

Abstract
Harnessing the bacterial clustered regularly interspaced short palindromic repeats (CRISPR) system for genome editing in eukaryotes has revolutionized basic biomedical research and the translational sciences. The ability to create targeted alterations of the genome through this easy to design system has presented unprecedented opportunities to treat inherited disorders and other diseases such as cancer through gene therapy. A major hurdle is the lack of an efficient and safe in vivo delivery system, limiting most of the current gene therapy efforts to ex vivo editing of extracted cells. Here we discuss the unique features of adenoviral vectors that enable tissue specific and efficient delivery of the CRISPR-Cas machinery for in vivo genome editing.
Keywords: CRISPR, Adenovirus, Gene Therapy, Genome Editing, Gene Delivery
CRISPR-CAS FOR GENE THERAPY
Initially conceptualized several decades ago, gene therapy aims to treat diseases by genetically altering cells, potentially resulting in life-long benefits from a single treatment. Several advances have allowed gene therapy research to accelerate in the past decade, with the FDA approving several drugs and multiple clinical trials ongoing [1]. Among the most promising of these advances is the discovery of the genome editing system known as CRISPR-Cas.
CRISPR-Cas is a diverse bacterial immune system, providing defense against invading DNA and RNA molecules. Since its initial discovery, two classes and six types have been characterized in various bacterial strains, but the simple Class 2 system, consisting of a single CRISPR associated protein (Cas) and CRISPR RNAs (crRNAs), remains among the most useful for biomedical research [2]. Cas is a nuclease directed to targeted genomic locations by crRNAs, which in modern systems are usually simplified into a single guide RNA (sgRNA or gRNA) [3]. Numerous Cas proteins have been characterized, but spCas9, discovered in Streptococcus pyogenes, remains the most widely studied and one of the most efficient [4,5].
Upon binding to a gRNA, a conformational change in Cas9 allows it to interact with a protospacer adjacent motif (PAM) sequence (a short DNA sequence such as NGG for Cas9, where N is any nucleotide), locally unwinding the dsDNA to allow the gRNA to invade and displace one strand and anneal with the other [6]. The Cas9/gRNA complex moves on to the next PAM site if the gRNA and target sequence do not anneal perfectly, whereas sufficient base pairing between the gRNA and the target triggers nuclease activity of the Cas9 and a double strand break (DSB) will be made near the PAM site [6].
Cleavage of the DNA target produces DSBs that are largely repaired by either the error-prone but highly efficient non-homologous end joining (NHEJ) pathway or the precise yet often less efficient homology directed repair (HDR) pathway [7]. NHEJ from a single DSB often produces small insertions and/or deletions (indels), while two gRNAs can be used to create a precise deletion between the two DSBs. Both mechanisms are useful in developing gene therapies - for example, creating a single small deletion around an intron/exon junction in the dystrophin gene has been shown to effectively cause exon “skipping”, restoring the function of mutated dystrophin and treating Duchenne’s Muscular Dystrophy (DMD) [8]. Similarly, the two-gRNA deletion strategy was used to physically remove an entire exon to achieve similar results [9]. Therapies relying on NHEJ are expected to be highly efficient and only require delivery of the CRISPR-Cas machinery (Cas protein and gRNA), and many of the ongoing CRISPR-Cas gene therapy programs utilize this strategy.
DSBs can also be repaired by the HDR pathway using either a sister chromatid or an exogenously provided DNA template, called a donor. A donor brings in the exact sequences to be introduced at the target site, from a point mutation to larger insertions, with flanking homology sequences to provide specificity [10]. HDR is usually significantly less efficient than NHEJ, with the majority of DSBs being repaired by the NHEJ pathway, especially in vivo [11,12]. In addition to the CRISPR-Cas machinery, the donor also must be delivered to the same cells at the same time - the larger the donor, the more challenging the delivery. The low hanging fruits for current CRISPR-Cas therapeutic programs are therefore mostly small corrections, such as single nucleotide polymorphisms. However, there is a clear need in gene therapy for HDR-mediated site-specific insertion of corrective genes for long term, stable production of therapeutic proteins. Improving HDR efficiency is an unmet need in both basic research and therapeutics.
The promises of CRISPR-Cas technologies have resulted in an explosion of preclinical and clinical studies in gene therapy. Academia and industry are exploring this technology for numerous medical applications, with a focus on inherited disorders and cancer.
Nearly all these clinical trials utilize ex vivo editing, or in a single trial subretinal injection of the CRISPR-Cas machinery using an adeno-associated virus (AAV). In ex vivo editing, a patient’s cells are extracted from the body and genetically edited in vitro, then returned to the patient, where they are potentially able to create therapeutic results due to their new properties. This process has shown great success with techniques such as CAR-T immunotherapy and hematopoietic stem cell (HSC) transplantation - in CAR-T immunotherapy, a patients T-cells are edited to contain a receptor capable of recognizing cancer antigens and activating the T cell to destroy them [28]. In HSC transplantation, HSCs can be edited to produce corrective proteins for the treatment of diseases such as sickle cell disease and beta-thalassemia [29]. Although ex vivo cellular editing has shown tremendous successes with these techniques, it remains a complex, costly and convoluted process likely limited to serious diseases where no other treatments have shown success [30].
DELIVERY OF CRISPR-CAS MACHINERY FOR IN VIVO GENE THERAPY
In vivo gene therapy could traverse several of the problems associated with ex vivo therapy. In this process, rather than removing cells from the patient, editing them and returning them to the patient, a vector is delivered to the patient’s cells in vivo. Conceptually, this process has the potential to create major advancements in gene therapy: a single dose delivered to the cells of interest could correct a disease for life. With the advent of CRISPR-Cas technology, it is now possible to permanently integrate genes of interest in a targeted manner, potentially solving one part of this challenge – lifelong genetic correction. However, efficient in vivo delivery of Cas9, gRNA(s) and donor DNA to the right cells remains a challenge. A vector suitable for in vivo delivery of the CRISPR-Cas machinery must surmount several difficulties, including gene transfer efficiency and tissue/organ targeting, all while maintaining a strong safety profile [1]. Numerous delivery systems are currently under development to deliver the CRISPR-Cas components as DNA, RNA or ribonucleoprotein, including viral (AAV, adenovirus) and non-viral (liposomes, lipid nanoparticles, polymeric carriers, etc) [31]. For example, the first phase I clinical trial with ex vivo engineered T cells used a combination of electroporation of Cas9/gRNA ribonucleoprotein complexes and lentiviral transgenes [32]. AAV has been a strong frontrunner in studies involving in vivo CRISPR-Cas delivery in a DNA format, but has significant drawbacks, as discussed further. The purpose of this review is to discuss the unique advantages of adenovirus for in vivo delivery of the CRISPR-Cas machinery, particularly in contrast to AAV – for reviews discussing various other systems, see [33–35].
AAV IN GENE THERAPY
Adeno-associated viruses (AAVs) are small non-enveloped viruses that require other viruses, namely adenovirus, to replicate [36]. They are considered non-pathogenic in humans and can integrate efficiently into the host genome at the AAVS1 site or can be engineered to ablate this integration [37]. Combined with the mild immune response to their administration, these features have made AAV vectors the de facto frontrunner for in vivo delivery of genetic material, including CRISPR-Cas. To date, AAV has been involved in over a hundred clinical trials and has resulted in FDA approval in several instances [38].
Despite this incredible track record, lingering safety issues with AAV remain. Although recombinant AAV has been considered non-integrative, reports indicate that integration events do occur, raising the potential issue of genotoxicity and oncogenicity [39–41] - see [42] for a comprehensive review. Additionally, AAV vectors suffer from a triangle of three issues – potency, immunogenicity, and manufacturing cost. Host immune responses to the vector reduces effective viral concentration, requiring higher vector doses to achieve meaningful therapeutic corrections [42]. Higher doses in turn drive further immune responses [42]. Additionally, given that much of the population has been exposed to AAV, pre-existing immunity can dramatically reduce the efficacy of AAV therapy [37]. Underlining these problems is the complexity and cost involved in producing clinical grade recombinant AAVs. These complex processes have resulted in AAV therapies being among the most expensive in the world, with Novartis’ Zolgensma costing $2.1 million USD/dose [43].
In addition to issues with safety, the native biology of AAV makes it a difficult vector for use with CRISPR-Cas systems. The small genomic size of AAV gives it a limited transgene capacity of less than 5kb [44]. Given that spCas9 is a 4.2kb cassette without promoter or polyadenylation signal, novel strategies have had to be implemented to utilize AAV for this technology, including novel Cas proteins with a smaller size and split Cas9 proteins, with varying degrees of success [35,44].
However, spCas9 is by far the most used Cas protein and has the most data regarding off target editing. Additionally, new technologies are usually first developed using spCas9, such as CRISPRa and CRISPRi, base editors, and Prime editors. CRISPRa and CRISPRi (CRISPR activation and CRISPR inhibition, respectively) are inactivated Cas9 fused with epigenetic modifiers such as promoter activators and silencers [10]. Base editors are fusions of Cas9 with proteins capable of altering nucleotides that can correct DNA by directly changing bases [45]. Prime editors are fusions of Cas9 and a reverse transcriptase that mediate small insertions without the need for donor DNA [46]. All of these require the efficient delivery of large transgene cassettes. The Prime editor 2 system described by David Liu’s group has a size of approximately 7.3kb for the whole cassette with promoter and polyadenylation signal – clearly beyond the capacity of AAV [47]. The need for the ability to carry a larger cargo on a single vector is clear, especially in the context of clinical translation.
ADENOVIRUS IN GENE THERAPY
Adenoviral (Ad) vectors have been used since the earliest days of gene therapy and represent one of the best studied viruses biologically and clinically. Adenoviruses are non-enveloped double-strand DNA viruses with a genome ~36kb in size, which does not integrate into the host genome, ablating the risk of oncogenicity or genotoxicity seen with other vectors [48]. The protein capsid encapsulating the genome is composed majorly of three proteins, the hexon, penton and fiber, all of which have proven amenable to genetic modification to alter the properties of the virus [49]. These features, combined with the proven safety record of adenovirus in clinical trials, make Ad an attractive candidate for in vivo delivery of advanced genome editing machinery such as CRISPR-Cas.
The large dsDNA genome of Ad endows it with a large transgene packaging capacity. First generation Ad vectors with deletions in the E1 and E3 genes rendering it replication incompetent have a packaging capacity of about 8.5kB – nearly double the capacity of AAV [54]. This capacity is large enough to carry many of the advanced editing machineries previously described. Additionally, third generation so-called gutless or helper-dependent Ads are completely devoid of viral genes and have a significantly expanded packaging capacity of theoretically approximately 37kB [55]. This capacity has been used by several groups to produce highly advanced vectors packaging multiple genes into a single viral particle [56–58]. Of particular note, many CRISPR-Cas delivery techniques for HDR transgene integration have utilized a two-vector system – one vector carriers the Cas9 gene and gRNA expression cassette, while the second vector carries the HDR template DNA. Successful integration of the template DNA requires co-infection of target cells with both vectors. Recent studies with gutless Ads have enabled the molecular design of a single vector carrying Cas9, the gRNA cassette and the donor template DNA, an approach with clear advantages over prior techniques [59]. This expanded packaging capacity has obvious utility with present technologies as well as advantages for future deployment of novel advanced genome editing technologies.
Additionally, the strong understanding of Ad structure and biology have enabled rational genetic design of vectors with altered features. Engineering the protein capsid has emerged as a successful strategy for altering how Ad particles behave in vivo after administration [49]. This has resulted in the development of vectors which have overcome several of the challenges with in vivo delivery of Ads, namely 1) sequestration of the virus in the liver 2) limited transduction of cells lacking the Ad receptor, and 3) pre-existing host immune responses to Ads.
Wild-type Adenovirus serotype 5 (Ad5), the best studied and most widely used serotype, is largely sequestered in the liver after intravenous administration. The Ad5 hexon protein is bound by blood coagulation factor X in the blood and transported directly to the liver [60]. As a result, gene transfer from Ad5 is mostly limited to the liver. Although this approach has shown utility in several diseases, ablating this targeting allows for potentially advantageous delivery to different tissues, such as the endothelium [61]. Additionally, ablating this targeting via hexon or fiber modifications allows for transduction of cells in the blood, such as mobilized hematopoietic stem cells, and potentially any other type of circulating cell given proper vector engineering [62]. Several strategies have been used to achieve this liver de-targeting, including the use of Ad serotypes with low or no binding FX (such as Ad35) and genetic modification of the Ad hexon protein [63,64]. Finally, the strong liver tropism of Ad5 can result in significant immune responses, resulting in hepatoxicity [65]. Designing vectors with altered tropism is therefore an important step in overcoming this issue with Ad5. The success of these strategies represents a critical step in realizing a central goal of gene therapy - delivering genetic corrections to specific cells or tissues. Avoidance of liver targeting is an important step in engineering vectors to achieve this goal.
The fiber protein is another popular target for Ad5 modification. The Ad5 fiber mediates binding to the coxsackie and adenovirus receptor (CAR), which is followed by internalization of the viral particle, initiating the first step in cell infection [66]. Although CAR is expressed on many tissues, its expression is low on cells of interest for gene therapy such as cancer cells and hematopoietic stem cells [67,68]. As such, alteration of Ad tropism has long been a goal of Ad engineering. Multiple strategies have been utilized to achieve this. Pseudotyping has been one of the most successful techniques towards this end – in this process, all or part of the Ad5 fiber is genetically removed and replaced with a fiber from a different Ad serotype, or even a non-human Ad (termed xenotyping). Through this process, the well-studied Ad5 biology is preserved, but cell targeting is altered [49]. This has been used to dramatically alter Ad5 tropism, such as expanding tropism towards hematopoietic stem cells using an Ad35 fiber targeting the CD46 receptor [68].
Although fiber swapping has been highly successful, it generally results in expanded or altered tropism without selectivity towards a single cell or tissue type. As cell and tissue specificity is one the of the goals of vector engineering, molecular adaptors and chimeric fiber fusion proteins have been explored. The molecular adaptor strategy consists of administering an Ad5 type vector along with a chimeric adaptor protein containing a binding domain for the Ad5 fiber on one end (such as the soluble CAR domain) and a binding domain for a selected surface protein present on the target cell type on the other end. This adaptor then allows binding between Ad5 and the target protein, creating a bridge to the target cell by binding to the designated receptor [69–71]. A strong advantage of this approach is the ability to utilize the large repertoire of antibodies and other binding proteins to easily design an adaptor which binds to selected surface proteins on the target cell type.
Although this strategy shows promise in targeting specific cell types, the design and implementation of adaptor molecules suitable for in vivo delivery is challenging, and clinical translation of a two-component system may further complicate the already difficult process of moving genetic therapies from bench to bedside [49,72]. As an alternative strategy, genetic modification of the Ad5 fiber protein has been pursued. Alteration of Ad tropism towards targeted cell surface proteins has been shown via the incorporation of peptide ligands, single domain antibodies and other targeting agents into the Ad5 fiber [73–75]. This complex and elegant strategy is a significant achievement in vector engineering, enabling genetic targeting of Ads to specific cell types. This represents a large step towards the goal of cell specific gene therapy. However, this technology is still in its infancy, and various questions regarding the in vivo efficacy of these constructs remain unanswered.
In addition to tropism alteration via incorporation of targeting agents, the Ad5 fiber protein has also been genetically altered to expand its tropism rather than restrict it. The Ad5 fiber has been modified to include cell binding motifs such as tripeptide Arg-Gly-Asp (RGD) or polylysine, which expands the tropism nonspecifically beyond the CAR receptor. This approach has particular utility for treating cancers, many of which are refractory to Ad5 infection due to low expression of CAR [76,77]. In total, these technologies demonstrate the feasibility of engineering Ads for use in targeted gene therapy. For a comprehensive review of these tissue targeting strategies see [49].
Finally, Ads possess a significant cost and scalability advantage over other vectors including AAV. Even the more complex gutless Ads are producible at scale for roughly 1/50th the cost of rAAVs [30]. This advantage is further demonstrated by Johnson & Johnson’s announcement that their lead candidate for vaccination against COVID19 is an adenovirus. This confidence that an Ad based vaccine can be scaled to global levels prove its utility in this realm [78].
Given the clear advantages of Ads for in vivo gene delivery, several reports have described the use and efficacy of these vectors for in vivo delivery of the CRISPR-Cas machinery, enabling correction of various diseases in mice.
ADENOVIRUS FOR INHERITED DISORDERS
The most widely used application of adenoviruses for in vivo delivery of CRISPR-Cas thus far has been the treatment of inherited disorders. These approaches have generally followed one of two strategies; utilizing either NHEJ to knock out mutated genes, or HDR to knock in corrected genes.
The earliest explorations of adenovirus for in vivo CRISPR-Cas delivery utilized NHEJ [79,80]. In these studies, the general strategy employed was to deliver a standard Ad5 vector to the liver with Cas9 and a gRNA designed to target a mutated gene. This approach was employed with varying degrees of success in several studies in mice, as shown in Table 2 below.
Table 2:
Ongoing clinical trials using CRISPR-Cas technology, via search of Clinicaltrials.gov [27]. Observational and diagnostic studies excluded.
| Title | Phase | Status | Conditions | Interventions | Sponsor/Location |
|---|---|---|---|---|---|
| Safety of Transplantation of CRISPR CCR5 Modified CD34+ Cells in HIV-infected Subjects with Hematological Malignancies | N/A | Recruiting | HIV-1 Infection | Infusion of HSPCs modified ex vivo to knockout CCR5 gene | Affiliated Hospital to Academy of Military Medical Sciences/China |
| Study of CRISPR-Cas9 Mediated PD-1 and TCR Gene-knocked Out Mesothelin-directed CAR-T Cells in Patients With Mesothelin Positive Multiple Solid Tumors | I | Recruiting | Mesothelin+ Solid Tumors | Infusion of anti-mesothelin CAR-T cells modified ex vivo to knockout PD-1 and TCR genes | Chinese PLA General Hospital/China |
| CRISPR (HPK1) Edited CD19-specific CAR-T Cells (XYF19 CAR-T Cells) for CD19+ Leukemia or Lymphoma | I | Recruiting | Leukemia or Lymphoma | Infusion of anti-CD19 CAR-T cells modified ex vivo to knockout HPK1 gene | Xijing Hospital/China |
| A Safety and Efficacy Study Evaluating CTX001 in Subjects With Transfusion-Dependent β-Thalassemia | I/II | Recruiting | Beta-Thalassemia | Infusion of HSPCs modified ex vivo to disrupt the BCL11A gene | Vertex Pharmaceuticals/USA |
| A Safety and Efficacy Study Evaluating CTX120 in Subjects With Relapsed or Refractory Multiple Myeloma | I | Recruiting | Multiple Myeloma | Infusion of anti-BCMA CAR-T cells modified ex vivo | CRISPR Therapeutics AG/USA |
| A Safety and Efficacy Study Evaluating CTX001 in Subjects With Severe Sickle Cell Disease | I/II | Recruiting | Sickle Cell Disease | Infusion of HSPCs modified ex vivo to disrupt the BCL11A gene | CRISPR Therapeutics/USA |
| A Safety and Efficacy Study Evaluating CTX110 in Subjects With Relapsed or Refractory B-Cell Malignancies | I/II | Recruiting | B-cell Malignancy, Non-Hodgkin Lymphoma, B-Cell Lymphoma | Infusion of anti-CD19 CAR-T cells modified ex vivo | CRISPR Therapeutics AG/USA |
| iHSCs With the Gene Correction of HBB Intervent Subjests With β-thalassemia Mutations | I | Not yet recruiting | Beta-Thalassemia | Infusion of iHSCs modified ex vivo to genetically correct the HBB gene | Allife Medical Science and Technology Co., Ltd./n/a |
| Study of PD-1 Gene-knocked Out Mesothelin-directed CAR-T Cells With the Conditioning of PC in Mesothelin Positive Multiple Solid Tumors | I | Recruiting | Mesothelin+ Solid Tumors | Infusion of anti-mesothelin CAR-T cells modified ex vivo to knockout PD-1 in combination with Paclitaxel and Cyclophosphamide | Chinese PLA General Hospital/China |
| A Study Evaluating UCART019 in Patients With Relapsed or Refractory CD19+ Leukemia and Lymphoma | I/II | Recruiting | B Cell Leukemia/Lymphoma | Infusion of anti-CD19 “universal” CAR-T cells modified ex vivo to disrupt TCR and B2M genes | Chinese PLA General Hospital/China |
| A Feasibility and Safety Study of Universal Dual Specificity CD19 and CD20 or CD22 CAR-T Cell Immunotherapy for Relapsed or Refractory Leukemia and Lymphoma | I/II | Recruiting | B Cell Leukemia/Lymphoma | Anti-CD19/CD20 or Anti-CD19-CD22 dual specificity CAR-T cells | Chinese PLA General Hospital/China |
| Cell Therapy for High Risk T-Cell Malignancies Using CD7-Specific CAR Expressed On Autologous T Cells | I | Not yet recruiting | T-cell Acute Lymphoblastic Leukemia/Lymphoma, T-non-Hodgkin Lymphoma | Infusion of anti-CD7 CAR-T cells modified ex vivo to remove the CD7 gene in combination with Fludarabine and Cytoxan | Baylor College of Medicine/USA |
| PD-1 Knockout Engineered T Cells for Metastatic Non-small Cell Lung Cancer | I | Active | Metastatic Non-small Cell Lung Cancer | Infusion of T cells modified ex vivo to knockout PD-1 in combination with Cyclophosphamide | Sichuan University/China |
| PD-1 Knockout EBV-CTLs for Advanced Stage Epstein-Barr Virus (EBV) Associated Malignancies | I/II | Recruiting | Stage IV Gastric Carcinoma, Stage IV Nasopharyngeal Carcinoma, Stage IV T-cell Lymphoma, Stage IV Adult Hodgkin Lymphoma, Stage IV Diffuse B-Cell Lymphoma | Infusion of cytotoxic T cells modified ex vivo to knockout PD-1 in combination with Fludarabine, Cyclophosphamide and Interleukin-2 | Yang Yang/China |
| Single Ascending Dose Study in Participants With LCA10 | I/II | Recruiting | Leber Congenital Amaurosis 10 | Sub-retinal injection of AAV5 carrying SaCas9 and two gRNAs for exon splicing mediated correction of disease | Allergan/USA |
One study utilized NHEJ but pursued an alternate method to correct Duchenne’s muscular dystrophy [83], in which mutations in the dystrophin gene cause a frame shift that results in the entire protein being misfolded. To correct this, the authors utilized a clever “exon skipping” strategy – due to the size of the dystrophin gene, an entire mutated exon was excised, resulting in correction of the disease-causing frame shift mutation. Impressively, intramuscular injections of Ad delivering Cas9 and two gRNAs to cut out the exon resulted in systemic improvement in the rat muscular dystrophy model used. This particular study highlights the strength of techniques that are able to utilize the NHEJ pathway rather than HDR – approaches that are less straight forward may be able to succeed due to the high efficiency of NHEJ.
Another study utilized a similar strategy to induce switching of hemoglobin production in erythrocytes from beta to gamma globin [81]. This strategy has proven useful for correcting hemoglobinopathies such as beta-thassalemia and sickle cell disease – the presence of the non-mutated gamma globin is able to correct the phenotypes of these diseases if it is present in sufficiently high levels. The use of CRISPR-Cas to generate indels at specific locations in the beta-globin locus control region has been proven to be an elegant and efficient approach to achieve this switch [84]. The authors demonstrated the use of this technique in vivo on mobilized hematopoietic stem cells and were able to achieve near corrective levels of gamma globin in the blood [81]. Importantly, the authors were able to achieve this gene editing using an advanced engineered Ad5 vector containing a fiber swap and other fiber mutations, demonstrating the utility of the aforementioned vector engineering strategies to achieve highly complex and challenging in vivo gene edits with CRISPR-Cas.
An alternative approach to NHEJ is to utilize HDR to knock in corrective genes – rather than trying to knock out regions of the beta-globin locus control region to generate gamma globin, knock in of the gamma globin gene could be used, for example. Although this approach could be generally less efficient than those based on NHEJ, it has the advantage of being a potentially universal treatment for diseases caused by deficiencies of plasma proteins [85]. If sufficient gene transfer and knock-in is achieved, producer cells could produce corrective amounts of protein for any such disease. This technology therefore represents a potential platform that could be realized via simple switching of the relevant gRNAs and donor DNA.
A smaller number of studies have utilized this technology. In the earliest study found for this review, the authors described successful integration of a gene corrective for hemophilia B into the livers of hemophilic mice [86]. The authors tested adenovirus alongside injected plasmid DNA and found that although adenovirus produced significantly higher rates of HDR, a corrective phenotype was not seen, potentially due to liver damage and inflammation from the vector. Interestingly, this result is at odds with the results seen in future studies, and could stem from vector design and quality, gRNA design, and differences in animal models. Regardless, this study does highlight the need to consider immune responses against the vector used in any gene transfer study.
Two other studies utilized a similar Ad5 system with liver transduction but achieved dramatically different results [85,87]. In these studies, a two-vector system was used to deliver Cas9 and a gRNA targeting the mouse ROSA26 safe-harbor locus, and template DNA for correction of either hemophilia B or alpha-1-antitrypsin deficiency. In both cases, high rates of HDR were observed without apparent long-term damage to the mouse livers. Remarkably, transgene expression was studied for > 200 days in each case, comparing the CRISPR-Cas system against standard episomal Ads, which demonstrated that the integrative systems persisted at higher levels than the episomal vectors throughout the course of the entire study. These studied showed that Ad5 transduction of hepatocytes and transgene integration with CRISPR-Cas could achieve corrective levels of protein in the blood for an extremely long timespan.
Lastly, a recent study further demonstrated the potential of advanced Ad vector design in combination with CRISPR-Cas [57]. In this study, mobilized HSCs were transduced with the same fiber swapped Ad5 vector previously described. A two-vector system was utilized delivering Cas9 and a gRNA targeting the AAVS1 locus in transgenic mice with donor template DNA containing gamma globin under the control of the beta-globin locus control region with an attached mgmt in vivo selection cassette. This regime resulted in integration of the entire donor DNA cassette at the AAVS1 locus in hematopoietic stem cells and allowed the authors to provide a proliferative stimulus to the edited HSCs via the mgmt gene, resulting in their multiplication in vivo. Briefly, mice were treated with a drug cocktail toxic to normal HSCs. Edited HSCs containing the mgmt gene were resistant to the drugs, allowing them to survive and replace the normal HSCs. Through this technique, the authors achieved potentially corrective levels of gamma globin in the blood, possibly enabling treatment of beta-thassalemia and sickle cell disease.
ADENOVIRUS FOR CANCER GENE THERAPY
Several studies in mice have also demonstrated the utility of Ads for delivering the CRISPR-Cas system in vivo for the treatment of cancer [88–91]. Ads have a strong history as oncolytic vectors with over 200 clinical trials ongoing at the time of writing [92]. In general, these therapies rely on the ability of replicating Ads to lyse infected cells. Tumor specific promoters driving the genes responsible for Ad replication have enabled the generation of so-called “conditionally replicative Ads”, which have the potential to selectively destroy tumor cells while sparing off-target tissues. The inherent immunogenicity of Ads further strengthens the immune response against cancer, and the packing capacity of Ad can be harnessed to deliver therapeutic compounds to the cancer cells at the same time [72].
The advent of CRISPR-Cas technology has added an additional potential utility to oncolytic Ads. In general, these studies aimed to leverage the targeted nature of CRISPR-Cas to knock-out or inhibit mutated oncogenes, such as EGFR or KRAS [88,91]. A strength of this approach was the ability to select PAM sites that allow Cas9 or inactivated Cas9 (dCas9) to bind to the mutated oncogene while sparing normally functioning copies of the gene. dCas9 is a mutated version of spCas9 without nuclease activity. As a result, dCas9 can bind to DNA but not introduce nicks or DSBs. This protein can be tied to a DNA activator or repressor domain (such as VP64 or KRAB, respectively) to selectively turn on or off transcription at targeted sites. This technology is also known as CRISPRa and CRISPRi, as described previously. Compared to traditional Cas9 knock-out techniques, using dCas9 to inhibit transcription may be safer due to the lack of introduced DSBs and lower chances for off-target effects [90]. However, one study did find that a Cas9 knockout was more effective in inhibiting tumor growth than dCas9-KRAB repression [91]. As always, the tradeoff between safety and efficiency must be considered when developing such therapies.
This CRISPR-Cas based approach to reduce the activity of mutated oncogenes increases the safety profile of such therapies and possesses significant advantages over conventional small-molecule or antibody inhibition-based strategies. Rather than inhibiting the product of various oncogenes, CRISPR-Cas technology directly targets the genes themselves, potentially enabling one-shot treatments that dramatically alter the tumor genome. Additionally, the CRISPR-Cas system is highly modular and flexible. Compared to the lengthy and costly process to develop new antibodies or small molecules inhibiting each target mutant protein, development of new gRNAs is trivial. Furthermore, multiplexing of gRNAs into a single vector and dose is possible, potentially targeting multiple mutated genes in one shot [56].
One unique utilization of more advanced genome engineering techniques to treat cancer was reported [89]. In this study, the authors developed a photoactivable CRISPR-Cas system. Cas9 was fused to two CIBN domains, while the DNA activating VP64 domain was fused to a CRY2 domain. In the presence of blue light, CIBN and CRY2 are capable of dimerization. Thus, upon activation of the system dCas9 binds to its target DNA sequence, and CIBN and CRY2 dimerize to link VP64 to dCas9 and activate gene transcription. The authors utilized this system to target the gene Dkk-3, which has been implicated as a tumor suppressor. Without blue light, dCas9 is bound to the target DNA but unable to influence transcription. In the presence of blue light, VP64 is brought close to the dCas9 binding site via CIBN/CRY2 binding and Dkk-3 transcription is activated. The authors demonstrated that this technology could be utilized to control Dkk-3 spatially and temporally, with tumor suppression observed after Dkk-3 activation [89].
This novel technology could be explored for spatiotemporal control of any CRISPR-Cas based gene therapy, potentially resulting in an increased safety profile. Uncontrolled Cas9 expression has been implicated with undesirable toxicities and off-target editing events, thus highlighting the need for control of these systems [93,94].
In contrast, most described systems thus far have been relatively simple – a single vector delivered intratumorally produces Cas9 with a gRNA targeting a mutant oncogene. The results obtained have been generally impressive, with studies often achieving significant reduction in tumor growth in mouse xenograft models. Of course, a limitation of this approach is the requirement to screen patients for the target mutations prior to administration, potentially requiring design and testing of multiple gRNAs to develop a product capable of treating a single type of cancer. However, as described previously this approach is significantly more feasible with CRISPR-Cas technology than with antibodies or small molecules, and genetic screening of patients with cancer is increasingly common [95].
Additionally, in reality many tumors are not homogenous – a solid tumor may contain cells harboring the targeted mutation and cells that lack it [96]. Knocking out of the target oncogene may reduce cancer growth initially but eventually lead to re-emergence of the cancer from spared cells lacking the mutation. Combining multiplexed CRISPR-Cas based therapeutic techniques with traditional oncolytic virotherapy approaches might be a solution. It is easy to envision a single conditionally replicative Ad vector delivering Cas9 and multiple gRNAs, along with a transgene promoting apoptosis or a pro-drug, potentially attacking the tumor from multiple angles.
Finally, all studies found at the time of writing this article relied on intratumoral injection of the vector. While this strategy is applicable to certain cancers, it is likely less applicable to more diffuse or metastatic cancers. It may be useful to develop Ads capable of targeting cancer cells after systemic administration. There are numerous reports in the literature of Ads with tropism towards specific tissues such as the lung, kidneys and of course liver [65,97]. This approach may be useful for cancers for which local administration is not feasible due to the tumor location or diffuse nature.
LESSONS LEARNED AND FUTURE OUTLOOK
Numerous studies have now shown the potential utility of Ad vectors for in vivo delivery of the CRISPR-Cas machinery. These studies have focused on and achieved success with various inherited disorders and cancer. However, several unanswered questions remain, namely 1) the extent and impact of off-target edits and 2) understanding and mitigating immune responses.
An increasing number of reports have discussed the potential for undesirable outcomes due to off-target editing by CRISPR-Cas systems. Although Cas9 is guided to a selected site by the gRNA, the match need not be perfect for binding, leading to DSBs outside the target site [98]. This can result in gene deletions or insertions at undesired locations in the genome or translocations when DSBs are created on different chromosomes [32], potentially producing deleterious effects. Awareness of this issue is especially critical for in vivo editing. Increasingly powerful profiling and purification technologies allow for selection of cells containing the desired traits when editing is conducted ex vivo, reducing the risk of introducing pathogenic mutations into the patient. However, this selection is impossible with in vivo editing. It is therefore of utmost importance to the translation of this technology to obtain a robust understanding of the extent and effects of off-target editing after CRISPR-Cas delivery.
One strategy which has been employed when developing HDR knock-in strategies has been the use of a genetic safe-harbor locus. Safe-harbor loci are considered areas of the genome where genetic edits are unlikely to introduce issues. A variety of requirements have been proposed, but in general a safe-harbor locus should be far from rapidly replicating genes, far from oncogenes and not involved in critical cellular processes [99]. Several human and mouse safe-harbor loci have been proposed and utilized, including AAVS1 in humans and ROSA26 in mice [57,100].
These loci can alleviate some of the risks encountered when utilizing HDR knock-in strategies by potentially reducing detrimental effects from NHEJ. For example, if correction of sickle cell disease was undertaken in vivo by using two gRNAs and Cas9 to attempt to cut out the defective beta-globin gene and knock-in a corrected one, even at relatively high HDR efficiencies most cells would contain an indel produced by NHEJ, rather than the corrected gene. The remaining cells would simply lack the beta-globin gene, likely causing serious issues. In contrast, if the corrected gene is knocked-in at a safe-harbor locus, NHEJ mediated edits would not interfere with important processes, while HDR edits would express the corrected gene. This technique therefore has potential to help reduce the risk involved in employing in vivo CRISPR-Cas therapies.
An additional technique which may be helpful in mediating the off-target effects of Cas9 as well as potential toxicities from the nuclease may be temporal control. Ideally, a CRISPR-Cas based therapy should consist of rapid expression of Cas9 and any template DNA in the cell of interest, DNA cutting and integration of donor DNA if applicable, followed rapidly by clearance of the nuclease. Continued expression of Cas9 after these events may result in increased numbers of off-target edits as Cas9 continues to roam the genome and search for gRNA matches [101]. Interestingly, the authors of one study detected the Cas9 gene over 200 days after they delivered it, further highlighting the need for temporal control [85]. Increasing reports around off-target effects of spCas9 and its immunogenicity and toxicity to mammalian cells highlight the need implement controls on the expression and activity of genome editing machineries [102,103]. A variety of approaches have been explored to achieve this, including anti-Cas9 peptides [94], self-destructing cassettes with gRNAs against the Cas9 gene [104], and others - for a comprehensive review, see [103]. Adenoviruses provide an ideal opportunity to leverage these technologies due to their large packing capacity and studying such systems will likely prove valuable.
Moving forward, this problem may also be addressed at the fundamental level by the development of novel genome editing machineries. Extensive research has been undertaken to engineer Cas9 variants with higher efficiencies and specificities. In parallel, work is ongoing to attempt to discover new Cas variants in bacteria with different properties than Cas9. This work has resulted in variants with smaller sizes for ease in packaging, different PAM sequences, the ability to target RNA rather than DNA and otherwise [10]. Additionally, as previously described novel fusion proteins such as base editors and Prime editors have been created. It will likely be highly advantageous for researchers pursuing in vivo editing to stay abreast of these developments and choose highly specific and efficient systems which reduce the potential for off-target editing events.
An understanding of the body’s immune responses is also critical to the translation of this technology. Several studies have noted immune responses to the Ad capsid, Cas9 nuclease, and introduced transgene [58,80,85]. This has implications for successful therapy. Responses against the Ad capsid and Cas9 nuclease have not been generally reported to be overly toxic, but they may limit potential dosages. Liver damage and immune responses to Ad5 vectors is a well-documented effect. Furthermore, antibody responses to the introduced transgene may limit the efficacy of the therapy, depending on the nature and number of antibodies produced. Liver targeted gene therapy generally does not have this issue due to the liver’s role as a tolerance inducing organ, but expansion of gene therapy outside the liver may require efforts to mediate these effects, such as utilizing promoters with activity in the target tissue and the liver [105].
CONCLUSIONS
As gene therapy continues to be explored scientifically and clinically, increasingly advanced systems have become possible. Capitalizing on the tremendous potential of CRISPR-Cas technology, adenoviral vectors have achieved potentially corrective levels of permanent genomic editing, either through gene knock-in or knock-out. Although ex vivo cell editing has resulted in several successes, its utilization is limited to high-cost diseases in the first world where advanced medical technology is available and cell types capable of being collected and expanded clonally outside the body. Adenoviruses are capable of being engineered rationally to achieve cell specific targeting with high levels of gene transfer, making them an ideal vector for in vivo delivery of the CRISPR-Cas machinery.
To capitalize on the advantages of gene therapy, a simplified approach is required. The in vivo approach will be especially valuable to leverage the power of CRISPR-Cas for applications such as vaccines, where the scale is too large for ex vivo editing, or diseases endemic to the third world where the infrastructure is lacking for ex vivo editing. Additionally, taking advantage of the ability to modify the tropism of Ads may yield advancements in targeting blood cells such as erythrocytes and leukocytes, as well as non-liver organs and tissues. Ads may also have a strong potential to treat viral diseases such as HIV after in vivo delivery of CRISPR-Cas targeting viral DNA. Future work with advanced vectors and genome editing technologies therefore has the potential to treat diseases previously beyond the reach of medical science and improve on human health.
Figure 1:
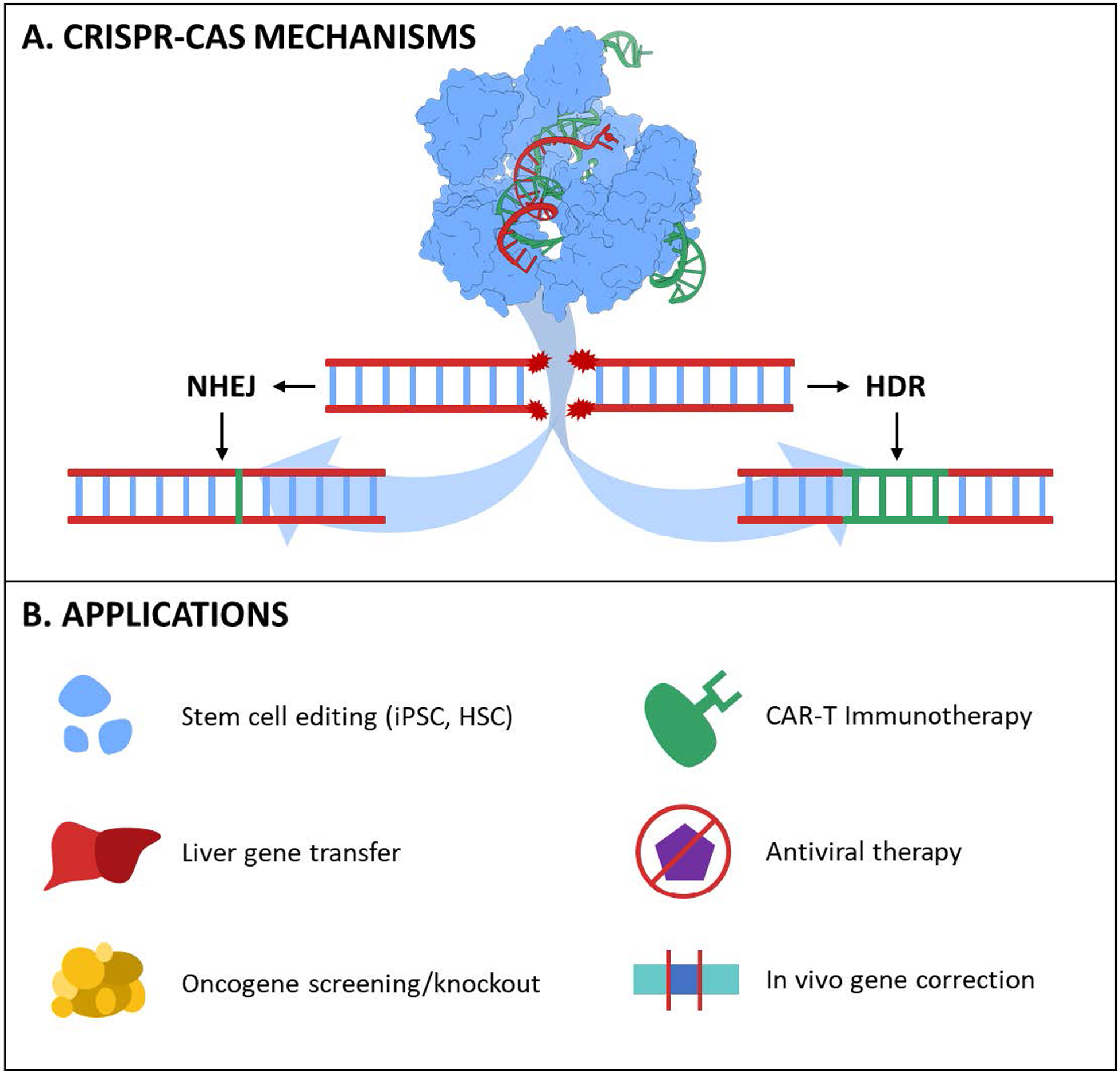
Overview of CRISPR-Cas technology for medicine. A) Cas nucleases create targeted DSBs in dsDNA via gRNA hybridization with complementary DNA. Shows a molecular graphic of spCas9 with guide RNA in complex with DNA. PDB ID: #4OO8 [24]. Rendered with UCSF ChimeraX [25]. B) Potential therapeutic applications of CRISPR-Cas [26,27].
Figure 2:
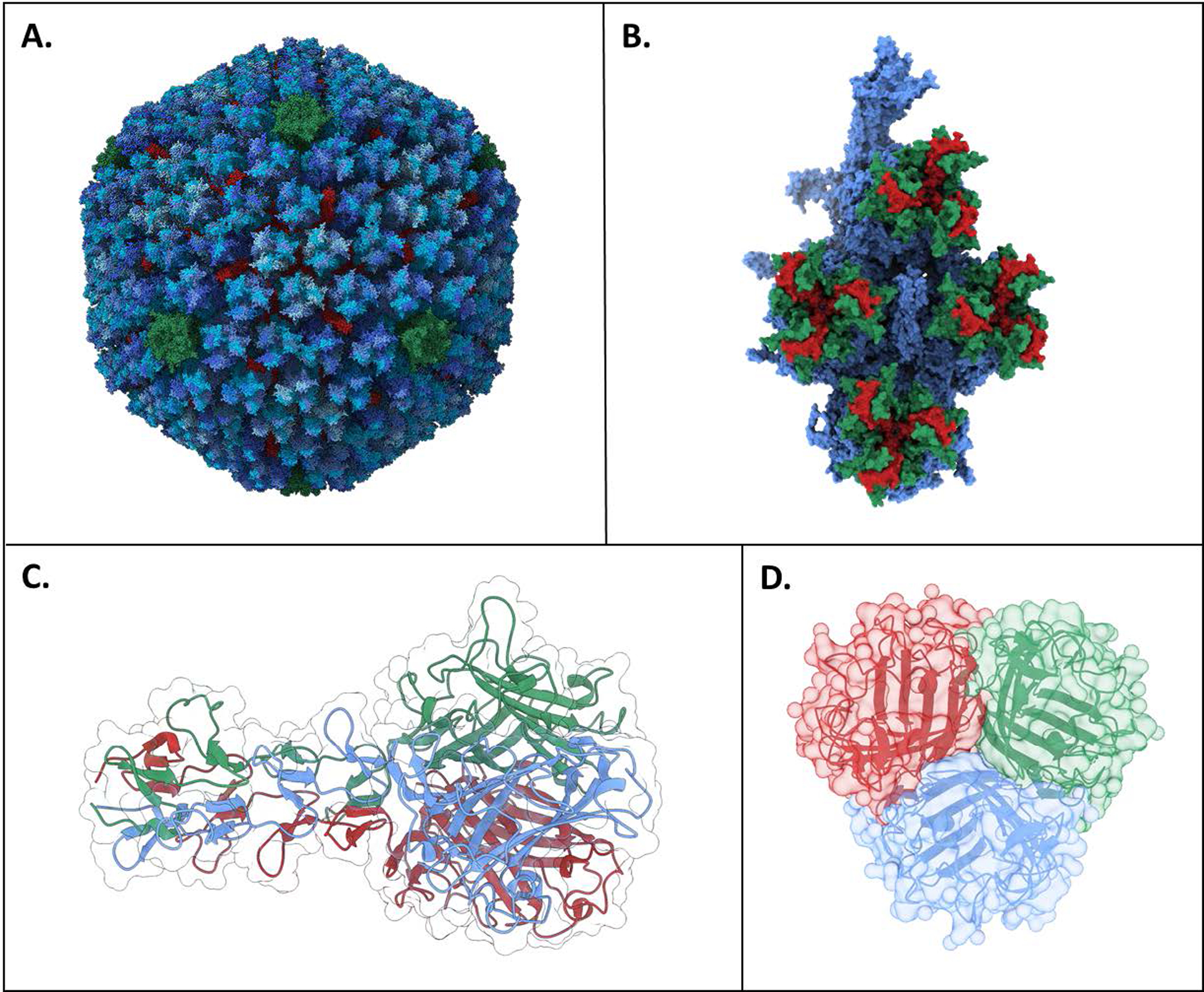
Adenovirus structure. A) Adenovirus capsid structure. The various hexon protein chains are colored in blues, penton protein in green, and hexon-interlacing protein in red. Trimer fiber inserts into green penton protein center (not shown). B) Hexon monomeric unit. Hypervariable loop 1 is shown on green and hypervariable loop 2 is shown in red. Alignment of hypervariable regions determined from [50] C) Side view of fiber knob from Ad2 (structurally similar to Ad5 and four shaft repeat motifs. D) Top view of fiber Ad5 fiber knob showing trimeric symmetry. PDB IDs: 6B1T (A, B) [51], 1QIU (C) [52], and 6HCN (D) [53]. Rendered with UCSF ChimeraX [25]. Print in color.
Figure 3:
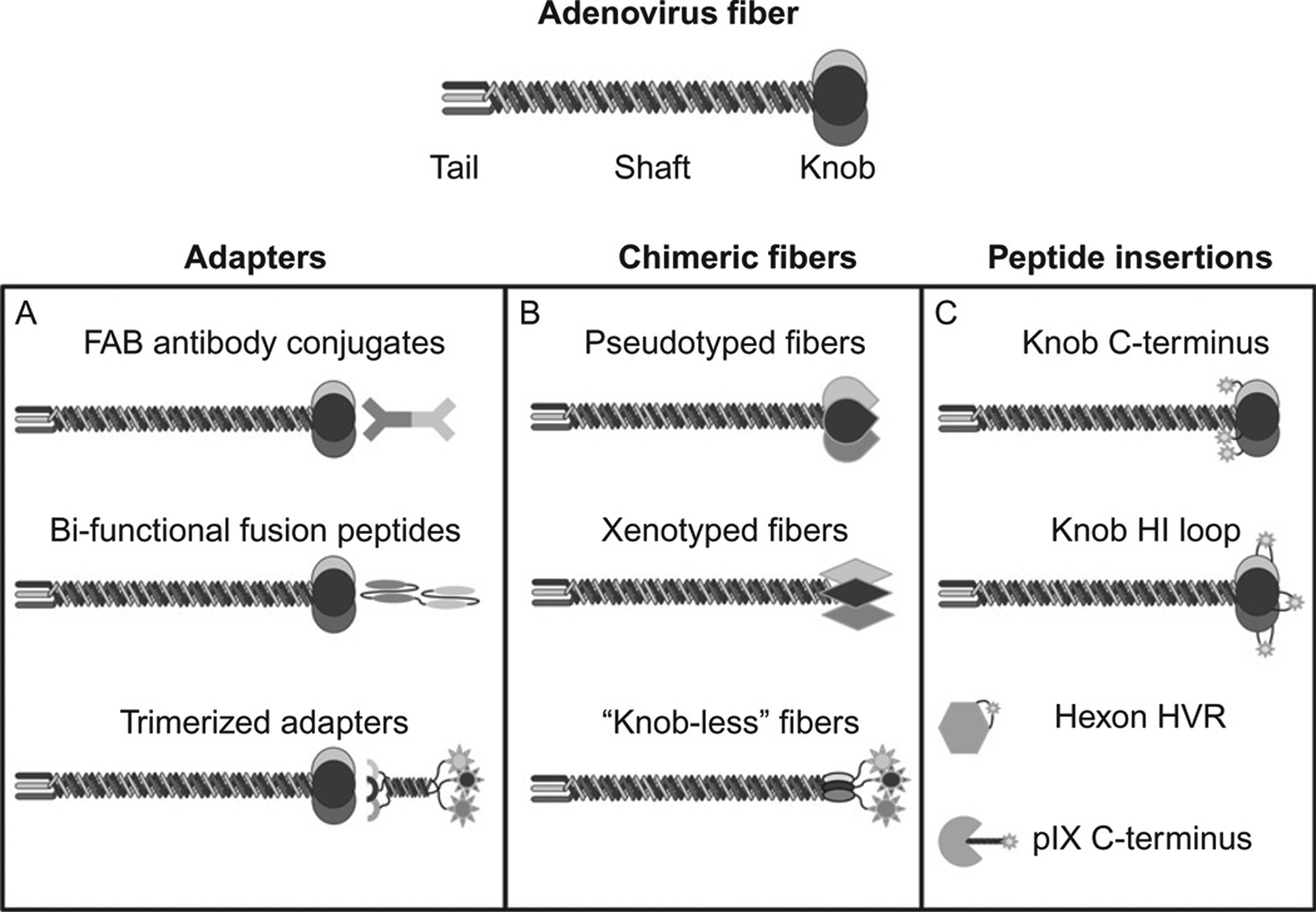
Strategies for modifying Ad5 tropism, including A) Molecular adaptors B) Fiber swaps and C) Fiber modifications. Re-used with permission from [49].
Figure 4:
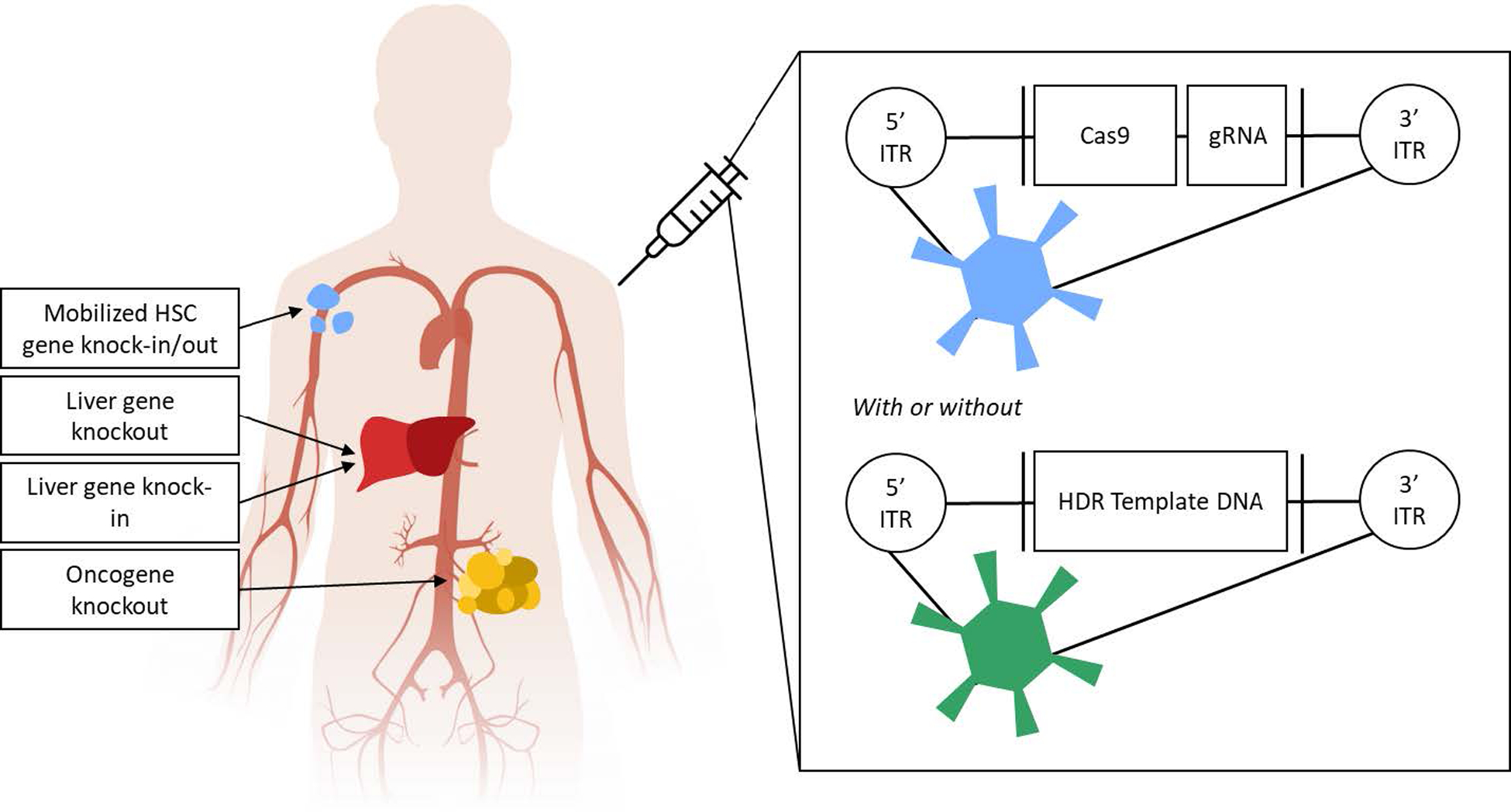
Overview of explored strategies for CRISPR-Cas mediated gene editing in vivo using Adenovirus. Ads delivering the Cas9 gene and gRNA can induce gene editing via NHEJ in the liver or in circulating hematopoietic stem cells (HSCs) after systemic administration. Intratumoral delivery can mediate NHEJ in tumor cells. Alternatively, a two-vector system delivering the Cas9 gene and gRNA in one vector and donor template DNA in another can achieve HDR in the liver or circulating HSCs.
Figure 5:
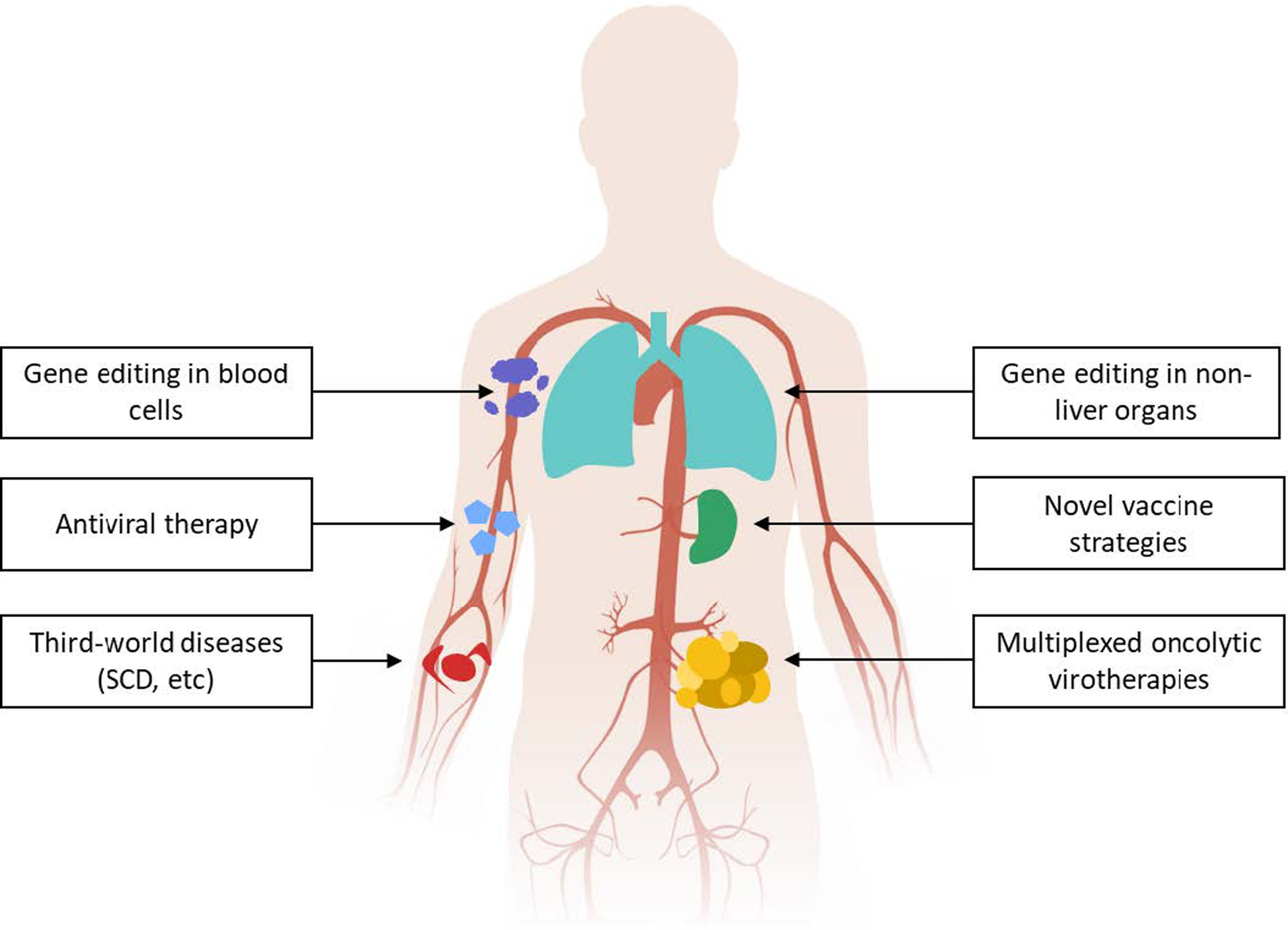
Potential future applications of in vivo adenoviral CRISPR-Cas delivery.
Table 1:
CRISPR-Cas based gene therapy programs currently pursued by several major companies, including CRISPR Therapeutics, Editas Medicine, Intellia Therapeutics and Beam Therapeutics [13–23].
| Company | Disease | Editing | Phase | Administration |
|---|---|---|---|---|
| CRISPR Therapeutics | β-thalassemia | NHEJ | Clinical | Ex vivo |
| Sickle cell disease | NHEJ | Clinical | Ex vivo | |
| Anti-CD19 allogeneic CAR-T | NHEJ & HDR | Clinical | Ex vivo | |
| Anti-BCMA allogeneic CAR-T | NHEJ & HDR | Clinical | Ex vivo | |
| Anti-CD70 allogeneic CAR-T | NHEJ & HDR | Clinical | Ex vivo | |
| Type I diabetes mellitus | NHEJ & HDR | Research | Ex vivo | |
| Glycogen storage disease type 1a | NHEJ & HDR | Research | In vivo | |
| Duchenne Muscular Dystrophy | NHEJ & HDR | Research | In vivo | |
| Myotonic dystrophy type 1 | NHEJ & HDR | Research | In vivo | |
| Cystic fibrosis | NHEJ & HDR | Research | In vivo | |
| Editas Medicine | Leber congenital amaurosis 10 | NHEJ | Clinical | In vivo |
| Usher syndrome 2A | NHEJ | Research | In vivo | |
| Autosomal dominant retinitis pigmentosa 4 | HDR | Research | In vivo | |
| Neurological Diseases | Unknown | Research | In vivo | |
| Sickle Cell Disease | NHEJ | Research | Ex vivo | |
| β-thalassemia | NHEJ | Research | Ex vivo | |
| Cancer – Healthy Donor NK Cells | NHEJ & HDR | Research | Ex vivo | |
| Cancer – iPSC NK Cells | NHEJ & HDR | Research | Ex vivo | |
| Cancer - γδ T cells | NHEJ & HDR | Research | Ex vivo | |
| Cancer αβ T cells | NHEJ & HDR | Research | Ex vivo | |
| Intellia Therapeutics | Transthyretin amyloidosis | NHEJ | Research | In vivo |
| Hereditary Angioedema | NHEJ | Research | In vivo | |
| Hemophilia A and B | HDR | Research | In vivo | |
| Undisclosed programs | NHEJ & HDR | Research | In vivo | |
| Sickle cell disease | NHEJ & HDR | Clinical | Ex vivo | |
| Acute myeloid leukemia | NHEJ & HDR | Research | Ex vivo | |
| Solid tumors | NHEJ & HDR | Research | Ex vivo | |
| Undisclosed programs | Unknown | Undisclosed | Ex vivo | |
| Beam Therapeutics | Sickle Cell Disease | Base Editing | Research | Ex vivo |
| β-thalassemia | Base Editing | Research | Ex vivo | |
| T-cell acute lymphoblastic leukemia | Base Editing | Research | Ex vivo | |
| Acute myeloid leukemia | Base Editing | Research | Ex vivo | |
| Alpha-1 antitrypsin deficiency | Base Editing | Research | In vivo | |
| Glycogen storage disorder 1a | Base Editing | Research | In vivo | |
| Undisclosed | Base Editing | Research | In vivo | |
| Stargardt disease | Base Editing | Research | In vivo |
Table 3:
Studies that used Adenovirus to achieve NHEJ gene knockout to potentially treat or study inherited disorders.
| Study and Reference | Year | Disease | Results Summary |
|---|---|---|---|
| Reactivation of γ-globin in adult β-YAC mice after ex vivo and in vivo hematopoietic stem cell genome editing [81] | 2018 | β-thassalemia/SCD | Advanced Ad5/35 fiber modified vector delivering Cas9 targeting BCL11A was administered intravenously targeting mobilized hematopoietic stem and progenitor cells. BCL11A was targeted as knockout mutations can induce switching from beta-globin to gamma-globin, potentially correct sickle cell disease and beta-thassalemia. After in vivo selection of edited HSPCs 13% of RBCs were positive for gamma-globin. |
| Therapeutic Genome Editing With CRISPR/Cas9 in a Humanized Mouse Model Ameliorates α1-antitrypsin Deficiency PhenotypAmeliorates α1-antitrypsin Deficiency Phenotype [82] | 2018 | Alpha-1-antitrypsin deficiency | Ad5 delivering Cas9 targeting mutant hSERPINA was delivered intravenously to knock-down levels of mutated alpha-1-antitrypsin (AAT) and reduce damage to the liver and lungs caused by the misfolded protein. Treated mice showed reduced liver damage and reduced levels of AAT. |
| CRISPR mediated Genome Editing Restores Dystrophin Expression and Function in mdx Mice [83] | 2016 | Duchenne muscular dystrophy | Ad5 delivering Cas9 and two gRNAs was delivered intramuscularly to excise a mutated exon from mdx mice and restore the reading frame of the dystrophin gene. Treated mice had significantly increased levels of functional dystrophin. |
| Adenovirus-Mediated Somatic Genome Editing of Pten by CRISPR/Cas9 in Mouse Liver in Spite of Cas9-Specific Immune Responses [80] | 2016 | Nonalcoholic steatohepatitis, Cancer | Ad5 delivering Cas9 targeting PTEN was administered intravenously. Reduced PTEN levels in the liver correlate with liver conditions such as nonalcohol steatohepatitis and liver cancers. Adult mice treated with the vector developed phenotypic characteristics similar to traditional PTEN knockout strains. |
| Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing [79] | 2014 | Coronary Heart Disease | Ad5 delivering Cas9 targeting PCSK9 was administered intravenously as a strategy to reduce heart disease. PCSK9 was targeted for knockout as mutations can cause severely increased LDL levels. Treated mice showed significant reductions in plasma LDL and PCSK9 levels. |
HIGHLIGHTS.
CRISPR-Cas gene editors provide unprecedented opportunities for gene therapy
In vivo editing of cells is advantageous but requires an efficient delivery vector
Adenoviral vectors possess unique advantages that enable in vivo editing
Adenoviral vectors have achieved successful in vivo editing in mouse disease models
ACKNOWLEDEMENTS
The authors thank Reka Lorincz for her valuable input editing this article.
FUNDING
Support for this review was provided by the National Institutes of Health (R01EB026468-02 and UG3TR002851-01 to David T. Curiel, T32HL007088-45 to Stephen Oh).
Footnotes
DECLARATIONS OF INTEREST:
Declarations of interest: none.
REFERENCES:
- [1].Dunbar CE, High KA, Joung JK, Kohn DB, Ozawa K, Sadelain M, Gene therapy comes of age, Science. 359 (2018). 10.1126/science.aan4672. [DOI] [PubMed] [Google Scholar]
- [2].Koonin E. v., Makarova KS, Origins and evolution of CRISPR-Cas systems, Philosophical Transactions of the Royal Society B: Biological Sciences. 374 (2019). 10.1098/rstb.2018.0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E, A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity, Science. 337 (2012) 816–821. 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Araldi RP, Khalil C, Grignet PH, Teixeira MR, de Melo TC, Módolo DG, Fernandes LGV, Ruiz J, de Souza EB, Medical applications of clustered regularly interspaced short palindromic repeats (CRISPR/Cas) tool: A comprehensive overview, Gene. 745 (2020) 144636. 10.1016/j.gene.2020.144636. [DOI] [PubMed] [Google Scholar]
- [5].Jacinto F. v., Link W, Ferreira BI, CRISPR/Cas9-mediated genome editing: From basic research to translational medicine, Journal of Cellular and Molecular Medicine. 24 (2020) 3766–3778. 10.1111/jcmm.14916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Jiang F, Doudna JA, CRISPR–Cas9 Structures and Mechanisms, Annual Review of Biophysics. 46 (2017) 505–529. 10.1146/annurev-biophys-062215-010822. [DOI] [PubMed] [Google Scholar]
- [7].Sander JD, Joung JK, CRISPR-Cas systems for editing, regulating and targeting genomes, Nature Biotechnology. 32 (2014) 347–350. 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Amoasii L, Hildyard JCW, Li H, Sanchez-Ortiz E, Mireault A, Caballero D, Harron R, Stathopoulou TR, Massey C, Shelton JM, Bassel-Duby R, Piercy RJ, Olson EN, Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy, Science. 362 (2018) 86–91. 10.1126/science.aau1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Moretti A, Fonteyne L, Giesert F, Hoppmann P, Meier AB, Bozoglu T, Baehr A, Schneider CM, Sinnecker D, Klett K, Fröhlich T, Rahman FA, Haufe T, Sun S, Jurisch V, Kessler B, Hinkel R, Dirschinger R, Martens E, Jilek C, Graf A, Krebs S, Santamaria G, Kurome M, Zakhartchenko V, Campbell B, Voelse K, Wolf A, Ziegler T, Reichert S, Lee S, Flenkenthaler F, Dorn T, Jeremias I, Blum H, Dendorfer A, Schnieke A, Krause S, Walter MC, Klymiuk N, Laugwitz KL, Wolf E, Wurst W, Kupatt C, Somatic gene editing ameliorates skeletal and cardiac muscle failure in pig and human models of Duchenne muscular dystrophy, Nature Medicine. 26 (2020) 207–214. 10.1038/s41591-019-0738-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Pickar-Oliver A, Gersbach CA, The next generation of CRISPR–Cas technologies and applications, Nature Reviews Molecular Cell Biology. 20 (2019) 490–507. 10.1038/s41580-019-0131-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Banan M, Recent advances in CRISPR/Cas9-mediated knock-ins in mammalian cells, Journal of Biotechnology. 308 (2020) 1–9. 10.1016/j.jbiotec.2019.11.010. [DOI] [PubMed] [Google Scholar]
- [12].Bischoff N, Wimberger S, Maresca M, Brakebusch C, Improving Precise CRISPR Genome Editing by Small Molecules: Is there a Magic Potion?, Cells. 9 (2020) 1318. 10.3390/cells9051318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].CRISPR Therapeutics, Pipeline | CRISPR, (2020). http://www.crisprtx.com/programs/pipeline (accessed June 28, 2020). [Google Scholar]
- [14].CRISPR Therapeutics, Creating transformative gene-based medicines for serious diseases: Corporate Overview, 2020. https://crisprtx.gcs-web.com/static-files/6a4a2db8-faf2-4fe9-a2b7-d81a05312e9f.
- [15].Editas Medicine, Research and Pipeline, (2017) 1–11. https://www.editasmedicine.com/gene-editing-pipeline/#s0 (accessed June 28, 2020).
- [16].Zuris Z, Margulies C, Viswanathan R, Edelstein J, Pattali R, Gareau K, Scott S, Lele S, Wilson A, Moon J, Dilmac N, Friedland A, Morgan R, Highly Efficient Multi-Gene Knockout and Transgene Knock-in using CRISPR-Cas12a in Induced Pluripotent Stem Cells for the Generation of Engineered Cell Immunotherapies, 2020. https://www.editasmedicine.com/wp-content/uploads/2020/05/20200515_Zuris_ASGCT_Scientific_Talk_FINAL.pdf. [Google Scholar]
- [17].Heath J, de Dreuzy E, Sousa P, Zuris JA, Viswanathan R, Scott S, da Silva J, Ta T, Wang T, An H, Monesmith T, Wilson CJ, Zhang K, Albright CF, Teixeira S, Chang K-H, EDIT-301: AN AUTOLOGOUS CELL THERAPY TO PROMOTE FETAL HEMOGLOBIN EXPRESSION FOR THE POTENTIAL TREATMENT OF SICKLE CELL DISEASE, 2020. https://www.editasmedicine.com/wp-content/uploads/2020/06/EDIT-301-presentation-EHA-2020.pdf. [Google Scholar]
- [18].Dass A, Diner B, Nayak R, Flinkstrom Z, Tallo T, Miller M, Dasilva J, Gotta G, Wang T, Marco E, Giannoukos G, Ramachandran S, de Erkenez A, Jin S, Albright CF, Reyon D, Dual AAV-based “Knock-out-and-replace” of RHO as a Therapeutic Approach to Treat RHO-associated Autosomal Dominant Retinitis Pigmentosa (RHO adRP) Acknowledgements Disclosures: Employees and shareholders of Editas Medicine: A, 2020. https://www.editasmedicine.com/wp-content/uploads/2020/05/Diner_Dass_ASGCT_2020-RhoADRP_Final.pdf. [Google Scholar]
- [19].Editas Medicine, Corporate Presentation, 2020. https://ir.editasmedicine.com/static-files/03350a40-33d7-413c-b2d6-c46f94ab273f.
- [20].Intellia Therapeutics, Pipeline - Intellia Therapeutics, (n.d.). https://www.intelliatx.com/pipeline-2/ (accessed June 28, 2020). [Google Scholar]
- [21].Schultes B, Developing Next-Generation Engineered TCR-T Cells with CRISPR, 2020. https://3o5c4w3neipl16yvhj3nfqam-wpengine.netdna-ssl.com/wp-content/uploads/Keystone_Engineered-Cell-Therapy_AML_02.10.2020-1.pdf. [Google Scholar]
- [22].Intellia Therapeutics, Corporate Overview, 2020. http://ir.intelliatx.com/static-files/a57908a6-aaca-404d-9072-32471cd3d41e.
- [23].Beam Therapeutics, Our Portfolio - Beam Therapeautics, (n.d.). https://beamtx.com/our-portfolio/ (accessed June 28, 2020). [Google Scholar]
- [24].Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, Dohmae N, Ishitani R, Zhang F, Nureki O, Crystal structure of Cas9 in complex with guide RNA and target DNA, Cell. 156 (2014) 935–949. 10.1016/j.cell.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, Ferrin TE, UCSF ChimeraX: Meeting modern challenges in visualization and analysis, Protein Science. 27 (2018) 14–25. 10.1002/pro.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Araldi RP, Khalil C, Grignet PH, Teixeira MR, de Melo TC, Módolo DG, Fernandes LGV, Ruiz J, de Souza EB, Medical applications of clustered regularly interspaced short palindromic repeats (CRISPR/Cas) tool: A comprehensive overview, Gene. 745 (2020) 144636. 10.1016/j.gene.2020.144636. [DOI] [PubMed] [Google Scholar]
- [27].Search of: crispr | Recruiting, Not yet recruiting, Active, not recruiting, Enrolling by invitation Studies - List Results - Clinicaltrials.gov, (n.d.). https://clinicaltrials.gov/ct2/results?term=crispr&Search=Apply&recrs=b&recrs=a&recrs=f&recrs=d&age_v=&gndr=&type=&rslt= (accessed May 29, 2020). [Google Scholar]
- [28].June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC, CAR T cell immunotherapy for human cancer, Science. 359 (2018) 1361–1365. 10.1126/science.aar6711. [DOI] [PubMed] [Google Scholar]
- [29].Morgan RA, Gray D, Lomova A, Kohn DB, Hematopoietic Stem Cell Gene Therapy: Progress and Lessons Learned, Cell Stem Cell. 21 (2017) 574–590. 10.1016/j.stem.2017.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Li C, Lieber A, Adenovirus vectors in hematopoietic stem cell genome editing, FEBS Letters. 593 (2019) 3623–3648. 10.1002/1873-3468.13668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Luther DC, Lee YW, Nagaraj H, Scaletti F, Rotello VM, Delivery approaches for CRISPR/Cas9 therapeutics in vivo: advances and challenges, Expert Opinion on Drug Delivery. 15 (2018) 905–913. 10.1080/17425247.2018.1517746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, Mangan PA, Kulikovskaya I, Gupta M, Chen F, Tian L, Gonzalez VE, Xu J, young Jung I, Joseph Melenhorst J, Plesa G, Shea J, Matlawski T, Cervini A, Gaymon AL, Desjardins S, Lamontagne A, Salas-Mckee J, Fesnak A, Siegel DL, Levine BL, Jadlowsky JK, Young RM, Chew A, Hwang WT, Hexner EO, Carreno BM, Nobles CL, Bushman FD, Parker KR, Qi Y, Satpathy AT, Chang HY, Zhao Y, Lacey SF, June CH, CRISPR-engineered T cells in patients with refractory cancer, Science. 367 (2020). 10.1126/science.aba7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Gong Y, Tian S, Xuan Y, Zhang S, Lipid and polymer mediated CRISPR/Cas9 gene editing, Journal of Materials Chemistry B. 8 (2020) 4369–4386. 10.1039/d0tb00207k. [DOI] [PubMed] [Google Scholar]
- [34].Rui Y, Wilson DR, Green JJ, Non-Viral Delivery To Enable Genome Editing, Trends in Biotechnology. 37 (2019) 281–293. 10.1016/j.tibtech.2018.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wang D, Zhang F, Gao G, CRISPR-Based Therapeutic Genome Editing: Strategies and In Vivo Delivery by AAV Vectors, Cell. 181 (2020) 136–150. 10.1016/j.cell.2020.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Carter BJ, Adeno-associated virus and the development of adeno-associated virus vectors: A historical perspective, Molecular Therapy. 10 (2004) 981–989. 10.1016/j.ymthe.2004.09.011. [DOI] [PubMed] [Google Scholar]
- [37].Naso MF, Tomkowicz B, Perry WL, Strohl WR, Adeno-Associated Virus (AAV) as a Vector for Gene Therapy, BioDrugs. 31 (2017) 317–334. 10.1007/s40259-017-0234-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wang D, Tai PWL, Gao G, Adeno-associated virus vector as a platform for gene therapy delivery, Nature Reviews Drug Discovery. 18 (2019) 358–378. 10.1038/s41573-019-0012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Nakai H, Montini E, Fuess S, Storm TA, Grompe M, Kay MA, AAV serotype 2 vectors preferentially integrate into active genes in mice, Nature Genetics. 34 (2003) 297–302. 10.1038/ng1179. [DOI] [PubMed] [Google Scholar]
- [40].Nault JC, Datta S, Imbeaud S, Franconi A, Mallet M, Couchy G, Letouzé E, Pilati C, Verret B, Blanc JF, Balabaud C, Calderaro J, Laurent A, Letexier M, Bioulac-Sage P, Calvo F, Zucman-Rossi J, Recurrent AAV2-related insertional mutagenesis in human hepatocellular carcinomas, Nature Genetics. 47 (2015) 1187–1193. 10.1038/ng.3389. [DOI] [PubMed] [Google Scholar]
- [41].Donsante A, Miller DG, Li Y, Vogler C, Brunt EM, Russell DW, Sands MS, AAV Vector Integration Sites in Mouse Hepatocellular Carcinoma, (n.d.). 10.1126/science.1142658. [DOI] [PubMed] [Google Scholar]
- [42].Colella P, Ronzitti G, Mingozzi F, Emerging Issues in AAV-Mediated In Vivo Gene Therapy, Molecular Therapy - Methods and Clinical Development. 8 (2018) 87–104. 10.1016/j.omtm.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Dolgin E, Gene therapy successes point to better therapies, Proceedings of the National Academy of Sciences of the United States of America. 116 (2019) 23866–23870. 10.1073/pnas.1918306116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Patel A, Zhao J, Duan D, Lai Y, Design of AAV vectors for delivery of large or multiple transgenes, in: Methods in Molecular Biology, Humana Press Inc., 2019: pp. 19–33. 10.1007/978-1-4939-9139-6_2. [DOI] [PubMed] [Google Scholar]
- [45].Rees HA, Liu DR, Base editing: precision chemistry on the genome and transcriptome of living cells, Nature Reviews Genetics. 19 (2018) 770–788. 10.1038/s41576-018-0059-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Anzalone A. v., Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, Chen PJ, Wilson C, Newby GA, Raguram A, Liu DR, Search-and-replace genome editing without double-strand breaks or donor DNA, Nature. 576 (2019) 149–157. 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Addgene: pCMV-PE2, (n.d.). https://www.addgene.org/132775/ (accessed May 29, 2020). [Google Scholar]
- [48].Lukashev AN, Zamayatnin AA Jr., Viral Vectors for Gene Therapy: Current State and Clinical Perspectives, Biochemistry (Moscow). 81 (2016) 700–708. 10.1134/S0006297916070063. [DOI] [PubMed] [Google Scholar]
- [49].Beatty MS, Curiel DT, Adenovirus Strategies for Tissue-Specific Targeting, in: Advances in Cancer Research, Academic Press Inc., 2012: pp. 39–67. 10.1016/B978-0-12-398342-8.00002-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Gall JGD, Crystal RG, Falck-Pedersen E, Construction and Characterization of Hexon-Chimeric Adenoviruses: Specification of Adenovirus Serotype, 1998. http://jvi.asm.org/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Dai X, Wu L, Sun R, Zhou ZH, Atomic Structures of Minor Proteins VI and VII in Human Adenovirus, Journal of Virology. 91 (2017). 10.1128/jvi.00850-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].van Raaij MJ, Mitrakl A, Lavigne G, Cusack S, A triple β-spiral in the adenovirus fibre shaft reveals a new structural motif for a fibrous protein, Nature. 401 (1999) 935–938. 10.1038/44880. [DOI] [PubMed] [Google Scholar]
- [53].Baker AT, Greenshields-Watson A, Coughlan L, Davies JA, Uusi-Kerttula H, Cole DK, Rizkallah PJ, Parker AL, Diversity within the adenovirus fiber knob hypervariable loops influences primary receptor interactions, Nature Communications. 10 (2019). 10.1038/s41467-019-08599-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B, A simplified system for generating recombinant adenoviruses, in: Proceedings of the National Academy of Sciences of the United States of America, National Academy of Sciences, 1998: pp. 2509–2514. 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Brunetti-Pierri N, Ng P, Helper-Dependent Adenoviral Vectors, in: Adenoviral Vectors for Gene Therapy: Second Edition, Elsevier Inc., 2016: pp. 423–450. 10.1016/B978-0-12-800276-6.00017-6. [DOI] [Google Scholar]
- [56].Schiwon M, Ehrke-Schulz E, Oswald A, Bergmann T, Michler T, Protzer U, Ehrhardt A, One-Vector System for Multiplexed CRISPR/Cas9 against Hepatitis B Virus cccDNA Utilizing High-Capacity Adenoviral Vectors, Molecular Therapy - Nucleic Acids. 12 (2018) 242–253. 10.1016/j.omtn.2018.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Li C, Mishra AS, Gil S, Wang M, Georgakopoulou A, Papayannopoulou T, Hawkins RD, Lieber A, Targeted Integration and High-Level Transgene Expression in AAVS1 Transgenic Mice after In Vivo HSC Transduction with HDAd5/35++ Vectors, Molecular Therapy. 27 (2019) 2195–2212. 10.1016/j.ymthe.2019.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Wang H, Liu Z, Li C, Gil S, Papayannopoulou T, Doering CB, Lieber A, High-level protein production in erythroid cells derived from in vivo transduced hematopoietic stem cells, Blood Advances. 3 (2019) 2883–2894. 10.1182/bloodadvances.2019000706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Palmer DJ, Turner DL, Ng P, A Single “All-in-One” Helper-Dependent Adenovirus to Deliver Donor DNA and CRISPR/Cas9 for Efficient Homology-Directed Repair, Molecular Therapy - Methods and Clinical Development. 17 (2020) 441–447. 10.1016/j.omtm.2020.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Waddington SN, McVey JH, Bhella D, Parker AL, Barker K, Atoda H, Pink R, Buckley SMK, Greig JA, Denby L, Custers J, Morita T, Francischetti IMB, Monteiro RQ, Barouch DH, van Rooijen N, Napoli C, Havenga MJE, Nicklin SA, Baker AH, Adenovirus Serotype 5 Hexon Mediates Liver Gene Transfer, Cell. 132 (2008) 397–409. 10.1016/j.cell.2008.01.016. [DOI] [PubMed] [Google Scholar]
- [61].Kaliberov SA, Kaliberova LN, Hong Lu Z, Preuss MA, Barnes JA, Stockard CR, Grizzle WE, Arbeit JM, Curiel DT, Retargeting of gene expression using endothelium specific hexon modified adenoviral vector, Virology. 447 (2013) 312–325. 10.1016/j.virol.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Richter M, Saydaminova K, Yumul R, Krishnan R, Liu J, Nagy EE, Singh M, Izsvák Z, Cattaneo R, Uckert W, Palmer D, Ng P, Haworth KG, Kiem HP, Ehrhardt A, Papayannopoulou T, Lieber A, In vivo transduction of primitive mobilized hematopoietic stem cells after intravenous injection of integrating adenovirus vectors, Blood. 128 (2016) 2206–2217. 10.1182/blood-2016-04-711580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Vogels R, Zuijdgeest D, van Rijnsoever R, Hartkoorn E, Damen I, de Béthune M-P, Kostense S, Penders G, Helmus N, Koudstaal W, Cecchini M, Wetterwald A, Sprangers M, Lemckert A, Ophorst O, Koel B, van Meerendonk M, Quax P, Panitti L, Grimbergen J, Bout A, Goudsmit J, Havenga M, Replication-Deficient Human Adenovirus Type 35 Vectors for Gene Transfer and Vaccination: Efficient Human Cell Infection and Bypass of Preexisting Adenovirus Immunity, Journal of Virology. 77 (2003) 8263–8271. 10.1128/jvi.77.15.8263-8271.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Alba R, Bradshaw AC, Parker AL, Bhella D, Waddington SN, Nicklin SA, van Rooijen N, Custers J, Goudsmit J, Barouch DH, McVey JH, Baker AH, Identification of coagulation factor (F)X binding sites on the adenovirus serotype 5 hexon: Effect of mutagenesis on FX interactions and gene transfer, Blood. 114 (2009) 965–971. 10.1182/blood-2009-03-208835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Lu ZH, Dmitriev IP, Brough DE, Kashentseva EA, Li J, Curiel DT, A New Gorilla Adenoviral Vector with Natural Lung Tropism Avoids Liver Toxicity and Is Amenable to Capsid Engineering and Vector Retargeting, Journal of Virology. 94 (2020). 10.1128/jvi.00265-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Luisoni S, Greber UF, Biology of Adenovirus Cell Entry: Receptors, Pathways, Mechanisms, in: Adenoviral Vectors for Gene Therapy: Second Edition, Elsevier Inc., 2016: pp. 27–58. 10.1016/B978-0-12-800276-6.00002-4. [DOI] [Google Scholar]
- [67].Reynolds PN, Dmitriev I, Curiel DT, Insertion of an RGD motif into the HI loop of adenovirus fiber protein alters the distribution of transgene expression of the systemically administered vector, Gene Therapy. 6 (1999) 1336–1339. 10.1038/sj.gt.3300941. [DOI] [PubMed] [Google Scholar]
- [68].Shayakhmetov DM, Papayannopoulou T, Stamatoyannopoulos G, Lieber A, Efficient Gene Transfer into Human CD34+ Cells by a Retargeted Adenovirus Vector, Journal of Virology. 74 (2000) 2567–2583. 10.1128/jvi.74.6.2567-2583.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Pereboev A. v., Nagle JM, Shakhmatov MA, Triozzi PL, Matthews QL, Kawakami Y, Curiel DT, Blackwell JL, Enhanced gene transfer to mouse dendritic cells using adenoviral vectors coated with a novel adapter molecule, Molecular Therapy. 9 (2004) 712–720. 10.1016/j.ymthe.2004.02.006. [DOI] [PubMed] [Google Scholar]
- [70].Dmitriev I, Kashentseva E, Rogers BE, Krasnykh V, Curiel DT, Ectodomain of Coxsackievirus and Adenovirus Receptor Genetically Fused to Epidermal Growth Factor Mediates Adenovirus Targeting to Epidermal Growth Factor Receptor-Positive Cells, Journal of Virology. 74 (2000) 6875–6884. 10.1128/jvi.74.15.6875-6884.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Bhatia S, O’Bryan SM, Rivera AA, Curiel DT, Mathis JM, CXCL12 retargeting of an adenovirus vector to cancer cells using a bispecific adapter, Oncolytic Virotherapy. Volume 5 (2016) 99–113. 10.2147/ov.s112107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Baker AT, Aguirre-Hernández C, Halldén G, Parker AL, Designer oncolytic adenovirus: Coming of age, Cancers. 10 (2018). 10.3390/cancers10060201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Magnusson MK, Kraaij R, Leadley RM, de Ridder CMA, van Weerden WM, van Schie KAJ, van der Kroeg M, Hoeben RC, Maitland NJ, Lindholm L, A transductionally retargeted adenoviral vector for virotherapy of her2/neu-expressing prostate cancer, Human Gene Therapy. 23 (2012) 70–82. 10.1089/hum.2011.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Piao Y, Jiang H, Alemany R, Krasnykh V, Marini FC, Xu J, Alonso MM, Conrad CA, Aldape KD, Gomez-Manzano C, Fueyo J, Oncolytic adenovirus retargeted to Delta-EGFR induces selective antiglioma activity, Cancer Gene Therapy. 16 (2009) 256–265. 10.1038/cgt.2008.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Sharma PK, Dmitriev IP, Kashentseva EA, Raes G, Li L, Kim SW, Lu ZH, Arbeit JM, Fleming TP, Kaliberov SA, Goedegebuure SP, Curiel DT, Gillanders WE, Development of an adenovirus vector vaccine platform for targeting dendritic cells, Cancer Gene Therapy. 25 (2018) 27–38. 10.1038/s41417-017-0002-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Wickham TJ, Tzeng E, Shears LL, Roelvink PW, Li Y, Lee GM, Brough DE, Lizonova A, Kovesdi I, Increased in vitro and in vivo gene transfer by adenovirus vectors containing chimeric fiber proteins., Journal of Virology. 71 (1997) 8221–9. http://www.ncbi.nlm.nih.gov/pubmed/9343173 (accessed May 30, 2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Belousova N, Krendelchtchikova V, Curiel DT, Krasnykh V, Modulation of Adenovirus Vector Tropism via Incorporation of Polypeptide Ligands into the Fiber Protein, Journal of Virology. 76 (2002) 8621–8631. 10.1128/jvi.76.17.8621-8631.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Johnson & Johnson Announces a Lead Vaccine Candidate for COVID-19; Landmark New Partnership with U.S. Department of Health & Human Services; and Commitment to Supply One Billion Vaccines Worldwide for Emergency Pandemic Use | Johnson & Johnson, (n.d.). https://www.jnj.com/johnson-johnson-announces-a-lead-vaccine-candidate-for-covid-19-landmark-new-partnership-with-u-s-department-of-health-human-services-and-commitment-to-supply-one-billion-vaccines-worldwide-for-emergency-pandemic-use (accessed May 30, 2020). [Google Scholar]
- [79].Ding Q, Strong A, Patel KM, Ng SL, Gosis BS, Regan SN, Cowan CA, Rader DJ, Musunuru K, Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing, Circulation Research. 115 (2014) 488–492. 10.1161/CIRCRESAHA.115.304351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Wang D, Mou H, Li S, Li Y, Hough S, Tran K, Li J, Yin H, Anderson DG, Sontheimer EJ, Weng Z, Gao G, Xue W, Adenovirus-Mediated Somatic Genome Editing of Pten by CRISPR/Cas9 in Mouse Liver in Spite of Cas9-Specific Immune Responses, Human Gene Therapy. 26 (2015) 432–442. 10.1089/hum.2015.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Li C, Psatha N, Sova P, Gil S, Wang H, Kim J, Kulkarni C, Valensisi C, David Hawkins R, Stamatoyannopoulos G, Lieber A, Reactivation of g-globin in adult b-YAC mice after ex vivo and in vivo hematopoietic stem cell genome editing, Blood. 131 (2018) 2915–2928. 10.1182/blood-2018-03-838540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Bjursell M, Porritt MJ, Ericson E, Taheri-Ghahfarokhi A, Clausen M, Magnusson L, Admyre T, Nitsch R, Mayr L, Aasehaug L, Seeliger F, Maresca M, Bohlooly-Y M, Wiseman J, Therapeutic Genome Editing With CRISPR/Cas9 in a Humanized Mouse Model Ameliorates α1-antitrypsin Deficiency Phenotype, EBioMedicine. 29 (2018) 104–111. 10.1016/j.ebiom.2018.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Xu L, Park KH, Zhao L, Xu J, el Refaey M, Gao Y, Zhu H, Ma J, Han R, CRISPR-mediated genome editing restores dystrophin expression and function in mdx mice, Molecular Therapy. 24 (2016) 564–569. 10.1038/mt.2015.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Wu Y, Zeng J, Roscoe BP, Liu P, Yao Q, Lazzarotto CR, Clement K, Cole MA, Luk K, Baricordi C, Shen AH, Ren C, Esrick EB, Manis JP, Dorfman DM, Williams DA, Biffi A, Brugnara C, Biasco L, Brendel C, Pinello L, Tsai SQ, Wolfe SA, Bauer DE, Highly efficient therapeutic gene editing of human hematopoietic stem cells, Nature Medicine. 25 (2019) 776–783. 10.1038/s41591-019-0401-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Stephens CJ, Lauron EJ, Kashentseva E, Lu ZH, Yokoyama WM, Curiel DT, Long-term correction of hemophilia B using adenoviral delivery of CRISPR/Cas9, Journal of Controlled Release. 298 (2019) 128–141. 10.1016/j.jconrel.2019.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Guan Y, Ma Y, Li Q, Sun Z, Ma L, Wu L, Wang L, Zeng L, Shao Y, Chen Y, Ma N, Lu W, Hu K, Han H, Yu Y, Huang Y, Liu M, Li D, CRISPR /Cas9‐mediated somatic correction of a novel coagulator factor IX gene mutation ameliorates hemophilia in mouse , EMBO Molecular Medicine. 8 (2016) 477–488. 10.15252/emmm.201506039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Stephens CJ, Kashentseva E, Everett W, Kaliberova L, Curiel DT, Targeted in vivo knock-in of human alpha-1-antitrypsin cDNA using adenoviral delivery of CRISPR/Cas9, Gene Therapy. 25 (2018) 139–156. 10.1038/s41434-018-0003-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Koo T, Yoon A-R, Cho H-Y, Bae S, Yun C-O, Kim J-S, Selective disruption of an oncogenic mutant allele by CRISPR/Cas9 induces efficient tumor regressionitle, Nucleic Acids Research. 45 (2017) 7897–7908. 10.1093/nar/gkx490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Takayama K, Mizuguchi H, Generation of Optogenetically Modified Adenovirus Vector for Spatiotemporally Controllable Gene Therapy, ACS Chemical Biology. 13 (2018) 449–454. 10.1021/acschembio.7b01058. [DOI] [PubMed] [Google Scholar]
- [90].Yoshida M, Yokota E, Sakuma T, Yamatsuji T, Takigawa N, Ushijima T, Yamamoto T, Fukazawa T, Naomoto Y, Development of an integrated CRISPRi targeting ▵Np63 for treatment of squamous cell carcinoma, Oncotarget. 9 (2018) 29220–29232. 10.18632/oncotarget.25678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Gao Q, Ouyang W, Kang B, Han X, Xiong Y, Ding R, Li Y, Wang F, Huang L, Chen L, Wang D, Dong X, Zhang Z, Li Y, Ze B, Hou Y, Yang H, Ma Y, Gu Y, Chao CC, Selective targeting of the oncogenic KRAS G12S mutant allele by CRISPR/Cas9 induces efficient tumor regression, Theranostics. 10 (2020) 5137–5153. 10.7150/thno.42325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Search of: adenovirus OR adv OR adenoviral OR ad | Recruiting, Not yet recruiting, Active, not recruiting, Enrolling by invitation Studies | Cancer NOT communicable - List Results - Clinicaltrials.gov, (n.d.). https://clinicaltrials.gov/ct2/results?cond=Cancer+NOT+communicable&term=adenovirus+OR+adv+OR+adenoviral+OR+ad&type=&rslt=&recrs=b&recrs=a&recrs=f&recrs=d&age_v=&gndr=&intr=&titles=&outc=&spons=&lead=&id=&cntry=&state=&city=&dist=&locn=&strd_s=&strd_e=&prcd_s=&prcd_e=&sfpd_s=&sfpd_e=&lupd_s=&lupd_e= (accessed May 30, 2020).
- [93].Chen Y, Liu X, Zhang Y, Wang H, Ying H, Liu M, Li D, Lui KO, Ding Q, A Self-restricted CRISPR System to Reduce Off-target Effects, (2016). 10.1038/mt.2016.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Li C, Psatha N, Gil S, Wang H, Papayannopoulou T, Lieber A, HDAd5/35++ Adenovirus Vector Expressing Anti-CRISPR Peptides Decreases CRISPR/Cas9 Toxicity in Human Hematopoietic Stem Cells, Molecular Therapy - Methods and Clinical Development. 9 (2018) 390–401. 10.1016/j.omtm.2018.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Bode AM, Dong Z, Precision oncology- the future of personalized cancer medicine?, Npj Precision Oncology. 1 (2017) 1–2. 10.1038/s41698-017-0010-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Marusyk A, Polyak K, Tumor heterogeneity: Causes and consequences, Biochimica et Biophysica Acta - Reviews on Cancer. 1805 (2010) 105–117. 10.1016/j.bbcan.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Nakayama M, Both GW, Banizs B, Tsuruta Y, Yamamoto S, Kawakami Y, Douglas JT, Tani K, Curiel DT, Glasgow JN, An adenovirus serotype 5 vector with fibers derived from ovine atadenovirus demonstrates CAR-independent tropism and unique biodistribution in mice, Virology. 350 (2006) 103–115. 10.1016/j.virol.2006.01.037. [DOI] [PubMed] [Google Scholar]
- [98].Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F, Rationally engineered Cas9 nucleases with improved specificity, Science. 351 (2016) 84–88. 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Papapetrou EP, Lee G, Malani N, Setty M, Riviere I, Tirunagari LMS, Kadota K, Roth SL, Giardina P, Viale A, Leslie C, Bushman FD, Studer L, Sadelain M, Genomic safe harbors permit high β-globin transgene expression in thalassemia induced pluripotent stem cells, Nature Biotechnology. 29 (2011) 73–81. 10.1038/nbt.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Chu VT, Weber T, Graf R, Sommermann T, Petsch K, Sack U, Volchkov P, Rajewsky K, Kühn R, Efficient generation of Rosa26 knock-in mice using CRISPR/Cas9 in C57BL/6 zygotes, BMC Biotechnology. 16 (2016) 4. 10.1186/s12896-016-0234-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Tsai SQ, Joung JK, Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases, Nature Reviews Genetics. 17 (2016) 300–312. 10.1038/nrg.2016.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Kosicki M, Tomberg K, Bradley A, Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements, Nature Biotechnology. 36 (2018) 765. 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Han HA, Pang JKS, Soh BS, Mitigating off-target effects in CRISPR/Cas9-mediated in vivo gene editing, Journal of Molecular Medicine. 98 (2020) 615–632. 10.1007/s00109-020-01893-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Palmer DJ, Turner DL, Ng P, Production of CRISPR/Cas9-Mediated Self-Cleaving Helper-Dependent Adenoviruses, Molecular Therapy - Methods and Clinical Development. 13 (2019) 432–439. 10.1016/j.omtm.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Jacobs F, Gordts SC, Muthuramu I, de Geest B, The liver as a target organ for gene therapy: State of the art, challenges, and future perspectives, Pharmaceuticals. 5 (2012) 1372–1392. 10.3390/ph5121372. [DOI] [PMC free article] [PubMed] [Google Scholar]