

Abstract
Xenopus egg extracts provide a cell-free system to analyze various aspects of chromatin biology. Here we describe a modified method of chromatin immunoprecipitation (ChIP) to detect the interaction of proteins with plasmid DNA incubated in extract. The combination of ChIP and Xenopus egg extracts provides a highly versatile and tractable approach to analyze dynamic protein-DNA interactions with great spatial and temporal detail.
Keywords: Xenopus egg extract, Nucleoplasmic extract, chromatin immunoprecipitation (ChIP), DNA repair, cell-free system
1. Introduction
Gilmour and Lis first described a method for detecting protein-DNA interactions in vivo in 1984 [1, 2]. As a proof of concept experiment, UV light was used to permanently couple proteins to DNA in Escherichia coli. The relative enrichment of RNA polymerase was then determined by immunoprecipitating the protein and hybridizing the associated DNA fragments to different DNA sequences. Current methods now utilize formaldehyde to form reversible crosslinks, allowing DNA fragments to be analyzed by quantitative PCR. Over time, chromatin immunoprecipitation (ChIP) has become an essential tool in molecular biology due to its simplicity and high sensitivity (reviewed in [3]).
Several approaches have been developed using Xenopus egg extracts to monitor various aspects of chromatin biology, including DNA replication and termination, transcription, DNA damage signaling and repair, and chromatin organization [4–7]. Protocols for different types of Xenopus egg extracts have been established, allowing preparation of different protein fractions from different stages of the cell cycle and oocyte development [8]. In this chapter, we describe a modified method of ChIP for analyzing protein interactions with plasmid DNA incubated in Xenopus egg extracts. By coupling ChIP with highly synchronized extract reactions, a detailed analysis of protein recruitment, retention, and displacement from DNA can be performed, revealing dynamic protein activity not easily observed in cells [9–12].
2. Materials
Prepare all solutions using analytical grade reagents and ultrapure water (prepared by purifying deionized water to a sensitivity of 18 MΩ-cm at 25°C).
2.1. Reagents
Protein A-Sepharose Fast Flow (PAS-FF) beads. Beads are washed with 1× PBS and stored as a 33% slurry with 0.01% Sodium Azide at 4°C.
0.5 and 1.5 mL siliconized Microfuge tubes
50 mL conical tubes
Bio-Rad Bio-Spin Columns
Antibodies (commercial or serum) (see Note 1)
Ultrafine pipette tips
2 mg/mL RNase
20 mg/mL Pronase
20 mg/mL Glycogen
Oligonucleotide primers (see Step 3.10-1)
384-well PCR plates and plate sealing films
2.2. Buffers and Solutions
1x Phosphate-Buffered Saline (PBS): 10 mM Na2HPO4, 1.8 mM KH2PO4, 137 mM NaCl, and 2.7 mM KCl in water.
1x Egg Lysis Buffer (ELB): 250 mM sucrose, 2.5 mM MgCl2, 50 mM KCl, 10mM HEPES pH 7.8. Filter sterilize and store at 4°C.
ATP Regeneration Mix (ARM): 64.5 mM ATP, 645 mM Phosphocreatine (PC), 2 μM Creatine Phosphokinase (CPK).
Formaldehyde Crosslinking Buffer: 2% formaldehyde in 1x ELB.
Glycine Buffer: 1.25 M glycine in water, pH 7.
PMSF solution: 100 mM Phenylmethylsulfonyl (PMSF) in 100% ethanol.
Lithium Chloride solution: 4 M LiCl in water. Dissolve slowly, as solution becomes very hot.
Aprotinin/Leupeptin: 5 mg/mL Aprotinin, 5 mg/mL Leupeptin.
2x Sonic Buffer: 40 mM Tris-HCl (pH 7.5), 300 mM NaCl, 4 mM EDTA, and 1% NP-40 in water.
1x Sonic Buffer: 50% 2x Sonic Buffer, 2 mM PMSF, 5 μg/mL Aprotinin/Leupeptin (see Note 2) in water.
1x Sonic Salts Buffer: 50% 2x Sonic Buffer, 2 mM PMSF, 5 μg/mL Aprotinin/Leupeptin (see Note 2), 650 mM NaCl, 100 mM KCl in water.
ChIP Wash Buffer: 10 mM Tris-HCl (pH 7.5), 0.25 M LiCl, 1 mM Ethylenediaminetetraacetic Acid (EDTA) pH 8, 0.5% NP-40, 0.5% SDS.
TE Buffer: 10 mM Tris-HCl (pH 7.5), 1 mM EDTA pH 8.
ChIP Elution Buffer: 50 mM Tris-HCl (pH 7.5), 10 mM EDTA pH 8, 1% SDS.
Sodium Acetate solution: 3 M NaAc (pH 5.2).
5M NaCl in water.
De-crosslinking Mix: 70 μL TE Buffer, 20 μL Pronase (20 mg/mL), 10 μL 5M NaCl in water.
1-to-1 Phenol/Chloroform solution: Prepare by carefully mixing 1.25 volumes of buffered phenol solution with 1 volume of chloroform. Once the two are mixed, the solution should settle into ~2 volumes of Phenol:Chloroform and 0.25 volumes of aqueous buffer.
100% and 70% Ethanol.
Applied Biosystems Power-Up SYBR Green master mix or similar (containing SYBR Green dye, DNA Polymerase, and dNTP mix).
2.3. Equipment
Diagenode Bioruptor UCD-600 TS or similar sonicator
Rotating wheel or rotisserie
Thermocycler (or heat block)
Bio-Rad Real-time PCR machine CFX-384 or similar
Horizontal centrifuge drum rotor (See Note 3)
Swinging platform microfuge to spin down PCR plates
3. Methods
3.1. Preparation
Aliquot 48 μL of Formaldehyde Crosslinking Buffer into 0.5 mL microfuge tubes for each sample. Cap tubes and store at room temperature (RT).
Prepare a Bio-Spin column with provided wash and collection tubes for each sample. According to the manufacturer’s instructions: Suspend resin by inverting each column several times. Loosen the cap of the column and then snap off the bottom to allow drainage of column storage buffer. Allow columns to drain into wash tube at 4°C until needed (see Step 3.3-3.
3.2. Replication Reactions
Replication of plasmid DNA in High-Speed Supernatant (HSS) and NucleoPlasmic Extract (NPE) can be performed as described (see [8]).
Thaw and prepare extracts on ice. Supplement HSS with ARM (0.6 μL ARM per 20 μL HSS; final concentration 18 mM PC, 1.8 mM ATP, 0.057 μM CPK) and 10 μM Nocodazole. Supplement NPE with ARM (0.6 μL ARM per 20 μL NPE; final concentration 18 mM PC, 1.8 mM ATP, 0.057 μM CPK), 10 μM DTT, and ELB. Add plasmid DNA to HSS at 7.5 ng/μL (see Note 4) and incubate at RT for 20 min to allow formation of pre-Replication Complexes (pre-RCs).
After pre-RC formation, Add 2 volumes of NPE mixture to 1 volume of HSS/plasmid DNA for a final concentration of 2.5 ng/μL. Mix thoroughly by pipetting up and down or gently flicking the tube several times. For most experiments, addition of NPE is considered to be the start of the reaction.
3.3. Formaldehyde Crosslinking
For each condition or time point to be analyzed by ChIP, remove 2 μL of reaction sample and add to a prepared 0.5 mL siliconized microfuge tube containing 48 μL of Formaldehyde Crosslinking Buffer (see Note 5) (Figure 1). Mix thoroughly by pipetting the Formaldehyde Crosslinking Buffer up and down or gently flicking the tube several times. Incubate samples in Formaldehyde Crosslinking Buffer at RT for exactly 10 min.
During the Formaldehyde Crosslinking Buffer incubation, finish preparing a Bio-Spin column for each sample being crosslinked. Decant flowthrough storage buffer from the wash tube and remove residual storage buffer by spinning the column at 1,000 RCF for 2 min. Discard the wash tube and place the dried column into a collection tube (see Note 6).
Immediately after 10 min Formaldehyde Crosslinking Buffer incubation, quench the crosslinking reaction with 5 μL of Glycine Buffer (see Note 7). After glycine addition, mix samples by pipetting up and down twice and apply to the resin of the dried Bio-Spin column.
Spin samples through the Bio-Spin column at 1,000 RCF for 4 min. Discard the column and add 950 μL of 1x Sonic Buffer (made fresh with PMSF and Aprotinin/Leupeptin) to the eluate, mix gently by inversion, and store on ice until sonication.
Figure 1.
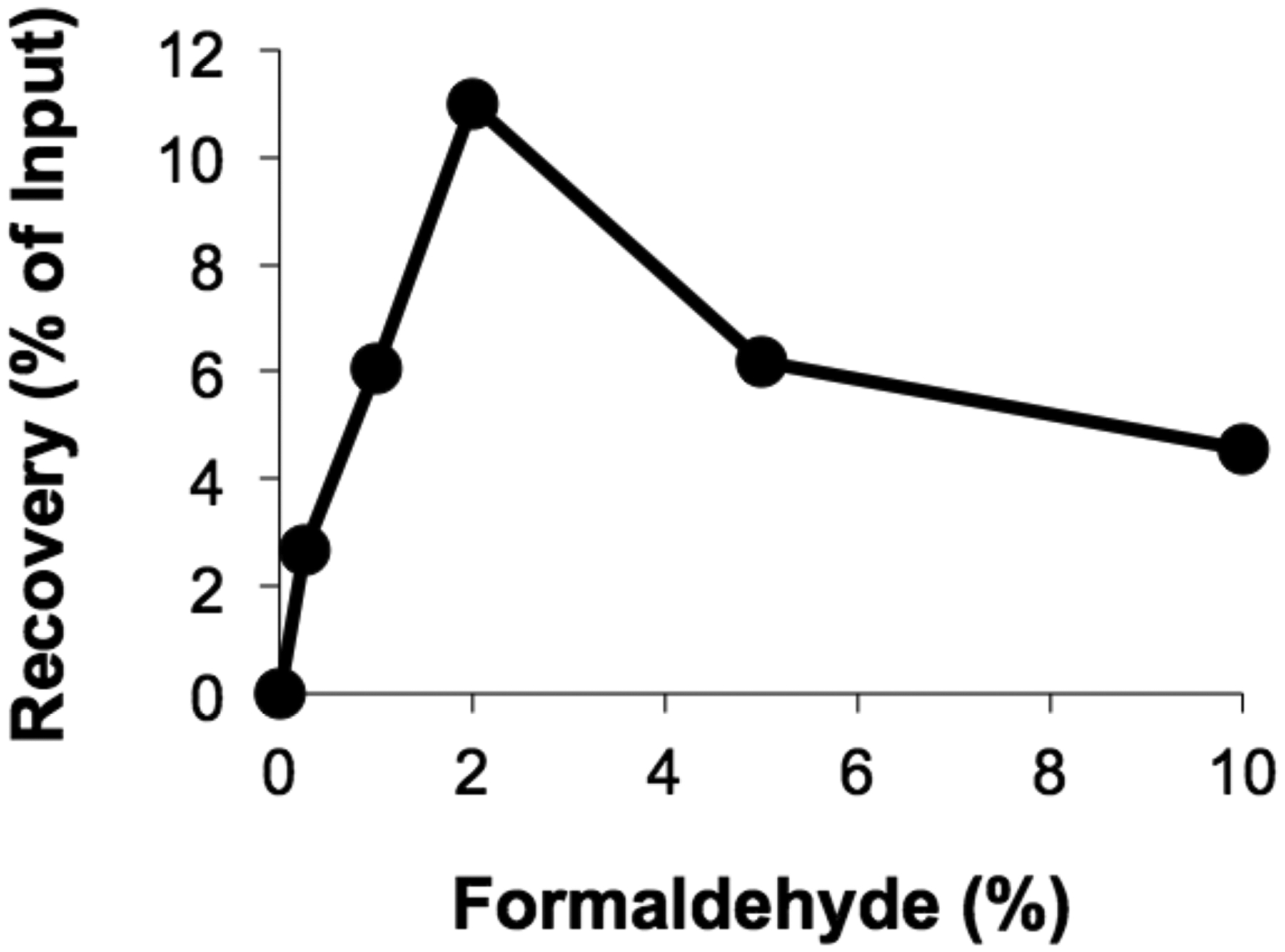
Crosslinked plasmid DNA was replicated for 60 minutes in HSS/NPE and then incubated with a range of formaldehyde concentrations in the Formaldehyde Crosslinking Buffer (see Step 3.3-1) prior to immunoprecipitation with anti-RPA antibodies [9]. Percent recovery of DNA near the crosslink is plotted for each formaldehyde concentration tested.
3.4. Sonication
Transfer 100 μL of each sample to a 0.5 mL siliconized microfuge tube for sonication. The rest of the sample may be aliquoted and snap frozen for storage at −80°C (See Note 8).
Briefly spin samples to remove bubbles and collect liquid from the sides of the tube.
Sonicate samples using the Diagenode Bioruptor or similar sonicator to produce DNA fragments ~300–500 bp in length (Figure 2). For the Diagenode Bioruptor, pre-chill the water bath to 4°C, and sonicate for 25 cycles: 30 seconds ON, 60 seconds OFF, at HIGH power. The total run time is about 35 min.
Figure 2.

250 ng of Plasmid DNA (5.2 kb in size) was sonicated using a Diagenode Bioruptor for increasing cycles of: 30 seconds ON, 60 seconds OFF, at HIGH power (see Step 3.4-3). Samples were then separated by 1% agarose gel electrophoresis and visualized by ethidium bromide staining. M, GeneRuler 1 KB plus DNA ladder.
3.5. Antibody Incubation
Transfer 10 μL from each sonicated sample into a separate 0.5 mL siliconized microfuge tube and store at 4°C for “INPUT” samples (see Step 3.7-4 below).
Add 0.5–2 μg of antibody to the remaining 90 μL of each sonicated sample for immunoprecipitation (IP) (see Note 9) (Figure 3).
Make sure all tubes are capped tightly and place them into a 50 mL conical or other container. A balled up tissue or paper towel can be used to fill extra space and prevent tubes from sliding around and possibly coming open.
Incubate IP samples at 4°C overnight using a rotating wheel or rotisserie.
Figure 3.
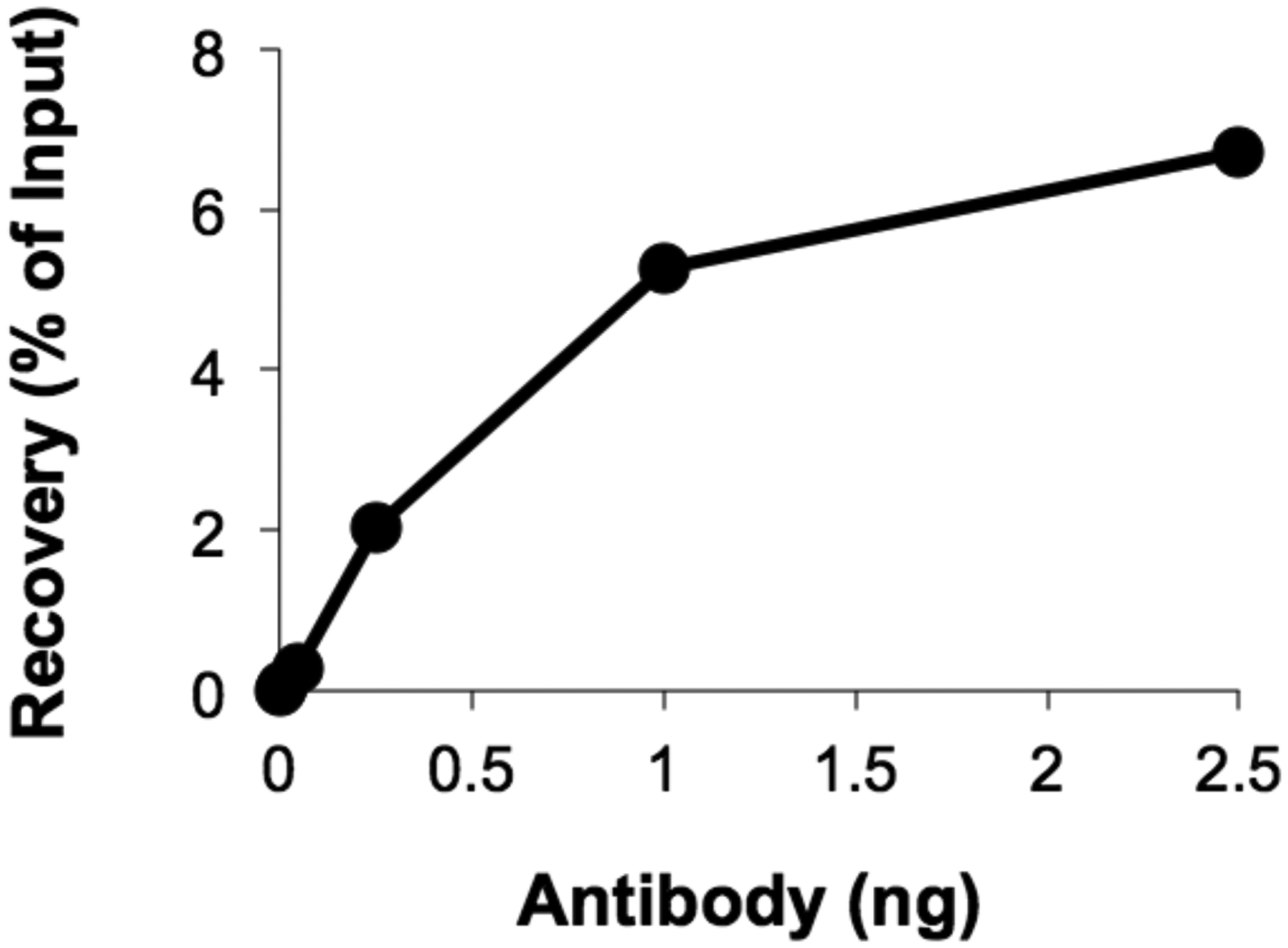
Crosslinked plasmid DNA was replicated for 60 minutes in HSS/NPE and then immunoprecipitated with an increasing amount of anti-RPA antibody (see Step 3.5-2). Percent recovery of DNA near the crosslink is plotted for each amount of antibody tested.
3.6. Antibody Coupling to Beads
Prepare 10 μL of beads for each IP by collectively washing ~1.2x the total amount of beads needed (see Note 10).
Resuspend 33% PAS-FF bead slurry by inverting the tube several times. For each IP, remove ~36 μL of slurry and transfer to a 1.5 mL microfuge tube using a wide-bore or cut pipette tip (e.g. For 6 IPs, remove 216 μL of 33% slurry for 72 μL of beads total).
Spin down beads at 1,500 RCF for 60 sec in a horizontal drum rotor. Remove the supernatant by aspirator or pipette, but do not completely dry beads throughout washes. Adding Sonic buffer to dry beads will create bubbles, making it difficult to aliquot.
Wash PAS-FF beads by resuspending with 1 mL 1x Sonic Buffer, spinning at 1,500 RCF for 60 sec, and removing supernatant. Repeat two times for three total washes.
After the final wash, add an equal volume of 1x Sonic Buffer to beads for a 50% bead slurry (e.g. For 6 IPs, add 72 μL of 1x Sonic Buffer to ~72 μL of washed beads).
Resuspend beads by pipetting up and down or inverting the tube several times. Using a fresh wide-bore or cut pipette tip to prevent clogging and inconsistent results, transfer 20 μL of 50% bead slurry (10 μL of beads) to each IP sample.
Make sure all tubes are capped tightly and place them into a 50 mL conical.
Incubate IP samples at RT for 2 hours using a rotating wheel or rotisserie (see Note 11).
3.7. Bead Washing and Elution
Spin down beads at 1,500 RCF for 60 sec in a horizontal drum rotor. Remove the supernatant by aspirator or pipette, but do not completely dry beads throughout washes.
- For each wash, resuspend PAS-FF beads with 450 μL of the appropriate buffer (see below). Cap tubes tightly, place them into a 50 mL conical, and rotate at RT for 5 min (see Note 12). Spin tubes at 1,500 RCF for 60 sec and remove supernatant. Wash once with each of the following buffers, totaling four washes:
- 1x Sonic Buffer (made fresh with PMSF and Aprotinin/Leupeptin)
- 1x Sonic Salts Buffer (made fresh with PMSF and Aprotinin/Leupeptin)
- 1x ChIP Wash Buffer
- TE Buffer
After the final wash, use an ultrafine tip to remove all traces of buffer from the beads and resuspend beads with 115 μL of ChIP elution buffer.
Add 90 μL of ChIP elution buffer to the INPUT samples from Step 3.5-1.
Incubate both IP and INPUT samples at 65°C for 20 min using a heat block or thermocycler.
Add 1 μL of RNase (2 mg/mL stock) to INPUT samples and incubate at 37°C for 30 min (see Note 13).
3.8. De-crosslinking
Aliquot 100 μL of de-crosslinking mix into a new 0.5 mL siliconized microfuge tube for each IP sample.
For IP samples, spin at 1,500 RCF for 1 min to pellet beads. Transfer 100 μL of the supernatant (eluted IP sample) to the prepared 0.5 mL siliconized microfuge tubes containing 100 μL of de-crosslinking mix.
For INPUT samples, add 100 μL of de-crosslinking mix directly to the tubes containing 10 μL of sonicated sample and 90 μL of ChIP elution buffer.
Incubate IP and INPUT samples at 42°C for 6 hours (protease digestion) and 70°C for 9 hours (de-crosslinking) using a thermocycler or heat block. Samples can be stored at 4°C for several days until proceeding to DNA extraction.
3.9. DNA Extraction and Precipitation
Add 150 μL of 1:1 Phenol:Chloroform solution to each IP and INPUT sample. Cap tubes tightly, mix by inverting the tube several times, and then spin in a microfuge at 16,000 RCF for 5 min.
For each sample, transfer as much of the aqueous (upper) layer as possible to a new 0.5 mL siliconized microfuge tube containing 150 μL of Chloroform. Cap tubes tightly, mix by inverting the tube several times, and then spin in a microfuge at 16,000 RCF for 5 min.
For each sample, transfer 180 μL of the aqueous (upper) layer to a new 1.5 mL microfuge tube containing 18 μL 3M NaOAc (pH 5.5) and 2 μL Glycogen (20 mg/mL) (see Note 14).
Add 525 μL of ice-cold 100% EtOH to each tube, mix by inverting the tube several times, and then incubate on ice for 15 min.
Spin samples in a microfuge at 16,000 RCF for 30 min at 4°C.
Remove all but ~25 μL of the supernatant by aspirator or pipette.
Add 300 μL of ice-cold 70% EtOH to each tube and spin in a microfuge at 16,000 RCF for 10 min.
Carefully remove all of the supernatant using a pipette without disturbing the pellet.
Air-dry the pellets at RT with tubes uncapped for 10 min to remove residual ethanol. Pellets will appear translucent when dry.
Add 40 μL of 10 mM Tris-HCl, pH 7.5 to each pellet. Resuspend pellets by gently flicking the tube several times, and then spin briefly in a microfuge to collect liquid from the sides of the tube. Store DNA samples at −20°C and avoid multiple freeze/thaw cycles to prevent degradation (see Note 15).
3.10. Quantitative PCR
- To analyze recovery of DNA by quantitative real-time PCR, prepare an RT Master Mix for each primer pair to be tested. In general, primers should be 17–25 bases in length, have a melting Temperature of ~60°C, a G/C content of ~50%, and produce an amplification product ~100 bp in size (see Note 16). For analysis using a 384-well plate, prepare mixes based on the following single reaction size volumes:
- 5 μL of 2X SYBR Green Master Mix
- 0.5 μL Forward primer (10 μM)
- 0.5 μL Reverse primer (10 μM)
- 2 μL water
First, aliquot 8 μL of RT Master Mix into each well needed to analyze: IP and INPUT samples, a dilution series of plasmid DNA (standard curve), and a no-template control. Each sample should be loaded in triplicate.
Next, add 2 μL of each DNA sample or 10 mM Tris-HCl, pH 7.5 for the no-template control.
Seal the plate using optically clear PCR plate sealing film and spin in a swinging platform microfuge at 100 RCF for 1 min to collect liquid from the sides of the well.
- Load the plate onto a quantitative real-time PCR machine and analyze using SYBR Detection (520 nm) with the following conditions for reaction cycle (Stage 1–2) and melting curve (Stage 3–4):
• Stage 1 95°C 10 min (Denature) • Stage 2 (x45 cycles) 95°C 10 sec (Denature) 60°C 15 sec (Annealing) 72°C 30 sec (Extension, Fluorescence Reading) • Stage 3 95°C 15 sec (Denature) • Stage 4 (x70 cycles) 60°C 15 sec (Annealing, Fluorescence Reading) +0.5°C each cycle
3.11. Results Analysis
Use a standard curve (Figure 4) to determine the absolute starting quantity of DNA recovered for each IP and INPUT reaction.
Determine the median starting quantity for each IP and INPUT sample set run in triplicate.
Calculate the recovery of IP samples as a percentage of INPUT: Percent of INPUT = (90 μL) × (median IP quantity) / (10 μL) × (median INPUT quantity) × 100 (Figure 5A and B).
To compare recovery between different IPs, percent of INPUT values can be normalized by setting the peak value as 100% (Figure 5C).
Figure 4.

Representative standard curves are shown using primer pairs adjacent to a DNA inter-strand crosslink (ICL) or on the opposite side of the plasmid (FAR). Plasmid DNA concentrations tested range from 2.5×10−1 to 2.5×10−7 μM. The quantitation cycle (Cq) indicates the cycle number where the curvature of the amplification curve is maximal.
Figure 5.

Crosslinked plasmid DNA was incubated in HSS/NPE and samples were withdrawn at different time points for analysis by ChIP with anti-RPA (A) and anti-Histone H3 (B) antibodies. Percent recovery of DNA is shown for both the ICL (dark red and blue traces) and FAR (light red and blue traces) regions of DNA. (C) Relative recovery of RPA and Histone H3 IPs was normalized based on peak values in each time course and graphed together for comparison.
4. Notes
Both commercial antibodies and antiserum can be used for immunoprecipitation. To achieve consistent results, total IgG antibodies should be purified from the antiserum.
PMSF, Aprotinin, and Leupeptin are added to buffers “fresh” and solutions should be kept on ice.
A standard microfuge can also be used, but does not pellet beads to the bottom of tubes. After spinning in a fixed angle microfuge, allow beads to settle before removing supernatant during wash steps.
Plasmid DNA concentration can be changed depending on the reaction conditions and experimental setup. Plasmids used here were isolated from bacterial cultures and purified by standard miniprep spin columns.
Larger reaction samples can be processed by scaling the sample and Formaldehyde Buffer incubation up to the capacity of the Bio-Spin column or by collecting additional samples for the same condition and processing them separately.
If collecting multiple time points in rapid succession, several spin columns can be decanted in advance. However, they should not sit dry for more than ~1 hour.
Consistency is crucial for formaldehyde incubations. Add glycine to cross-linked samples exactly 10 min after they are added to formaldehyde. For extremely rapid time points, multiple samples can also be treated with Glycine successively and then stored at RT for a short time (up to ~20 min) before being processed together. Incomplete mixing or differences in the duration of Formaldehyde incubation will change the efficiency of crosslinking and sample recovery.
Samples may degrade with repeated freeze-thaw cycles, which will reduce recovery by immunoprecipitation.
Less sample can be used for antibodies that have good recovery efficiency. In general, the amount of antibody and sample required for efficient IP must be tested empirically.
10 μL of PAS-FF beads are used per IP to improve recovery after washes. The binding capacity of 10 μL of PAS-FF beads greatly exceeds what is needed for antibody binding.
Incubation times will change the level of background observed, so be sure to incubate each “set” of IPs together.
The 5 min bead washing steps can be adjusted anywhere from 3–10 min, allowing you to wash 2–3 batches of samples consecutively. However, bead wash times should be kept consistent between samples.
RNA is removed from IP samples during wash steps and does not typically impact qPCR. However, RNAse can be added to all samples if desired.
By removing a fixed volume from each extraction sample, results are more precise and reproducible.
For studies of inter-strand crosslink (ICL) repair, resuspended DNA fragments are digested with BbsI, which cuts immediately adjacent to the crosslink, to allow efficient amplification of DNA near the ICL.
Compared to cell lysates, Xenopus egg extracts are relatively free of nucleic acids, so designing specific primers is straightforward. However, each primer pair must be tested empirically to ensure specific amplification of the DNA region of interest in prepared samples.
Acknowledgements
This work was supported by NIH grant R35GM119512 to D.T.L. and T32 CA193201 to K.B.
References
- 1.Gilmour DS and Lis JT (1985) In vivo interactions of RNA polymerase II with genes of Drosophila melanogaster. Mol Cell Biol 5(8): 2009–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gilmour DS and Lis JT (1984) Detecting protein-DNA interactions in vivo: distribution of RNA polymerase on specific bacterial genes. Proc Natl Acad Sci U S A 81(14): 4275–4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Collas P (2010) The current state of chromatin immunoprecipitation. Mol Biotechnol 45(1): 87–100. [DOI] [PubMed] [Google Scholar]
- 4.Bernis C and Forbes DJ (2014) Analysis of nuclear reconstitution, nuclear envelope assembly, and nuclear pore assembly using Xenopus in vitro assays. Methods Cell Biol 122: 165–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Halpin D, et al. (2011) Mitotic spindle assembly around RCC1-coated beads in Xenopus egg extracts. PLoS Biol 9(12): e1001225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gillespie PJ, Gambus A, and Blow JJ (2012) Preparation and use of Xenopus egg extracts to study DNA replication and chromatin associated proteins. Methods 57(2): 203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eisenhardt N, Schooley A, and Antonin W (2014) Xenopus in vitro assays to analyze the function of transmembrane nucleoporins and targeting of inner nuclear membrane proteins. Methods Cell Biol 122: 193–218. [DOI] [PubMed] [Google Scholar]
- 8.Lebofsky R, Takahashi T, and Walter JC (2009) DNA replication in nucleus-free Xenopus egg extracts. Methods Mol Biol 521: 229–252. [DOI] [PubMed] [Google Scholar]
- 9.Long DT, et al. (2014) BRCA1 promotes unloading of the CMG helicase from a stalled DNA replication fork. Mol Cell 56(1): 174–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park J, et al. (2013) The MCM8-MCM9 complex promotes RAD51 recruitment at DNA damage sites to facilitate homologous recombination. Mol Cell Biol 33(8): 1632–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Long DT, et al. (2011) Mechanism of RAD51-dependent DNA interstrand cross-link repair. Science 333(6038): 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fullbright G, et al. (2016) p97 Promotes a Conserved Mechanism of Helicase Unloading during DNA Cross-Link Repair. Mol Cell Biol 36(23): 2983–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]