

Abstract
Introduction:
The transcription factor IKAROS and IKAROS family members are critical for the development of lymphocyte and other blood cell lineages. Germline heterozygous IKZF1 mutations have been described in primary immunodeficiency as well as in human hematologic malignancies, affecting both B and T cells. Depending on the allelic variants of IKZF1 mutations (haploinsufficiency and dominant negative) clinical phenotypes vary from bacterial, viral, or fungal infection to autoimmune disease and malignancy.
Areas covered:
In this review, the authors provide an overview of genotype-phenotype correlation and clinical manifestations in patients with IKZF1 mutations. The importance of accurate diagnosis and monitoring immunological changes are also discussed for the management of these complex and rare diseases. IKZF1/IKAROS, immunodeficiency, and CVID were used as the search terms in PubMed and Google Scholar.
Expert opinion:
Over the past 5 years both genetic and molecular studies have unveiled surprisingly diverse roles of IKZF1 mutations in primary immunodeficiency. While an increasing number of novel IKZF1 variants are being reported, limited and complex laboratory testing is necessary to verify the mutation’s pathogenicity. Therefore, the combination of understanding mechanistic concepts and clinical and immunological follow-up is necessary to increase our knowledge of IKAROS-associated diseases.
Keywords: IKZF1, IKAROS, primary immunodeficiency, common variable immunodeficiency (CVID), combined immunodeficiency (CID), autoimmunity, malignancy
1. Introduction
Ikaros, encoded by Ikzf1 and first studied in mouse models, is the founding member of a family of zinc finger transcription factors (Ikaros or Ikzf1, Helios or Ikzf2, Aiolos or Ikzf3, Eos or Ikzf4, and Pegasus or Ikzf5). [1–7]. Ikaros is a hematopoietic-specific transcription factor involved in the regulation of lymphocyte and other cell lineages including myeloid, megakaryocyte, and erythroid differentiation [8–13]. Several Ikaros isoforms are generated through alternative splicing. Full-length Ikaros (isoform 1) has six highly conserved C2H2 zinc-finger domains, four at N-terminus involved in DNA binding and two at C-terminus required for dimerization between Ikaros isoforms (homodimerization) or with other Ikaros family members (heterodimerization)[14]. The DNA binding and dimerization domains are the main drivers of Ikaros functions. Ikaros has been shown to regulate gene expression during T- and B-cell differentiation through association with transcriptional complexes or transcription factors including the nucleosome remodeling and deacetylase (NuRD) complex, signal transducer and activator of transcription (STAT) family members as well as interferon regulatory factor 4 (IRF4), among others[15–17]. Mouse models and human studies have revealed that decreased IKAROS protein expression or function leads to profound lymphocyte differentiation defects, as well as leukemic transformation of T and/or B cell precursors [10,18–21]. Recently, several studies have demonstrated that germline heterozygous IKZF1 mutations are associated with primary immunodeficiencies (PID)/inborn errors of immunity (IEI) [22–38].
In this review, we will overview the recent findings of human germline IKZF1 mutations in PID/IEI and discuss the mechanisms underlying phenotype heterogeneity, clinical manifestations, and laboratory findings.
Literature search was performed using PubMed and Google Scholar with no date limits and last updated in December 2020. The search keywords ‘IKZF1 or IKAROS’ and/or ‘immunodeficiency’ or ‘CVID’ were used. Reference lists of relevant articles were also reviewed.
2. IKZF1 mutations biology, genotypes and phenotypes
To date, more than 30 different germline heterozygous IKZF1 mutations have been reported in patients with PID/IEI (Figure 1A) [22–38]. Among these variants, almost half are located within ZF2 and ZF3 which mediate direct interaction with the specific IKAROS DNA binding sequence (GGGAA)[14]. Other mutations map to the C-terminal region, harboring the dimerization domain. In terms of protein expression, most patients carrying missense mutations affecting either the DNA-binding or dimerization domains express comparable levels of IKAROS protein in lymphocytes, whereas patients carrying an early N-terminal stop codon or large deletions encompassing the entire IKZF1 gene express ~half of wild type (WT) IKAROS protein compared to healthy controls (i.e., S46fs and large 4.7Mb)[24,31]. A similar situation applies when a stop codon renders the protein devoid of the dimerization domain-containing C-terminus region and an IKAROS C-terminus-specific antibody is used for protein detection [22].
Figure 1.
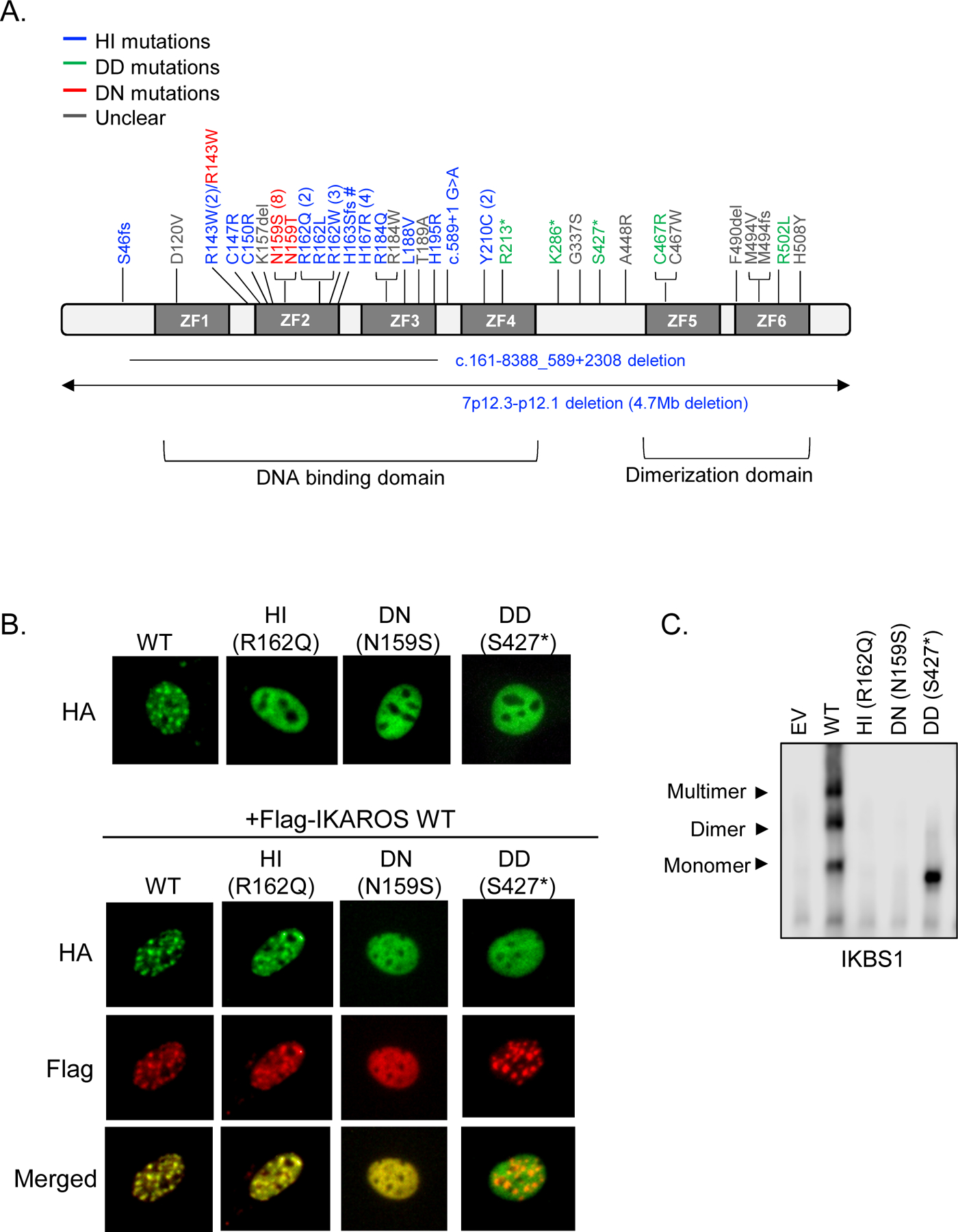
Functional assessments for IKZF1 allelic variants. (A) Schematic diagram of IKAROS isoform 1 (NM_006060) with mutations reported in patients with PID/IEI. ZF indicates zinc finger. Different IKZF1 allelic variants are shown in blue (haploinsufficiency, HI), green (dimerization defective, DD), red (dominant negative, DN), and gray color (unclear mechanism; e.g., variants in which functional experimental data is unknown/inconclusive). Numbers next to the mutation indicate the number of families carrying the same mutation. Three groups reported R143W variants, two as a HI mutation (one validated by functional assays and the other identified by whole-exome sequencing without functional validation [32,36]) and one as a partial DN mutation [35]. # No IKAROS functional data is available but the mutation predicts the loss of DNA binding due to the partial to full loss of ZF2 and ZF3, respectively. (B) Pericentromeric targeting of IKAROS wild type (WT) and a representative mutant of each allelic variant. PC-HC localization of HA-tagged WT or mutants alone, or co-expression with Flag-tagged WT in NIH3T3 cells. (C) HEK293T cells were transfected with a vector expressing IKAROS WT, mutants or empty vector control (EV). Nuclear extracts were prepared and used for the EMSA assay using an IKBS1 probe containing the high affinity IKAROS binding site. Arrows indicate the monomer, dimer, and multimer forms of IKAROS binding to the probe.
WT IKAROS homo- and heterodimerizes as dimers or multimers with IKAROS family members, localizes to pericentromeric heterochromatin (PC-HC) and regulates (mostly represses but can also activate) the expression of its target genes [39,40]. Several studies have demonstrated that the DNA binding ability of the IKAROS protein to its target sequence is essential for its localization to PC-HC [41]. Our group and others have shown that when IKZF1 mutants affecting the DNA-binding domain were expressed in vitro (e.g., HEK 293 or NIH3T3 transfected cells), they completely abrogated DNA binding and displayed a diffuse PC-HC pattern, in contrast to a punctate staining pattern indicative of normal PC-HC localization. Co-expression of mutant and WT protein, mimicking the heterozygous condition found in the patients carrying heterozygous IKZF1 mutations, revealed that amino acid N159 mutations but not others exerted a dominant negative effect over WT IKAROS’ pericentromeric targeting [23]. Variants located to or disrupting ZF5 and/or ZF6 impaired homo-and hetero dimerization and also led to abnormal DNA binding and PC-HC localization without a dominant effect on WT function (Figure 1B and C) [22].
Based on IKZF1 mutants expression, functional defects (i.e., DNA binding, PC-HC targeting ability, and dimerization) and their impact on IKAROS WT protein, we classified human germline IKZF1 mutations found in PID/IEI patients into three groups: 1) haploinsufficiency (HI) caused by either the reduction of IKAROS WT protein expression (by large deletions encompassing IKZF1 gene or early truncating mutations affecting protein expression) or the loss of IKAROS function primarily due to disruption of DNA binding and PC-HC targeting, but not affecting the WT protein; 2) dimerization defective (DD), also acting by haploinsufficiency (no impact on the WT protein), but caused primarily by disruption of the dimerization domain; DD mutants are unable to dimerize with WT IKAROS protein but are still able to bind IKAROS target sequences as monomers but not as dimers (or multimers); and 3) dominant negative (DN) primarily affecting DNA binding as well as PC-HC targeting, and adversely affecting the PC-HC targeting of WT IKAROS proteins.
Most germline heterozygous IKZF1 mutations found in patients presenting with common variable immunodeficiency (CVID) are HI (either by the complete lack of one WT allele or presence of missense loss-of-function mutations in one allele)[24–34,38]. Patients with DN mutations mostly presented with early onset combined immunodeficiency [23]. The HI and DN variants mainly affect ZF2 and ZF3 at the DNA binding domain. More recently, patients presenting with more hematologic (benign and malignant) than infectious manifestations were reported to carry DD variants. The DD variants primarily affect ZF5 and/or ZF6, leading to impaired homo-and hetero dimerization as well as abnormal DNA binding and PC-HC localization [22].
As described above, testing the ability of IKAROS mutant to bind regulatory elements of its target genes and subcellular localization, as well as their capacity to homo- and heterodimerize are critically important steps to verify the functional impact of IKZF1 variants that in turn will mostly associate a genotype to particular phenotypes. The functional impact, immunological phenotype as well as predominant clinical manifestations of each IKZF1 allelic variants are summarized in Table 1.
Table 1.
Clinical and immunological phenotypes of patients with germline heterozygous IKZF1 mutations. Allelic variants associated with IKZF1 mutations and their impact on IKAROS functions (DNA binding, pericentromeric-heterochromatin [PC-HC] localization, and dimerization). Frequencies of clinical manifestations were calculated based on the total number of mutation carriers in each group. Immunologic phenotype proportions were calculated based on available data.
ALL: acute lymphoblastic leukemia, CID: combined immunodeficiency, CVID: common variable immune deficiency, DD: dimerization defective, DN: dominant negative, HI: haploinsufficiency, mis: missense mutation, NA: not applicable, null: no mutant IKAROS protein expression by a large deletion or early truncation, PC-HC: pericentromeric heterochromatin, WT: wild type
| Allelic variants | Haploinsufficiency through DNA binding defect (WT/null or WT/mis expression) | Haploinsufficiency through Dimerization defect | Dominant Negative (N159S/T) through DNA binding defect |
|---|---|---|---|
| Mutant protein expression in primary cells | No (null)/Yes (mis) | Yes (reduction in truncated mutants) | Yes |
| Functional assessments | |||
| Mutant DNA binding | NA (null)/ No (mis) | Impaired at multimer sites | No |
| Mutant Dimerization with WT | NA (null)/ Normal (mis) | Complete defect or Partial defect | Normal |
| Mutant PC-HC localization | NA (null)/ Diffused (mis) | Complete defect: diffused Partial defect: normal |
Diffused |
| Inhibition of WT’s PC-HC localization | NA (null)/ No (mis) | No | Yes |
| Predominant Clinical phenotypes | |||
| Predominant Clinical phenotype | CVID | Hematologic manifestations | CID |
| Bacterial Infection | +++ | + | ++++ |
| Other infections | + (Mycobacterial, Viral, Parasitic) |
- | ++++/+++ (Fungal and Viral) + (Mycobacterial, Parasitic) |
| Autoimmunity/ Immune dysregulation | ++ | ++ | - |
| Malignancy | + (B-ALL, pancreas tumor) |
+ (B-ALL, T-ALL, Burkitt lymphoma) |
+ (T-ALL) |
| Immunologic phenotypes | |||
| Low IgG | +++ | ++++ | ++++ |
| Low IgM | ++++ | +++ | ++++ |
| Low IgA | ++++ | +++ | ++++ |
| Low B cell number | +++ | ++ | ++++ |
| CD3 T cell number | Normal ++++ High + Low + |
Normal ++++ High + Low - |
Normal ++ High +++ Low + |
| CD4 T cell number | Normal ++++ High + Low + |
Normal ++++ High + Low + |
Normal + High ++ Low ++ |
| CD8 T cell number | Normal +++ High ++ (mostly in mis) Low + |
Normal ++++ High + Low - |
Normal +++ High + Low + |
| Low CD4/CD8 ratio | +++ | +++ | + |
| NK cell number | Normal ++++ High + Low + (mostly in mis) |
Normal +++ High - Low ++ |
Normal +++ High - Low ++ |
1–25%: +
26–50%:++
51–75%: +++
76–100%:++++
3. The role of IKAROS in the lymphoid lineage
The Ikaros gene was first discovered as a factor binding to regulatory elements of lymphocyte-specific genes Dntt and Cd3d [1,2]. Since then, Ikaros has been extensively studied due to its role in hematopoiesis and tumor suppression. Mice with Ikzf1 null mutations showed absence of B, NK, and fetal T cells and developed T-cell leukemia/lymphoma with a high penetrance [8]. Mice lacking the DNA binding domain (Ikzf1 DN−/−dominant negative mutation, deletion of ZF1–3) showed more severe lymphocyte defects with a complete lack of T, B, and NK cells along with early lymphoid progenitors [10,42]. Heterozygous mice with the DN mutation have normal lymphocyte numbers at birth, but also develop T cell malignancies a few months after birth [10]. Similar to the mouse models, most patients with germline heterozygous IKZF1 mutations showed severe defects in lymphocyte development, particularly in B lymphocytes. Our group showed Pro-B cells and early B- cell precursors were markedly decreased in patients with missense HI mutations, but plasma cells were present in the bone marrow, indicating that some precursors were able to differentiate into B cells [24]. Hoshino et al. showed reduced common lymphoid progenitors (CLP) with normal development of pro-B to pre-B cells in the bone marrow of patients with missense HI mutations [25]. Bogaert et.al showed partial arrest at the multipotent progenitors (MPP) and the B-lineage stage and moderate expansion of CLP in the bone marrow of patients with an early frame shift mutation[31]. Among patients with IKZF1 mutations, patients with DN mutations had the most severe defect on B cell development. Bone marrow aspirates from patients with DN mutations showed a severe reduction in pre-pro-B cells, pro-B cells, and mature B cells as well as the absence of plasma cells. This result provides evidence that the arrest of early B cell development results in the low peripheral B cell number in patients with DN mutations [23].
DD mutations showed less impact on B cell numbers than the other allelic variants as 36% of DD patients showed low B cell numbers in the peripheral blood compared to ~70% in HI patients and 100% in patients with DN mutations [22–34]. Although the impact of IKZF1 mutations on B cell development varies among the three allelic variants, hypogammaglobulinemia was observed in more than 70% of patients in all three allelic variants of germline IKZF1 mutations, indicating that B cell dysfunction is highly prevalent in patients with IKZF1-associated diseases.
Patients carrying IKZF1 mutations also showed an abnormal T cell phenotype. Among the allelic variants of IKZF1 mutations, patients with DN mutations appeared to have the most severe lymphocyte phenotypes including T and B cells. Increased T cell numbers were observed in about half of patients with DN mutations, but the T cell repertoire was unaffected in patients who were tested [23]. Moreover, T cells from DN patients exhibited an increased naïve phenotype. In most patients, over 90% of CD4 T cells were CD45RA+CD31+, the phenotype of recent thymic emigrants (RTE), in the periphery. The function of T lymphocytes is also affected in patients with DN mutations, as indicated by defects of proliferation (upon low dose TCR stimulation), signaling (IL-2-induced STAT5 phosphorylation), and Th polarization to Th1/Th2/Th17/Treg[23]. Increased CD8 T cells were more profound in patients with HI mutations affecting DNA binding (about 40%) than patients with other IKAROS allelic variants (~25% in DD and DN patients) [22–34].
In addition to B cell defects, NK cell deficiency is one of the main features of Ikaros transgenic mice[8,10]. In humans, whereas 26–38% of patients with DD, DN and missense HI mutations showed low NK cell numbers, patients with HI defects associated with reduced IKAROS expression due to a large deletion or early truncation, had normal NK cell counts [22–34]. Further NK functional testing and detailed phenotypes will help in the understanding of IKAROS role in NK cell development and function in humans.
4. The role of IKAROS in the myeloid lineage
Variable abnormalities in myeloid and dendritic cell differentiation have been reported in Ikaros mouse studies. Ikaros null mice showed a severe reduction in thymic dendritic cells (DC), but mice with reduced Ikaros expression, Ikzf1L/L, displayed a specific defect in plasmacytoid DC (pDC) [8,43]. While Ikaros DN−/− mice produced normal erythroid and myeloid lineages, they also showed a complete lack of DC in lymphoid organs (thymus and spleen), demonstrating the important role of Ikaros in DC development[9,42]. In humans, abnormal DC differentiation was also observed in patients with IKZF1 mutations. Patients with HI mutations had decreased pDC numbers and expansion of conventional DC1 (cDC1), and patients with DN variants showed a mild reduction in pDC, while one patient showed a severe reduction in myeloid DC (mDC) [23,44]. Moreover, patients with DN mutations showed myeloid abnormalities including eosinopenia and neutropenia[23]. The function of monocytes was also affected in patients with DN mutations, as indicated by defects of TLR-induced cytokine generation[23]. Transcriptome analysis also supported an abnormal myeloid function as patients with DN mutations displayed abnormal gene expression patterns when compared to normal controls or patients with HI mutations in LPS-stimulated monocytes [23].
5. IKAROS variants in infections
Increased susceptibility to infections (severe, recurrent and/or opportunistic) is shared among all allelic variants of germline IKZF1 mutations although with markedly variable penetrance and severity (Figure 2). Patients with HI mutations present with recurrent infections in 55% of cases [24–34,38]. Sinopulmonary infections are the most common and Streptococcus pneumoniae is the most frequently isolated agent. Viral infections among these patients are neither frequent nor usually severe (3 cases of herpes simplex labialis, 2 cases of warts due to papillomavirus and 1 case of mumps meningitis). Among patients carrying DD mutations even a smaller subset of them present with recurrent infections, typically non-invasive and non-severe (20%)[22,32]. On the other hand, most of patients with DN mutations present with earlier onset (before 2 years of age) and a broad spectrum of mostly severe and invasive infectious complications. Bacterial infections were reported in 78% of patients and viral infections, including respiratory syncytial virus (RSV), herpes simplex virus (HSV), adenovirus, influenza, and molluscum, were equally prevalent [23,38,45]. Remarkably, eight out of nine patients with DN mutations reported had Pneumocystis jirovecii pneumonia (PJP) early in life (from 6 months to 2 years), with repeated episodes in some patients.
Figure 2.
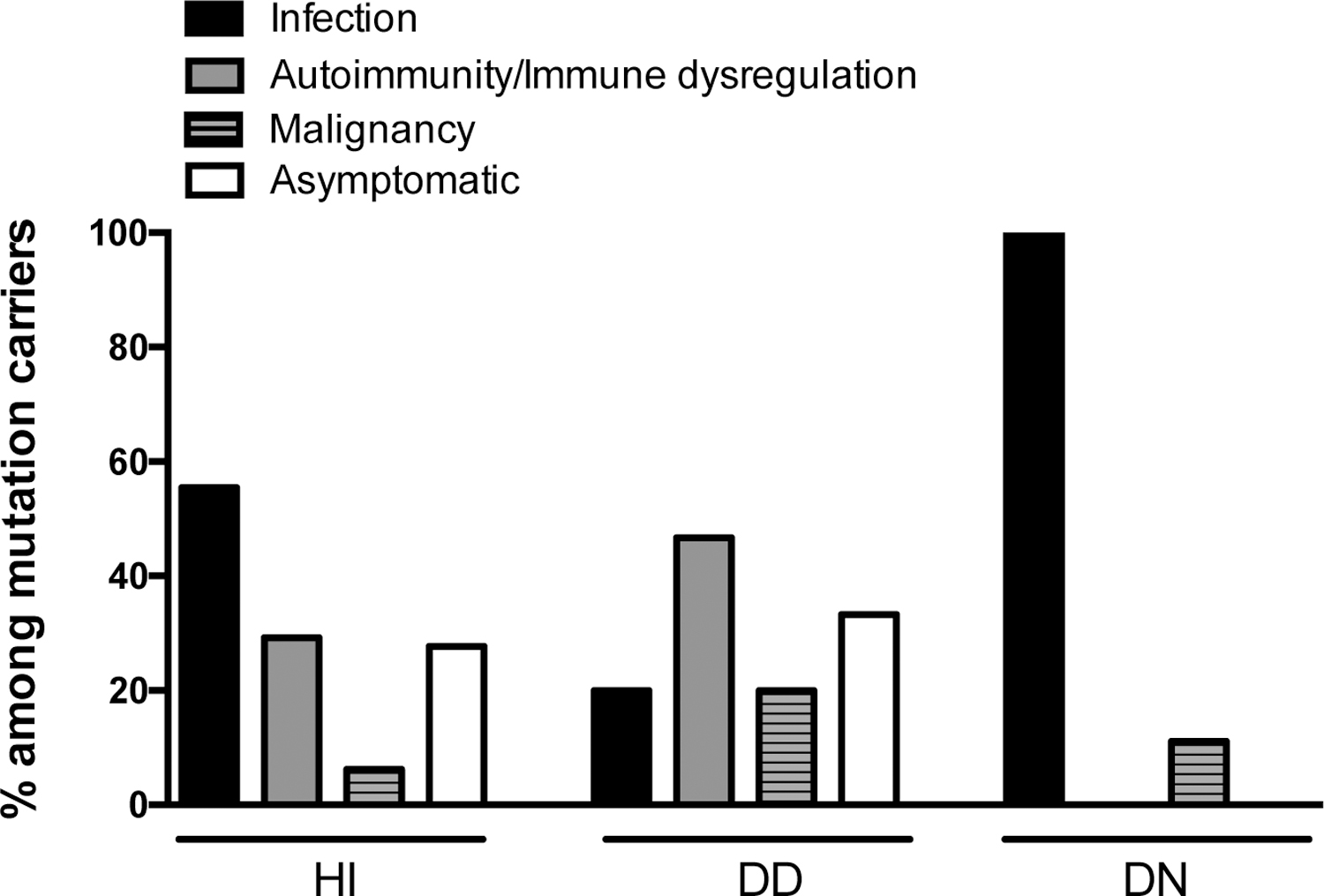
Clinical manifestations in patients with germline heterozygous IKZF1 mutations.
HI: haploinsufficiency; DD: dimerization defective; DN: dominant negative
6. IKAROS variants in autoimmune diseases
Several genome-wide association studies (GWAS) have recognized IKZF1 as risk loci for autoimmunity, particularly for systemic lupus erythematosus (SLE) [46,47]. As GWAS studies predicted, patients with HI and DD IKZF1 mutations, except DN mutations, developed autoimmunity which includes immune thrombocytopenia (ITP), arthritis, SLE, antiphospholipid syndrome and others (reviewed in [48]). Interestingly, patients carrying DN variants and mostly lacking B cells, plasma cells, immunoglobulins, eosinophils, Th2 cells, T regulatory cells, and memory T cells (both central memory and effector memory) did not develop autoimmune diseases. Among patients with IKAROS-associated diseases and autoimmune manifestations, DD patients showed a higher incidence rate (47%) of autoimmune disease when compared to patients with HI mutations (~30%) (Figure 2). Our recent study demonstrated that DD mutants, but not HI and DN mutants, altered IKAROS posttranslational modification (i.e., Sumoylation) and recruitment of the NuRD complex which led to abnormal transcription regulation in NuRD and Sin3 associated genes [22]. Chromatin deregulation by IKZF1 DD mutations may cause an imbalanced histone acetylation or methylation, resulting in impaired B cell tolerance and predisposing to autoimmune diseases but further studies in humans are needed to support this hypothesis. In mouse studies, targeted deletion of Ikzf1 demonstrated that Ikaros played an important role in autoimmunity [49,50]. Loss of Ikaros expression in B cells promote systemic autoimmunity due to a failure of induction of B cell anergy and hyper-reactivity of TLR signaling[49]. This finding provides an evidence of Ikaros role in maintaining tolerance through controlling B cell anergy and TLR signaling [49].
7. IKAROS variants in malignancies
Several Ikaros murine models have demonstrated that the loss of Ikaros expression or functions results in a high incidence of T-cell leukemia and lymphoma (reviewed in [18,51]). In contrast to the mouse models, IKZF1 somatic mutations were rarely reported in T-ALL in humans (<5%)[52,53]. However, IKZF1 somatic deletions and missense mutations have been shown to be associated with high-risk-B-ALL in humans for both pediatric and adult B-ALL, particularly in cases with BCR-ABL1 translocation (up to 80%) [21,54]. More recently, germline IKZF1 variants were reported in 0.9% of a large cohort of pediatric B-ALL patients [20]. In this study, 28 germline IKZF1 variants in 45 children with ALL were described: two variants affecting the DNA binding domain (R162P and H163Y), three truncate and one missense variants affecting or located in the dimerization domain (D186fs, M306*, C394*, and M476T) at the N-terminal region, and other variants clustered outside of the dimerization domain at the C-terminal region. This contrasts with the pattern of heterozygous germline IKZF1 mutations found in patients with CVID (mostly HI), CID (mostly DN), and hematologic manifestations (mostly DD) that cluster and directly affect the DNA binding or the dimerization domains.
Malignancies were also reported in all three groups of patients with PID/IEI; four cases in patient with HI mutations (3 B-ALL and 1 pancreatic tumor), three cases in DD patients (1 B-ALL, 1 T-ALL, and 1 Burkitt lymphoma) and one case in DN patients (1 T-ALL). Among the three allelic variants, DD patients showed a relatively higher incidence (20%) of malignancy than the other groups (6.2% in HI patients and 11.1% in DN patients) (Figure 2). Recently, our group showed that T-cell blasts from patients with DD and DN mutations presented different gene transcription patterns compared to normal controls and a patient with a HI mutation [22]. A higher number of malignancy-associated genes were upregulated in the DD and DN mutation groups. Functions related with T cell development and differentiation were largely dysregulated in patients with HI and DN mutations, but not in patients with DD mutations. Dysregulation of malignancy and/or T cell development pathways in the three allelic variants may explain, at least in part, their different clinical and immunological phenotypes.
Of note, one patient with a DN mutation who developed T-ALL, also carried a somatic NOTCH1 mutation. Similar to IKZF1, NOTCH1 is a tumor suppressor gene, and deregulation of NOTCH1 activities has also been associated with development of hematological malignancies [55]. In this case, combined loss of IKAROS function and activation of NOTCH1 may have contributed to the development of T-ALL.
8. Treatment of IKAROS-associated diseases
Treatment for patients with IKAROS-associated diseases could be divided, as with most PID/IEI, into therapeutic, prophylactic and curative approaches. As a general rule, standard of care practices for each type of complication should be followed. For patients with absent B cells or B-cell dysfunction, hypogammaglobulinemia and impaired antibody responses, IgG replacement therapy (intravenous or subcutaneous) has been used without specific complications.
For infectious diseases in patients with HI mutations presenting with CVID, S. pneumoniae and H. influenzae B should be considered as frequent microorganisms associated with upper and lower respiratory infections, meningitis and sepsis; therefore, macrolide prophylaxis should be contemplated. In patients with DN mutations presenting with a more severe CID phenotype, PJP has to be highly suspected in lower respiratory infections and TMP-SMX (or other forms of PJP prophylaxis) is highly recommended. Besides, other fungal (e.g., Candida spp. and Aspergillus spp.), bacterial (as S. pneumoniae, Klebsiella spp, and Pseudomonas spp.), severe viral (including RSV, adenovirus and different herpes virus infections), as well as parasitic Cryptosporidium spp. infections have also been associated with patients carrying this allelic variant. In this context, hygienic and adequate prophylactic measures should be considered on a case-by-case basis.
Immune dysregulation complications in patients with IKAROS-associated diseases most commonly involved hematologic cytopenias. Immune thrombocytopenia has been the most frequently recorded, but also autoimmune hemolytic anemia and neutropenia are described. These complications were treated with steroids, high dose IgG, rituximab, mycophenolate mofetil (MMF) or sirolimus, with in general a good success rate. Other forms of immunodysregulatory manifestations include arthritis (e.g., reactive, juvenile idiopathic or seronegative), systemic lupus erythematous, antiphospholipid syndrome, IgA vasculitis, Hashimoto’s disease, myasthenia gravis, and autoimmune hepatitis, and were reported to be treated following a “standard of care” approach.
When focused on curative options, hematopoietic stem cell transplantation (HSCT) has been successfully performed (with or without a previously defined IKAROS-associated disease diagnosis) in patients with DN (the most severe form of the disease) and HI allelic variants with severe clinical presentations [23,24,28,35,45,55].
In the HI group, HSCT has been indicated in the context of congenital pancytopenia in 1 infant, and bone marrow suppression in a 6-year-old child following treatment for B-ALL [24,28]. In both cases, donors were HLA-matched siblings. The first patient passed away due to multi-organ failure 40 days after transplantation whereas the second patient was alive, free of infections and off immunoglobulin replacement at the time of reporting, 11 years after HSCT.
In the DN group, Kellner et. al., described outcomes of HSCT in 4 patients, one of whom had previously rejected an unconditioned CD34+ haploidentical transplant and underwent a second transplant after a T-ALL diagnosis [45]. Yilmaz et. al., additionally reported HSCT results in a 3-month-old patient presenting with pancytopenia (including lymphopenia), hemophagocytosis and carrying a variant in IKZF1 (R143W) which was initially described as HI but was later shown to behave in a partial DN manner [32,35]. Among the 5 patients, one had a matched sibling donor while the other 4 had unrelated donors. All patients achieved complete chimerism (>99% donor). Two patients developed acute graft-versus-host disease (GVHD), a grade II -skin stage III and gastrointestinal tract stage I- was seen in one patient while the other presented with grade II, -skin stage III and gastrointestinal tract stage II- disease and both responded well to standard of care treatment; chronic GVHD has not been reported. Transplant-associated microangiopathy was described in one patient and managed with MMF and plasma exchange. Other post-transplant complications included cytomegalovirus (CMV) viremia (3 patients), norovirus infection, Epstein-Barr virus (EBV) reactivation, and Mycobacterium avium complex lymphadenitis (reported in one patient each). Survival in this small, HSCT-heterogeneous group was 60%, with a follow-up time of surviving patients ranging from 1 to 7 years. Among the deceased patients, 1 died of a respiratory infection and pulmonary hemorrhage on day +194 post HSCT [35], and another patient who had a history of Cryptosporidium spp. cholangitis passed away 1 year after transplantation due to liver failure [45]. All surviving patients were able to stop immunoglobulin replacement as well as antimicrobial prophylaxis.
9. Conclusions
Based on the experience gained from more than 90 PID/IEI published patients carrying IKZF1 germline heterozygous mutations, the role of IKAROS in human immunity of lymphoid and myeloid cells has been extensively demonstrated (Table 1 and Figure 3). The immunological, and in its turn clinical phenotype of patients carrying IKZF1 mutations depend on the protein domain primarily affected (i.e., DNA binding or dimerization) and the pathophysiology mechanism involved (i.e., haploinsufficiency or dominant negative). In terms of lineage-specific impact, a reduction of B cell numbers can be detected among the three allelic variants reported so far (severity DN>HI>DD). T cells on the other hand, are heavily impacted by DN mutations and to a lesser extent by HI and DD mutations, as reflected by their clinical presentations: while T-cell dependent opportunistic infections are the rule in patients with DN variants (i.e., pneumocystis pneumonia), they are extremely rare in the other two allelic variants. Moreover, myeloid involvement is evident in patients with DN, DD and HI variants, through different mechanisms and with different penetrance and expressivity levels. The above-described genotype/phenotype correlation may also represent a gene dosage effect by this transcription factor where “stronger” mutations (i.e., DN) have a more penetrant, immunologically broader and clinically more severe impact, opposed to “milder” variants (e.g., null mutations deleting a full allele but allowing the expression of ~ 50% of WT IKAROS) presenting with a less penetrant, more limited immune and clinical disease.
Figure 3.
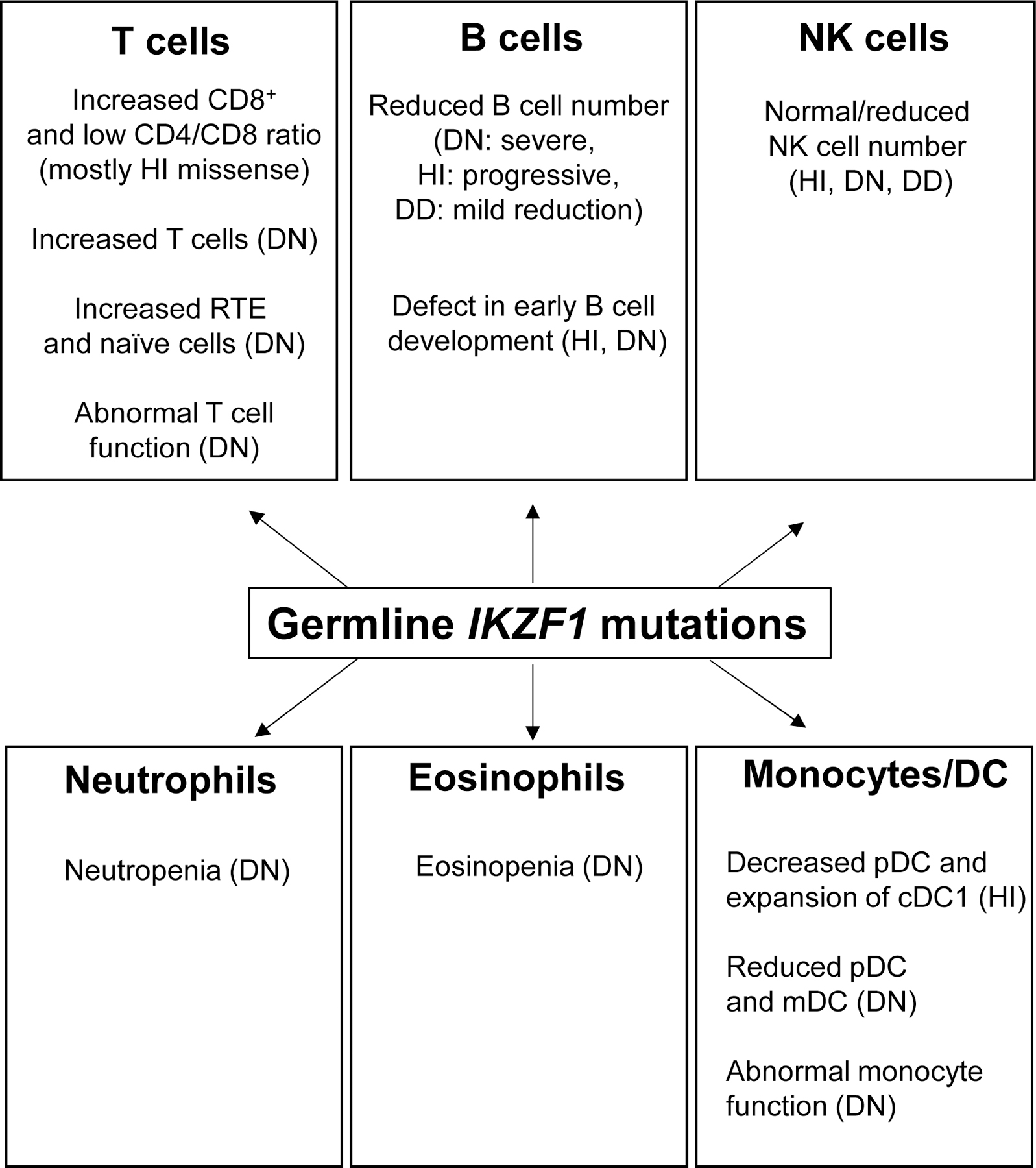
Impact of germline IKZF1 mutations on lymphocyte and myeloid phenotypes in patients with PID/IEI. The overview above was compiled from data reported in patients with IKZF1 mutations (HI: haploinsufficiency, DD: dimerization defective, DN: dominant negative). The detailed explanation of the phenotype is described in sections 3 and 4 (the role of IKAROS in the lymphoid (3) and the myeloid (4) lineage) cDC: conventional dendritic cells, mDC: myeloid dendritic cells, pDC: plasmacytoid dendritic cells, RTE: recent thymic emigrant.
In summary, IKAROS transcription factor was well recognized as having a promiscuous biologic behavior based on its interaction with other IKAROS family members (i.e., AIOLOS, HELIOS and PEGASUS), either by homo and/or heterodimerization. This quality, not surprisingly, also applies to the clinical spectrum of diseases associated to the mutations affecting this gene.
10. Expert opinion
IKAROS-associated diseases have long been suspected and more recently confirmed to be associated with PID/IEI, including different degrees of increased susceptibility to infectious disease, autoimmunity, immune dysregulation and cancer[22–38,56]. While different phenotypes (e.g., CVID, CID, mostly hematologic or immune deregulation/autoimmunity manifestations) and disease expressivity spectra are broad, a genotype-phenotype correlation can be suspected upon functional in vitro testing, but not easily predicted when new variants are detected.
Although scientifically fascinating, this represents a daily challenge for clinical practitioners when facing patients with a defined clinical phenotype carrying variants of unknown significance (VUS) in IKZF1. In this setting, the Who Wants to Be a Millionaire “Phone-a-friend” option is probably the most efficient way to go, as laboratories experienced and actively doing research in IKZF1 (or other particular genes) are likely to have tested some of these unreported VUS themselves and have the know-how and expertise to functionally test them when justified.
Another hurdle patients and clinicians have to face in terms of getting an IKAROS-associated disease genetic diagnosis, is related to IKZF1 inclusion/exclusion from particular commercially available next generation sequencing (NGS) PID/IEI panels. While IKZF1 is a well-established disease-causing gene, not all NGS PID/IEI panels available in the United States test for this gene. IKAROS, together with HELIOS, AIOLOS, EOS and PEGASSUS, belongs to a family of transcription factors sharing a high genetic homology. Whereas this could be technically challenging for genetic testing, several but not all laboratories were able to overcome those difficulties and include IKZF1 in their NGS PID/IEI offered panels. Importantly to point out, clinicians using NGS PID/IEI panels as their primary genetic diagnostic approach and studying patients with CVID, CID or other immune phenotypes fitting IKAROS-associated diseases, should be aware that reports not including variants in IKZF1 (or other genes not tested in those particular panels) do not exclude the disease and only mean that the gene was not tested. Moreover, this issue together with the incomplete penetrance and clinical heterogeneity of the disease, likely contributes to IKAROS-associated diseases underdiagnosis. The more accessible and frequent use of whole exome sequencing (WES) as a genetic diagnostic approach for PID/IEI, lowers the chances of missing this diagnosis.
Therapeutically speaking, IKAROS-associated diseases could be cured by HSCT as has been already shown in patients with somatic defects, as well as in those carrying HI and DN germline allelic variants. On the other hand, most of these patients and their complications are symptomatically treated, either prophylactically or therapeutically. When focused on gene-specific medical treatments oriented to restore a function that is lost (as in IKAROS HI, DN and DD patients), this aim proved to be more difficult than trying to ameliorate an excessive function, as shown in patients with gain-of-function defects in STAT1 and the use of Janus kinase (JAK)-inhibitors controlling the signaling in this pathway [57]. On the same token, if IKAROS gain-of-function defects are eventually detected, FDA-approved and commercially available thalidomide analogs known to promote IKAROS degradation via increased ubiquitination and degradation [58] may become useful as part of the medical armamentarium for such allelic variants and disease. While gene-therapy for IKZF1 defects and other germline dominant monogenic diseases seems like a plausible goal to achieve in the future, the very precise and fine-tuned gene dosage regulation of IKZF1/IKAROS is subjected to, adds another layer of complexity to this already ambitious task.
IKAROS represents a prototypical example of modern genetics and disease association where both germline and somatic variants acting through different mechanisms of actions can be associated to a broad spectrum of human diseases. If the experience gained from the past can serve as predictive of the future, we can still expect more allelic variants and new phenotypes to be associated with IKZF1 mutations, as well as novel therapeutic approaches to modulate its expression and function to help patients with IKAROS-associated diseases.
Article highlights.
While somatic IKZF1 alterations are associated with increased risk of B-progenitor acute lymphoblastic leukemia (B-ALL), autosomal dominant heterozygous germline mutations in IKZF1 are associated with immunodeficiency as well as B-ALL.
Heterozygous germline IKZF1 mutations can be categorized into three allelic variants acting by haploinsufficiency (HI), dominant-negative (DN), or dimerization defective (DD).
Patients with germline HI variants mostly present with a CVID phenotype which includes hypogammaglobulinemia, defective vaccine responses, progressive loss of B cell numbers, recurrent bacterial infections, and increased risk of autoimmunity/immune dysregulation and malignancy.
Patients carrying DN mutations present with an early onset (<2 years) CID phenotype with severe immunological and clinical manifestations including opportunistic infections (i.e., Pneumocystis jirovecii) and increased leukemia susceptibility.
Patients with DD variants show a higher incidence of autoimmune diseases/immune dysregulation and malignancy compared to the other allelic variants; recurrent or severe infections are less frequently seen in patients within this allelic variant.
Acknowledgments
This work was supported by the Intramural Research Program, National Institutes of Health Clinical Center. The content of this article does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US government.
Funding
This work was supported by the Intramural Research Program, National Institutes of Health Clinical Center.
Footnotes
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Papers of special note have been highlighted as:
* of interest
** of considerable interest
- 1.Lo K, Landau NR, Smale ST. LyF-1, a transcriptional regulator that interacts with a novel class of promoters for lymphocyte-specific genes. Mol Cell Biol, 11(10), 5229–5243 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Georgopoulos K, Moore DD, Derfler B. Ikaros, an early lymphoid-specific transcription factor and a putative mediator for T cell commitment. Science, 258(5083), 808–812 (1992). [DOI] [PubMed] [Google Scholar]
- 3.Morgan B, Sun L, Avitahl N et al. Aiolos, a lymphoid restricted transcription factor that interacts with Ikaros to regulate lymphocyte differentiation. EMBO J, 16(8), 2004–2013 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelley CM, Ikeda T, Koipally J et al. Helios, a novel dimerization partner of Ikaros expressed in the earliest hematopoietic progenitors. Curr Biol, 8(9), 508–515 (1998). [DOI] [PubMed] [Google Scholar]
- 5.Honma Y, Kiyosawa H, Mori T et al. Eos: a novel member of the Ikaros gene family expressed predominantly in the developing nervous system. FEBS Lett, 447(1), 76–80 (1999). [DOI] [PubMed] [Google Scholar]
- 6.Perdomo J, Holmes M, Chong B, Crossley M. Eos and pegasus, two members of the Ikaros family of proteins with distinct DNA binding activities. J Biol Chem, 275(49), 38347–38354 (2000). [DOI] [PubMed] [Google Scholar]
- 7.Hahm K, Cobb BS, McCarty AS et al. Helios, a T cell-restricted Ikaros family member that quantitatively associates with Ikaros at centromeric heterochromatin. Genes Dev, 12(6), 782–796 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang JH, Nichogiannopoulou A, Wu L et al. Selective defects in the development of the fetal and adult lymphoid system in mice with an Ikaros null mutation. Immunity, 5(6), 537–549 (1996). [DOI] [PubMed] [Google Scholar]
- 9.Wu L, Nichogiannopoulou A, Shortman K, Georgopoulos K. Cell-autonomous defects in dendritic cell populations of Ikaros mutant mice point to a developmental relationship with the lymphoid lineage. Immunity, 7(4), 483–492 (1997). [DOI] [PubMed] [Google Scholar]
- 10.Winandy S, Wu P, Georgopoulos K. A dominant mutation in the Ikaros gene leads to rapid development of leukemia and lymphoma. Cell, 83(2), 289–299 (1995). [DOI] [PubMed] [Google Scholar]
- 11.Malinge S, Thiollier C, Chlon TM et al. Ikaros inhibits megakaryopoiesis through functional interaction with GATA-1 and NOTCH signaling. Blood, 121(13), 2440–2451 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Francis OL, Payne JL, Su RJ, Payne KJ. Regulator of myeloid differentiation and function: The secret life of Ikaros. World J Biol Chem, 2(6), 119–125 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dijon M, Bardin F, Murati A, Batoz M, Chabannon C, Tonnelle C. The role of Ikaros in human erythroid differentiation. Blood, 111(3), 1138–1146 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Molnar A, Georgopoulos K. The Ikaros gene encodes a family of functionally diverse zinc finger DNA-binding proteins. Mol Cell Biol, 14(12), 8292–8303 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Powell MD, Read KA, Sreekumar BK, Oestreich KJ. Ikaros Zinc Finger Transcription Factors: Regulators of Cytokine Signaling Pathways and CD4(+) T Helper Cell Differentiation. Front Immunol, 10, 1299 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ochiai K, Kondo H, Okamura Y et al. Zinc finger-IRF composite elements bound by Ikaros/IRF4 complexes function as gene repression in plasma cell. Blood Adv, 2(8), 883–894 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim J, Sif S, Jones B et al. Ikaros DNA-binding proteins direct formation of chromatin remodeling complexes in lymphocytes. Immunity, 10(3), 345–355 (1999). [DOI] [PubMed] [Google Scholar]
- 18.Heizmann B, Kastner P, Chan S. The Ikaros family in lymphocyte development. Curr Opin Immunol, 51, 14–23 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Mullighan CG, Miller CB, Radtke I et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature, 453(7191), 110–114 (2008).• Somatic IKZF1 deletions (dominant-negative IKZF1 isoform, loss of IKZF1 expression) in patients with BCR-ABL1-ALL
- 20.Churchman ML, Qian M, Te Kronnie G et al. Germline Genetic IKZF1 Variation and Predisposition to Childhood Acute Lymphoblastic Leukemia. Cancer Cell, 33(5), 937–948 e938 (2018).•• Germline IKZF1 variants in familial and sporadic acute lymphoblastic leukemia.
- 21.Kastner P, Dupuis A, Gaub MP, Herbrecht R, Lutz P, Chan S. Function of Ikaros as a tumor suppressor in B cell acute lymphoblastic leukemia. Am J Blood Res, 3(1), 1–13 (2013). [PMC free article] [PubMed] [Google Scholar]
- 22.Kuehn HS, Niemela JE, Stoddard J et al. Germline IKAROS dimerization haploinsufficiency causes hematologic cytopenias and malignancies. Blood, (2020).•• IKAROS dimerization defects associated with hematologic cytopenias and malignancies
- 23.Boutboul D, Kuehn HS, Van de Wyngaert Z et al. Dominant-negative IKZF1 mutations cause a T, B, and myeloid cell combined immunodeficiency. J Clin Invest, 128(7), 3071–3087 (2018).•• The description of dominant negative IKZF1 variants (N159S/T) in combined immunodeficiency.
- 24.Kuehn HS, Boisson B, Cunningham-Rundles C et al. Loss of B Cells in Patients with Heterozygous Mutations in IKAROS. N Engl J Med, 374(11), 1032–1043 (2016).•• The first report of germline IKZF1 variants acting by haploinsufficiency in patients with common variable immunodeficiency (CVID)
- 25.Hoshino A, Okada S, Yoshida K et al. Abnormal hematopoiesis and autoimmunity in human subjects with germline IKZF1 mutations. J Allergy Clin Immunol, 140(1), 223–231 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Groth DJ, Lakkaraja MM, Ferreira JO, Feuille EJ, Bassetti JA, Kaicker SM. Management of Chronic Immune Thrombocytopenia and Presumed Autoimmune Hepatitis in a Child with IKAROS Haploinsufficiency. J Clin Immunol, 40(4), 653–657 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Dieudonne Y, Guffroy A, Vollmer O, Carapito R, Korganow AS. IKZF1 Loss-of-Function Variant Causes Autoimmunity and Severe Familial Antiphospholipid Syndrome. J Clin Immunol, 39(4), 353–357 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Goldman FD, Gurel Z, Al-Zubeidi D et al. Congenital pancytopenia and absence of B lymphocytes in a neonate with a mutation in the Ikaros gene. Pediatr Blood Cancer, 58(4), 591–597 (2012).• The first report of an IKZF1 variant in a patient with immunodeficiency.
- 29.Chen QY, Wang XC, Wang WJ, Zhou QH, Liu DR, Wang Y. B-cell Deficiency: A De Novo IKZF1 Patient and Review of the Literature. J Investig Allergol Clin Immunol, 28(1), 53–56 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Van Nieuwenhove E, Garcia-Perez JE, Helsen C et al. A kindred with mutant IKAROS and autoimmunity. J Allergy Clin Immunol, 142(2), 699–702 e612 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bogaert DJ, Kuehn HS, Bonroy C et al. A novel IKAROS haploinsufficiency kindred with unexpectedly late and variable B-cell maturation defects. J Allergy Clin Immunol, 141(1), 432–435 e437 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eskandarian Z, Fliegauf M, Bulashevska A et al. Assessing the Functional Relevance of Variants in the IKAROS Family Zinc Finger Protein 1 (IKZF1) in a Cohort of Patients With Primary Immunodeficiency. Front Immunol, 10, 568 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Banday AZ, Jindal AK, Kaur A et al. Cutaneous IgA vasculitis-presenting manifestation of a novel mutation in the IKZF1 gene. Rheumatology (Oxford), (2020). [DOI] [PubMed] [Google Scholar]
- 34.Sriaroon P, Chang Y, Ujhazi B et al. Familial Immune Thrombocytopenia Associated With a Novel Variant in IKZF1. Front Pediatr, 7, 139 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yilmaz E, Kuehn HS, Odakir E et al. Common Variable Immunodeficiency, Autoimmune Hemolytic Anemia, and Pancytopenia Associated With a Defect in IKAROS. J Pediatr Hematol Oncol, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Okano T, Imai K, Naruto T et al. Whole-Exome Sequencing-Based Approach for Germline Mutations in Patients with Inborn Errors of Immunity. J Clin Immunol, 40(5), 729–740 (2020). [DOI] [PubMed] [Google Scholar]
- 37.Belot A, Rice GI, Omarjee SO. Contribution of rare and predicted pathogenic gene variants to childhood-onset lupus: a large, genetic panel analysis of British and French cohorts (vol 2, pg e99, 2020). Lancet Rheumatol, 2(11), E664–E664 (2020). [DOI] [PubMed] [Google Scholar]
- 38.Thaventhiran JED, Lango Allen H, Burren OS et al. Publisher Correction: Whole-genome sequencing of a sporadic primary immunodeficiency cohort. Nature, 584(7819), E2 (2020). [DOI] [PubMed] [Google Scholar]
- 39.Brown KE, Guest SS, Smale ST, Hahm K, Merkenschlager M, Fisher AG. Association of transcriptionally silent genes with Ikaros complexes at centromeric heterochromatin. Cell, 91(6), 845–854 (1997). [DOI] [PubMed] [Google Scholar]
- 40.Koipally J, Heller EJ, Seavitt JR, Georgopoulos K. Unconventional potentiation of gene expression by Ikaros. J Biol Chem, 277(15), 13007–13015 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Cobb BS, Morales-Alcelay S, Kleiger G, Brown KE, Fisher AG, Smale ST. Targeting of Ikaros to pericentromeric heterochromatin by direct DNA binding. Genes Dev, 14(17), 2146–2160 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Georgopoulos K, Bigby M, Wang JH et al. The Ikaros gene is required for the development of all lymphoid lineages. Cell, 79(1), 143–156 (1994). [DOI] [PubMed] [Google Scholar]
- 43.Allman D, Dalod M, Asselin-Paturel C et al. Ikaros is required for plasmacytoid dendritic cell differentiation. Blood, 108(13), 4025–4034 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cytlak U, Resteu A, Bogaert D et al. Ikaros family zinc finger 1 regulates dendritic cell development and function in humans. Nat Commun, 9(1), 1239 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kellner ES, Krupski C, Kuehn HS et al. Allogeneic Hematopoietic Stem Cell Transplant Outcomes for Patients with Dominant-Negative IKFZ1/IKAROS Mutations. J Allergy Clin Immunol, (2019).•• Outcome of allogenic HSCT in patients with a dominant-negative IKZF1 N159S mutation.
- 46.Han JW, Zheng HF, Cui Y et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat Genet, 41(11), 1234–1237 (2009). [DOI] [PubMed] [Google Scholar]
- 47.Cunninghame Graham DS, Morris DL, Bhangale TR et al. Association of NCF2, IKZF1, IRF8, IFIH1, and TYK2 with systemic lupus erythematosus. PLoS Genet, 7(10), e1002341 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nunes-Santos CJ, Kuehn HS, Rosenzweig SD. IKAROS Family Zinc Finger 1-Associated Diseases in Primary Immunodeficiency Patients. Immunol Allergy Clin North Am, 40(3), 461–470 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schwickert TA, Tagoh H, Schindler K, Fischer M, Jaritz M, Busslinger M. Ikaros prevents autoimmunity by controlling anergy and Toll-like receptor signaling in B cells. Nat Immunol, 20(11), 1517–1529 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Macias-Garcia A, Heizmann B, Sellars M et al. Ikaros Is a Negative Regulator of B1 Cell Development and Function. J Biol Chem, 291(17), 9073–9086 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marke R, van Leeuwen FN, Scheijen B. The many faces of IKZF1 in B-cell precursor acute lymphoblastic leukemia. Haematologica, 103(4), 565–574 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marcais A, Jeannet R, Hernandez L et al. Genetic inactivation of Ikaros is a rare event in human T-ALL. Leuk Res, 34(4), 426–429 (2010). [DOI] [PubMed] [Google Scholar]
- 53.Payne KJ, Dovat S. Ikaros and tumor suppression in acute lymphoblastic leukemia. Crit Rev Oncog, 16(1–2), 3–12 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mullighan CG, Su X, Zhang J et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med, 360(5), 470–480 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yoshida N, Sakaguchi H, Muramatsu H et al. Germline IKAROS mutation associated with primary immunodeficiency that progressed to T-cell acute lymphoblastic leukemia. Leukemia, 31(5), 1221–1223 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Ng S, Fanta C, Okam M, Bhatt AS. NK-cell and B-cell deficiency with a thymic mass. N Engl J Med, 364(6), 586–588 (2011). [DOI] [PubMed] [Google Scholar]
- 57.Higgins E, Al Shehri T, McAleer MA et al. Use of ruxolitinib to successfully treat chronic mucocutaneous candidiasis caused by gain-of-function signal transducer and activator of transcription 1 (STAT1) mutation. J Allergy Clin Immunol, 135(2), 551–553 (2015). [DOI] [PubMed] [Google Scholar]
- 58.Fink EC, Ebert BL. The novel mechanism of lenalidomide activity. Blood, 126(21), 2366–2369 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]