

Abstract
We describe mechanistically-distinct enzymes, i.e., a kinase, a guanosine triphosphatase and a ubiquitin protein hydrolase, which function in disparate biochemical pathways, that can also act in concert to mediate a series of redox reactions. Each enzyme manifests a second, noncanonical function—transnitrosylation—that triggers a pathological biochemical cascade in mouse models and in humans with Alzheimer’s disease (AD). The resulting series of transnitrosylation reactions contributes to synapse loss, the major pathological correlate to cognitive decline in AD. We conclude that enzymes with distinct primary reaction mechanisms can form a completely separate network for aberrant transnitrosylation. This network operates in the post-reproductive period, so natural selection against such abnormal activity may be decreased.
One Sentence Summary:
Transnitrosylation reactions among seemingly unrelated signaling proteins cause synaptic damage in neurodegenerative disease.
Excessive nitric oxide (NO) can engender nitrosative stress in the nervous system, contributing to neurodegenerative damage (1, 2). In Alzheimer’s disease (AD), oligomerized amyloid-β peptide (Aβ), neuronal hyperexcitability, and neuroinflammation associated with aging can each trigger NO generation and resultant S-nitrosylation (donation of an NO group most likely as a nitrosonium cation intermediate, NO+) to specific cysteine thiol (or more properly thiolate anion, -S−) (3–14). One such cysteine is located on dynamin-related protein 1 (Drp1), a guanosine triphosphatase (GTPase) that mediates mitochondrial fission (15). This posttranslational redox reaction, forming SNO-Drp1, hyperactivates the enzyme, leading to mitochondrial fragmentation and bioenergetic compromise, with consequent synapse loss, in part because of the great demand for energy during neurotransmission (15, 16). Because reduction in synapse number is closely correlated with cognitive decline in AD (17, 18), this nitrosylation reaction may contribute to disease pathogenesis (4). S-Nitrosylated cyclin-dependent kinase 5 (Cdk5) (19) acts to pass NO to Drp1 to form SNO-Drp1 via a reaction termed transnitrosylation (20). However, additional members of this pathological cascade are yet to be determined. Characterizing this aberrant series of reactions may further reveal its contribution to the pathogenesis of AD and possible therapeutic implications of the pathway. We used both in vitro and in vivo models of AD as well as human AD postmortem brains to show that enzymes with disparate catalytic activities from different biochemical pathways can form a transnitrosylation cascade that contributes to synapse loss in AD. We demonstrate that the deubiquitinylating enzyme, Uch-L1 (Ubiquitin carboxyl-terminal hydrolase isozyme-L1), contributes to a transnitrosylation cascade leading to synaptic damage in AD that involves transfer of a NO group from Uch-L1 to Cdk5 and then to Drp1. Although other potential members of the cascade remain to be determined, our results show pathways mediating a series of aberrant transnitrosylation reactions may be important in the etiology of neurodegenerative disorders.
S-Nitrosylation of Uch-L1 in cell lines and AD transgenic mice by biotin switch and mass spectrometry
We examined upstream events to S-nitrosylation of Cdk5 (forming SNO-Cdk5), which in turn leads to SNO-Drp1 formation and consequent Aβ-related synaptic spine loss in AD (15, 20). The ubiquitin hydrolase Uch-L1 can potentiate cyclin-dependent kinase activity by an unknown reaction mechanism not involving its ubiquitin hydrolase activity (21), and we found that this mechanism involves transnitrosylation to Cdk5.
Aberrant S-nitrosylation of a number of proteins that affect their canonical enzymatic activity has been demonstrated in AD and other neurologic diseases (4), including members of the ubiquitin pathway such as the E3 ligase Parkin and the ubiquitin hydrolase Uch-L1 (22–24). However, we discovered that aberrant and seemingly unrelated S-nitrosylated enzymes are linked by multiple transnitrosylation steps between them that result in a wholly disparate cascade of biochemical pathological activity. We exposed human neural SH-SY5Y cells to a physiological NO donor, S-nitrosocysteine (SNOC). We then made cell lysates and analyzed them for S-nitrosylated Uch-L1. Using the biotin-switch method (25), we found evidence for the formation of SNO-Uch-L1 under these conditions (Fig. 1A). Additionally, endogenous NO, generated by activation of stably-expressed neuronal NO synthase (nNOS) in human embryonic kidney (HEK) cells (designated HEK-nNOS cells), also resulted in formation of SNO-Uch-L1 (Fig. 1B); nNOS activation by the calcium ionophore A23187 led to a ~2-fold increase in the relative abundance of SNO-Uch-L1 in replicate experiments. The absence of ascorbate in the reaction mixture abrogated detection of the increase in SNO-Uch-L1 via the biotin-switch assay because, under these conditions, NO was not removed and replaced by biotin (Fig. 1, A and B). Of potential pathological relevance, we found increased SNO-Uch-L1 compared to wild-type (WT) littermates in cerebrocortices of AD transgenic mouse models overexpressing human amyloid precursor protein (hAPP-J20 and Tg2576), which produce Aβ oligomers (Fig. 1C and fig. S1). The very high abundance of Uch-L1 in the brain might favor its S-nitrosylation, not necessarily requiring a catalyst for the reaction.
Fig. 1. S-Nitrosylation of Uch-L1 at Cys152.
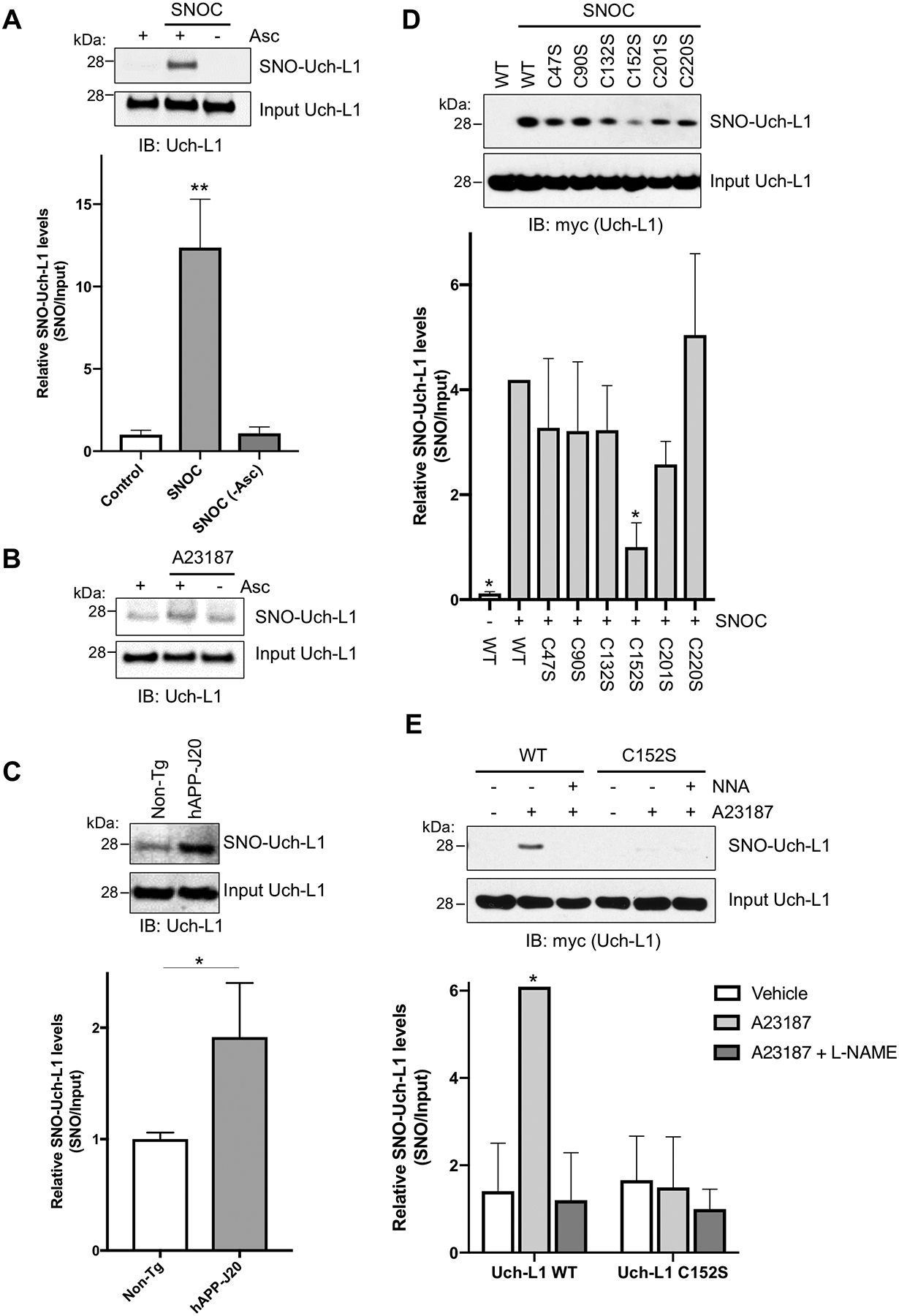
(A) SNO-Uch-L1 formation by biotin-switch assay in SH-SY5Y cells exposed to 100 μM SNOC. As a negative control, ascorbate (Asc) was omitted. Values are expressed as mean + SEM (n = 3 biological replicates per group). **P < 0.01 by ANOVA. (B) SNO-Uch-L1 formation by biotin-switch assay in HEK-nNOS cells exposed to calcium ionophore A23187. (C) Presence of SNO-Uch-L1 in the cerebrocortex of 3–6 mos-old hAPP-J20 AD transgenic mice by biotin-switch assay. Values are mean + SEM (n = 6 per group). *P < 0.05 by Student’s t test. (D) S-Nitrosylation of Uch-L1 at Cys152. HEK cells expressing myc-tagged WT Uch-L1 or cysteine mutants of Uch-L1 were exposed to SNOC. Lack of a band by biotin-switch assay for Uch-L1(C152S) indicates that it is the predominant site of S-nitrosylation. Values are expressed as mean + SEM (n = 3–4 per group). *P < 0.05 compared to ‘WT Uch-L1 + SNOC’ by ANOVA. (E) S-Nitrosylation of Uch-L1 at Cys152 by biotin-switch assay in HEK-nNOS cells. HEK-nNOS cells expressing myc-tagged Uch-L1 WT or C152S mutant were exposed to A23187 to increase intracellular Ca2+ and thus activate nNOS to generate endogenous NO. The NOS inhibitor NG-nitro-l-arginine (NNA, 1 mM) was used to inhibit NO generation as a control. Values are expressed as mean + SEM (n = 3 per group). *P < 0.05 by ANOVA. Uncropped immunoblots are included in fig. S7.
We used site-directed mutagenesis in HEK-293 cells to investigate which cysteine residue was S-nitrosylated in Uch-L1. Mutation of Cys152 decreased formation of SNO-Uch-L1, indicating that it is a major site of S-nitrosylation after exposure to exogenous SNOC (Fig. 1D) or endogenous NO in HEK-nNOS cells (Fig. 1E). Additionally, the NOS inhibitor NG-nitro-l-arginine (NNA) prevented S-nitrosylation of WT Uch-L1, confirming that NO was indeed the reactant (Fig. 1E). We used mass spectrometry-based S-nitrosoproteomics analysis to confirm the predominant site of S-nitrosylation as Cys152 (Fig. 2, A and B and table S1). Moreover, we identified an acidic (Glu)-basic (Arg) region as a candidate motif for S-nitrosylation surrounding the critical Cys residue in the atomic resolution model of Uch-L1 (Fig. 2C) (12, 26, 27).
Fig. 2. Mass spectrometry (MS) identification of S-nitrosylated Cys residue in Uch-L1 and atomic resolution model.
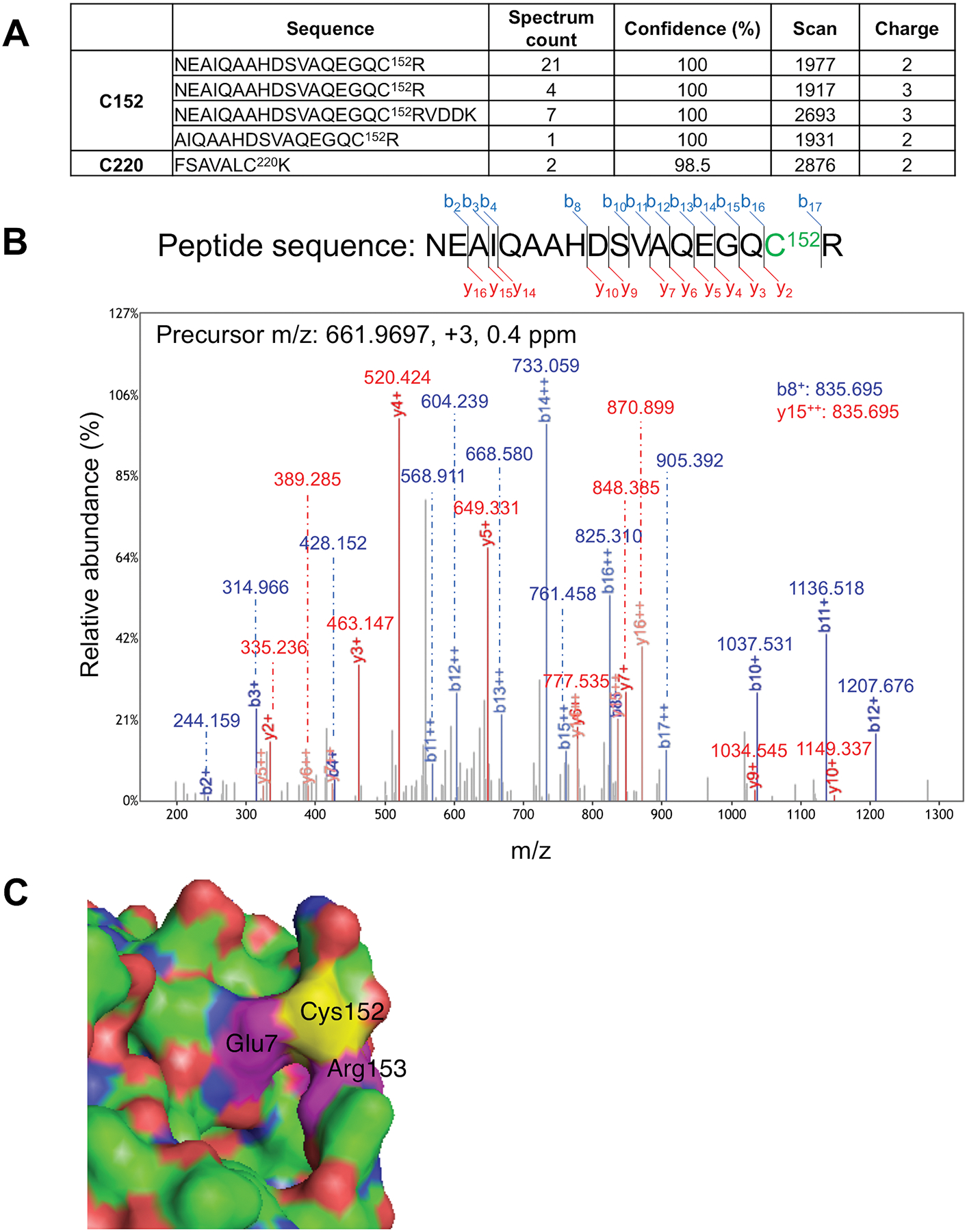
(A) Table summarizing S-nitrosylated peptides for Uch-L1 by MS analysis. Four peptides were identified containing Cys152, and one peptide with Cys220. Spectral counting clearly demonstrated that Cys152 is the predominant S-nitrosylation site on Uch-L1 (total spectral counts 33 for Cys152 vs. 2 for Cys220). (B) Tandem mass spectrum of a Uchl-L1 peptide identifying S-nitrosylation at Cys152. The measured mass, charge state, and measurement accuracy of the precursor ion are displayed. The b (blue) and y (red) fragment ions are annotated with the measured mass. (C) Crystal structure of Uch-L1 (PDB ID: 2ETL) near Cys152 (yellow). Magenta: Glu7 and Arg153. Molecular visualization and graphics handling were performed using PyMol.
Effect of S-nitrosylation on Uch-L1 deubiquitinylation activity in cell-based models and AD transgenic mice
To determine the effect of formation of SNO-Uch-L1, we monitored Uch-L1 binding to ubiquitin and Uch-L1 deubiquitinylation activity. Exposure to SNOC decreased Uch-L1 associated with ubiquitin and decreased deubiquitinylation activity in both recombinant and cell-based assays (fig. S2, A to C). As a control, non-nitrosylatable mutant Uch-L1(C152S) was not affected by SNOC. Additionally, in the hAPP-J20 transgenic mouse model of AD, we observed a decrease in the active form of Uch-L1, i.e., Uch-L1 bound to ubiquitin on immunoblots (fig. S2D). Conversely, treatment with ascorbate (to reductively remove NO from SNO-protein) resulted in an increase in the active form of Uch-L1, consistent with increased ubiquitinylation activity due to denitrosylation of Uch-L1.
Decreased Uch-L1 activity is observed in human brains with AD as well as in AD mouse models (28, 29). Hence, SNO-Uch-L1 formation in AD brain, with consequent inhibition of Uch-L1 activity, might contribute to this effect. Overexpression of Uch-L1 decreases Aβ production, increases activity of protein kinase A (PKA) and cAMP response element-binding protein (CREB), and improves synaptic function and memory performance in AD transgenic mice (29–31). Therefore, Uch-L1 activity appears to be important in the pathogenesis of the disease.
Oligomeric Aβ triggers SNO-Uch-L1 formation and synapse loss in AD models in vitro and in vivo
We reasoned that if the therapeutic action of Uch-L1 overexpression was mediated by decreasing endogenous Aβ production, then the pathological effects on synapses of adding exogenous Aβ oligomers might not be greatly affected by Uch-L1 overexpression. On the other hand, if the beneficial effect of Uch-L1 occurred, at least in part, through another function of the enzyme, we should observe this therapeutic action in the presence of exogenous Aβ oligomers. Indeed, S-nitrosylation of Uch-L1 occurred in a neuronal cell-based model upon exposure to Aβ oligomers -- incubation of primary rat cerebrocortical cultures in 500 nM Aβ1–42 oligomers resulted in formation of SNO-Uch-L1 as determined by the biotin-switch assay (Fig. 3A and fig. S3A).
Fig. 3. Aβ-induced S-nitrosylation of Uch-L1 contributes to AD-related synapse loss.
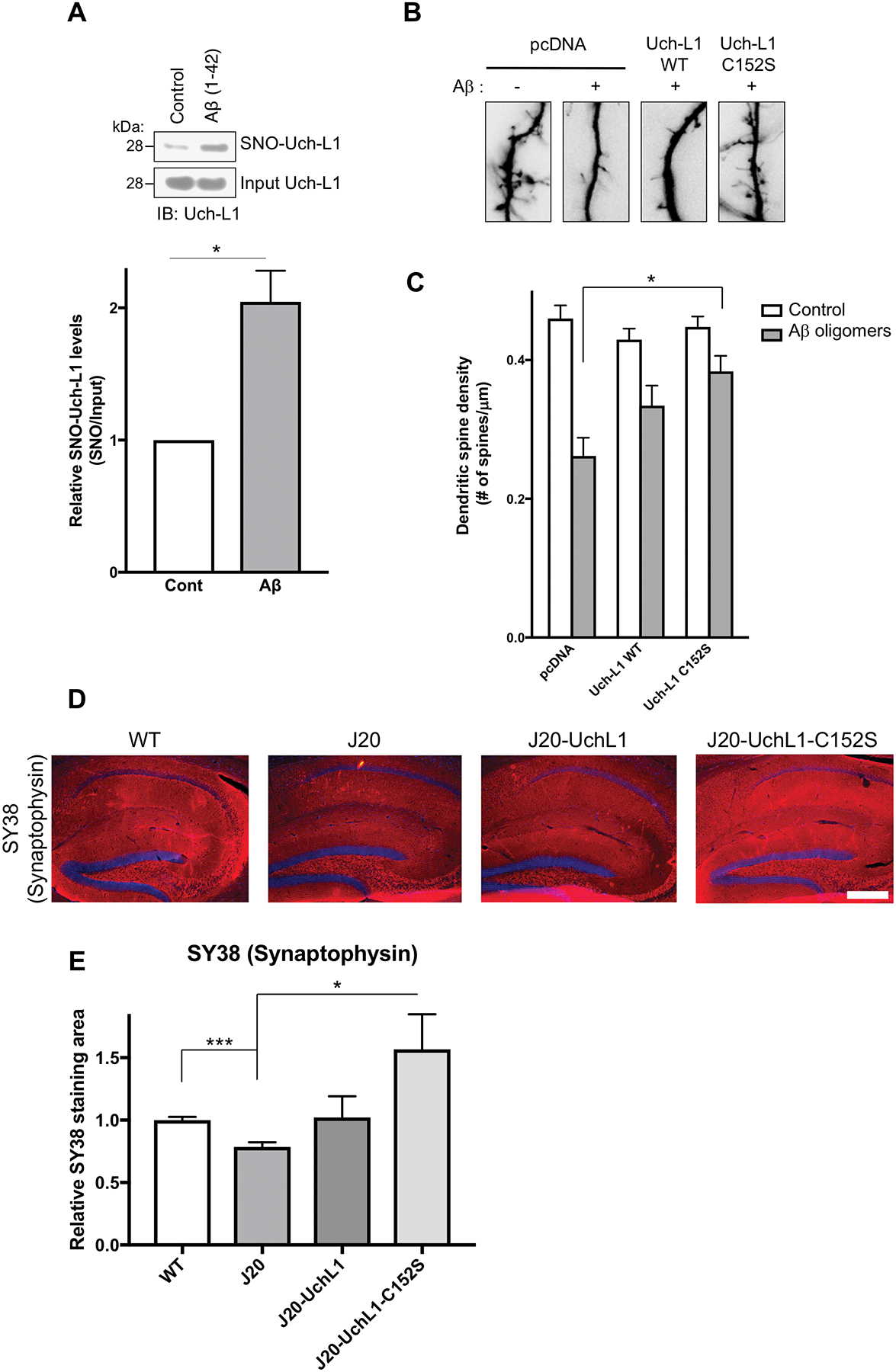
(A) SNO-Uch-L1 formation in primary rat cerebrocortical neurons in culture after 4.5 h exposure to 500 nM oligomerized Aβ1–42 assayed by the biotin-switch method (upper panel). Quantification of biotin-switch blots (lower panel). Values are mean + SEM (n = 3 per group). *P < 0.05 by Student’s t test. (B and C) Exposure of rat cerebrocortical neurons in vitro to Aβ oligomers (500 nM) for 24 h resulted in decreased dendritic spines, visualized by GFP transfection (B). Transfection with non-nitrosylatable mutant Uch-L1(C152S) abrogated Aβ-induced dendritic spine damage in a statistically significant manner, while WT Uch-L1 manifested a lesser, nonsignificant effect. Values are mean + SEM (spines scored for n = 25 neurons). *P < 0.05 by ANOVA with Bonferroni correction. (D and E) Lentiviral expression of Uch-L1(C152S) in vivo prevents loss of presynaptic marker in hAPP-J20 mice. Representative immunostaining of hippocampal sections with synaptophysin antibody (SY38; pseudo-colored red). Scale bar, 200 μm (D). Quantification of hippocampal immunohistochemistry from WT and hAPP-J20 mice injected with WT Uch-L1, mutant Uch-L1(C152S), or empty lentiviral vectors (E). Immunohistochemistry was performed with synaptophysin antibody (SY38). Values are mean + SEM (n = 18 mice injected). *P < 0.05, ***P < 0.001 by Student’s t test with Bonferroni correction.
Additionally, after exposure of rat cerebrocortical neurons to 500 nM Aβ1–42 oligomers, approximately half of the dendritic spines were lost (Fig. 3, B and C) (15, 20, 32–34); loss of synaptic spines was dependent on NO, as preincubation in NNA abrogated the effect (15, 20). Transfection with non-nitrosylatable mutant Uch-L1, but not with WT Uch-L1, decreased the Aβ-induced loss of dendritic spines in a statistically significant manner, although there was also a trend toward improvement with WT Uch-L1 (Fig. 3C and fig. S3B). This trend with WT Uch-L1 may reflect overexpression of WT protein, which can act as a sink for NO (allowing substantial WT protein to remain un-nitrosylated) and therefore potentially offer some benefit (15). However, non-nitrosylatable mutant protein is more effective and showed statistically significant synaptic protection.
To determine if S-nitrosylated Uch-L1 also contributes in vivo to synapse loss, we used the hAPP-J20 transgenic mouse model of AD, which overexpresses oligomeric Aβ in the brain (35). We generated lentiviral vectors expressing tdTomato fluorescent protein and Uch-L1 (WT or C152S), and injected them into the dentate gyrus of hAPP-J20 AD mice (fig. S4, A and B). Two months later, we evaluated the histological effects on presynaptic integrity. Quantitative unbiased confocal immunohistochemistry of hippocampal sections with an antibody to the presynaptic marker synaptophysin (SY38) (36, 37) revealed that expression of Uch-L1(C152S), but not that of WT Uch-L1, significantly increased SY38 signal compared to that obtained from vehicle-treated hAPP-J20 AD mice (Fig. 3, D and E). Although overexpression of WT protein afforded a trend toward presynaptic protection, probably because it acted as a sink for NO (15), only non-nitrosylatable mutant protein resulted in statistically significant protection of the presynaptic marker. These findings are consistent with the notion that SNO-Uch-L1 contributes to synapse loss in AD models. Because synapse loss is a close neuropathological correlate to cognitive decline in human AD, this finding supports biological relevance to the disease state.
Transnitrosylation from Uch-L1 to Cdk5 to Drp1
Aβ oligomer-induced S-nitrosylation of Drp1 results in mitochondrial fragmentation, bioenergetic compromise, and consequent synapse loss (15, 16, 38). Studies with purified recombinant proteins support direct transfer of an NO group (representing transnitrosylation) to Drp1 from SNO-Cdk5 (20), although the upstream effector linking Aβ-induced NO generation to S-nitrosylation of Cdk5 was unknown. Given our new evidence for S-nitrosylation of Uch-L1 in AD transgenic mouse brain and the fact that Uch-L1 potentiates cyclin-dependent kinase activity by a reaction mechanism not involving its ubiquitin hydrolase activity (21), we tested whether Uch-L1 might contribute to an NO-mediated pathway to synapse loss, leading to a series of transnitrosylation steps among these enzymes from diverse biochemical pathways (NO group transfer from Uch-L1 to Cdk5 to Drp1).
We performed kinetic, cell-based S-nitrosylation assays to monitor possible transfer of an NO group from SNO-Uch-L1 to Cdk5 and then to Drp1. Using the biotin-switch assay, we found evidence for formation of both SNO-Uch-L1 and SNO-Cdk5 in HEK-293 cells within 10 min of exposure to SNOC; with increasing formation of SNO-Drp1 by 25 min, concurrently with a relative decrease in the amount of SNO-Cdk5 (Fig. 4, A to D). The fact that RNAi depletion of Uch-L1, compared to control siRNA (siCTL), partially prevented formation of SNO-Cdk5 under these conditions at 10 min is consistent with the notion that transnitrosylation occurs from upstream SNO-Uch-L1 to Cdk5 (Fig. 4, A to C). Similarly, RNAi depletion of Uch-L1 also partially prevented the subsequent formation of SNO-Drp1 (Fig. 4D). Although RNAi depletion of Uch-L1 decreased SNO-Cdk5 and SNO-Drp1 generation, it did not eliminate it, presumably because SNOC is a non-enzymatic donor of NO, producing super-physiological amounts of protein S-nitrosylation that would be expected to continue undeterred when competing with transnitrosylation among proteins. To clarify further, this experiment indicates that donation of NO from SNOC to SNO-Uch-L1 occurred, but SNOC also S-nitrosylated Cdk5 and Drp1. The fact that knockdown of Uch-L1 decreased to a statistically significant degree the formation of SNO-Cdk5 (at 10 min) and SNO-Drp1 (at 10 min and even moreso at 25 min) under these conditions argues that SNO-Uch-L1 triggered downstream transnitrosylation reactions (20, 39).
Fig. 4. Transnitrosylation from SNO-Uch-L1 to Cdk5 on biotin-switch assays.
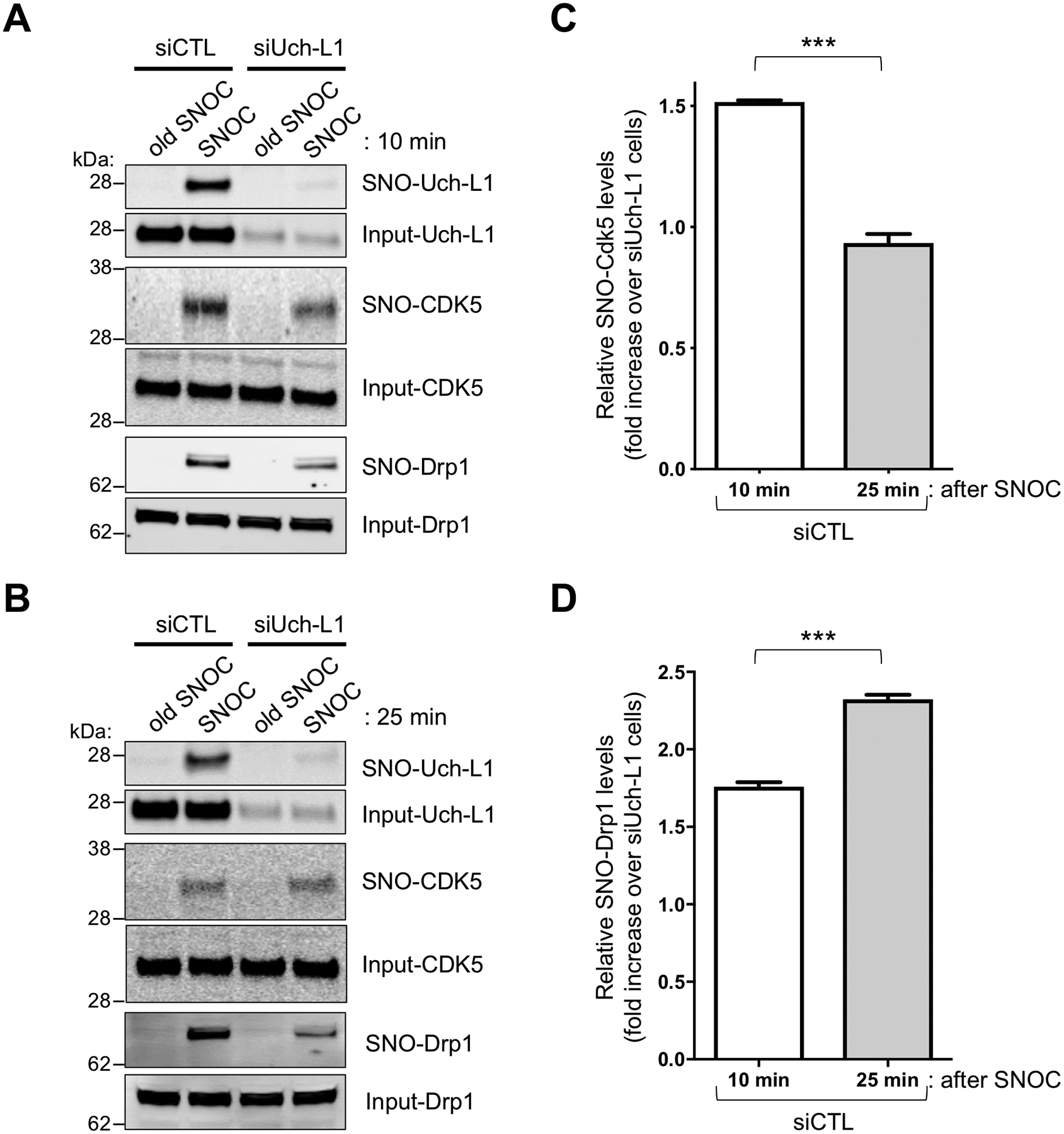
(A) SNO-Uch-L1 formed 10-min after exposure of HEK-293 cells to 100 μM SNOC, but not after exposure to control represented by ‘old’ SNOC from which the NO group had been dissipated, although this solution might still contain nitrite and disulfides. Formation of SNO-Cdk5 was also observed, and this was partially abrogated by siRNA knockdown of Uch-L1 (siUch-L1) compared to control siRNA (siCTL) cells. (B) Knockdown of Uch-L1 largely prevented formation of SNO-Drp1, which first appeared 25 min after exposure to SNOC. (C) Quantification of SNO-Cdk5 on biotin-switch blots. Relative SNO-Cdk5 levels (SNO-protein/Input protein) in siCTL cells were increased 10 min after exposure to SNOC vs. 25 min when compared to siUch-L1 cells exposed to SNOC. Values are mean + SEM (n = 3 per group). ***P < 0.001 by two-tailed Student’s t test. (D) Similar type of quantification of SNO-Drp1 on biotin-switch blots. In this case, relative SNO-Drp1 levels were increased 25 min after exposure to SNOC vs. 10 min. Values are mean + SEM (n = 3 per group). ***P < 0.001 by two-tailed Student’s t test.
By graphing the difference of relative SNO-Cdk5 and SNO-Drp1 concentrations, as calculated from control (siCTL) values compared to those in cells lacking Uch-L1 (siUch-L1), we can subtract the effect of S-nitrosylation due to SNOC and quantify the effect of transnitrosylation between the enzymes (Fig. 4, C and D). The temporal delay in appearance of SNO-Drp1 relative to SNO-Cdk5 is consistent with the notion that SNO-Cdk5, in turn, may transnitrosylate Drp1.
To characterize this putative transnitrosylation pathway from SNO-Cdk5 to Drp1, we used in vitro nitrosylation assays. We transfected SH-SY5Y neural cells with WT Uch-L1 or mutant Uch-L1(C152S), each tagged with V5 (derived from a small epitope found in paramyxovirus proteins), prepared cell lysates, and performed immunoprecipitation with antibody to V5. Immunoprecipitated Uch-L1 protein (WT or C152S mutant) was then exposed to SNOC and incubated for a sufficient period of time to ensure near total dissipation of NO from SNOC (20, 39). The WT or C152S mutant Uch-L1 immunoprecipitates were then added to cell lysates containing hemagglutinin (HA)-tagged Cdk5 (Fig. 5, A to E). We monitored possible transnitrosylation reactions by probing for formation of SNO-V5-Uch-L1, SNO-HA-Cdk5, and endogenous SNO-Drp1 by biotin-switch assay. SNO-Cdk5 and SNO-Drp1 formed in the WT Uch-L1-immunoprecipitated lysates after SNOC exposure but significantly less was found in immunoprecipitates from mutant Uch-L1(C152S) (Fig. 5, A to E). Although immunoprecipitate experiments do not rule out the possibility that additional members of the cascade participate in these transnitrosylation reactions, they support participation of Uch-L1 and Cdk5 in the reaction sequence leading to the generation of SNO-Drp1.
Fig. 5. Triple transnitrosylation from SNO-Uch-L1 to SNO-Cdk5 to SNO-Drp1.
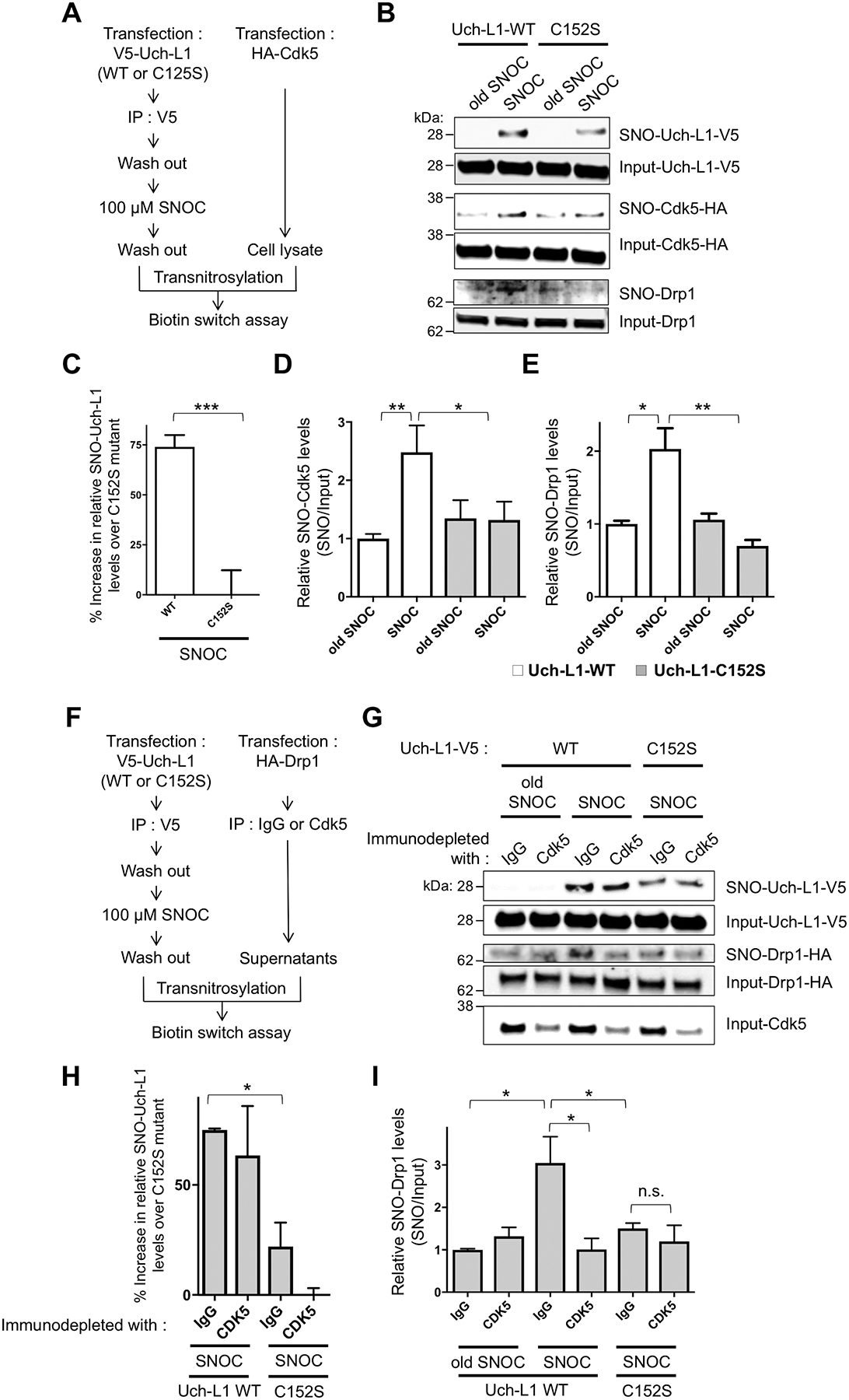
(A) Schematic diagram of in vitro transnitrosylation assay. (B) Transnitrosylation occurs from WT SNO-Uch-L1 to Cdk5 to Drp1 but significantly less with mutant Uch-L1(C152S). WT Uch-L1-V5 or mutant Uch-L1(C152S)-V5 from SH-SY5Y cell lysates was immunoprecipitated with anti-V5 antibody and exposed to 100 μM SNOC. These immunoprecipitates were then added to whole cell lysates expressing HA-Cdk5, which were subjected to the biotin-switch assay to assess transnitrosylation. Compilation of representative blots shown. (C and D) Quantification of Uch-L1 and Cdk5 biotin-switch blots. Values are mean + SEM (n = 4 per group). ***P < 0.001 by two-tailed Student’s t test, **P < 0.01, *P < 0.05 by ANOVA. (E) Quantification of Drp1 biotin-switch blots. Values are mean + SEM (n = 3 per group). *P < 0.05, **P < 0.01 by ANOVA. (F) Schematic diagram of in vitro transnitrosylation assay after immunoprecipitation to decrease the amount of endogenous Cdk5. (G) SNO-Uch-L1 nitrosylation of Drp1 is affected by the level of endogenous Cdk5. WT Uch-L1-V5 or Uch-L1(C152S)-V5 from SH-SY5Y cell lysates was immunoprecipitated with anti-V5 antibody, and the immunoprecipitates exposed to 100 μM SNOC or old SNOC. The immunoprecipitates containing SNOC-exposed Uch-L1 proteins were then incubated with SH-SY5Y cell lysates expressing Drp1-HA after substantial immunodepletion of endogenous Cdk5 with anti-Cdk5 antibody (or IgG control antibody). Samples were subjected to biotin-switch assay to assess transnitrosylation. (H and I) Quantification of Uch-L1and Drp1 biotin-switch blots. Values are mean + SEM (n = 3 per group). **P < 0.01, *P < 0.05 by ANOVA.
The relative degree of transnitrosylation of Drp1 resulting from S-nitrosylated Uch-L1 was dependent on the amount of endogenous Cdk5. When Cdk5 was depleted by immunoprecipitation, the amount of transnitrosylated SNO-Drp1 decreased (Fig. 5, F to I). The degree of immunodepletion of endogenous Cdk5 corresponded with the decrement in SNO-Drp1 formation in the presence of WT Uch-L1 but not in the presence of C152S mutant Uch-L1 (Fig. 5G). This finding confirms that SNO-Cdk5, rather than SNO-Uch-L1, predominantly mediates transnitrosylation to Drp1 under these conditions. This type of experiment is also consistent with the notion that these enzymes transnitrosylate with a preference of one substrate over another. Mechanistically, the particular environment of the thiolate anion can kinetically favor one NO donor over another.
In cell lysates expressing either HA-Cdk5 or V5-Uch-L1, we found transnitrosylation from SNO-Uch-L1 to Cdk5, and then to Drp1, but lack of NO group transfer from SNO-Cdk5 to Uch-L1 (Fig. 6A). These experiments, although lacking formal derivation of rate constants, turnover number (kcat) or substrate affinity (inversely related to Km) to support an enzymatic process (40), do provide additional evidence for specific transnitrosylation reactions because of relative substrate preference. Thus, the entire pathway of NO transfer from SNO-Uch-L1 to SNO-Cdk5 to SNO-Drp1 had to be present in order to observe significant SNO-Drp1 formation under these conditions. Nonetheless, other proteins present in the immunoprecipitates may participate in this series of transnitrosylation events. To show more definitively that SNO-Uch-L1 directly transfers NO to Cdk5, we performed an in vitro assay with purified recombinant proteins. SNO-Uch-L1 could directly transfer an NO group to Cdk5 (fig. S5).
Fig. 6. Redox blot quantifying transnitrosylation from Uch-L1 to Cdk5.
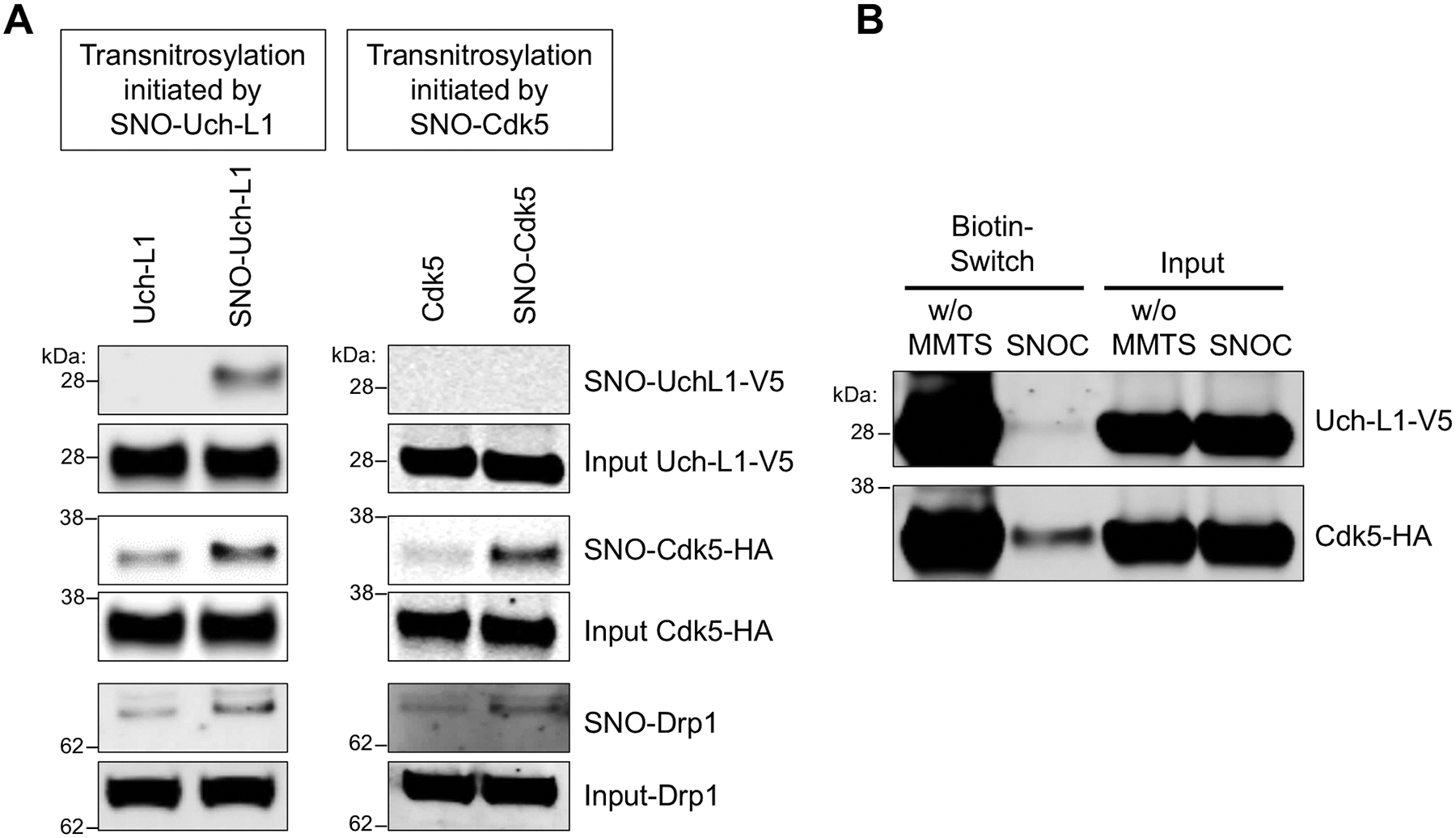
(A) Transnitrosylation occurs predominantly from SNO-Uch-L1 to Cdk5 to Drp1 rather than in the opposite direction from SNO-Cdk5 to Uch-L1. Expression of Uch-L1-V5 and Cdk5-HA increased the formation of SNO-Drp1, as evidenced on biotin-switch assay. Lysates of SH-SY5Y cells were prepared by V5- or HA-immunoprecipitation and exposed to 100 μM SNOC. These immunoprecipitates were then added to whole-cell lysates expressing either HA-Cdk5 or Uch-L1-V5, which were subjected to the biotin-switch assay to assess transnitrosylation. (B) SH-SY5Y cells were transfected with Uchl-L1-V5 and Cdk5-HA tagged constructs, and exposed to 50 μM SNOC at room temperature. After 30 min, cell lysates were prepared, incubated to achieve steady state (~1 h, by which time SNOC, a short-lived NO donor, had completely dissipated), and then subjected to the biotin-switch assay. Methyl-methanethiosulfonate (MMTS, 25 mM) was used to block free thiols during the assay for S-nitrosylated protein. Chemically-reduced thiol protein represents the relative amount of total protein obtained by the biotin-switch assay performed in the absence of MMTS (w/o MMTS). SNO-protein represents the relative amount of S-nitrosylated protein obtained by the biotin-switch assay. The relative concentration of protein in a redox pair in the S-nitrosylated (oxidized) form and the reduced form can be measured by quantitative densitometry of their respective bands on the gels (n = 4).
Because of differences in methods between standard immunoblotting and the biotin-switch method, one cannot compare the density of the bands representing the total input protein and the S-nitrosylated protein for evaluation of the absolute concentration of the SNO-protein. However, we can compare the ratio of SNO-protein to total (input) protein to obtain the relative amount of S-nitrosylated protein, and we can then compare this ratio across various preparations (20, 39, 41). Therefore, more critical to the overall hypothesis of a series of transnitrosylation events than the actual magnitude of an individual SNO-protein band generated is the fact that we could detect significant differences in their relative abundance. This ratio concept is also amenable to precise quantitative analysis of electron transfer and associated energetics of transnitrosylation reactions under steady-state conditions, as described below.
Nernst equations to predict transnitrosylation pathways and Gibbs free energy at steady state
In general, biological redox reactions often do not go to completion and depend on the relative redox potential of the pair of proteins that interact. Thus, SNO-protein bands observed in biotin-switch assays are not expected to be completely abated when individual upstream or downstream components are disrupted. Accordingly, we devised a quantitative method based on the Nernst equation to characterize these transnitrosylation reactions thermodynamically (20, 39). For this analysis, we make the assumption that these reactions go to steady state, as might be reasonably expected to occur in a chronic disease. The Nernst equation quantifies the electromotive force for net electron movement between a redox pair. To calculate the difference in the electromotive force (ΔE) between two redox pairs, e.g., for transnitrosylation between Uch-L1 and Cdk5, we calculated the difference in the values of their respective Nernst equations:
| Eqn. 1 |
| Eqn. 2 |
where ‘SNO’ represents the S-nitrosylated (or oxidized) protein and ‘red’ represents the chemically reduced protein, as obtained from densitometric analysis of redox immunoblots (Fig. 6B). Because the Nernst equation uses log values, the difference in Nernst equations represents the natural log of the ratio of effective concentrations of the reduced and oxidized forms of the protein. Thus, the absolute concentrations of the reduced protein and oxidized (in this case, S-nitrosylated) protein are not needed for the evaluation of the blots or for the resulting calculation; only the relative ratio is required. Hence, we can treat the reactions quantitatively without actually knowing the exact protein concentrations.
Using this method, we found that transnitrosylation from SNO-Uch-L1 to Cdk5 is predicted to be energetically very favorable under steady-state conditions in intact cells. Evidence for this was obtained by obtaining the relative redox potential for the reaction (Eqn. 1, ΔEo’ = 74.02 ± 4.60 mV [n = 4]), which in turn allowed us to calculate the associated change in Gibbs free energy (Eqn. 2, ΔGo’ = −18.35 ± 1.14 kJ/mol [n = 4]), predicting the reaction will proceed spontaneously. Our previous results indicated that the subsequent transnitrosylation from SNO-Cdk5 to Drp1 was also energetically favorable (20). Collectively, these findings lead us to propose that an NO group is transferred from SNO-Uch-L1 to Cdk5, which leads to subsequent transfer from SNO-Cdk5 to Drp1 in an energetically-favorable manner.
S-Nitrosylation of Uch-L1 in human AD brains
To assess the potential pathophysiological relevance of our findings in human AD, we obtained human postmortem brain tissue (table S2). Abundance of SNO-UchL1 was significantly increased in human brains with advanced stages of AD compared to that in control brains (Fig. 7A and fig. S6). Amounts of SNO-Uch-L1 were extremely low by these methods in control human brains, consistent with the notion that S-nitrosylation of Uch-L1 represents an aberrant nitrosylation event occurring in the diseased state only. To determine whether accumulation of SNO-Uch-L1 in human AD brain might be of pathophysiological significance, we calculated the ratio of SNO-Uch-L1 (by biotin-switch assay) to total Uch-L1 (as quantified from immunoblots), using methods previously described (20, 39, 41). In human AD brain this ratio was comparable to or greater than that encountered in our neuronal cell-based models exhibiting Aβ/SNO-Uch-L1—induced dendritic spine loss (Fig. 3A and Fig. 7B) or in the brains of hAPP-J20 AD mice (Fig. 1C and Fig. 7B), which exhibit synapse loss (42). Taken together, these findings indicated that the aberrant transnitrosylation cascade from SNO-Uch-L1 to Cdk5 to Drp1 may contribute to the synaptic pathology observed in AD (Fig. 7C).
Fig. 7. S-Nitrosylation of Uch-L1 in human AD brains and schema of transnitrosylation in the pathophysiology of synapse loss in AD.
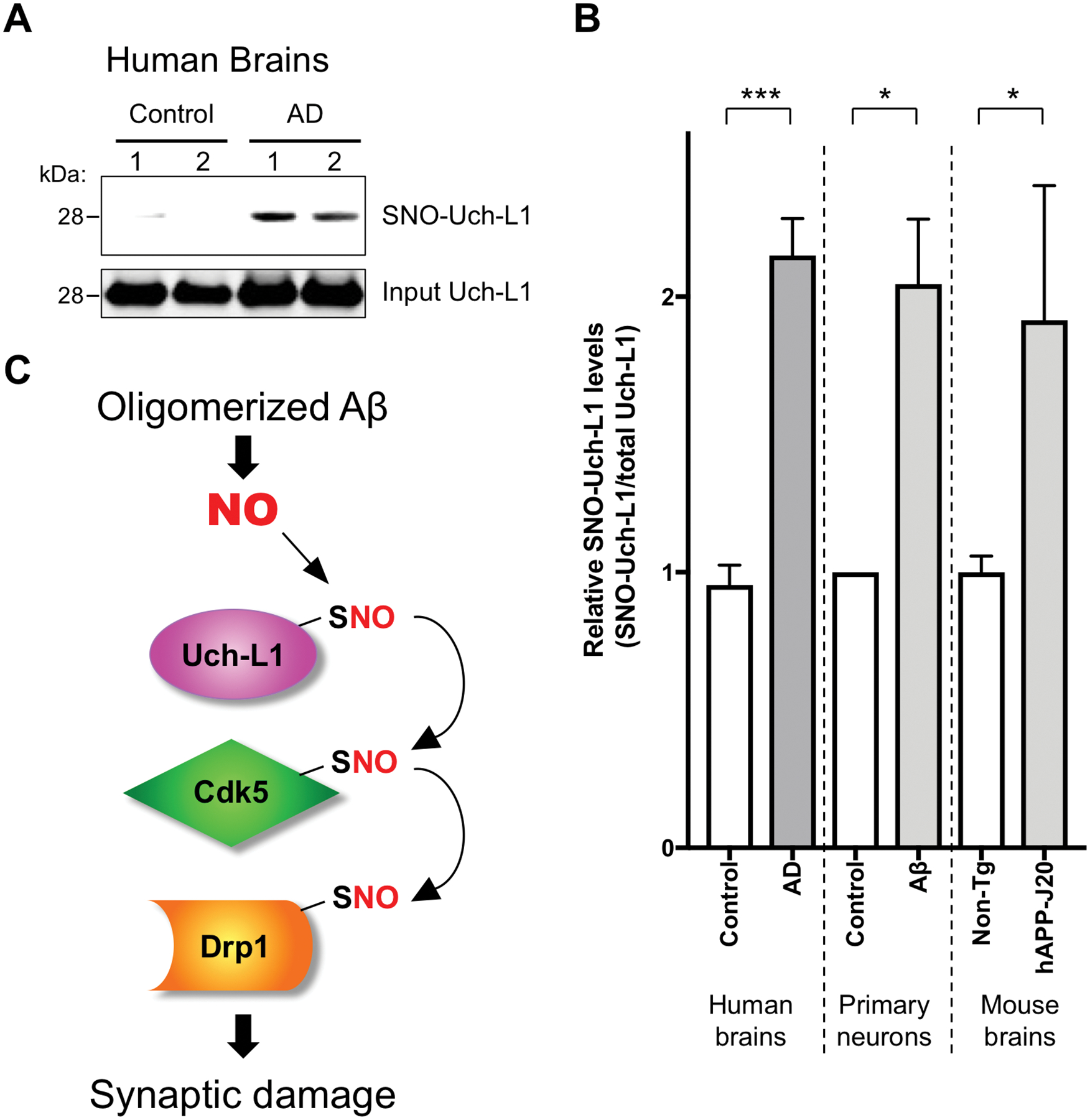
(A) Human brain tissues from control or AD patients were subjected to the biotin-switch assay to detect SNO-Uch-L1. (B) Quantification of the ratio of SNO-Uch-L1 to total Uch-L1 in human brains and in cell-based assays. Biotin-switch assays and immunoblot analyses were quantified by densitometry, and the relative ratio of SNO-Uch-L1 to total Uch-L1 was calculated for the following conditions: In vitro in cerebrocortical neurons after Aβ exposure (as shown in Figure 3A), in vivo in the hAPP-J20 mouse model of AD (as shown in Figure 1C), and in human AD brains vs. control human brains. Values are mean + SEM (n = 6 for control human brains, n = 7 samples from 5 AD human brains, n = 3 for each group in primary neuron experiments, n = 6 for each group in mouse brain experiments). ***P < 0.001, *P < 0.05 by Student’s t test. (C) Biochemical schema of transnitrosylation pathway leading to synaptic damage and consequent memory loss in AD. Note that these transnitrosylation reactions may be direct or indirect, with additional, as yet unknown, members of the transnitrosylation network still to be discovered.
Discussion
The abundance of Uch-L1 protein is decreased in human brains with sporadic AD (28). On the other hand, overexpression of Uch-L1 delays AD progression in vivo in animal models (29). This is thought to occur, at least in part, because of decreased Aβ production (30, 31). In the continued presence of exogenous oligomeric Aβ, however, Uch-L1 still modulates synaptic damage in AD models, implicating a second mechanism of Uch-L1 action. Along these lines, in in vivo experiments, expression of mutant Uch-L1(C152S) in mice is neuroprotective (43), implicating the possible importance of redox reactions at the critical Cys residue 152 of Uch-L1.
We found that Cys152 of Uch-L1 is S-nitrosylated in human AD brain to a similar degree as in our cell-based and animal models of AD. This reaction forms SNO-Uch-L1, which in turn, transnitrosylates Cdk5, an enzyme that functions in neuronal maturation and migration, among other processes. Next, SNO-Cdk5 transnitrosylates Drp1, a GTPase that mediates mitochondrial fission. Not only do these transnitrosylation reactions occur in cell-based immunoprecipitates, but also in in vitro transnitrosylation assays using purified/recombinant enzymes, showing that the donation of the NO group proceeds directly from one protein to another. Hence, the data show that there is overall donation of an NO group via a series transnitrosylation reactions from Uch-L1 to Cdk5 to Drp1 (Fig. 7C). The S-nitrosylation of Drp1 triggers excessive mitochondrial fission/fragmentation with consequent bioenergetic loss and synaptic damage (15, 20). This finding may influence progression of AD, as synapse loss is a major neuropathological correlate of cognitive decline in AD (17, 18).
The proteins that participate in this transnitrosylation cascade (Uch-L1/Cdk5/Drp1) also function in totally disparate pathways with different mechanisms of enzymatic action (ubiquitin hydrolase, kinase, and GTPase, respectively). One reason that this second non-canonical function of transnitrosylation may occur is that the catalytic sites of many types of enzymes contain cysteine residues, and their thiol groups (in fact, thiolates) are nucleophiles that can perform a reversible nucleophilic attack on the nitroso nitrogen via an S-nitrosylation reaction scheme (44–46). This reaction is particularly favored under conditions of high NO-induced nitrosative stress, as found in neurodegenerative diseases associated with aging.
In conclusion, we have discovered sequential NO transfer among unrelated enzymes that is not accounted for by the canonical catalytic activity originally discovered for the enzymes. This concept of ‘non-canonical transnitrosylation networks’ may represent a set of biochemical reactions not currently enveloped in standard bioinformatics packages such as gene ontology (GO) enrichment or KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analysis, and thus this finding could change our concept of pathway analysis and selection pressure on such cascades. Although it is unusual for a deleterious biochemical reaction network to develop because of natural selection, the fact that these aberrant reactions occur in the setting of neurodegenerative diseases in aged individuals, far beyond their reproductive years, may explain why they are subject to less evolutionary selection pressure (47).
Our mass spectrometry screening method uncovered a large number of potentially S-nitrosylated or transnitrosylated proteins in the brain (table S1). In the future, as mass spectrometry methods continue to advance, additional members of this non-canonical transnitrosylation network and additional transnitrosylation pathways in various disease states may be discovered. In summary, we find that enzymes previously thought to be in distinct biochemical cascades are actually linked by pathological crosstalk via transnitrosylation under disease conditions that may contribute in a causal manner to the disease process. The transnitrosylation activity of these enzymes may represent a therapeutic target for AD.
Materials and methods
Cell lines
SH-SY5Y cells (ATCC; CRL-2266) and HEK cells stably expressing nNOS (HEK-nNOS cells, gift from Drs. David Bredt and Solomon Snyder) were maintained in DMEM (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum, 2 mM l-glutamine (Thermo Fisher Scientific), 50 IU/ml penicillin (Omega Scientific), and 50 μg/ml streptomycin (Omega Scientific). For HEK-nNOS cell culture, 100 μg/ml geneticin (Thermo Fisher Scientific) was also included in the culture medium to maintain the expression of nNOS, and nNOS activation was achieved by the incubation with the calcium ionophore, A23187 (10 μM, EMD Millipore). Primary rat cortical neurons were isolated and cultured as described previously (15, 39, 41, 48).
E. coli strain for purification of recombinant Uch-L1
BL21 (DE3) E. coli (Agilent Technology) were transformed with a pET28a vector containing His-tagged Uch-L1 (WT or C152S mutant) and grown in 2XYT medium until the optical density measured at 600 nm (OD 600) reached 0.5. Expression of Uch-L1 was induced by 0.5 mM IPTG for 16 h, and E. coli were collected by centrifugation. The bacterial pellets were re-suspended in PBS, frozen down at −80 °C for 20 min, and then thawed at room temperature for cell disruption. Cells were further disrupted by sonication on ice, followed by incubation in 1% Triton X-100 at 4 °C for 30 min. The samples were centrifuged for 15 min at 10,000 x g, collected as supernatants, and then centrifuged again to completely remove bacterial debris. Recombinant Uch-L1 protein was purified using Ni-NTA agarose (Qiagen) or TALON metal affinity resin (Takara) at room temperature for 1 h. Imidazole (50 mM) was used for protein elution, and excess imidazole removed by PD-10 columns (GE Healthcare Life Sciences).
Animals and patient brain samples
We used two mouse models of AD (3–6 months old): 1) Tg2576, overexpressing a mutant form of human (h)APP with the Swedish mutation (KM670/671NL) (Taconic Biosciences, Model 1349) (49), and 2) hAPP-J20, expressing hAPP with two familial AD-linked mutations (the Swedish and Indiana [V717F] mutations) (The Jackson Laboratory, 34836-JAX) (42). Age and sex matched samples were randomly chosen and assigned to different experimental groups. All experiments were performed in accordance with the guidelines outlined by the Institutional Animal Care and Use Committees at The Sanford Burnham Prebys Medical Discovery Institute, the Scintillon Institute, or The Scripps Research Institute.
Postmortem brain tissues from AD or age-matched control patients (age <95) were collected at the University of California, San Diego, School of Medicine from subjects whose age, postmortem interval, and gender are described in table S2. Human brain samples were analyzed with institutional permission under the state of California and NIH guidelines. Informed consent was obtained according to procedures approved by the Institutional Review Board at the University of California, San Diego, School of Medicine.
NO donors, Aβ oligomer preparation, cell cultures, plasmids, and transfections
For induction of S-nitrosothiol formation using exogenous NO donors, the rapidly-decaying NO donor, SNOC was synthesized as described previously (50) and employed for short-term additions of NO. Oligomerized Aβ1–42 was prepared as we described previously (36). HEK-nNOS cells and rat primary neurons were transfected with Lipofectamine 2000 (Thermo Fisher Scientific) as per the manufacturer’s instruction. Lipofectamine LTX with PLUS reagent (Thermo Fisher Scientific) was used for transfection of SH-SY5Y cells. Measurements of dendritic spine density in rat primary cortical neuron cultures were performed in a masked fashion, as we previously described (15, 36, 48).
For exogenous expression of Uch-L1 in mammalian cells, the human cDNA sequence for Uch-L1 containing N-terminal V5 tag or C-terminal myc tag was subcloned into pcDNA3.1 vector (Thermo Fisher Scientific). For knockdown experiments, HEK-293 cells were transfected with siUch-L1 (Ambion, #4390824, ID: s14616) or Silencer™ Select Negative Control No. 1 siRNA (Ambion, #4390843) using Lipofectamine™ RNAiMAX Transfection Reagent (Thermo Fisher Scientific) according to the manufacturer’s protocol and incubated for 72 hours to achieve Uch-L1 knockdown. For bacterial expression, the Uch-L1 sequence was inserted into pET28a, adding a His tag to its N-terminus. C47S, C90S, C132S, C152S, C201S, C220S Uch-L1 mutants were generated using the QuikChange Lightning Multi Site-Directed Mutagenesis Kit (Agilent Technologies) according to the manufacturer’s protocol.
Biotin-switch assays and immunoblots
For analysis of S-nitrosylated proteins, we performed the biotin-switch assay as previously described (15, 39, 41). In brief, cell or tissue homogenates were incubated with the MMTS (S-methyl methanethiosulfonate, Sigma-Aldrich) blocking buffer (10 mM MMTS, 2.5% SDS in HEN [100 mM Hepes, pH 7.4, 1 mM EDTA, 0.1 mM neocuproine] buffer) to block free thiol groups. After specific reduction of S-nitrosothiols by 20 mM ascorbate, the newly generated free thiol groups were biotinylated with 1 mM biotin-HPDP (Thermo Fisher Scientific or Soltec Ventures). For control groups, ascorbate was omitted from the procedure to ensure the specificity of biotinylation. The biotinylated proteins were pulled down using High Capacity NeutrAvidin agarose (Thermo Fisher Scientific), and the purified proteins were subjected to immunoblot analysis. For the loading control, 5% input was saved before conducting a NeutrAvidin pull-down.
Protein samples were resolved by Bolt Bis-Tris Plus gels (Thermo Fisher Scientific) and transferred to Immobilon-FL PVDF membranes (EMD Millipore). After blocking with Odyssey TBS blocking buffer (Li-Cor), the membranes were incubated with primary antibodies against Uch-L1 (1:1000, Sigma-Aldrich, U5258), myc (1:1000, Cell Signaling Technology, 2276), V5 (1:5000, Thermo Fisher Scientific, R960–25), HA (1:1000, Santa Cruz Biotechnology, sc-805), Cdk5 (1:1000, Santa Cruz Biotechnology, sc-173 or Cell Signaling Technology, 12134), or Drp1 (1:1000, BD Biosciences, 611113). IR-dye 680LT-conjugated goat anti-mouse IgG (1:15000, Li-Cor, 926–68020) and IR-dye 800CW-conjugated goat anti-rabbit IgG (1:15000, Li-Cor, 926–32211) were used as secondary antibodies. For visualization of target proteins, the membranes were scanned with an infrared-based Odyssey imaging system (Li-Cor), and quantification of each band was performed with Image Studio software (Li-Cor).
Resin-assisted capture of S-nitrosylated proteins (SNO-RAC) for unbiased mass spectrometry analysis
To purify S-nitrosylated proteins, including SNO-Uch-L1, SNO-RAC was performed as previously described (51) with minor modifications. In brief, mouse brain cerebrocortical lysates were prepared in cold HENTS (1% Triton X-100, 0.1% SDS in HEN [100 mM Hepes, pH 7.4, 1 mM EDTA, 0.1 mM neocuproine]) buffer, assayed for protein concentration using the Bio-Rad Protein Assay Dye Reagent Concentrate (BioRad), and diluted with HEN buffer to a concentration of 1 μg/μl. SNOC (final concentration 25 μM) was added to 250 μl of mouse brain lysate, and the mixture was left at room temperature for 25 min in the dark to allow formation of S-nitrosylated (SNO)-proteins. Protein thiols were blocked with MMTS (50 mM) at 50 °C for 30 min, and excess MMTS removed by acetone precipitation. S-Nitrosothiols were then converted to free thiols with ascorbate (50 mM), and then captured by thiopropyl sepharose 6B resin (GE Healthcare Life Sciences) with rotation at room temperature for 2 h. The samples were washed twice with HENS buffer (HEN containing 1% SDS), twice with HENS/NaCl (HENS containing 400 mM NaCl), twice with HENS/10 (1:10 HENS), and then once with trypsin digestion buffer (50 mM NH4HCO3, 1 mM EDTA). Trypsin digestion was conducted on resin at 37 °C for 12 h, and the eluate was subjected to analysis by mass spectrometry (MS).
Digested peptide mixtures were pressure-loaded onto a fused silica capillary column of 250-μm internal diameter (I.D.) packed with 3 cm of 5 μm Partisphere strong cation exchanger (SCX, Whatman) and 3 cm of 10 μm Jupiter reversed-phase C12 material (Phenomenex); the SCX was end fritted with immobilized Kasil 1624 (PQ Corp.). After desalting, a 100 μm I.D. capillary with a 5 μm pulled tip packed with 15 cm of 4 μm Jupiter C12 material was attached to a zero-dead-volume union, and the entire split-column was placed in line with an Eksigent nano HPLC pump (Eksigent) and analyzed using modified, four-step multi-dimensional protein identification technology (MudPIT), as described previously (52). The eluted peptides were electrosprayed directly into a hybrid LTQ-Orbitrap mass spectrometer (ThermoFinnigan) with a distal spray voltage of 2.4 kV. A cycle of one full-scan mass spectrum (400–1400 m/z) followed by 6 data-dependent MS/MS scans were repeated continuously. Mass spectrum data were collected using Thermo XCalibur 3.1 (Thermo Fisher Scientific). The MS/MS spectra were searched with the ProLucid (a SEQUEST-based software) algorithm (53) against the EBI mouse IPI database concatenated to a decoy database in which the sequence for each entry in the original database was reversed. No modification on cysteine was allowed to reflect the reduced SNO thiols. The search results were assembled and filtered using the DTASelect program (54) with a peptide false-positive rate of 1%. All peptides identified were at least half tryptic, and precursor ion mass tolerance was set to 20 parts per million (ppm).
Ubiquitin pull-down assays
To prepare resin-bound SNO-Uch-L1 in vitro, recombinant Uch-L1 bound to Ni-NTA agarose was mixed with SNOC (100 μM) or ‘old’ SNOC (from which NO had been dissipated), and left at room temperature for 30 min in the dark with occasional mixing. The samples were washed once with PBS, and resuspended in PBS containing 0.5% Triton X-100. Recombinant Ubiquitin (Ub) (Sigma) was then mixed with resin-bound SNO-Uch-L1 (or a reduced form of Uch-L1) and incubated at room temperature for 1 h with rotation. After incubation, the resin was washed twice with PBS containing 0.5% Triton X-100. The samples were resuspended in NuPAGE LDS sample buffer (Thermo Fisher Scientific), boiled at 95 °C for 3 min, and loaded onto NuPAGE Bis-Tris gel. The bands were visualized by the standard Coomassie staining method.
Uch-L1 deubiquitinylation activity assays
Deubiquitinylation activity of recombinant Uch-L1 was fluorometrically assayed using Ub-7-amido-4-methylcoumarin (Ub-AMC, Boston Biochem) as described (29) with minor modification. In brief, 0.25 μM Ub-AMC and 0.02 μM Uch-L1 (or SNO-Uch-L1) were mixed in a final volume of 100 μl in reaction buffer (50 mM Tris-HCl, pH 7.6, 0.5 mM EDTA), and incubated for 10 min at 25 °C. The amount of AMC released from the Ub-AMC substrate was monitored using SpectraMax M2 (Molecular Devices) with excitation and emission wavelengths of 380 nm and 460 nm, respectively.
Activity of Uch-L1 in cells or tissue lysates was assessed with the activity-based probe, ubiquitin-vinylmethyl ester (Ub-VME), irreversibly modifying the “active” form of ubiquitin C‐terminal hydrolases and thus causing a shift in electrophoretic mobility on immunoblots (55, 56). For the Ub-VME assay, cell or tissue lysate was freshly prepared in PBS containing 0.1% Triton X-100. The reaction was initiated by incubating 2 μg of lysate with 0.2 μM Ub-VME (Boston Biochem), incubated for 5 min at room temperature, and stopped by adding NuPAGE LDS sample buffer (Thermo Fisher Scientific). Proteins were resolved by NuPAGE Bis-Tris gel followed by western blotting.
Lentivirus production, stereotaxic injection, and quantitative immunohistochemistry
Uch-L1 (WT or C152S mutant) was subcloned into lentiviral pCDH-EF1-dTomato-T2A-Kpn21 (System Biosciences) and used for preparation of active lentiviral particles for animal injections (57). hAPP-J20 AD transgenic mice or non-transgenic littermate control mice at the age of 3–4 months received bilateral, stereotactic injections of lentiviral vectors into the dentate gyrus at a rate of 1 μl per 2 min for a total of 2 μl of lentivirus infused into each hemisphere. Coordinates used for viral injection were as follows (35): AP-2.1, ML±1.8, and DV-2.0. Eight weeks after lentiviral injection, mice were anesthetized, perfused transcardially with saline (0.9% NaCl) containing 20 mU/ml Heparin (Sigma), followed by 4% PFA perfusion. Brains were then removed and fixed in 4% PFA for 48 h at 4 °C and kept in 1% PFA until analyzed. Quantitative immunohistochemistry was performed as described (36, 37). Immunolabeling was performed using mouse monoclonal antibody against Synaptophysin (Clone SY38, Millipore). Analysis of co-localization was performed on the whole dentate gyrus image using 3 separate images per condition by Fiji (ImageJ) software using the Squassh plugin as previously described (58, 59). All procedures described here were approved by the Institutional Animal Care and Use Committees at the Scintillon Institute and The Scripps Research Institute, where the work was performed.
Transnitrosylation assays
SH-SY5Y cells were transfected with Uch-L1-V5 or Cdk5-HA. After 1 day, Uch-L1-V5 expressing cells were lysed in RIPA buffer (50 mM Tris–HCl, pH 8.0, 150 mM NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 0.1% sodium dodecyl sulfate) and immunoprecipitated overnight at 4 °C with anti-V5 antibody conjugated to Dynabeads Protein G (Thermo Fisher Scientific), and then washed 3 times with high-salt RIPA buffer (50 mM Tris–HCl, pH 8.0, 500 mM NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 0.1% sodium dodecyl sulfate). Immunoprecipitated Uch-L1-V5 was exposed to decayed or freshly prepared 100 μM SNOC in lysis buffer (PBS containing 0.1% Triton X-100) for 30 min at room temperature in the dark with gentle shaking. Concurrently, cell lysates were prepared in lysis buffer (PBS containing 1% Triton X-100) from SH-SY5Y cells expressing Cdk5-HA. Immunoprecipitated Uch-L1-V5 exposed to SNOC (or as a control ‘old’ SNOC, from which NO had been dissipated) was then added to SH-SY5Y whole-cell lysates and incubated for 1 h at room temperature in the dark with gentle shaking. These reaction mixtures were subsequently subjected to biotin-switch assay after addition of MMTS blocking buffer. Dynabeads used for immunoprecipitation were removed by magnet rack prior to acetone precipitation.
For transnitrosylation assays using Cdk5-immunodepleted cell lysates, SH-SY5Y cells were transfected with WT Uch-L1-V5, mutant Uch-L1(C152S)-V5, or Drp1-HA. After 1 day, Uch-L1-V5—expressing cells were lysed in lysis buffer (PBS containing 1% Triton X-100) and immunoprecipitated with anti-V5 antibody. SH-SY5Y cells expressing Drp1-HA were also lysed in lysis buffer and immunodepleted overnight at 4 °C with anti-Cdk5 antibody (C-8, Santa Cruz Biotechnology, sc-173); or rabbit IgG (Santa Cruz, SC-2027) was used as a control antibody. Immunoprecipitated Uch-L1-V5 was washed in high-salt RIPA buffer (50 mM Tris–HCl, pH 8.0, 500 mM NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 0.1% sodium dodecyl sulfate) and exposed to freshly prepared or ‘old’ SNOC and then mixed with SH-SY5Y cell lysates immunodepleted of Cdk5. Transnitrosylation was assessed by biotin-switch assay. To calculate the redox potential and change in Gibbs free energy for the transnitrosylation reaction, Uch-L1-V5 and CDK5-HA co-transfected SH-SY5Y cells were exposed to 50 μM SNOC; after 30 min, cell lysates were subjected to biotin-switch assay in the absence or presence of MMTS free-thiol blocking agent, allowing us to observe total protein (both the reduced and S-nitrosylated forms) and oxidized protein (the S-nitrosylated forms), respectively. Subsequently, immunoblotted bands were quantified by densitometry, and the values used in a ratio of Nernst equations, as described in the manuscript text.
For in vitro transnitrosylation assays with recombinant proteins to detect direct transfer of the NO group from one protein to another, we first exposed purified Uch-L1 (10 μM) to 200 μM SNOC and then incubated for 30 min to generate SNO-Uch-L1 while allowing NO to dissipate from SNOC. Residual SNOC or cysteine remaining after NO dissipation was removed with a Zeba Spin Desalting Column (Thermo Fisher Scientific), and the resulting purified SNO-Uch-L1 was incubated with recombinant Cdk5/p25 (2 μM, Abcam, ab60761) for an additional 45 min to facilitate transnitrosylation from Uch-L1 to Cdk5. Note that p25 was included for stabilization of Cdk5, as we and others had previously shown that Cdk5 was the S-nitrosylated species (19). The biotin-switch assay was then performed as described above.
Quantification and statistical analysis
Fiji (ImageJ) and Image Studio were used for quantification of immunohistochemistry and immunoblot data, respectively. A Power Analysis of our prior data was used to determine the number of replicates needed for statistical purposes. The number of replicates or experiments are indicated in the individual figure legends. Data are expressed as mean + SEM. Differences between experimental groups were evaluated using an ANOVA followed by a post hoc test for multiple comparisons (Dunnett’s or Bonferroni’s) or a Student’s t test for comparison between two groups. A P value < 0.05 was considered significant. Statistical analyses were performed using GraphPad Prism software.
Supplementary Material
ACKNOWLEDGMENTS
We thank Drs. David Bredt and Solomon Snyder (Johns Hopkins) for HEK-nNOS cells, Traci Fang-Newmeyer (Sanford Burnham Prebys Medical Discovery Institute) and Weiping Tan (The Scripps Research Institute) for preparing cultures, and Scott R. McKercher (The Scripps Research Institute) for help with manuscript critical review and preparation.
Funding:
This work was supported in part by NIH grants RF1 AG057409, R01 AG056259, R01 DA048882, R01 NS086890, P30 NS076411, P01 ES016738 and DP1 DA041722 (to S.A.L.), R01 AG061845 (to T.N.), P30 AG062429 and R01 AG018440 (to R.A.R), and P41 GM103533 (to J.R.Y.); by a Distinguished Investigator Award from the Brain & Behavior Research Foundation (to S.A.L.); by a New Investigator Award from the Alzheimer’s Association (to T.N.); by the Michael J. Fox Foundation (to S.A.L. and T.N.); and by the Step Family Foundation (to S.A.L.).
Footnotes
Competing Interests: The authors declare no competing interests with regard to this work.
Data and materials availability: All data is available in the main text or the supplementary materials. All plasmids generated in this study will be available under a material transfer agreement.
Supplementary Materials:
Figures S1-S7
Tables S1-S2
REFERENCES AND NOTES:
- 1.Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH, Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc. Natl. Acad. Sci. U.S.A 88, 6368–6371 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hara MR et al. , S-Nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat. Cell Biol 7, 665–674 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Lipton SA et al. , A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature 364, 626–632 (1993). [DOI] [PubMed] [Google Scholar]
- 4.Nakamura T et al. , Aberrant protein S-nitrosylation in neurodegenerative diseases. Neuron 78, 596–614 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS, Protein S-nitrosylation: purview and parameters. Nat. Rev. Mol. Cell Biol 6, 150–166 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Ignarro LJ, Nitric oxide. A novel signal transduction mechanism for transcellular communication. Hypertension 16, 477–483 (1990). [DOI] [PubMed] [Google Scholar]
- 7.Seneviratne U et al. , S-Nitrosation of proteins relevant to Alzheimer’s disease during early stages of neurodegeneration. Proc. Natl. Acad. Sci. U.S.A 113, 4152–4157 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doulias PT, Tenopoulou M, Greene JL, Raju K, Ischiropoulos H, Nitric oxide regulates mitochondrial fatty acid metabolism through reversible protein S-nitrosylation. Sci. Signal 6, rs1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hao G, Derakhshan B, Shi L, Campagne F, Gross SS, SNOSID, a proteomic method for identification of cysteine S-nitrosylation sites in complex protein mixtures. Proc. Natl. Acad. Sci. U.S.A 103, 1012–1017 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wijasa TS et al. , Quantitative proteomics of synaptosome S-nitrosylation in Alzheimer’s disease. J. Neurochem 152, 710–726 (2020). [DOI] [PubMed] [Google Scholar]
- 11.Lee YI et al. , Protein microarray characterization of the S-nitrosoproteome. Mol. Cell. Proteomics 13, 63–72 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jia J et al. , Target-selective protein S-nitrosylation by sequence motif recognition. Cell 159, 623–634 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marino SM, Gladyshev VN, Structural analysis of cysteine S-nitrosylation: a modified acid-based motif and the emerging role of trans-nitrosylation. J. Mol. Biol 395, 844–859 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kornberg MD et al. , GAPDH mediates nitrosylation of nuclear proteins. Nat. Cell Biol 12, 1094–1100 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cho DH et al. , S-Nitrosylation of Drp1 mediates β-amyloid-related mitochondrial fission and neuronal injury. Science 324, 102–105 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakamura T, Cieplak P, Cho DH, Godzik A, Lipton SA, S-Nitrosylation of Drp1 links excessive mitochondrial fission to neuronal injury in neurodegeneration. Mitochondrion 10, 573–578 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeKosky ST, Scheff SW, Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann. Neurol 27, 457–464 (1990). [DOI] [PubMed] [Google Scholar]
- 18.Terry RD et al. , Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol 30, 572–580 (1991). [DOI] [PubMed] [Google Scholar]
- 19.Zhang P et al. , S-Nitrosylation of cyclin-dependent kinase 5 (cdk5) regulates its kinase activity and dendrite growth during neuronal development. J. Neurosci 30, 14366–14370 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qu J et al. , S-Nitrosylation activates Cdk5 and contributes to synaptic spine loss induced by β-amyloid peptide. Proc. Natl. Acad. Sci. U.S.A 108, 14330–14335 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kabuta T et al. , Ubiquitin C-terminal hydrolase L1 (UCH-L1) acts as a novel potentiator of cyclin-dependent kinases to enhance cell proliferation independently of its hydrolase activity. J. Biol. Chem 288, 12615–12626 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chung KK et al. , S-Nitrosylation of parkin regulates ubiquitination and compromises parkin’s protective function. Science 304, 1328–1331 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Yao D et al. , Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc. Natl. Acad. Sci. U.S.A 101, 10810–10814 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumar R et al. , S-Nitrosylation of UCHL1 induces its structural instability and promotes α-synuclein aggregation. Sci. Rep 7, 44558 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH, Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat. Cell Biol 3, 193–197 (2001). [DOI] [PubMed] [Google Scholar]
- 26.Stomberski CT, Hess DT, Stamler JS, Protein S-nitrosylation: determinants of specificity and enzymatic regulation of S-nitrosothiol-based signaling. Antioxid. Redox Signal 30, 1331–1351 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stamler JS, Toone EJ, Lipton SA, Sucher NJ, (S)NO signals: translocation, regulation, and a consensus motif. Neuron 18, 691–696 (1997). [DOI] [PubMed] [Google Scholar]
- 28.Choi J et al. , Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson’s and Alzheimer’s diseases. J. Biol. Chem 279, 13256–13264 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Gong B et al. , Ubiquitin hydrolase Uch-L1 rescues β-amyloid-induced decreases in synaptic function and contextual memory. Cell 126, 775–788 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Zhang M et al. , Control of BACE1 degradation and APP processing by ubiquitin carboxyl-terminal hydrolase L1. J. Neurochem 120, 1129–1138 (2012). [DOI] [PubMed] [Google Scholar]
- 31.Zhang M, Cai F, Zhang S, Zhang S, Song W, Overexpression of ubiquitin carboxyl-terminal hydrolase L1 (UCHL1) delays Alzheimer’s progression in vivo. Sci. Rep 4, 7298 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shankar GM et al. , Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci 27, 2866–2875 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Calabrese B et al. , Rapid, concurrent alterations in pre- and postsynaptic structure induced by naturally-secreted amyloid-β protein. Mol. Cell. Neurosci 35, 183–193 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wei W et al. , Amyloid β from axons and dendrites reduces local spine number and plasticity. Nat. Neurosci 13, 190–196 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cisse M et al. , Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature 469, 47–52 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Talantova M et al. , Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc Natl Acad Sci U.S.A 110, E2518–2527 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lipton SA et al. , Therapeutic advantage of pro-electrophilic drugs to activate the Nrf2/ARE pathway in Alzheimer’s disease models. Cell Death Dis 7, e2499 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barsoum MJ et al. , Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J 25, 3900–3911 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakamura T et al. , Transnitrosylation of XIAP regulates caspase-dependent neuronal cell death. Mol. Cell 39, 184–195 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mitchell DA, Marletta MA, Thioredoxin catalyzes the S-nitrosation of the caspase-3 active site cysteine. Nat Chem Biol 1, 154–158 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Uehara T et al. , S-Nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 441, 513–517 (2006). [DOI] [PubMed] [Google Scholar]
- 42.Mucke L et al. , High-level neuronal expression of Aβ1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J. Neurosci 20, 4050–4058 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu H et al. , Role of UCHL1 in axonal injury and functional recovery after cerebral ischemia. Proc. Natl. Acad. Sci. U.S.A 116, 4643–4650 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lancaster JR Jr., How are nitrosothiols formed de novo in vivo? Arch. Biochem..Biophys 617, 137–144 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Seth D et al. , A multiplex enzymatic machinery for cellular protein S-nitrosylation. Mol. Cell 69, 451–464.e456 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Talipov MR, Timerghazin QK, Protein control of S-nitrosothiol reactivity: interplay of antagonistic resonance structures. J. Phys. Chem. B 117, 1827–1837 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Reznick D, Bryant M, Holmes D, The evolution of senescence and post-reproductive lifespan in guppies (Poecilia reticulata). PLoS Biol 4, e7 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Akhtar MW et al. , Elevated glucose and oligomeric β-amyloid disrupt synapses via a common pathway of aberrant protein S-nitrosylation. Nat. Commun 7, 10242 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hsiao K et al. , Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 274, 99–102 (1996). [DOI] [PubMed] [Google Scholar]
- 50.Lei SZ et al. , Effect of nitric oxide production on the redox modulatory site of the NMDA receptor-channel complex. Neuron 8, 1087–1099 (1992). [DOI] [PubMed] [Google Scholar]
- 51.Forrester MT et al. , Proteomic analysis of S-nitrosylation and denitrosylation by resin-assisted capture. Nat. Biotechnol 27, 557–559 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Washburn MP, Wolters D, Yates JR 3rd, Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol 19, 242–247 (2001). [DOI] [PubMed] [Google Scholar]
- 53.Eng JK, McCormack AL, Yates JR 3rd, An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom 5, 976–989 (1994). [DOI] [PubMed] [Google Scholar]
- 54.Tabb DL, McDonald WH, Yates JR 3rd, DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J. Proteome Res 1, 21–26 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Borodovsky A et al. , A novel active site-directed probe specific for deubiquitylating enzymes reveals proteasome association of USP14. EMBO J 20, 5187–5196 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cartier AE et al. , Regulation of synaptic structure by ubiquitin C-terminal hydrolase L1. J. Neurosci 29, 7857–7868 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reuter JD, Fang X, Ly CS, Suter KK, Gibbs D, Assessment of hazard risk associated with the intravenous use of viral vectors in rodents. Comp. Med 62, 361–370 (2012). [PMC free article] [PubMed] [Google Scholar]
- 58.Rizk A et al. , Segmentation and quantification of subcellular structures in fluorescence microscopy images using Squassh. Nat. Protoc 9, 586–596 (2014). [DOI] [PubMed] [Google Scholar]
- 59.Spencer B, Rank L, Metcalf J, Desplats P, Identification of insulin receptor splice variant B in neurons by in situ detection in human brain samples. Sci Rep 8, 4070 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.