genotypic and phenotypic correction of a chronic granulomatous disease mutation in human iPS cells. Exp. Hematol. 2015;43:838–848.e3. doi: 10.1016/j.exphem.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.De Ravin S.S., Li L., Wu X., Choi U., Allen C., Koontz S., Lee J., Theobald-Whiting N., Chu J., Garofalo M. CRISPR-Cas9 gene repair of hematopoietic stem cells from patients with X-linked chronic granulomatous disease. Sci. Transl. Med. 2017;9:3480. doi: 10.1126/scitranslmed.aah3480. [DOI] [PubMed] [Google Scholar]
- 50.Zhang F., Cox D.B.T., Marraffini L., Bikard D.O., Jiang W., Sanjana N.E. 2017. CRISPR-Cas component systems, methods and compositions for sequence manipulation. US patent 9822372, filed January 7, 2016, and granted November 21, 2017. [Google Scholar]
- 51.Konermann S., Trevino A., Brigham M., Ran F., Hsu P., Lin C., Nureki O., Nishimasu H., Ishitani R., Zhang F. 2015. Systems, methods and compositions for sequence manipulation with optimized functional CRISPR-Cas systems. US patent 10550372, filed December 12, 2014, and published June 18, 2015. [Google Scholar]
- 52.Zetsche B., Zhang F. 2021. CRISPR-Cas systems and methods for altering expression of gene products. US patent 8945839, filed May 8, 2019, and published January 12, 2021. [Google Scholar]
- 53.Economos N.G., Oyaghire S., Quijano E., Ricciardi A.S., Saltzman W.M., Glazer P.M. Peptide Nucleic Acids and Gene Editing: Perspectives on Structure and Repair. Molecules. 2020;25:E735. doi: 10.3390/molecules25030735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ricciardi A.S., McNeer N.A., Anandalingam K.K., Saltzman W.M., Glazer P.M. Targeted genome modification via triple helix formation. Methods Mol. Biol. 2014;1176:89–106. doi: 10.1007/978-1-4939-0992-6_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bahal R., Ali McNeer N., Quijano E., Liu Y., Sulkowski P., Turchick A., Lu Y.C., Bhunia D.C., Manna A., Greiner D.L. In vivo correction of anaemia in β-thalassemic mice by γPNA-mediated gene editing with nanoparticle delivery. Nat. Commun. 2016;7:13304. doi: 10.1038/ncomms13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ricciardi A.S., Quijano E., Putman R., Saltzman W.M., Glazer P.M. Peptide Nucleic Acids as a Tool for Site-Specific Gene Editing. Molecules. 2018;23:E632. doi: 10.3390/molecules23030632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.El-Beshlawy A., El-Ghamrawy M. Recent trends in treatment of thalassemia. Blood Cells Mol. Dis. 2019;76:53–58. doi: 10.1016/j.bcmd.2019.01.006. [DOI] [PubMed] [Google Scholar]
- 58.Breda L., Casu C., Gardenghi S., Bianchi N., Cartegni L., Narla M., Yazdanbakhsh K., Musso M., Manwani D., Little J. Therapeutic hemoglobin levels after gene transfer in β-thalassemia mice and in hematopoietic cells of β-thalassemia and sickle cells disease patients. PLoS ONE. 2012;7:e32345. doi: 10.1371/journal.pone.0032345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gambari R., Fibach E. Medicinal chemistry of fetal hemoglobin inducers for treatment of beta-thalassemia. Curr. Med. Chem. 2007;14:199–212. doi: 10.2174/092986707779313318. [DOI] [PubMed] [Google Scholar]
- 60.Fibach E., Bianchi N., Borgatti M., Zuccato C., Finotti A., Lampronti I., Prus E., Mischiati C., Gambari R. Effects of rapamycin on accumulation of alpha-, beta- and gamma-globin mRNAs in erythroid precursor cells from beta-thalassaemia patients. Eur. J. Haematol. 2006;77:437–441. doi: 10.1111/j.1600-0609.2006.00731.x. [DOI] [PubMed] [Google Scholar]
- 61.Fibach E., Bianchi N., Borgatti M., Prus E., Gambari R. Mithramycin induces fetal hemoglobin production in normal and thalassemic human erythroid precursor cells. Blood. 2003;102:1276–1281. doi: 10.1182/blood-2002-10-3096. [DOI] [PubMed] [Google Scholar]
- 62.Lampronti I., Bianchi N., Zuccato C., Dall’Acqua F., Vedaldi D., Viola G., Potenza R., Chiavilli F., Breveglieri G., Borgatti M. Increase in gamma-globin mRNA content in human erythroid cells treated with angelicin analogs. Int. J. Hematol. 2009;90:318–327. doi: 10.1007/s12185-009-0422-2. [DOI] [PubMed] [Google Scholar]
- 63.Fibach E., Prus E., Bianchi N., Zuccato C., Breveglieri G., Salvatori F., Finotti A., Lipucci di Paola M., Brognara E., Lampronti I. Resveratrol: Antioxidant activity and induction of fetal hemoglobin in erythroid cells from normal donors and β-thalassemia patients. Int. J. Mol. Med. 2012;29:974–982. doi: 10.3892/ijmm.2012.928. [DOI] [PubMed] [Google Scholar]
- 64.Reid M.E., El Beshlawy A., Inati A., Kutlar A., Abboud M.R., Haynes J., Jr., Ward R., Sharon B., Taher A.T., Smith W. A double-blind, placebo-controlled phase II study of the efficacy and safety of 2,2-dimethylbutyrate (HQK-1001), an oral fetal globin inducer, in sickle cell disease. Am. J. Hematol. 2014;89:709–713. doi: 10.1002/ajh.23725. [DOI] [PubMed] [Google Scholar]
- 65.Zuccato C., Breda L., Salvatori F., Breveglieri G., Gardenghi S., Bianchi N., Brognara E., Lampronti I., Borgatti M., Rivella S., Gambari R. A combined approach for β-thalassemia based on gene therapy-mediated adult hemoglobin (HbA) production and fetal hemoglobin (HbF) induction. Ann. Hematol. 2012;91:1201–1213. doi: 10.1007/s00277-012-1430-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lechauve C., Keith J., Khandros E., Fowler S., Mayberry K., Freiwan A., Thom C.S., Delbini P., Romero E.B., Zhang J. The autophagy-activating kinase ULK1 mediates clearance of free α-globin in β-thalassemia. Sci. Transl. Med. 2019;11:eaav4881. doi: 10.1126/scitranslmed.aav4881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Luther D.C., Lee Y.W., Nagaraj H., Scaletti F., Rotello V.M. Delivery approaches for CRISPR/Cas9 therapeutics in vivo: advances and challenges. Expert Opin. Drug Deliv. 2018;15:905–913. doi: 10.1080/17425247.2018.1517746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xu X., Wan T., Xin H., Li D., Pan H., Wu J., Ping Y. Delivery of CRISPR/Cas9 for therapeutic genome editing. J. Gene Med. 2019;21:e3107. doi: 10.1002/jgm.3107. [DOI] [PubMed] [Google Scholar]
- 69.Zhao W., Hou X., Vick O.G., Dong Y. RNA delivery biomaterials for the treatment of genetic and rare diseases. Biomaterials. 2019;217:119291. doi: 10.1016/j.biomaterials.2019.119291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lattanzi A., Meneghini V., Pavani G., Amor F., Ramadier S., Felix T., Antoniani C., Masson C., Alibeu O., Lee C. Optimization of CRISPR/Cas9 Delivery to Human Hematopoietic Stem and Progenitor Cells for Therapeutic Genomic Rearrangements. Mol. Ther. 2019;27:137–150. doi: 10.1016/j.ymthe.2018.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li L., Hu S., Chen X. Non-viral delivery systems for CRISPR/Cas9-based genome editing: Challenges and opportunities. Biomaterials. 2018;171:207–218. doi: 10.1016/j.biomaterials.2018.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vertex Pharmaceuticals . 2021. A Long-term Follow-up Study of Subjects With β-thalassemia or Sickle Cell Disease Treated With Autologous CRISPR-Cas9 Modified Hematopoietic Stem Cells (CTX001). ClinicalTrials.gov: NCT04208529. First posted December 23, 2019, and last update posted February 15, 2021. [Google Scholar]
- 73.Cosenza L.C., Breda L., Breveglieri G., Zuccato C., Finotti A., Lampronti I., Borgatti M., Chiavilli F., Gamberini M.R., Satta S. A validated cellular biobank for β-thalassemia. J. Transl. Med. 2016;14:255. doi: 10.1186/s12967-016-1016-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Van der Auwera G.A., Carneiro M.O., Hartl C., Poplin R., del Angel G., Levy-Moonshine A., Jordan T., Shakir K., Roazen D., Thibault J. From fastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinformatics. 2013;43:11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Broad Institute . 2019. “Picard Toolkit.” GitHub Repository.http://broadinstitute.github.io/picard/ [Google Scholar]
- 76.Chen S., Zhou Y., Chen Y., Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–i890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Koboldt D.C., Zhang Q., Larson D.E., Shen D., McLellan M.D., Lin L., Miller C.A., Mardis E.R., Ding L., Wilson R.K. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bae S., Park J., Kim J.S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.