

In this review, Pilger et al. summarize the current understanding of the molecular mechanisms underlying DNA damage-triggered immune responses, including cytosolic DNA sensing via the cGAS/STING pathway. They also highlight the implications of DDR components for therapeutic outcomes of immune checkpoint inhibitors or their use as biomarkers.
Keywords: DNA damage response, DNA repair, immunotherapy, PARP inhibitors, PD-1, PD-L1, STING, cGAS
Abstract
The DNA damage response (DDR) fulfils essential roles to preserve genome integrity. Targeting the DDR in tumors has had remarkable success over the last decade, exemplified by the licensing of PARP inhibitors for cancer therapy. Recent studies suggest that the application of DDR inhibitors impacts on cellular innate and adaptive immune responses, wherein key DNA repair factors have roles in limiting chronic inflammatory signaling. Antitumor immunity plays an emerging part in cancer therapy, and extensive efforts have led to the development of immune checkpoint inhibitors overcoming immune suppressive signals in tumors. Here, we review the current understanding of the molecular mechanisms underlying DNA damage-triggered immune responses, including cytosolic DNA sensing via the cGAS/STING pathway. We highlight the implications of DDR components for therapeutic outcomes of immune checkpoint inhibitors or their use as biomarkers. Finally, we discuss the rationale for novel combinations of DDR inhibitors with antagonists of immune checkpoints and current hindrances limiting their broader therapeutic applications.
Targeting DNA damage response factors for cancer therapy
The genome of every cell is constantly exposed to endogenously-arising and exogenous sources of DNA damage. To ensure genome stability and faithful replication and transmission of the genetic material, various DNA repair pathways have evolved to allow cellular and organism survival. This complex network of DNA damage sensor, mediator, and effector proteins is known as the DNA damage response (DDR) (Ciccia and Elledge 2010). The DDR exhibits tight spatiotemporal control, ensuring the precise and proper actions and coordination of repair enzymes in a DNA lesion-specific manner. Unscheduled or uncontrolled activity of DNA processing and repair factors can themselves generate DNA damage, thereby posing threats to genome integrity and cell survival. Consequently, dysregulation and mutations in such DDR factors and their regulators have implications for human health and disease (Jackson and Bartek 2009). Moreover, in recent years, DDR components have become accepted as attractive therapeutic targets for cancer therapy, with numerous small molecule inhibitors targeting DNA repair enzymes now being explored clinically and with inhibitors of the DNA-repair enzyme PARP being approved for treating various tumor types (Piliè et al. 2019). Due to the frequent loss or deregulation of DDR mechanisms and high levels of DNA replication stress caused by oncogene activation, cancer cells often display elevated levels of endogenous DNA lesions while simultaneously relying on certain DNA repair pathways for survival (Hanahan and Weinberg 2011). This enhanced dependence on certain DDR components—sometimes via the concept of synthetic lethality, where loss of one cellular pathway leads to reliance on an alternative pathway—came into the spotlight with the development of poly-(ADP-ribose) polymerase (PARP) inhibitors as precision medicines for certain cancers harboring defects in the DDR mechanism of homologous recombination (Lord and Ashworth 2017). PARP enzymes (mainly PARP1 and PARP2) fulfill various functions during DNA repair, but in particular, PARP1 is important for the effective repair of DNA single-strand breaks (SSBs). Notably, alongside inhibition of SSB repair, PARP inhibitors compete with the cofactor NAD+, thereby preventing poly-ADP-ribosylation (PARylation) of PARP1 itself and various other proteins. Because auto-PARylation promotes the release of PARP1 from DNA, PARP inhibitors result in PARP1 becoming “trapped” on DNA. Importantly, much of the cytotoxicity of PARP inhibitors has been attributed to when DNA replication forks encounter these trapped PARP–SSB complexes, leading to replication fork collapse and generation of DNA double-strand breaks (DSBs), specifically single-ended DSBs (a DSB end with no associated DNA end to be ligated to). The potential to exploit this scenario became apparent when two independent studies reported dramatic cytotoxicity of PARP inhibitors in the context of deficiencies in the tumor suppressor genes BRCA1 and BRCA2 (breast cancer susceptibility genes 1 and 2) (Bryant et al. 2005; Farmer et al. 2005). BRCA1 and BRCA2 have crucial functions during homologous recombination (HR)-mediated repair of DSBs (Fig. 1). While PARP inhibition is dispensable for cellular survival in HR-proficient cells, it becomes extremely toxic in the context of BRCA1/2 deficiency, highlighting the key role of HR in repairing PARP inhibitor-induced damage (Lord and Ashworth 2016). So far, four different PARP inhibitors (olaparib, niraparib, rucaparib, and talazoparib) have been approved by the FDA for use as single-agent chemotherapeutics (Table 1).
Figure 1.
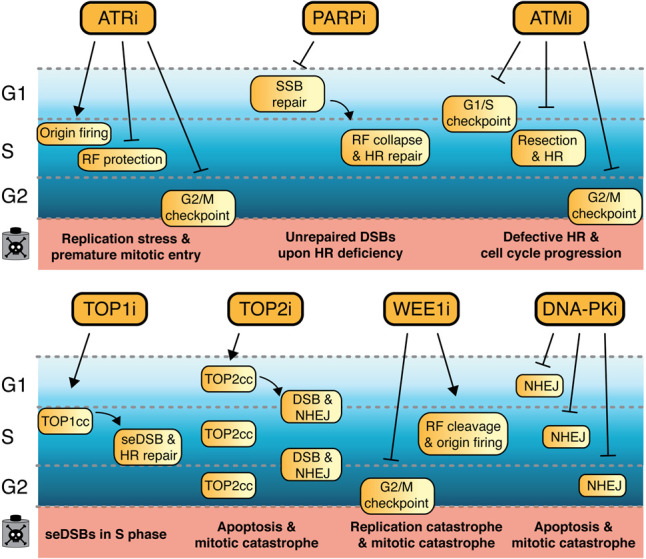
DDR inhibitors and their impacts on DSB repair and cell cycle progression. In mammalian cells, four main DSB pathways exist, which operate dependent on the stage of the cell cycle: nonhomologous end-joining (NHEJ), alternative end-joining (alt-EJ), homologous recombination (HR), and single-strand annealing (SSA) (Scully et al. 2019). TOP2i induced DSBs are predominantly repaired via NHEJ, which while being described as “error-prone,” ensures effective repair of broken DNA ends particularly during G0 and in the G1 phase of the cell cycle. DNA-PK is crucial for effective NHEJ, and DNA-PK inhibitors impair DSB repair via NHEJ. TOP1 inhibitors generate single-ended DSBs (seDSBs) in S phase, which require HR for accurate repair. HR is a form of DNA recombination where DNA homology and synthesis can accurately regenerate the sequence surrounding the DSB, facilitated by a sister chromatid as template and therefore restricted to S or G2 (Karanam et al. 2012). DNA damage arising from PARP inhibition also requires HR, since spontaneously occurring SSBs are converted into seDSBs during DNA replication upon PARP inhibitors (PARPis). In addition, PARPi cause mitotic catastrophe and induce DNA replication stress through altering DNA replication fork speed. A fundamental step during HR-mediated repair is DNA end resection, generating ssDNA overhangs that are rapidly covered by RPA and consequently by RAD51. ATR is activated by ssDNA as a result of DNA end resection or DNA polymerase uncoupling from helicase activity during DNA replication. Consequently, ATR inhibitors primarily have impacts during S and G2 phases of the cell cycle. Additionally, ATRi overrides the G2/M cell cycle checkpoint, therefore causing premature entry into mitosis. Importantly, ATM inhibition affects efficient HR, alongside its impact on G1/S and G2/M cell cycle checkpoints in response to DNA damage. Analogous to ATRi and ATMi, WEE1 inhibitors affect the G2/M cell cycle checkpoint. Moreover, WEE1i cause replication stress through dysregulated origin firing and cleavage of DNA replication forks, resulting in DSBs.
Table 1.
Inhibitors targeting the DNA damage response and their clinical development

The clinical successes of PARP inhibitors have nurtured efforts to target other components of the DNA repair network for therapeutic applications. Thus, the three apical PI3K-like kinases ATM, ATR, and DNA-PK, in addition to the S-phase checkpoint kinase CHK1 and G2 checkpoint kinase WEE1, have emerged as promising targets for small molecule inhibitors, with several of these compounds currently undergoing clinical development (Blackford and Jackson 2017; Forment and O'Connor 2018). The justification for targeting ATR in cancer cells centers around ATR's functions in protecting stalled DNA replication forks, regulating replication origin firing, and controlling the transition of cells from G2 phase into mitosis by enforcing the G2/M cell cycle checkpoint (Saldivar et al. 2017). Due to their proliferative nature and being subject to oncogenic forces that either lead to heightened levels of reactive oxygen species or deregulated S-phase entry and/or progression, cancer cells are particularly vulnerable to ATR inhibition, experiencing extensive DNA and chromosomal damage. Furthermore, it has been found that ATR inhibitors are selectively toxic to cancer cells harboring mutations in the ATM tumor suppressor, in part because ATM and ATR share some overlapping targets (Kantidze et al. 2018). Additionally, ATM loss impairs DSB repair by HR and also weakens the G1/S cell cycle checkpoint, thereby generating more replicative stress (Lecona and Fernandez-Capetillo 2018). Several ATR inhibitors are currently undergoing clinical development, with their application mainly aiming to exploit high levels of replication stress in tumors. Since CHK1 is the effector kinase of ATR, several cellular functions are shared between these kinases, and the rationales behind exploring CHK1 inhibitors for cancer therapy are hence similar to those for ATR inhibitors (Table 1; Forment and O'Connor 2018).
WEE1 is involved in responses to DNA damage, whereby it enforces a cell cycle arrest/checkpoint at the transition from S phase into M phase in response to DNA damage or replication stress. WEE1 phosphorylates and inhibits cyclin-dependent kinases (CDKs) CDK1 and CDK2, thus counteracting cell cycle progression and unscheduled replication-origin firing (Elbæk et al. 2020). One of the concepts for the clinical application of WEE1 inhibitors is that WEE1 inhibition should potentiate the effects of certain other DNA damage-inducing chemotherapeutics, since cells undergoing WEE1 inhibition would enter mitosis with DNA lesions or underreplicated DNA, thereby causing mitotic catastrophe and ensuing cell death. Indeed, the efficacy of WEE1 inhibition in combination with other DNA-damaging agents such as platinum drugs or irradiation has been observed in preclinical model systems (Hirai et al. 2009; PosthumaDeBoer et al. 2011).
DNA-PK and ATM are key protein kinases that function in the DDR to promote DNA nonhomologous end-joining (NHEJ) and HR, respectively, with ATM also playing major roles in regulating signaling cascades in response to irradiation and other DNA-damaging agents (Blackford and Jackson 2017). Because their absence or inhibition sensitizes cells to irradiation, PARP inhibitors, and various DNA-damaging chemotherapeutic agents, there is the potential for using ATM or DNA-PK inhibition to enhance antitumor efficacies of radiotherapy and certain chemotherapies, although their potential to also accentuate toxic side effects in patients will have to be carefully managed in such contexts (Table 1).
In contrast to DDR enzyme inhibitors, other classes of drugs possess the potential to generate DNA damage and therefore are classed as genotoxic agents. Among them, topoisomerase inhibitors are well-established chemotherapeutics for a variety of cancers. The topoisomerase enzymes TOP1 and TOP2 generate transient DNA breaks in order to resolve topological stresses during DNA replication and transcription, using transesterification reactions to break the DNA phosphodiester backbone while at the same time forming a transient covalent bond between the enzyme's catalytic tyrosine (Tyr) and the DNA, known as the topoisomerase cleavage complex (TOPcc). Exploiting the potential of topoisomerases to induce DNA damage, especially in highly replicating and transcribing cells, underpinned the development of several classes of TOP1 and TOP2 inhibitors. Among others, molecules that reversibly stabilize TOPccs to block the religation reaction have had dramatic impacts as chemotherapeutic agents. Since these molecules act as interfacial inhibitors (functioning via stabilizing a reaction intermediate), DNA damage arises from TOP1 or TOP2 that has become trapped in the state where the DNA backbone is already cut and the enzyme cross-linked to DNA, but the reverse reaction is abrogated (Hsiang et al. 1985; Covey et al. 1989; Pommier 2009). In the context of TOP1, trapped TOP1cc becomes particularly toxic in replicating cells when DNA replication forks encounter these lesions, leading to replication fork arrest, fork collapse, and eventually DNA DSB formation (Hsiang et al. 1989; Strumberg et al. 2000; Furuta et al. 2003). Although camptothecin was among the first TOP1 inhibitors to be identified, its clinical application was not pursued further, and the camptothecin derivatives topotecan and irinotecan instead gained regulatory approval (Table 1). Clinically approved TOP2 inhibitors, such as etoposide and doxorubicin (Table 1), function as interfacial inhibitors by trapping the TOP2ccs on DNA and preventing religation of the DNA backbone, analogous to the actions of camptothecin derivatives on TOP1 (Wu et al. 2011, 2013a). However, due to TOP2's dimeric mode of action, key aspects of TOP2 inhibitors’ mechanism are their ability to directly induce DSBs and consequently yield such toxic lesions in all stages of the cell cycle (Holm et al. 1989).
Platinum-based chemotherapeutics represent another class of genotoxic agents, which have found broad application in cancer therapy. Initially, cisplatin was FDA approved for the treatment of testicular and ovarian cancers in 1978, whereas nowadays cisplatin and its derivates carboplatin and oxaliplatin are established chemotherapeutic agents for various tumor types (Kelland 2007). Platinum-based agents create DNA monoadducts and DNA cross-links, which impair cellular processes such as DNA replication and transcription and require specific DNA repair pathways for their resolution. As a consequence, platinum agents are strong inducers of cell cycle arrest and apoptosis (Rottenberg et al. 2021). Mechanistically, cisplatin is activated intracellularly through the aquation of the chloride group(s) and subsequently chemically cross-link DNA molecules, preferably at N7 or O6 in purines. Notably, platinum agents can cross-link proteins to DNA, although the occurrence of this type of lesion is much rarer compared with DNA intra-strand and inter-strand cross-links (Siddik 2003). Alongside topoisomerase inhibitors and platinum drugs, radiation therapy can be classified as a genotoxic treatment and represents the most frequently applied cancer therapy aside from surgery. The idea of radiation therapy is based on the locally targeted induction of DNA damage in order to deprive cancer cells of their proliferative potential by forcing them into senescence, apoptosis, or other forms of cell death (Bernier et al. 2004). Since radiation therapy induces a variety of DNA lesions, including DSBs, SSBs, base damage, and cross-links, the application of various DDR enzyme inhibitors can further potentiate the cytotoxic effects of radiation therapy (Moding et al. 2013).
Sensing damaged DNA by components of the innate immune system
Alongside the immediate cytotoxic effects and persistent DNA damage caused by certain DDR inhibitors (DDRis), induction of innate immune responses has frequently been observed in such settings. Unrepaired DNA lesions and/or impaired chromosome segregation during mitosis contribute to the formation of endogenous cytosolic DNA, often in the form of micronuclei. Although their origin is distinct, DNA damage-induced cytosolic DNA shares common downstream effects and cellular responses with those observed upon viral or bacterial infections. In mammalian cells, multiple factors and enzymes have the capability to sense cytosolic DNA to activate appropriate immune responses. The induction of innate immune responses by the presence of cytosolic DNA not only is crucial for the host organism's first-lines of defense against invading pathogens but also fulfils key oncosuppressive functions through elimination of damaged cells (Vanpouille-Box et al. 2018). Intensive research in recent years has led to the discovery and characterization of the cGAS/STING system as a major component of cells’ intrinsic immune response to the occurrence of cytosolic oligonucleotides. cGAS is a cytosolic nucleotidyltransferase that, upon binding DNA, catalyzes the synthesis of cyclic GMP-AMP (cGAMP) from ATP and GTP, which subsequently acts as a messenger molecule for the adaptor protein STING (Ablasser et al. 2013; Sun et al. 2013; Wu et al. 2013b). cGAS binds double-stranded DNA (dsDNA) in a sequence-nonspecific manner through interaction with the sugar-phosphate backbone, although a preference for specific DNA length (>45 bp) has been observed (Civril et al. 2013; Li et al. 2013; Herzner et al. 2015; Zhou et al. 2018). Upon cGAMP binding, STING, originally located in the endoplasmic reticulum (ER) membrane, translocates to the Golgi apparatus via the ER–Golgi intermediate compartment, while activating at least three distinct kinases—TBK1, IKK, and NIK (TANK-binding kinase 1, IκB kinase, and NFκB-inducing kinase, respectively)—during this process. The activation of these kinases and concomitant phosphorylation of interferon regulatory factor 3 (IRF3) and IκBα transduces the initial stimulus of cytosolic DNA into IRF3 and NFκB transcriptional programs, which control the balance between cellular survival and controlled cell death (Fitzgerald et al. 2003; Ishikawa and Barber 2008; Ishikawa et al. 2009; Tanaka and Chen 2012).
In order to prevent the accumulation of cytosolic DNA and potentially persistent proinflammatory signaling, several enzymes and factors constantly survey and eliminate nucleic acids appearing in the cytosol. One of these, TREX1, is a 3′–5′ exonuclease that acts on either single-stranded DNA (ssDNA) or dsDNA and comprises the predominant enzyme that degrades cytosolic DNA in mammalian cells (Yang et al. 2007; Stetson et al. 2008; Lindahl et al. 2009). Mutations in TREX1 result in dysregulation of type I IFN production and are associated with the autoimmune disorders Aicardi-Goutières syndrome (AGS) and familial chilblain lupus (Crow et al. 2006a; Rice et al. 2007). Type I IFN responses observed in TREX1-deficient cells are dependent on the cGAS/STING system, which presumably recognizes elevated cytosolic DNA arising in the absence of its TREX1-mediated clearance. In line with these observations, inactivation of cGAS/STING abrogates type I IFN signaling in TREX1-deficient cells and rescues the embryonic lethality of Trex1−/− mice (Stetson et al. 2008; Ablasser et al. 2014; Gray et al. 2015). Notably, individuals with mutations in the RNase H2 complex, consisting of RNASEH2A, RNASEH2B, and RNASEH2C, show clinical pathologies that resemble those observed in TREX1 mutated patients, including aberrant type I IFN signaling along with AGS and systemic lupus erythematosus. Although this maladaptive immune response upon impaired RNase H2 function relies on cGAS/STING, as is the case for TREX1 dysfunction, it is unlikely that RNase H2 directly eliminates cytosolic DNA (Crow et al. 2006b; Mackenzie et al. 2016). Rather, RNase H2 functions inside the nucleus by resolving DNA–RNA hybrids and removing ribonucleotides during DNA replication, thereby preserving genome integrity by restricting cytosolic accumulation of broken DNA molecules (Reijns et al. 2011, 2012). Notably, DNA–RNA hybrids harbor the potential to directly stimulate cGAS and, consequently, cGAMP production in vitro (Mankan et al. 2014). Although the induction of cGAMP synthesis in the presence of DNA–RNA hybrids is orders of magnitude lower compared with cGAMP synthesis upon dsDNA stimulation, it may be that DNA–RNA hybrids represent direct stimuli for IFN responses upon RNase H2 dysfunction in certain settings. Recently, it became apparent that the simple model of cGAS being exclusively localized in the cytoplasm might not represent the full picture. Several reports indicated nuclear functions for cGAS, suggesting that a subfraction of the cellular cGAS pool resides within the nucleus (Liu et al. 2018a; Jiang et al. 2019). The concomitant conflict of autoreactivity against self-DNA is reported to be overcome by the tethering of cGAS to the acid patch of nucleosomes via its DNA binding domains, thus blocking DNA binding and activity toward chromatinized DNA (Boyer et al. 2020; Kujirai et al. 2020; Michalski et al. 2020; Pathare et al. 2020; Zhao et al. 2020). This mechanism may also give a plausible explanation to the long-standing question of how cGAS activity is suppressed in mitosis, when the nuclear envelope breaks down and chromosomes are exposed to cytosolic cGAS (Zierhut et al. 2019).
Impacting cellular immune responses through DDR inhibitors
The importance of cytosolic DNA-stimulated immune responses in the context of cancer therapy is highlighted by the contribution of cGAS/STING to antitumor immunity in response to radiotherapy. Longstanding reports have indicated that, besides generating cytotoxic DNA damage, radiotherapy also induces a tumor-specific immune response that contributes to the efficacy of this therapeutic modality (McBride et al. 2004). Secretion of inflammatory cytokines such as TNF-α, IL-1α, and IL-6 has been observed in response to ionizing radiation, which ultimately leads to adaptive immune responses via CD8+ T cells (Hallahan et al. 1989; McBride et al. 2004; Lee et al. 2009; Di Maggio et al. 2015). Recent reports have established that cGAS/STING plays a key role in linking innate and adaptive immune responses after irradiation. Specifically, cGAS/STING mediates increased IFN-β production in the tumor environment to promote antitumor responses upon irradiation. Impaired type I IFN signaling resulting from cGAS/STING inactivation can be bypassed by exogenous supplementation of cGAMP to induce the antitumor efficacy of radiotherapy, further supporting the idea that in response to radiotherapy, cGAS/STING is an initial sensor and signal transducer of antitumor immune responses (Burnette et al. 2011; Woo et al. 2014; Wu et al. 2014; Deng et al. 2014b). From the above, one can assume that the stimulus that activates cGAS/STING after irradiation must originate from cytosolic DNA. Indeed, micronuclei, resulting from irradiation and ensuing incomplete DNA repair, are recognized by cGAS/STING to activate type I IFN signaling (Fig. 2). Importantly, micronuclei formation requires progression through mitosis following DNA-damage induction, wherein the dissolution and ensuing reformation of the nuclear membrane end up enclosing DNA fragments in a micronuclear envelope (Harding et al. 2017; Mackenzie et al. 2017). Although this envelope shares similarities with the normal nuclear envelope, its assembly remains rudimentary, reflecting its fragility that frequently results in the breakdown of envelope integrity and cytosolic exposure of genomic DNA (Liu et al. 2018b). This phenomenon is particularly important in explaining cGAS/STING activation in response to micronucleation, since cGAS recognizes cytosolic DNA rather than the outer structure of a micronucleus (Mackenzie et al. 2017). Interestingly, TREX1 antagonizes immunogenicity of cancer cells following radiation via its own up-regulation and the consequent enhanced degradation of cytosolic DNA. However, continuous low-dose irradiation circumvents TREX1 engagement and promotes cGAS/STING-dependent IFN-β secretion for antitumor immunity (Vanpouille-Box et al. 2017). In addition, TREX1 inhibits the transfer of dsDNA from irradiated tumor cells to dendritic cells (DCs) via tumor-derived exosomes, resulting in dampened cGAS/STING activation and IFN-β production in DCs (Diamond et al. 2018).
Figure 2.
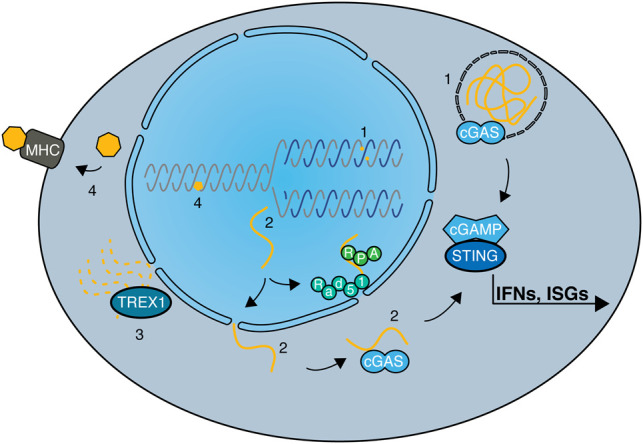
DNA repair defects and their impacts on cellular immune responses. Defects in DNA repair pathways or DDR components affect innate and adaptive immune responses in various ways. (1) The induction of DSBs via chemotherapeutics or irradiation can lead to micronuclei formation and consequent recognition of cytosolic DNA by cGAS/STING. Deficiencies in genes encoding proteins such as BRCA2, RNase H2, or ATM further augment these effects. (2) Cytosolic DNA can also be a result of aberrant processing of DNA replication intermediates, with several DDR factors limiting either the generation (SAMHD1) or the translocation (RAD51 and RPA) of cytosolic DNA from the nucleus, or degrading DNA once it is present in the cytosol (TREX1). (3) Activation of the cGAS-cGAMP-STING cascade leads to IRF3 and NFκB transcriptional programs, resulting in expression of IFN and ISGs, therefore inducing strong innate immune responses. (4) In contrast, MMR defects can lead to adaptive immune responses through increased somatic mutations and consequent synthesis of neoantigens. When presented by MHC molecules at the cell surface, neoantigens elicit a strong T-cell response, dependent on the immunogenicity of the neoantigen.
Besides radiation-induced antitumor responses, conventional chemotherapy has the potential to stimulate the innate and adaptive immune systems. As described above, HR deficiency (HRD) is a predictor of PARP inhibitor efficacy and PARP inhibitors are particularly cytotoxic in tumors displaying mutations in BRCA1/2. In this regard, it is worthwhile mentioning that beside the generation of DNA damage, induction of a STING-dependent antitumor immunity is a considerable feature of PARP inhibitor cancer therapy. Specifically, PARPi-mediated trapping of PARP on DNA lesions appears to be influential for the innate immune response, as the extent of PARP trapping correlates with the magnitude of innate immune signaling (Kim et al. 2020). PARP inhibitor-mediated immunogenicity depends on the activation of cGAS/STING to elicit a cytotoxic T-cell response. Intriguingly, this immune response is augmented upon BRCA deficiency, since Brca1-proficient mouse tumors show mitigated immunogenicity in response to PARP inhibitors as compared with Brca1 mutated ones (Ding et al. 2018; Pantelidou et al. 2019). In line with these observations, PARP inhibition led to a strong induction of interferon-stimulated genes (ISGs) in BRCA2-deficient cells compared with their BRCA2-proficient counterpart (Reislander et al. 2019). However, it has been reported that PARP inhibitors have the potential to initiate cGAS/STING-dependent type I IFN production in human cancer cells independent of their BRCA gene status (Shen et al. 2019), presumably due to the mitotic defects or increased replication stress caused by PARPi affecting replication fork speed (Maya-Mendoza et al. 2018; Slade 2019). Future studies are required to illuminate the underlying determinants for the differential responses in BRCA-proficient and BRCA-deficient contexts.
Other chemotherapeutic agents that generate DNA damage also affect cellular immune responses in various ways, often depending on the DNA damage-induced release of cytosolic DNA. For example, the topoisomerase II inhibitor teniposide induces STING-dependent type I IFN signaling and NF-κB activation in a mouse tumor model, with the consequent DC and T-cell activation promoting antitumor responses, including increased immune cell infiltration (Wang et al. 2019). Furthermore, the TOP2 inhibitors doxorubicin and daunorubicin induce IFN-β production in human cancer cell lines via a mechanism shown to be dependent on STING function (Luthra et al. 2017). Notably, doxorubicin treatment enhances CD8+ T-cell amplification and infiltration, as well as IFN-γ production, in tumor environments in mice (Mattarollo et al. 2011). Similar to TOP2 inhibitors, topoisomerase I inhibitors trigger potential immunogenicity, exemplified by DC and CD8+ T-cell activation in mice treated with topotecan (Kitai et al. 2017).
Despite lacking clear evidence for affecting the cellular immune system, ATR inhibitors have been shown to potentiate immune stimulations in response to radiotherapy. Thus, combinatorial treatment of radiotherapy and ATR inhibition was found to induce type I/II IFN-based gene expression changes and CD8+ T-cell infiltration in a manner dependent on cGAS/STING (Vendetti et al. 2018; Dillon et al. 2019; Sheng et al. 2020). While ATR inhibitors do not damage DNA directly, it can be assumed that the increased immunogenicity in irradiated tumors in the context of ATRi results from overriding the G2/M cell cycle checkpoint, with an increased proportion of cells with unrepaired DNA lesions entering mitosis, leading to DNA fragmentation and micronuclei formation capable of triggering innate immune responses (Ruiz et al. 2016; Harding et al. 2017). In line with this model, it has been observed that inhibition of the ATR effector kinase CHK1 abrogates the G2/M checkpoint post irradiation, leading to micronuclei formation and type I IFN signaling in cancer cells (Chao et al. 2020). Moreover, increased CD8+ T-cell infiltration and tumor volume reduction was observed in mice treated with a combination of radiotherapy and the CHK1 inhibitor AZD7762 compared with treatments with these agents individually (Chao et al. 2020). Similar to ATR inhibitors, pharmacologic inhibition of ATM in combination with radiotherapy was found to induce type I IFN signaling, which notably occurred independent of cGAS/STING but was reliant on TBK1 (Zhang et al. 2019).
Consequences of DDR defects for immune responses
The involvement of DDR factors and their inhibition in the induction of innate immune responses has been highlighted by the consequences of RNase H2 dysfunction in the autoimmune disorder AGS. While certain DDR factors such as TREX1 participate in the sensing of extranuclear DNA, others influence cellular immunity via more indirect mechanisms. The tumor suppressor BRCA2 has a pivotal role in HR-mediated repair and in the protection of stalled replication forks, while its absence is accompanied by genome instability and chromosome breakage (Prakash et al. 2015). Because BRCA2 loss is highly detrimental to cell viability, BRCA2-deficient cancer cells have invariably undergone adaptation processes in order to survive, exemplified by the inactivation of p53 functions. Importantly, alongside rewiring of DNA repair processes, the absence of BRCA2 causes enhanced phosphorylation and therefore activation of STAT1-IRF3, followed by concomitant up-regulation of ISGs. Mechanistically, this adaptive immune response is dependent on cGAS/STING, with BRCA2-deficient cells exhibiting an increase in cGAS-positive micronuclei compared with BRCA2-proficient cells, presumably as result of the DNA repair defects and chromosome instability observed in the absence of BRCA2 function (Reislander et al. 2019). In line with these observations, elevated secretion of proinflammatory cytokines, such as TNF-α, arises as a consequence of BRCA2 loss, resulting from cytosolic DNA sensing by cGAS/STING and ensuing interferon responses (Heijink et al. 2019). Additionally, it has been shown that in the absence of BRCA2, RNase H2 recruitment to DSBs is impaired, resulting in increased DNA–RNA hybrid formation and thus providing and additional explanation for cGAS/STING activation in BRCA2-deficient cells (D'Alessandro et al. 2018). Importantly, the abrogation of TNF-α signaling improves viability of BRCA2-deficient cells, indicative of TNF-α signaling promoting cell death when BRCA2 function is impaired (Heijink et al. 2019). Besides BRCA2 deficiency causing induction of interferon responses and an increase in TNF-α signaling per se, it also further sensitizes cells to autocrine TNF-α signaling.
Notably, depletion of BRCA1 or the inter-strand DNA cross-link (ICL) repair factor FANCD2 sensitizes cells to recombinant TNF-α, suggesting that the general impairment of HR repair or replication stress arising from compromised ICL repair harbors the potential to make cells more susceptible to interferon responses (Heijink et al. 2019). The idea of unprotected replication forks comprising an entry point for aberrant DNA processing and consequent leakage into the cytosol is further supported by the report that mutations in SAMHD1 are causative for the autoimmune disorder AGS (Crow and Manel 2015). Besides its role as a dNTPase (deoxynucleotide triphosphohydrolase), SAMHD1 promotes the controlled degradation of newly synthesized DNA at stalled replication forks via the exonuclease MRE11. In the absence of SAMHD1, nascent DNA at stalled replication forks is displaced by the RECQ1 helicase and translocates to the cytosol, where it activates the cGAS/STING pathway and ensuing type I IFN responses (Coquel et al. 2018).
In order to avoid chronic proinflammatory signaling as occurs in AGS patients, several cellular mechanisms have evolved to counteract or minimize leakage of DNA fragments into the cytosol. The DNA repair proteins RPA and RAD51 both have the capability to directly bind ssDNA, which is crucial for their functions during DNA replication and DNA repair (Bhat and Cortez 2018). In addition, they prevent the accumulation of cytosolic DNA by binding and therefore retaining ssDNA within the nucleus, thereby working in cooperation with nuclear membrane-bound TREX1, which normally swiftly degrades any DNA leaking into the cytosol (Fig. 2). Exhaustion of the available RPA/RAD51 pool in the absence of TREX1, or upon depletion of RPA or RAD51, results in cGAS/STING-dependent type I IFN signaling (Wolf et al. 2016). Additionally, RAD51 protects newly synthesized DNA from aberrant processing by MRE11 and consequent cytosolic DNA translocation and ensuing cGAS/STING activation (Bhattacharya et al. 2017). Notably, the functions of MRE11 as a nuclease that processes DNA replication fork intermediates can be seen as a double-edged sword. On one hand, MRE11 prevents cGAS/STING activation through trimming of replication intermediates, stimulated by SAMHD1. On the other hand, excessive processing of unprotected, stalled replication forks in the absence of RAD51 by MRE11 generates cytosolic DNA and consequently stimulates cGAS/STING. Whether or in which form RAD51 cooperates with SAMDH1 for fine-tuning of this process is an interesting area for future research. Besides MRE11-dependent degradation of nascent DNA at stalled or collapsed replication forks, other DNA nucleases can process DNA structures in such contexts, subsequently leading to activation of innate immune responses via cytosolic DNA sensing. In particular, the actions of the structure-specific endonuclease MUS81 can bring about the accumulation of cytosolic DNA via MUS81-dependent cleavage of stalled replication forks, leading to cGAS/STING activation and induction of type I IFNs, as observed in various prostate cancer cell lines (Ho et al. 2016). Moreover, the MUS81–STING axis is responsible for a prostate cancer cell-specific T-cell response in mice, thus highlighting a likely role for MUS81 in promoting antitumor immunity. In addition, endogenously arising DSBs are capable of stimulating innate immune responses in situations where accurate DNA repair is compromised. Accordingly, deficiency in ATM induces type I IFNs in unchallenged conditions, indicative of spontaneously arising DNA DSBs being the stimulus of this immune response. Furthermore, in the absence of ATM, ssDNA resulting from unrepaired DNA lesions accumulates in the cytoplasm and activates cGAS/STING-dependent type I IFN signaling (Härtlova et al. 2015). Notably, this DDR defect primes type I IFNs to enhance the innate immune response toward invading pathogens, highlighting the fact that damaged DNA serves as a danger signal for cellular homeostasis and prepares the innate immune system for a rapid response in the face of bacterial challenges (Härtlova et al. 2015).
Mismatch repair deficiency and adaptive immunity
Deficiencies in DNA repair pathways can also affect adaptive immune responses, which are key for immunogenicity of tumors and have clinical implications in the context of cancer immunotherapy. Additionally, DNA damage responses are crucial during the development of the immune system and maturation of immune cells, as exemplified by the controlled induction and concomitant repair of DSBs during V(D)J recombination and class switch recombination in lymphocytes (for an extensive review, see Bednarski and Sleckman 2019). Furthermore, over recent years, a connection between defects in the DNA mismatch repair (MMR) pathway and tumor immunogenicity has been observed both in preclinical model systems and in cancer patients. The MMR machinery detects and replaces base mismatches resulting from erroneous DNA replication or repair and, in particular, plays key roles in correcting small insertion or deletions (indels) arising at repetitive sequences in the genome, so-called microsatellite instability (Kunkel and Erie 2015). Tumors harboring mutations in genes encoding the core MMR factors MLH1, MSH2, MSH6, or PMS2 are characterized by microsatellite instability (MSI), a hypermutator phenotype specified by large numbers of single-nucleotide variants (SNVs) and indels, thus leading to a high mutational burden. Additionally, specific DNA proofreading mutations in the replicative DNA polymerases POLE and POLD1 drive hypermutation phenotypes in some cancers without causing MSI (Campbell et al. 2017). MSI predominantly occurs in endometrial, gastric, and colorectal cancers, suggesting that such tissue types or the tumor environment and extracellular influences affect MSI development (Hause et al. 2016; Cortes-Ciriano et al. 2017). The correlation between MSI colorectal tumors and high numbers of tumor-infiltrating lymphocytes has long been acknowledged. Colorectal MSI tumors frequently display high infiltration of CD8+ cytotoxic T cells, type 1 helper (Th1) cells, and memory T cells, alongside the up-regulated expression of interferon γ (IFN-γ) and immune checkpoint molecules PD-1, PD-L1, and CTLA4 (Fig. 3; Llosa et al. 2015; Mlecnik et al. 2016).
Figure 3.

Innate and adaptive immune responses in cancer. In recent years, the concept of cancer immunoediting evolved from traditional views of the immune system constantly surveying and eliminating transformed cells in order to counteract cancer development (immunosurveillance) (Keast 1970). Cancer immunoediting unifies observations of the immune system promoting tumor outgrowth with reports of immunosurveillance, highlighting the dual functions of the immune system during tumor development (Schreiber et al. 2011). The cancer immunoediting concept consists of three phases: elimination, equilibrium, and escape. In the elimination phase, components of both the innate and adaptive immune response recognize and destroy cells undergoing oncogenesis. Elimination is promoted by a number of signaling molecules such as type I and type II IFNs and is executed via the interplay of a subset of immune cells such as CD8+ T cells, dendritic cells (DCs), natural killer cells (NKs), natural killer T cells (NKTs), proinflammatory (“M1”) macrophages, and others (Mittal et al. 2014). Notably, the DDR participates in this process, since DNA damage induction in tumors cells results in up-regulation of ligands for the receptors NKG2D and DNAM-1, therefore stimulating cytotoxicity of NK and CD8+ T cells in addition to IFN-γ secretion (Gasser et al. 2005; Croxford et al. 2013). Moreover, radiotherapy-induced DNA damage, and consequent cell death due to uncomplete DNA repair, stimulates cross-presentation by dendritic cells and increased lymphocyte influx, thus further contributing to cancer cell elimination (Deng et al. 2014b; Samstein and Riaz 2018; Cornel et al. 2020; Cheng et al. 2021). Paradoxically, TNF-α has both antitumor and tumor-promoting activity. When secreted by macrophages and innate immune cells, TNF-α induces cancer cell elimination, whereas chronic inflammation promoted by TNF-α signaling can drive carcinogenesis (Balkwill 2009; Charles et al. 2009). In the equilibrium phase, the adaptive immune system holds the tumor in a dormant state with cancer cells resisting constant immune recognition through genetic and epigenetic changes in antigen presentation and immunosuppressive pathways. Cancer cells achieve immune evasion by various mechanisms, including loss of tumor antigens or factors involved in antigen presentation, such as type I HLA (MHC) function, expression of inhibitory ligands (e.g., PD-L1 and CTLA-4), secretion of immunosuppressive cytokines (IL-10, TGF-β), and recruitment of tumor-associated macrophages (TAMs) and regulatory T cells (Tregs). These scenarios result in the inability of innate and adaptive immune cells to recognize and appropriately respond to oncogenic cells, therefore facilitating tumor progression (escape phase) (Vinay et al. 2015).
MSI endometrial tumors also exhibit increased numbers of tumor-infiltrating lymphocytes and elevated expression of PD-1/PD-L1 compared with microsatellite stable (MSS) tumors (Howitt et al. 2015). Recently, this connection has been attributed to the increase in neoantigens due to the high mutational burden in these settings (Tougeron et al. 2009; Maby et al. 2015). To a certain degree, it seems reasonable to assume that the more mutations in the genome, the more likely it is that neoantigens are formed and in due course recognized by the immune system (Fig. 2; Turajlic et al. 2017). In line with this idea, increased mutational burden correlates with improved survival in colorectal cancer patients (Giannakis et al. 2016). Interestingly, additional tumor types such as melanomas and lung cancers, despite being microsatellite stable (MSS), exhibit an increased number of overall mutations compared with others, most likely due to exposure to exogenous mutagens, including ultraviolet light and tobacco smoke components, respectively, suggesting that these may also promote antitumor immune responses (Alexandrov et al. 2013). In accord with this idea, a recent study by Bardelli and colleagues (Germano et al. 2017) demonstrated that colorectal, breast, and pancreatic mouse cancer cell lines, where MMR was genetically inactivated, grew significantly slower when transplanted into immunocompetent mice compared with the isogenic MMR-proficient cancer cell lines, indicating rejection by the host immune system. Enhanced immunosurveillance was accompanied by accumulation of neoantigens over time in MMR-deficient cells, wherein the amount of neoantigens remained stable in MMR-proficient cells, implicating neoantigens generated by MMR deficiency as a direct cause for immune system-mediated elimination of cancer cells (Germano et al. 2017). Importantly, fully established MMR-deficient tumors are often sensitive to immune checkpoint inhibitors in people, which points toward MMR deficiency as an important determinant of immune checkpoint blockade (Fig. 4) efficacy, at least in certain cancer types (Le et al. 2015).
Figure 4.
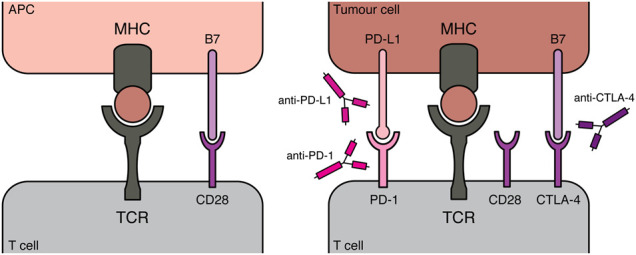
Principles of immune checkpoint inhibitors for cancer therapy. As described in Figure 3, during the escape phase of the immunoediting concept, tumor cells evade immune recognition and destruction by active immunosuppression in the tumor. A milestone for the field of immune checkpoint inhibitors was the report in which melanoma patients treated with an antibody targeting the T-cell checkpoint protein CTLA-4 showed significantly improved survival compared with the control group (Hodi et al. 2010). This suggested that targeting suppressive immune checkpoints can improve overall survival in melanomas, indicating that a patient's immune system has capabilities to control tumor growth once immunosuppressive signals are overcome. Since this pharmacological approach targets the patient's immune system rather than the tumor itself, a new field for clinical research arose. CTLA-4 is a surface receptor on T cells. To acquire effector function, a T-cell recognizes its compatible antigen, presented by MHC molecules of an antigen-presenting cell (APC), via its T-cell receptor (TCR). However, this initial recognition is insufficient, with binding of the CD28 T cell receptor to B7 molecules (CD80 or CD86 ligand) on APCs serving as crucial costimulatory signals to adequately prime T cells. CTLA-4 translocates to the cell surface once T cells are activated, where it binds CD80 and CD86 with higher affinity than CD28, therefore dampening T-cell activation (Walunas et al. 1994; Krummel and Allison 1995). Moreover CTLA-4 expression by Tregs is crucial for their immune suppressive functions, potently binding CD80/CD86 ligands on APCs and therefore preventing T-cell activation (Takahashi et al. 2000; Wing et al. 2008). Following its initial success in clinical trials, the CTLA-4 antibody ipilimumab was FDA approved in 2011 for treating melanomas. PD-1 represents another inhibitory receptor present on T cells, while its ligands PD-L1 and PD-L2 can be expressed by various cell types, including APCs and malignant cells, predominantly after exposure to inflammatory cytokines such as IFN-γ. Engagement of PD-L1 with its receptor PD-1 interferes with TCR signaling, therefore limiting T-cell responses toward tumor cells (Freeman et al. 2000; Dong et al. 2002). Following this rational, antibodies targeting PD-1/PD-L1 have provoked clinical benefits in various types of cancers, warranting FDA approval of pembroluzimab and nivolumab (both PD-1 antagonists) in 2014. Unlike CTLA-4, PD-1/PD-L1 does not interfere with costimulation during the T-cell activation, suggesting that combination therapy of CTLA-4 and PD-1/PD-L1 antibodies could have synergistic therapeutic effects. Regaining T-cell activation, by blocking inhibitory signals during costimulation via CTLA-4 antibodies, could drive increased PD-L1 expression in tumor cells, making them particularly susceptible to PD-1/PD-L1 checkpoint blockade (Sharma and Allison 2015).
Combined targeting of DDR and immune checkpoints in cancer therapy
Immune checkpoint inhibition (or immune checkpoint blockade [ICB]) has experienced considerable success in recent years as a promising therapeutic strategy for a subset of cancers, presenting an alternative to irradiation therapy or classical chemotherapies (Fig. 4; Ribas and Wolchok 2018). In particular, their potential to counteract immune suppressive signals in the tumor microenvironment to overcome T-cell exhaustion has been proven to be beneficial in clinical settings (Fig. 3). ICBs target immune checkpoints, which in normal settings are important to accurately regulate T-cell activation and T-cell receptor signaling, thus preventing chronic (or inappropriate) immune responses. In cancers, these checkpoints, often engaged via cell surface ligands or receptors, are repurposed to dampen antitumor immune responses and create an immune-suppressive tumor microenvironment. ICB agents, in the form of antibodies, binding to these ligands/receptors (anti-CTLA-4 and anti-PD-1/anti-PD-L1) can overcome inhibitory signaling and reactivate T-cell engagement toward the tumor (Fig. 4). In order to broaden the spectrum of applications for ICB, extensive efforts have centered around the identification of suitable biomarkers for predicting ICB efficacy, as well as exploring the potential of combination therapies with conventional irradiation or chemotherapy. Increased mutational load is a promising indicator of ICB responsiveness, and indeed, patients harboring mutations in MMR genes and displaying MSI showed strong responsiveness to PD-1 antagonists (Rizvi et al. 2015; Le et al. 2017; Overman et al. 2017). Importantly, the clinical benefits of blocking the PD-1/PD-L1 immune checkpoint occurred across various tumor types, including melanomas, colorectal, and non-small cell lung cancers (NSCLCs), highlighting the likelihood that increased mutational burden was the common denominator of ICB efficacy between these tumors. In line with this premise, progression free survival and overall survival upon PD-1 blockade was further augmented when combined with a CTLA-4 antagonist in small cell lung cancers, which exhibit a high load of somatic mutations (Hellmann et al. 2018).
Synergy of PARP inhibitors with immune checkpoint inhibition
In addition to being used to treat tumors with a high mutational burden, ICBs have also been shown to be effective when applied in combination with DDR inhibition (Table 2). An important feature of PARP inhibition is that it is associated with increases in CD8+ T-cell infiltration and IFN-γ production in the tumor, which occurs in mouse cancer models independent of their Brca1/2 status. Moreover, tumor regression was further increased upon combination therapy of PARPi with anti-PD-1 antibodies compared with the respective monotherapies (Ding et al. 2018; Shen et al. 2019). Initial results of ensuing clinical trials indicate beneficial effects in a subset of patients treated with ICB and PARPi, with results from the MEDIOLA trial suggesting increased antitumor activity in germline BRCA mutated ovarian cancers treated with the combination therapy of olaparib and durvalumab (PD-L1 inhibitor) (Domchek et al. 2020).
Table 2.
Registered clinical trials using DDR and immune checkpoint inhibitors

Another clinical trial, TOPACIO, investigated the efficacy of the PARPi niraparib and the PD-L1 inhibitor pembrolizumab in platinum-resistant advanced breast cancers and recurrent ovarian cancers, independent of their HR proficiency. Notably, patient response toward the combination therapy in ovarian cancers exceeded expectations based on monotherapy efficacy, while HRD-associated mutational signature 3 or a positive immune score were reliable indicators of responsiveness (Konstantinopoulos et al. 2019; Färkkilä et al. 2020). Although mutational signature 3 and therefore HR proficiency predicted efficacy of this combination therapy, therefore implying an application for niraparib and pembrolizumab in BRCA-deficient tumors, it was also encouraging that PD-L1 presence and interferon priming of immune cells present in the tumor microenvironment can be used to estimate responsiveness to the combination therapy independent of HRD. Indeed, patient responses to PARPi and PD-L1 inhibition have been observed in tumors displaying a functional HR pathway (Färkkilä et al. 2020). Additionally, a proof-of-concept clinical study further confirmed immune activation in ovarian cancers treated with olaparib and the anti-PD-L1 antibody durvalumab regardless of their BRCA status, highlighted by increased IFN-γ production and tumor-infiltrating lymphocytes (Lampert et al. 2020). Notably, in this study no significant increase in tumor mutational burden was observed (not even in BRCA-deficient tumors), which is in line with previous observations in ovarian cancers and suggests that immune stimulation with concomitant PARPi treatment is unlikely to result from the exposure of neoantigens (Chan et al. 2019). However, increased proinflammatory cytokine expression and T-cell activation was associated with clinical benefits following olaparib plus durvalumab treatment, implying a potential application of this therapy for a wider spectrum of cancers. Currently, numerous clinical trials are evaluating the efficacy of PARP inhibitors in combination with ICB (Table 2). Although a significant proportion of these studies are preselecting patients based on potential DDR defects, it will be valuable to compare outcomes with trials where patients received the combination therapy independent of DDR deficiencies. Clarifying whether and to what extent DDR deficiency promotes responses to PARPi + ICB, and whether mutations in DDR genes (or mutational signatures characteristic for DDR deficiencies) can serve as reliant biomarkers for predicting PARPi + ICB efficacy will be crucial for the clinical prospects of this combination therapy approach.
Potentiating immune checkpoint inhibitor efficacy via DDR inhibitors
Beyond PARP inhibitors, combinations of DNA-damage inducing agents with ICB have shown encouraging preclinical/clinical results, as exemplified by the recent FDA approval of atezolizumab (anti-PD-L1) in combination with carboplatin and etoposide for extensive-stage small cell lung cancers (SCLCs). Prior to this approval, the Impower133 clinical trial had shown significantly longer overall and progression-free survival in small cell lung cancer (SCLC) patients subjected to this combination therapy compared with chemotherapy alone (Horn et al. 2018). Interestingly, in this study there was no indication of higher mutational burden being beneficial for patient response, opposing preclinical studies suggesting positive correlation between mutational load and ICB monotherapy (Hellmann et al. 2018). Since the Impower133 trial cohort represented patients with advanced disease, it is possible that tumor progression selects against strong immunogenic neoantigens. Consequently, the efficacy of combination therapy could result from chemotherapy-induced DNA damage and induction of inflammatory responses in the tumor microenvironment. Other DDR inhibitors being assessed for synergistic effects with immune modulating agents are still at the stage of preclinical investigations. In this regard, it is notable that inhibiting PARP or CHK1 in the presence of anti-PD-L1 antibodies caused tumor regression in SCLC mouse models, which was accompanied by CD8+ T-cell infiltration (Sen et al. 2019b). Further analysis revealed that PARP inhibition caused a drastic increase in PD-L1 and IFN-β expression in tumors, while the antitumor effect of either CHK1i + anti-PD-L1 or PARPi + anti-PD-L1 was dependent on functional cGAS/STING. Additionally, combining the TOP1 inhibitor irinotecan with PD-1/PD-L1 antibody mildly reduced tumor growth in cancer mouse models (Iwai et al. 2018; McKenzie et al. 2018; Sen et al. 2019a).
As we described above, radiation therapy has multiple impacts on immune responses in cancer cells, resulting in cGAS/STING activation via cytosolic DNA, leading to type I IFN responses. Therefore, it appears that ICBs have the potential to augment anti-immune responses upon radiotherapy of tumors. Indeed, complementing radiotherapy with anti-PD-L1 antibody treatment extended the efficacy of radiotherapy in mouse tumor models via cytotoxic T-cell activation, accompanied by reduced infiltration of immune suppressive myeloid cells (Deng et al. 2014a). Furthermore, a phase I clinical trial revealed major tumor regression in a group of metastatic melanoma patients treated with radiotherapy and an anti-CTLA-4 antibody (Twyman-Saint Victor et al. 2015). Importantly, this effect was reproduced in mouse models, although resistance was observed after a certain time, likely caused by up-regulation of PD-L1 expression in melanoma cells, leading to T-cell exhaustion. In line with these observations, patients displaying high levels of PD-L1 did not response to radiotherapy + anti-CTLA-4 therapy, but inhibition of the PD-1/PD-L1 axis in these tumors re-engaged exhausted T cells and improved the efficacy of radiotherapy + anti-CTLA-4 (Twyman-Saint Victor et al. 2015). Moreover, a phase III study in non-small cell lung cancer (NSCLC) patients revealed increased progression-free survival when radiotherapy was combined with the PD-L1 inhibitor durvalumab compared with the control group (Antonia et al. 2017). An important feature of radiotherapy is its potential to induce tumor regression distant from the site of radiation, which depends on the host's immune system; termed the “abscopal effect.” Although this effect occurs only sporadically, there is a consensus that immune checkpoint inhibitors have the potential to systematically promote abscopal effects in tumors (Ngwa et al. 2018). In mouse models, sensing of cytosolic DNA via cGAS/STING was shown to be required for the abscopal effect observed in response to radiotherapy and CTLA-4 inhibitors (Harding et al. 2017). In line with these observations, combining radiotherapy with ipilimumab (anti-CTLA-4) induced systemic antitumor immune responses in NSCLC patients, indicative of abscopal effects. Increased IFN-β secretion and clonal expansion of tumor-derived T-cell receptors (TCRs) predicted responsiveness to this combination therapy (Formenti et al. 2018), thereby highlighting the potential of radiotherapy to induce type I IFN responses promoting innate antitumor immunogenicity. In conclusion, innate immunity is fundamental to creating an adaptive immune response in immunosuppressive tumors, and the induction of DNA damage via DDRis or radiotherapy represents a potent trigger of this response.
Concluding remarks
It is becoming increasingly clear that DNA damage and activation of DNA damage responses can result in profound stimulation of aspects of the immune system. Defects in DNA repair pathways such as HR as well as DNA replication stress and concomitant aberrant processing of DNA replication forks can trigger cytosolic DNA sensing via cGAS/STING, leading to robust interferon responses. Furthermore, DDR inhibitors harbor the potential to activate antitumor immune responses and contribute to their therapeutic efficacy. The abrogation of cytosolic DNA sensing via cGAS/STING and proinflammatory signaling diminishes PARPi cytotoxicity in mouse tumor models, indicative of uncompleted DNA repair products being the initial stimulus of this immune response. Since comparable immune stimulation from PARPi can be observed in clinical scenarios, particularly when their application is combined with immune checkpoint inhibitors, it will be crucial to define and understand the exact circumstances wherein DNA lesions can activate antitumor immunity. Our current understanding suggests that in some scenarios, DDR inhibition or radiotherapy induces proinflammatory signaling events in tumor-suppressive microenvironments, which, when combined with ICB, promote T-cell-mediated destruction of tumor cells. It is tempting to speculate that the sheer presence of aberrant DNA repair products, rather than mutational burden and resulting neoantigens, is the key stimulus for these immune responses. Importantly, this could imply efficacy of DDR inhibitors and ICB treatment combinations in advanced disease settings, where a strong immune suppression of the tumor milieu frequently suppresses antitumor immunity. Ongoing and future clinical trials will shed light on these issues and may identify biomarkers for predicting a patient's responsiveness to various potential therapies, thereby helping tailor therapies to individual patients and impacting on the future clinical success of combining DDR inhibitors with ICBs. Importantly, in the context of MSI tumors, DDR defects can be a source for mutational burden and the consequent generation of neoantigens in tumor cells. The prevailing clinical success of ICB in MSI or other high mutational burden tumors opens the therapeutic avenue of combining ICB with MMR inhibitors for targeted application in patients independent of the mutational load in the tumor. Finally, the development and clinical exploration of inhibitors of DDR components such as ATR, ATM, and DNA-PK will shed more light on how these inhibitors potentially affect cellular immune responses and how these might be exploited in combinations with immune-targeting therapies. Furthermore, inhibitors targeting DNA repair polymerases, such as POLθ, represent an exciting arena for future studies, not only in the context of DDR alone but also in regards to their potential interplay with the cellular immune systems (Schrempf et al. 2020). In this regard, it is of note that preclinical studies with POLθ inhibitors have indicated promising efficacies in tumor models, reminiscent of the initial studies when PARP inhibitors were being developed (Zhou et al. 2020). Given the widespread efforts in both the DDR inhibitors and ICB arenas and the interest in their synergies, it seems likely that we will soon learn of other contexts where their combination will enhance the health and well-being of cancer patients.
Acknowledgments
We thank Kate Dry, Rebecca Lloyd, and Guido Zagnoli-Vieira for critical reading of the manuscript. Work in the Jackson laboratory is funded by Cancer Research UK (CRUK) program grant C6/A18796 and core grant C6946/A24843, and Wellcome investigator award 206388/Z/17/Z and core grant 203144. S.P.J. receives salary from the University of Cambridge. D.P. is funded by Cancer Research UK studentship C6/A21454. L.W.S is supported by Cancer Research UK (grant C552/A17720).
Footnotes
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.348314.121.
Freely available online through the Genes & Development Open Access option.
Competing interest statement
S.P.J. declares that he is founder and shareholder of Mission Therapeutics Ltd., Adrestia Therapeutics Ltd., Ahren Innovation Capital LLP, and Carrick Therapeutics Ltd. L.W.S. holds equity in PsiOxus Therapeutics Ltd. The other authors declare no competing interests.
References
- Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Röhl I, Hopfner KP, Ludwig J, Hornung V. 2013. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature 498: 380–384. 10.1038/nature12306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ablasser A, Hemmerling I, Schmid-Burgk JL, Behrendt R, Roers A, Hornung V. 2014. TREX1 deficiency triggers cell-autonomous immunity in a cGAS-dependent manner. J Immunol 192: 5993–5997. 10.4049/jimmunol.1400737 [DOI] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. 2013. Signatures of mutational processes in human cancer. Nature 500: 415–421. 10.1038/nature12477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, Yokoi T, Chiappori A, Lee KH, de Wit M, et al. 2017. Durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. N Engl J Med 377: 1919–1929. 10.1056/NEJMoa1709937 [DOI] [PubMed] [Google Scholar]
- Balkwill F. 2009. Tumour necrosis factor and cancer. Nat Rev Cancer 9: 361–371. 10.1038/nrc2628 [DOI] [PubMed] [Google Scholar]
- Bednarski JJ, Sleckman BP. 2019. At the intersection of DNA damage and immune responses. Nat Rev Immunol 19: 231–242. 10.1038/s41577-019-0135-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernier J, Hall EJ, Giaccia A. 2004. Radiation oncology: a century of achievements. Nat Rev Cancer 4: 737–747. 10.1038/nrc1451 [DOI] [PubMed] [Google Scholar]
- Bhat KP, Cortez D. 2018. RPA and RAD51: fork reversal, fork protection, and genome stability. Nat Struct Mol Biol 25: 446–453. 10.1038/s41594-018-0075-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S, Srinivasan K, Abdisalaam S, Su F, Raj P, Dozmorov I, Mishra R, Wakeland EK, Ghose S, Mukherjee S, et al. 2017. RAD51 interconnects between DNA replication, DNA repair and immunity. Nucleic Acids Res 45: 4590–4605. 10.1093/nar/gkx126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackford AN, Jackson SP. 2017. ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol Cell 66: 801–817. 10.1016/j.molcel.2017.05.015 [DOI] [PubMed] [Google Scholar]
- Boyer JA, Spangler CJ, Strauss JD, Cesmat AP, Liu P, McGinty RK, Zhang Q. 2020. Structural basis of nucleosome-dependent cGAS inhibition. Science 370: 450–454. 10.1126/science.abd0609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. 2005. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434: 913–917. 10.1038/nature03443 [DOI] [PubMed] [Google Scholar]
- Burnette BC, Liang H, Lee Y, Chlewicki L, Khodarev NN, Weichselbaum RR, Fu YX, Auh SL. 2011. The efficacy of radiotherapy relies upon induction of type I interferon-dependent innate and adaptive immunity. Cancer Res 71: 2488–2496. 10.1158/0008-5472.CAN-10-2820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell BB, Light N, Fabrizio D, Zatzman M, Fuligni F, de Borja R, Davidson S, Edwards M, Elvin JA, Hodel KP, et al. 2017. Comprehensive analysis of hypermutation in human cancer. Cell 171: 1042–1056.e10. 10.1016/j.cell.2017.09.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan TA, Yarchoan M, Jaffee E, Swanton C, Quezada SA, Stenzinger A, Peters S. 2019. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol 30: 44–56. 10.1093/annonc/mdy495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao HH, Karagounis IV, Thomas C, François NB, Facciabene A, Koumenis C, Maity A. 2020. Combination of CHEK1/2 inhibition and ionizing radiation results in abscopal tumor response through increased micronuclei formation. Oncogene 39: 4344–4357. 10.1038/s41388-020-1300-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles KA, Kulbe H, Soper R, Escorcio-Correia M, Lawrence T, Schultheis A, Chakravarty P, Thompson RG, Kollias G, Smyth JF, et al. 2009. The tumor-promoting actions of TNF-α involve TNFR1 and IL-17 in ovarian cancer in mice and humans. J Clin Invest 119: 3011–3023. 10.1172/JCI39065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JN, Luo W, Sun C, Jin Z, Zeng X, Alexander PB, Gong Z, Xia X, Ding X, Xu S, et al. 2021. Radiation-induced eosinophils improve cytotoxic T lymphocyte recruitment and response to immunotherapy. Sci Adv 7: eabc7609. 10.1126/sciadv.abc7609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. 2010. The DNA damage response: making it safe to play with knives. Mol Cell 40: 179–204. 10.1016/j.molcel.2010.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civril F, Deimling T, de Oliveira Mann CC, Ablasser A, Moldt M, Witte G, Hornung V, Hopfner KP. 2013. Structural mechanism of cytosolic DNA sensing by cGAS. Nature 498: 332–337. 10.1038/nature12305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coquel F, Silva MJ, Técher H, Zadorozhny K, Sharma S, Nieminuszczy J, Mettling C, Dardillac E, Barthe A, Schmitz AL, et al. 2018. SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature 557: 57–61. 10.1038/s41586-018-0050-1 [DOI] [PubMed] [Google Scholar]
- Cornel AM, Mimpen IL, Nierkens S. 2020. MHC class I downregulation in cancer: underlying mechanisms and potential targets for cancer immunotherapy. Cancers 12: 1760. 10.3390/cancers12071760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes-Ciriano I, Lee S, Park WY, Kim TM, Park PJ. 2017. A molecular portrait of microsatellite instability across multiple cancers. Nat Commun 8: 15180. 10.1038/ncomms15180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covey JM, Jaxel C, Kohn KW, Pommier Y. 1989. Protein-linked DNA strand breaks induced in mammalian cells by camptothecin, an inhibitor of topoisomerase I. Cancer Res 49: 5016–5022. [PubMed] [Google Scholar]
- Crow YJ, Manel N. 2015. Aicardi–Goutières syndrome and the type I interferonopathies. Nat Rev Immunol 15: 429–440. 10.1038/nri3850 [DOI] [PubMed] [Google Scholar]
- Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van Bokhoven H, Brunner HG, Hamel BC, et al. 2006a. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutières syndrome at the AGS1 locus. Nat Genet 38: 917–920. 10.1038/ng1845 [DOI] [PubMed] [Google Scholar]
- Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, Ali M, Semple C, Aicardi J, Babul-Hirji R, et al. 2006b. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutières syndrome and mimic congenital viral brain infection. Nat Genet 38: 910–916. 10.1038/ng1842 [DOI] [PubMed] [Google Scholar]
- Croxford JL, Tang ML, Pan MF, Huang CW, Kamran N, Phua CM, Chng WJ, Ng SB, Raulet DH, Gasser S. 2013. ATM-dependent spontaneous regression of early Eμ-myc-induced murine B-cell leukemia depends on natural killer and T cells. Blood 121: 2512–2521. 10.1182/blood-2012-08-449025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Alessandro G, Whelan DR, Howard SM, Vitelli V, Renaudin X, Adamowicz M, Iannelli F, Jones-Weinert CW, Lee M, Matti V, et al. 2018. BRCA2 controls DNA:RNA hybrid level at DSBs by mediating RNase H2 recruitment. Nat Commun 9: 5376. 10.1038/s41467-018-07799-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, Fu YX. 2014a. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest 124: 687–695. 10.1172/JCI67313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li XD, Mauceri H, Beckett M, Darga T, et al. 2014b. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 41: 843–852. 10.1016/j.immuni.2014.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JM, Vanpouille-Box C, Spada S, Rudqvist NP, Chapman JR, Ueberheide BM, Pilones KA, Sarfraz Y, Formenti SC, Demaria S. 2018. Exosomes shuttle TREX1-sensitive IFN-stimulatory dsDNA from irradiated cancer cells to DCs. Cancer Immunol Res 6: 910–920. 10.1158/2326-6066.CIR-17-0581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon MT, Bergerhoff KF, Pedersen M, Whittock H, Crespo-Rodriguez E, Patin EC, Pearson A, Smith HG, Paget JTE, Patel RR, et al. 2019. ATR inhibition potentiates the radiation-induced inflammatory tumor microenvironment. Clin Cancer Res 25: 3392–3403. 10.1158/1078-0432.CCR-18-1821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Kim HJ, Wang Q, Kearns M, Jiang T, Ohlson CE, Li BB, Xie S, Liu JF, Stover EH, et al. 2018. PARP inhibition elicits STING-dependent antitumor immunity in Brca1-deficient ovarian cancer. Cell Rep 25: 2972–2980.e5. 10.1016/j.celrep.2018.11.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Maggio FM, Minafra L, Forte GI, Cammarata FP, Lio D, Messa C, Gilardi MC, Bravatà V. 2015. Portrait of inflammatory response to ionizing radiation treatment. J Inflamm 12: 14. 10.1186/s12950-015-0058-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domchek SM, Postel-Vinay S, Im SA, Park YH, Delord JP, Italiano A, Alexandre J, You B, Bastian S, Krebs MG, et al. 2020. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): an open-label, multicentre, phase 1/2, basket study. Lancet Oncol 21: 1155–1164. 10.1016/S1470-2045(20)30324-7 [DOI] [PubMed] [Google Scholar]
- Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, et al. 2002. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med 8: 793–800. 10.1038/nm730 [DOI] [PubMed] [Google Scholar]
- Elbæk CR, Petrosius V, Sørensen CS. 2020. WEE1 kinase limits CDK activities to safeguard DNA replication and mitotic entry. Mutat Res 819-820: 111694. 10.1016/j.mrfmmm.2020.111694 [DOI] [PubMed] [Google Scholar]
- Färkkilä A, Gulhan DC, Casado J, Jacobson CA, Nguyen H, Kochupurakkal B, Maliga Z, Yapp C, Chen YA, Schapiro D, et al. 2020. Immunogenomic profiling determines responses to combined PARP and PD-1 inhibition in ovarian cancer. Nat Commun 11: 1459. 10.1038/s41467-020-15315-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. 2005. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434: 917–921. 10.1038/nature03445 [DOI] [PubMed] [Google Scholar]
- Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. 2003. IKKε and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol 4: 491–496. 10.1038/ni921 [DOI] [PubMed] [Google Scholar]
- Forment JV, O'Connor MJ. 2018. Targeting the replication stress response in cancer. Pharmacol Ther 188: 155–167. 10.1016/j.pharmthera.2018.03.005 [DOI] [PubMed] [Google Scholar]
- Formenti SC, Rudqvist NP, Golden E, Cooper B, Wennerberg E, Lhuillier C, Vanpouille-Box C, Friedman K, Ferrari de Andrade L, Wucherpfennig KW, et al. 2018. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat Med 24: 1845–1851. 10.1038/s41591-018-0232-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, et al. 2000. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 192: 1027–1034. 10.1084/jem.192.7.1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta T, Takemura H, Liao ZY, Aune GJ, Redon C, Sedelnikova OA, Pilch DR, Rogakou EP, Celeste A, Chen HT, et al. 2003. Phosphorylation of histone H2AX and activation of Mre11, Rad50, and Nbs1 in response to replication-dependent DNA double-strand breaks induced by mammalian DNA topoisomerase I cleavage complexes. J Biol Chem 278: 20303–20312. 10.1074/jbc.M300198200 [DOI] [PubMed] [Google Scholar]
- Gasser S, Orsulic S, Brown EJ, Raulet DH. 2005. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 436: 1186–1190. 10.1038/nature03884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germano G, Lamba S, Rospo G, Barault L, Magrì A, Maione F, Russo M, Crisafulli G, Bartolini A, Lerda G, et al. 2017. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 552: 116–120. 10.1038/nature24673 [DOI] [PubMed] [Google Scholar]
- Giannakis M, Mu XJ, Shukla SA, Qian ZR, Cohen O, Nishihara R, Bahl S, Cao Y, Amin-Mansour A, Yamauchi M, et al. 2016. Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep 15: 857–865. 10.1016/j.celrep.2016.03.075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray EE, Treuting PM, Woodward JJ, Stetson DB. 2015. Cutting edge: cGAS is required for lethal autoimmune disease in the Trex1-deficient mouse model of Aicardi-Goutières syndrome. J Immunol 195: 1939–1943. 10.4049/jimmunol.1500969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallahan DE, Spriggs DR, Beckett MA, Kufe DW, Weichselbaum RR. 1989. Increased tumor necrosis factor α mRNA after cellular exposure to ionizing radiation. Proc Natl Acad Sci 86: 10104–10107. 10.1073/pnas.86.24.10104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144: 646–674. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. 2017. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548: 466–470. 10.1038/nature23470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Härtlova A, Erttmann SF, Raffi FA, Schmalz AM, Resch U, Anugula S, Lienenklaus S, Nilsson LM, Kröger A, Nilsson JA, et al. 2015. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 42: 332–343. 10.1016/j.immuni.2015.01.012 [DOI] [PubMed] [Google Scholar]
- Hause RJ, Pritchard CC, Shendure J, Salipante SJ. 2016. Classification and characterization of microsatellite instability across 18 cancer types. Nat Med 22: 1342–1350. 10.1038/nm.4191 [DOI] [PubMed] [Google Scholar]
- Heijink AM, Talens F, Jae LT, van Gijn SE, Fehrmann RSN, Brummelkamp TR, van Vugt M. 2019. BRCA2 deficiency instigates cGAS-mediated inflammatory signaling and confers sensitivity to tumor necrosis factor-α-mediated cytotoxicity. Nat Commun 10: 100. 10.1038/s41467-018-07927-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmann MD, Callahan MK, Awad MM, Calvo E, Ascierto PA, Atmaca A, Rizvi NA, Hirsch FR, Selvaggi G, Szustakowski JD, et al. 2018. Tumor mutational burden and efficacy of nivolumab monotherapy and in combination with ipilimumab in small-cell lung cancer. Cancer Cell 33: 853–861.e4. 10.1016/j.ccell.2018.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzner AM, Hagmann CA, Goldeck M, Wolter S, Kubler K, Wittmann S, Gramberg T, Andreeva L, Hopfner KP, Mertens C, et al. 2015. Sequence-specific activation of the DNA sensor cGAS by Y-form DNA structures as found in primary HIV-1 cDNA. Nat Immunol 16: 1025–1033. 10.1038/ni.3267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai H, Iwasawa Y, Okada M, Arai T, Nishibata T, Kobayashi M, Kimura T, Kaneko N, Ohtani J, Yamanaka K, et al. 2009. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol Cancer Ther 8: 2992–3000. 10.1158/1535-7163.MCT-09-0463 [DOI] [PubMed] [Google Scholar]
- Ho SS, Zhang WY, Tan NY, Khatoo M, Suter MA, Tripathi S, Cheung FS, Lim WK, Tan PH, Ngeow J, et al. 2016. The DNA structure-specific endonuclease MUS81 mediates DNA sensor STING-dependent host rejection of prostate cancer cells. Immunity 44: 1177–1189. 10.1016/j.immuni.2016.04.010 [DOI] [PubMed] [Google Scholar]
- Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. 2010. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363: 711–723. 10.1056/NEJMoa1003466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm C, Covey JM, Kerrigan D, Pommier Y. 1989. Differential requirement of DNA replication for the cytotoxicity of DNA topoisomerase I and II inhibitors in Chinese hamster DC3F cells. Cancer Res 49: 6365–6368. [PubMed] [Google Scholar]
- Horn L, Mansfield AS, Szczesna A, Havel L, Krzakowski M, Hochmair MJ, Huemer F, Losonczy G, Johnson ML, Nishio M, et al. 2018. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N Engl J Med 379: 2220–2229. 10.1056/NEJMoa1809064 [DOI] [PubMed] [Google Scholar]
- Howitt BE, Shukla SA, Sholl LM, Ritterhouse LL, Watkins JC, Rodig S, Stover E, Strickland KC, D'Andrea AD, Wu CJ, et al. 2015. Association of polymerase e-mutated and microsatellite-instable endometrial cancers with neoantigen load, number of tumor-infiltrating lymphocytes, and expression of PD-1 and PD-L1. JAMA Oncol 1: 1319–1323. 10.1001/jamaoncol.2015.2151 [DOI] [PubMed] [Google Scholar]
- Hsiang YH, Hertzberg R, Hecht S, Liu LF. 1985. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J Biol Chem 260: 14873–14878. 10.1016/S0021-9258(17)38654-4 [DOI] [PubMed] [Google Scholar]
- Hsiang YH, Lihou MG, Liu LF. 1989. Arrest of replication forks by drug-stabilized topoisomerase I-DNA cleavable complexes as a mechanism of cell killing by camptothecin. Cancer Res 49: 5077–5082. [PubMed] [Google Scholar]
- Ishikawa H, Barber GN. 2008. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455: 674–678. 10.1038/nature07317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Ma Z, Barber GN. 2009. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461: 788–792. 10.1038/nature08476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai T, Sugimoto M, Wakita D, Yorozu K, Kurasawa M, Yamamoto K. 2018. Topoisomerase I inhibitor, irinotecan, depletes regulatory T cells and up-regulates MHC class I and PD-L1 expression, resulting in a supra-additive antitumor effect when combined with anti-PD-L1 antibodies. Oncotarget 9: 31411–31421. 10.18632/oncotarget.25830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, Bartek J. 2009. The DNA-damage response in human biology and disease. Nature 461: 1071–1078. 10.1038/nature08467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Xue X, Panda S, Kawale A, Hooy RM, Liang F, Sohn J, Sung P, Gekara NO. 2019. Chromatin-bound cGAS is an inhibitor of DNA repair and hence accelerates genome destabilization and cell death. EMBO J 38: e102718. 10.15252/embj.2019102718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantidze OL, Velichko AK, Luzhin AV, Petrova NV, Razin SV. 2018. Synthetically lethal interactions of ATM, ATR, and DNA-PKcs. Trends Cancer 4: 755–768. 10.1016/j.trecan.2018.09.007 [DOI] [PubMed] [Google Scholar]
- Karanam K, Kafri R, Loewer A, Lahav G. 2012. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol Cell 47: 320–329. 10.1016/j.molcel.2012.05.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keast D. 1970. Immunosurveillance and cancer. Lancet 296: 710–712. 10.1016/S0140-6736(70)91973-2 [DOI] [PubMed] [Google Scholar]
- Kelland L. 2007. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer 7: 573–584. 10.1038/nrc2167 [DOI] [PubMed] [Google Scholar]
- Kim C, Wang XD, Yu Y. 2020. PARP1 inhibitors trigger innate immunity via PARP1 trapping-induced DNA damage response. Elife 9: e60637. 10.7554/eLife.60637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitai Y, Kawasaki T, Sueyoshi T, Kobiyama K, Ishii KJ, Zou J, Akira S, Matsuda T, Kawai T. 2017. DNA-containing exosomes derived from cancer cells treated with topotecan activate a STING-dependent pathway and reinforce antitumor immunity. J Immunol 198: 1649–1659. 10.4049/jimmunol.1601694 [DOI] [PubMed] [Google Scholar]
- Konstantinopoulos PA, Waggoner S, Vidal GA, Mita M, Moroney JW, Holloway R, Van Le L, Sachdev JC, Chapman-Davis E, Colon-Otero G, et al. 2019. Single-arm phases 1 and 2 trial of niraparib in combination with pembrolizumab in patients with recurrent platinum-resistant ovarian carcinoma. JAMA Oncol 5: 1141–1149. 10.1001/jamaoncol.2019.1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummel MF, Allison JP. 1995. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med 182: 459–465. 10.1084/jem.182.2.459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujirai T, Zierhut C, Takizawa Y, Kim R, Negishi L, Uruma N, Hirai S, Funabiki H, Kurumizaka H. 2020. Structural basis for the inhibition of cGAS by nucleosomes. Science 370: 455–458. 10.1126/science.abd0237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel TA, Erie DA. 2015. Eukaryotic mismatch repair in relation to DNA replication. Annu Rev Genet 49: 291–313. 10.1146/annurev-genet-112414-054722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampert EJ, Zimmer A, Padget M, Cimino-Mathews A, Nair JR, Liu Y, Swisher EM, Hodge JW, Nixon AB, Nichols E, et al. 2020. Combination of PARP inhibitor olaparib, and PD-L1 inhibitor durvalumab, in recurrent ovarian cancer: a proof-of-concept phase II study. Clin Cancer Res 26: 4268–4279. 10.1158/1078-0432.CCR-20-0056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al. 2015. PD-1 Blockade in tumors with mismatch-repair deficiency. N Engl J Med 372: 2509–2520. 10.1056/NEJMoa1500596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, et al. 2017. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357: 409–413. 10.1126/science.aan6733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecona E, Fernandez-Capetillo O. 2018. Targeting ATR in cancer. Nat Rev Cancer 18: 586–595. 10.1038/s41568-018-0034-3 [DOI] [PubMed] [Google Scholar]
- Lee Y, Auh SL, Wang Y, Burnette B, Wang Y, Meng Y, Beckett M, Sharma R, Chin R, Tu T, et al. 2009. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood 114: 589–595. 10.1182/blood-2009-02-206870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Shu C, Yi G, Chaton CT, Shelton CL, Diao J, Zuo X, Kao CC, Herr AB, Li P. 2013. Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity 39: 1019–1031. 10.1016/j.immuni.2013.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl T, Barnes DE, Yang YG, Robins P. 2009. Biochemical properties of mammalian TREX1 and its association with DNA replication and inherited inflammatory disease. Biochem Soc Trans 37: 535–538. 10.1042/BST0370535 [DOI] [PubMed] [Google Scholar]
- Liu H, Zhang H, Wu X, Ma D, Wu J, Wang L, Jiang Y, Fei Y, Zhu C, Tan R, et al. 2018a. Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature 563: 131–136. 10.1038/s41586-018-0629-6 [DOI] [PubMed] [Google Scholar]
- Liu S, Kwon M, Mannino M, Yang N, Renda F, Khodjakov A, Pellman D. 2018b. Nuclear envelope assembly defects link mitotic errors to chromothripsis. Nature 561: 551–555. 10.1038/s41586-018-0534-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, Blosser RL, Fan H, Wang H, Luber BS, et al. 2015. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov 5: 43–51. 10.1158/2159-8290.CD-14-0863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord CJ, Ashworth A. 2016. BRCAness revisited. Nat Rev Cancer 16: 110–120. 10.1038/nrc.2015.21 [DOI] [PubMed] [Google Scholar]
- Lord CJ, Ashworth A. 2017. PARP inhibitors: synthetic lethality in the clinic. Science 355: 1152–1158. 10.1126/science.aam7344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthra P, Aguirre S, Yen BC, Pietzsch CA, Sanchez-Aparicio MT, Tigabu B, Morlock LK, García-Sastre A, Leung DW, Williams NS, et al. 2017. Topoisomerase II inhibitors induce DNA damage-dependent interferon responses circumventing ebola virus immune evasion. MBio 8: e00368-17. 10.1128/mBio.00368-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maby P, Tougeron D, Hamieh M, Mlecnik B, Kora H, Bindea G, Angell HK, Fredriksen T, Elie N, Fauquembergue E, et al. 2015. Correlation between density of CD8+ T-cell infiltrate in microsatellite unstable colorectal cancers and frameshift mutations: a rationale for personalized immunotherapy. Cancer Res 75: 3446–3455. 10.1158/0008-5472.CAN-14-3051 [DOI] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Lettice L, Tarnauskaite Z, Reddy K, Dix F, Revuelta A, Abbondati E, Rigby RE, Rabe B, et al. 2016. Ribonuclease H2 mutations induce a cGAS/STING-dependent innate immune response. EMBO J 35: 831–844. 10.15252/embj.201593339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, et al. 2017. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548: 461–465. 10.1038/nature23449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankan AK, Schmidt T, Chauhan D, Goldeck M, Höning K, Gaidt M, Kubarenko AV, Andreeva L, Hopfner KP, Hornung V. 2014. Cytosolic RNA:DNA hybrids activate the cGAS-STING axis. EMBO J 33: 2937–2946. 10.15252/embj.201488726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattarollo SR, Loi S, Duret H, Ma Y, Zitvogel L, Smyth MJ. 2011. Pivotal role of innate and adaptive immunity in anthracycline chemotherapy of established tumors. Cancer Res 71: 4809–4820. 10.1158/0008-5472.CAN-11-0753 [DOI] [PubMed] [Google Scholar]
- Maya-Mendoza A, Moudry P, Merchut-Maya JM, Lee M, Strauss R, Bartek J. 2018. High speed of fork progression induces DNA replication stress and genomic instability. Nature 599: 279–284. 10.1038/s41586-018-0261-5 [DOI] [PubMed] [Google Scholar]
- McBride WH, Chiang CS, Olson JL, Wang CC, Hong JH, Pajonk F, Dougherty GJ, Iwamoto KS, Pervan M, Liao YP. 2004. A sense of danger from radiation. Radiat Res 162: 1–19. 10.1667/RR3196 [DOI] [PubMed] [Google Scholar]
- McKenzie JA, Mbofung RM, Malu S, Zhang M, Ashkin E, Devi S, Williams L, Tieu T, Peng W, Pradeep S, et al. 2018. The effect of topoisomerase I inhibitors on the efficacy of T-cell-based cancer immunotherapy. J Natl Cancer Inst 110: 777–786. 10.1093/jnci/djx257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalski S, de Oliveira Mann CC, Stafford CA, Witte G, Bartho J, Lammens K, Hornung V, Hopfner KP. 2020. Structural basis for sequestration and autoinhibition of cGAS by chromatin. Nature 587: 678–682. 10.1038/s41586-020-2748-0 [DOI] [PubMed] [Google Scholar]
- Mittal D, Gubin MM, Schreiber RD, Smyth MJ. 2014. New insights into cancer immunoediting and its three component phases—elimination, equilibrium and escape. Curr Opin Immunol 27: 16–25. 10.1016/j.coi.2014.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlecnik B, Bindea G, Angell HK, Maby P, Angelova M, Tougeron D, Church SE, Lafontaine L, Fischer M, Fredriksen T, et al. 2016. Integrative analyses of colorectal cancer show immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity 44: 698–711. 10.1016/j.immuni.2016.02.025 [DOI] [PubMed] [Google Scholar]
- Moding EJ, Kastan MB, Kirsch DG. 2013. Strategies for optimizing the response of cancer and normal tissues to radiation. Nat Rev Drug Discov 12: 526–542. 10.1038/nrd4003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngwa W, Irabor OC, Schoenfeld JD, Hesser J, Demaria S, Formenti SC. 2018. Using immunotherapy to boost the abscopal effect. Nat Rev Cancer 18: 313–322. 10.1038/nrc.2018.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, Desai J, Hill A, Axelson M, Moss RA, et al. 2017. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol 18: 1182–1191. 10.1016/S1470-2045(17)30422-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantelidou C, Sonzogni O, De Oliveria Taveira M, Mehta AK, Kothari A, Wang D, Visal T, Li MK, Pinto J, Castrillon JA, et al. 2019. PARP inhibitor efficacy depends on CD8+ T-cell recruitment via intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast cancer. Cancer Discov 9: 722–737. 10.1158/2159-8290.CD-18-1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathare GR, Decout A, Gluck S, Cavadini S, Makasheva K, Hovius R, Kempf G, Weiss J, Kozicka Z, Guey B, et al. 2020. Structural mechanism of cGAS inhibition by the nucleosome. Nature 587: 668–672. 10.1038/s41586-020-2750-6 [DOI] [PubMed] [Google Scholar]
- Piliè PG, Tang C, Mills GB, Yap TA. 2019. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat Rev Clin Oncol 16: 81–104. 10.1038/s41571-018-0114-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y. 2009. DNA topoisomerase I inhibitors: chemistry, biology, and interfacial inhibition. Chem Rev 109: 2894–2902. 10.1021/cr900097c [DOI] [PMC free article] [PubMed] [Google Scholar]
- PosthumaDeBoer J, Würdinger T, Graat HC, van Beusechem VW, Helder MN, van Royen BJ, Kaspers GJ. 2011. WEE1 inhibition sensitizes osteosarcoma to radiotherapy. BMC Cancer 11: 156. 10.1186/1471-2407-11-156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash R, Zhang Y, Feng W, Jasin M. 2015. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol 7: a016600. 10.1101/cshperspect.a016600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijns MA, Bubeck D, Gibson LC, Graham SC, Baillie GS, Jones EY, Jackson AP. 2011. The structure of the human RNase H2 complex defines key interaction interfaces relevant to enzyme function and human disease. J Biol Chem 286: 10530–10539. 10.1074/jbc.M110.177394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijns MA, Rabe B, Rigby RE, Mill P, Astell KR, Lettice LA, Boyle S, Leitch A, Keighren M, Kilanowski F, et al. 2012. Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell 149: 1008–1022. 10.1016/j.cell.2012.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reislander T, Lombardi EP, Groelly FJ, Miar A, Porru M, Di Vito S, Wright B, Lockstone H, Biroccio A, Harris A, et al. 2019. BRCA2 abrogation triggers innate immune responses potentiated by treatment with PARP inhibitors. Nat Commun 10: 3143. 10.1038/s41467-019-11048-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A, Wolchok JD. 2018. Cancer immunotherapy using checkpoint blockade. Science 359: 1350–1355. 10.1126/science.aar4060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice G, Newman WG, Dean J, Patrick T, Parmar R, Flintoff K, Robins P, Harvey S, Hollis T, O'Hara A, et al. 2007. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutières syndrome. Am J Hum Genet 80: 811–815. 10.1086/513443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. 2015. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348: 124–128. 10.1126/science.aaa1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottenberg S, Disler C, Perego P. 2021. The rediscovery of platinum-based cancer therapy. Nat Rev Cancer 21: 37–50. 10.1038/s41568-020-00308-y [DOI] [PubMed] [Google Scholar]
- Ruiz S, Mayor-Ruiz C, Lafarga V, Murga M, Vega-Sendino M, Ortega S, Fernandez-Capetillo O. 2016. A genome-wide CRISPR screen identifies CDC25A as a determinant of sensitivity to ATR inhibitors. Mol Cell 62: 307–313. 10.1016/j.molcel.2016.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldivar JC, Cortez D, Cimprich KA. 2017. The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol 18: 622–636. 10.1038/nrm.2017.67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samstein RM, Riaz N. 2018. The DNA damage response in immunotherapy and radiation. Adv Radiat Oncol 3: 527–533. 10.1016/j.adro.2018.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber RD, Old LJ, Smyth MJ. 2011. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science 331: 1565–1570. 10.1126/science.1203486 [DOI] [PubMed] [Google Scholar]
- Schrempf A, Slyskova J, Loizou JI. 2020. Targeting the DNA repair enzyme polymerase theta in cancer therapy. Trends Cancer 7: 98–111. 10.1016/j.trecan.2020.09.007 [DOI] [PubMed] [Google Scholar]
- Scully R, Panday A, Elango R, Willis NA. 2019. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol 20: 698–714. 10.1038/s41580-019-0152-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen T, Della Corte CM, Milutinovic S, Cardnell RJ, Diao L, Ramkumar K, Gay CM, Stewart CA, Fan Y, Shen L, et al. 2019a. Combination treatment of the oral CHK1 inhibitor, SRA737, and low-dose gemcitabine enhances the effect of programmed death ligand 1 blockade by modulating the immune microenvironment in SCLC. J Thorac Oncol 14: 2152–2163. 10.1016/j.jtho.2019.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen T, Rodriguez BL, Chen L, Corte CMD, Morikawa N, Fujimoto J, Cristea S, Nguyen T, Diao L, Li L, et al. 2019b. Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov 9: 646–661. 10.1158/2159-8290.CD-18-1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Allison JP. 2015. The future of immune checkpoint therapy. Science 348: 56–61. 10.1126/science.aaa8172 [DOI] [PubMed] [Google Scholar]
- Shen J, Zhao W, Ju Z, Wang L, Peng Y, Labrie M, Yap TA, Mills GB, Peng G. 2019. PARPi triggers the STING-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCAness. Cancer Res 79: 311–319. 10.1158/0008-5472.CAN-18-1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng H, Huang Y, Xiao Y, Zhu Z, Shen M, Zhou P, Guo Z, Wang J, Wang H, Dai W, et al. 2020. ATR inhibitor AZD6738 enhances the antitumor activity of radiotherapy and immune checkpoint inhibitors by potentiating the tumor immune microenvironment in hepatocellular carcinoma. J Immunother Cancer 8: e000340. 10.1136/jitc-2019-000340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddik ZH. 2003. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene 22: 7265–7279. 10.1038/sj.onc.1206933 [DOI] [PubMed] [Google Scholar]
- Slade D. 2019. Mitotic functions of poly(ADP-ribose) polymerases. Biochem Pharmacol 167: 33–43. 10.1016/j.bcp.2019.03.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetson DB, Ko JS, Heidmann T, Medzhitov R. 2008. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134: 587–598. 10.1016/j.cell.2008.06.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strumberg D, Pilon AA, Smith M, Hickey R, Malkas L, Pommier Y. 2000. Conversion of topoisomerase I cleavage complexes on the leading strand of ribosomal DNA into 5′-phosphorylated DNA double-strand breaks by replication runoff. Mol Cell Biol 20: 3977–3987. 10.1128/MCB.20.11.3977-3987.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, Chen ZJ. 2013. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339: 786–791. 10.1126/science.1232458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, Mak TW, Sakaguchi S. 2000. Immunologic self-tolerance maintained by CD25+CD4+ regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med 192: 303–310. 10.1084/jem.192.2.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Chen ZJ. 2012. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal 5: ra20. 10.1126/scisignal.2002521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tougeron D, Fauquembergue E, Rouquette A, Le Pessot F, Sesboüé R, Laurent M, Berthet P, Mauillon J, Di Fiore F, Sabourin JC, et al. 2009. Tumor-infiltrating lymphocytes in colorectal cancers with microsatellite instability are correlated with the number and spectrum of frameshift mutations. Mod Pathol 22: 1186–1195. 10.1038/modpathol.2009.80 [DOI] [PubMed] [Google Scholar]
- Turajlic S, Litchfield K, Xu H, Rosenthal R, McGranahan N, Reading JL, Wong YNS, Rowan A, Kanu N, Al Bakir M, et al. 2017. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol 18: 1009–1021. 10.1016/S1470-2045(17)30516-8 [DOI] [PubMed] [Google Scholar]
- Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, Benci JL, Xu B, Dada H, Odorizzi PM, et al. 2015. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 520: 373–377. 10.1038/nature14292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, Inghirami G, Coleman CN, Formenti SC, Demaria S. 2017. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun 8: 15618. 10.1038/ncomms15618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanpouille-Box C, Demaria S, Formenti SC, Galluzzi L. 2018. Cytosolic DNA sensing in organismal tumor control. Cancer Cell 34: 361–378. 10.1016/j.ccell.2018.05.013 [DOI] [PubMed] [Google Scholar]
- Vendetti FP, Karukonda P, Clump DA, Teo T, Lalonde R, Nugent K, Ballew M, Kiesel BF, Beumer JH, Sarkar SN, et al. 2018. ATR kinase inhibitor AZD6738 potentiates CD8+ T cell-dependent antitumor activity following radiation. J Clin Invest 128: 3926–3940. 10.1172/JCI96519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, Lichtor T, Decker WK, Whelan RL, Kumara H, et al. 2015. Immune evasion in cancer: mechanistic basis and therapeutic strategies. Semin Cancer Biol 35 Suppl: S185–S198. 10.1016/j.semcancer.2015.03.004 [DOI] [PubMed] [Google Scholar]
- Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, Thompson CB, Bluestone JA. 1994. CTLA-4 can function as a negative regulator of T cell activation. Immunity 1: 405–413. 10.1016/1074-7613(94)90071-X [DOI] [PubMed] [Google Scholar]
- Wang Z, Chen J, Hu J, Zhang H, Xu F, He W, Wang X, Li M, Lu W, Zeng G, et al. 2019. cGAS/STING axis mediates a topoisomerase II inhibitor-induced tumor immunogenicity. J Clin Invest 129: 4850–4862. 10.1172/JCI127471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, Sakaguchi S. 2008. CTLA-4 control over Foxp3+ regulatory T cell function. Science 322: 271–275. 10.1126/science.1160062 [DOI] [PubMed] [Google Scholar]
- Wolf C, Rapp A, Berndt N, Staroske W, Schuster M, Dobrick-Mattheuer M, Kretschmer S, Konig N, Kurth T, Wieczorek D, et al. 2016. RPA and Rad51 constitute a cell intrinsic mechanism to protect the cytosol from self DNA. Nat Commun 7: 11752. 10.1038/ncomms11752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, Duggan R, Wang Y, Barber GN, Fitzgerald KA, et al. 2014. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41: 830–842. 10.1016/j.immuni.2014.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CC, Li TK, Farh L, Lin LY, Lin TS, Yu YJ, Yen TJ, Chiang CW, Chan NL. 2011. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science 333: 459–462. 10.1126/science.1204117 [DOI] [PubMed] [Google Scholar]
- Wu CC, Li YC, Wang YR, Li TK, Chan NL. 2013a. On the structural basis and design guidelines for type II topoisomerase-targeting anticancer drugs. Nucleic Acids Res 41: 10630–10640. 10.1093/nar/gkt828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ. 2013b. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339: 826–830. 10.1126/science.1229963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CY, Yang LH, Yang HY, Knoff J, Peng S, Lin YH, Wang C, Alvarez RD, Pai SI, Roden RB, et al. 2014. Enhanced cancer radiotherapy through immunosuppressive stromal cell destruction in tumors. Clin Cancer Res 20: 644–657. 10.1158/1078-0432.CCR-13-1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YG, Lindahl T, Barnes DE. 2007. Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell 131: 873–886. 10.1016/j.cell.2007.10.017 [DOI] [PubMed] [Google Scholar]
- Zhang Q, Green MD, Lang X, Lazarus J, Parsels JD, Wei S, Parsels LA, Shi J, Ramnath N, Wahl DR, et al. 2019. Inhibition of ATM increases interferon signaling and sensitizes pancreatic cancer to immune checkpoint blockade therapy. Cancer Res 79: 3940–3951. 10.1158/0008-5472.CAN-19-0761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Xu P, Rowlett CM, Jing T, Shinde O, Lei Y, West AP, Liu WR, Li P. 2020. The molecular basis of tight nuclear tethering and inactivation of cGAS. Nature 587: 673–677. 10.1038/s41586-020-2749-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Whiteley AT, de Oliveira Mann CC, Morehouse BR, Nowak RP, Fischer ES, Gray NS, Mekalanos JJ, Kranzusch PJ. 2018. Structure of the human cGAS-DNA complex reveals enhanced control of immune surveillance. Cell 174: 300–311.e11. 10.1016/j.cell.2018.06.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Gelot C, Pantelidou C, Li A, Yücel H, Davis RE, Farkkila A, Kochupurakkal B, Syed A, Shapiro GI, et al. 2020. Polymerase θ inhibition kills homologous recombination deficient tumors. bioRxiv 10.1101/2020.05.23.111658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zierhut C, Yamaguchi N, Paredes M, Luo JD, Carroll T, Funabiki H. 2019. The cytoplasmic DNA sensor cGAS promotes mitotic cell death. Cell 178: 302–315.e23. 10.1016/j.cell.2019.05.035 [DOI] [PMC free article] [PubMed] [Google Scholar]