

SUMMARY
Glaucoma is a neurodegenerative disease that features the death of retinal ganglion cells (RGCs) in the retina, often as a result of prolonged increases in intraocular pressure. We show that preventing the formation of neuroinflammatory reactive astrocytes prevents the death of RGCs normally seen in a mouse model of glaucoma. Further, we show that these spared RGCs are electrophysiologically functional and thus still have potential value for the function and regeneration of the retina. Finally, we demonstrate that the death of RGCs is dependent on a combination of both injury to the neurons as well as the presence of reactive astrocytes, suggesting a model that may explain why reactive astrocytes are toxic in some circumstances but not in others. Together, these findings highlight reactive astrocytes as drivers of RGC death in a chronic neurodegenerative disease of the eye.
Keywords: astrocytes, glia, glaucoma, neurodegeneration, neuroprotection, optic nerve crush, astrogliosis, reactive astrocyte
eTOC Blurb
Guttenplan et al. show that dampening the formation of reactive astrocytes prevents neuronal death following an acute optic nerve crush or in a chronic model of glaucoma, with spared neurons remaining electrophysiologically functional. More generally, they show that injury is required to induce neurons to become susceptible to astrocyte-mediated toxicity.
Graphical Abstract
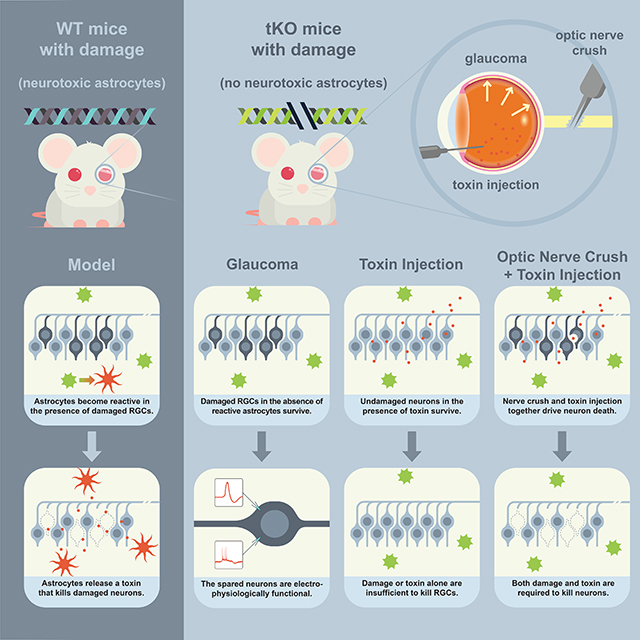
Introduction
Glaucoma is a neurodegenerative disease marked by axonal damage of retinal ganglion cells (RGCs) that results in RGC death and vision loss. While we have long known the identity of the cells that degenerate in the disease, we still do not understand how or why these cells die. Considerable work into the mechanisms of glaucoma has been conducted in rodent models, with most leveraging an increase in fluid volume in the eye to increase intraocular pressure (Almasieh & Levin, 2017, Moon et al., 2005). These models include inherited models, such as DBA/2 mice with mutations in Tyrp1 and Gpnmb that reduce aqueous outflow (John et al., 1998), and a variety of experimental approaches that physically obstruct the fluid channels of the eye (Morrison et al., 1997, Ueda et al., 1998, Sappington et al., 2010; Chen et al., 2011, El-Danaf & Huberman, 2015). The resulting increase in intraocular pressure (IOP) leads to RGC death and axonopathy, pathology that closely mimics the progression and cell-type specificity of human glaucoma. As in many neurodegenerative diseases, dysfunction in mouse models of glaucoma is coupled with a response of the surrounding glial cells in the retina and optic nerve known as gliosis, usually measured by increases in the protein and transcript of glial fibrillary acidic protein (GFAP; Inman & Horner, 2007, Sun et al., 2017, Wang et al., 2017). Importantly, this response increases with disease progression (Bosco et al., 2016), suggesting a mechanistic link between gliosis and degenerative pathology.
We previously reported that microglia and astrocytes interact in response to acute injury to the optic nerve and in chronic neurodegenerative disease to produce a neurotoxic reactive astrocyte phenotype (Liddelow et al., 2017). This response by astrocytes is largely mediated by the microglial release of interleukin 1 alpha (IL-1α), tumor necrosis factor alpha (TNFα), and the classical complement component C1q. Together, these three factors are necessary and sufficient to induce neuroinflammatory reactive astrocytes, which can then release of an as-of-yet-unidentified toxin to specifically kill neurons and mature oligodendrocytes. We have also shown that crushing the optic nerve is sufficient to induce both a pro-inflammatory microglial state and this neuroinflammatory reactive astrocyte phenotype. Blocking the activation of these astrocytes (while maintaining an otherwise ‘normal’ microglia response) using either neutralizing antibodies to IL-1α, TNFα, and C1q or an Il1a−/−Tnf−/−C1qa−/− triple knock-out mouse line preserves RGC viability following axotomy. Thus, neurotoxic reactive astrocytes appear to contribute to neuronal death following acute injury in the optic system.
Here we investigate whether astrocytes drive neuronal death in chronic neurodegenerative diseases of the eye as they do in response to acute axotomy of RGCs. We show that the bead-injection model of glaucoma induces a neurotoxic reactive astrocyte phenotype that is paired with loss of RGCs. We also show that blocking this reactive phenotype is sufficient to preserve RGC numbers. Given recent advances in encouraging the regrowth of axotomized RGCs (Park et al., 2008), we also ask whether neurons preserved from death in this model are still electrically active and thus of potential benefit to the regeneration of the retina. We perform electrophysiological and morphological analysis of RGCs protected from death following a glaucoma model and find that they are altered but still largely functional, with generally appropriate dendritic stratification and electrical response properties. Together, these findings suggest that reactive astrocytes are a potentially powerful target for therapeutic intervention in both acute and chronic injuries to the eye, and that preventing the death of RGCs may provide a useful platform for the regeneration of the retina.
Results
Retinal ganglion cells die following acute axonal injury
We first investigated the timing of RGC death following acute axotomy using the retro-orbital optic nerve crush (ONC) model (Frank & Wolburg, 1996; Tang et al., 2011). We previously reported that ONC induces reactive astrogliosis and that reducing astrogliosis prevents RGC death in this model (Liddelow et al., 2017). To better understand the mechanism of cell death following axotomy and to assess cell death at a time point comparable to mouse models of glaucoma, we performed a more detailed time-course of RGC death following ONC in both mouse and rat. In both animal models, RGC death began at 7 days post injury and progressively worsened through 28 days post injury (Figure 1).
FIGURE 1. Reactive astrocytes drive death of retinal ganglion cells.

A. RBPMS (RNA-binding protein with multiple splicing, an RGC marker) immunostaining of whole-mount mouse retinas showing decreased number of retinal ganglion cells (RGCs) following optic nerve crush (ONC) at 28 days, which was prevented in Il1a−/−Tnf−/−C1qa−/− (tKO) mice. B-C. Quantification of RGC survival 1,3, 7,14, and 28 days following optic nerve crush in wild-type (B) and tKO (C) mice. D. RBPMS immunostaining of whole-mount rat retinas showing decreased number of RGCs 28 days following ONC and IgG control antibody injection (E) which was prevented with Il-1α, TNFα, and C1q neutralizing antibody treatment (F). Data normalized to contralateral un-manipulated eye. * P < 0.05, one-way ANOVA followed by Tukey’s multiple comparisons test. Individual data points are plotted and represent individual animals, while bars represent mean ± s.e.m. Scale bar is 20 μm for all micrographs in A/D.
To confirm the role of reactive astrocytes in the death of RGCs after axotomy, we performed ONC in the absence of IL-1⍺, TNF⍺, and C1q, three factors secreted from microglia that are necessary to induce neuroinflammatory astrocyte reactivity (Liddelow et al., 2017). In mouse, we performed ONCs in an Il1a−/−Tnf−/−C1qa−/− triple knockout (tKO) generated in a previous study, as these mice fail to produce neurotoxic reactive astrocytes following activation of microglia (Figure 1A-C, Liddelow et al., 2017). In rat, we performed ONCs while simultaneously injecting neutralizing antibodies against IL-1⍺, TNF⍺, and C1q or IgG control antibodies into the vitreous of the eye (Figure 1D-F). In both animal models, RGC viability was maintained in the absence of functional IL-1⍺, TNF⍺, and C1q signaling at all time points tested, further highlighting reactive astrocytes as drivers of neuronal death seen following acute injury (Figure 1).
The bead injection glaucoma model induces gliosis
We next asked whether astrocytes drive neuronal death following chronic central nervous system (CNS) injury as they do following acute injury. A sustained increase in intraocular pressure, the pathogenic mechanism in glaucoma, leads to the progressive death of retinal ganglion cells in rodent models. In microbead occlusion models of glaucoma, ⩽15μM beads are injected into the anterior chamber of the eye, disrupting aqueous outflow and increasing intraocular pressure (IOP). We performed microbead occlusions in WT and tKO mice to compare the response of the retina to prolonged increases in IOP in the presence or absence of neuroinflammatory reactive astrocytes.
Two major responses of ocular pressure were observed in microbead occlusion retinas. Some animals showed a sustained increase in IOP, with average daily pressures significantly higher than the uninjected contralateral control eye at 7 days post injection (Figure 2A,B). Other animals showed a temporary increase in IOP that quickly resolved to baseline by 7 days post injection (Figure 2C,D). Importantly, maximum increases in IOP were similar in both WT and Il1a−/−Tnf−/−C1qa−/− mice and in mice with both sustained and transient changes in pressure (Figure 2E, Supp. Figure S1).
FIGURE 2. Bead injection glaucoma model increases intraocular pressure and causes astrocyte-dependent RGC loss.

A-D. Bead injection glaucoma produced increases in intraocular pressure (IOP) in wildtype (WT, A) and triple knock-out mice (tKO, Il1a−/−Tnf−/−C1qa−/−, B). Data in C/D are normalized around the day of peak (P) IOP increase. Refer to Supp. Figure S1 for individual animal plots. E. IOP maximum increase was approximately 20–30% in both wildtype (WT) and tKO mice. Similar maximal levels were reported in mice that had a sustained (S) increase in IOP (A/B), or transient (T) increase (C/D). F. Representative RBPMS+ staining of whole mount retinas from WT and tKO animals following sustained and transient IOP increase. Quantified in (G/I). G. IOP increase paired with death of RBPMS+ retinal ganglion cells in WT but not tKO mice following bead occlusion (compared to contralateral eye). There was a particularly large drop in SMI-32+ retinal ganglion cells (H). I. Mice with transient increase in IOP (C/D) had no loss of RGCs. J. Heat map of z-scores from microfluidic qPCR analysis highlights enrichment of reactive astrocyte transcripts in retina, optic nerve head, and optic nerve of bead-injected but not PBS-injected eyes of WT mice (largely absent in bead-injected eyes from tKO mice). K-N. Analysis of average fold induction for all reactive astrocyte transcripts (taken from J) highlights retinas from mice with transient increase in IOP that returned to baseline (see C) showed upregulated reactive astrocyte transcripts (same level as sustained IOP increase in the retina (K/L), optic nerve head (M), or optic nerve (N), see also Figure S2, S3). See also Supplemental Figures S1-S3. Abbreviations: tKO, triple knock out mice (Il1a−/−Tnf−/−C1qa−/−, neuroinflammatory reactive astrocyte deficient); S, sustained (IOP increase); T, transient (IOP increase); WT, wildtype. * P < 0.05, one-way ANOVA followed by Tukey’s multiple comparisons test. Individual data points representative of individual animals are plotted, while bars represent mean ± s.e.m. Scale bar is 20 μm for all micrographs in F.
While there was a ~25% decrease in RGCs 28 days after injection in WT mice with sustained increases in IOP compared to uninjected control eyes (as quantified by the RGC marker RBPMS), there was no change in RGC number in mice lacking the inducers of inflammatory astrogliosis (Figure 2G). SMI-32+ ɑ-RGCs were particularly vulnerable to increased IOP, with a ~50% decrease in cell number that was also completely prevented in the absence of IL-1⍺, TNF⍺, and C1q (Figure 2H). Retinas in which IOP only increased transiently showed no change in RGC number in WT or tKO mice (Figure 2I), confirming that sustained increases in pressure are required to induce cell death.
These results suggest that reactive astrocytes are required for the death of RGCs following sustained increases in IOP. We next characterized the reactive response of astrocytes in the retina, optic nerve head, and optic nerve to the microbead occlusion model to better understand how astrocytes participate in degeneration. An inflammatory astrocyte response was observed in the retina, optic nerve head, and optic nerve of injected WT mice with sustained increases in IOP relative to uninjected controls, and these responses were largely abated in tKO mice (Figure 2J-N, Supp. Figures S2, S3). Unexpectedly, while RGCs did not die in retinas with transient changes in IOP, WT retinas with transient changes in IOP still showed robust astrogliosis at the level of the retina (Figure J, Supp. Figures S2, S3). These data suggest that both acute and chronic injuries to the eye are sufficient to drive formation of neurotoxic reactive astrocytes, and that this activation is mediated by IL-1α, TNFα, and C1q. Surprisingly, these data also suggest that the presence of neurotoxic astrocytes alone is not sufficient to induce death of RGCs.
Only damaged neurons are susceptible to reactive astrocyte toxin-induced death
In the case of transient increases in IOP, astrogliosis occurs robustly at the level of the retina but does not induce RGC death. A similar phenomenon occurs in systemic LPS injection, where neuroinflammatory astrogliosis occurs throughout the brain without the widespread death of neurons (Castaño et al., 1998; Zamanian et al., 2012; Liddelow et al., 2017). We hypothesized that the observed discrepancy could be explained by the requirement of a second signal, such as an injury, to induce the target neurons to become vulnerable to the toxicity of reactive astrocytes. To test this hypothesis, we directly injected astrocyte conditioned media (ACM) from quiescent or reactive immunopanned astrocytes (Liddelow et al., 2017) into the retinas of WT mice. The toxicity of the reactive ACM was already tested in in vitro assays using both primary neurons and oligodendrocytes (data not shown), and only ACM that was validated as toxic was used in further experiments. Injecting equal protein concentrations of either control or reactive ACM into the vitreous of an uninjured WT eye failed to induce cell death (Figure 3). Thus, toxic factors secreted from reactive astrocytes alone are not sufficient to induce the death of uninjured RGCs. As described previously (Figure 1), tKO mice lack inflammatory reactive astrocytes and do not suffer the normal death of RGCs after axotomy by ONC, presumably because the neurons are injured by the nerve crush but the absence of reactive astrocytes leads to a lack of production of the astrocyte-derived toxic factors (Figure 2, Supp. Figure S2, S3). If, however, we injected reactive ACM into the retinas of Il1a−/−Tnf−/−C1qa−/− mice following axotomy and thus restore the presence of astrocyte-derived toxic factors to the injured retina, the death of RGCs normally seen after ONC is restored to WT levels (Figure 3). Together, these experiments suggest that neurons of the retina are normally resistant to astrocyte-mediated toxicity, but become vulnerable after axotomy. A similar mechanism may explain the lack of cell death seen in the retinas of mice with transient IOP, where reactive astrogliosis occurs but in the absence of the prolonged changes in ocular pressure necessary to induce neurons into a vulnerable state.
FIGURE 3. Retinal ganglion cells require both damage and astrocyte-derived toxins for targeted removal.

Two weeks following optic nerve crush (ONC) in wild type (WT) mice, retinal ganglion cell number was decreased approximately 75%. This death was not seen in ONC of neuroinflammatory reactive astrocyte-deficient tKO mice, or in mice that had intact optic nerves but received an injection of toxic reactive astrocyte conditioned media (neurotoxic ACM) which was otherwise toxic in in vitro assays. Triple knockout mice that would normally not show loss of RGCs following ONC had loss of cells when paired with injection of neurotoxic ACM. RGC numbers normalized to contralateral control eye. * P < 0.05, one-way ANOVA followed by Tukey’s multiple comparisons test. Individual data points are plotted and represent individual animals, while bars represent mean ± s.e.m.
Do spared neurons have altered morphology?
While RGCs can be spared from death following bead-induced glaucoma, certain types of RGCs undergo dendritic remodeling following transient increases in IOP, potentially losing their normal circuit connectivity and function and thus their value to the regeneration of the retina (El-Danaf & Huberman, 2015). In order to determine if spared neurons resemble uninjured neurons from the same mouse, we first tested whether OFF-S RGCs (RGCs with sustained rather than transient responses to the removal of a light stimulus and that undergo dendritic remodeling after transient IOP increases) retain their stereotyped dendritic morphology. We dye-filled randomly selected OFF-S RGCs from tKO mice 30–35 days after increasing IOP and performed confocal imaging of their dendritic arbors through the depth of the inner plexiform layer (IPL). We chose this time as several studies show changes in RGC numbers from 2–6 weeks using this model (Cone et al., 2010; Sappington et al., 2010; El-Danaf & Huberman, 2015; Sabharwal et al., 2017) and we wanted to study chronic effects of IOP increase on RGC maintenance. Counterstaining for choline acetyltransferase (ChAT) allowed for the position of the RGC dendrites within the IPL to be measured and compared between animals (Figure 4A-B). To quantify dendritic localization within the IPL, the normalized fluorescence intensity of the dendritic signal was plotted relative to that of the ChAT signal in a small region of the IPL (Figure 4C). This normalized measure was used to quantify the depth of the dendritic stratification within the retina as well as the total width of the dendritic arborization (Figure 4D). Although the depth at which the dendritic arbors stratified in the IPL did not change, the width of those arbors significantly increased (Figure 4D,E). Thus, while the dendrites of spared RGCs are likely receiving presynaptic input from neurons in the correct region of the IPL (i.e. OFF versus ON), it is possible for them to receive input from a wider range of presynaptic neurons.
FIGURE 4. Dendrites of OFF-S RGCs in Il1a−/−Tnf−/−C1qa−/− KO mice stratify more broadly in the IPL following elevated IOP.

A. Example z-projection of a stack of confocal images taken from an OFF-S RGC from Il1a−/−Tnf−/−C1qa−/− mice under control conditions showing the level of RGC dendrites (red) and ChAT bands (cyan). B. Same as in (A) but from OFF-S RGCs from Il1a−/−Tnf−/−C1qa−/− mice under elevated IOP conditions. Dendrites stratify in approximately the correct location, but more broadly. C. The position and width of the dendrites in the IPL can be quantified. The normalized fluorescence intensity of the dendritic signal relative to that of the ChAT bands across a portion of the IPL (denoted by yellow box in (B)) is shown for a single RGC. By plotting the position of the dendrites relative to the normalized position of the ChAT bands, the position and width of the dendritic stratification can be quantified. D. Quantification of average stratification position within the IPL of OFF-S RGCs from Il1a−/−Tnf−/−C1qa−/− mice under control and bead-injected retinas. E. The dendrites of RGCs from bead-injected Il1a−/−Tnf−/−C1qa−/− mice stratified across a broader region of the IPL than RGCs from control retinas. F. Representative dye-filled RGC used for electrophysiological recordings. G-H. Ray-trace images of dye-fills pictured in (F) shown from the perspective of a flat retina (z-projection) to visualize the radial branching of RGCs (G) and from a side view(y-projection) to visualize dendritic position across layers of the retina (H). I. Sholl analysis of RGC process branching in control or bead-injected eyes. J-L. Quantification of number of dendritic branch points per cell (J), cell radius (K), and total neurite length (J) for RGCs in spared vs control retinas. All quantification performed on mice with sustained increases in IOP. * P < 0.05, unpaired t-test. Individual data points are plotted and represent individual cells from 4 animals (control) and 9 animals (bead-injected), while bars represent mean ± s.e.m. Scale bar is 100 μm for all micrographs in F.
In addition to stratifying at stereotypical depths within the IPL to ensure proper connectivity with presynaptic partners, the dendrites of RGCs also extend radially in conjunction with their spatial summation properties. We performed 3D reconstruction of dye-filled OFF-S RGCs to analyze their radial dendritic arborization (Figure 4F-H). Example images of reconstructed dendritic arbors in control or elevated IOP Il1a−/−Tnf−/−C1qa−/− retinas show that spared RGCs retain high levels of dendritic branching (Figure 4G), and side views of the reconstructed dendrites recapitulate the increase in width of stratification within their target layer found by confocal IHC (Figure 4H). Indeed, Scholl analysis suggests that the average radius of an RGC from the elevated IOP mice is significantly smaller than those from uninjured retinas from the same mouse, but the peak level of branching is largely similar in both cases (Figure 4I,J). While there was no difference in branching complexity, spared RGCs had slightly smaller dendritic arbors, with a decrease in the total cell radius (maximum dendrite extension) in spared RGCs (Figure 4K). We also measured the total neurite length of RGCs from control and bead-injected eyes and found no change in the total dendritic arborization (Figure 4L)
Together, these data suggest that OFF-S RGCs spared from death following sustained increases in IOP undergo dendritic remodeling, but the overall targeting and branching characteristics of the dendritic arbor are largely maintained when compared to undamaged tKO RGCs.
Are spared neurons still functional?
The light response properties of RGCs arise in part from the patterns of input they receive from their presynaptic partners. This presynaptic partner choice is dictated primarily by the level of the IPL at which they stratify their dendrites. The changes in morphology and stratification of spared OFF-S RGCs following sustained increases in IOP in tKO mouse retina raised the possibility that the light response properties of RGCs might be altered. To address this possibility, we investigated whether spared RGCs were functionally similar to uninjured tKO RGCs. We analyzed the activity of OFF-S RGCs by recording whole-cell current-clamp responses to visual stimuli in tKO mice following 30–35 days of elevated IOP. OFF-S RGCs have been well-characterized and can be targeted reliably based on cell soma size and light response properties (Krieger et al., 2017). All spared OFF-S RGCs from which we recorded remained light-responsive, although their light-responses were qualitatively different compared to control cells (Figure 5A). To quantify these changes, light response kinetics were compared. Spared OFF-S RGCs had a significantly decreased peak spike rate, but latency and decay of the light response as well as the Final/Peak ratio were unchanged (Figure 5B-C). These data indicate that neurons spared from astrocyte-targeted death following increased IOP are still active and light responsive.
FIGURE 5. OFF-S RGCs in Il1a−/−Tnf−/−C1qa−/− KO mice show subtle changes in light response properties following elevated IOP.

A. Response of an OFF-S RGC from Il1a−/−Tnf−/−C1qa−/− mice to a one second long negative contrast pulse under control conditions (top) and after elevated IOP following bead injection (bottom). B. Mean normalized spike rate histogram from a population of OFF-S RGCs from Il1a−/−Tnf−/−C1qa−/− mice. Inset shows components of the spiking response used to analyze response properties in (C). C. Analysis of various light response properties of OFF-S RGCs. The peak spike rate (left) was significantly reduced in OFF-S RGCs from Il1a−/−Tnf−/−C1qa−/− mice that experienced elevated IOP. Other measurements of light response properties of OFF-S RGCs (decay, latency, and final/peak ratio) remained unchanged following bead-occlusion. D. Mean normalized responses of OFF-S RGCs to spots of increasing diameter in Il1a−/−Tnf−/−C1qa−/− KO mice under elevated IOP (red) or control (grey) conditions. Elevated IOP significantly reduced responses to spot sizes below 600 microns. E. Mean normalized responses of a single OFF-S RGC to spots of increasing size (left). Spot size response function was fit (smooth line) and the spot size that produced a half maximal response (dashed lines) was calculated. Half maximal responses for a population of OFF-S RGCs from control and bead-injected Il1a−/−Tnf−/− C1qa−/− mice (right). The spots size that produced a half-maximal response was significantly larger in bead-injected mice. All quantification performed on mice with sustained increases in IOP. * P < 0.05, unpaired t-test or one-way ANOVA as appropriate. Individual data points are plotted and represent individual cells from 4 animals (control) and 9 animals (bead-injected), while bars represent mean ± s.e.m.
The altered kinetics of spared OFF-S RGCs suggests that these cells might be integrating input from presynaptic partners differently. To test whether the integration of spatial information might be altered in OFF-S RGCs, we next probed the size tuning of cells by recording responses to spots of negative contrast of increasing size. Although the spike rates of spared OFF-S RGCs increased with spot diameter, they were significantly lower than control cells for smaller spots, before reaching equivalent spike rates at the largest spot diameters tested (Figure 5D). These results suggest that the ability of spared RGCs to integrate spatial information was altered. To better quantify this change, normalized size tuning functions were calculated for each cell (Figure 5E and see Methods) and used to determine the spot size that produced a half-maximal response in spared OFF-S RGCs (Figure 5E). These findings indicate an impairment in the ability of spared OFF-S RGCs to integrate spatial information and are consistent with changes in the presynaptic input they receive.
The changes in light response kinetics and spatial integration of spared OFF-S RGCs could arise if these cells received input from novel presynaptic partners. We next tested if this was the case by probing the spatial summation properties of spared OFF-S RGCs. Spatial summation properties, including the nonlinear subunit structure within the receptive field, are known to be generated by inputs from specific populations of bipolar cells (Borghuis, et al. 2013; Demb, et al., 1999). To probe spatial summation, RGCs were presented with visual stimuli consisting of contrast reversing gratings at a range of spatial frequencies and temporal phases. This stimulus produced robust spiking responses in spared OFF-S RGCs that modulated mainly at the temporal frequency of the stimulus (F1 component; Figure 6A). The F1 component of the response was also highly phase-dependent with null responses at two spatial phases separated by 180°, indicating that spared cells could encode specific features of the stimuli used (Figure 6B). To more completely quantify the spatial summation response properties, we calculated the F1 and F2 Fourier harmonics as a function of spatial frequency (Petrusca, et al., 2007). Both spared and control OFF-S RGCs showed similar subunit structures that were dominated by the F1 harmonic at all spatial frequencies tested (Figure 6C). There was also an F2 component, but it was not strongly spatial-frequency dependent, suggesting that both populations of cells integrated spatial information linearly. We also quantified the level of non-linearity of spatial summation (F2/F1) and found that there were no significant differences between spared and control OFF-S RGCs (Figure 6D).
FIGURE 6. Spatial summation properties of OFF-S RGCs in Il1a−/−Tnf−/−C1qa−/− mice are unchanged following elevated IOP.

A. Response of an OFF-S RGC from an Il1a−/−Tnf−/−C1qa−/− mice after elevated IOP to contrast reversing gratings (CRGs) presented for four seconds at 2 Hz. B. Typical normalized post stimulus time histograms (PSTHs) from one cycle of the CRG stimulus of an OFF-S RGC from Il1a−/−Tnf−/−C1qa−/− mice after elevated IOP to one cycle of CRGs at a single spatial frequency at four spatial phases. The response modulates primarily at the temporal frequency of the stimulus. C. Spatial frequency tuning curves for the first Fourier harmonic (F1; black) and the second Fourier harmonic (F2; red) for OFF-S RGCs from Il1a−/−Tnf−/−C1qa−/− mice under control (left) and after elevated IOP (right). The F2 component of the response is small at all spatial frequencies tested. D. Spatial summation nonlinearities of OFF-S RGCs from Il1a−/−Tnf−/−C1qa−/− mice under control and elevated IOP conditions. The nonlinearity index is defined as the maximum value of the ratio between the second and the first harmonics (F2/F1) over all spatial frequencies tested. All quantification performed on mice with sustained increases in IOP. n.s. P > 0.05, unpaired t-test. Individual data points are plotted and represent individual cells from 4 animals (control) and 9 animals (bead-injected), while bars represent mean ± s.e.m.
Collectively these data indicate that, although spared OFF-S RGCs stratify their dendrites over a broader region of the IPL and integrate spatial information differently compared to the uninjured RGCs of the contralateral eye, they likely do not connect to novel presynaptic partners because the nonlinear subunit structure of their receptive fields does not change.
Discussion
Here we demonstrate the contribution of reactive astrocytes to the death of retinal ganglion cells both in acute and chronic injuries of the retina. By inhibiting or knocking out IL-1⍺, TNF⍺, and C1q, the factors secreted from microglia that induce neurotoxic reactive astrocytes, we can largely prevent the death of RGCs induced following optic nerve crush or following prolonged increases in IOP. The neurotoxicity of reactive astrocytes in glaucoma, combined with studies from other groups highlighting the contribution of this astrocytic response to models of Parkinson’s Disease, Alzheimer’s Disease, and Huntington’s Disease (Shi et al., 2017; Yun et al., 2018; Diaz-Castro et al., 2019; Joshi et al., 2019) suggests that neurotoxic reactive astrocytes may be a common feature in the complex and heterogeneous milieu of the CNS’s response to prolonged injury. Interestingly, this response stands in stark contrast to the growing literature that other forms of astrocyte reactivity, such as those governed by the transcription factor STAT3, can be beneficial to CNS recovery after injury, including in the case of glaucoma (Qu & Jakobs, 2013; Anderson et al., 2016; Sun et al., 2017). Further, eliminating neurotoxic reactive astrocytes in a model of prion infection actually worsened outcomes (Hartmann et al., 2019), suggesting that these phenotypes have highly contextual functions. The divergent impact of different reactivity states on cell health and regeneration highlights the need to carefully manipulate and consider astrogliosis as a potentially heterogeneous response, and it is still unclear what mechanisms link the increase in IOP to the activation of glial cells or how homogeneous/heterogeneous the glial response is in space and time. This will no doubt be the focus of future studies across many disease paradigms, especially with the proliferation of techniques to analyze cell responses at the single cell level (Menon et al., 2019; Tran et al., 2019).
In addition to showing that RGCs can be spared from death following acute axotomy and prolonged IOP, we found that spared RGCs are still functional with many electrophysiological and morphological features similar to those of the uninjured retina of the contralateral eye. There are many ways to prevent cell death by eliminating final executioners of the cell death process. Unfortunately, surviving cells are sometimes irreversibly changed by the intracellular signaling processes that would normally lead to cell death or, in the case of neurons, may feature dramatically altered electrophysiological processes that make the surviving cell more detrimental to the normal function of a circuit than if the cell had simply died. Thus, our observation that the electrophysiological and morphological features of RGCs spared from cell death were similar to those of the uninjured retina suggests that eliminating the factors that induce reactive astrocytes prevents many pathological changes that often occur before eventual cell death. There were noticeable changes in certain properties in spared compared to uninjured RGCs, such as the decrease in peak firing rate or the increase in width of dendritic arborization. However, the fact that these cells retained largely normal light response properties, which are governed by the cells’ presynaptic inputs, suggests that they retained many correct presynaptic inputs and suggests that damaged or diseased neurons, if encouraged to fully regenerate their pre- and post-synaptic connections, might be able to reconnect into normal circuitry.
The exact mechanism of the dendritic morphology changes that do occur is unclear at this point. While previous studies have highlighted C1q and TNF⍺ directly in synaptic and dendritic remodeling (Stellwagen and Malenka, 2006; Stevens et al., 2007), we believe that the observed changes are not directly linked to the lack of these proteins as such dendritic changes occur in mouse models of glaucoma even when these factors are still present (El-Danaf & Huberman, 2015) and because our previous studies have implicated reactive astrocytes more directly in synaptic remodeling separate from the direct influence of these signaling molecules (Liddelow et al., 2017). Future experiments should use alternative methods for increasing intraocular pressure that allow for the analysis of visual acuity to determine if tKO mice also regain the components of vision normally lost following RGC death, as well as to determine if there are other compensatory changes in the spared neurons or their pre/postsynaptic partners that would help reestablish normal vision after changes to spike rate or dendritic patterning. Alternatively, investigations into similar microglia-astrocyte derived neurotoxicity models in other brain regions may highlight other important components of this complex cascade.
Finally, observations from mice with transient rather than prolonged increases in intraocular pressure, as well as experiments combining optic nerve crushes with the injection of toxic or control astrocyte conditioned media from primary rodent astrocyte culture, have suggested that the presence of astrocyte secreted toxic factors or the injury of RGCs alone are not sufficient to induce RGC death. Rather, both toxin and injury are required together to induce cell death, which has led us to a model where cellular injury makes the target cell susceptible to the eventual cell death signal provided by the astrocytes. Another potential interpretation of the lack of in vivo neuron death seen when injecting toxin into the retina without optic nerve crush is that Il-1⍺, TNF⍺, and C1q induce changes to other cells in the visual system that are required for the neurons to die rather than a cell-intrinsic change in RGCs themselves. However, we have previously demonstrated that RGCs are vulnerable to the toxic factor when cultured in the absence of other cell types and in the presence of full trophic support (Liddelow et al., 2017). Further, we greatly reduce the abundance of these cytokines via the size-dependent concentration of media prior to injection, which renders the proteins undetectable by mass spectrometry as well as makes the media incapable of further activating astrocytes in vitro (results not shown). Together, these results suggest that the changes that make a neuron susceptible are cell intrinsic, but experiments should be performed to further test this hypothesis. In this model, we predict that cultured neurons are injured during their removal from the brain, an event that necessarily causes axotomy, and this damage induces the neurons to become susceptible to the toxin. This model also aligns with observations from systemic injection of LPS, where widespread astrogliosis occurs throughout the CNS in the absence of the cataclysmic death of all neurons and mature oligodendrocytes. In these large systemic insults, reactive astrocytes may be present and secreting factors toxic to neurons, but neurons are not susceptible due to a lack of conditioning injury. Future experiments should take advantage of techniques such as ATAC-seq to try and identify the cellular changes that make cells susceptible to astrocyte-mediated death, as these changes would be powerful therapeutic targets in a range of injuries and diseases. Regardless of whether therapies to stop this process are found, this model adds a satisfying logic to the otherwise precarious presence of toxic astrocytes in the CNS, as well as cautions a thoughtful approach to stopping neuronal cell death in some contexts as injured neurons may be more detrimental to the system alive than dead in certain diseases.
METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Shane Liddelow (shane.liddelow@nyulangone.org).
Materials Availability
This study did not generate new unique reagents. However, there are restrictions to the availability of the Il1a−/−Tnf−/−C1qa−/− mouse line due to MTAs. Please contact Shane Liddelow (shane.liddelow@nyulangone.org) for more information.
Data and Code Availability
This study did not generate or utilize any dataset or code that requires distribution. The data that support the findings of this study are available from the lead contact upon reasonable request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal ethics
Mice
All animal procedures were conducted in accordance with guidelines from the National Institute of Health and Stanford University’s Administrative Panel on Laboratory Animal Care. All mice were housed with food and water available ad libitum in a 12-h light/dark environment.
Rats
All animal procedures were conducted in accordance with guidelines from the National Institute of Health and Stanford University’s Administrative Panel on Laboratory Animal Care. All rats were housed with food and water available ad libitum in a 12-h light/dark environment.
Animals
Mice
Wild type (WT) C57Bl/6J mice were obtained from Jackson Laboratories. Triple knockout (Il1a−/−Tnf−/−C1qa−/−) neuroinflammatory reactive astrocyte-deficient mice (tKO mice) were from a previous study in our laboratory (Liddelow et al., 2017). All lines were maintained by breeding with C57Bl/6J mice. Animals were randomly assigned numbers and thereafter evaluated blind (to both experimental condition and genotype). Only female mice were used for glaucoma studies.
Rats
Sprague Dawley rats were from obtained from Charles River. Mixed genders were used for both optic nerve crush studies as well as primary cell culture studies.
Cell culture and conditioned media collection
Astrocytes were purified by immunopanning from P5 rat forebrains and cultured as previously described (Foo et al., 2011). Briefly, cortices were dissected and enzymatically digested by papain at 37°C and 10% CO2. Tissue was then mechanically triturated using a 5ml serological pipette at room temperature and the suspension filtered through a 70μm nitex filter to generate a single-cell suspension. The suspension was negatively panned for microglia (CD45), endothelial cells (BSLI), and oligodendrocyte-lineage cells (O4) followed by positive panning for astrocytes (Itgb5). Astrocytes were removed from the final positive selection plate by brief digestion with 0.025% trypsin and plated on poly-D-lysine coated 10cm tissue culture plates. Astrocytes were cultured in defined, serum-free medium containing 50% neurobasal, 50% DMEM, 100 U/mL penicillin, 100 μg/mL streptomycin, 1 mM sodium pyruvate, 292 μg/mL L-glutamine, 1× SATO (neurobasal media supplemented with 100 μg/mL transferrin, 100 μg/mL BSA, 16 μg/ml putrescine, 60 ng/mL progesterone, 40 ng/mL sodium selenite), 5 μg/mL of N-acetyl-L-cysteine, and 5ng/mL HBEGF (Peptrotech, 100–47).
After 6 days in culture, astrocytes were treated with IL-1α (3 ng ml−1, Sigma, I3901), TNFα (30 ng ml−1, Cell Signaling Technology, 8902SF) and C1q (400 ng ml−1, MyBioSource, MBS143105) for 24hrs to generate neuroinflammatory reactive astrocytes or treated with an identical volume of PBS (control). Conditioned media was removed after 24hrs, supplemented with complete protease inhibitor cocktail (Roche), and concentrated with 30-kDa size exclusion Vivaspin ultrafiltration units (Sartorious). Control and reactive conditioned media were concentrated to the same final volume, and equivalent final protein concentration confirmed by Bradford protein concentration analysis (BioRad, 500–0006). In indicated experiments, 1 ul of either control or reactive ACM of equivalent protein concentration was injected into the vitreous of the eye using a Hamilton syringe (Hamilton Company, 87919) in either unmanipulated rats or rats that received an optic nerve crush (see below) immediately prior to the injection.
METHODS DETAILS
Retro-orbital optic nerve crush
Postnatal day 21 wildtype (WT, C57Bl/6J) and tKO mice or postnatal day Sprague Dawley rats (Charles River) were anaesthetized with 2.5% inhaled isoflurane in 2.0 L O2 per min. Without incision to the orbital rim, the supero-external orbital contents were blunt-dissected, the superior and lateral rectus muscles teased apart, and the left optic nerve exposed. The nerve was crushed for 3–5 s approximately 2 mm distal to the lamina cribrosa. After surgery, the eye fundi were checked to ensure retinal blood flow was intact. 1, 3, 7, 14, and 28 days following crush retinas were collected for immunofluorescence to determine viability of retinal ganglion cells. In a separate set of experiments, WT and tKO mice had optic nerves crushed, retinas removed, and microfluidic qPCR analyses conducted. These mice had expected levels of astrocyte reactivity as reported previously (Liddelow et al., 2017), and this was similar to activation seen in experimental glaucoma via bead injection at 28 days (data not shown). In rat optic nerve crush experiments, some rats received a 2 μl intravitreal injection of neutralizing antibodies to Il-1α (150 μg μl−1, Abcam, ab9614), TNF (150 μg μl−1, Cell Signaling Technology, 7321), and C1q (Quidel, A301), or rabbit IgG control (150 μg μl−1, Abcam, ab27472) at day 0 (the time of optic nerve crush) and every 7 days until retinas were collected.
Microbead injection and intraocular pressure measurements
The microbead occlusion model was used to achieve intraocular pressure (IOP) elevation applying previously described protocols (Sappington et al., 2010; Chen et al., 2011, El-Danaf and Huberman, 2015). Mice were anesthetized with a mixture of ketamine/xylazine and a small hole was made in the cornea using a glass micropipette attached to a picospritzer. A 1–2 μl volume of polyesterene microbeads (10 μm diameter; Invitrogen F-8834) was injected into the anterior chamber of one eye. The other eye served as an internal control, in which a hole was made and no beads were delivered (El-Danaf & Huberman, 2015). In some experiments, control animals received an equal volume (1–2 μl) of sterile PBS. IOPs were measured daily, around the same time every day (± 60 min), using a rebound tonometer (TonoLab, Colonial Medical Supply). The reported IOP for each day consisted of an average reading of 10 consecutive IOP measurements. Baseline IOP values were obtained by monitoring the pressure for 2–3 days prior to bead injections. Pressures were then measured for 4–6 days and at 28 days following the bead injections to detect any elevations in IOP. Mice showing no IOP elevation resulting from bead leakage were omitted from the study.
Microfluidic quantitative PCR
Total RNA was extracted from whole retinas using the RNeasy Plus kit (Qiagen) and cDNA synthesis performed using the High-Capacity RNA-to-cDNA™ Kit (Applied Biosystems™) according to supplier protocols. Microfluidic qPCR was performed using a 96.96 Dynamic Array chip (Fluidigm Corporation) run on a BioMark Real-Time PCR System (Fluidigm) using a cycling program of 10 min at 95 °C followed by 40 cycles of 95 °C for 15 s, 60 °C for 30 s and 72 °C for 30 s, as described previously (Zhang et al., 2014; Liddelow et al., 2017; Clarke et al., 2018). Data were collected using BioMark Data Collection Software 2.1.1 build 20090519.0926 (Fluidigm). Three reference genes Aldh1l1, Gapdh and Rplp0 were used to normalize transcript expression depending on cell type. Data preprocessing and analysis was completed using Fluidigm Melting Curve Analysis Software 1.1.0 build 20100514.1234 (Fluidigm) and Real-time PCR Analysis Software 2.1.1 build 20090521.1135 (Fluidigm) to determine valid PCR reactions. Reactive astrocyte primers were used as described previously (Liddelow et al., 2017).
Electrophysiology
Electrophysiological procedures were similar to those described previously (Stafford et al., 2014; Osterhout et al., 2015). Briefly, retinas were harvested and dissected in gassed (95% O2 and 5% CO2) Ames medium (Sigma) under infrared illumination. A piece of ventral retina was placed in a chamber on an upright microscope and superfused (~5ml/min) with gassed Ames medium heated to 33–35°C. RGCs with large somas were targeted under IR illumination to bias recordings toward OFF-S cells for recording. A glass electrode (tip resistance, 4–6 MΩ) was filled with Ames’ medium for loose-patch recording and the type of RGC was determined by measuring light responses. Once a presumed OFF-S cell was located, a second glass electrode was filled with intracellular solution and the same cell was targeted for whole-cell recording. The intracellular solution contained (in mM): 120 K-methanesulphonate 10 HEPES, 5 NaCl, 0.1 EGTA, 2 ATP-Mg2+, and 0.3 GTP-Na, titrated to pH 7.3. Chemicals were purchased from Sigma-Aldrich or Tocris. Custom programs in MATLAB were used for spike thresholding and analysis of spiking parameters. For analysis of normalized spiking responses as a function of spot size, the average normalized response from at least three trials was fit with a Naka-Rushton equation to determine the spot size that generated a half maximal response.
Patterned light stimuli were generated by custom software developed in Psychophysics Toolbox and MATLAB. Stimuli were projected onto the retina using a custom DLP projector (EKB Technologies; E4500MKII) custom fitted with a UV LED (final emission, 385 nm), attenuated by a neutral density 1.0 filter, and focused using a 10X objective to the level of rod and cone outer segments. The mean luminance generated 2.6 × 105 R*/S-cone/sec. The receptive field center was mapped by recording responses to square-wave modulations of a 300 μm diameter spot presented at eight positions. In subsequent experiments, stimuli were presented as a contrast pulse (100% Weber contrast), 100–800 μm in diameter or as a patch of contrast reversing gratings (800 μm patch diameter) modulated at 2 Hz at 5 spatial frequencies and 12 spatial phases. Responses to drifting square-wave gratings (500 μm/cycle; 100% Michelson contrast) modulated at 1 Hz against a background mean luminance were also recorded from a subset of cells.
Following recordings, pieces of retina were fixed in 4% PFA for one hour at room temperature, transferred to a blocking solution containing 1x PBS, 0.5% Triton-X-100, and 10% donkey serum for two hours at room temperature, then incubated with antibodies against lucifer yellow (1:1000; Invitrogen A-5750) and choline acetyltransferas (ChAT, 1:100, Millipore AB-144P) diluted in blocking solution while rocking for three days at 4°C. Following washing and staining with appropriate fluorescently conjugated secondary antibodies, retinas were mounted on coverslips for confocal imaging. Fluorescent signals from the filled RGC and ChAT staining were imaged through the entire depth of the retina from a region that covered the full dendritic field of the filled cell. Morphological analyses of elecrophysiologically recorded cells was conducted by determining the position of the dendrites relative to the ChAT bands as described previously (Manookin, et al., 2008). Briefly, confocal z-stacks were analyzed in small regions that adequately reflected the stratification of the RGC dendrites and where the dendritic signal and ChAT signals displayed minimal tissue warping/compression. A maximum projection of each fluorescent signal in these regions was generated by averaging the stacks across the z-dimension (Figure 4A-B). The fluorescent signals were analyzed as a function of distance across the IPL, the boundaries of which were defined by the ChAT cell bodies in the INL and GCL (Figure 4B, yellow box). Third-order polynomials were fitted around six to eight points (3–4 μm) at the fluorescent peaks associated with the ChAT signal to determine the percentage depth of the two ChAT bands in the IPL. The dendritic signal was analyzed and the width of the dendritic signal was determined as follows: the positions corresponding to the first time the dendritic signal rose above, and the last time it dropped below, 0.5 were identified (Figure 4C). For all cells, these positions were manually checked by eye to confirm they captured the extent of the dendritic signal. These positions were then normalized relative to the two fitted peaks of the ChAT signal in the same tissue where the On ChAT band = 0, and the Off ChAT band = 1. The stratification width of the dendrites was considered the difference between these two positions. This normalization procedure allows the stratification position and width of dendrites to be compared from cells in tissue from different animals.
Immunohistochemistry
Mice were euthanized by inhaled CO2 and cervical dislocation. Whole eyes were dissected and placed in ice-cold 4% paraformaldehyde for 10 min, and then washed in DPBS before dissecting the retina away from the rest of the eye and post-fixing in 4% paraformaldehyde overnight a 4°C. Retinas were blocked with 10% goat serum (MP Biomedicals, 0219135680) and stained with rabbit anti-RBPMS (1:500, PhosphoSolutions 1830-RBPMS) diluted in phosphate buffered saline and visualized with appropriate secondary antibodies conjugated with Alexa fluorophores (Invitrogen). Immunofluorescent images were collected using a Keyence BZ-X700 Fluorescence Microscope (Keyence) and final multi-fluorescent images prepared using FIJI (Schindelin et al., 2012). Z-stacked dye-filled RGCs were reconstituted in FIJI (Schindelin et al., 2012)
Sholl analysis
Z-stacked images of dye-field RGCs from tKO mice injected with either microbeads or control preparations were uploaded to FIJI (Schindelin et al., 2012) for Sholl analysis (Ferreira et al., 2014). Each RGC neurite was traced by an evaluator blind to experimental conditions using the FIJI Simple Neurite Tracer (SNT) plugin (Longair, et al., 2011). Sholl analysis was performed on each traced RGC using a 10μm radius step size. The radius from the center of the soma to the terminal trace of the longest neurite was used as a marker of total RGC radius. Traces were skeletonized using the SNT render/skeletonize function then oriented and collapsed along the +YZ plane using the FIJI 3D viewer (Schmid et al., 2010).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were completed using GraphPad Prism 7.00 software. Most data were analyzed by one-way ANOVA followed by Dunnett’s multiple post hoc test for comparing more than three samples, and two-sample unpaired t-tests for comparing two samples with 95% confidence. Power calculations were performed using G* Power Software V 3.1.9.2 (Faul et al., 2007). Group sizes were calculated to provide at least 80% calculable power with the following parameters: probability of type I error (0.05), conservative effect size (0.25). Four to ten individual animals per treatment group with multiple measurements were obtained per replicate. Further details on the exact tests and data presentation for each experiment can be found in its accompanying figure legend.
KEY RESOURCES TABLE
Supplementary Material
FIGURE S1. Intraocular pressure measurements for individual animals, Related to Figure 2. In wildtype (WT), and tKO (Il1a−/−Tnf−/−C1qa−/−) mice intraocular pressure (IOP) measurements were taken each day. Mice were split into two groups, those with sustained IOP increase (where IOP increased and remained high), and those with a transient increase (where IOP increased temporarily but then returned to normal levels). Each colored line represents an individual animal. Arrows highlight peak IOP increase in transient groups.
FIGURE S2. Induction of reactive astrocyte transcripts in the retina following bead-injection glaucoma model in the mouse, Related to Figure 2. A-F Microfluidic qPCR analysis of reactive astrocyte transcripts in whole retinas. n = 6-8 individual animals per group. All data are mean ± s.e.m.
FIGURE S3. Induction of reactive astrocyte transcripts in the optic nerve head and optic nerve following induction of bead-injection glaucoma model in the mouse, Related to Figure 2. A-F Microfluidic qPCR analysis of reactive astrocyte transcripts in whole tissue sample of optic nerve head (A-D) and optic nerve (excluding optic nerve head, E-H). n = 6-8 individual animals per group. All data are mean ± s.e.m.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-TNF-α neutralizing antibody | Cell Signaling Technology | Cat# 7321S |
| Anti-C1q neutralizing antibody | Quidel | Cat# A301 |
| Anti-IL-1α neutralizing antibody | abcam | Cat# ab9614 |
| Bandeiraea Simplicifolia Lectin I | Vector Labs | Cat# B-1105 |
| Donkey anti-Goat Alexa Fluor 647 secondary antibody | Thermo Fisher | Cat# A21447 |
| Donkey anti-Rabbit Alexa Fluor 488 secondary antibody | Thermo Fisher | Cat# A21206 |
| Goat ant-mouse IgM μ-chain | Jackson ImmunoResearch | Cat# 115-005-02 |
| Goat anti-mouse IgG+IgM (H+L) | Jackson ImmunoResearch | Cat# 115-005-044 |
| Goat anti-rat IgG (H+L) | Jackson ImmunoResearch | Cat# 112-005-167 |
| Goat polyclonal antibody against ChAT | Sigma | Cat# AB144P |
| Mouse anti-human Integrin beta 5 | eBioscience | Cat# 14-0497-82 |
| O4 hybridoma supernatant | Sommer and Schachner, 1981 | N/A |
| Rabbit polyclonal antibody against Lucifer Yellow | Invitrogen | Cat# A-5750 |
| Rabbit polyclonal antibody against RBPMS | PhosphoSolutions | Cat# 1830-RBPMS |
| Rat anti-mouse CD45 | BD Pharmingen | Cat# 550539 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ames’ medium | Sigma | Cat# A1420 |
| Apo-transferrin | Sigma | Cat# T1147 |
| ATP-magnesium salt | Sigma | Cat# A9187 |
| Bovine serum albumin (BSA) | Sigma | Cat# A4161 |
| DMEM (high glucose) | GIBCO | Cat# 11960-044 |
| DNase I | Worthington | Cat# DPRFS |
| Donkey serum | EMD Millipore | Cat# S30-100ML |
| Earle’s balanced salt solution | Sigma | Cat# E7510 |
| EDTA-free Protease Inhibitor Cocktail | Sigma | Cat# 04693159001 |
| EGTA | Sigma | Cat# E3889 |
| Fetal calf serum (FCS) | Gibco | Cat# 10437-028 |
| FluoSpheres™ Polystyrene Microspheres, 10 μm, red fluorescent (580/605) | Invitrogen | Cat# F8834 |
| Goat serum | MP Biomedicals | Cat# 0219135680 |
| GTP-sodium salt | Sigma | Cat# G8877 |
| HEPES | Sigma | Cat# H3375 |
| L-cysteine hydrochloride monochloride | Sigma | Cat# C7880 |
| L-glutamine | GIBCO | Cat# 25030-081 |
| N-acetyl cysteine | Sigma | Cat# A9165 |
| Native hC1q | Mybiosource | Cat# MBS143105 |
| Neurobasal | GIBCO | Cat# 21103-049 |
| Ovomucoid trypsin inhibitor | Worthington | Cat# LS003086 |
| Papain | Worthington | Cat# LS003126 |
| Paraformaldehyde Aqueous Solution | Electron Microscopy Sciences | Cat# 15710 |
| Penicillin/ Streptomycin | GIBCO | Cat# 15140-122 |
| Poly-D-lysine hydrobromide | Sigma | Cat# P6407 |
| Potassium methanesulfonate | Sigma | Cat# 83000 |
| Progesterone | Sigma | Cat# P-8783 |
| Putrescine | Sigma | Cat# P-5780 |
| Recombinant HB-EGF | PeproTech | Cat# 100-47 |
| Recombinant hTNFa | Cell Signaling Technology | Cat# 8902 |
| Recombinant II-1a | Sigma | Cat# I3901 |
| Sodium chloride | Sigma | Cat# S9888 |
| Sodium pyruvate | Sigma | Cat# P5280 |
| Sodium selenite | Sigma | Cat# S-5261 |
| SYBR Green PCR Master Mix | Thermo Fisher | Cat# 4385612 |
| TEA-Cl | Sigma | Cat# T2265 |
| Triton X-100 | Sigma | Cat# T8787 |
| Trypsin | Sigma | Cat# T9935 |
| Critical Commercial Assays | ||
| 96.96 Dynamic Array™ IFC for Gene Expression (includes Control Line Fluid, 2x Assay Loading Reagent, 20x GE Sample Loading Reagent, PreAmp Master Mix, RT Master Mix) | Fluidigm | Cat# 101-0349 |
| Bradford Assay | Bio-Rad | Cat# 500-0006 |
| High-Capacity RNA-to-cDNA Kit | Thermo Fisher Scientific | Cat# 4387406 |
| RNeasy Micro Kit | QIAGEN | Cat# 74004 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57Bl/6J | Jackson Laboratory | Strain Code 000664 |
| Mouse: II1a-/-Tnf-/-C1qa-/- | Liddelow et al., 2017 | N/A |
| Rat: Sprague Dawley | Charles River | Strain Code 400 |
| Software and Algorithms | ||
| FIJI/ImageJ | NIH | N/A |
| MATLAB | MathWorks | N/A |
| Psychophysics Toolbox | https://github.com/kleinerm/Psychtoolbox-3 | |
| Fluidigm Melting Curve Analysis Software 1.1.0 build 20100514.1234 | Fluidigm | N/A |
| BioMark Data Collection Software 2.1.1 build 20090519.0926 | Fluidigm | N/A |
| Real-time PCR Analysis Software 2.1.1 build 20090521.1135 | Fluidigm | N/A |
| pClamp 11.0 | Molecular Devices | N/A |
| Other | ||
| 1.0 Neutral Density Filter | Chroma | Cat# ND 1.0 |
| 30 kDa MWCO centrifugal filter unit | Sartorius | Cat# VS2022 |
| 5μl Hamilton Syringe | Hamilton Company | Cat# 87919 |
| DLP® LightCrafter™ E4500 MKII™ Fiber Couple | EKB Technologies | Cat# E4500MKII |
| icare® TONOLOAB rebound tonometer | icare | Cat# TV02 |
| IR (830 nm) LED Spotlight | Edmunds Optics | Cat# 66-860 |
| Picospritzer III - Intracellular Microinjection Dispense Systems | Parker Precision Fluidics | Cat# 051-0500-900 |
| Poly-D-Lysine coated coverslips (12 mm) | Neuvitro | Cat# NC0565504 |
| Polystyrene Microspheres, 10 μm | Invitrogen | Cat# F-8834 |
| UV DLP Projector | EKB Technologies | Cat# DPM-E4500UVGOAMKII |
Highlights.
Astrogliosis occurs following acute optic nerve injury and in a mouse model of glaucoma
Reducing astrogliosis stops neuron death after optic nerve crush and a glaucoma model
Neurons spared from death remain electrophysiologically functional
Injury is required for neurons to become susceptible to astrocyte-mediated toxicity
ACKNOWLEDGEMENTS
We thank all members of the Liddelow lab at NYU School of Medicine, and the Barres and Huberman labs at Stanford University. In particular, we thank Ben Barres for his mentorship and support. SAL was supported by NYU School of Medicine, generous anonymous donors, the Cure Alzheimer’s Foundation, a postdoctoral fellowship from the Australian National Health and Medical Research Council (GNT1052961), and the Glenn Foundation Glenn Award. This work was supported by The Gilbert Vision Restoration Initiative (ADH), NIH R01 EY026100 (ADH), NIH R01 EY027713 and Stanford Vision Core NIH P30 EY026877. KAG was supported by a Stanford Interdisciplinary Graduate Fellowship Affiliated with the Wu Tsai Neurosciences Institute. We thank V. Coates and S. Coates for their generous support.
Footnotes
DECLARATION OF INTERESTS
SAL is an academic founder of AstronauTx Ltd.
REFERENCES
- Almasieh M, Levin LA (2016). Neuroprotection in glaucoma: animal models and clinical trials. Annu. Rev. Vis. Sci 3, 1–30. [DOI] [PubMed] [Google Scholar]
- Anderson MA, Burda JE, Ren Y, Ao Y, O’Shea TM, Kawaguchi R, Coppola G, Khakh BS, Deming TJ, and Sofroniew MV (2016). Astrocyte scar formation aids central nervous system axon regeneration. Nature 532, 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borghuis BG, Marvin JS, Looger LL, Demb JB (2013). Two-photon imaging of nonlinear glutamate release dynamics at bipolar cell synapses in the mouse retina. J. Neurosci 33(27), 10972–10985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco A, Breen KT, Anderson SR, Steele MR, Calkins DJ, Vetter ML (2016). Glial coverage in the optic nerve expands in proportion to optic axon loss in chronic mouse glaucoma. Exp. Eye Res 150, 34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castaño A, Herrera AJ, Cano J, Machado A (1998). Lipopolysaccharide intranigral injection induces inflammatory reaction and damage in nigrostriatal dopaminergic system. J. Neurochem 70, 1584–1592. [DOI] [PubMed] [Google Scholar]
- Chen H, Wei X, Cho K-S, Chen G, Sappington R, Calkins DJ, and Chen DF (2011). Optic neuropathy due to microbead-induced elevated intraocular pressure in the mouse. Invest Ophth Vis Sci 52, 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke LE, Liddelow SA, Chakraborty C, Münch AE, Heiman M, Barres BA (2018). Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. U. S. A. 115(8), E1896–E1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cone FE, Gelman SE, Son JL, Pease ME, Quigley HA (2010). Differential susceptibility to experimental glaucoma among 3 mouse strains using bead and viscoelastic injection. Exp. Eye. Res 91(3), 415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demb JB, Haarsma L, Freed MA, Sterling P (1999). Functional circuitry of the retinal ganglion cell’s nonlinear receptive field. J. Neurosci 19(22), 9756–9767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Castro B, Gangwani MR, Yu X, Coppola G, Khakh BS (2019). Astrocyte molecular signatures in Huntington’s disease. Sci. Transl. Med 16, 11. [DOI] [PubMed] [Google Scholar]
- Dumitrescu ON, Pucci FG, Wong KY, Berson DM (2009). Ectopic retinal ON bipolar cell synapses in the OFF inner plexiform layer: contacts with dopaminergic amacrine cells and melanopsin ganglion cells. J. Comp. Neurol 517, 226–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Danaf RN, Huberman AD (2015). Characteristic patterns of dendritic remodeling in early-stage glaucoma: evidence from genetically identified retinal ganglion cell types. J. Neurosci 35(6), 2329–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faul F, Erdfelder E, Lang A-GG & Buchner A G* Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav. Res. Methods 39, 175–191 (2007). [DOI] [PubMed] [Google Scholar]
- Ferreira TA, Blackman AV, Oyrer J, Jayabal S, Chung AJ, Watt AJ, Sjostrom PJ, van Meyel DJ (2014). Neuronal morphometry directly from bitmap images. Nat. Methods 11, 982–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foo LC, Allen NJ, Bushong EA, Ventura PB, Chung W-S, Zhou L, Cahoy JD, Daneman R, Zong H, Ellisman MH, et al. (2011). Development of a Method for the Purification and Culture of Rodent Astrocytes. Neuron 71, 799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank M, Wolburg H (1996). Cellular reactions at the lesion site after crushing of the rat optic nerve. Glia 16(3), 227–240. [DOI] [PubMed] [Google Scholar]
- John S, Smith R, Savinova O, Hawes N, Chang B, Turnbull D, Davisson M, Roderick T, Heckenlively J (1998). Essential iris atrophy, pigment dispersion, and glaucoma in DBA/2J mice. Invest. Ophth. Vis. Sci. 39, 951–962. [PubMed] [Google Scholar]
- Hartmann K, Sepulveda-Falla D, Rose IVL, Madore C, Muth C, Matschke J, Butovsky O, Liddelow S, Glatzel M, Krasemann S (2019). Complement 3+-astrocytes are highly abundant in prion diseases, but their abolishment led to an accelerated disease course and early dysregulation of microglia. Acta Neuropathol. Commun. 7(1), 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inman DM, Horner PJ (2007). Reactive nonproliferative gliosis predominates in a chronic mouse model of glaucoma. Glia 55(9), 942–953. [DOI] [PubMed] [Google Scholar]
- Joshi AU, Minhas PS, Liddelow SA, Haileselassie B, Andreasson KI, Dorn GW 2nd, Mochly-Rosen D (2019). Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat. Neurosci 22(10), 1635–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger B, Qiao M, Rousso DL, Sanes JR, Meister M (2017). Four alpha ganglion ecll types in mouse retina: function, structure, and molecular signatures. PLoS One 12(7), e0180091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung W-S, Peterson TC, et al. (2017). Activated microglia induce neurotoxic reactive astrocytes via Il-1α, TNFα, and C1q. Nature 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longair MH, Baker DA, Armstrong JD (2011). Simple Neurite Tracer: open source software for reconstruction, visualization and analysis of neuronal processes. Bioinformatics 27, 2453–2454. [DOI] [PubMed] [Google Scholar]
- Manookin MB, Beaudoin DL, Ernst ZR, Flagel LJ, Demb JB (2008). Disinhibition combines with excitation to extend the range of the OFF visual pathway in daylight. J. Neurosci 28(16), 4136–4150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon M, Mohammadi S, Davila-Velderrain J, Goods BA, Cadwell TD, Xing Y, Stemmer-Rachamimov A, Shalek AK, Love J, Kellis M, et al. (2019). Single-cell transcriptomic atlas of the human retina identifies cell types associated with age-related macular degeneration. Nat. Commun 10(1), 4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon JI, Kim IB, Gwon JS, Park MH, Kang TH, Lim EJ, Choi KR, Chun MH (2005). Changes in retinal neuronal populations in the DBA/2J mouse. Cell Tissue Res. 320(1), 51–59. [DOI] [PubMed] [Google Scholar]
- Morrison JC, Moore CG, Deppmeier L, Gold BG, Charles K, Johnson EC (1997). A rat model of chronic pressure-induced optic nerve damage. Exp. Eye Res. 64, 85–96. [DOI] [PubMed] [Google Scholar]
- Park K, Liu K, Hu Y, Smith PD, Wang C, Cai B, Xu B, Connolly L, Kramvis I, Sahin M, He Z (2008). Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 322, 963–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrusca D, Grivich MI, Sher A, Field GD, Gauthier JL, Greschner M, Shlens J, Chichilinisky EJ, Litke AM (2007). Identification and characterization of a Y-like primate retinal ganglion cell type. J. Neurosci. 27(41), 11019–11027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu J, Jakobs TC (2013). The time course of gene expression during reactive gliosis in the optic nerve. PLoS One 8(6), e67094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabharwal J, Seilheimer RL, Tao X, Cowan CS, Frankfort BJ, Wu SM (2017). Elevated IOP alters the space-time profiles in the center and surround of both ON and OFF RGCs in mouse. Proc. Natl. Acad. Sci. U.S.A. 114(33), 8859–8864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sappington RM, Carlsson BJ, Crish SD, Calkins DJ (2010) The microbead occlusion model: a paradigm for induced ocular hypertension in rats and mice. Invest. Ophthalmol. Vis. Sci. 51(1), 207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9(7), 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid B, Schindelin J, Cardona A, Longair M, Heisenberg M (2010). A high-level 3D visualization API for Java and ImageJ. B. M. C. Bioinformatics 11, 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, Tsai R, Spina S, Grinberg L, Rojas J, et al. (2017). ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer I, and Schachner M (1981). Monoclonal antibodies (O1 to O4) to oligodendrocyte cell surfaces: An immunocytological study in the central nervous system. Dev Biol 83, 311–327. [DOI] [PubMed] [Google Scholar]
- Stafford BK, Manookin MB, Singer JH, Demb JB (2014). NMDA and AMPA receptors contribute similarly to temporal processing in mammalian retinal ganglion cells. J. Physiol 592, 4877–4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen D, and Malenka RC (2006). Synaptic scaling mediated by glial TNF-α. Nature 440, 1054–1059. [DOI] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, et al. (2007). The Classical Complement Cascade Mediates CNS Synapse Elimination. Cell 131, 1164–1178. [DOI] [PubMed] [Google Scholar]
- Sun D, Moore S, and Jakobs TC (2017). Optic nerve astrocyte reactivity protects function in experimental glaucoma and other nerve injuries. J Exp Medicine 214, 1411–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Zhang S, Lee C, Kumar A, Arjunan P, Li Y, Zhang F, Li X (2011). An optic nerve crush injury murine model to study retinal ganglion cell survival. J. Vis. Exp. 50, 2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran NM, Shekhar K, Whitney IE, Jacobi A, Benhar I, Hong G, Yan W, Adiconis X, Arnold MW, Lee JM, et al. (2019). Single-cell profiles of retinal ganglion cells differing in resilience to injury reveal neuroprotective genes. Neuron 104(6), 1039–1055.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda J, Sawaguchi S, Hanyu T, Yaoeda K, Fukuchi T, Abe H, Ozawa H (1998). Experimental glaucoma model in the rat induced by laser trabecular photocoagulation after an intracameral injection of India ink. Jpn. J. Ophthalmol. 42, 337–344. [DOI] [PubMed] [Google Scholar]
- Wang R, Seifert P, and Jakobs TC (2017). Astrocytes in the Optic Nerve Head of Glaucomatous Mice Display a Characteristic Reactive Phenotype. Investigative Opthalmology Vis Sci 58, 924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun SP, Kam TI, Panicker N, Kim S, Oh Y, Park JS, Kwon SH, Park YJ, Karuppagounder SS, Park H, et al. , (2018). Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nature Med. 24(7), 931–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. , (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci 34(36), 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1. Intraocular pressure measurements for individual animals, Related to Figure 2. In wildtype (WT), and tKO (Il1a−/−Tnf−/−C1qa−/−) mice intraocular pressure (IOP) measurements were taken each day. Mice were split into two groups, those with sustained IOP increase (where IOP increased and remained high), and those with a transient increase (where IOP increased temporarily but then returned to normal levels). Each colored line represents an individual animal. Arrows highlight peak IOP increase in transient groups.
FIGURE S2. Induction of reactive astrocyte transcripts in the retina following bead-injection glaucoma model in the mouse, Related to Figure 2. A-F Microfluidic qPCR analysis of reactive astrocyte transcripts in whole retinas. n = 6-8 individual animals per group. All data are mean ± s.e.m.
FIGURE S3. Induction of reactive astrocyte transcripts in the optic nerve head and optic nerve following induction of bead-injection glaucoma model in the mouse, Related to Figure 2. A-F Microfluidic qPCR analysis of reactive astrocyte transcripts in whole tissue sample of optic nerve head (A-D) and optic nerve (excluding optic nerve head, E-H). n = 6-8 individual animals per group. All data are mean ± s.e.m.
Data Availability Statement
This study did not generate or utilize any dataset or code that requires distribution. The data that support the findings of this study are available from the lead contact upon reasonable request.