

Multilocus sequence typing (MLST) provides allele-based characterization of bacterial pathogens in a standardized framework. However, classical MLST schemes for Bordetella pertussis, the causative agent of whooping cough, seldom reveal diversity among the small number of gene targets and thereby fail to delineate population structure.
KEYWORDS: Bordetella pertussis, whooping cough, wgMLST, surveillance, genomics, whole-genome sequencing
ABSTRACT
Multilocus sequence typing (MLST) provides allele-based characterization of bacterial pathogens in a standardized framework. However, classical MLST schemes for Bordetella pertussis, the causative agent of whooping cough, seldom reveal diversity among the small number of gene targets and thereby fail to delineate population structure. To improve the discriminatory power of allele-based molecular typing of B. pertussis, we have developed a whole-genome MLST (wgMLST) scheme from 225 reference-quality genome assemblies. Iterative refinement and allele curation resulted in a scheme of 3,506 coding sequences and covering 81.4% of the B. pertussis genome. This wgMLST scheme was further evaluated with data from a convenience sample of 2,389 B. pertussis isolates sequenced on Illumina instruments, including isolates from known outbreaks and epidemics previously characterized by existing molecular assays, as well as replicates collected from individual patients. wgMLST demonstrated concordance with whole-genome single nucleotide polymorphism (SNP) profiles, accurately resolved outbreak and sporadic cases in a retrospective comparison, and clustered replicate isolates collected from individual patients during diagnostic confirmation. Additionally, a reanalysis of isolates from two statewide epidemics using wgMLST reconstructed the population structures of circulating strains with increased resolution, revealing new clusters of related cases. Comparison with an existing core genome (cgMLST) scheme highlights the stable gene content of this bacterium and forms the initial foundation for necessary standardization. These results demonstrate the utility of wgMLST for improving B. pertussis characterization and genomic surveillance during the current pertussis disease resurgence.
INTRODUCTION
Whooping cough (pertussis) is a respiratory disease with its highest rates of morbidity and mortality in young infants that continues to resurge in the United States and many other countries. Vaccines against pertussis were introduced in the 1940, leading to a dramatic reduction in reported disease incidence. However, the switch to acellular formulations in the 1990s was followed by increased reporting among all age groups in the decades since, despite high or increasing coverage with pertussis-containing vaccines among industrialized countries (1). While not fully understood, reported resurgence likely results from multiple factors, including heightened awareness, expanded surveillance, improved diagnostics, shifting transmission dynamics, and pathogen evolution (1–4). Waning protection conferred by acellular vaccines is likely also responsible for increased disease among vaccinated individuals (5, 6).
Increased pertussis in the United States manifests in local outbreaks but also in cyclical, statewide and national epidemics (7). Past molecular study of epidemics has been challenged by the low genetic diversity of Bordetella pertussis, frequently described as a “monomorphic” pathogen (8). Traditional multilocus sequence typing (MLST) targeting selected housekeeping genes provides very little discriminatory power, and isolates of B. pertussis are often genotyped according to alleles of key vaccine immunogen-encoding genes, which resolve most isolates into only few sequence types (9–12). Quantifying specific repeat content with multilocus variable-number tandem-repeat analysis (MLVA) similarly reveals few discrete types (13). Alternatively, pulse-field gel electrophoresis (PFGE) has proven more useful for molecular typing, owing to the structural plasticity of the B. pertussis chromosome (14–16), but lacks throughput and standardization across laboratories (17, 18). The molecular study of polyclonal epidemics, as well as broader geographic or temporal dynamics, has required a time-consuming combination of various methodologies, as no single assay can sufficiently identify linked case clusters or describe the molecular characteristics of circulating B. pertussis to guide prevention and control efforts (9, 16, 19, 20).
Applications of high-throughput sequencing have transformed public health microbiology by exploiting the profound resolution of pathogen genomics for effective investigation and surveillance of infectious disease (21–24). A number of recent whole-genome sequencing (WGS) analyses of circulating B. pertussis have successfully reconstructed the accumulation of single nucleotide polymorphisms (SNPs) to reveal the bacterium’s population structure, geographic dispersion, and phylogenetic history with new depth (3, 25–29). However, allele-based molecular fingerprints may provide a more viable implementation of genome-based strain typing that can be standardized for routine use in public health laboratories (30–32). Whole-genome MLST (wgMLST) schemes, which capture the full complement of protein-coding genes in the genome, have been successfully applied to microbial pathogens for molecular epidemiology and food source attribution (22, 31, 33). More restrictive core genome MLST (cgMLST) schemes, which evaluate only highly conserved genes, have also been implemented, including for B. pertussis (22, 34).
A growing number of recent B. pertussis clinical isolates recovered worldwide have been sequenced, including many closed assemblies. In this study, we leveraged a collection of annotated, reference-quality genome assemblies to develop a standardized genome-based B. pertussis strain typing system using wgMLST and evaluated its performance with 2,389 sequenced isolates, primarily recovered from U.S. pertussis cases. The curated scheme includes 3,506 protein-coding gene sequences, covering 81.4% of the average B. pertussis genome, and reproduced the population structure concordant with SNPs in retrospective analyses. These results highlight that the genomic stability of this bacterium perhaps makes wgMLST well suited for routine genome-based strain typing by public health institutions and pertussis researchers.
MATERIALS AND METHODS
Strain selection.
The Centers for Disease Control and Prevention’s (CDC) collection includes U.S. B. pertussis isolates gathered through routine surveillance and during outbreaks. In total, sequence data from a convenience sample of 2,389 isolate genomes were included in the current study based on availability, and most were selected for sequencing as part of previous studies (see Table S2 in the supplemental material). Many isolates were obtained through the Enhanced Pertussis Surveillance/Emerging Infection Program Network (EPS) (35), including sets of 2 to 7 replicate isolates (average = 5) recovered from 153 patients during diagnostic culture confirmation.
Genomic DNA preparation and sequencing.
Isolates were cultured on Regan-Lowe agar without cephalexin for 72 h at 37°C. Genomic DNA isolation and purification were performed using the Gentra Puregene yeast/bacterium kit (Qiagen, Valencia, CA), with slight modification (36). Briefly, two aliquots of approximately 1 × 109 bacterial cells were harvested and resuspended in 500 μl of 0.85% sterile saline and then pelleted by centrifugation for 1 min at 16,000 × g. Recovered genomic DNA was resuspended in 100 μl of DNA hydration solution. Aliquots were quantified using a NanoDrop 2000 instrument (Thermo Fisher Scientific Inc., Wilmington, DE). Whole-genome shotgun libraries were prepared using the NEB Ultra Library Prep kit (New England BioLabs, Ipswich, MA) for sequencing on either the MiSeq or HiSeq platform (Illumina, San Diego, CA). Sequencing reads from 10 isolates were randomly selected and subsampled without replacement, yielding 5 replicate samples at each of 7 coverage depths (9×, 14×, 21×, 31×, 46×, 70×, and 105×).
wgMLST scheme design.
The initial wgMLST scheme was developed from all protein-coding genes predicted in a convenience sample of closed, reference-quality genome assemblies from 225 B. pertussis isolates (Table S1). Multicopy genes and paralogs (e.g., insertion sequence element [ISE] transposases) with >95% sequence identity were detected and removed, excluding 240 to 260 coding sequences (CDS) (6.5%) per genome. The remaining CDS were clustered into 3,681 orthologous loci. Each locus was further evaluated based on consensus allele call frequency and errors using custom scripts from Applied Maths, as well as manual inspection of allele alignments in BioNumerics. Loci were manually removed from the scheme based on criteria such as low frequency, low-complexity sequence repeats, homopolymeric tracts, length discrepancy, or variable allele calling between replicates. The process of locus curation was repeated twice, first with an initial input set of 214 CDC genomes and then with a larger collection of 614 isolates, leaving 3,506 loci in the final scheme.
wgMLST allele calling and strain comparison.
Allele calling was performed with the BioNumerics (v7.6.3) Calculation Engine. Imported sequencing reads were quality trimmed and filtered (minimum average read quality = 25, minimum read tail quality score = 15, and minimum read length = 35 bp) before de novo assembly using Spades v3.7.1 (careful mode; minimum contig length = 300 bp) (37). Consensus allele calls were derived from the combination of read mapping (assembly free [AF]) and reference alignment to the assembled contigs (assembly based [AB]). Assembly-free allele calls were determined by screening kmer profiles (k = 35 bp) of the reference alleles in the input sequencing reads, during which the quality and frequency (forward and reverse) of matching reads were recorded. Alleles were considered present if all their kmers were detected at least once in each direction and three times in total (i.e., minimum coverage = 3×, minimum forward = 1×, and minimum reverse = 1×). Assembly-based allele calls were determined by alignment of assembled contigs to a reference database containing one wgMLST allele per locus using discontinuous MegaBLAST (dc-megablast, k = 11, minimum similarity = 95%; gapped alignment allowed). Only alignment hits with intact reading frames between start and stop codons were retained. Hits with >70% length overlap were further filtered, retaining only the aligned allele with highest sequence identity and longest length and without internal stop codons. An assembly-based allele call was made only if the hit included a proper CDS structure with no ambiguous bases and there were no secondary hits. Resulting assembly-free and assembly-based allele calls for each locus were compared to assign a consensus call according to the following union principle: (i) if both methods called the same allele, the call was retained; (ii) if only one method called an allele, the call was retained; and (iii) if multiple possible alleles were detected by either method, no call was made. In short, the agreement between the two methods was required to be perfect: the set of alleles found by each of the two calling methods had to have a nonempty intersection.
Allele pattern comparisons were performed by selecting all sequencing read sets with at least 3,000 consensus allele calls and filtering out any monomorphic loci. Pairwise distances were determined using a simple cluster analysis based on categorical differences and UPGMA (unweighted pair group method using average linkages) hierarchical clustering in BioNumerics. Minimum-spanning trees were calculated in BioNumerics using the advanced cluster analysis for categorical data.
SNP detection.
SNP variation among sequenced isolate genomes was determined with the exported de novo assemblies from BioNumerics using kSNP3 with k of 23 (38). Pairwise distances were calculated from all variable SNPs shared between each pair of sequenced isolates.
Comparison to Institut Pasteur’s cgMLST scheme.
The cgMLST scheme and allele definitions developed at Institut Pasteur (34) were kindly provided by Sylvain Brisse and Valérie Bouchez. Ortholog matching between the cgMLST and wgMLST schemes was performed by reciprocal best-match alignment using BLASTn (minimum of 95% identity and 90% length match). Unmatched loci were further compared to identify overlapping sequence content with relaxed alignment parameters (minimum of 90% identity and 50% length match).
The cgMLST scheme was loaded into a local database in BioNumerics, and allele calling was performed with selected isolates using the same parameters as indicated above for wgMLST. A representative subset of sequenced isolates was derived from the collection of 2,039 read sets with at least 3,000 wgMLST consensus allele calls by clustering isolates with 0 or 1 pairwise SNP using mcl (39). One isolate was selected from each of the resulting clusters and combined with all unclustered (unique) isolates, as well as isolates from a retrospective high school outbreak and replicates from individual patients, into a data set of 379 isolates representing the phylogenetic breadth of the larger collection.
Data availability.
The whole-genome shotgun sequences are available from the NCBI Sequence Read Archive, organized under BioProject accession number PRJNA279196.
RESULTS
Locus curation.
The wgMLST scheme was developed from the protein-coding genes predicted in closed, reference-quality genome assemblies from 214 B. pertussis isolates in the CDC collection combined with 11 publicly available genome sequences (Table S1). All multicopy genes, paralogs, and ISE transposases were excluded resulting in 3,681 orthologous loci captured in the initial version, the majority of which exhibited one allele. Each locus was evaluated with a larger set of raw sequencing reads to identify any systematic errors in allele calling due to either coding sequence (CDS) disruption (i.e., indels, gene truncations) or non-ACGT bases (i.e., Ns), as determined in BioNumerics. Locus reliability was further evaluated by confirming matching allele calls in sequencing reads from pairs of isolates which were independently confirmed to differ by ≤1 SNP using kSNP, and discrepant alleles were inspected manually for insertion or deletion variation. The whole process of locus curation, described in detail in Materials and Methods, was repeated twice and removed 175 problematic loci. The final scheme included 3,506 loci, covering >3.3 Mb (81.4%) of the B. pertussis genome and an average 76.8% of protein-coding nucleotides (Fig. 1). Details of each locus are available in Data Set S1.
FIG 1.
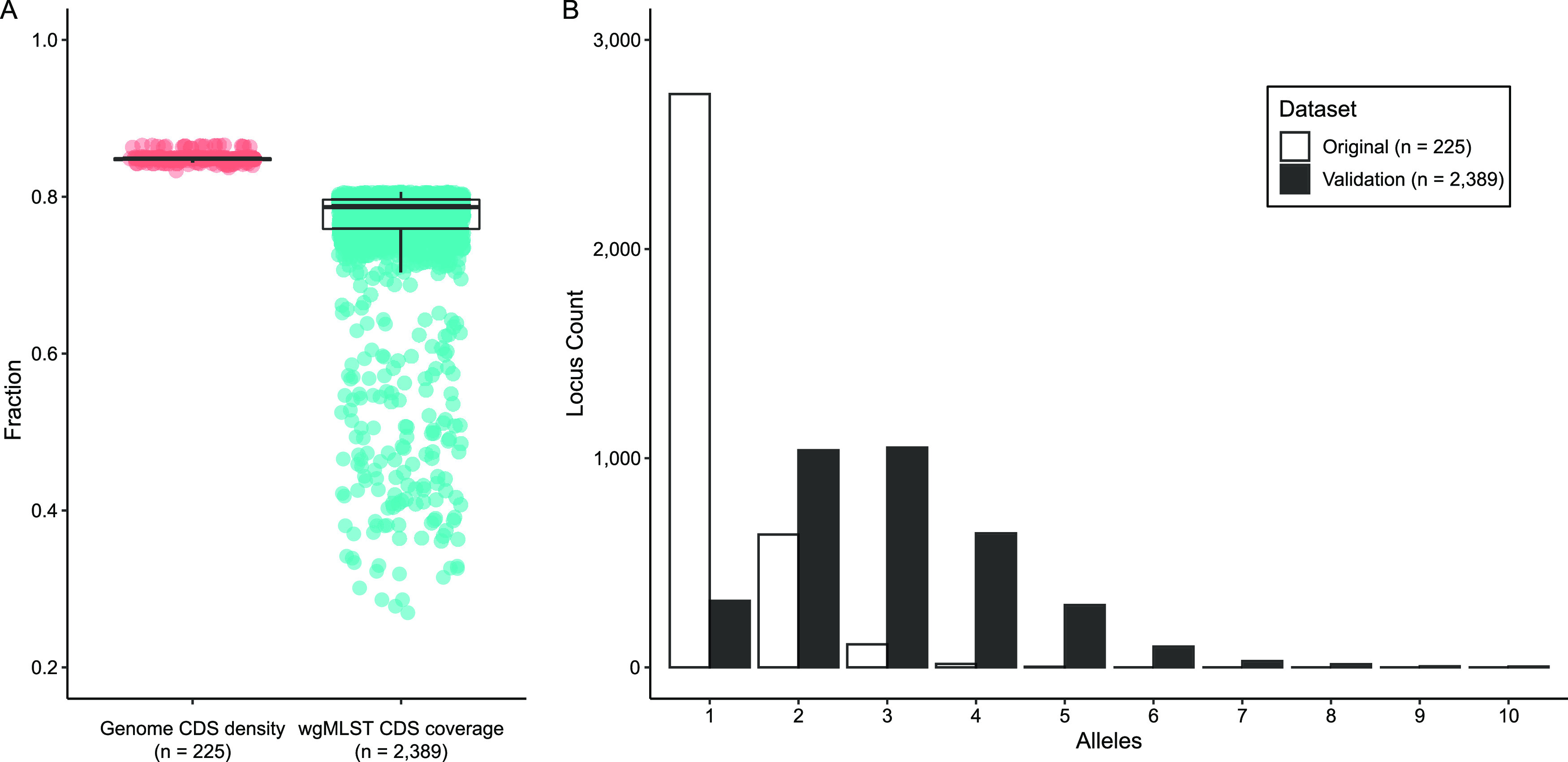
wgMLST scheme statistics. (A) The scheme was developed from 225 complete genome assemblies, which had an average coding density of 84.5%, and the scheme loci captured an average 76.8% of all protein-coding nucleotides for characterizing a collection of 2,389 sequenced isolates. (B) The distribution of unique alleles observed at each locus shifted as incorporating additional isolates revealed more diverse allele sequences at many gene loci.
Performance testing.
The process of allele calling in BioNumerics combines independent read mapping against a database (assembly free [AF]) and reference alignment to de novo assemblies (assembly based [AB]) to produce consensus allele calls. Performance was assessed across various metrics using sequencing reads from 2,389 isolates, including the 214 used for initial scheme development, to determine potential variations in allele calling due to instruments, read lengths, coverage depth, or average read quality. Assembly quality proved a good indicator of allele calling, regardless of sequencing instrument or format, as better assemblies yielded more consensus allele calls (Fig. S1). Read lengths influenced allele call performance more than the sequencing instrument, likely due to improved assembly as seen with 250-bp reads from either the MiSeq or HiSeq platform (Fig. S1). Read quality and coverage depth also impact assembly and were important for allele calling, as expected. Accordingly, decreased allele calling primarily resulted from non-ACGT errors that corresponded to the number of ambiguous bases in the contigs, further illustrating the dependence on de novo assembly (Fig. S1 and S2). Failed allele calling due to CDS disruption did not depend on sequencing instrument or read length but rather reflected the known accumulation of pseudogenes present in B. pertussis genomes (40) (Fig. S1 and S2). Locus-centric assessment of allele calling also indicated that the highest failure rates were due to CDS disruption in a small set of frequent pseudogenes, while others were comparably sporadic. Based on these results, a minimum cutoff of 3,000 consensus allele calls per genome was used for all subsequent analyses.
Allele profile differences among the 2,239 isolates with at least 3,000 allele calls were compared to pairwise SNPs distances predicted independently with kSNP to assess agreement between the two approaches for sequence-based strain typing. As expected, there was strong concordance between pairwise allele and pairwise SNP distances (Fig. 2). Most sequenced isolates of B. pertussis differed by <200 SNPs but exhibited more allelic differences due to variations not linked to single base substitutions, which are not detectable with kSNP.
FIG 2.

Concordance of pairwise distances. SNP and allele distances between all pairwise combinations of 2,389 sequenced isolates were concordant. Distances measured by wgMLST were consistently greater than measures of SNPs, reflecting the many types of sequence variation among the alleles. Distances are plotted as density according to the key, and dashed lines indicate perfect correlation. Most isolates differed by <200 alleles, and a small number of very distant isolates resembled strain 18323 that differed from the majority by >1,000 alleles (inset).
Reproducibility testing.
To assess allele calling reproducibility and determine a minimum coverage depth cutoff, 10 sequencing read sets were randomly subsampled at seven coverage depths (9×, 14×, 21×, 31×, 46×, 70×, and 105×), with five replicates. Each subsample was imported into BioNumerics and the total numbers of consensus allele calls, as well as their accuracies compared to the full read set, were compared across replicates. The total number of consensus allele calls remained above 3,000 for most replicates with average coverage depths of >25× before dropping quickly (Fig. 3A and Fig. S3). As coverage decreased the consensus allele calls remained accurate, even as the total number of calls declined, and errors were only observed in two replicates at 9× depth (Fig. 3B). Both errors were traced back to a single miscalled nucleotide in their respective loci. A minimum cutoff for average sequencing coverage depth was set at 30× for all subsequent analyses.
FIG 3.

Technical replicates from subsampled read sets. At decreasing coverage depths (9×, 14×, 21×, 31×, 46×, 70×, and 105×), replicated subsamples from select HiSeq and MiSeq read sets produced fewer consensus allele calls (A) but largely still made accurate allele calls compared to the full read set (B). Replicates for each read set are plotted separately in Fig. S3.
Consistency among biological replicates was also evaluated using a collection of multiple isolates recovered from 152 individual patients. Selected participating surveillance laboratories pick “sets” of colonies (average = 5; range = 2 to 7) during culture confirmation and submission to the CDC for characterization, including whole-genome sequencing (Fig. S4). Sequence variation among isolates within each set was quantified as both SNPs and alleles. While 49 sets exhibited no differences by either measure, many sets included 1 SNP and pairwise distances up to 5 SNPs or 12 alleles were observed (Fig. 4). Some allele differences resulted from mutations other than single base substitutions, such as indels.
FIG 4.

Biological replicates from individual patients. Sets of replicate isolates recovered from surveillance cases during culture confirmation were frequently not all identical, with a maximum pairwise distance within the set often equaling 1 or 2 SNPs or alleles. The inset shows sets with ≤1 SNP or allele maximum distance.
Taken together, these replicates indicate that allele calling results are reproducible. Allelic and SNP variation detected within replicate isolate sets from individual patients suggest that genetic diversification occurs during infection. Therefore, the resolution of outbreak clustering may be limited to approximately 2 allele differences, as most cases are represented by a single isolate and results could vary depending on which colony is selected during laboratory isolation.
Retrospective outbreak cluster detection.
To test the utility of the wgMLST scheme for studying the molecular epidemiology of pertussis, 12 isolates from epidemiologically linked cases associated with an outbreak occurring at a high school during a 2-month period in 2016 were characterized. The case isolates were compared to 83 contemporaneous, sporadic isolates, which together represented 22% of cases reported from the surveillance catchment area that included the outbreak. The 12 outbreak cases shared an identical allele profile and were discretely clustered in a minimum spanning tree calculated from 157 variable loci (Fig. 5). Comparing allele profiles also identified potential links to 3 sporadic case isolates recovered from infants, two of whom were siblings, which predated the outbreak. All other contemporaneous isolates differed from the outbreak cluster by at least 2 alleles, some forming clusters of their own, demonstrating the effectiveness of wgMLST to delineate linked cases and potentially complement epidemiological investigation of localized B. pertussis outbreaks.
FIG 5.
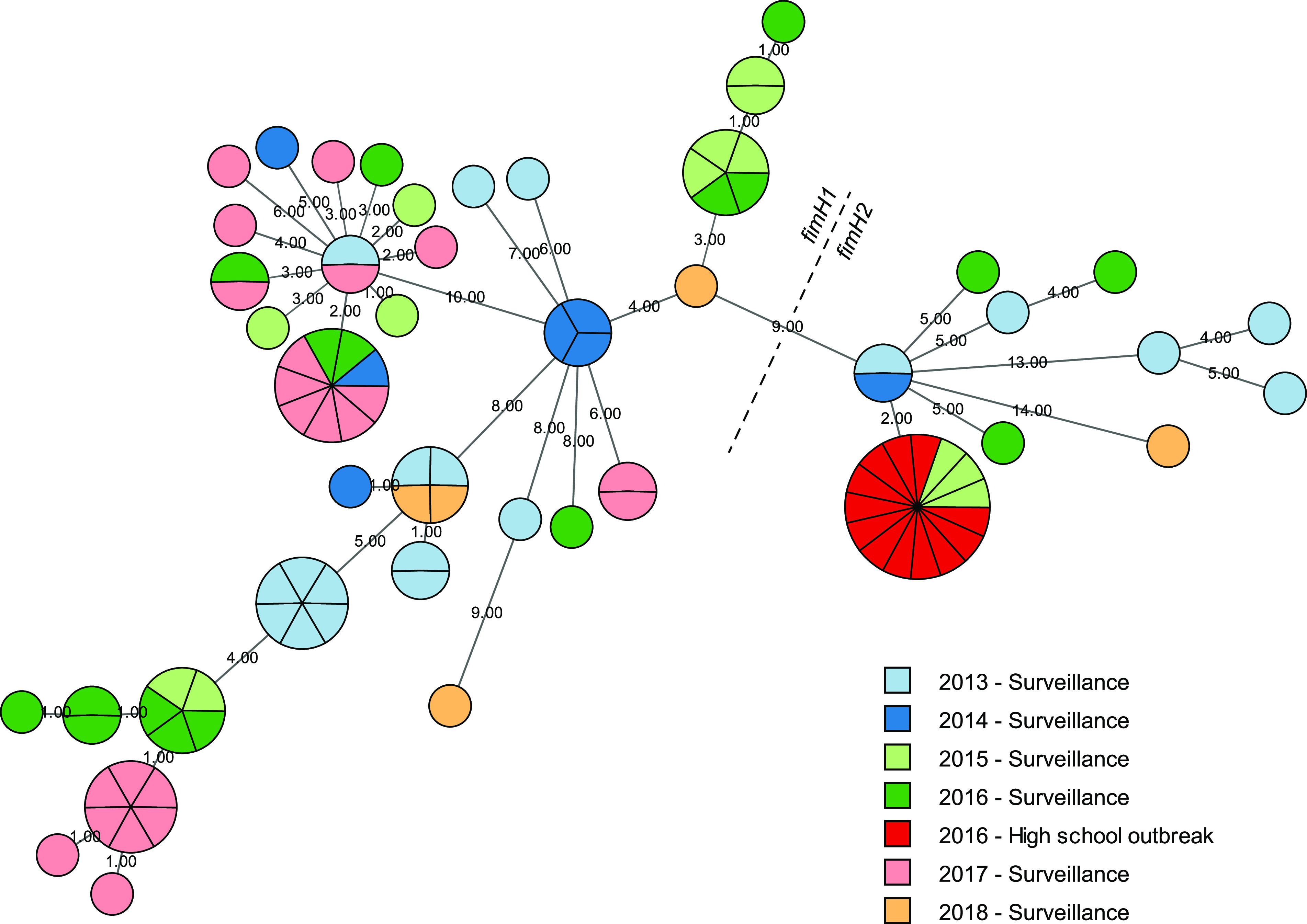
Molecular epidemiology of a high school outbreak. A minimum spanning tree calculated from 157 polymorphic loci clustered 12 case isolates from a high school outbreak, distinguishing them from 83 contemporaneous sporadic case isolates collected through routine surveillance in a retrospective comparison. Outbreak and sporadic case isolates are indicated according to the key and detailed in Table S2. All isolates carried the ptxP3 allele, and divergence between isolates with the fimH1 (fim3-1) and fimH2 (fim3-2) alleles is indicated by the dashed line. Node size indicates abundance and connecting lines are numbered according to allele distance.
Population structure of statewide epidemics.
Periods of increased disease have been reported across geographically defined regions (7), such as U.S. states, and the test data here included sequenced B. pertussis isolates recovered from two such epidemics in Washington (9) and Vermont (36, 41). wgMLST revealed discrete population structures within each epidemic, confirming the polyclonal nature of each while identifying putative clusters of transmission among linked cases in a minimum spanning tree calculated from variable loci (Fig. 6). In both states, some of the genotypes present during the epidemic were also detected among surveillance isolates collected in subsequent years. Combining the isolates from both state epidemics further confirmed that each included circulation of common genotypes, despite >3,700 km of physical separation (Fig. S5).
FIG 6.
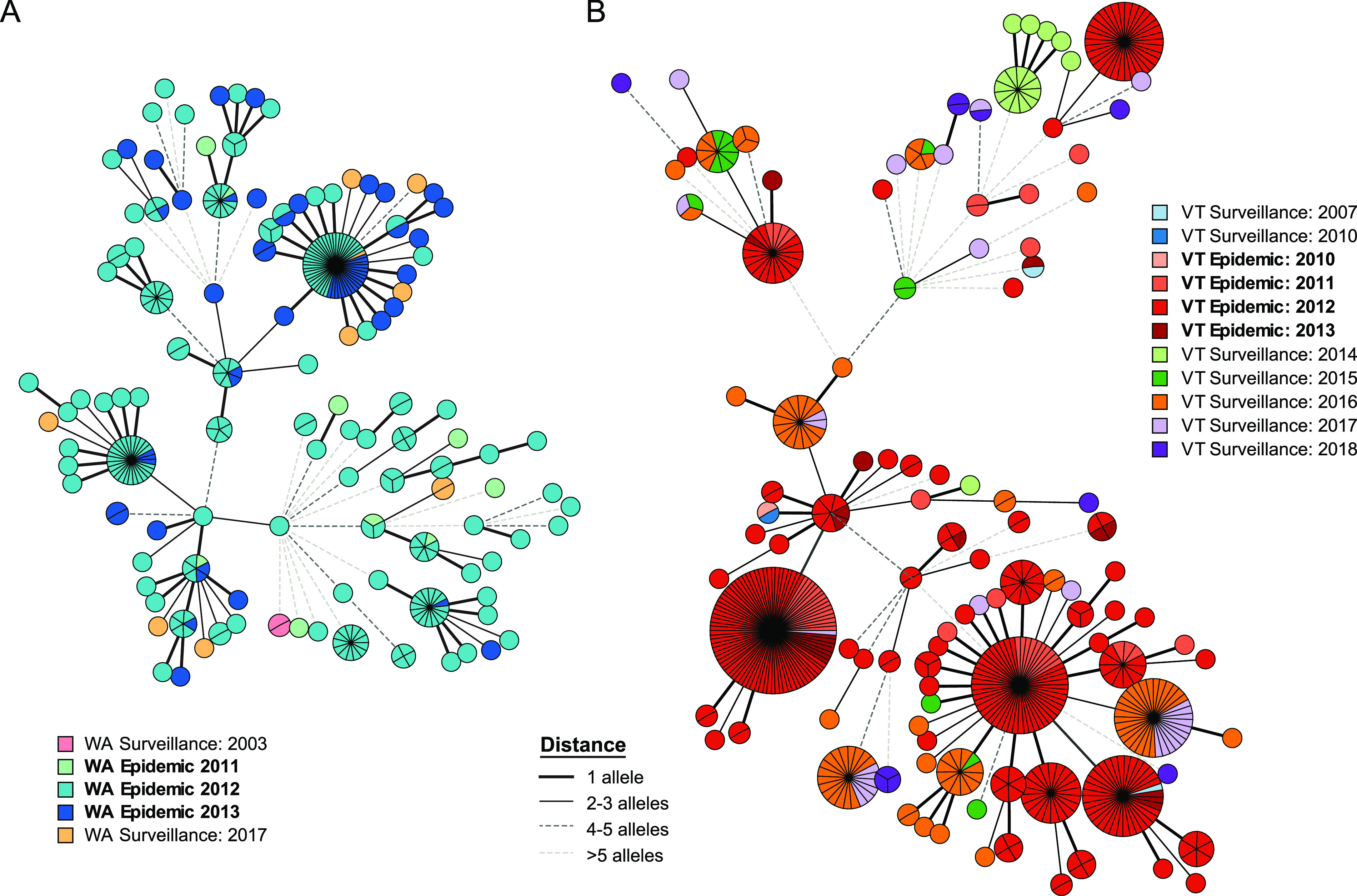
Molecular epidemiology of state-wide epidemics. Minimum spanning trees of 296 WA isolates calculated from 298 polymorphic loci (A) or 536 VT isolates calculated from 277 polymorphic loci (B) are shown. Epidemic and routine surveillance case isolates are indicated according to the key in each panel. Node size indicates abundance, and connecting lines are weighted according to allele distance as indicated in the key. Trees comparing isolates from both epidemics can be found in the supplemental material (Fig. S5).
Comparison to Institut Pasteur cgMLST scheme.
A similar core genome MLST (cgMLST) scheme for B. pertussis was recently developed at Institut Pasteur that includes 2,038 gene loci, or 1.75 Mbp (42.7%) of the average B. pertussis genome (34). The overlapping gene content shared between that cgMLST scheme and the wgMLST scheme in this study was determined by reciprocal BLASTn alignment. The two schemes shared 1,822 common loci, defined as >95% nucleotide sequence identity and >90% length overlap. Some loci in each scheme could not be directly linked, likely because the annotated genome inputs used for developing the two schemes relied on different gene prediction algorithms. Relaxing the minimum length overlap allowed matching an additional 108 shared loci. However, some predicted protein-coding gene loci in one scheme were split into two smaller genes in the other scheme. After accounting for these gene prediction artifacts, there were 1,583 unique wgMLST (45.2%) and 108 unique cgMLST (5.3%) loci that could not be matched. Thirty-three of these cgMLST loci did match predicted CDS in the input genomes in this study but were removed from the wgMLST scheme during curation. Many others aligned to predicted pseudogenes, all of which were excluded from the wgMLST scheme. Identified overlaps and unique gene loci are detailed in Data Set S1.
The resolution of cgMLST, implemented within BioNumerics to ensure consistent allele calling methodology, was tested using the same collection of high school outbreak and sporadic surveillance isolates as mentioned above. A minimum spanning tree calculated from 76 polymorphic cgMLST loci (Fig. 7) exhibited a topology similar to that determined using wgMLST (Fig. 5), with subtle differences, as expected. However, cgMLST clustered the 12 outbreak isolates with an additional three surveillance isolates that differed by up to seven alleles according to wgMLST. The two schemes were further compared using pairwise allele distances among a subset of 379 sequenced isolates selected to represent the phylogenetic breadth of the larger collection. Similarly, the schemes were concordant but wgMLST identified more allelic differences among isolates reflecting the added resolution provided by the additional loci, as expected (Fig. S6). The difference in resolution was particularly evident at shorter distances (Fig. S6C) relevant for pertussis outbreak cluster delineation, consistent with observed clustering in the retrospective analysis (for an example, see Fig. 7).
FIG 7.
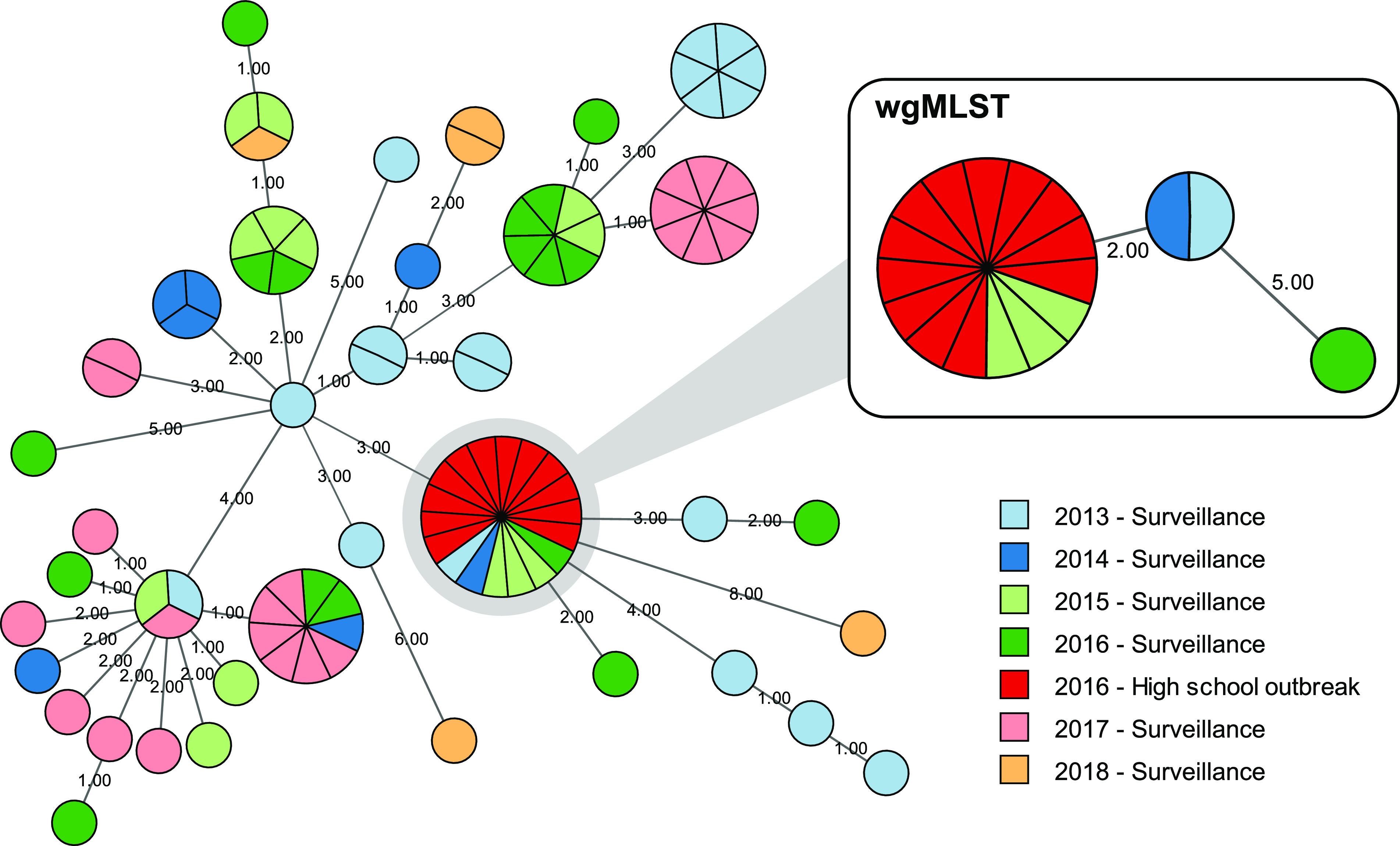
Typing resolution of cgMLST versus wgMLST. A minimum spanning tree from 76 polymorphic cgMLST loci clustered 12 case isolates from a high school outbreak with additional sporadic case isolates that were differentiated by wgMLST (inset). Outbreak and sporadic case isolates are indicated according to the key. Node size indicates abundance and connecting lines are numbered according to allele distance.
DISCUSSION
Here, we present the development and validation of a wgMLST scheme for B. pertussis, the primary agent of whooping cough. Traditional molecular methods provide little support for pertussis epidemiology, and multiple assays used in combination have been needed to identify linkages among contemporaneous cases with only limited resolution (9, 12, 16). Through retrospective analyses, the results presented here demonstrate the utility of wgMLST for strain characterization using a single, genome-based assay within a standardized platform suitable for local and state public health laboratories. Widespread implementation of WGS and wgMLST for clinical B. pertussis can promote genomic surveillance, enhance understanding of the epidemiology of pertussis, and further empower the study of pertussis resurgence.
The species B. pertussis has been frequently described as “monomorphic” for its limited genome sequence diversity and nearly fixed accessory gene content (8). While such characteristics have made the traditional molecular characterization of clinical isolates challenging, the design approach of gene-by-gene allele typing may benefit from the large core fraction and lack of detectible recombination in the B. pertussis population. As a result, the wgMLST scheme developed in this study captures the majority of protein-coding nucleotides not associated with insertion sequence element (ISE) transposases, of which the B. pertussis genome harbors >250 (∼7% of all CDS). Proliferation of these ISEs, particularly the >240 copies of IS481, facilitated genome reduction during the speciation of B. pertussis from the closely related “classic bordetellae” (40, 42, 43). Such repetitive sequences also obstruct draft genome assembly from short-read sequencing data, which critically influences allele call performance by bioinformatic tools like BioNumerics when using read formats of 100 bp or less, regardless of average sequencing depth or read quality. Accordingly, appropriate wgMLST scheme curation considers the impact of both technical variables and microbe-specific biological idiosyncrasies, such as pseudogenes, repeat polymorphisms, and ISEs in the case of B. pertussis, as well as their intersection.
The economization of high-throughput sequencing and resolution of genome-based molecular typing have revolutionized characterization of numerous microbes associated with animal and human disease (21, 23). Successfully translating these technologies into application for molecular (genomic) epidemiology requires both standardization and portability. Perhaps the most successful example is the widespread implementation of cgMLST/wgMLST, supplanting PFGE, for surveillance of foodborne pathogens (22, 44). Reported SNP phylogenetic reconstructions of B. pertussis clinical isolates have provided clear delineation of branching lineages and divergence from vaccine reference strains within the recent genomic history of B. pertussis (3, 15, 25). SNP rates in B. pertussis are low and this wgMLST scheme, like other allele-based approaches, does sacrifice some genomic resolution by excluding intergenic regions and pseudogenes. The data here highlight that wgMLST may provide sufficient resolution by capturing allele variants not resulting from single base substitutions and, therefore, provide a powerful single assay for strain typing and molecular epidemiology of B. pertussis.
A similar cgMLST scheme for B. pertussis was recently developed and reported by Institut Pasteur (34). That scheme targets genes present in nearly all isolates (“core”) in contrast to the larger wgMLST scheme in this study. Comparing the two schemes revealed that they differed beyond the number of loci and the cgMLST scheme was not simply a subset of wgMLST. Differences in input data and gene prediction algorithms used to develop the two schemes produced CDS discrepancies, highlighting subtle differences in popular gene finding approaches (45, 46). Accurate locus detection and subsequent allele calling, not just in BioNumerics, benefits from conserved start and stop codon positions (31). The comparison here suggests that wgMLST does provide additional resolution in pairwise measurements, particularly among closely related B. pertussis isolates separated by distances relevant for outbreak cluster delineation. Broad application of allele-based typing for molecular epidemiology and genomic surveillance would benefit from scheme harmonization, which will require careful modification of loci in both schemes, starting with those which overlap only under relaxed alignment parameters. Similarly, standardization of locus detection and allele assignment algorithms across software platforms is also needed for consistency. Such efforts would surely be rewarded with a more thorough database of observed, circulating allelic variation for use by varied public health institutions and researchers, including those focused on developing future pertussis vaccines.
An allele-based approach to strain characterization cannot resolve chromosome structure variation, which provides a significant source of genomic diversity among circulating B. pertussis strains (14, 15, 47). Genomes of B. pertussis clinical isolates exhibit frequent rearrangement, most often as large inversions but, more recently, also amplifications (28, 48). It remains unclear whether such structural forms of genomic variation yield phenotypes, such as varied transmission or clinical disease presentation, but observed patterns among circulating isolates compared to common reference strains suggest that they are under selection (49). These types of genomic structural features remain largely intractable, particularly by short-read sequencing platforms widespread in public health settings. However, the example retrospective data sets presented here, and previously (34), demonstrate the utility of allele-based strain typing for linking cases based on inferred ancestral relationships among recovered B. pertussis isolates. Previous comparative study of rearrangement variation among closed genome assemblies has revealed that many chromosome structures are phylogenetically restricted (15, 49), suggesting that reconstructing B. pertussis populations from polymorphic SNPs, or alleles, still captures meaningful relationships among case isolates.
Perhaps the largest barrier to widespread implementation of wgMLST (or cgMLST) for genomic surveillance of B. pertussis is the continued decline of diagnostic culture. All the data included here were derived from whole-genome sequencing of cultured isolates, but on average, fewer than 3.5% of U.S. cases captured annually by the Enhanced Pertussis Surveillance/Emerging Infections Program (EPS) yield isolates (35). In principle, wgMLST can be applied to data derived from direct—“metagenomic”—sequencing of clinical nasopharyngeal specimens, but it will likely require careful modification. Limited observation of allelic variation among replicate isolates in this study highlights that application of wgMLST to direct sequencing data will need to evaluate polymorphic loci. For example, at least some replicate isolates recovered from individual patients differed by more alleles than were used to delineate a retrospective high school outbreak. Defining cluster cutoffs for sequence-based typing is a common problem (22, 30, 32), made more challenging by within-patient sequence variability of an organism with so little diversity. Solving these challenges and successful interoperability of wgMLST and direct sequencing are likely the only way for this, or any other method of genomic surveillance, to advance the study of pertussis resurgence. Hopefully the results facilitate production of sufficient data sets to enable large-scale, integrated analysis of genomic and epidemiological data.
Supplementary Material
ACKNOWLEDGMENTS
We thank Pam Cassiday and Tami Skoff (CDC), Lingzi Xiaoli and Matt Cole (IHRC, Inc.), the CDC Biotechnology Core Facilities Branch Genome Sequencing Laboratory, The Enhanced Pertussis Surveillance/Emerging Infection Program Network, and Sylvain Brisse and Valérie Bouchez (Institut Pasteur).
This work was made possible through support from the CDC’s Advanced Molecular Detection (AMD) program.
The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention. Use of trade names and commercial sources is for identification only and does not imply endorsement by the Centers for Disease Control and Prevention, the Public Health Service, or the U.S. Department of Health and Human Services.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Clark TA. 2014. Changing pertussis epidemiology: everything old is new again. J Infect Dis 209:978–981. 10.1093/infdis/jiu001. [DOI] [PubMed] [Google Scholar]
- 2.Ausiello CM, Cassone A. 2014. Acellular pertussis vaccines and pertussis resurgence: revise or replace? mBio 5:e01339-14. 10.1128/mBio.01339-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bart MJ, Zeddeman A, van der Heide HG, Heuvelman K, van Gent M, Mooi FR. 2014. Complete genome sequences of Bordetella pertussis isolates B1917 and B1920, representing two predominant global lineages. Genome Announc 2:e01301-14. 10.1128/genomeA.01301-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bento AI, King AA, Rohani P. 2018. A simulation study on the relative role of age groups under differing pertussis transmission scenarios. bioRxiv 10.1101/247007. [DOI]
- 5.Burdin N, Handy LK, Plotkin SA. 2017. What is wrong with pertussis vaccine immunity? The problem of waning effectiveness of pertussis vaccines. Cold Spring Harb Perspect Biol 9:a029454. 10.1101/cshperspect.a029454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Warfel JM, Edwards KM. 2015. Pertussis vaccines and the challenge of inducing durable immunity. Curr Opin Immunol 35:48–54. 10.1016/j.coi.2015.05.008. [DOI] [PubMed] [Google Scholar]
- 7.Skoff TH, Hadler S, Hariri S. 2019. The epidemiology of nationally reported pertussis in the United States, 2000–2016. Clin Infect Dis 68:1634–1640. 10.1093/cid/ciy757. [DOI] [PubMed] [Google Scholar]
- 8.Mooi FR. 2010. Bordetella pertussis and vaccination: the persistence of a genetically monomorphic pathogen. Infect Genet Evol 10:36–49. 10.1016/j.meegid.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 9.Bowden KE, Williams MM, Cassiday PK, Milton A, Pawloski L, Harrison M, Martin SW, Meyer S, Qin X, DeBolt C, Tasslimi A, Syed N, Sorrell R, Tran M, Hiatt B, Tondella ML. 2014. Molecular epidemiology of the pertussis epidemic in Washington State in 2012. J Clin Microbiol 52:3549–3557. 10.1128/JCM.01189-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diavatopoulos DA, Cummings CA, Schouls LM, Brinig MM, Relman DA, Mooi FR. 2005. Bordetella pertussis, the causative agent of whooping cough, evolved from a distinct, human-associated lineage of B. bronchiseptica. PLoS Pathog 1:e45. 10.1371/journal.ppat.0010045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Loo IHM, Heuvelman KJ, King AJ, Mooi FR. 2002. Multilocus sequence typing of Bordetella pertussis based on surface protein genes. J Clin Microbiol 40:1994–2001. 10.1128/jcm.40.6.1994-2001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barkoff A-M, He Q. 2019. Molecular epidemiology of Bordetella pertussis. Adv Exp Med Biol 1183:19–33. 10.1007/5584_2019_402. [DOI] [PubMed] [Google Scholar]
- 13.Schouls LM, van der Heide HG, Vauterin L, Vauterin P, Mooi FR. 2004. Multiple-locus variable-number tandem repeat analysis of Dutch Bordetella pertussis strains reveals rapid genetic changes with clonal expansion during the late 1990s. J Bacteriol 186:5496–5505. 10.1128/JB.186.16.5496-5505.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stibitz S, Yang MS. 1999. Genomic plasticity in natural populations of Bordetella pertussis. J Bacteriol 181:5512–5515. 10.1128/JB.181.17.5512-5515.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weigand MR, Peng Y, Loparev V, Batra D, Bowden KE, Burroughs M, Cassiday PK, Davis JK, Johnson T, Juieng P, Knipe K, Mathis MH, Pruitt AM, Rowe L, Sheth M, Tondella ML, Williams MM. 2017. The history of Bordetella pertussis genome evolution includes structural rearrangement. J Bacteriol 199:e00806-16. 10.1128/JB.00806-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Advani A, Van der Heide HG, Hallander HO, Mooi FR. 2009. Analysis of Swedish Bordetella pertussis isolates with three typing methods: characterization of an epidemic lineage. J Microbiol Methods 78:297–301. 10.1016/j.mimet.2009.06.019. [DOI] [PubMed] [Google Scholar]
- 17.Cassiday PK, Skoff TH, Jawahir S, Tondella ML. 2016. Changes in predominance of pulsed-field gel electrophoresis profiles of Bordetella pertussis isolates, United States, 2000–2012. Emerg Infect Dis 22:442–448. 10.3201/eid2203.151136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barkoff A-M, Mertsola J, Pierard D, Dalby T, Hoegh SV, Guillot S, Stefanelli P, van Gent M, Berbers G, Vestrheim DF, Greve-Isdahl M, Wehlin L, Ljungman M, Fry NK, Markey K, Auranen K, He Q. 2018. Surveillance of circulating Bordetella pertussis strains in Europe during 1998 to 2015. J Clin Microbiol 56:e01998-17. 10.1128/JCM.01998-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mir-Cros A, Moreno-Mingorance A, Martín-Gómez MT, Codina G, Cornejo-Sánchez T, Rajadell M, Van Esso D, Rodrigo C, Campins M, Jané M, Pumarola T, Fàbrega A, González-López JJ. 2019. Population dynamics and antigenic drift of Bordetella pertussis following whole cell vaccine replacement, Barcelona, Spain, 1986–2015. Emerg Microbes Infect 8:1711–1720. 10.1080/22221751.2019.1694395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rocha EL, Leite D, Camargo CH, Martins LM, Silva RSN, Martins VP, Campos TA. 2017. The characterization of Bordetella pertussis strains isolated in the Central-Western region of Brazil suggests the selection of a specific genetic profile during 2012–2014 outbreaks. Epidemiol Infect 145:1392–1397. 10.1017/S0950268816003332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Armstrong GL, MacCannell DR, Taylor J, Carleton HA, Neuhaus EB, Bradbury RS, Posey JE, Gwinn M. 2019. Pathogen genomics in public health. N Engl J Med 381:2569–2580. 10.1056/NEJMsr1813907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Besser JM, Carleton HA, Trees E, Stroika SG, Hise K, Wise M, Gerner-Smidt P. 2019. Interpretation of whole-genome sequencing for enteric disease surveillance and outbreak investigation. Foodborne Pathog Dis 16:504–512. 10.1089/fpd.2019.2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Black A, MacCannell DR, Sibley TR, Bedford T. 2020. Ten recommendations for supporting open pathogen genomic analysis in public health. Nat Med 26:832–841. 10.1038/s41591-020-0935-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacCannell D. 2019. Platforms and analytical tools used in nucleic acid sequence-based microbial genotyping procedures. Microbiol Spectr 7:AME-0005-2018. 10.1128/microbiolspec.AME-0005-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Octavia S, Maharjan RP, Sintchenko V, Stevenson G, Reeves PR, Gilbert GL, Lan R. 2011. Insight into evolution of Bordetella pertussis from comparative genomic analysis: evidence of vaccine-driven selection. Mol Biol Evol 28:707–715. 10.1093/molbev/msq245. [DOI] [PubMed] [Google Scholar]
- 26.van Gent M, Bart MJ, van der Heide HG, Heuvelman KJ, Mooi FR. 2012. Small mutations in Bordetella pertussis are associated with selective sweeps. PLoS One 7:e46407. 10.1371/journal.pone.0046407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weigand MR, Peng Y, Cassiday PK, Loparev VN, Johnson T, Juieng P, Nazarian EJ, Weening K, Tondella ML, Williams MM. 2017. Complete genome sequences of Bordetella pertussis isolates with novel pertactin-deficient deletions. Genome Announc 5:e00973-17. 10.1128/genomeA.00973-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weigand MR, Williams MM, Peng Y, Kania D, Pawloski LC, Tondella ML, CDC Pertussis Working Group.. 2019. Genomic survey of Bordetella pertussis diversity, United States, 2000–2013. Emerg Infect Dis 25:780–783. 10.3201/eid2504.180812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu Y, Liu B, Grondahl-Yli-Hannuksila K, Tan Y, Feng L, Kallonen T, Wang L, Peng D, He Q, Wang L, Zhang S. 2015. Whole-genome sequencing reveals the effect of vaccination on the evolution of Bordetella pertussis. Sci Rep 5:12888. 10.1038/srep12888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moura A, Criscuolo A, Pouseele H, Maury MM, Leclercq A, Tarr C, Björkman JT, Dallman T, Reimer A, Enouf V, Larsonneur E, Carleton H, Bracq-Dieye H, Katz LS, Jones L, Touchon M, Tourdjman M, Walker M, Stroika S, Cantinelli T, Chenal-Francisque V, Kucerova Z, Rocha EPC, Nadon C, Grant K, Nielsen EM, Pot B, Gerner-Smidt P, Lecuit M, Brisse S. 2016. Whole genome-based population biology and epidemiological surveillance of Listeria monocytogenes. Nat Microbiol 2:16185. 10.1038/nmicrobiol.2016.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jolley KA, Maiden MC. 2014. Using multilocus sequence typing to study bacterial variation: prospects in the genomic era. Future Microbiol 9:623–630. 10.2217/fmb.14.24. [DOI] [PubMed] [Google Scholar]
- 32.Schürch AC, Arredondo-Alonso S, Willems RJL, Goering RV. 2018. Whole genome sequencing options for bacterial strain typing and epidemiologic analysis based on single nucleotide polymorphism versus gene-by-gene–based approaches. Clin Microbiol Infect 24:350–354. 10.1016/j.cmi.2017.12.016. [DOI] [PubMed] [Google Scholar]
- 33.Besser J, Carleton HA, Gerner-Smidt P, Lindsey RL, Trees E. 2018. Next-generation sequencing technologies and their application to the study and control of bacterial infections. Clin Microbiol Infect 24:335–341. 10.1016/j.cmi.2017.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bouchez V, Guglielmini J, Dazas M, Landier A, Toubiana J, Guillot S, Criscuolo A, Brisse S. 2018. Genomic sequencing of Bordetella pertussis for epidemiology and global surveillance of whooping cough. Emerg Infect Dis 24:988–994. 10.3201/eid2406.171464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Skoff TH, Baumbach J, Cieslak PR. 2015. Tracking pertussis and evaluating control measures through Enhanced Pertussis Surveillance, Emerging Infections Program, United States. Emerg Infect Dis 21:1568–1573. 10.3201/eid2109.150023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bowden KE, Weigand MR, Peng Y, Cassiday PK, Sammons S, Knipe K, Rowe LA, Loparev V, Sheth M, Weening K, Tondella ML, Williams MM. 2016. Genome structural diversity among 31 Bordetella pertussis isolates from two recent U.S. whooping cough statewide epidemics. mSphere 1:e00036-16. 10.1128/mSphere.00036-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gardner SN, Slezak T, Hall BG. 2015. kSNP3.0: SNP detection and phylogenetic analysis of genomes without genome alignment or reference genome. Bioinformatics 31:2877–2878. 10.1093/bioinformatics/btv271. [DOI] [PubMed] [Google Scholar]
- 39.Van Dongen S. 2008. Graph clustering via a discrete uncoupling process. SIAM J Matrix Anal Appl 30:121–141. 10.1137/040608635. [DOI] [Google Scholar]
- 40.Parkhill J, Sebaihia M, Preston A, Murphy LD, Thomson N, Harris DE, Holden MT, Churcher CM, Bentley SD, Mungall KL, Cerdeno-Tarraga AM, Temple L, James K, Harris B, Quail MA, Achtman M, Atkin R, Baker S, Basham D, Bason N, Cherevach I, Chillingworth T, Collins M, Cronin A, Davis P, Doggett J, Feltwell T, Goble A, Hamlin N, Hauser H, Holroyd S, Jagels K, Leather S, Moule S, Norberczak H, O’Neil S, Ormond D, Price C, Rabbinowitsch E, Rutter S, Sanders M, Saunders D, Seeger K, Sharp S, Simmonds M, Skelton J, Squares R, Squares S, Stevens K, Unwin L, Whitehead S, Barrell BG, Maskell DJ. 2003. Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. Nat Genet 35:32–40. 10.1038/ng1227. [DOI] [PubMed] [Google Scholar]
- 41.Martin SW, Pawloski L, Williams M, Weening K, DeBolt C, Qin X, Reynolds L, Kenyon C, Giambrone G, Kudish K, Miller L, Selvage D, Lee A, Skoff TH, Kamiya H, Cassiday PK, Tondella ML, Clark TA. 2015. Pertactin-negative Bordetella pertussis strains: evidence for a possible selective advantage. Clin Infect Dis 60:223–227. 10.1093/cid/ciu788. [DOI] [PubMed] [Google Scholar]
- 42.Linz B, Ivanov YV, Preston A, Brinkac L, Parkhill J, Kim M, Harris SR, Goodfield LL, Fry NK, Gorringe AR, Nicholson TL, Register KB, Losada L, Harvill ET. 2016. Acquisition and loss of virulence-associated factors during genome evolution and speciation in three clades of Bordetella species. BMC Genomics 17:767. 10.1186/s12864-016-3112-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park J, Zhang Y, Buboltz AM, Zhang X, Schuster SC, Ahuja U, Liu M, Miller JF, Sebaihia M, Bentley SD, Parkhill J, Harvill ET. 2012. Comparative genomics of the classical Bordetella subspecies: the evolution and exchange of virulence-associated diversity amongst closely related pathogens. BMC Genomics 13:545. 10.1186/1471-2164-13-545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nadon C, Van Walle I, Gerner-Smidt P, Campos J, Chinen I, Concepcion-Acevedo J, Gilpin B, Smith AM, Kam KM, Perez E, Trees E, Kubota K, Takkinen J, Nielsen EM, Carleton H, Panel F-NE. 2017. PulseNet International: vision for the implementation of whole genome sequencing (WGS) for global food-borne disease surveillance. Euro Surveill 22:30544. 10.2807/1560-7917.ES.2017.22.23.30544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stein L. 2001. Genome annotation: from sequence to biology. Nat Rev Genet 2:493–503. 10.1038/35080529. [DOI] [PubMed] [Google Scholar]
- 46.Lomsadze A, Gemayel K, Tang S, Borodovsky M. 2018. Modeling leaderless transcription and atypical genes results in more accurate gene prediction in prokaryotes. Genome Res 28:1079–1089. 10.1101/gr.230615.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weigand MR, Williams MM, Otero G. 2019. Temporal patterns of Bordetella pertussis genome sequence and structural evolution, p 144–165. In Rohai P, Scarpino SV (ed), Pertussis: epidemiology, immunology, & evolution. Oxford University Press, Oxford, United Kingdom. [Google Scholar]
- 48.Abrahams JS, Weigand MR, Ring N, MacArthur I, Peng S, Williams MM, Bready B, Catalano AP, Davis JR, Kaiser MD, Oliver JS, Sage JM, Bagby S, Tondella ML, Gorringe AR, Preston A. 2020. Duplications drive diversity in Bordetella pertussis on an underestimated scale. bioRxiv 10.1101/2020.02.06.937284:2020.02.06.937284. [DOI]
- 49.Weigand MR, Peng Y, Batra D, Burroughs M, Davis JK, Knipe K, Loparev VN, Johnson T, Juieng P, Rowe LA, Sheth M, Tang K, Unoarumhi Y, Williams MM, Tondella ML. 2019. Conserved patterns of symmetric inversion in the genome evolution of Bordetella respiratory pathogens. mSystems 4:e00702-19. 10.1128/mSystems.00702-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The whole-genome shotgun sequences are available from the NCBI Sequence Read Archive, organized under BioProject accession number PRJNA279196.