

Abstract
7SK small nuclear RNA (snRNA) is an abundant and ubiquitously expressed noncoding RNA that functions to modulate the activity of RNA Polymerase II (RNAPII) in part by stabilizing distinct pools of 7SK-protein complexes. Prevailing models suggest that the secondary structure of 7SK is dynamically remodeled within its alternative RNA-protein pools such that its architecture differentially regulates the exchange of cognate binding partners. The nuclear hnRNP A1/A2 proteins influence the biology of 7SK snRNA via processes that require an intact stem loop (SL) 3 domain; however, the molecular details by which hnRNPs assemble onto 7SK snRNA are yet to be described. Here, we have taken an integrated approach to present a detailed description of the 7SK-hnRNP A1 complex. We show that unbound 7SK snRNA adopts at least two major conformations in solution, with significant structural differences localizing to the SL2–3 linker and the base of SL3. Phylogenetic analysis indicates that this same region is the least genetically conserved feature of 7SK snRNA. By performing DMS modifications with the presence of excess protein, we reveal that hnRNP A1 binds with selectivity to SL3 through mechanisms that increase the flexibility of the RNA adjacent to putative binding sites. Calorimetric titrations further validate that hnRNP A1-SL3 assembly is complex with the affinity of discrete binding events modulated by the surrounding RNA structure. To interpret this context-dependent binding phenomenon, we determined a 3D model of SL3 to show that it folds to position minimal hnRNP A1/A2 binding sites (5’-Y/RAG-3’) within different local environments. SL3-protein complexes resolved by SEC-MALS-SAXS confirm that up to four hnRNP A1 proteins bind along the entire surface of SL3 via interactions that preserve the overall structural integrity of this domain. In sum, the collective results presented here reveal a specific role for a folded SL3 domain to scaffold hnRNP A1/A2–7SK assembly via mechanisms modulated by the surrounding RNA structure.
Introduction
Eukaryotic gene expression is tightly regulated with control points positioned throughout the lifespan of cellular transcripts. An important checkpoint occurs at the step where RNA Polymerase II (RNAPII) transitions from an initiating polymerase into a processive elongating complex. This step of gene expression involves the positive transcription elongation factor b (P-TEFb), a complex that minimally consists of the cyclin-dependent kinase 9 (CDK9) and its regulatory partner cyclin T1 (CycT1). P-TEFb phosphorylates the C-terminal domain of RNAPII to facilitate the switch to processive elongation and the coordination of co-transcriptional processing of nascent transcripts1. Given the importance of P-TEFb to modulating the function of RNAPII, its kinase activity is regulated by storing the heterodimer within RNA-protein (RNP) pools that contain the ubiquitous 7SK small nuclear RNA (snRNA).
Human 7SK snRNA is an RNAPIII transcript that is 330–332 nucleotides (nts) in length and is present at ~2 x 105 copies per cell. The established function of 7SK snRNA is to regulate the homeostatic levels of P-TEFb, and it accomplishes this by using its structure to dynamically assemble RNPs that cycle among at least three different pools, each containing methylphosphate capping enzyme (MePCE) and La-related protein 7 (LARP7) constitutively bound to the 5’ and 3’ ends of 7SK, respectively2–5 (Figure 1). The pool containing P-TEFb also has a dimer of the host accessory factor, hexamethylene bis-acetamide inducible protein 1/2 (just referred to as HEXIM throughout) bound to form the 7SK-HEXIM-(P-TEFb) snRNP complex, which inhibits the kinase activity of the CDK9 subunit to in turn regulate transcription6–9. Liberation of P-TEFb from the HEXIM-associated 7SK snRNA pool relieves the kinase inhibition of CDK9 to ultimately catalyze phosphorylation of the RNAPII CTD. 7SK snRNA then cycles to RNP pools consisting of several hnRNP proteins (Figure 1), including A1/A2 and Q/R in mutually exclusive 7SK complexes10. The hnRNP pools are believed to block the back association of HEXIM and P-TEFb with 7SK snRNA, thus indirectly affecting RNAPII-dependent transcription and associated RNA processing events11–13.
Figure 1. A 7SK-Protein Equilibrium Distribution Regulates P-TEFb Availability.
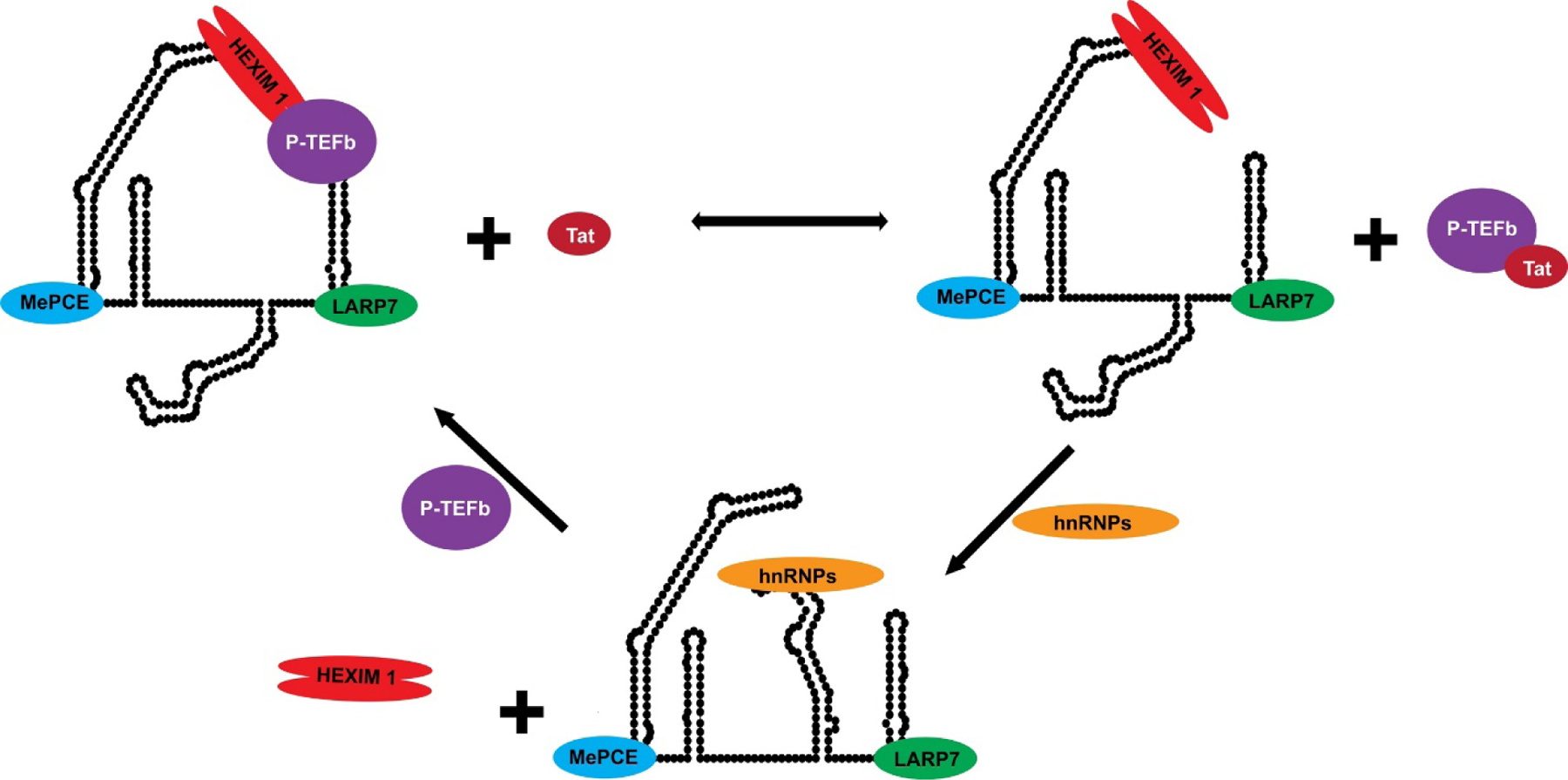
An equilibrium of active and inactive pools of P-TEFb is regulated by the dynamic assembly and disassembly of distinct 7SK-protein complexes, including complexes formed with members of the hnRNP protein family.
Given the central role of 7SK snRNA to eukaryotic gene expression, its structure has been widely studied by different methods2, 3, 14–18. Wassarman and Steitz reported the first model of 7SK snRNA, which was mapped using classical chemo-enzymatic methods14. More recent secondary structural models determined by SHAPE-based chemistries report that 7SK snRNA adopts two major conformations (closed, with long-range 5’-3’ pairing vs open with no 5’-3’ pairing) consisting of four major stem loops (SL) and at least two minor SLs2. Reconstitution studies suggest that 7SK snRNA changes its structure as it cycles through its different RNP pools; however, the effects of hnRNPs on the 7SK architecture has not been described2, 12. In addition to the secondary structural models of intact 7SK snRNA, high-resolution NMR or X-ray crystallographic structures have been reported for its isolated SL 1 and 4 domains3, 16–18. Three-dimensional structures of the remaining SLs have yet to be reported.
The 7SK-HEXIM-(P-TEFb) complex has been studied extensively within the context of normal RNAPII-dependent transcription control and when hijacked by HIV Tat2, 9, 12, 13, 19, 20. These studies have produced different models explaining the mechanisms by which P-TEFb is extracted from its HEXIM-associated 7SK snRNA complex, and evidence indicates that the RNA undergoes changes to its structure in a protein-dependent way. By comparison, very little work has been done to characterize the nature of the 7SK-hnRNP pools despite their biological role in preventing the re-formation of the 7SK-HEXIM-P-TEFb complex10, 11, 21. By interacting with 7SK snRNA, the hnRNP proteins regulate the equilibrium distribution of P-TEFb to indirectly influence its enzymatic activity on RNAPII. Therefore, details of complex formation between 7SK snRNA and hnRNPs are equally important to describing mechanisms by which P-TEFb availability is regulated and to inform on strategies to modulate the biology 7SK-protein complexes.
Since hnRNP A1/A2 proteins are components within the 7SK-protein pools, we endeavored to address two broadly unanswered questions pertaining to the biochemistry of this important 7SK snRNA complex: by what mechanism(s) do hnRNP A1/A2 proteins assemble onto 7SK snRNA, and what are the nature of the interactions within the 7SK-hnRNP A1/A2 complex? Answers to these questions are essential to understanding how 7SK-hnRNP interactions modulate the availability of P-TEFb (Figure 1). Here, we describe an approach that integrates data from DMS-MaPseq, NMR spectroscopy, computational modeling, calorimetry and SAXS to provide initial insights into the molecular mechanisms by which hnRNPs modulate the 7SK-protein equilibrium distribution (Figure 1). We first determined that free 7SK snRNA adopts at least two conformations in solution with structural differences localized to the SL2–3 linker and the base of SL3. Local mutations introduced to stabilize the major conformation of the SL2–3 linker surprisingly impacted the global folding of 7SK snRNA, indicating that the conformational dynamics of 7SK snRNA are cooperative. We further determined that the SL2–3 linker region is the least phylogenetically conserved genetic feature of 7SK snRNA. We then determined that hnRNP A1 assembles onto 7SK snRNA by interacting selectively with its SL3 domain via site-specific interactions that are differentially modulated by the surrounding RNA structure. Hybrid 3D models of isolated SL3 were calculated to reveal that hnRNP A1/A2 consensus motifs (5’-R/YAG-3’) reside within noncanonical structures positioned along the entire body of SL3. By resolving SL3 complexes by SEC-MALS-SAXS, we confirm that up to four hnRNP A1 proteins bind stably to distinct sites along SL3 through contacts that preserve the overall structural integrity of the RNA. Thus, the collective results presented here reveal a specific role for a folded SL3 domain to scaffold hnRNP A1/A2 assembly as 7SK snRNA cycles through its different pools to modulate the equilibrium distribution of P-TEFb (Figure 1). The work also shows that integration of chemical probing and solution biophysics produces a deeper understanding of the complex mechanisms by which proteins assemble along conformationally dynamic RNA structures, and it also provides guidelines on how to design 7SK snRNA constructs and complexes for higher-resolution structural studies.
Materials and Methods
RNA Synthesis and Purification
The full-length 7SK snRNA was sub-cloned into pUc19 vector (gift from Dr. Jonathan Karn). The plasmid template was linearized with EcoRI-HF (NEB), and then desalted using centrifugal filter (Millipore Amicon). The plasmid template for SL3 (97-nt) was sub-cloned into pUc19 vector using gBlock gene fragment (IDT), and linearized with PstI-HF (NEB). For SL3S (57-nt) and SL3M (75-nt), synthetic DNA oligos (IDT) were used as templates for in vitro Transcription. Uniformly 15N/13C-labeled uridine (UTP) and guanidine (GTP) (Cambridge Isotope Laboratories), and fully protonated adenosine (ATP) and cytidine (CTP) (Sigma-Aldrich) were used to prepare the labeled SL3S and SL3M RNA samples for SOFAST-HSQC titrations, while the RNA samples for all other studies were prepared with fully protonated NTPs (Sigma-Aldrich). Transcription reactions were optimized in individual trials following published protocol22. The SL3 constructs were purified to homogeneity by 8–10% Urea-PAGE and eluted in Tris-Borate-EDTA (TBE) buffer. The RNA samples were desalted and annealed freshly before proceeding with each experiment. The annealed samples were concentrated using centrifugation filtration system (Amicon) and purified using size exclusion column (SEC), exchanging into the buffer specifically made for each following application: for NMR, including NOESY and HSQC, 10 mM K2HPO4, 50 mM KCl, pH 5.5, and 10% D2O; for ITC, 10 mM K2HPO4, 120 mM KCl, 1 mM TCEP, 0.5 mM EDTA, pH 6.5; and for SEC-MALS-SAXS, 5 mM MES, 50 mM KCl, pH 6.5.
UP1 Purification
The C-terminal (His)6-tagged UP1 protein (residues 1–196) was prepared as previously described, and where we demonstrated that the presence of the tag does not affect RNA binding23, 24. In short, the UP1 construct was overexpressed in BL21(DE3) (NEB) cells and purified using nickel affinity chromatography on Hi-Trap columns (GE Biosciences) using high salt binding, washing and elution buffers (1.2 M NaCl, 20 mM Na2HPO4, pH 7.5) containing 10 mM, 20 mM and 250 mM imidazole, respectively. Once eluted, the UP1 construct was exchanged into ITC buffer (120 mM KCl, 10 mM K2HPO4, 1 mM TCEP and 0.5 mM EDTA, pH 6.5) using a HiPrep 16/60 sephacryl S-100 column (Pharmacia Biotech) FPLC. The final sample was tested for purity using a 10% SDS-PAGE and stored at 4 °C until use.
DMS-MaPseq of 7SK snRNA
5 μL (1 μg) of purified full-length 7SK snRNA was heated to 95 °C for 15 sec and flash cooled on ice for 2 min. 95 μL of DMS modification buffer (100 mM Sodium Cacodylate, 140 mM KCl, +/- 3 mM MgCl2, pH 7.5) was added into the RNA sample for 30 min incubation at room temperature. 2–5% of DMS was added and the sample was kept incubating at 37 °C with 500 rpm shaking for 5 to 10 min. The methylation reaction was terminated with 60 μL of BME (Sigma-Aldrich). The modified RNA sample was cleaned using RNA cleanup and concentrator-5 column (Zymo Research) to recover the RNA > 200 nt.
Methylated RNA was reverse transcribed with specific reverse primer (RV, reverse complement to nts 296–320 of 7SK snRNA sequence) using thermostable group II intron reverse transcriptase, 3rd generation (TGIRT-III, InGex). As followed, the RNA templates were digested using RNase H (NEB) for 20 min at 37 °C. The reverse-transcribed DNA was sequentially PCR amplified with specific primer set (FW nts 1–19; RV nts 296–320) using Phusion DNA polymerase (NEB). The PCR began with initial denaturing for 30 sec at 98 °C, followed by 25 PCR cycles, including denaturing for 5 sec at 98 °C, annealing for 10 sec at 65 °C, and extension for 15 sec at 72 °C, in order; then the final extension is set for 5 min at 72 °C. The PCR product (nts 1–320) was desalted using DNA cleanup and concentrator-5 column kit (Zymo Research). The homogeneity of the amplified sample was checked using agarose gel before sending for sequencing.
The sequencing was performed on an Illumina HiSeq 2000 system, which uses cluster generation and sequencing by synthesis (SBS) chemistry. The sequencing result of 7SK snRNA was aligned using methods developed in the Rouskin group25. The minimal mutational signals of 5’ and 3’ primer regions and signals from T and G in the sequence were determined to be null. Signals from A and C in the target region (nts 20–295) were 95% Winsorized and normalized to the highest to generate DMS reactivity index, which were used to calculate the secondary structural model of 7SK snRNA.
Next-Generation Sequencing (NGS) technologies are based on an initial count of the total number of reads mapping to each transcript. In the study, several steps were taken to validate the data, particularly to avoid 3’ bias of sequencing. First, TGIRT-III was used to generate a cDNA pool of full-length transcripts, instead of regular Reverse Transcriptase (RT). TGIRT induces a random mutation as a signal of DMS modified A or C during the RT, while regular reverse transcriptase induces a stop when extending to a DMS modified A or C, which yields an early terminated transcript. Thus, the mechanism of action of TGIRT-III reduces 3’ bias because the enzyme processively introduces mutations at the sites of modification instead of terminating. Second, the PCR products (nts 1–320) were tested with 1% agarose electrophoresis before sending for sequencing to ensure sample homogeneity. In addition, during the data analysis, read coverage plots and mutational fraction plots were used as stringent quality controls of each dataset. The read coverage directly counts the reads aligning to the selected region (for 7SK snRNA, nts 20–295 were set as target region to exclude the primer regions). The percentile value of each nucleotide shows the coverage fraction of all aligned reads at each nucleotide. Mutational fraction plots were used to ensure effective DMS modification and low background GU reactivity. DMS nontreated (NT) samples were also tested to rule out other sequencing abnormalities. All datasets used in the study have over 90% read coverage throughout entire target region without any significant difference of coverage between 5’ and 3’ ends.
RNA Structure Derivation Using DREEM Pipeline
DREEM (Detection of RNA folding Ensembles using Expectation-Maximization clustering) is a computational tool to detect alternative structures formed by RNA molecules developed by the Rouskin Group26. The analysis of 7SK snRNA consisted of 3 steps: 1. The raw sequencing files were aligned to the sequence of 7SK snRNA; 2. The aligned read files were converted to “bit vectors” consisting of “0”s (matches) and “1”s (mismatches and deletions), which is a computation friendly format for processing; 3. Clustering of the bit vectors based on their mutational profiles using an Expectation-Maximization (EM) clustering algorithm. For 7SK snRNA, the clustering region was set to nts 20–295 (excluding the primer overlap regions for sequencing) so only the actual sequencing data were processed. The Maximum number of Clusters was set to 2 and the minimum iterations of the EM algorithm was set to 500. The output files of the pipeline include DMS reactivity indices of each cluster for further processing.
DREEM uses a very stringent statistical test: the Bayesian information criterion (BIC) to penalize the number of parameters and prevent overfitting. The platform has been carefully validated using both simulations and crystalized RNAs to benchmark the DREEM algorithm. The BIC test is very stringent that conformations will be penalized if it is not abundant enough or not different enough. As a result, the alternative conformations will not pass the test and be filtered out.
Secondary Structure Model Visualization of 7SK snRNA
The DMS reactivity indices of the population average model and clusters were generated as described above. The files were used as pseudoenergy restraints to guide the secondary structure folding of 7SK snRNA using RNAStructure27. The final structural models were drawn using VARNA and individual nucleotides were further color coded based on the normalized DMS reactivity indices.
Differential DMS-MaPseq Titrations of UP1–7SK Complex
For complex sample, 1μg (stocked at 200 ng/μL) of native 7SK snRNA was refolded before adding buffers and UP1. The final concentration of the RNA was adjusted to 100 nM (1 μg/ 100 μL). In each sample, 10 μL of UP1 (covering a 25-fold concentration range), 5 μL of RNA solution and 80 μL of DMS buffer were added to form the complexes. The titration samples were examined using EMSA to ensure complex formation. The complexes were incubated in the DMS modification buffer at room temperature for 30 min before proceeding with the conventional DMS-MaPseq method. All the DMS-MaPseq profiles were processed using the conventional protocol including internal normalization as described above. To calculate differential DMS reactivities, z-score test was done for the unbound 7SK snRNA and each UP1 titration dataset to normalize the overall DMS reactivity under each condition. The normalized DMS reactivity indices of unbound 7SK snRNA were subtracted from the ones of UP1–7SK snRNA complexes (up to 25:1) to yield differential DMS reactivities.
NMR Data Acquisition, Processing and Analysis
All NMR experiments were performed on Advance 900/800/700 MHz high-field NMR spectrometer (Bruker) equipped with cryogenically cooled HCN triple resonance probes and a z-axis pulsed-field gradient accessory. After collection, all data was processed using NMRpipe/NMRDraw28 and analyzed/assigned using NMRViewJ/NMRFAM-Sparky29, 30. All NMR experiments of SL3 constructs were conducted in the NMR buffer. Exchangeable 1H spectra were measured at 283 K with the Watergate NOESY (τm = 200ms) pulse sequence on fully protonated SL3 constructs. 1H-15N HSQC spectra were collected to verify imino assignment using a selective 15N/13C-labeled G and U (fully protonated A and C) SL3S and SL3M samples. Assignments of imino protons were carried out following well-established procedure31, 32. SOFAST-HSQC titrations were carried out using selective 15N/13C-labeled 7SK snRNA SL3S construct and unlabeled UP1. Spectra were collected at four different [UP1]:[SL3S] molar ratios: 0.25:1, 0.5:1, 0.75:1 and 1.25:1, in the buffer condition of 10 mM K2HPO4, 50 mM KCl, pH 5.5, at 288 K.
Sixteen Residual dipolar couplings (RDCs) were measured using 1H-15N TROSY and HSQC for both isotropic and anisotropic (ASLA, Pf1 bacteriophage at ~7 mg/ml) condition with 15N-selectively labeled 7SK snRNA SL3M constructs in the buffer condition of 10 mM K2HPO4, 50 mM KCl, pH 5.5 and 10% D2O, at 283 K33. Phage concentration was verified via 2H splitting (~14.5 Hz) at 900 MHz. Residual dipolar coupling values were determined by taking the difference in 1JNH couplings under anisotropic and isotropic conditions.
HSQC titrations were conducted using selective 15N-labeled C-terminal UP1 construct and unlabeled 7SK SL3S. HSQC spectra were collected at six different [UP1]:[SL3S] molar ratios: 0.1, 0.3:1, 0.5:1, 0.7:1, 1:1 and 2:1, in the buffer condition of 10mM K2HPO4, 120mM KCl, 280mM NaCl, pH 6.5, at 303K.
Phylogenetic Analysis of 7SK snRNA
The 7SK snRNA sequence used for phylogenetic comparisons and generation of the consensus logo plot were obtained from chicken (AJ890101), zebrafish (AJ890102), Tetraodon nigrovidis (AJ890103) and Takifugu rubripes (AJ890104), Rattus norvegicus (K02909), Mus musculus (M63671), human (NR_001445)6.
Calorimetric Titrations of UP1
Calorimetric titration studies were performed at 25 °C using VP-ITC calorimeter (MicroCal, LLC) with the 7SK snRNA SL3 and SL3S constructs. Each RNA sample was prepared by diluting to a concentration of ~5 μM in binding buffer (10mM K2HPO4, 120mM KCl, 0.5mM EDTA, 1mM TCEP, pH 6.5). UP1 protein was prepared for the titration studies by exchanging it in the same binding buffer as used for RNA sample preparation using Ultra-4 centrifugal filter devices (Amicon). The UP1 protein (~80 μM) was titrated into 1.4 ml of 5 μM RNA over 36 injections of 8ul each. To minimize the accumulation of experimental error associated with batch-to-batch variation, titrations were performed in triplicate. The three titration data sets were input for the global fitting using AFFINImeter 34. Error bars on individual data points are automatically calculated in AFFINImeter and correspond to the uncertainty associated to the integral calculation of each peak, including the noise in the baseline and noise in the peak35. Besides the simple 1:1 binding model, the titration data were also fit to an independent sets of site model where the number of sites were set equal to the number of partially exposed 5’-AG-3’ motifs in each construct. The independent sets of sites model allow determination of site-specific thermodynamic parameters, while holding the stoichiometry constan.
SAXS Data Acquisition and Analysis
The SL3M RNA construct that was used for size exclusion chromatography in line with multiple angle light scattering and small angle X-ray scattering (SEC-MALS-SAXS) were prepared as described above using fully protonated unlabeled rNTPs. SEC-MALS-SAXS experiments were performed at BioCAT (Beamline 18-ID) at the Advanced Photon Source (Argonne National Laboratory; Lemont, IL) and set up was the same as previously described36. To minimize any non-negligible structure effects such as aggregation or repulsion, SAXS experiments were performed in the buffer condition of 5 mM MES, 50 mM KCl, pH 6.5, at a concentration of ~3 mg/mL in 200 μL load volume for SEC. The SAXS data were collected at 0.5 s exposures every 3 s for the duration of the SEC run. 10 points coinciding with a single SEC peak were taken as sample + buffer and 100 points coinciding with the SEC baseline trace directly prior to sample peak were taken as buffer only. Buffer only scattering was subtracted from buffer plus sample scattering to obtain the solution scattering from the RNA. After initial processing, Primus from the ATSAS suite of small angle X-ray scattering programs was used to visualize the data37.Guinier fitting (Rg × q < 1.3) was used to check for non-negligible structure factors (aggregation or repulsion) and determine the Radius of Gyration (Rg). GNOM was used to fit the SAXS data and generate the pairwise-distance distribution (P(r)) to determine the maximum particle dimension (Dmax). The molecular envelope of the SL3M construct was determined using DAMMIF in fast mode. In short, the ab initio models were generated from the fitting model that was determined by the SAXS data using GNOM. The models were then averaged using DAMAVER, and the most populated models were determined using DAMFILT. The overall Normalized Spatial Discrepancy (NSD) for all 32 models was ~ 0.6. For SL3M, the final ab initio molecular envelope was overlaid onto atomic model for visualization using SUPCOMB by allowing for enantiomers and fitting in fast mode. Raw SAXS scattering data were used to generate the molecular density envelope using the DAMMIF module of ATSAS 2.6.0 on the Rider cluster. The envelope was written to a PDB file and introduced to AMBER structures by way of the EMAP module38. Fitness and validity of models were assessed based on the linearity of their AMBER back-calculated RDC values with respect to measured data, alongside the SAXS Chi-square score as given by the Crysol function of ATSAS39. Crysol scores were calculated with a truncated SAXS profile from points 10–700, a solvent density of 0.55, and allowing for constant subtraction.
Structural Modeling
10,000 structures were generated using the FARFAR2 RNA folding algorithm for ROSETTA. These were then filtered with SAXS data to select 10 models for refinement in AMBER. 50 ns AMBER simulations were conducted with NOE restraints in implicit solvent, followed by a 1 ns refinement with RDC restraints activated within the Born solvation model. All simulations were performed using the High Performance Computing Resource in the Core Facility for Advanced Research Computing at Case Western Reserve University.
Based upon the SL3M (75-nt) construct and its experimentally determined secondary structure, sequence and base pairing data were input to FARFAR240. RNA de novo structure prediction was performed using the FARFAR2 module of Rosetta 3.11 with the sequence and secondary structure as input. The RNA tools package within Rosetta was used to generate idealized helices for base paired regions of the construct as reference for structure prediction. FARFAR2 was run until 10,000 pdbs were generated by output. In the 10,000 pdb pool, the SAXS data of 7SK snRNA SL3M was used as filter to select global conformation using crysol in batch model, allowing for constant subtraction and a maximum q value of 0.23 Å−1. The resulting χ2 value range from SAXS fitting in the 10,000 pdb pool were from 2.97 to 41.18. The 10 lowest χ2 value structures (χ2 lower than 3.95) were selected from SAXS filter for further refinement in AMBER.
Each of the 10 starting structures selected by crysol scoring were then prepared for AMBER using the ff99bscOL3 force field in tLEaP41, 42. Structures were then each minimized with sander. In the first round of minimization, the RNA was held fixed with arbitrary 10.0 kcal/mol Å restraints and minimized for 500 steps of steepest descent followed by 500 steps of conjugate gradient43. Next, the restraints were lifted and a longer round of minimization was conducted with 1000 steps of steepest descent and 1500 steps of conjugate gradient. A 10.0 Å cutoff was used for calculation of Born radii through each minimization.
Molecular Dynamics Simulations with Residual Dipolar Couplings
Final refinement in AMBER made use of restraints from NOE data, DMS-MaPseq information, and 1H-15N Residual Dipolar Couplings (RDCs). The NOE data restraints were made for the base paring region based on the NMR H2O NOESY assignment at the imino group for the SL3M. The RNA was heated from 0 to 300 K over 100 ps and simulated for the remainder of the 50 ns simulation at the 300 K while writing the trajectory every 100ps. After the 50 ns of simulation, structures were written from the trajectory files using the cpptraj44, providing a structure for every 100 ps of simulation (500 per starting structure). Only the structure running longer than 5 ns were included in the pool with a total 4500 structures. These structures were filtered by SAXS using Crysol mode and 20 structures best fit with lowest χ2 were chosen for the next stage of RDC refinement.
20 structures selected from 50 ns simulation trajectory were fit to the RDC dataset individually to determine alignment tensors and generate the dipole restraint file45. The RDC data were used for N3H3 atoms in the Uridine nucleotides and N1H1 atoms in the Guanine nucleotides. Refinement with RDC restraints added was conducted in AMBER using the sander executable. Structures were heated linearly from 0 to 300 K over 100 ps, simulated at 300 K for 1 ns, then cooled linearly to 0K over 100 ps. The final 10 lowest energy structures of RDC refinement simulation was extracted yielding an ensemble of 10 structures.
Note on the Different Buffer Conditions used in This Study
Given the experimental nature of this study, it was necessary to use buffer conditions that ranged in ionic strength and pH. Nevertheless, we observed very high agreement (R2 > 0.9) of the normalized DMS reactivity indices for intact 7SK snRNA under each condition studied, and the NMR-determined secondary structures of isolated SL3M and SL3S fragments are very consistent with the SL3 structure as probed by DMS for intact 7SK snRNA. Lastly, the RDC-refined structure of SL3M shows good agreement with the SAXS-derived experimental parameters and molecular envelope. Therefore, the different buffer conditions used here do not complicate the interpretation of the results.
Results
DMS-MaPseq Reveals free 7SK snRNA Adopts at Least Two Conformations that Differ in the SL2–3 Linker and the Base of SL3
Different secondary structures of 7SK snRNA have been described that show variations in its base pair (bp) configurations, including bps within SL3, the presumptive binding domain for hnRNPs2, 14. To characterize the solution properties of 7SK snRNA under our conditions, we in vitro transcribed, purified and treated the intact RNA with DMS in buffer +/- 3 mM Mg2+, which is within the physiological concentration range46. Chemical modifications were performed in three individual experiments, and each sample was further processed for sequencing as described in the Methods section. For each dataset, read coverage plots (Figure S1A) and mutational fraction plots (Figure S1B) were used to ensure high quality of the dataset (not shown for each dataset). We observed a robust correlation (R2>0.95, Figure S2A) in the degree of modification across all three replicates throughout the target region (nts 20–295) of full length 7SK (Figure S2B), which reflects the reproducibility by which DMS modifies exposed adenosines and cytosines within our 7SK snRNA samples and the homogeneity of our folded RNA. Comparison of DMS reactivity indices of 7SK snRNA samples probed +/- 3 mM Mg2+ show that the addition of Mg2+ does not significantly alter the global modification profile (Figure S3), consistent with previous observations using SHAPE-based chemistry where Mg2+ addition affected only the localized folding of SL12. Of note, DMS reactivity indices for the first ~19 nt of 7SK snRNA were not obtained here due to the design of the sequencing primers (see Methods) so we were unable to detect any magnesium-dependent differences for this portion of SL1; however, the addition of Mg2+ showed minimal effect to the DMS reactivity indices and structure similarity throughout the target region picked up by our sequencing primers (Figure. S3A and S3C).
Figure 2A reveals that the population averaged structure of 7SK snRNA, in the presence of 3 mM Mg2+, folds into 4 major stem loops (SL1, SL2, SL3 and SL4) and two minor stem loops (SL2B and SL2C). Of note, the folding of SL4 is entirely driven by the thermodynamic parameters since we are missing reactivity data due to the design of sequencing primers. By comparison to the Wassarman and Steitz model14, the 7SK snRNA secondary structure determined here by DMS-MaPseq shows a high degree of similarity in SLs 1 and 4; however, there are significant differences in SLs 2 and 3 (highlighted in yellow, Figure S4A). For example, the DMS-MaPseq reactivity profile for nts 140–200 show that many residues within this region have moderate to low reactivity indices consistent with the formation of secondary structure unlike that observed in the Steitz model. The model of 7SK snRNA determined here agrees more favorably with the secondary structure proposed by Brogie and Price (Figure S4B), which was determined by SHAPE-based chemistry2. Both models fold 7SK snRNA into 4 major stem loops (SL1, SL2, SL3 and SL4) and two minor stem loops (SL2B and SL2C). The majority of bps between the two models are the same, but the most notable structural differences are observed in the apical loop region of SL3 (highlighted in green, Figure S4B). Under our conditions, C229-A231 have medium to strong DMS reactivities (>0.5) and are therefore determined to be unpaired in SL3.
Figure 2. Secondary Structural Model of Full Length 7SK snRNA as Determined by DMS-MaPseq.
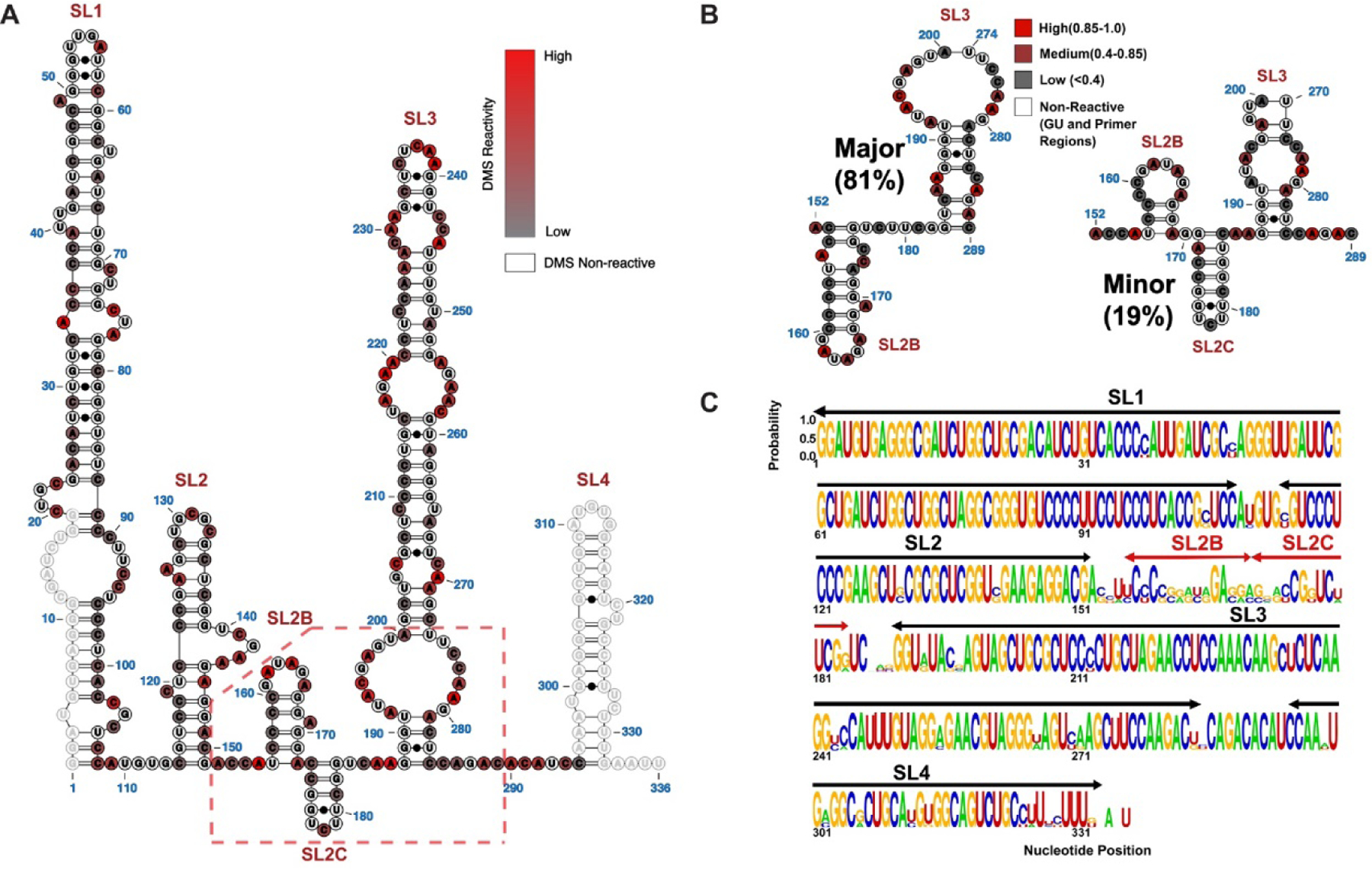
(A) The population averaged secondary structure of 7SK snRNA as determined by incorporating DMS reactivity indices as pseudoenergy restraints. The normalized indices are superimposed as a scaled color-code from 0 (low reactivity) to 1 (high reactivity) for each modified AC nucleotide. Primer regions and GU nucleotides are shown as open circles with no color code. (B) Two major 7SK snRNA conformers were derived from DREEM clustering of DMS-MaPseq reactivity indices. The color code is according to DMS reactivities of the nucleotides, coded as low (Grey), medium (Maroon) and high (Red). The significant structural differences localize to the SL2–3 linker and the base of SL3. (C) Logo plot analysis depicting the phylogenetic conservation of 7SK snRNA elements by aligning sequences from human, Mus musculus (M63671), Rattus norvegicus (K02909), chicken(AJ890101), zebra fish (AJ890102), Tetraodon nigrovidis (AJ890103) and Takifugu rubripes (AJ890104).
Given the variation in reported 7SK snRNA structures, we reasoned that 7SK snRNA might exist as a mixture of distinct conformers. Recent advancements in DMS-MaPseq data analysis allow for Detection of RNA folding Ensembles using Expectation-Maximization (DREEM), which reveals alternative conformations assumed by the same RNA sequence26. DREEM groups sequencing reads issued for each structure into distinct clusters by exploiting information contained in the observation of multiple DMS modifications on single molecules26. DREEM revealed that 7SK snRNA has the potential to adopt at least two distinct conformations, which differ specifically within the linker connecting SL2-SL3 and the base of SL3 (Figure 2B). The major conformer folds an additional SL2B domain within the SL2–3 linker characterized by a 6-nt GAUAGA apical loop and an imperfect helix composed of 7-bps (Left, Figure 2B). The base of SL3 is slightly remodeled in the major conformer relative to the population average structure to include a longer helix with a 2X2 internal loop. By comparison, the minor conformer folds two additional SL2 domains (SL2B and SL2C) within the SL2–3 linker (Right, Figure 2B). SL2B in the minor conformer consists of an 8-nt CCGAUAGA apical loop and a 3-bp helix, whereas SL2C adopts a 6-bp helix capped by a UCU triloop. The large internal loop in the lower portion of SL3 refolds in the minor conformer to include two additional WC bps.
To test if the specific alternating structures truly exist, we prepared a 7SK snRNA mutant construct where we replaced the 6-nt GAUAGA apical loop of the major conformer with a GAGA tetraloop with the expectation that this mutation would “lock” SL2B to prevent its refolding into the structure observed in the minor conformer. Comparison of the DMS reactivity indices of the wildtype and 7SK snRNA mutant constructs show only a 71% agreement (Figure S5A), which indicates this mutation perturbs the overall 7SK snRNA conformational ensemble. To more precisely map the changes induced by the GAGA mutation, we plotted a sliding window (80-nts) average of the R2 correlation coefficient of the reactivity indices of wildtype 7SK snRNA vs the mutant to produce a structural similarity plot (Figure S5B). Inspection of the structural similarity reveals that the most significant changes (R2 < ~0.6) induced by the mutation localize to the SL2–3 linker region as anticipated; however, we also observed a lower than expected correlation coefficient for SL1 (R2 ~0.8). The most straightforward interpretation of this observation is that the introduction of the GAGA mutation within the dynamic SL2–3 region negatively influences the cooperative folding of 7SK snRNA. This result highlights the complexities of RNA folding and the potential complications of introducing mutations within conformationally labile regions of a large RNA structure to lock putative structures. Given the intrinsic dynamics observed for the SL2–3 linker, we decided to determine the extent to which this region is phylogenetically conserved relative to the remaining 7SK snRNA sequence. We aligned representative (see Methods) 7SK snRNA sequences from seven different organisms to observe that nts 150–190 are the least conserved genetic feature, and the next least conserved feature corresponds to nts 181–198 which form the 5’-half of the lower SL3 structure (Figure 2C). Taken together, these data show that 7SK snRNA encodes localized conformational dynamics within the SL2–3 linker and the base of SL3.
HnRNP A1/A2 Proteins Interact Selectively with SL3 Through Both Specific and Nonspecific Interactions
Prevailing models suggest that 7SK snRNA undergoes conformational changes as it cycles through its different RNP pools; however, this has only been characterized following release of P-TEFb and HEXIM, after which hnRNP proteins (A1/A2 and Q/R) are thought to assemble onto a new 7SK structure2, 12. The specific hnRNP binding sites on 7SK are not known, although deletion studies revealed SL3 is necessary for hnRNP A1/A2 to dynamically modulate the 7SK-protein equilibrium distribution11. To determine where along 7SK snRNA hnRNP A1/A2 proteins assemble, we carried out DMS modification of 7SK snRNA while titrating the UP1 domain of hnRNP A1 (Figure S6). The UP1 domain, which consists of the tandem RNA Recognition Motifs, is a good surrogate to determine potential hnRNP A1/A2 binding sites since it imparts specificity for single strand 5’-R/YAG-3’ motifs within a range of different structural contexts47, 48. Moreover, the sequence specificities of hnRNP A1/A2 proteins are identical and they complement each other in many biological contexts49. Figure 3A shows that 7SK snRNA contains 14 partially single strand 5’-R/YAG-3’ motifs with 8 found within or in the immediate vicinity of SL3 alone.
Figure 3. HnRNP A1/A2 Proteins Bind Selectively to Stem Loop 3 of 7SK snRNA as Revealed by Differential DMS-MaPseq.
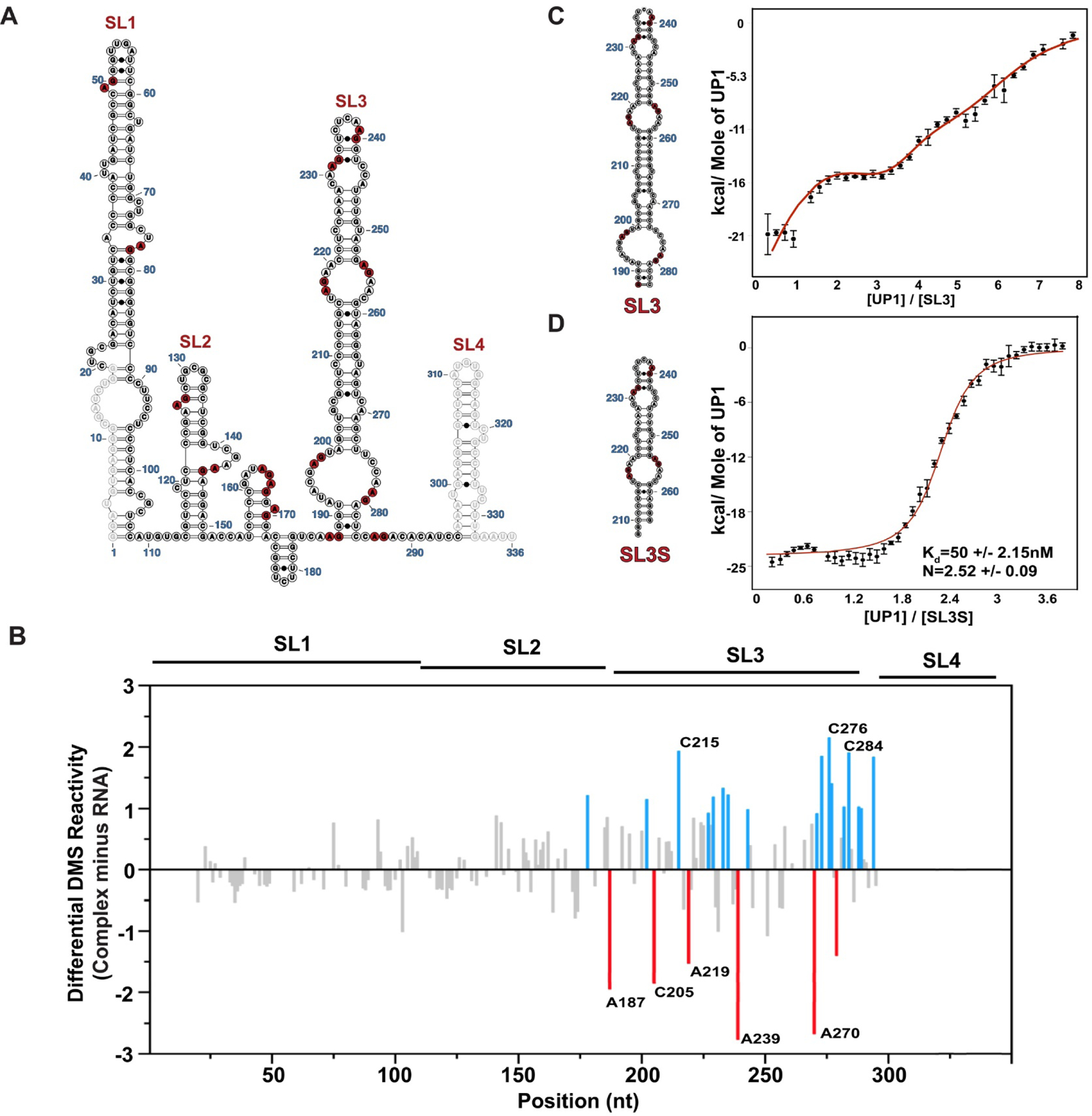
(A) The population average secondary structure of 7SK snRNA with 14 partially single strand high affinity (5’-R/YAG-3’) hnRNP A1/A2 consensus binding motifs depicted in red. (B) The differential DMS-MaPseq signal calculated by subtracting reactivity indices of free 7SK snRNA (100 nM) from that of the UP1–7SK complex with 25 fold excess of UP1 (2.5 μM). Delta indices above and below zero correspond to sites that become more and less reactive, respectively within the complex. The blue and red bars show the delta DMS reactivity indices larger than one standard deviation from the average. Calorimetric titration isotherms of UP1 titrated into (C) SL3 and (D) SL3S ITC data were collected in the buffer condition of 10mM K2HPO4,120mM KCl, 0.5mM EDTA, 1mM TCEP pH 6.5, at 25 °C. The red curves in the ITC isotherms fit to an independent sets of site model as in panel C or a single-site model as in panel D. Error bars on individual data points are automatically calculated in AFFINI meter and correspond to the uncertainty associated to the integral calculation of each peak, including the noise in the baseline and noise in the peak
To identify potential hnRNP A1/A2 binding sites or induced structural changes, we performed stepwise titrations of increasing concentrations of UP1 into 7SK snRNA followed by DMS treatment (Figure S6). Using the structural similarity plots as a metric for saturation, we observed the most significant differences when UP1 was added at a 25-fold molar ratio (Figure S6A). Under this condition, we proceeded to calculate a differential DMS-MaPseq signal by subtracting normalized reactivity indices of free 7SK snRNA from the indices of 7SK snRNA bound in the presence of 25-fold excess UP1 (Figure S6B). Figure 3B shows that the majority of the UP1-induced changes in chemical reactivity localize to SL3 with delta indices both greater and lesser than zero. This observation clearly indicates that UP1 has selectivity for SL3 within the context of the intact 7SK snRNA, in agreement with previous data showing that SL3 is necessary for hnRNP A1/A2 proteins to modulate the 7SK-protein distribution11. The results further suggest that hnRNP A1 makes specific contacts with SL3 since the delta indices of A187, C205, A219 and A239 all decrease within the complex. We interpret a decrease in the delta DMS indices of a given site as becoming more protected in the presence of 25-fold excess UP1. This new “protection” could manifest from direct protein binding or indirectly by induced conformational change; regardless of the mechanism the data are robust. A187 and A239 are both located within single strand 5’-R/YAG-3’ motifs, while C205 and A219 are proximal to similar motifs (Figure 3A). In addition, several sites show an increase in delta indices when UP1 is bound. These sites are adjacent to those that show a decrease in reactivity and likely reflect a local increase in flexibility of the RNA structure. Interestingly, the delta indices from nts 275 to 295 reveal that these residues are globally more reactive when UP1 is bound. This region corresponds to the 3’-side of the large internal loop at the base of SL3 and the linker region that connects to SL4. One straightforward interpretation of these results is that some residual structure exists within this stretch of free 7SK snRNA. Indeed, the reactivity indices of the population-averaged structure are not uniform across this region of the RNA with some “unpaired” nucleotides exhibiting low reactivity (Figure 2A), which is also consistent with results from clustering the DMS reactivities (Figure 2B).
To independently test for specific binding of hnRNP A1 to SL3, we performed calorimetric titrations with UP1 into different SL3 constructs. We first carried out titrations using the entire SL3 region (nts 188–284) to demonstrate that UP1 binds to multiple independent sites along 7SK snRNA with apparent KD values that range from low double-digit nM to single-digit μM (Figure 3C and S7A). This observation is consistent with our differential DMS-MaPseq results that show UP1-dependent protection patterns within multiple and distinct regions along SL3 (Figure 3B). We next performed titrations using a shorter SL3 construct, referred to as SL3S (nts 210–264), which contains four partially single-stranded 5’-R/YAG-3’ motifs. Interestingly, the binding isotherm for the UP1-SL3S titration is monophasic, unlike the complex isotherm observed for UP1-SL3 titration (Figure 3D and S7B). Fitting the UP1-SL3S titration to a simple 1:1 model shows that two or more UP1 proteins bind SL3S with high affinity (KD = 50 +/- 2.2 nM); however, electrophoretic mobility shift assays performed with UP1 and hnRNP A1 reveal that the binding equilibria may be more complicated since complexes of intermediate stoichiometries are visualized on the gel (Figure S8). In order to quantitate the affinity for potential intermediates, we also fit the UP1-SL3S calorimetric titration data to an independent sites (n = 4) model in AFFINImeter to obtain apparent site-specific KD values that ranged from ~40–800 nM. In sum, the collective results presented here clearly demonstrate that hnRNP A1/A2 proteins selectively assemble along the SL3 domain of 7SK snRNA via context dependent mechanisms that include both specific (primary) and non-specific (secondary) interactions.
HnRNP A1/A2 Binding Sites reside within Distinct Non-canonical Regions Along the Surface of SL3
Having established that hnRNP A1/A2 proteins bind selectively to SL3, we proceeded to determine a three-dimensional structural model of this stem loop in order to observe how the UP1-dependent protection patterns map to the surface of this 7SK snRNA domain. 3D structures of SLs 1 and 2 have been reported elsewhere4, 16–18. We first attempted to solve the structure of intact SL3 (nts 188–284); however, the NMR spectra of the imino region (base paired) were broad under several buffer and temperature conditions (not shown). We reasoned that the large internal loop at the base of SL3 may undergo intermediate conformational exchange on the NMR timescale. To that point, cluster analysis of our DMS reactivities show that this internal loop indeed exists in at least two conformations within the context of 7SK snRNA (Figure 2B). By contrast, the SL3M (nts 200–274) construct showed sharp and well-resolved NMR signals within the imino region, which allowed us to proceed with NMR analysis.
We assigned the imino chemical shifts using a divide-and-conquer strategy by collecting 1H-1H NOESY and 1H-15N HSQC data sets for the smaller SL3S construct (Figure S9). According to the DMS-derived secondary structure of 7SK snRNA, SL3S is expected to fold with 16 bps consisting of 5 AU bps, 8 GC bps, and 3 GU bps. Indeed, the 1H-15N HSQC spectra of SL3S reveal a well-dispersed set of correlation peaks and chemical shifts consistent with the expected base pair composition. Moreover, we observed sequential NOE cross peak patterns that allowed partial assignment for each of the expected helical stretches of SL3S, which were cross-validated to the G and U chemical shifts from the 1H-15N HSQC. Overall,13 of the expected 16 base pairs of SL3S were uniquely confirmed by NMR experiments. Next, we collected the 1H-1H NOESY spectrum of SL3M and compared it to SL3S to observe that the majority of the NOE assignments were readily transferable to SL3M (Figure 4A and S10). According to the DMS-derived 7SK snRNA structure, SL3M folds with two additional short helices (bps 200–203:271–274 and 206–208:266–268) relative to SL3S, and indeed we observed additional NOE cross peak patterns and 1H-15N correlation signals consistent with this. We were able to partially assign the additional imino signals by starting with the NOE cross peak patterns and characteristic chemical shifts of the two extra GU wobble base pairs (G200:U274 and G206:U268) of SL3M. For example, G206 H1 gives a medium intensity NOE to U268 H3 and both bases show NOE connectivity patterns to G267 H1 (Figure 4A). Albeit weak, we also observed NOE evidence for the terminal G200:U274 base pair and these signals were verified in the corresponding 1H-15N HSQC spectrum. In addition, we were able to trace a sequential NOE walk from G200 H1 to U203 H3 thereby confirming the lower helix of SL3M. Altogether, we were able to assign ~90% of the base pairs of SL3M using this divide-and-conquer approach and to validate that central region of SL3 folds into a phylogenetically conserved structure.
Figure 4. The Apical Loop of SL3 Folds into a Stable 3D Structure to Position HnRNP A1/A2 Binding Surfaces.
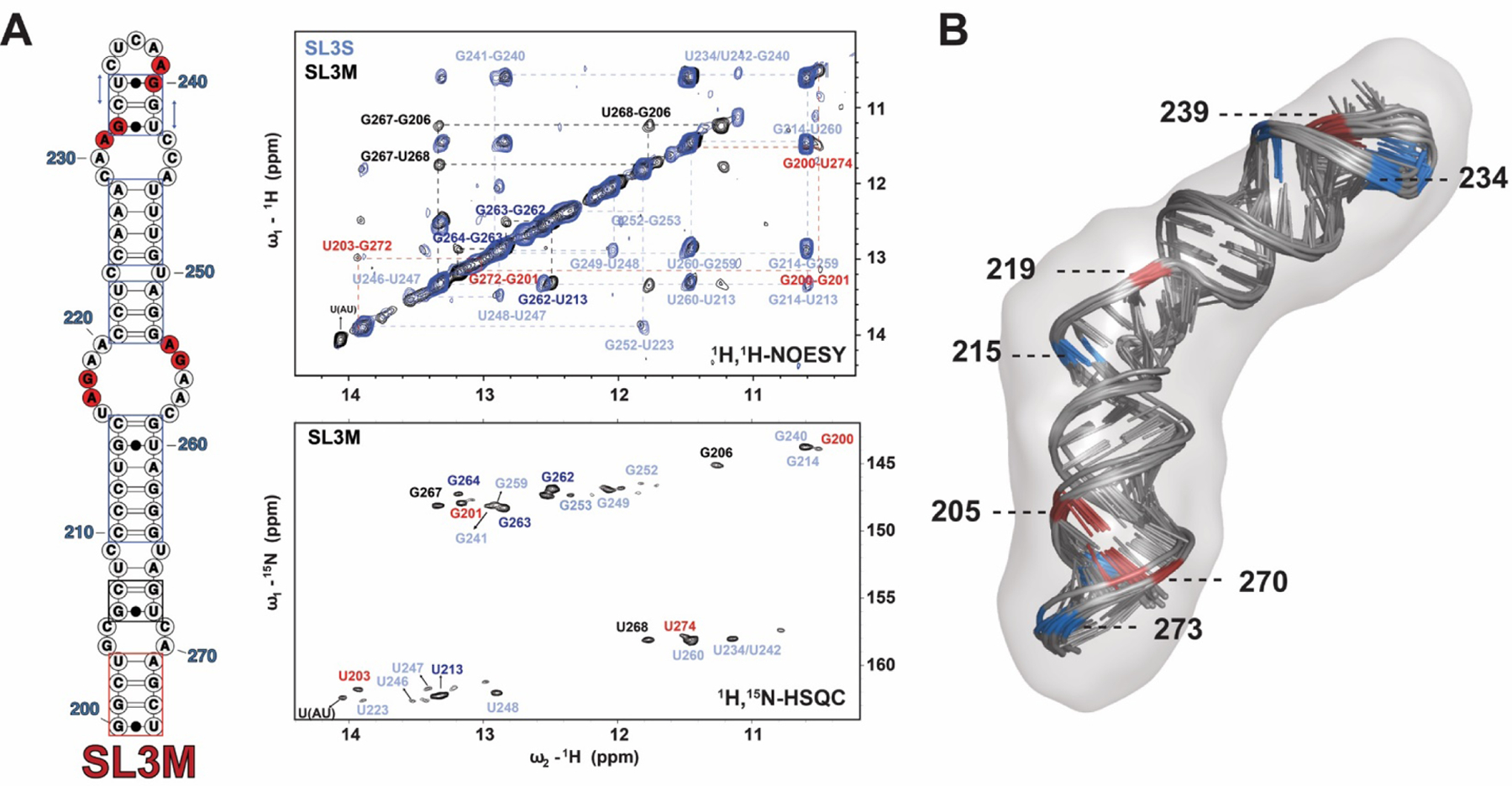
(A) Left, secondary structure of the isolated SL3M construct (75-nt) used to verify that this element is an independently folded 7SK snRNA domain. In this construct, A200 was mutated to G200 to form 5’-GG- for in vitro T7 transcription. Middle upper,1H-1H2O NOESY spectrum of SL3M collected in the buffer condition of 10 mM K2HPO4, 50 mM KCl, pH 5.5 and 10% D2O, at 283K. The vertical and horizontal dashed lines trace the NOE stacking pattern for each stable helical region demonstrating that SL3M is an independently folded domain. Middle lower, 1H-15N HSQC spectrum of SL3M collected in the buffer condition of 10 mM K2HPO4, 50 mM KCl, pH 5.5 and 10% D2O, at 283K confirms the1H-1H H2O NOESY assignments. The assignments in spectrum with light blue color are transferred from SL3S assignment and those one with dark blue are from the middle stem of SL3M. (B) Ten lowest energy structures selected from Farfar2 RNA prediction algorithm were further refined in AMBER. After RDC (16 1H-15N) refinement, the ten 10 lowest energy structures were aligned to the SAXS envelope data . Residues were color coded in red and blue according to their delta DMS reactivity indices (Figure 3D) to indicate there approximate stereochemical environments.
Because we used the DMS-derived structure of 7SK snRNA to design the SL3 constructs, we were able to compare the NMR-determined hydrogen bonding patterns (G/U nts) of SL3M to the DMS-derived reactivity indices (A/C nts). Figure S11 shows that this comparison provides compelling evidence that these independent techniques for probing RNA structure are highly complementary and validate that the central region of SL3 indeed adopts a well-folded structure. Furthermore, the SAXS-derived Kratky profile of free SL3M has the characteristic inverted bell shape further supporting its overall conformation (Figure S12A).
Building on these observations, we proceeded to determine a hybrid NMR-SAXS 3D structural model of SL3M. Ten thousand de novo SL3M structures were generated in ROSETTA40 using bp restraints jointly derived from DMS-MaPseq and NMR. From this large ensemble, 10 models with best fit (lowest χ2) with SAXS envelope by Crysol were selected for further refinement in AMBER47 against A-form distance restraints for all stable base-paired regions (as determined by DMS and NMR), sparse (16) 1H-15N Residual Dipolar Couplings (RDCs) for N3H3 atoms in the Uridine and N1H1 atoms in the Guanine, and a SAXS molecular envelope, included during different stages of the simulation (full details described in Methods). Figure 4B shows that the final ensemble of 10 lowest energy AMBER refined structures (RMSD = 1.34 Å) were generated with good agreement with the experimental RDCs (R2=0.84, Figure S13), and that the AMBER refinement improved the correlation when compared to the starting ROSETTA models (R2=0.53) 1.34 Å. Figure 4B shows the ten lowest energy SL3M models superposed into the SAXS molecular reconstruction. Inspection of these structures reveal that all four of the minimal (5’-R/YAG-3’) hnRNP A1/A2 binding sites localize to distinct structural environments (apical and internal loops) positioned along the entire length of SL3M. The model also reveals that those residues with positive delta reactivity indices are spatially nearby sites with negative indices, which overlap consensus hnRNP A1/A2 binding sites (Figure 3B and 4B). Based on this observation, we posit that the different structural environments determine the accessibility of the otherwise degenerate 5’- R/YAG-3’ motifs and positions them to direct the functional assembly of hnRNP A1/A2– 7SK snRNA complexes.
Biophysical Description of the HnRNP A1–7SK snRNA Complex
To gain insights into the binding mode of hnRNP A1/A2 proteins on 7SK snRNA, we first characterized complexes of UP1 and SL3M by SEC-MALS-SAXS as previously described43, 50. The 2D structure of SL3M reveals four possible binding sites and the measured stoichiometry with SL3S (which contains all 4 sites) indicates that two or more UP1 proteins bind with comparable affinities (Figure 3D). The Gunnier plots of the SEC-MALS-SAXS resolved UP1-SL3M complexes are of high-quality up to a 2:1 stoichiometric ratio and reveal that the overall radius of gyration increases from ~32 Å for free SL3M to ~38 Å for the 2:1 UP1-SL3M complex (Figure S12B). By comparison, the Guinier plot for the 4:1 UP1-SL3M complex is of lower quality, likely a result of partial aggregation as the complex travels along the SEC column. Since the MALS setup is in-line with the SEC column, we were able to estimate molecular masses of the samples prior to exposing to the x-ray beam. Figure S14 shows that the experimentally determined molecular mass for free SL3M (~25 kDa) is in excellent agreement with its theoretical value (23 kDa) and that the molecular masses linearly increase for its 1:1, 2:1 and the 4:1 UP1-SL3M complexes. The experimental molecular masses for the 1:1 and 2:1 complexes are ~20–30% larger than the expected values indicating mixed stoichiometric heterogeneity within these samples. Despite the reduced SAXS quality of the 4:1 complex, the molecular mass as estimated by MALS (~113 kDa) is in good agreement with its theoretical value (118 kDa).
Inspection of the P(r) pair distribution functions for SL3M, and its resolved 1:1 and 2:1 UP1 complexes reveal how binding of UP1 changes the gross structural features (shape and size) of SL3M while preserving a true 3D architecture as reflected in ab initio molecular reconstructions for free SL3M, its 1:1 and its 2:1 UP1 complexes (Figure 5B). Consistent with the Guinier analysis, the P(r) pair distribution function shows a gradual increase in the radius of gyration and maximum length (Dmax = 119–158 Å) of SL3M when titrated with increasing amounts of UP1 (Figure 5A and Table 1). When considered together, these data provide solid evidence that at least four hnRNP A1/A2 proteins assemble onto the upper surface of SL3 by forming specific interactions within the context of a folded RNA structure.
Figure 5. HnRNP A1/A2 Proteins Bind Stably to the Upper Surface of SL3 to Form a Specific Complex.
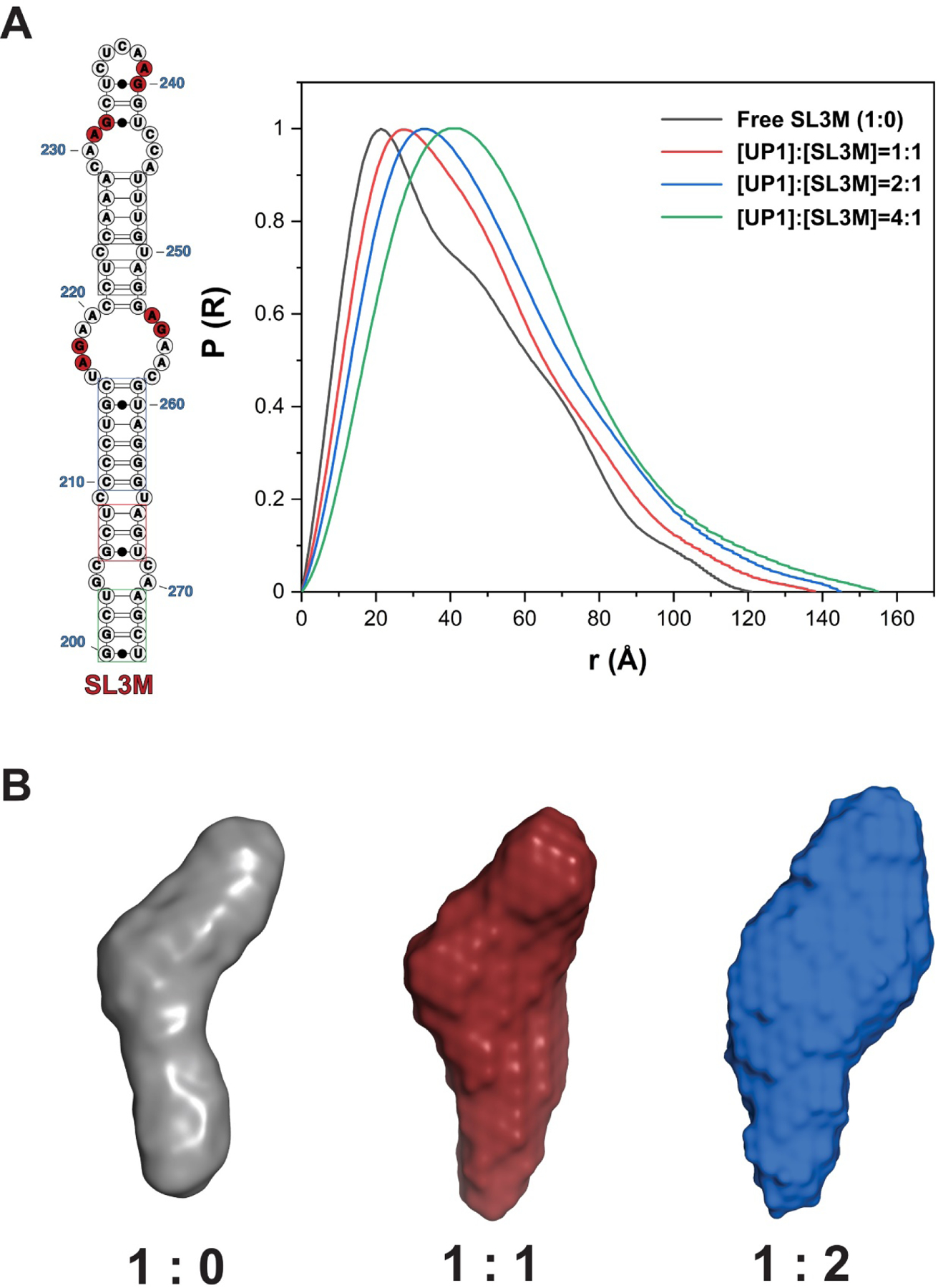
(A) Comparison of the pair distance distribution P(r) functions of free SL3M (Black) and stoichiometric 1:1 (Red), 2:1 (Blue), and 4:1 (Green) UP1-SL3M complexes resolved by inline SEC-MALS-SAXS reveal that hnRNP A1/A2 proteins interact stably with the upper surface of SL3. (B) Comparison of the SAXS molecular reconstructions for free SL3M (Purple), its stoichiometric 1:1 (Red) and 2:1 (Blue) UP1-SL3M complexes.
Table 1.
Physical Parameters of UP1-SL3M Complexes Derived from SEC-MALS-SAXS
| Rg (Å) | Dmax (Å) | MW (exp/the, kDa) | |
|---|---|---|---|
| Free SL3M | 32.1±0.1 | 118.7 | 23.11/25 |
| [UP1]:[SL3M]=1:1 | 34.6±0.1 | 137.1 | 45.53/60 |
| [UP1]:[SL3M]=2:1 | 38.4±0.5 | 145.2 | 70.59/82 |
| [UP1]:[SL3M]=4:1 | 40.94±0.16 | 158.2 | 115.3/113 |
Rg here were derived from Guinier analysis and the data was used with sRg limit smaller than 1.3.
Experimental molecular weights derived from MALS.
To better understand the nature by which hnRNP A1 binds SL3, we performed 1H-15N HSQC titrations of unlabeled UP1 into a 15N(G/U)-labeled SL3S construct (contains all four binding sites). Figure S15A shows the HSQC of the 15N(G/U) correlation signals (imino region) of free SL3S overlaid with the signals of a 1.25:1 UP1 complex. At this molar ratio, we observe evidence of a specific UP1-SL3S complex since the signals of U234/U242 iminos broaden beyond detection and the intensity of the signal of G252 is significantly attenuated. Of interest, U234 and U242 are proximal to A239, which showed the most “protection” (negative index) in the differential DMS-MaPseq titration, and G252 is adjacent to the large internal loop that contains a “protected” A219 (Figure 3B). We observed complete loss of all signals at higher molar ratios, likely due to the large size (~65 kDa) of the 1:2 complex. We next mapped the SL3 binding interface on hnRNP A1 by titrating RNA into a 15N-labeled UP1 construct (Figure S15B). The titration shows broadening of a subset of 1H-15N correlation signals at low molar ratios of RNA to protein, with near complete loss of signals with 2-fold excess of SL3S is titrated (Figure S15B). These results are compatible with there being multiple hnRNP A1/A2 binding sites on SL3S.
Discussion
Human 7SK snRNA regulates transcription in part by storing P-TEFb within an inhibitory complex that also includes HEXIM1/2, MePCE and LARP7. Once released by cellular or viral factors, 7SK snRNA binds hnRNP proteins to block the back association of P-TEFb. In HeLa cells, only a fraction of the nuclear pool of 7SK snRNA stably associates with P-TEFb, with the remainder being bound by other RNA binding proteins, including members from the hnRNP family. The mechanisms by which hnRNPs assemble onto 7SK snRNA to regulate its RNA biology and P-TEFb availability are not well understood; however, deletion studies indicated that the SL3 domain is necessary for hnRNP A1/A2 proteins to dynamically modulate the extent of cyclin T1 bound to endogenously expressed 7SK snRNA.
In this study, we addressed two broadly unanswered questions that pertain to the structural biochemistry of complexes formed between 7SK snRNA and hnRNP A1/A2 proteins: by what mechanism(s) do hnRNP A1/A2 proteins assemble onto 7SK snRNA, and what are the nature of the interactions within the 7SK-hnRNP A1/A2 complex? We first revisited the secondary structure of full-length 7SK by applying the relatively new DMS-MaPseq method26 to reveal that free 7SK snRNA adopts at least two stable conformations with structural similarities preserved in both conformers at SL1 and the majority of SL3 (Figure 2A and 2B). SL4 is also folded in our model but its structure is primarily driven by thermodynamics. By contrast, the SL2 region and part of SL3 are structurally different within each conformer. It is believed that 7SK snRNA cycles through different structures within its unique RNP complexes such that protein-induced conformational changes regulate P-TEFb availability2, 12. Our results show that in the absence of protein partners, in vitro transcribed 7SK snRNA adopts more than one stable conformation. If present within the cellular environment, these intrinsically distinct 7SK snRNA conformers might function as adaptable scaffolds to assemble unique RNP complexes to regulate 7SK snRNA biology. Along those lines, no proteins have been reported to bind the SL2 region of 7SK snRNA, and our results indicate that this region is the least phylogenetically conserved and it forms alternating SL structures that present potentially species-specific binding surfaces for cognate proteins. Alternatively, the 7SK snRNA conformers might have different molecular shapes (due to the differential numbers of SLs) that modulate protein-RNA or RNA-RNA interactions. Either of these possibilities allows for regulatory control of 7SK snRNA biology by mechanisms that include changes in its protein composition or subcellular localization as observed in complexes with hnRNP R21.
Our results revealed that hnRNP A1/A2 proteins bind selectively to the SL3 domain of 7SK snRNA by making specific and non-specific contacts that we posit are tuned by the surrounding structural context of each minimal 5’-R/YAG-3’ motif. The differential DMS-MapSeq titration further showed that hnRNP A1/A2 binding induces local changes to the SL3 structure that increases flexibility of the nucleotides mostly 3’ to the binding site (Figure 3B). Calorimetric titrations of the UP1 domain into different SL3 constructs indicate the interactions are driven by favorable changes in enthalpy and the affinities span two orders of magnitude; however, we only observed a monophasic isotherm using the SL3S construct, which contains four possible hnRNP A1/A2 binding sites (Figure 3C and 3D). Two of the four most “protected” adenosines (A219 and A239) in the differential DMS-MaPseq experiment are located within the upper apical loop region of SL3. SEC-MALS-SAXS studies showed that up to four UP1 molecules stably associate with the apical loop surface of SL3M (nts 200–274). These collective observations lead to a model whereby the SL3 domain functions as a scaffold to assemble multiple hnRNP A1/A2 molecules through binding surfaces that are specific and non-specific (Figure 6). The binding events increase flexibility in a 3’ direction which further relaxes residual structure to open weaker hnRNP A1/A2 binding sites. Such complex binding behavior might be generalized as being promiscuous, but it likely represents mechanisms by which genetically-encoded RNA structural dynamics promote assembly of hnRNPs with unique compositions and architectures that in turn provide additional regulation of 7SK snRNA biology. Our results suggest a specific role for SL3 to function as a scaffold that positions protein-binding surfaces to assemble unique RNP complexes. The surrounding stereochemical context of a given surface can then be tuned by local physicochemical features, including dynamics. A notable caveat is that the UP1 domain lacks the C-terminal intrinsically disordered region of full-length hnRNP A1/A2 proteins, which is known to mediate protein- protein interactions. It is thus plausible that hnRNP-7SK snRNA assembly is further modulated through biochemical cooperativity. Finally, this work also highlights the importance of integrative approaches to reveal mechanisms of dynamic RNA-protein complexes that must adapt over time to respond to the ever-changing cellular environment. We posit that these types of dynamic interactions are a manifestation of genetically encoded instructions to assemble unique RNA-protein complexes.
Figure 6. A Conceptual Model to Interpret hnRNP A1/A2–7SK Assembly.
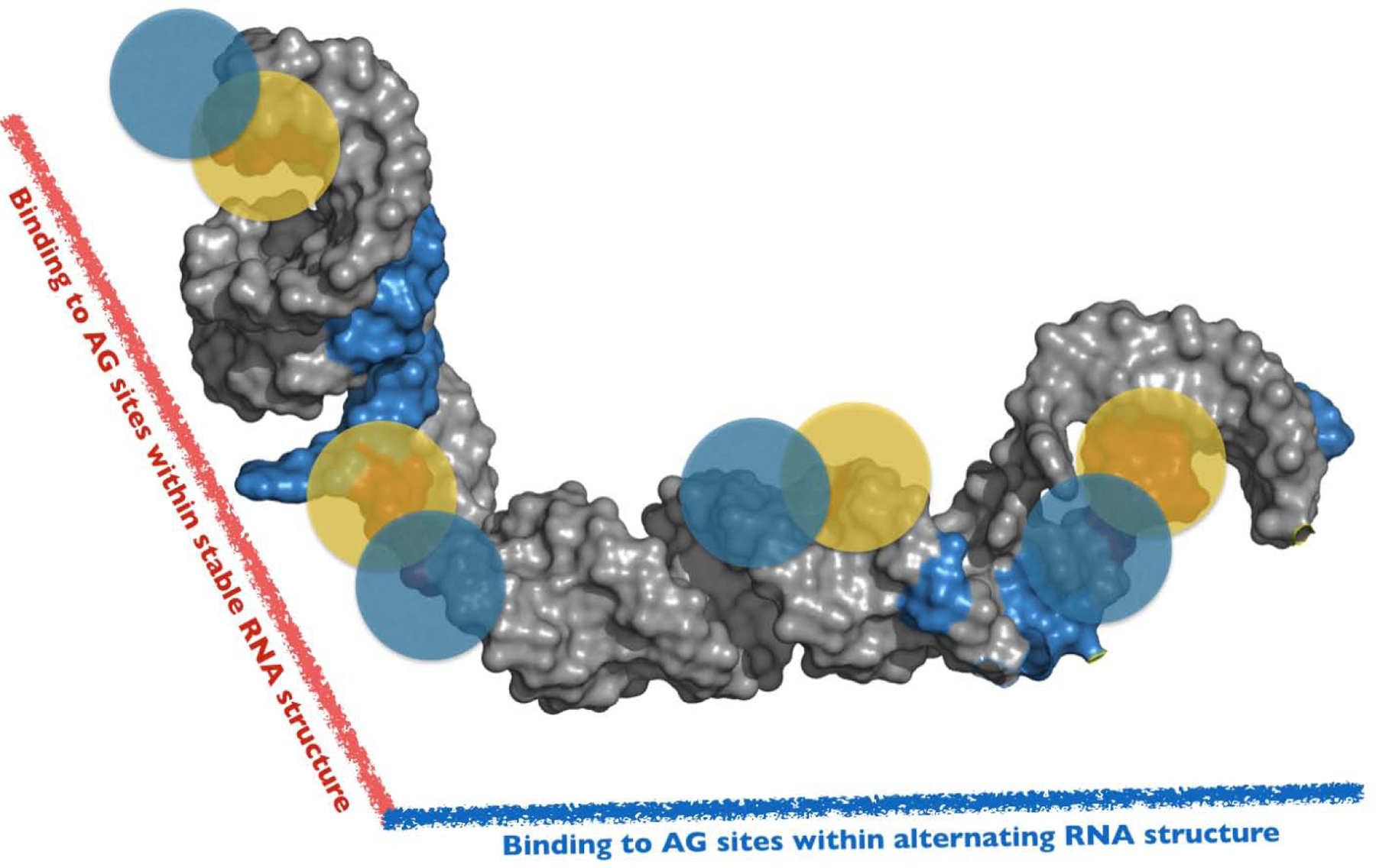
A 3D conceptual framework to interpret the complexities by which hnRNP A1/A2 proteins assemble along the SL3 domain of 7SK snRNA. The model is consistent with independent data sets derived from DMS-MaPseq, NMR, Calorimetry and SEC-SAXS measurements. The model posits that hnRNP A1/A2 proteins bind to specific 5’-Y/RAG-3’ sites positioned within distinct structural environments along the entire surface of SL3. Accordingly, the physicochemical properties of these local sites differentially modulate hnRNP A1/A2 interactions to form a functional 3D complex. The full-length SL3 (nts 188–184) structure shown here was determined using the FARFAR2 module of Rosetta 3.11 and (note this is not the experimentally-determined structural model of the shorter SL3M construct). Blue and Yellow circles represent the UP1 domain of hnRNP A1.
Supplementary Material
Highlights.
The secondary structure of 7SK snRNA was probed using DMS-MaPseq to reveal the RNA adopts two alternate conformers with differences localized to the SL2–3 linker and the base of SL3
Differential DMS-MaPseq probing demonstrates that hnRNP A1/A2 proteins bind selectively to the SL3 domain of 7SK snRNA to induce local structural changes 3’ to distinct binding sites.
The 3D structure of SL3 shows that the upper apical loop folds to position multiple hnRNP A1/A2 consensus (5’-R/YAG-3’) binding motifs within different stereochemical environments.
Biophysical characterizations provide evidence that up to four hnRNP A1/A2 proteins bind with high affinity and specificity through the upper surface of SL3.
A 3D conceptual model is proposed to interpret complex binding mechanisms by which hnRNP A1/A2 proteins assemble onto 7SK snRNA.
Acknowledgements
This work was funded by National Institutes of Health grants U54AI50470 (the Center for HIV RNA Studies, SR) and R01AI150830 (BST). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02–06CH11357. This project was supported by grant 9 P41 GM103622 from the National Institute of General Medical Sciences of the National Institutes of Health. Use of the Pilatus 3 1M detector was provided by grant1S10OD018090–01 from NIGMS.
The authors would like to thank the BioCAT (Beamline 18-ID) scientist, Srinivas Chakravarthy, for assistance with analyzing MALS. The authors would like to also thank Joseph Yesselman for helpful discussions with using Rosetta FARFAR and Andrea Berman for intellectual contributions and reading the manuscript. This study also made use of the Campus Chemical Instrument Center NMR facility at the Ohio State University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
BST – conceptualization, funding acquisition, project management and drafting the manuscript
LL, LYC, AS – Data curation, formal analysis, methodology, and drafting the manuscript SR - Resources
Reference
- 1.Zhou Q; Li T; Price DH, RNA polymerase II elongation control. Annu Rev Biochem 2012, 81, 119–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brogie JE; Price DH, Reconstitution of a functional 7SK snRNP. Nucleic Acids Res 2017, 45 (11), 6864–6880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eichhorn CD; Yang Y; Repeta L; Feigon J, Structural basis for recognition of human 7SK long noncoding RNA by the La-related protein Larp7. Proc Natl Acad Sci U S A 2018, 115 (28), E6457–E6466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eichhorn CD; Chug R; Feigon J, hLARP7 C-terminal domain contains an xRRM that binds the 3’ hairpin of 7SK RNA. Nucleic Acids Res 2016, 44 (20), 9977–9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.C Quaresma AJ; Bugai A; Barboric M, Cracking the control of RNA polymerase II elongation by 7SK snRNP and P-TEFb. Nucleic Acids Res 2016, 44 (16), 7527–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Egloff S; Van Herreweghe E; Kiss T, Regulation of polymerase II transcription by 7SK snRNA: two distinct RNA elements direct P-TEFb and HEXIM1 binding. Mol Cell Biol 2006, 26 (2), 630–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barboric M; Kohoutek J; Price JP; Blazek D; Price DH; Peterlin BM, Interplay between 7SK snRNA and oppositely charged regions in HEXIM1 direct the inhibition of P-TEFb. EMBO J 2005, 24 (24), 4291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Molle D; Maiuri P; Boireau S; Bertrand E; Knezevich A; Marcello A; Basyuk E, A real-time view of the TAR:Tat:P-TEFb complex at HIV-1 transcription sites. Retrovirology 2007, 4, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Michels AA; Fraldi A; Li Q; Adamson TE; Bonnet F; Nguyen VT; Sedore SC; Price JP; Price DH; Lania L; Bensaude O, Binding of the 7SK snRNA turns the HEXIM1 protein into a P-TEFb (CDK9/cyclin T) inhibitor. EMBO J 2004, 23 (13), 2608–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barrandon C; Bonnet F; Nguyen VT; Labas V; Bensaude O, The transcription-dependent dissociation of P-TEFb-HEXIM1–7SK RNA relies upon formation of hnRNP-7SK RNA complexes. Mol Cell Biol 2007, 27 (20), 6996–7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Herreweghe E; Egloff S; Goiffon I; Jady BE; Froment C; Monsarrat B; Kiss T, Dynamic remodelling of human 7SK snRNP controls the nuclear level of active P-TEFb. EMBO J 2007, 26 (15), 3570–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krueger BJ; Varzavand K; Cooper JJ; Price DH, The mechanism of release of P-TEFb and HEXIM1 from the 7SK snRNP by viral and cellular activators includes a conformational change in 7SK. PLoS One 2010, 5 (8), e12335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peterlin BM; Brogie JE; Price DH, 7SK snRNA: a noncoding RNA that plays a major role in regulating eukaryotic transcription. Wiley Interdiscip Rev RNA 2012, 3 (1), 92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wassarman DA; Steitz JA, Structural analyses of the 7SK ribonucleoprotein (RNP), the most abundant human small RNP of unknown function. Mol Cell Biol 1991, 11 (7), 3432–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flynn RA; Do BT; Rubin AJ; Calo E; Lee B; Kuchelmeister H; Rale M; Chu C; Kool ET; Wysocka J; Khavari PA; Chang HY, 7SK-BAF axis controls pervasive transcription at enhancers. Nat Struct Mol Biol 2016, 23 (3), 231–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bourbigot S; Dock-Bregeon AC; Eberling P; Coutant J; Kieffer B; Lebars I, Solution structure of the 5’-terminal hairpin of the 7SK small nuclear RNA. RNA 2016, 22 (12), 1844–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pham VV; Salguero C; Khan SN; Meagher JL; Brown WC; Humbert N; de Rocquigny H; Smith JL; D’Souza VM, HIV-1 Tat interactions with cellular 7SK and viral TAR RNAs identifies dual structural mimicry. Nat Commun 2018, 9 (1), 4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martinez-Zapien D; Legrand P; McEwen AG; Proux F; Cragnolini T; Pasquali S; Dock-Bregeon AC, The crystal structure of the 5΄ functional domain of the transcription riboregulator 7SK. Nucleic Acids Res 2017, 45 (6), 3568–3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muniz L; Egloff S; Ughy B; Jády BE; Kiss T, Controlling cellular P-TEFb activity by the HIV-1 transcriptional transactivator Tat. PLoS Pathog 2010, 6 (10), e1001152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.D’Orso I; Jang GM; Pastuszak AW; Faust TB; Quezada E; Booth DS; Frankel AD, Transition step during assembly of HIV Tat:P-TEFb transcription complexes and transfer to TAR RNA. Mol Cell Biol 2012, 32 (23), 4780–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Briese M; Saal-Bauernschubert L; Ji C; Moradi M; Ghanawi H; Uhl M; Appenzeller S; Backofen R; Sendtner M, hnRNP R and its main interactor, the noncoding RNA 7SK, coregulate the axonal transcriptome of motoneurons. Proc Natl Acad Sci U S A 2018, 115 (12), E2859–E2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chillon I; Marcia M; Legiewicz M; Liu F; Somarowthu S; Pyle AM, Native Purification and Analysis of Long RNAs. Methods Enzymol 2015, 558, 3–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levengood JD; Rollins C; Mishler CH; Johnson CA; Miner G; Rajan P; Znosko BM; Tolbert BS, Solution structure of the HIV-1 exon splicing silencer 3. J Mol Biol 2012, 415 (4), 680–98. [DOI] [PubMed] [Google Scholar]
- 24.Davila-Calderon J; Patwardhan NN; Chiu LY; Sugarman A; Cai Z; Penutmutchu SR; Li ML; Brewer G; Hargrove AE; Tolbert BS, IRES-targeting small molecule inhibits enterovirus 71 replication via allosteric stabilization of a ternary complex. Nat Commun 2020, 11 (1), 4775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tomezsko P; Swaminathan H; Rouskin S, Viral RNA structure analysis using DMS-MaPseq. Methods 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tomezsko PJ; Corbin VDA; Gupta P; Swaminathan H; Glasgow M; Persad S; Edwards MD; Mcintosh L; Papenfuss AT; Emery A; Swanstrom R; Zang T; Lan TCT; Bieniasz P; Kuritzkes DR; Tsibris A; Rouskin S, Determination of RNA structural diversity and its role in HIV-1 RNA splicing. Nature 2020, 582 (7812), 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bellaousov S; Reuter JS; Seetin MG; Mathews DH, RNAstructure: Web servers for RNA secondary structure prediction and analysis. Nucleic Acids Res 2013, 41 (Web Server issue), W471–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Delaglio F; Grzesiek S; Vuister GW; Zhu G; Pfeifer J; Bax A, NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 1995, 6 (3), 277–93. [DOI] [PubMed] [Google Scholar]
- 29.Johnson BA; Blevins RA, NMR View: A computer program for the visualization and analysis of NMR data. J Biomol NMR 1994, 4 (5), 603–14. [DOI] [PubMed] [Google Scholar]
- 30.Lee W; Tonelli M; Markley JL, NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 2015, 31 (8), 1325–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takor G; Morgan CE; Chiu L-Y; Kendrick N; Clark E; Jaiswal R; Tolbert BS, Introducing Structure–Energy Concepts of RNA at the Undergraduate Level: Nearest Neighbor Thermodynamics and NMR Spectroscopy of a GAGA Tetraloop. Journal of Chemical Education 2020. [Google Scholar]
- 32.Fürtig B; Richter C; Wöhnert J; Schwalbe H, NMR spectroscopy of RNA. Chembiochem 2003, 4 (10), 936–62. [DOI] [PubMed] [Google Scholar]
- 33.Kontaxis G; Clore GM; Bax A, Evaluation of cross-correlation effects and measurement of one-bond couplings in proteins with short transverse relaxation times. J Magn Reson 2000, 143 (1), 184–96. [DOI] [PubMed] [Google Scholar]
- 34.Burnouf D; Ennifar E; Guedich S; Puffer B; Hoffmann G; Bec G; Disdier F; Baltzinger M; Dumas P, kinITC: a new method for obtaining joint thermodynamic and kinetic data by isothermal titration calorimetry. J Am Chem Soc 2012, 134 (1), 559–65. [DOI] [PubMed] [Google Scholar]
- 35.Piñeiro Á; Muñoz E; Sabín J; Costas M; Bastos M; Velázquez-Campoy A; Garrido PF; Dumas P; Ennifar E; García-Río L; Rial J; Pérez D; Fraga P; Rodríguez A; Cotelo C, AFFINImeter: A software to analyze molecular recognition processes from experimental data. Anal Biochem 2019, 577, 117–134. [DOI] [PubMed] [Google Scholar]
- 36.Penumutchu SR; Chiu LY; Meagher JL; Hansen AL; Stuckey JA; Tolbert BS, Differential Conformational Dynamics Encoded by the Linker between Quasi RNA Recognition Motifs of Heterogeneous Nuclear Ribonucleoprotein H. J Am Chem Soc 2018, 140 (37), 11661–11673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Franke D; Petoukhov MV; Konarev PV; Panjkovich A; Tuukkanen A; Mertens HDT; Kikhney AG; Hajizadeh NR; Franklin JM; Jeffries CM; Svergun DI, ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J Appl Crystallogr 2017, 50 (Pt 4), 1212–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu X; Subramaniam S; Case DA; Wu KW; Brooks BR, Targeted conformational search with map-restrained self-guided Langevin dynamics: application to flexible fitting into electron microscopic density maps. J Struct Biol 2013, 183 (3), 429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Svergun D; Barberato C; Koch MHJ, CRYSOL– a Program to Evaluate X-ray Solution Scattering of Biological Macromolecules from Atomic Coordinates. Journal of Applied Crystallography 1995, 28 (6), 768–773. [Google Scholar]
- 40.Das R; Karanicolas J; Baker D, Atomic accuracy in predicting and designing noncanonical RNA structure. Nat Methods 2010, 7 (4), 291–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pérez A; Marchán I; Svozil D; Sponer J; Cheatham TE; Laughton CA; Orozco M, Refinement of the AMBER force field for nucleic acids: improving the description of alpha/gamma conformers. Biophys J 2007, 92 (11), 3817–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zgarbová M; Otyepka M; Sponer J; Mládek A; Banáš P; Cheatham TE; Jurečka P, Refinement of the Cornell et al. Nucleic Acids Force Field Based on Reference Quantum Chemical Calculations of Glycosidic Torsion Profiles. J Chem Theory Comput 2011, 7 (9), 2886–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tolbert M; Morgan CE; Pollum M; Crespo-Hernandez CE; Li ML; Brewer G; Tolbert BS, HnRNP A1 Alters the Structure of a Conserved Enterovirus IRES Domain to Stimulate Viral Translation. J Mol Biol 2017, 429 (19), 2841–2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roe DR; Cheatham TE, PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J Chem Theory Comput 2013, 9 (7), 3084–95. [DOI] [PubMed] [Google Scholar]
- 45.Tsui V; Zhu L; Huang TH; Wright PE; Case DA, Assessment of zinc finger orientations by residual dipolar coupling constants. J Biomol NMR 2000, 16 (1), 9–21. [DOI] [PubMed] [Google Scholar]
- 46.Jahnen-Dechent W; Ketteler M, Magnesium basics. Clin Kidney J 2012, 5 (Suppl 1), i3–i14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jain N; Lin HC; Morgan CE; Harris ME; Tolbert BS, Rules of RNA specificity of hnRNP A1 revealed by global and quantitative analysis of its affinity distribution. Proc Natl Acad Sci U S A 2017, 114 (9), 2206–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Levengood JD; Tolbert BS, Idiosyncrasies of hnRNP A1-RNA recognition: Can binding mode influence function. Semin Cell Dev Biol 2019, 86, 150–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jean-Philippe J; Paz S; Caputi M, hnRNP A1: the Swiss army knife of gene expression. Int J Mol Sci 2013, 14 (9), 18999–9024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morgan CE; Meagher JL; Levengood JD; Delproposto J; Rollins C; Stuckey JA; Tolbert BS, The First Crystal Structure of the UP1 Domain of hnRNP A1 Bound to RNA Reveals a New Look for an Old RNA Binding Protein. J Mol Biol 2015, 427 (20), 3241–3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.