

Abstract
Innate immune responses to Gram-negative bacteria depend on the recognition of lipopolysaccharide (LPS) by a receptor complex that includes CD14 and TLR4. In dendritic cells (DCs), CD14 enhances the activation not only of TLR4 but also that of the NFAT family of transcription factors, which suppresses cell survival and promotes the production of inflammatory mediators. NFAT activation requires Ca2+ mobilization. In DCs, Ca2+ mobilization in response to LPS depends on phospholipase C γ2 (PLCγ2), which produces inositol 1,4,5-trisphosphate (IP3). Here, we showed that the IP3 receptor 3 (IP3R3) and ITPKB, a kinase that converts IP3 to inositol 1,3,4,5-tetrakisphosphate (IP4), were both necessary for Ca2+ mobilization and NFAT activation in mouse and human DCs. A pool of IP3R3 was located on the plasma membrane of DCs, where it colocalized with CD14 and ITPKB. Upon LPS binding to CD14, ITPKB was required for Ca2+ mobilization through plasma membrane-localized IP3R3 and for NFAT nuclear translocation. Pharmacological inhibition of ITPKB in mice reduced both LPS-induced tissue swelling and the severity of inflammatory arthritis to a similar extent as that induced by the inhibition of NFAT using nanoparticles that delivered an NFAT-inhibiting peptide specifically to phagocytic cells. Our results suggest that ITPKB may represent a promising target for anti-inflammatory therapies that aim to inhibit specific DC functions.
INTRODUCTION
Innate immune myeloid cells sense the presence of microbes or microbial products through pattern recognition receptors (PRRs). Among the PRRs, Toll-like receptors (TLRs) are widely present on innate immune myeloid cells such as dendritic cells (DCs) and macrophages, both of which serve as sentinels of the immune system (1). Once TLRs are activated, adaptor proteins containing Toll-interleukin receptor (TIR) domains are recruited and initiate multiple downstream pathways for the activation of transcription factors, including nuclear factor κB (NF-κB), activator protein 1, and the interferon regulatory factors. In addition to these potent proinflammatory transcription factors, the nuclear factor of activated T cells (NFAT) is also activated in phagocytes either directly (in the case of TLR9) or indirectly (through the co-receptor CD14 in the case of TLR4) downstream of TLRs (2, 3). Other PRRs that can efficiently activate the NFAT family of transcription factors are the C-type lectin-like receptors, Dectin-1 and Dectin-2 (4, 5). Key functions of the NFAT signaling pathway in innate immunity have emerged, including that it protects against fungal and bacterial infections (6–10), maintains intestinal homeostasis (11), promotes vasodilation through prostaglandin E2 (PGE2) production (12), and is proapoptotic in DCs in response to Gram-negative bacteria or lipopolysaccharide (LPS) (3, 13). Moreover, during chronic inflammation, deregulation of the NFAT signaling pathway in macrophages causes hyperinflammation and exacerbates disorders such as inflammatory bowel disease and rheumatoid arthritis (14, 15). We have previously shown that acute exposure of DCs to LPS causes CD14, in a TLR4-independent manner, to induce the activation of NFAT by activating Src family kinases (SFKs) and phospholipase C γ2 (PLCγ2), which, in turn, induces a rapid monophasic Ca2+ influx and calcineurin activation (3).
Ca2+ mobilization is one of the first events in the activation of the NFATc1 to NFATc4, the four main members of the NFAT pathway. In electrically nonexcitable cells, the main Ca2+ entry pathway is promoted by the depletion of intracellular Ca2+ stores, coupled with the opening of specific plasma membrane channels, a mechanism called store-operated Ca2+ entry (SOCE) (16, 17). In immune cells, SOCE is the only pathway described for Ca2+ mobilization that leads to NFAT activation. SOCE is initiated by the binding of antigen to the clonotypic receptors of T and B cells, resulting in activation of PLC and the production of inositol 1,4,5-trisphosphate (IP3). This second messenger binds to the IP3 receptors (IP3Rs) located on the membrane of the endoplasmic reticulum (ER) and causes a rapid Ca2+ release from the ER, followed by the opening of Ca2+ release-activated Ca2+ (CRAC) channels at the cell surface. This process generates a sustained increase of intracellular Ca2+ concentration that is necessary for the activation of NFATc family members, NFATc target gene expression, and the acquisition of effector functions (18). However, Ca2+ entry into the cells across the plasma membrane can also occur through two additional mechanisms: receptor-operated Ca2+ entry (in which Ca2+ influx is directly activated by receptor occupation) (19) and second-messenger-operated Ca2+ entry (in which Ca2+ channels in the plasma membrane open in response to the binding of intracellular second messengers, such as products released in response to PLCγ activation) (20).
IP3Rs (21) are important for initiating Ca2+ signaling and are critical for regulating immune responses in health and disease (22). Three different isoforms (IP3R1, IP3R2, and IP3R3) have been identified that have differential expression patterns and gating capacities (21, 23). IP3R1 is most abundant in the nervous system (24), and it is important for embryonic development (25), but it is also present in peripheral tissues, although to a lesser extent (24). In addition, IP3R2 and IP3R2 are widely distributed and prominent in specific cell types and organs, with IP3R2 prevalent in cardiomyocytes and hepatocytes (26) and IP3R3 abundant in the spleen (27). Patterns of Ca2+ signaling mediated by IP3Rs have been determined using genetically engineered B cells that express either single or combined IP3R subtypes (28). IP3R1 generates regular Ca2+ oscillations; IP3R2 generates robust, long-lasting, and regular Ca2+ oscillations; and IP3R3 is associated with monophasic single Ca2+ transients. Given its gating properties, IP3R3 is believed to play a key role in initiating Ca2+ signaling (29, 30).
In DCs, CD14-induced Ca2+ mobilization depends on the activation of SFKs and PLCγ2 (3), which could suggest that SOCE is activated downstream of CD14. Nevertheless, the elimination of this pathway in DCs by deleting the genes encoding the ER-localized Ca2+ sensors stromal interaction molecule 1 (STIM1) and STIM2 does not alter the responses to LPS (31). Therefore, the molecular mechanism of Ca2+ entry for NFAT activation and the channels and molecules involved remain to be determined. Given the importance of this pathway in inflammatory responses, the identification of key factors is biologically and pharmacologically relevant. Here, we found that, downstream of CD14, intracellular Ca2+ increases required IP3 kinase B (ITPKB), which converts IP3 to inositol 1,3,4,5-tetrakisphosphate (IP4). ITPKB was required for the activation of the IP3R3 that are colocalized on the plasma membrane with CD14 and ITPKB. The consequent direct influx of Ca2+ across the plasma membrane led to the nuclear translocation of NFAT that, in turn, promoted key aspects of the inflammatory process elicited by LPS.
RESULTS
LPS induces a direct Ca2+ influx across the plasma membrane
We investigated the mechanism of Ca2+ mobilization that is induced by LPS. To define the dynamics of Ca2+ entry into DCs that were stimulated with LPS, we compared the Ca2+ transients triggered by LPS to a well-known SOCE process that occurs in DCs stimulated with adenosine 5’-triphosphate (ATP) (32, 33). We used the mouse D1 cell line, which closely mimics splenic DCs (sDCs) (34). ATP and other nucleotides bind to specific plasma membrane purinergic receptors called P2Rs, which are subdivided into several classes, including the P2Xa family of ligand-gated ion channels and the G-protein-coupled P2Y receptors (32). In DCs, ATP signaling predominantly occurs through P2Y receptors (33) and leads to phosphatidylinositol hydrolysis, the release of Ca2+ from intracellular stores, and an influx of Ca2+ across the plasma membrane through CRAC channels (22).
Ca2+ mobilization was measured by flow cytometry because the cells respond synchronously to the stimulus (fig. S1A). LPS-stimulated cells showed a rapid monophasic Ca2+ transient (Fig. 1A) that was completely inhibited upon addition of the Ca2+ chelator EGTA to the medium to inhibit extracellular Ca2+ entry (Fig. 1A). The same results were obtained in Ca2+-free medium and by analyzing the cells with confocal microscopy using a ratiometric dye [fig. S1, B and C, and (3)]. ATP induced the expected biphasic Ca2+ entry in D1 cells: a rapid Ca2+ transient (ER store depletion), followed by a CRAC-mediated long-term plateau (Fig. 1A). Extracellular EGTA selectively impaired only the delayed inward Ca2+ influx, but not the first Ca2+ wave (Fig. 1A). The same effect was observed when D1 cells were pretreated with the potent and selective CRAC channel inhibitor N-[4–3,5-bis(trifluromethyl)pyrazol-1-yl]-4-methyl-1,2,3-thiadiazole-5-carboxamide (YM-58483) (35–38) before the ATP stimulation (Fig. 1A). In contrast, YM-58483 did not inhibit Ca2+ mobilization in DCs that were treated with LPS (Fig. 1A), indicating that in this case, CRAC channels were not involved.
Fig. 1. Modalities of Ca2+ mobilization induced by LPS in DCs.
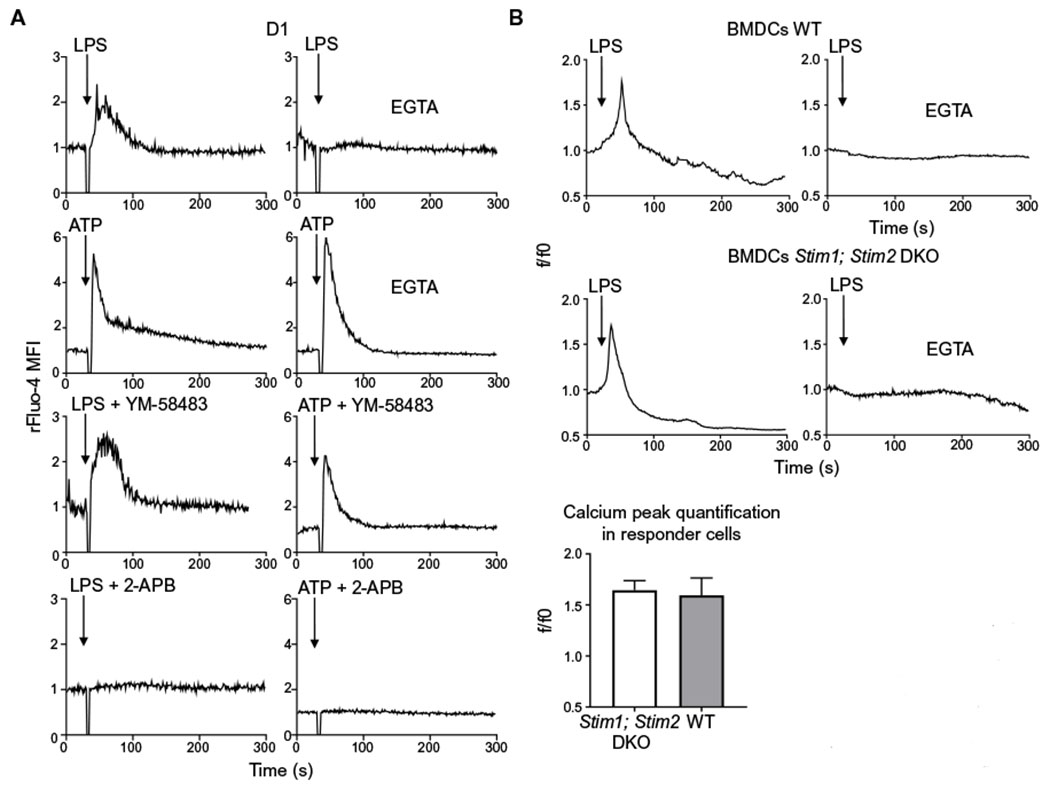
(A) Representative Ca2+ transients in mouse D1 cells treated with LPS or ATP in the presence or absence of the Ca2+ chelator EGTA, the CRAC inhibitor YM-58483, or the general ion channels inhibitor 2-APB. Arrows indicate the time (30 s) of LPS or ATP addition. Ca2+ fluctuations were evaluated by FACS on bulk populations as changes in Fluo-4 fluorescence in response to the stimuli and normalized to the value during the 30 s before LPS or ATP addition (rFluo-4 MFI). Traces are representative of three independent experiments. (B) Representative Ca2+ transients in wild-type (WT) and Stim1; Stim2 double-deficient mouse BMDCs. Ca2+ mobilization was measured by confocal microscopy in the presence or absence of EGTA. Arrows indicate the time (30 s) of LPS administration. Ca2+ fluctuations were evaluated as changes in Fluo-4 fluorescence in response to the stimuli and normalized over the first 30 s of analysis (f/f0). A minimum number of 100 cells in each group was analyzed. The quantification analysis shows the increase of fluorescence intensity in the peak interval for 25 responder cells. One experiment representative of two independent experiments is shown. DKO, double-knockout.
When we pretreated D1 cells with 2-aminoethoxydiphenyl borate (2-APB), a nonselective modulator of several ion channels including IP3Rs, we observed that LPS- and ATP-induced Ca2+ mobilizations were completely blocked (Fig. 1A). In keeping with the results obtained with these inhibitors, Ca2+ fluxes were not affected in bone marrow (BM)-derived DCs (BMDCs) lacking both Stim1 and Stim2, which showed a response similar to wild-type (WT) BMDCs (Fig. 1B). Because BMDCs are heterogeneous (39), with only about 20 to 30% responding asynchronously to LPS (fig. S1D) (3), and therefore not suitable for bulk cytofluorimetric measurements, we analyzed Ca2+ mobilization in BMDCs by confocal microscopy. These results confirm our previous observation (3) that LPS induces a direct Ca2+ influx across the plasma membrane of DCs.
IP3R3 channels on the DC plasma membrane mediate Ca2+ entry after LPS stimulation
After LPS stimulation, PLCγ2 activation mediated by SFKs is required for Ca2+ mobilization (3). This suggests that IP3, generated after the hydrolysis of phosphatidylinositol 4,5-bisphosphate by PLCγ2, could be the second messenger involved in this pathway, but in contrast to T cells and according to our previous results, IP3Rs operate Ca2+ entry directly from the plasma membrane in DCs. Thus, on the basis of our prediction, we asked whether IP3Rs in DCs are present not only at the ER membrane but also on the plasma membrane, as has been shown for B cells (40). For this analysis, we used total internal reflection fluorescence (TIRF), confocal, and electron microscopy. Results of TIRF microscopy were compatible with a localization of IP3R3, but not IP3R1 and IP3R2, on the plasma membrane of D1 cells (Fig. 2A). Confocal analysis confirmed this observation. Sites of overlapping staining with the plasma membrane were observed for IP3R3, whereas IP3R1 and IP3R2 localized to the ER and nuclear membranes (Fig. 2B). Transmission electron microscopy (TEM) analysis confirmed that IP3R3 was present not only on intracellular membranes (presumably the ER) but also on the plasma membrane of D1 cells (Fig. 2C and fig. S2A). Last, the localization of IP3R3 on the plasma membrane was confirmed also by surface protein biotinylation and immunoprecipitation assay (Fig. 2D and fig. S2B).
Fig. 2. IP3R3 channels are present on the plasma membranes of DCs.
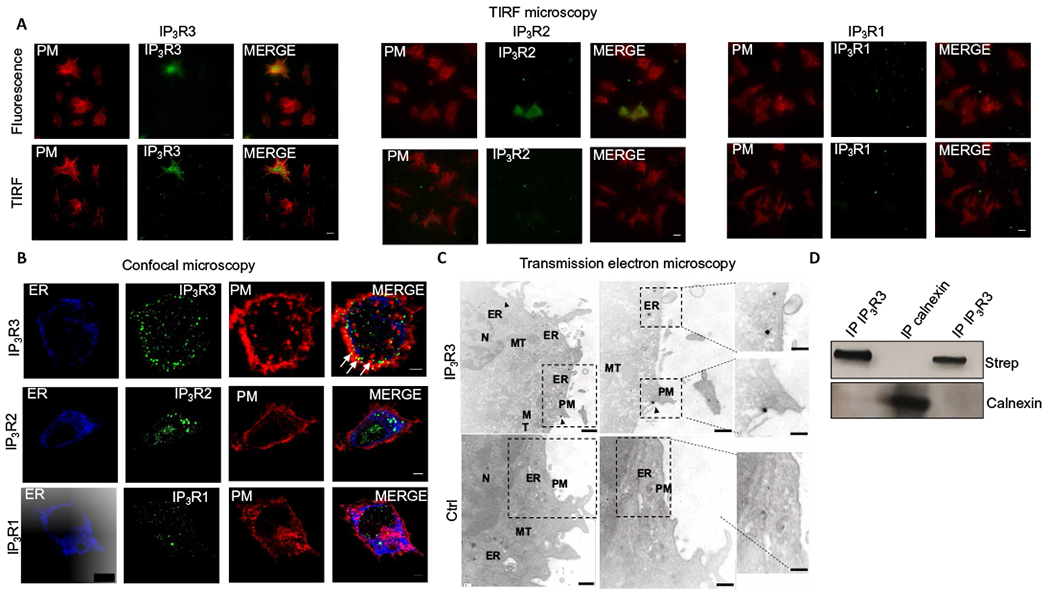
(A) TIRF imaging for plasma membrane (PM) glycoproteins (red) and the indicated IP3Rs (green) in D1 cells. Scale bars, 10 μm. Images are representative of three independent experiments. (B) Confocal microscopy of D1 cells labeled to show the endoplasmic reticulum (ER) marker calnexin (blue), the indicated IP3Rs (green), and the plasma membrane (red). Scale bars, 3 μm. Images are representative of three independent experiments. Arrows indicate sites of IP3R3 and plasma membrane colocalization. (C) TEM showing IP3R3 localization in D1 cells stained with a primary antibody specific for IP3R3 and nanogold-conjugated secondary antibody (IP3R3) or with only nanogold-conjugated secondary antibody as a control (control). Arrows indicate IP3R3 localized at the plasma membrane. ER, mitochondria (MT), and nucleus (N) are also indicated. Scale bars, 200 nm (main) and 50 nm (insets). Images are representative of three independent experiments. (D) D1 cells were surface-biotinylated, and the lysates subjected to immunoprecipitation with two different antibodies specific for IP3R3 (lanes 1 and 3) or an antibody specific for calnexin (lane 2) as a control. After SDS-polyacrylamide gel electrophoresis and transfer to nitrocellulose, membranes were probed with streptavidin or calnexin antibody.
To determine the involvement of IP3R3 in activating the CD14-Ca2+-NFAT pathway in DCs stimulated by LPS, we silenced IP3R3 expression in DCs by RNA interference (fig. S2, C and D). IP3R3 knockdown completely abrogated Ca2+ transients (Fig. 3A and fig. S3A) in response to LPS. We also evaluated the impact of IP3R3 knockdown in the regulation of the transcription of the NFAT-dependent genes il2 and Ptges1 (12). As expected, IP3R3 silencing strongly diminished the capacity of LPS-treated DCs to increase il2 and Ptges1 expression, whereas the induction of Tnfa, which is NF-κB dependent and NFAT independent, was unaffected (Fig. 3B). These data demonstrate that IP3R3 is expressed not only on the ER membrane but also at the cell surface and is the Ca2+ channel involved in Ca2+ mobilization in response to LPS.
Fig. 3. IP3R3 channels are required for LPS-induced Ca2+ mobilization and NFAT activation in DCs.
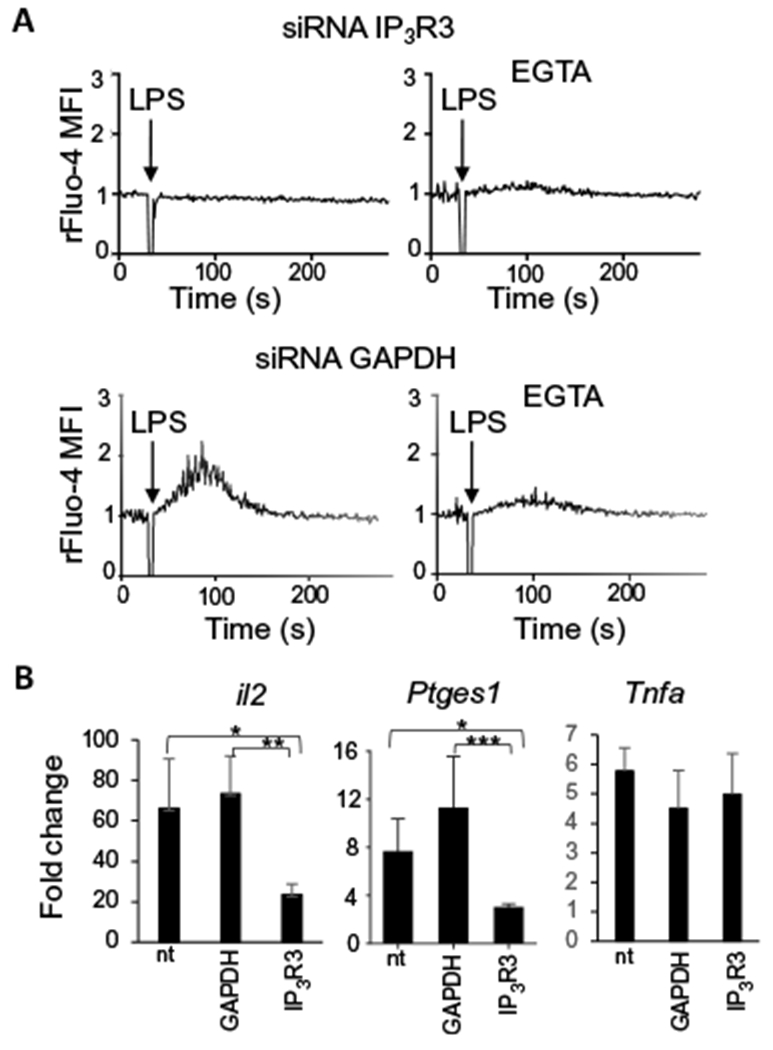
(A) Ca2+ transients induced by LPS in D1 cells, 48 hours after the knockdown of IP3R3 with specific siRNAs in the presence or absence of the extracellular calcium chelator EGTA. Knockdown of GAPDH with specific siRNA was used as negative control. Arrows indicate the time (30 s) of LPS administration. Ca2+ fluctuations were evaluated by FACS on bulk populations as changes in the Fluo-4 fluorescence, in response to the stimuli, and were normalized to the value during the 30 s before LPS addition (rFluo-4 MFI). Traces are representative of three independent experiments. (B) Real-time polymerase chain reaction (PCR) analysis of the increase in il2, Ptges1, and Tnfa expression after 4 hours of LPS treatment in D1 cells transfected or not (nt) with siRNAs specific for GAPDH (control) or IP3R3. Values indicate the mean ± SEM from six independent experiments. Statistical significance was determined with one-way analysis of variance, followed by Tukey’s multiple comparisons test, *P < 0.05, **P < 0.01 and ***P < 0.001.
IP3R3 and CD14 colocalize at the plasma membrane
After LPS stimulation, Ca2+ influx in DCs totally depends on CD14 and IP3R3 (Fig. 2C). We thus hypothesized that CD14 and IP3R3 colocalize and evaluated this by using two different approaches: (i) stimulated emission depletion (STED) microscopy, and (ii) proximity ligation assay (PLA). Both STED microscopy and PLA showed IP3R3 and CD14 colocalization (Fig. 4, A and B). Both D1 cells and sDCs were used for the PLA analysis, and colocalization was quantified (Fig. 4B). The colocalization of CD14 with TLR4 after LPS treatment was used as a positive control, and colocalization of CD14 with major histocompatibility complex class II (MHCII) or IP1R1 was used as a negative control (Fig. 4B). As an additional control, we measured CD14 and IP3R3 colocalization after having altered the organization of the plasma membrane by cyclodextrin-mediated cholesterol depletion. The elimination of cholesterol abrogated the colocalization (Fig. 4C). Accordingly, treating DCs with cyclodextrin also impaired Ca2+ mobilization in response to LPS but not in response to ATP (Fig. 4D and fig. S3B).
Fig. 4. IP3R3 and CD14 colocalize in lipid rafts at the plasma membrane in mouse and human DCs.
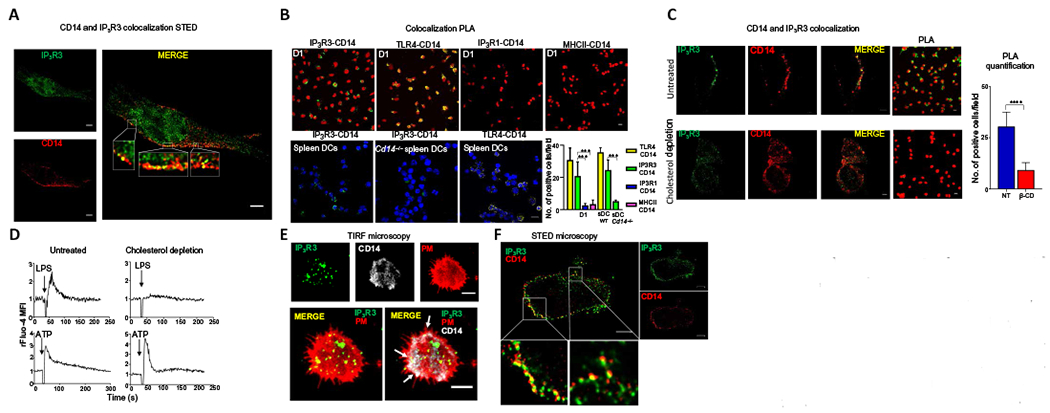
(A) STED microscopy showing localization of IP3R3 and CD14 in mouse D1 cells. Scale bars, 2 μm. Images are representative of three independent experiments. (B) PLA visualized by confocal microscopy for the indicated pairs of proteins in LPS-stimulated D1 cells or splenic DCs (sDCs) from WT or Cd14−/− mice. Sites of colocalization between CD14 and IP3R3 appear green. Sites of CD14 and TLR4 colocalization appear yellow (positive control). PLA between CD14 and IP3R1 (blue) and between CD14 and MHCII (pink) are negative controls. Nuclei are shown in red (top) or in blue (bottom). Scale bar, 10 μm. Images are representative of three independent experiments. Numbers of cells showing IP3R3-CD14, TLR4-CD14, IP3R1-CD14, and MHCII-CD14 colocalization were quantified. Statistical significance was determined with Sidak’s multiple comparisons test, ***P < 0.001, n = 10 fields of cells analyzed from three independent experiments. (C) Immunofluorescence and PLA showing IP3R3 (green) and CD14 (red) in untreated D1 cells and after lipid raft disruption by β-cyclodextrin (cholesterol depletion). Scale bars, 3 μm. Images are representative of three independent experiments. Numbers of cells showing IP3R3-CD14 colocalization in untreated (NT) or treated (β-CD) cells are also shown. Statistical significance was determined with unpaired Student’s t test, ****P < 0.0001, n = 8 fields of cells analyzed from three independent experiments. (D) Ca2+ transients in D1 cells after lipid raft disruption. Cells were treated (cholesterol depletion) or not (untreated) with β-cyclodextrin for 30 min before LPS activation. Arrows indicate the time of LPS or ATP (100 μM) administration. Ca2+ fluctuations were evaluated by FACS on the bulk population as changes in Fluo-4 fluorescence in response to the stimuli and normalized over the first 30 s of analysis (rFluo-4 MFI). Traces are representative of three independent experiments. (E) TIRF microscopy showing IP3R3 and CD14 in human CD1c+CD14+ cells. PM, plasma membrane (red). Scale bars, 2 μm. Images are representative of cells from five different donors. (F) STED microscopy showing IP3 R3 and CD14 in human CD1c+CD14+ cells. Scale bars, 2 μm. Images are representative of cells from three different donors.
DC3 cells are a subpopulation of type 2 DCs that are CD1c+CD14+ and present in human blood (41,42). We isolated DC3s by first purifying CD1c+ cells by magnetic-activated cell sorting (MACS) and then using fluorescence-activated cell sorting (FACS) to isolate CD14+ DCs from the CD1c+ population (fig. S4). This population differs from monocytes in the amount of CD1c on the cell surface, with monocytes being CD1clow (43). In line with the results obtained with mouse DCs, TIRF analysis confirmed the presence of IP3R3 on the plasma membrane of CD1c+CD14+ DC3 cells (Fig. 4E), and TIRF and STED analyses showed colocalization of CD14 and IP3R3 (Fig. 4, E and F). Collectively, these findings demonstrate that CD14 and IP3R3 colocalized in mouse DCs and in a subpopulation of human DCs and suggest that their colocalization is required for the increase in intracellular [Ca2+] in DCs after LPS stimulation.
IP4 is a second messenger required for Ca2+ mobilization
To understand why IP3R3s in the plasma membrane are preferentially activated relative to those in the ER, we reasoned that IP3 may be generated in such low amounts by plasma membrane-associated PLC that it is effective only locally. IP3 is quickly converted to IP4 by two different classes of enzymes: ITPKs (44) and inositol polyphosphate multikinase. In addition, IP3 can be converted to IP2 by IP3 phosphatases. We reasoned that inhibiting the metabolic conversion of IP3 to IP4 isomers and IP2 should lead to increased amounts of IP3 and possibly to the activation of IP3R3 on the ER. The inhibition of ITPKs with either of two different inhibitors that target all three ITPK isoforms (A, B, and C), TNP [N2-(m-(trifluoromethyl)benzyl) N6-(p-nitrobenzyl)purine] and GNF362 (45), completely abrogated LPS-induced Ca2+ mobilization in D1 cells (Fig. 5A and fig. S5A). This indicated that the generation of IP4 was a prerequisite for inducing Ca2+ entry.
Fig. 5. IP4 is a second messenger required for Ca2+ mobilization in DCs after LPS stimulation.
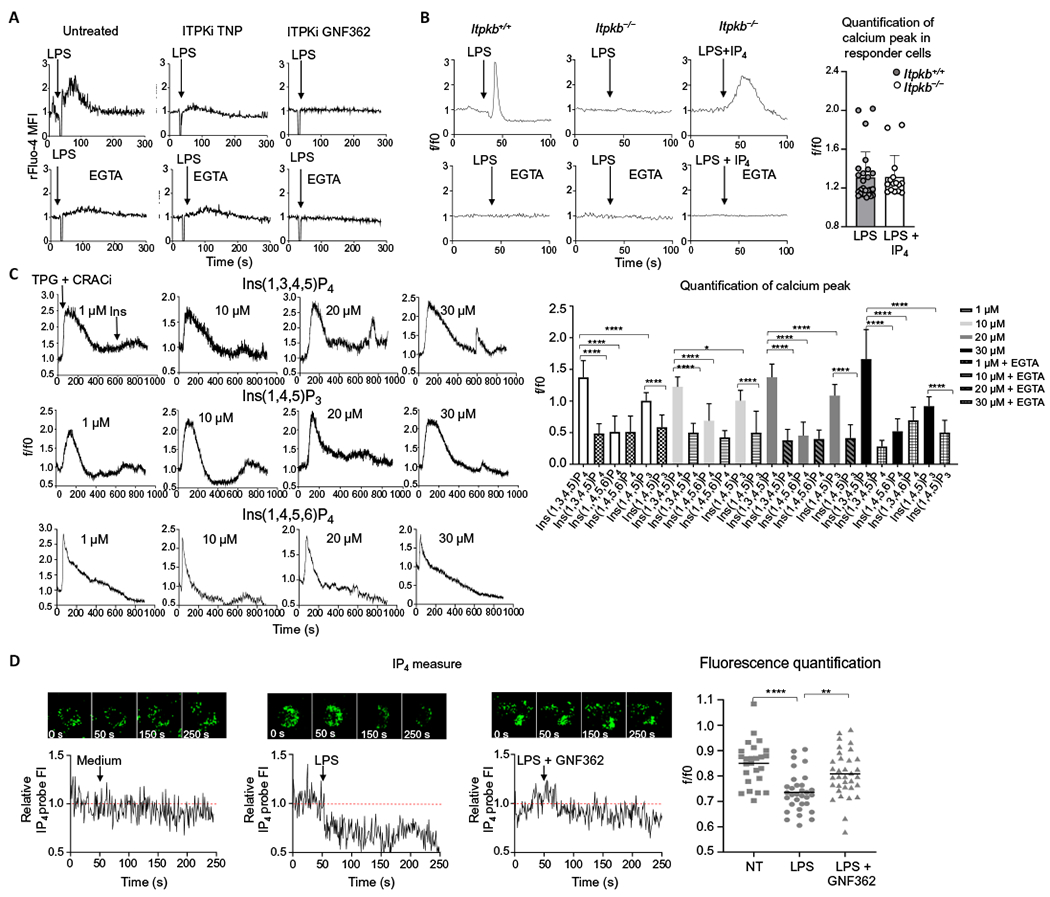
(A) Ca2+ transients in mouse D1 cells that were untreated or pretreated with the ITPK inhibitors (ITPKi) TNP or GNF362 before being stimulated with LPS in the absence or presence or EGTA, as indicated. Arrows indicate LPS administration at 30 s. Ca2+ fluctuations were evaluated by FACS on the bulk population as changes in the Fluo-4 fluorescence in response to the stimuli and normalized over the first 30 s of analysis (rFluo-4 MFI). Traces are representative of three independent experiments. (B) Representative Ca2+ transients in BMDCs from WT (Itpkb+/+) and Itpkb−/− mice in the presence or absence of EGTA. Arrows indicate the addition of LPS or LPS plus IP4 at 40 s. Ca2+ fluctuations were measured by confocal microscopy as changes in Fluo-4 fluorescence in response to the stimuli and normalized over the first 40 s of analysis (rFluo-4 MFI). A minimum number of 100 cells in each group was analyzed. The quantification analysis shows the increase of fluorescence intensity in the peak interval in n = 25 responder cells. (C) Representative Ca2+ profiles of D1 cells pretreated with CRAC inhibitor (CRACi) YM-58483 and then treated with thapsigargin (TPG) to deplete the intracellular Ca2+ stores and, last, with cell-permeant Ins(1,3,4,5,)P4, Ins(1,4,5)P3, or Ins(1,4,5,6)P4 at the indicated doses (see also fig. S5 for Ca2+ plots in the presence of EGTA). Thapsigargin was added after 30 s and inositols after 600 s of data acquisition. Ca2+ fluctuations were evaluated as changes in Fluo-4 fluorescence in response to the stimuli and normalized over the first 30 s of analysis (rFluo-4 MFI). The quantification analysis shows the increase of fluorescence intensity in the peak interval with respect to the first 30 s for responder cells or the increase of fluorescence intensity after the addition of inositols (600 s) until the end of the experiment (900 s) for nonresponder cells. Statistical significance was determined using one-way analysis of variance, followed by Sidak’s multiple comparisons test, ****P < 0.0001 and *P < 0.05, n = 20 responder cells. Data are representative of three independent experiments. (D) Measure of IP4 increase revealed by confocal microscopy in D1 cells transfected with a fluorescent probe that is quenched by IP4 binding and pretreated (or not) with GNF362. Arrows indicate administration of plain medium or LPS at 50 s. Fluctuations in IP4 amounts were evaluated as changes in the IP4 probe fluorescence in response to the stimuli and normalized over the first 50 s of analysis (relative IP4 probe FI). Traces show a representative profile of a single cell with the corresponding images at the indicated time points. The quantification analysis shows the mean of the intensities of IP4 probe after LPS administration, normalized over the first 50 s of analysis (f/f0). Statistical significance was determined using one-way analysis of variance, followed by Tukey’s multiple comparisons test, ****P < 0.0001 and **P < 0.01, n = 25 cells. Data are representative of three independent experiments.
Among the three ITPK isoforms, ITPKA is restricted to the central nervous system, whereas ITPKB and ITPKC are detected in multiple organs, although ITPKC is largely restricted to epithelial cells (46–48). We thus focused our attention on ITPKB, which is active in immune cells (49–51) and is the only isoform that can associate with the plasma membrane (52, 53) and tested whether it was the isoform that was required for Ca2+ mobilization in DCs. We first confirmed the presence of ITPKB in WT BMDCs by Western blot (fig. S5B). Then, we measured Ca2+ mobilization in BMDCs derived from ITPKB-deficient mice, which demonstrated that Ca2+ influxes in response to LPS were completely abrogated but rescued by the addition of cell-permeant IP4 (Fig. 5B).
To confirm that IP4 facilitates Ca2+ influxes through the plasma membrane through IP3Rs at the cell surface, we pretreated D1 DCs with thapsigargin to deplete the intracellular Ca2+ stores in the presence of a CRAC inhibitor to prevent Ca2+ influx and then treated the cells with cell-permeant IP3 or IP4. A Ca2+ transient was reproducibly observed in single cells with IP3 or IP4 treatment after intracellular store depletion, and it was inhibited by the addition of EGTA to the medium (Fig. 5C and fig. S5C), indicating that IP3 and IP4 could induce a Ca2+ influx through the plasma membrane. At all concentrations tested, IP4 was more efficient than IP3 in allowing Ca2+ entry across the plasma membrane (Fig. 5C). An alternative IP4 isomer [Ins(1,4,5,6)P4] did not induce any Ca2+ flux (Fig. 5C and fig. S5C) under these conditions.
To evaluate IP4 generation in DCs stimulated with LPS, we used a biosensor that enables real-time monitoring of IP4 (54). A clear ITPK-dependent increase in the amount of IP4, assessed as a decrease in the presence of the free probe, occurred in D1 cells exposed to LPS (Fig. 5D). Consistent with the notion that Ca2+ fluxes are compartmentalized, we also found sites of colocalization of IP3R3 and ITPKB by STED microscopy (fig. S6A). Accordingly, STED analysis revealed regions of colocalization of the three key molecules CD14, IP3R3, and ITPKB in human CD1c+CD14+ DC3 cells (fig. S6B). Together, these data indicate that the IP4 that is generated by activation of ITPKB when DCs are exposed to LPS is a second messenger required for Ca2+ mobilization.
IP4 is required for LPS-induced nuclear translocation of NFAT in vitro and in vivo
An increase of intracellular Ca2+ concentration and calcineurin activation are early events in the pathway leading to NFAT activation when SOCE is involved. To investigate whether Ca2+ mobilization and calcineurin activation were required for the nuclear translocation of NFAT also in our system, DCs were activated with LPS in the presence or absence of EGTA and FK506 (tacrolimus) to chelate the extracellular Ca2+ and to inhibit the phosphatase activity of calcineurin, respectively. For this experiment, we focused on NFATc2, which is an NFAT family member that is highly abundant in DCs (3). Both extracellular Ca2+ and calcineurin were required for the LPS-induced translocation of NFAT into the nucleus (Fig. 6A). Given that CRAC channels are not required for Ca2+ mobilization in LPS-activated DCs, we also blocked CRAC channels with YM-58483 to confirm that their activation was not required for the translocation of NFAT. In according with this prediction, LPS induced the nuclear translocation of NFAT when CRAC channels were inhibited, confirming that they are not necessary for NFAT activation (Fig. 6A).
Fig. 6. ITPKs are essential for LPS-dependent activation of the NFAT signaling pathway in vitro and in vivo.
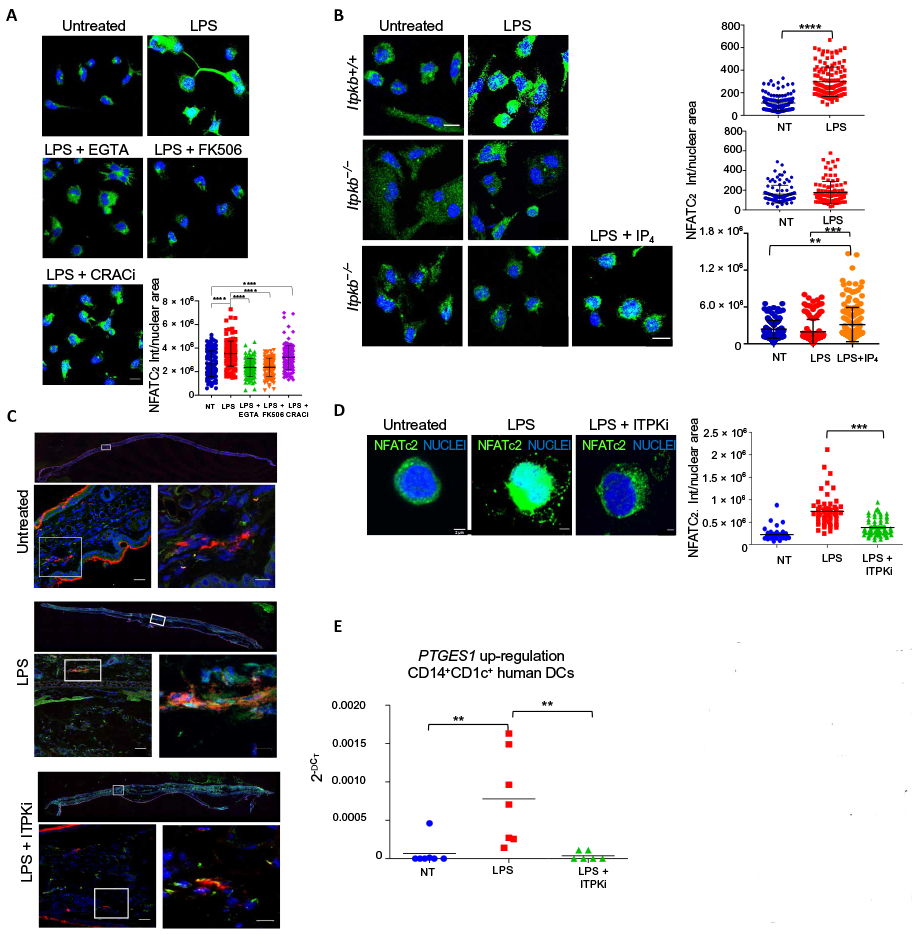
(A) Representative confocal immunofluorescence images showing NFATc2 in mouse BMDCs treated (or not) with LPS 1.5 hours in the presence or absence of EGTA, the calcineurin inhibitor FK506, or the CRAC inhibitor YM-58483 (CRACi). Nuclei were counterstained blue. Scale bar, 10 μm. Quantification of NFATc2 nuclear signal is shown. Statistical significance was determined using one-way analysis of variance, followed by Tukey’s multiple comparisons test, ****P < 0.0001, n = 105 cells from three independent experiments. (B) Representative confocal immunofluorescence images showing NFATc2 in BMDCs isolated from Itpkb+/+ or Itpkb−/− mice and treated (or not) with LPS for 1.5 hours in the presence or absence of cell-permeant IP4. Nuclei were counterstained blue. Scale bar, 10 μm. Quantification of NFATc2 nuclear signal is shown. Statistical significance was determined using one-way analysis of variance, followed by Tukey’s multiple comparisons test or using unpaired student’s t test. ****P < 0.0001 and **P < 0.01, n = 100 cells from three independent experiments. (C) Confocal immunofluorescence images showing NFATc2 (green) and MHCII (red) in ear sections of mice 1.5 hours after subcutaneous injection of PBS (untreated), LPS, or LPS plus the ITPK inhibitor TNP (ITPKi). Nuclei were counterstained blue. Images are representative of three independent experiments. Insets of each panel represent higher magnifications of the selected areas. Scale bars, 25 μm (lower magnification inset), 10 μm (higher magnification inset). ***P < 0.001. (D) Immunofluorescence showing NFATc2 in human CD1c+CD14+ cells, treated (or not) with LPS for 1.5 hours. Where indicated, cells were preincubated with the ITPK inhibitor TNP (ITPKi). Nuclei are counterstained blue. Scale bars, 2 μm. Quantification of the NFATc2 nuclear signal is shown. Statistical analysis was performed with one-way analysis of variance followed by Bonferroni’s multiple comparisons test, ***P < 0.001, n = 60 cells from three different donors. (E) Real-time PCR analysis of PTGES1 expression in human CD1c+CD14+ cells treated (or not) for 4 hours with LPS in the presence or absence of ITPK inhibitor TNP (ITPKi). n = 7 donors. Statistical significance was performed with Wilcoxon test. **P < 0.01.
We next analyzed the involvement of ITPKs in activating the NFAT signaling pathway. NFAT translocation in response to LPS was evaluated in vitro in BMDCs derived from WT or ITPKB-deficient mice. We also measured NFAT translocation in vivo in MHCII+ cells after intradermal LPS administration in the presence or absence of the ITPK inhibitor TNP. In agreement with the results on Ca2+ mobilization (Fig. 5, A and B), LPS-induced nuclear NFATc2 translocation was absent in ITPKB-deficient DCs and was rescued by the addition of IP4 to the cultures (Fig. 6B). NFATc2 translocation was also observed in vivo, where it was prevented by the pharmacological inhibition of ITPKs (Fig. 6C).
We then assessed NFAT nuclear translocation in human CD1c+CD14+ DCs. In agreement with the results obtained with mouse DCs, NFAT nuclear translocation was induced by LPS stimulation and inhibited upon ITPK inhibition (Fig. 6D). Furthermore, the induction of the expression of the NFAT-dependent gene PTGES1 in human CD1c+CD14+ DCs depended on ITPKs, implying that the mechanism initiated by CD14 in mouse DCs also operates in human DCs (Fig. 6E). Overall, these observations indicate that ITPKB is required for NFAT translocation downstream of LPS-induced Ca2+ mobilization in both human and mouse DCs.
Inhibition of ITPKs reduces LPS-induced, NFAT-dependent inflammatory events in vivo
We next explored the role of ITPKB in the NFAT signaling pathway in vivo, during LPS-mediated inflammation. We have previously shown that the increase of vascular permeability during the inflammatory process induced by LPS in vivo depends on NFAT activation in DCs (12). We thus evaluated whether the absence of ITPKB or ITPK inhibition interfered with the vascular leakage induced by LPS. Mice received an intradermal injection of LPS in the ear, after which the vascular permeability was evaluated by Evans blue extravasation. Extravasation was significantly reduced in ITPKB-deficient mice and in mice treated with the ITPK inhibitor TNP (Fig. 7A and fig. S7A). Analogously, vascular permeability was also reduced in mice treated with small interfering RNAs (siRNAs) targeting IP3R3 (Fig. 7A and fig. S7, A and B). Vascular leakage was restored by adoptive transfer of with WT, but not ITPKB-deficient, DCs into the ears of the ITPKB-deficient mice (Fig. 7B). Vascular leakage was also restored also injecting PGE2 into the ear (Fig. 7A and fig. S7A). NFAT activation in DCs increases vascular permeability through the induction of Ptges1, which encodes microsomal prostaglandin E synthase-1, an enzyme required for generation of the efficient vasodilator PGE2 (12). When the IP3R3-ITPKB pathway was inhibited, Ptges1 was also dampened, whereas the transcription of Tnfa was unaffected (fig. S7C). The observation that ITPK inhibition selectively affected the NFAT pathway was further confirmed in vitro. The induction of Ptges1 in DCs after LPS treatment was inhibited by the ITPK inhibitor, whereas the expression of proinflammatory myeloid differentiation primary response 88-dependent (Tnfa) and TRIF-dependent (Viperin) genes (fig. S7D), as well as TLR4 internalization (fig. S7E), were not affected by the presence of the ITPK inhibitor.
Fig. 7. ITPK inhibition in vivo reduces the inflammatory events promoted by NFAT.
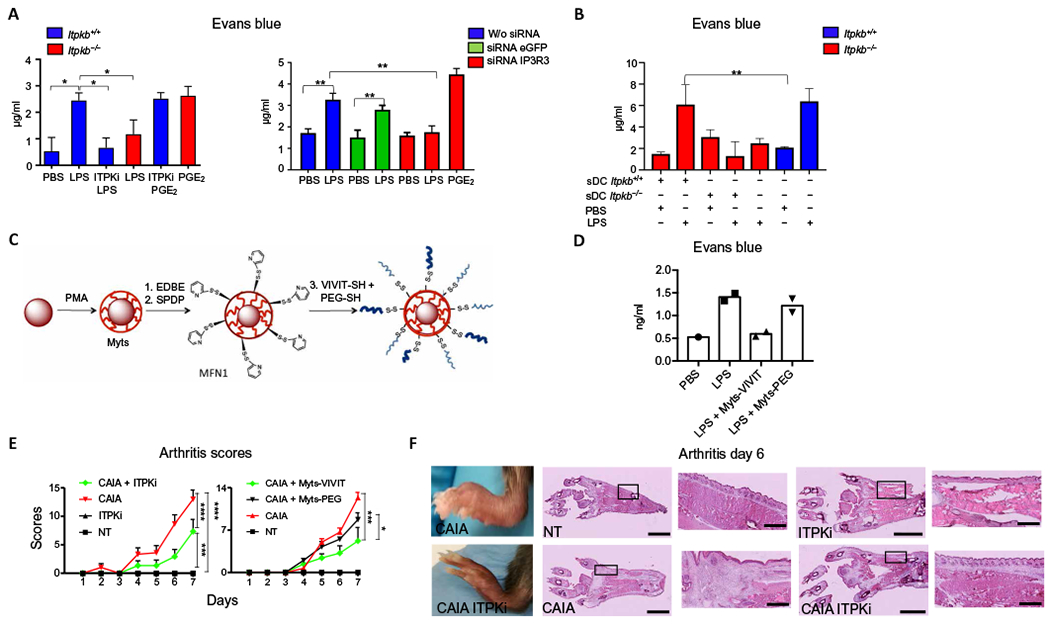
(A) Quantification of Evans blue extravasation in the ears of mice. WT (Itpkb+/+) and Itpkb−/− mice were simultaneously injected intravenously with Evans blue and subcutaneously with PBS or the indicated combinations of LPS, PGE2, and the ITPK inhibitor TNP (ITPKi). Mice treated with siRNAs targeting IP3R3 or eGFP or with vehicle alone (w/o siRNA) 24 hours before the assay were also injected with PBS or LPS. Data represent Evans blue quantification from excised ears 30 min after challenge. Values indicate mean ± SD from three mice. Statistical significance was determined with Mann-Whitney test, ***P< 0.001, **P< 0.01, and *P< 0.05. (B) Vascular leakage assay as in (A). Itbk−+/+ or Itbkb−/− DCs were subcutaneously injected (sDC) into the ears of Itbk−+/+ or Itbkb−/− mice before PBS or LPS treatment. The values indicate mean ± SD from three mice for each experimental condition. Statistical significance was determined with one-way analysis of variance, followed by Sidak’s multiple comparisons test, **P < 0.01. (C) Schematic representation of Myts nanoparticles functionalized with thiol-reactive groups and then conjugated with a mixture of VIVIT-SH peptide and PEG-SH, to yield Myts-VIVIT. SPDP, N-succinimidyl-3-[2-pyridylthio]-propionate. MFN1, Myts functionalized with thiol-reactive group. (D) Vascular leakage assay as in (A). Mice were treated with Myts-VIVIT or control Myts-PEG nanoparticles intravenously before LPS treatment. Each symbol represents an individual mouse. (E) CAIA clinical scores evaluated at the indicated time points. Mice were treated with ITPKi every other day starting from day −1 or with Myts-VIVIT or Myts-PEG nanoparticles every other day starting from day −1. Values represent mean ± SD. Statistical significance was performed with one-way analysis of variance, followed by Tukey’s multiple comparisons test. The one-way analysis of variance revealed a variation over time (P < 0.0001), differences between groups (P < 0.0001), and interactions between groups (P < 0.0001). ****P < 0.0001, ***P < 0.001, and *P < 0.05. n = 6 mice for each treatment regimen. (F) Images of intact and hematoxylin and eosin-stained sections of hindlimbs of mice 6 days after CAIA induction that were untreated (NT) or treated with ITPKi. Images are representative of six animals for each treatment group. Scale bars, 2.5 mm (whole sections), 0.5 mm (insets).
The inhibition of vascular permeability observed with ITPK inhibition was recapitulated by the inhibition of NFAT activation in vivo with use of the VIVIT peptide. The VIVIT peptide is a 16-mer oligopeptide that has high affinity for the NFAT-binding site of calcineurin, and it effectively inhibits the activation of NFAT without affecting the activation of NF-κB (55). To use the VIVIT peptide in vivo, we conjugated it to nanoparticles to protect it from degradation. We selected superparamagnetic iron nanoparticles (termed Myts nanoparticles) for their high colloidal stability (56, 57). The core of the Myts nanoparticles consists of an iron oxide nanocrystal that is tightly wrapped by an amphiphilic polymer poly(isobutylenealt-maleic anhydride)-graft-dodecylamine (PMA) (58), which provides colloidal stability in aqueous solution, and has multiple functional groups suitable for straightforward bioconjugation (59). We functionalized Myts nanoparticles with the VIVIT peptide (Fig. 7C) using a conjugation strategy that exploits a disulfide bridge-bearing linker between the peptide and the nanoparticle (Fig. 7C). Nanoparticles were developed and functionalized partly with the VIVIT oligopeptide and partly with polyethylene glycol (PEG) to further increase the colloidal stability in biological fluids (Fig. 7C).
We evaluated the efficacy of Myts-VIVIT in blocking NFAT activation in DCs in vitro. Myts-VIVIT nanoparticles effectively blocked nuclear translocation of NFAT, but not of NF-κB (fig. S8A), and inhibited the production of interleukin-2 (IL-2), but not of tumor necrosis factor (TNF) (fig. S8B). The advantage of using nanoparticles was not limited to the protection of the peptide from degradation, because nanoparticles also favored VIVIT peptide uptake almost exclusively by phagocytes in skin spleen and lymph nodes, thus confining the effect of the NFAT inhibition to these cells (fig. S8C). We next used the Myts-VIVIT nanoparticles for the subsequent experiments in vivo. Myts-VIVIT nanoparticles blocked LPS-induced vascular leakage similarly to ITPK inhibition (Fig. 7D), indicating that the direct inhibition of NFAT or ITPK has the same biological effect.
The relevance of ITPK-dependent NFAT activation was also tested in the mouse collagen antibody-induced arthritis (CAIA) model in which arthritis is induced by collagen-specific antibodies and enhanced by the injection of LPS (60). This model of arthritis offers some advantages with respect to the classic collagen-induced arthritis model. First, it is induced by LPS and independent of adaptive immunity, therefore suitable for our studies. Second, the treatment induces arthritis within a few days, whereas arthritis induced by immunization with type II collagen requires weeks to develop. Arthritis was induced in the presence or not of the ITPK inhibitor TNP or with injection of Myts-VIVIT (or control) nanoparticles to block the NFAT signaling pathway in phagocytes. The inhibition of ITPK and the direct inhibition of NFAT activation significantly dampened arthritis severity (Fig. 7E). In particular, arthritis developed in an asymmetric way, as already shown for NFATc2-deficient mice (61), and presented with a significantly decreased severity in mice treated with the ITPK inhibitor, as assessed by both clinical disease score and histological examination of inflammatory infiltrates (Fig. 7, E and F). In summary, the abrogation of IP4 generation by ITPK inhibition in vivo blocked LPS-induced activation of the NFAT signaling pathway and NFAT-mediated inflammatory events.
DISCUSSION
We have shown that, after LPS stimulation, Ca2+ mobilization in mouse and human DCs depended on activation of ITPKB, generation of IP4, and the opening of IP3R3 Ca2+ channels that colocalized with CD14 at the cell surface, thus facilitating the influx of Ca2+ directly through the plasma membrane. Because this Ca2+ entry was necessary for the activation of NFAT, functional events promoted by NFAT LPS-induced inflammation therefore depended on ITPKB.
This mechanism of Ca2+ mobilization that is induced by LPS and leads to NFAT activation in DCs is highly unusual. In most other leukocytes, in response to stimuli such as antigens, chemokines, agonists of Fce receptor, and others, Ca2+ mobilization for NFAT activation occurs through a SOCE mechanism (62). We cannot exclude that Ca2+ mobilization for NFAT activation occurs through a SOCE mechanism in myeloid cells downstream of TLRs or TLR co-receptors in other experimental conditions. We have previously shown that other LPS species induce Ca2+ mobilization from intracellular stores (63).
IP3R3 mediates Ca2+ mobilization in DCs and must colocalize with CD14 at the cell surface for Ca2+ influxes to occur in response to LPS (Fig. 4C). IP3Rs are commonly considered to be intracellular receptors, although some evidence exists for IP3R localization on the plasma membrane. The presence of sialylated IP3Rs on the plasma membrane has been documented in lobster olfactory receptor neurons (64), human T lymphocytes (65), and rat liver cells (66, 67). Even when the receptors are intentionally overexpressed, very few of the molecules localize to the cell surface (68). More recently, functional plasma membrane IP3R3s, accounting for part of the Ca2+ mobilization induced by antigen exposure, have been described in B cells (40). Nevertheless, the implication of this mechanism of Ca2+ entry into cells exposed to antigens has remained elusive. Here, we demonstrated that Ca2+ influx through membrane IP3R3 activated the NFAT signaling pathway. On the basis of our data, we hypothesize that, after DC exposure to smooth LPS, IP3 is rapidly metabolized to IP4 that, in turn, promotes the opening of plasma membrane IP3R3. Ca2+ entry through plasma membrane IP3R3 then favors NFAT activation. A single, highly transient spike of increased intracellular Ca2+ concentration was sufficient to initiate the NFAT pathway (Figs. 1, A and B, and 6A). This diverges from the common interpretation (based on studies of T and B cells) that a sustained increase in intracellular Ca2+ concentration is required for NFAT activation (62). Our data support the existence of a second noncanonical model wherein Ca2+ influx from the membrane, rather than a sustained Ca2+ mobilization, is responsible for calcineurin activation.
Because IP3Rs have been found on the plasma membrane of B cells and thymocytes, it will be interesting to dissect this second pathway and investigate whether it is also active in these cells and what are the circumstances in which it is activated, especially given that recognition of low-avidity antigens has been shown to induce transient rather than sustained Ca2+ mobilization (69). It would be interesting to investigate whether these Ca2+ transients can lead to NFAT translocation. For Ca2+ influxes to occur through the plasma membrane, IP3R3 and CD14 must colocalize in microdomains at the cell surface (Fig. 4C). Coclustering of IP3R3 with the signaling complex occurs also during TCR signaling events induced by the antigen (70). It remains to be determined whether IP4 itself is capable of opening the IP3R3 channels located on the cell surface or whether IP4 indirectly promotes the gating functions of IP3R3.
In lymphocytes, the absence of ITPKB results in a sustained SOCE (71), which makes T and B cells anergic and susceptible to apoptosis through the stimulation of the TNF family member FAS ligand and the B cell lymphoma 2 family member B cell lymphoma (Bcl)-2-like protein 11 (45). This may occur because IP4 can act as an inhibitor of Ca2+ entry through calcium release-activated calcium channel protein 1 (50), the pore component of the plasma membrane CRAC channels required for SOCE (62), and may also act by restricting the availability of IP3. In DCs, IP4 favored Ca2+ influxes, and this is in line with the observation that Ca2+ increases generated by LPS stimulation do not depend on SOC channels (Fig. 1A). Although inward Ca2+ currents through plasma membrane IP3R3 can occur in antigen-activated B cells, whether these currents depend on IP4 remains unclear; research efforts are focused on resolving this issue. IP4 may have multiple additional functions in innate immune cells, including inhibition of the Akt and mechanistic target of rapamycin signaling pathway (72). This will be another interesting issue to investigate in future work.
In innate myeloid cells, NFAT promotes the production of the vasoactive mediator PGE2 (12). Accordingly, inhibition of IP4 generation in vivo strongly reduced the vascular permeability induced by LPS (Fig. 7A). Vasodilation and the increase in vascular permeability are among the first events in the inflammatory process. Thus, inhibition of vasodilation is an efficient way to reduce local inflammation. In a model of inflammatory arthritis induced by collagen-specific antibodies and LPS, inhibition of IP4 generation in vivo significantly reduced inflammatory cell recruitment and reduced the severity of the disease (Fig. 7F). The direct inhibition of NFAT activation using the VIVIT peptide and the inhibition of ITPKs had overlapping results (Fig. 7E), in agreement with ITPKB being required downstream of CD14 for NFAT activation (Fig. 6B).
Last, our Myts-VIVIT nanoconjugates that targeted innate immune phagocytes and selectively blocked NFAT activation proved to be effective tools for anti-inflammatory treatments. The uptake of nanoparticles by mononuclear phagocytes is a major obstacle to the use of nanoparticles in nanomedicine, because phagocytes hamper the efficient conveyance of nanodrugs to the desired target area (73). Here, we exploited this intrinsic property of nanoparticles to preferentially deliver active compounds to phagocytic innate immune cells.
This opens the possibility of using similar nanomedicines for the treatment of inflammatory diseases. Our results also showed that such nanoconjugates can serve as new tools for the investigation of signaling pathways activated in phagocytes in vivo.
In conclusion, our data show that in mouse and in CD14+ human DCs, Ca2+ entry downstream of LPS-engaged CD14 required ITPKB, which mediated the production of IP4 that served as a second messenger for promoting the entry of Ca2+ through IP3Rs. After the increase of intracellular [Ca2+], activation of the NFAT pathway in DCs had clear inflammatory consequences in vivo. Our results also highlight the efficacy of using colloidal nanoparticles as effective carriers for delivering active compounds to phagocytes and strongly support ITPKB as a potential target for anti-inflammatory therapies aimed at inhibiting defined DC functions.
materials and methods
Reagents
LPS (Escherichia coli, O55:B5) was purchased from Enzo Life Sciences. 2-APB, YM-58483, EGTA, ATP, dynasore hydrate, β-cyclodextrin, and thapsigargin were purchased from Sigma-Aldrich. Tacrolimus/ FK506 was purchased from Fujisawa Pharmaceutical. TNP was purchased from Tocris. Ins(1,4,5)P3-AM, Ins(1,3,4,5)P4-AM, and Ins(1,4,5,6)P4-AM were purchased from Sichem. PGE2 was purchased by Cayman Chemical. GNF362 was provided by Novartis Institutes for BioMedical Research.
Mice
WT C57BL/6 were purchased from Envigo, Italy. CD14−/− mice were originally obtained from CNRS d’Orléans and kept in pathogen-free conditions. Itpkb−/− and Itpkb+/+ (74) were provided by S. Schurmans. All mice were used at 6 to 8 weeks of age.
Cell culture and purification
BMDCs from Stim1; Stim2 double-knockout mice were provided by S. Feske (75). BMDCs were generated from WT or mutant mice by culturing for 8 days BM precursors, flushed from femurs, in Iscove’s modified Dulbecco’s medium (IMDM) (Euroclone) containing 10% heat-inactivated fetal bovine serum (Euroclone), 100 IU of penicillin, streptomycin (100 μg/ml), 2 mM L-glutamine (Euroclone), and granulocyte-macrophage colony-stimulating factor (GM-CSF) (10 to 20 μg/ml) for BMDC. sDCs were purified from WT or mutant mice. The spleen unicellular suspensions were stained with CD11c antibody and positively selected using MACS beads according to the manufacturer’s instructions (Miltenyi Biotec). For the adoptive transfer experiments, 2 × 106 sDCs plus 30 μg of LPS or 30 μl of phosphate-buffered saline (PBS) were injected subcutaneously in the ear of WT or mutant mice. D1 cells generated as previously described (34) and cultured in IMDM (Euroclone) containing 10% heat-inactivated fetal bovine serum (Euroclone), 100 IU of penicillin, streptomycin (100 μg/ml), 2 mM L-glutamine (Euroclone), and 30% of supernatants from NIH3T3 fibroblasts (transfected with GM-CSF); the final GM-CSF in the medium was 10 to 20 ng/ml. Human BDCA1 cells were purified from peripheral blood mononuclear cell (PBMC) extracts from buffy coat of healthy donors (provided by Niguarda Hospital blood bank) by Ficoll-Paque density gradient centrifugation. Briefly, blood was stratified on Ficoll-Paque PLUS (GE Healthcare) in 3:4 ratio and centrifuged at 1500 rpm for 30 min without brake. PBMCs were washed twice and collected, and BDCA1 cells were purified using MACS beads according to the manufacturer’s instructions (Miltenyi Biotec). For some experiments, BDCA1 were stained with anti-CD14 Alexa Fluor 647 (BioLegend, clone M5E2) and sorted using BD FACSAria III cell sorter. Cells were cultured for no more than 24 hours in RPMI 1640 medium (Euroclone) containing 10% heat-inactivated fetal bovine serum (Euroclone), 100 IU of penicillin, streptomycin (100 μg/ml), 2 mM L-glutamine (Euroclone), and GM-CSF (2 ng/ml; Miltenyi Biotec).
Ca2+ measurement
In some experiments, Ca2+ fluctuations were determined by a ratiometric technique. Cells were loaded with 2 μM indo-1 (Molecular Probes) by incubation at 37°C for 20 min. Cells were then washed three times with PBS to allow for intracellular de-esterification of indo-1. A direct optical microscope (Olympus, BX51) with a two-photon Ti-Sapphire laser source (720-nm wavelength; Mai Tai, Spectra-Physics) was used for indo-1 excitation. The fluorescence signals emitted by indo-1-loaded cells were digitized at 200 Hz and recorded every 0.5 to 0.8 s. The ratio of fluorescence emissions at 400/40-nm bandpass to those at 500/20-nm bandpass was recorded (R400/500) and used as an index of [Ca2+]i.
Ca2+ measurements in BMDCs were performed with confocal microscopy. A total of 5 × 105 cells were seeded on glass coverslip, loaded with 5 μM Fluo-4 acetoxymethyl ester (Fluo-4 AM) (Invitrogen) and incubated for 30 min at 37°C in complete medium containing probenecid. Cells were then washed twice with complete medium plus probenecid and immediately analyzed. Cells were excited with a laser at 488 nm, and the intensity of the fluorescence between 505 and 550 nm was measured as the Fluo-4 signal. The change in Ca2+ signal was determined by the change in Fluo-4 fluorescence. The relative mean fluorescence intensity (MFI) was calculated considering the MFI mean measured 30 s before the stimuli were added.
For some experiments, Ca2+ fluctuation was detected with flow cytometry. A total of 5 × 105 cells were loaded with 5 μM Fluo-4 AM (Invitrogen) and incubated for 30 min at 37°C in complete medium containing probenecid. Cells were then washed twice with complete medium plus probenecid and immediately analyzed by flow cytometry. The above-described fluorescence setting was applied for the acquisition.
IP4 measurement
A total of 5 × 105 D1 cells were allowed in suspension in 1 ml of media without serum and transfected with 10 μg of 15F-IP4 fluorescent probe [described in (54) and provided by T.M.]. PULSin (Polyplus-transfection) was used for IP4 probe transfection following the manufacturer’s protocol. Cells were then plated 2.5 × 105 on Nunc glass-based dish (Thermo Fisher Scientific) in 2 ml of complete media and allowed to adhere for 2 hours. Imaging was performed on a Nikon A1+ confocal microscope with a 63× oil objective. Cells were excited with a laser at 488 nm, and the intensity of the fluorescence between 505 and 550 nm was measured as the IP4 signal. Images were acquired at 1.2-s intervals.
TNFα and IL-2 measurement
Concentrations of IL-2 and TNFα in culture supernatants were assessed by enzyme-linked immunosorbent assay kits purchased from R&D Systems.
Transmission electron microscopy
For electron microscopy analysis, DCs were seeded on glass coverslip previously coated with l-polylisine (Sigma-Aldrich) for 30 min at 37°C. After adhesion, cells were fixed for 1 hour and 30 min in a 0.1% glutaraldehyde and 4% paraformaldehyde solution prepared in 0.1 M phosphate buffer (PB). After several washes in PB, cells were incubated in tris-HCl-buffered saline solution (TBS) containing 10% bovine serum albumin (BSA) and 0.2% saponin for 45 min and then stained overnight at 4°C with anti-IP3R3 antibody diluted 1:50 in a solution of 10% BSA and 0.04% saponin in TBS. After several washes in 1% BSA in TBS, samples were incubated with anti-mouse 1.4-nm gold-labeled antibody (Nanoprobe) diluted 1:100 in a solution of 1% BSA in TBS for 3 hours at room temperature, and then washed overnight in TBS. Samples were subsequently fixed again with 1% glutaraldehyde in 0.1 M PB for 10 min at room temperature, washed in double-distilled water, and last, silver enhancement was performed for 10 min under a red light (HQ SILVER, Nanoprobes). After several washes in cold distilled water, samples were post-fixed in osmium tetroxide 1% in 0.1 M PB for 1 hour and stained ON at 4°C with 1% uranyl acetate aqueous solution. Subsequently, samples were dehydrated with series of alcohols, infiltrated, and embedded in Epon Resin. Hardened samples were cut in 70-nm-thick slices and observed with JEOL JEM 1220 transmission electron microscope (JEOL, Japan), operating at 80-kV acceleration voltage and equipped with a Lheritier LH72WA digital camera.
IP3R3 silencing
D1 cells (7 × 105) were cultured on six-well plates 2 hours before transfection in 1 ml of complete medium and transfected with iBONi siRNA plus (riboxx) for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (guide, 5’-ACAAUCUCCACUUUGCCACCCCC-3’ and passenger, 5’-GGGGGUGGCAAAGUGGAGAUUGU-3’) and IP3R3 (guide, 5’-AUCUUCUCGUUCUUGGUGCCCCC-3’ and passenger, 5’-GGGGGCACCAAGAACGAGAAGAU-3’). INTERFERin (Polyplus-transfection) was used for siRNA transfection following the manufacturer’s protocol. The final concentrations of siRNA transfected were 1 to 10 nM. Cells were harvested 48 hours after siRNA transfection for subsequent analysis. For in vivo IP3R3 silencing, 3 μg of siRNA against IP3R3 [mission endoribonuclease-prepared siRNA, Sigma-Aldrich, catalog no. EMU164061] or enhanced green fluorescent protein (eGFP) (mission siRNA, Sigma-Aldrich, catalog no. EHUEGFP) were subcutaneously injected in the ears of WT C57BL/6 mice with 9 μl of jet-peg transfection reagent (Polyplus-transfection). After 24 hours, mice were used for subsequent analysis.
Vascular permeability assay
Mice were injected subcutaneously in the ears with the indicated stimuli and then immediately intravenously with 100 μl of Evans blue. After 30 min, mice were euthanized; ears were explanted and incubated overnight in formalin (Sigma-Aldrich) at 55°C. Evans blue in formalin solution was quantified by measuring the absorbance at 650 nm.
Immunofluorescence analysis
The PLA was performed using Duolink kit (Sigma-Aldrich). Cells were seeded on glass coverslip or cultured in complete media for overnight adhesion and then fixed in 4% paraformaldehyde for 10 min at room temperature, washed twice with PBS, and kept in blocking solution (2% BSA in PBS) for 30 min at room temperature. Samples were incubated with a couple of primary antibodies: goat anti-mouse CD14 (Santa Cruz Biotechnology, T-19), mouse anti mouse IP3R3 (BD Biosciences, clone 2), rabbit anti-mouse TLR4 (Santa Cruz Biotechnology, M-300), mouse anti-MHCII (BioLegend, clone 10-3.6), and rabbit anti-IP3R1 (Santa Cruz Biotechnology, E-20), diluted in blocking solution overnight at 4°C. After two washes in buffer A in gentle agitation for 5 min, cells were incubated with complementary PLA probe (anti-mouse PLUS and anti-goat MINUS or anti-mouse PLUS and anti-rabbit MINUS) diluted in blocking solution; the incubation was performed at 37°C for 1 hour. Subsequently, sample was washed twice with buffer A in gentle agitation for 5 min, and then, the ligase solution was added for 30 min at 37°C. Cells were again washed twice with buffer A in gentle agitation for 2 min. Then, the polymerase solution and the fluorescent deoxynucleotide triphosphates were added to the sample for 100 min at 37°C. Cells were staining with 4’,6-diamidino-2-phenylindole (1 μg/ml) for 5 min at room temperature and lastly mounted using FluorSave Reagent (Calbiochem). Images were acquired with Nikon A1+ confocal microscope system and analyzed with ImageJ software.
For immunocytochemistry, cells were seeded on glass coverslip for overnight adhesion. For D1 cells acquisition with TIRF microscopy, the glasses were coated with l-polylisine (Sigma-Aldrich) for 30 min at 37°C, and cells were allowed to adhere for 30 min at 37°C. For the immunocytochemistry of BDCA1 cells, the glasses were coated for 45 min at 37°C with 0.01% of Alcian blue (prepared from a stock solution of 1% in acetic acid; Sigma-Aldrich). BDCA1 cells were then allowed to adhere overnight at 37°C. Plasma membrane staining was performed by incubated live cells with wheat germ agglutinin Alexa Fluor 555 (Invitrogen) for 10 min at 37°C. Cells were fixed with paraformaldehyde solution 4% PBS for 10 min at room temperature, permeabilized with 0.2% BSA and 0.1% Triton X-100 in PBS for 10 min, and subsequently kept in blocking solution [2% BSA in PBS plus rat anti-mouse CD16/CD32 (BioLegend) or human Fc blocking (BioLegend)] for 45 min. Incubation with first antibodies was performed overnight in blocking solution at 4°C. Incubation with second antibodies was performed 1 hour in PBS at room temperature. Samples were mounted in FluorSave Reagent (Calbiochem) and were acquired with Leica TCS SP8-STED, Leica SP2-TIRF, and Nikon A1+ confocal microscope system. ImageJ software was used for image analysis and processing.
The following antibodies were used: anti-mouse CD14 (Santa Cruz Biotechnology, clone T-19), anti-human/mouse IP3R3 (BD Biosciences, clone 2/IP3R-3), anti-mouse calnexin (Abcam, ab22595), anti-human/ mouse IP3R1 (Santa Cruz Biotechnology, clone E-20), anti-human/ mouse IP3R2 (Santa Cruz Biotechnology, clone C-20), anti-human CD14 (BioLegend, clone M5E2), anti-human/mouse ITPKB (ProteinTech), anti-human/mouse NFATc2 (ImmunoGlobe, clone IG-209), anti-mouse Alexa Fluor 488, anti-rabbit Alexa Fluor 647, and anti-goat Alexa Fluor 555. For STED microscope acquisition, anti-goat ATTO647N, anti-mouse ATTO488, and anti-mouse 594 were used. The quantitative analysis of NFAT nuclear translocation has been done blindly by biophysicist experts in microscopy and optics of the physics department.
For immunohistochemistry, mice were injected subcutaneously in ear with the indicated stimuli for 1 hour and 30 min. Explanted skin ears were embedded in optimal cutting temperature freezing media (Bio-Optica). Sections (5 μm) were cut on a cryostat, adhered to a Superfrost Plus slide (Thermo Fisher Scientific), fixed with acetone, and blocked with PBS/5% BSA 30 min at room temperature. Sections were then stained overnight at 4°C with primary antibody anti-human/mouse NFATc2 (ImmunoGlobe, clone IG-209) and anti-mouse MHCII Alexa Fluor 488 (BioLegend, clone M5) and then washed twice with PBS and incubated 30 min at room temperature with anti-rabbit Alexa Fluor 555. Nuclei were stained with DRAQ5, 1:1000 in dH2O, for 5 min at room temperature. Sections were then mounted in FluorSave Reagent (Calbiochem) and acquired with Leica SP5 confocal microscopy.
Quantitative reverse transcription polymerase chain reaction
Mouse ears or cell samples were homogenized in TRIzol reagent, and then, RNA was extracted using Quick-RNA Miniprep or Microprep kits (Zymo Research, catalog nos. R2050 and R1051, respectively). Single-strand complementary DNA (cDNA) was synthesized using High-Capacity cDNA Reverse Transcription kits (catalog no. 4368814, Applied Biosystems). The NanoDrop (Thermo Fisher Scientific) was used to titer mRNA. RNA from BDCA1 sample was amplified using the QuantiTect Whole Transcriptome Kit (Qiagen) according to the manufacturer’s instructions. cDNA amplification was performed using the TaqMan Gene Expression Master Mix (catalog no. 4369016, Applied Biosystems) and TaqMan probes: ITPR1 Mm_00439907_m1, ITPR2 Mm_00444937_m1, ITPR3 Mm_01306070_m1, IL-2 Mm_00434256_m1 or Hs_00174114_m1, TNFα Mm_004432258_m1 or Hs_00174128_m1, Ptges1 Mm_00452105_ m1, GAPDH Mm_99999915_g1, RPS13 Hs_01011487_g1, 18S, and Mm_03928990_g1. cDNA amplification of Viperin was performed using SYBR Green Master Mix (Applied Biosystems) and the following primers: 5’-CTTCAACGTGGACGAAGACA-3’ (forward) and 5’-GACGCTCCAAGAATGTTTCA-3’ (reverse). Relative mRNA expression was calculated using the ▵Ct method, using 18S or RPS13 as a reference gene.
Western blotting
Cells were lysed with a buffer containing 50 mM tris-HCl (pH 7.4), 150 mM NaCl, 10% glycerol, and 1% NP-40 supplemented with protease and phosphatase inhibitor cocktails (Roche). Cell debris were removed by centrifugation at 16,000g for 15 min (4°C). Thymus was smashed and homogenized in 1 ml of lysis buffer 50 mM tris-HCl (pH 7.4), 150 mM NaCl, 10% glycerol, and 1% NP-40 supplemented with protease and phosphatase inhibitor cocktails (Roche) using a TissueLyser (full speed for 20 min; Qiagen). The tissue was then maintained in constant agitation for 30 min at 4°C. Sample was centrifuged for 20 min at 13,000g at 4°C, and supernatant was collected. Both cell and thymus proteins were quantified using a bicinchoninic acid assay (Euroclone). Subsequently, 50 μg of cell lysates was run on an 8% polyacrylamide gel, and SDS-polyacrylamide gel electrophoresis was performed following standard procedures. After protein transfer, nitrocellulose membranes (Thermo Fisher Scientific) were incubated with anti-human/mouse ITPKB (ProteinTech), anti-human/mouse GAPDH [CST (Cell Signaling Technology), 14C10], or anti-human/mouse vinculin (E1E9V) antibody and developed using an enhanced chemiluminesence (ECL) substrate reagent (Thermo Fisher Scientific).
Protein biotinylation and IP3R3 immunoprecipitation
A total of 1 × 107 D1 cells were biotinylated with 1 mg of biotin (EZ-Link sulfo-NHS-SS-biotin, Thermo Fisher Scientific) for 30 min at 37°C. Cells were then lysed with a buffer containing 50 mM tris-HCl (pH 7.4), 150 mM NaCl, 10% glycerol, and 1% NP-40 supplemented with protease and phosphatase inhibitor cocktails (Roche). Cell debris were removed by centrifugation at 16,000g for 15 min (4°C). The immunoprecipitations were performed using Dynabeads Protein G Immunoprecipitation Kit according to the manufacturer’s instructions. For IP3R3 immunoprecipitation, 10 μg of anti-IP3R3 (BD Biosciences, mouse clone 2, catalog no. 610312) or 10 μg of anti-IP3R3 (Millipore, rabbit polyclonal, catalog no. ab9076) was used. For calnexin immunoprecipitation, 10 μg of anti-calnexin antibody (Abcam, rabbit polyclonal, catalog no. ab2259) was used. The samples were run on a 10% polyacrylamide gel, and SDS-polyacrylamide gel electrophoresis was performed following standard procedures. After protein transfer, nitrocellulose membrane was incubated with streptavidin to reveal biotin-IP3R3 and with anti-calnexin antibody and developed using an ECL substrate reagent (Thermo Fisher Scientific).
TLR4 internalization
After 30 min of LPS treatment, BMDCs were collected and stained with anti-mouse phycoerythrin-conjugated TLR4 (BioLegend, clone SA15-21) antibody for 20 min at 4°C.
Cells were then washed, and TLR4 surface expression was evaluated by Gallios flow cytometer (Beckman Coulter).
Generation of VIVIT-functionalized nanoparticles (Myts-VIVIT)
A Myts solution (10 mg in 1 ml) was reacted with 0.05 M 2,2’-(ethylenedioxy)bis(ethylamine) (EDBE) (3 μl in deionized water), in the presence of 0.1 M 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (8 μl) and left under shaking for 2 hours. The nanoparticle dispersion was washed with water to remove unreacted EDBE and concentrated by centrifugation. The nanoparticles were diluted and stirred for 4 hours after adding N-succinimidyl-3-[2-pyridylthio]-propionate (300 μl, 10 mg/ml in dimethyl sulfoxide) and then washed with water. Last, the peptide having sequence HS-CGGGKMAGPVIVITGPHEE-COOH (13 μl, 10 mg/ml) and PEG-SH (molecular weight ≈ 500 Da, 10 μl, 10 mg/ml) were added to the mixture. Myts-VIVIT were washed with water, and the final concentration was determined by ultraviolet measurements. Nanoparticles uptake were evaluated in mice spleen, lymph nodes, and skin after 2 hour of intraperitoneal injection (100 μg per mouse) with flow cytometry.
Collagen antibody-induced arthritis
CAIA was induced in WT C57BL/6 female mice using a cocktail of five monoclonal antibodies (mAbs) (Chondrex). Briefly, mice were injected intraperitoneally with 5 mg of cocktail Abs per mouse on day 0 and with 50 μg of LPS per mouse at day 3. Where indicated, mice were also treated with 200 μM TNP every other day starting from day −1. According to the “Chondrex Mouse Arthritis Scoring System” criteria, clinical disease score (redness, thickness, and swelling of wrist, ankle, individual digits, and entire paw) was evaluated daily by a blinded observer until day 7. Then, mice were euthanized, and limb was explanted in optimal cutting temperature freezing media (Bio-Optica). For histopathological analysis, paw section of 5 μm was stained with Meyer’s hematoxylin solution for 8 min and then washed in warm running tap water for 5 min. Sections were stained with eosin Y solution for 1 min, washed in warm running tap water for 5 min, rinsed in distilled water, and then dehydrated through passages in 95% and absolute alcohol. After dehydration, stained slides were cleared in xylene and mounted with Eukitt. Images were acquired with the NanoZoomer (Hamamatsu).
Statistical analysis
Means were compared by either unpaired parametric t tests or one-way or two-way analysis of variance (ANOVA). Data are expressed and plotted as means ± SDM or ± SEM values. Sample sizes for each experimental condition are provided in the figure legends. All P values were calculated using Prism (GraphPad). Differences were considered significant if P ≤ 0.05.
Ethical approvals
Animal studies have been performed after the approval from the Italian Ministry of Health, and the animals’ care was in accordance with institutional guidelines. Human samples have been analyzed after approval from the internal Ethical Committee.
Supplementary Material
Fig. S1. Ca2+ fluxes in D1 cells in response to LPS stimulation measured by flow cytometry or with a ratiometric method.
Fig. S2. IP3R3 localization on the plasma membrane and IP3R3 knockdown.
Fig. S3. Flow cytometry analysis of Ca2+ fluxes.
Fig. S4. Sorting strategy of DC3 cells.
Fig. S5. ITPKB in DCs and role in Ca2+ mobilization in response to LPS.
Fig. S6. Colocalization of IP3R3, CD14, and ITPKB in DCs.
Fig. S7. ITPKB is required for LPS-induced vascular leakage.
Fig. S8. Inhibition of nuclear NFAT translocation by Myts-VIVIT nanoparticles.
Acknowledgments:
We thank S. Feske (NYU School of Medicine) for providing Stim1 and Stim2 double-deficient BM cells.
Funding:
This work was supported by the NIH grant 1R01AI121066–01A1, HDDC P30 DK034854 grant, Harvard Medical School Milton Fund, CCFA Senior Research Awards, Associazione Italiana per la Ricerca sul Cancro (IG 2019Id.23512), Fondazione Regionale per la Ricerca Biomedica, FRRB (IANG-CRC - CP2_12/2018), Italian Ministry of Health (RF-2018–12367072), and Intramural Research Program of the NIH, National Institute of Environmental Health Sciences.
Footnotes
Competing interests: I.Z. is a consultant for Implicit Biosciences, which commercializes the humanized anti-CD14 antibody IC14. D.P., F.G., M.C., and I.Z. are inventors on patent application PCT/IB2013/055943, WO2014013473 A1 submitted by University of Milano-Bicocca that covers the use of nanoparticles targeting NFAT.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
REFERENCES AND NOTES
- 1.Ostuni R, Zanoni I, Granucci F, Deciphering the complexity of Toll-like receptor signaling. Cell. Mol. Life Sci . 67, 4109–4134 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Herbst S, Shah A, Mazon Moya M, Marzola V, Jensen B, Reed A, Birrell MA, Saijo S, Mostowy S, Shaunak S, Armstrong-James D, Phagocytosis-dependent activation of a TLR9-BTK-calcineurin-NFAT pathway co-ordinates innate immunity to Aspergillus fumigatus. EMBO Mol. Med . 7, 240–258 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zanoni I, Ostuni R, Capuano G, Collini M, Caccia M, Ronchi AE, Rocchetti M, Mingozzi F, Foti M, Chirico G, Costa B, Zaza A, Ricciardi-Castagnoli P, Granucci F, CD14 regulates the dendritic cell life cycle after LPS exposure through NFAT activation. Nature 460, 264–268 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Goodridge HS, Simmons RM, Underhill DM, Dectin-1 stimulation by Candida albicans yeast or zymosan triggers NFAT activation in macrophages and dendritic cells. J. Immunol . 178, 3107–3115 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Robinson MJ, Osorio F, Rosas M, Freitas RP, Schweighoffer E, Gross O, Verbeek JS, Ruland J, Tybulewicz V, Brown GD, Moita LF, Taylor PR, Reis e Sousa C, Dectin-2 is a Syk-coupled pattern recognition receptor crucial for Th17 responses to fungal infection. J. Exp. Med . 206, 2037–2051 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vandewalle A, Tourneur E, Bens M, Chassin C, Werts C, Calcineurin/NFAT signalling and innate host defence: A role for NOD1-mediated phagocytic functions. Cell Commun. Sig . 12, 8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greenblatt MB, Aliprantis A, Hu B, Glimcher LH, Calcineurin regulates innate antifungal immunity in neutrophils. J. Exp. Med . 207, 923–931 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Santus W, Barresi S, Mingozzi F, Broggi A, Orlandi I, Stamerra G, Vai M, Martorana AM, Polissi A, Kohler JR, Liu N, Zanoni I, Granucci F, Skin infections are eliminated by cooperation of the fibrinolytic and innate immune systems. Sci. Immunol. 2, eaan2725 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zelante T, Wong AYW, Mencarelli A, Foo S, Zolezzi F, Lee B, Poidinger M, Ricciardi-Castagnoli P, Fric J, Impaired calcineurin signaling in myeloid cells results in downregulation of pentraxin-3 and increased susceptibility to aspergillosis. Mucosal Immunol . 10, 470–480 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Pang Z, Junkins RD, MacNeil AJ, McCormick C, Cheng Z, Chen WM, Lin TJ, The calcineurin-NFAT axis contributes to host defense during Pseudomonas aeruginosa lung infection. J. Leukoc. Biol . 102, 1461–1469 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Mencarelli A, Khameneh HJ, Fric J, Vacca M, El Daker S, Janela B, Tang JP, Nabti S, Balachander A, Lim TS, Ginhoux F, Ricciardi-Castagnoli P, Mortellaro A, Calcineurin-mediated IL-2 production by CD11chighMHCII+ myeloid cells is crucial for intestinal immune homeostasis. Nat. Commun . 9, 1102 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zanoni I, Ostuni R, Barresi S, Di Gioia M, Broggi A, Costa B, Marzi R, Granucci F, CD14 and NFAT mediate lipopolysaccharide-induced skin edema formation in mice. J. Clin. Invest . 122, 1747–1757 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zanoni I, Granucci F, Regulation and dysregulation of innate immunity by NFAT signaling downstream of pattern recognition receptors (PRRs). Eur. J. Immunol . 42, 1924–1931 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Liu Z, Lee J, Krummey S, Lu W, Cai H, Lenardo MJ, The kinase LRRK2 is a regulator of the transcription factor NFAT that modulates the severity of inflammatory bowel disease. Nat. Immunol. 12, 1063–1070 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yarilina A, Xu K, Chen J, Ivashkiv LB, TNF activates calcium-nuclear factor of activated T cells (NFAT)c1 signaling pathways in human macrophages. Proc. Natl. Acad. Sci. U.S.A. 108, 1573–1578 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prakriya M, Lewis RS, Store-operated calcium channels. Physiol. Rev. 95, 1383–1436 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shaw PJ, Feske S, Regulation of lymphocyte function by ORAI and STIM proteins in infection and autoimmunity. J. Physiol. 590, 4157–4167 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Srikanth S, Gwack Y, Orai1-NFAT signalling pathway triggered by T cell receptor stimulation. Mol. Cells 35, 182–194 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G, Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 397, 259–263 (1999). [DOI] [PubMed] [Google Scholar]
- 20.Putney JW, PLC-gamma: An old player has a new role. Nat. Cell Biol. 4, E280–E281 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Patterson RL, Boehning D, Snyder SH, Inositol 1,4,5-trisphosphate receptors as signal integrators. Annu. Rev. Biochem. 73, 437–465 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Feske S, Calcium signalling in lymphocyte activation and disease. Nat. Rev. Immunol. 7, 690–702 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Nakagawa T, Okano H, Furuichi T, Aruga J, Mikoshiba K, The subtypes of the mouse inositol 1,4,5-trisphosphate receptor are expressed in a tissue-specific and developmentally specific manner. Proc. Natl. Acad. Sci. U.S.A. 88, 6244–6248 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ross CA, Danoff SK, Schell MJ, Snyder SH, Ullrich A, Three additional inositol 1,4,5-trisphosphate receptors: Molecular cloning and differential localization in brain and peripheral tissues. Proc. Natl. Acad. Sci. U.S.A. 89, 4265–4269 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matsumoto M, Nagata E, Type 1 inositol 1,4,5-trisphosphate receptor knock-out mice: Their phenotypes and their meaning in neuroscience and clinical practice. J. Mol. Med. (Berl) 77, 406–411 (1999). [DOI] [PubMed] [Google Scholar]
- 26.Vervloessem T, Yule DI, Bultynck G, Parys JB, The type 2 inositol 1,4,5-trisphosphate receptor, emerging functions for an intriguing Ca2+-release channel. Biochim. Biophys. Acta 1853, 1992–2005 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tamura T, Hashimoto M, Aruga J, Konishi Y, Nakagawa M, Ohbayashi T, Shimada M, Mikoshiba K, Promoter structure and gene expression of the mouse inositol 1,4,5-trisphosphate receptor type 3 gene. Gene 275, 169–176 (2001). [DOI] [PubMed] [Google Scholar]
- 28.Miyakawa T, Maeda A, Yamazawa T, Hirose K, Kurosaki T, Iino M, Encoding of Ca2+ signals by differential expression of IP3 receptor subtypes. EMBO J. 18, 1303–1308 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mak D-O, McBride S, Foskett JK, Regulation by Ca2+ and inositol 1,4,5-trisphosphate (InsP3) of single recombinant type 3 InsP3 receptor channels. Ca2+ activation uniquely distinguishes types 1 and 3 insp3 receptors. J. Gen. Physiol. 117, 435–446 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hagar RE, Burgstahler AD, Nathanson MH, Ehrlich BE, Type III InsP3 receptor channel stays open in the presence of increased calcium. Nature 396, 81–84 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vaeth M, Zee I, Concepcion AR, Maus M, Shaw P, Portal-Celhay C, Zahra A, Kozhaya L, Weidinger C, Philips J, Unutmaz D, Feske S, Ca2+ signaling but not store-operated Ca2+ entry is required for the function of macrophages and dendritic cells. J. Immunol. 195, 1202–1217 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Di Virgilio F, Purinergic mechanism in the immune system: A signal of danger for dendritic cells. Purinergic Signal. 1, 205–209 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hsu S, O’Connell PJ, Klyachko VA, Badminton MN, Thomson AW, Jackson MB, Clapham DE, Ahern GP, Fundamental Ca2+ signaling mechanisms in mouse dendritic cells: CRAC is the major Ca2+ entry pathway. J. Immunol. 166, 6126–6133 (2001). [DOI] [PubMed] [Google Scholar]
- 34.Winzler C, Rovere P, Rescigno M, Granucci F, Penna G, Adorini L, Zimmermann VS, Davoust J, Ricciardi-Castagnoli P, Maturation stages of mouse dendritic cells in growth factor-dependent long-term cultures. J. Exp. Med. 185, 317–328 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ishikawa J, Ohga K, Yoshino T, Takezawa R, Ichikawa A, Kubota H, Yamada T, A pyrazole derivative YM -58483, potently inhibits store-operated sustained Ca2+ influx and IL-2 production in T lymphocytes. J. Immunol. 170, 4441–4449 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Yoshino T, Ishikawa J, Ohga K, Morokata T, Takezawa R, Morio H, Okada Y, Honda K, Yamada T, YM-58483, a selective CRAC channel inhibitor, prevents antigen-induced airway eosinophilia and late phase asthmatic responses via Th2 cytokine inhibition in animal models. Eur. J. Pharmacol. 560, 225–233 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Ohga K, Takezawa R, Arakida Y, Shimizu Y, Ishikawa J, Characterization of YM-58483/ BTP2, a novel store-operated Ca2+ entry blocker, on T cell-mediated immune responses in vivo. Int. Immunopharmacol. 8, 1787–1792 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Takezawa R, Cheng H, Beck A, Ishikawa J, Launay P, Kubota H, Kinet JP, Fleig A, Yamada T, Penner R, A pyrazole derivative potently inhibits lymphocyte Ca2+ influx and cytokine production by facilitating transient receptor potential melastatin 4 channel activity. Mol. Pharmacol. 69, 1413–1420 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Helft J, Bottcher J, Chakravarty P, Zelenay S, Huotari J, Schraml BU, Goubau D, Reis e Sousa C, GM-CSF mouse bone marrow cultures comprise a heterogeneous population of CD11c+MHCII+ macrophages and dendritic cells. Immunity 42, 1197–1211 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Dellis O, Dedos SG, Tovey SC, Taufiq-Ur-Rahman R, Dubel SJ, Taylor CW, Ca2+ entry through plasma membrane IP3 receptors. Science 313, 229–233 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, Griesbeck M, Butler A, Zheng S, Lazo S, Jardine L, Dixon D, Stephenson E, Nilsson E, Grundberg I, McDonald D, Filby A, Li W, De Jager PL, Rozenblatt-Rosen O, Lane AA, Haniffa M, Regev A, Hacohen N, Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 356, eaah4573 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dutertre CA, Becht E, Irac SE, Khalilnezhad A, Narang V, Khalilnezhad S, Ng PY, van den Hoogen LL, Leong JY, Lee B, Chevrier M, Zhang XM, Yong PJA, Koh G, Lum J, Howland SW, Mok E, Chen J, Larbi A, Tan HKK, Lim TKH, Karagianni P, Tzioufas AG, Malleret B, Brody J, Albani S, van Roon J, Radstake T, Newell EW, Ginhoux F, Single-cell analysis of human mononuclear phagocytes reveals subset-defining markers and identifies circulating inflammatory dendritic cells. Immunity 51, 573–589.e8 (2019). [DOI] [PubMed] [Google Scholar]
- 43.Rusconi M, Gerardi F, Santus W, Lizio A, Sansone VA, Lunetta C, Zanoni I, Granucci F, Inflammatory role of dendritic cells in amyotrophic lateral sclerosis revealed by an analysis of patients’ peripheral blood. Sci. Rep. 7, 7853 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takazawa K, Perret J, Dumont JE, Erneux C, Molecular cloning and expression of a new putative inositol 1,4,5-trisphosphate 3-kinase isoenzyme. Biochem. J. 278 (Pt 3), 883–886 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller AT, Dahlberg C, Sandberg ML, Wen BG, Beisner DR, Hoerter JA, Parker A, Schmedt C, Stinson M, Avis J, Cienfuegos C, McPate M, Tranter P, Gosling M, Groot-Kormelink PJ, Dawson J, Pan S, Tian S-S, Seidel HM, Cooke MP, Inhibition of the inositol kinase Itpkb augments calcium signaling in lymphocytes and reveals a novel strategy to treat autoimmune disease. PLOS ONE 10, e0131071 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wen BG, Pletcher MT, Warashina M, Choe SH, Ziaee N, Wiltshire T, Sauer K, Cooke MP, Inositol (1,4,5) trisphosphate 3 kinase B controls positive selection of T cells and modulates Erk activity. Proc. Natl. Acad. Sci. U.S.A. 101, 5604–5609 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jia Y, Loison F, Hattori H, Li Y, Erneux C, Park SY, Gao C, Chai L, Silberstein LE, Schurmans S, Luo HR, Inositol trisphosphate 3-kinase B (InsP3KB) as a physiological modulator of myelopoiesis. Proc. Natl. Acad. Sci. U.S.A. 105, 4739–4744 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scoumanne A, Molina-Ortiz P, Monteyne D, Perez-Morga D, Erneux C, Schurmans S, Specific expression and function of inositol 1,4,5-trisphosphate 3-kinase C (ITPKC) in wild type and knock-out mice. Adv. Biol. Regul. 62, 1–10 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Pouillon V, Marechal Y, Frippiat C, Erneux C, Schurmans S, Inositol 1,4,5-trisphosphate 3-kinase B (Itpkb) controls survival, proliferation and cytokine production in mouse peripheral T cells. Adv. Biol. Regul. 53, 39–50 (2013). [DOI] [PubMed] [Google Scholar]
- 50.Miller AT, Beisner DR, Liu D, Cooke MP, Inositol 1,4,5-trisphosphate 3-kinase B is a negative regulator of BCR signaling that controls B cell selection and tolerance induction. J. Immunol. 182, 4696–4704 (2009). [DOI] [PubMed] [Google Scholar]
- 51.Marechal Y, Queant S, Polizzi S, Pouillon V, Schurmans S, Inositol 1,4,5-trisphosphate 3-kinase B controls survival and prevents anergy in B cells. Immunobiology 216, 103–109 (2011). [DOI] [PubMed] [Google Scholar]
- 52.Hascakova-Bartova R, Pouillon V, Dewaste V, Moreau C, Jacques C, Banting G, Schurmans S, Erneux C, Identification and subcellular distribution of endogenous Ins(1,4,5)P(3) 3-kinase B in mouse tissues. Biochem. Biophys. Res. Commun. 323, 920–925 (2004). [DOI] [PubMed] [Google Scholar]
- 53.Dewaste V, Moreau C, De Smedt F, Bex F, De Smedt H, Wuytack F, Missiaen L, Erneux C, The three isoenzymes of human inositol-1,4,5-trisphosphate 3-kinase show specific intracellular localization but comparable Ca2+ responses on transfection in COS-7 cells. Biochem. J. 374, 41–49 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sakaguchi R, Tainaka K, Shimada N, Nakano S, Inoue M, Kiyonaka S, Mori Y, Morii T, An in vivo fluorescent sensor reveals intracellular ins(1,3,4,5)P4 dynamics in single cells. Angew. Chem. Int. Ed. Engl. 49, 2150–2153 (2010). [DOI] [PubMed] [Google Scholar]
- 55.Aramburu J, Yaffe MB, Lopez-Rodriguez C, Cantley LC, Hogan PG, Rao A, Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science 285, 2129–2133 (1999). [DOI] [PubMed] [Google Scholar]
- 56.Fiandra L, Mazzucchelli S, De Palma C, Colombo M, Allevi R, Sommaruga S, Clementi E, Bellini M, Prosperi D, Corsi F, Assessing the in vivo targeting efficiency of multifunctional nanoconstructs bearing antibody-derived ligands. ACS Nano 7, 6092–6102 (2013). [DOI] [PubMed] [Google Scholar]
- 57.Colombo M, Sommaruga S, Mazzucchelli S, Polito L, Verderio P, Galeffi P, Corsi F, Tortora P, Prosperi D, Site-specific conjugation of ScFvs antibodies to nanoparticles by bioorthogonal strain-promoted alkyne-nitrone cycloaddition. Angew. Chem. Int. Ed. Engl. 51, 496–499 (2012). [DOI] [PubMed] [Google Scholar]
- 58.Lin C-A, Sperling RA, Li JK, Yang T-Y, Li P-Y, Zanella M, Chang WH, Parak WJ, Design of an amphiphilic polymer for nanoparticle coating and functionalization. Small 4, 334–341 (2008). [DOI] [PubMed] [Google Scholar]
- 59.Mazzucchelli S, Colombo M, Verderio P, Rozek E, Andreata F, Galbiati E, Tortora P, Corsi F, Prosperi D, Orientation-controlled conjugation of haloalkane dehalogenase fused homing peptides to multifunctional nanoparticles for the specific recognition of cancer cells. Angew. Chem. Int. Ed. Engl. 52, 3121–3125 (2013). [DOI] [PubMed] [Google Scholar]
- 60.Nandakumar KS, Holmdahl R, Efficient promotion of collagen antibody induced arthritis (CAIA) using four monoclonal antibodies specific for the major epitopes recognized in both collagen induced arthritis and rheumatoid arthritis. J. Immunol. Methods 304, 126–136 (2005). [DOI] [PubMed] [Google Scholar]
- 61.Gerth AJ, Pham CT, Peng SL, Regulation of the symmetry and intensity of immune complex-mediated synovitis by nuclear factor of activated T cells. Arthritis Rheum . 50, 3392–3395 (2004). [DOI] [PubMed] [Google Scholar]
- 62.Feske S, Wulff H, Skolnik EY, Ion channels in innate and adaptive immunity. Annu. Rev. Immunol . 33, 291–353 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zanoni I, Bodio C, Broggi A, Ostuni R, Caccia M, Collini M, Venkatesh A, Spreafico R, Capuano G, Granucci F, Similarities and differences of innate immune responses elicited by smooth and rough LPS. Immunol. Lett . 142, 41–47 (2012). [DOI] [PubMed] [Google Scholar]
- 64.Fadool DA, Ache BW, Plasma membrane inositol 1,4,5-trisphosphate-activated channels mediate signal transduction in lobster olfactory receptor neurons. Neuron 9, 907–918 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Khan AA, Steiner JP, Klein MG, Schneider MF, Snyder SH, IP3 receptor: Localization to plasma membrane of T cells and cocapping with the T cell receptor. Science 257, 815–818 (1992). [DOI] [PubMed] [Google Scholar]
- 66.Guillemette G, Balla T, Baukal AJ, Catt KJ, Characterization of inositol 1,4,5-trisphosphate receptors and calcium mobilization in a hepatic plasma membrane fraction. J. Biol. Chem . 263, 4541–4548 (1988). [PubMed] [Google Scholar]
- 67.Dargemont C, Hilly M, Claret M, Mauger JP, Characterization of Ca2+ fluxes in rat liver plasma-membrane vesicles. Biochem. J . 256, 117–124 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dellis O, Rossi AM, Dedos SG, Taylor CW, Counting functional inositol 1,4,5-trisphosphate receptors into the plasma membrane. J. Biol. Chem . 283, 751–755 (2008). [DOI] [PubMed] [Google Scholar]
- 69.Leitenberg D, Boutin Y, Constant S, Bottomly K, CD4 regulation of TCR signaling and T cell differentiation following stimulation with peptides of different affinities for the TCR. J. Immunol . 161, 1194–1203 (1998). [PubMed] [Google Scholar]
- 70.deSouza N, Cui J, Dura M, McDonald TV, Marks AR, A function for tyrosine phosphorylation of type 1 inositol 1,4,5-trisphosphate receptor in lymphocyte activation. J. Cell Biol . 179, 923–934 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Miller AT, Sandberg M, Huang YH, Young M, Sutton S, Sauer K, Cooke MP, Production of Ins(1,3,4,5)P4 mediated by the kinase Itpkb inhibits store-operated calcium channels and regulates B cell selection and activation. Nat. Immunol . 8, 514–521 (2007). [DOI] [PubMed] [Google Scholar]
- 72.Sauer K, Cooke MP, Regulation of immune cell development through soluble inositol-1,3,4,5-tetrakisphosphate. Nat. Rev. Immunol . 10, 257–271 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Prosperi D, Colombo M, Zanoni I, Granucci F, Drug nanocarriers to treat autoimmunity and chronic inflammatory diseases. Semin, Immunol . 34, 61–67 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pouillon V, Hascakova-Bartova R, Pajak B, Adam E, Bex F, Dewaste V, Van Lint C, Leo O, Erneux C, Schurmans S, Inositol 1,3,4,5-tetrakisphosphate is essential for T lymphocyte development. Nat. Immunol . 4, 1136–1143 (2003). [DOI] [PubMed] [Google Scholar]
- 75.Oh-Hora M, Yamashita M, Hogan PG, Sharma S, Lamperti E, Chung W, Prakriya M, Feske S, Rao A, Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat. Immunol . 9, 432–443 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Ca2+ fluxes in D1 cells in response to LPS stimulation measured by flow cytometry or with a ratiometric method.
Fig. S2. IP3R3 localization on the plasma membrane and IP3R3 knockdown.
Fig. S3. Flow cytometry analysis of Ca2+ fluxes.
Fig. S4. Sorting strategy of DC3 cells.
Fig. S5. ITPKB in DCs and role in Ca2+ mobilization in response to LPS.
Fig. S6. Colocalization of IP3R3, CD14, and ITPKB in DCs.
Fig. S7. ITPKB is required for LPS-induced vascular leakage.
Fig. S8. Inhibition of nuclear NFAT translocation by Myts-VIVIT nanoparticles.