

In this review, Cossa et al. discuss the current knowledge and outstanding questions about phosphatases in the context of the RNAPII transcription cycle.
Keywords: cyclin-dependent kinases (CDKs), phosphoprotein phosphatases, promoter-proximal pausing, protein phosphatase 1 (PP1), protein phosphatase 2A (PP2A), protein phosphatase 4 (PP4), protein phosphorylation, RNA polymerase II (RNAPII), RNAPII C-terminal domain (CTD)
Abstract
The transcription cycle of RNA polymerase II (RNAPII) is governed at multiple points by opposing actions of cyclin-dependent kinases (CDKs) and protein phosphatases, in a process with similarities to the cell division cycle. While important roles of the kinases have been established, phosphatases have emerged more slowly as key players in transcription, and large gaps remain in understanding of their precise functions and targets. Much of the earlier work focused on the roles and regulation of sui generis and often atypical phosphatases—FCP1, Rtr1/RPAP2, and SSU72—with seemingly dedicated functions in RNAPII transcription. Decisive roles in the transcription cycle have now been uncovered for members of the major phosphoprotein phosphatase (PPP) family, including PP1, PP2A, and PP4—abundant enzymes with pleiotropic roles in cellular signaling pathways. These phosphatases appear to act principally at the transitions between transcription cycle phases, ensuring fine control of elongation and termination. Much is still unknown, however, about the division of labor among the PPP family members, and their possible regulation by or of the transcriptional kinases. CDKs active in transcription have recently drawn attention as potential therapeutic targets in cancer and other diseases, raising the prospect that the phosphatases might also present opportunities for new drug development. Here we review the current knowledge and outstanding questions about phosphatases in the context of the RNAPII transcription cycle.
Transcription in eukaryotes relies on RNA polymerases I, II, and III to synthesize different types of RNA molecules encoded in the nuclear genome. RNA polymerase II (RNAPII) transcribes the majority of genes, including those encoding proteins, long noncoding RNAs (lncRNAs), microRNAs (miRNAs), enhancer RNAs (eRNAs), and most small nuclear RNAs (snRNAs) (Hsin and Manley 2012; Cramer 2019a). Transcription by RNAPII can be modeled as a cycle comprising discrete stages of initiation, elongation, 3′ end formation and termination, and, finally, recycling of RNAPII to restart the process (Fig. 1). Transcription is strictly regulated, as befitting its critical role in determining levels of gene expression and, thus, virtually all aspects of cellular identity, fate, and metabolism. Initiation was long considered the major, rate-limiting step in transcriptional regulation—a paradigm first established in prokaryotic systems and seemingly validated by the large numbers and important roles of transcriptional activators that help recruit RNAPII to eukaryotic promoters. A more complex picture has now emerged, in which elongation, termination, and recycling are also subject to regulation, which in many cases is determinative of transcriptional outputs (Sansó and Fisher 2013; Cramer 2019b).
Figure 1.
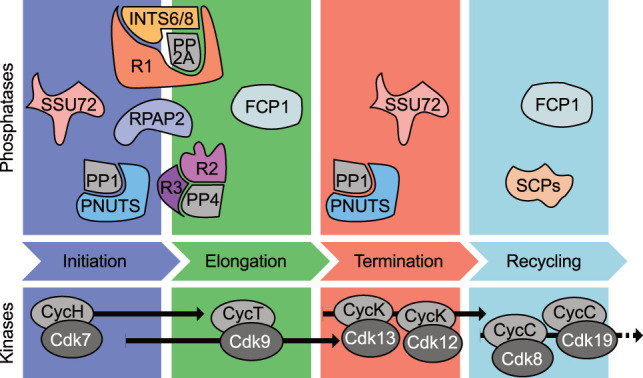
Protein phosphatases and kinases involved in the RNAPII transcription cycle. The protein phosphatases and cyclin-dependent kinases (CDKs) with their cognate cyclins (Cyc) associated with the sequential stages of the RNAPII transcription cycle are depicted. Horizontal arrows indicate specific intervals in which individual cyclin/CDKs act; analogous assignments of phosphatases remain imprecise. For PPPs, catalytic subunits are depicted in gray and regulatory subunits in colors.
The control of transcription initiation by RNAPII generally involves the regulation of DNA accessibility by chromatin-modifying and remodeling enzymes, binding to exposed regulatory elements by sequence-specific transcriptional activators, engagement of a multisubunit coactivator complex called the Mediator, and the stepwise association of general transcription factors (GTFs) and RNAPII to assemble a competent preinitiation complex (PIC) at the transcription start site (TSS) (Cramer 2019b; Schier and Taatjes 2020). In budding yeast, initiation is generally rate-limiting for the productive transcription of protein-coding genes. At a majority of genes in metazoans, however, transcription by RNAPII, once initiated, is paused ∼50–100 nt downstream from the TSS and requires additional signaling to resume processive elongation (Adelman and Lis 2012; Core and Adelman 2019). Both modes of transcription by RNAPII—initiation-limited and pause-regulated—are coordinated by the phosphorylation of RNAPII and its accessory proteins.
Protein phosphorylation is the reversible, covalent addition of a phosphate group to side chains of phospho-acceptor amino acid residues—predominantly serine, threonine, or tyrosine in eukaryotes. Phosphorylation at specific residues can alter the function of a protein by directly (i.e., electrostatically) promoting or disrupting intermolecular interactions or by allosterically altering conformation of the modified protein and thereby influencing its activity, positively or negatively (Beltrao et al. 2013). High-throughput, mass spectrometric analyses mapped nearly 300,000 phosphorylation sites in the human proteome, whereas ubiquitylation and acetylation—the second and third most common post-translational modifications (PTMs) readily detectable by mass spectrometry—occur at ∼110,000 and ∼40,000 sites, respectively (Hornbeck et al. 2015). Protein phosphorylation is also perhaps the most ancient PTM, ubiquitous throughout evolution and in all kingdoms of life, and is believed to have played an important role in the first living organisms (Beltrao et al. 2013).
Phosphorylation is dynamic and, like other PTMs, is controlled by the coordinated action of “writers” and “erasers”—the protein kinases and phosphatases, respectively. For reasons we discuss below, the kinases that place phosphorylation marks have received more attention than the phosphatases that remove them, but both classes of enzymes are, obviously, equally important (Gelens et al. 2018). Pathways regulated by phosphorylation need phosphatase activity to maintain signaling proteins in their unmodified state in the absence of the appropriate signal and to reset the system after activation so that it can respond again when restimulated. Moreover, because signaling proteins are commonly regulated by multisite phosphorylation, with readouts that depend on precise, combinatorial modification patterns, phosphorylation and dephosphorylation must be executed with similar levels of precision (Smoly et al. 2017).
Cotranscriptional phosphorylation of RNAPII and its accessory factors is predominantly catalyzed by the cyclin-dependent kinases (CDKs) (for a recent review, see Parua and Fisher 2020). In a current model, different CDKs are recruited or activated in a stage-specific manner to phosphorylate substrates in, or associated with, RNAPII transcription complexes (Fig. 1). CDK-dependent phosphorylation generally acts after initiation to promote transitions between phases of the transcription cycle and to coordinate these transitions with cotranscriptional events such as RNA-processing and chromatin modification (Buratowski 2009; Hsin and Manley 2012; Sansó and Fisher 2013; Cramer 2019b). This is in many ways analogous to the cell division cycle, another process driven largely by CDK activity (Swaffer et al. 2016; Örd and Loog 2019). As discussed below, this resemblance goes beyond broad similarities of regulatory logic and includes examples of entire signaling modules being duplicated or repurposed in cell division and transcriptional machineries (Malumbres 2014).
Although the functions and relevant protein substrates of CDKs in transcription are still being deciphered, work in diverse systems has provided a robust framework for ongoing investigations. Importantly, the temporal sequence in which different CDKs act during the RNAPII cycle appears to be conserved in evolution, so that we have a good idea of which steps in transcription are affected when one or another CDK is disabled (Parua and Fisher 2020). There is less clarity regarding the phosphatases (Fig. 1). This is largely due to impediments to the study of dephosphorylating enzymes, which we discuss below. Over the last several years, however, there has been renewed interest in the transcriptional phosphatases, with concomitant development of new tools for their genetic and biochemical analysis. Here we review insights revealed by these studies, from which a more complete picture is beginning to emerge of kinases and phosphatases working in concert.
The biology of protein phosphatases
Protein kinases are among the most intensely studied enzyme classes, owing to their near ubiquity in cellular signaling pathways and their potential for therapeutic targeting in diseases including cancer. Phosphatases help determine the balance between phosphorylation and dephosphorylation of myriad proteins in vivo but are relatively refractory to many of the genetic and biochemical tools used to dissect and characterize the kinome. Much of this difficulty stems from peculiarities and complexities of phosphatase biology and regulation, for which a specialized toolkit is still being developed.
Kinases vs. phosphatases: a contrast in regulatory style
Functional diversity among protein kinases has evolved mainly through iterative gene duplication and specialization of kinase catalytic subunits, such that genetic ablation or chemical inhibition of any one member typically affects relatively few signaling pathways in vivo (although there are important exceptions to this generalization). It has been possible, moreover, to achieve some measure of kinase selectivity with active site-directed small-molecule inhibitors that discriminate among subtle variants of the ATP-binding pocket and surrounding surface features (Wu et al. 2015; Parua and Fisher 2020). In contrast, genes encoding catalytic subunits of phosphatases are relatively few in number but are heavily transcribed and translated to maintain an overall abundance similar to that of kinases (Gelens et al. 2018). Moreover, the catalytic subunits of many phosphatases have little intrinsic substrate selectivity and are instead directed to dephosphorylate specific substrates and eschew others by interacting with accessory proteins. Although kinases can also be targeted to specific substrates or subcellular locations by binding to regulatory subunits such as cyclins, the catalytic subunits themselves typically have strong preferences for conserved motifs within their substrates. This difference has spawned a misconception that protein phosphatases are promiscuous in comparison to kinases; on the contrary, phosphatases can achieve exquisite substrate specificity in vivo through their quaternary interactions with regulatory partners (Virshup and Shenolikar 2009). Relative to kinases, however, it has been more difficult to identify protein substrates of specific phosphatases or to reconstitute and characterize the selective phosphatase holoenzymes that exist in cells.
The human genome encodes ∼500 kinases (Manning et al. 2002) and 189 protein phosphatases (Chen et al. 2017), but ∼90% of cellular dephosphorylation events are catalyzed by the phosphoprotein phosphatase (PPP) subclass, composed of 13 pSer/pThr phosphatases including the broadly active PP1 and PP2A (Fig. 2; Heroes et al. 2013). Consistent with its heavy workload in cells, PP1 is an abundant protein, with a cellular concentration of ∼0.2 µM (Verbinnen et al. 2017). PP2A is similarly abundant, reportedly accounting for up to 1% of total cellular protein and the majority of pSer/pThr phosphatase activity in some tissues (Shi 2009; Sangodkar et al. 2016). Strikingly, the abundance of PPPs is typically orders of magnitude higher than those of transcription-related, non-PPP phosphatases, as determined by proteomic analyses (Fig. 2). In aggregate, the intracellular abundance of phosphatases has been estimated to be twice that of kinases (Smoly et al. 2017). The disproportionate numbers of kinase and phosphatase isoforms are evolutionarily conserved: The genome of the budding yeast Saccharomyces cerevisiae encodes 129 kinase catalytic subunits but only 30 phosphatases, of which 13 are the PPPs (Breitkreutz et al. 2010; Chen et al. 2017). Similar numbers have been reported in fission yeast, mouse, fly, and plant systems (Morrison et al. 2000; Schweighofer and Meskiene 2015; Smoly et al. 2017).
Figure 2.

Human and yeast protein phosphatases implicated in RNAPII transcription. (Top) Hierarchical classification of 189 described human protein phosphatases (Chen et al. 2017). The “human gene” column lists genes encoding phosphatases involved in RNAPII transcription. For these genes, the corresponding homologs in S. cerevisiae or S. pombe are reported. For PPPs, only genes encoding catalytic subunits are included. In parentheses we list the numbers of phosphatases belonging to the respective human family or superfamily. In brackets we group paralogous genes. Layout is adapted from http://phosphatome.net. Homologous genes in different species are reported according to Homologene (http://ncbi.nlm.nih.gov/homologene) or Chen et al. (2017). (Bottom) Phosphoprotein phosphatases (PPPs) are typically more abundant than non-PPPs in human and yeast proteomes. Proteins are grouped in bins based on their abundance in the proteome. Bins containing transcription-related phosphatases (see top panel) are colored based on whether they are PPPs or not. Proteomic data were obtained and histogram layout was adapted from http://pax-db.org. (ppm) Parts per million.
Kinases and phosphatases differ in other important ways. Whereas kinases often undergo large fluctuations in expression and activity in response to stimuli, phosphatase holoenzymes, with some exceptions, tend to be stable in vivo and more constant in their specific activity. This may reflect the canonical roles of dephosphorylation in kinase-dependent signaling: setting thresholds for initial pathway activation, ensuring timely shutoff to prevent tonic activation, and buffering noise by suppressing activation in response to weak or transient stimuli (Ceulemans and Bollen 2004; Smoly et al. 2017).
How phosphatases achieve specificity
PPPs attain a target specificity comparable to that of kinases by acting as obligate heteromers in vivo (Fig. 3). For example, despite its abundance, PP1 is probably never present in the cell as a monomer (Virshup and Shenolikar 2009); physiologically relevant, active PP1 holoenzymes are composed of catalytic and regulatory subunits. In vivo, relatively few PP1 catalytic subunits achieve functional diversity by combining with a multitude of regulators that confer distinct localizations and substrate specificities (Bertolotti 2018).
Figure 3.

PPPs act as obligate heteromers. In order to specifically catalyze a large number of reactions, the catalytic subunit of a PPP (“C”) must interact with one or more regulatory subunits. C subunits are encoded by paralogous genes and therefore are depicted as invariable (gray). PP1 C subunits can interact with a large range of PP1-interacting proteins (PIPs) in both heterodimeric and higher-order complexes. In canonical PP2A holoenzymes, C subunits always interact with one scaffold A subunit (encoded by two similar genes, also depicted as invariable; gray) and one B subunit (encoded by multiple genes). Non-PPPs generally act as monomers.
PP1 holoenzymes are composed of a catalytic subunit encoded in humans by one of three paralogous genes, PPP1CA, PPP1CB, and PPP1CC (encoding PP1α, PP1β, and PP1γ, respectively), with a large exposed surface able to bind one or more PP1-interacting proteins (PIPs), giving rise to both dimeric and higher-order complexes (Fig. 3). Identified PIPs currently number >200 and are relatively divergent, apart from short motifs that mediate binding to the catalytic subunit. Two such motifs contain the consensus sequences RVxF and S/GILK, both found in a large number of PIPs (Moorhead et al. 2008; Verbinnen et al. 2017). In contrast, PP2A holoenzymes are composed of a core heterodimer, including one catalytic (“C”) subunit encoded by the paralogous PPP2CA and PPP2CB genes and one “A” scaffold subunit (also known as PR65 and encoded by the PPP2R1A and PPP2R1B genes), to which a third regulatory partner, the B subunit, can bind to generate functional diversity (Fig. 3; Kauko et al. 2020). Compared with PIPs, PP2A B subunits are more structurally related and can be clustered in four families (B/B55, B′/B56, B′′, or B′′′). B subunits are also fewer in number (two to five per family), potentially giving rise to ∼80 canonical PP2A holoenzymes (Sangodkar et al. 2016). PP2B, PP4, and PP6 share a similar assembly strategy, although the known interactors for these phosphatases are fewer. Other phosphatases covered in this review belong to classes distinct from the PPPs and function as monomers (Fig. 3). FCP1 (encoded in humans by the CTDP1 gene) and the related small CTD phosphatases (SCPs) were founding members of a new class of enzymes belonging to a broader superfamily of phosphotransferases and phosphohydrolases (Kamenski et al. 2004). SSU72 and Rtr1 (and its human ortholog RPAP2) also have active sites that are unique among pSer/pThr phosphatases (Meinhart et al. 2003; Irani et al. 2016). Detailed descriptions of their modes of action or catalytic mechanisms can be found elsewhere (Pons et al. 2014; Mayfield et al. 2016).
Specialized phosphatases help drive RNAPII transcription
The importance of protein phosphorylation in governing the RNAPII transcription cycle first became apparent with the identification and characterization of the C-terminal domain (CTD) of RPB1, the largest subunit of RNAPII (encoded by the POLR2A gene in humans). The RPB1-CTD is composed of variable numbers of heptad repeats (52 in humans, 26 in S. cerevisiae, 29 in S. pombe) of the consensus sequence Tyr1-Ser2-Pro3-Thr4-Ser5-Pro6-Ser7 (Corden 1990). Most or all residues in the CTD are subject to PTM—most commonly phosphorylation but also glycosylation, prolyl isomerization, and, in nonconsensus repeats containing Lys or Arg substitutions at position 7, methylation (Sims et al. 2011)—in stereotypical patterns correlated with transcription cycle stage (Fig. 4). These patterns have been proposed to constitute a “CTD code” that is read by RNAPII-interacting factors to coordinate transcription with cotranscriptional events (Buratowski 2003; Egloff and Murphy 2008; Schwer and Shuman 2011). There are examples in which a specific mark (“letter”) or combination of marks (“word”) is known to elicit a particular downstream effect, but CTD lexicography remains a work in progress. The most extensively investigated modifications are phosphorylations of the Ser5 (pSer5) and Ser2 (pSer2) positions, which predominate in RNAPII complexes sampled early and late in transcription, respectively (Fig. 4).
Figure 4.

Phosphatases target the RNAPII CTD and SPT5 throughout the transcription cycle. (Top) Typical profiles of indicated RNAPII and SPT5 phosphorylation marks along the bodies of genes in higher eukaryotes. Due to the high variability of pTyr1 profiles across species, the one depicted is based on the distribution reported in metazoans (Descostes et al. 2014). (Bottom) For each mark, proposed phosphatases are indicated. For PPPs, only catalytic subunits are reported. (TSS) Transcription start site, (PAS) polyadenylation signal.
Multiple kinases, including a subset of the CDK family comprising at least seven members in humans, have been implicated in CTD phosphorylation (Parua and Fisher 2020). It is now clear that many of these kinases phosphorylate components of the RNAPII machinery besides the CTD and can thus influence the transcription cycle through diverse mechanisms beyond recruitment of factors to the RNAPII complex (Larochelle et al. 2006; Poss et al. 2016; Sansó et al. 2016; Rimel et al. 2020). For example, the major substrate of positive transcription elongation factor b (P-TEFb, a complex of CDK9 and cyclin T1) is SPT5, the essential subunit of the conserved SPT4/SPT5 complex also known in metazoans as DRB sensitivity-inducing factor (DSIF). CDK9 phosphorylates SPT5 on multiple sites (Fig. 4) to regulate RNAPII pause release and processive elongation, as described in more detail below (Yamada et al. 2006; Booth et al. 2018; Vos et al. 2018a,b). Similarly, while many of the phosphatases that act during transcription were first identified by virtue of activity toward the RPB1-CTD, new targets and functions of these enzymes are emerging. Self-evidently, all phosphorylations of transcription-related proteins must be removed, at some point, by one or more phosphatases. For example, loss of PP1 results in increased phosphorylation of multiple residues of the transcription factor and proto-oncogene product MYC (Dingar et al. 2018). Nevertheless, we focus here on phosphatases that work on the core transcription machinery to regulate progression through the RNAPII cycle, as opposed to accessory factors such as activators or coactivators. We begin with the specialized or dedicated enzymes that were identified in targeted biochemical and genetic studies, and conclude with the PPP family, the transcriptional roles of which were initially obscured by their functional pleiotropy.
FCP1 and family: the original CTD phosphatases
The emergence of RPB1-CTD phosphorylation as a key regulator of transcription set off hunts for the kinases and phosphatases involved. Purification of the major CTD phosphatase detectable in whole-cell extracts led to the identification of budding yeast Fcp1. Mass spectrometric characterization of Fcp1-containing complexes revealed an interaction with the Rap74 subunit of the GTF TFIIF, which functions in both initiation and elongation; a yeast two-hybrid interaction screen with TFIIF subsequently identified human FCP1 (Chambers and Dahmus 1994; Chambers et al. 1995; Chambers and Kane 1996; Archambault et al. 1997, 1998). Fcp1 is encoded by an essential gene in budding yeast, and thermal inactivation of a temperature-sensitive mutant variant produced decreases in mRNA levels and RNAPII occupancy on chromatin consistent with transcription failure (Kobor et al. 1999). More than two decades later, the mechanistic basis for this requirement remains only partly understood. Early studies in vitro indicated a role for human FCP1 in reinitiation by RNAPII after completing a round of transcription; in a defined system consisting of purified factors, the addition of FCP1 allowed the formation of a competent PIC when the only source of RNAPII was the hyperphosphorylated II0 form, consistent with an ability of FCP1 to dephosphorylate the CTD of RNAPII while it is off chromatin (Cho et al. 1999). Similarly, Fcp1 depletion by RNA interference (RNAi) in Drosophila S2 cells selectively decreased RNAPII density and mRNA synthesis at the heavily transcribed heat shock genes; phosphorylation increased on both Ser2 and Ser5 residues, specifically in the chromatin-free fraction (Fuda et al. 2012). These and other studies suggest that an important function of FCP1 is to ensure a supply of unphosphorylated RNAPII (IIA form) to initiate at promoters of highly expressed genes.
Multiple studies suggest additional, albeit less clearly defined, roles for FCP1 during the elongation phase. In vitro, human FCP1 stimulated elongation, although this effect was independent of its phosphatase activity (Cho et al. 1999; Mandal et al. 2002). In vivo, FCP1 can be cross-linked to chromatin throughout the bodies of genes in both yeast and metazoan systems (Fuda et al. 2012; Zhang et al. 2012). In budding yeast (as in other systems), pSer5 plays a conserved, essential role in recruitment of the mRNA 5′ end capping enzymes, and Fcp1 is needed to promote CTD dephosphorylation and, possibly as a consequence, the timely release of capping enzymes from the elongating RNAPII complex (Schroeder et al. 2000). In another study in budding yeast, shifting an fcp1-ts mutant to restrictive temperature diminished total RNAPII occupancy but selectively increased pSer2—a mark associated with elongation—on chromatin of active genes, suggesting a continued requirement for Fcp1 activity even after initiation (Cho et al. 2001). That effect of Fcp1 inactivation on pSer2 was unchanged by mutation of kin28 or srb10 (encoding the CDK7 or CDK8 ortholog, respectively) but was abolished by the deletion of ctk1 (CDK12 ortholog). Interestingly, Ctk1, like Fcp1, cross-links along the lengths of gene coding regions, suggesting that the lack of spatial correlation between Ctk1 and its product pSer2, which increases toward 3′ ends (Fig. 4), might be due to Fcp1 removing this mark as quickly as it is placed by Ctk1 in more upstream regions.
A preference for pSer2 over pSer5 as a substrate is determined by unique structural features of FCP1. Budding yeast Fcp1 contains at its C terminus a BRCA1 C-terminal (BRCT) domain, which is essential for viability and required for activity in vitro (Kobor et al. 2000). Typically, BRCT domains occur in tandem pairs that form phosphoprotein-binding motifs (Manke et al. 2003). In the X-ray crystal structure of fission yeast Fcp1, the single BRCT domain forms one wall of a deep canyon with the active site at its floor (Ghosh et al. 2008) and confers a strong, albeit not absolute, substrate preference for pSer2-containing repeats. Amino acid substitutions in Fcp1 that enhance this preference and virtually abolish the ability of the enzyme to dephosphorylate pSer5-containing substrates had no obvious phenotypic effects in S. cerevisiae (Ghosh et al. 2008) but were lethal in S. pombe and suppressed by mutations that shorten the Rpb1-CTD (Schwer et al. 2015). Neither an rpb1-S2A mutation that replaces all Ser2 positions with Ala (and is viable in fission yeast) nor an rpb1-S5A-MCE mutation in which the lethality of the Ser5 → Ala substitution is suppressed by fusion of Rpb1 to mammalian capping enzyme obviated the essential requirement for Fcp1 function. Therefore, lethality due to loss of Fcp1 in fission yeast cannot be ascribed solely to dysregulation of either pSer2 or pSer5. FCP1 has also been implicated in dephosphorylation of pThr4 of the CTD in human cells (Hsin et al. 2014) and might have transcriptionally relevant substrates apart from RNAPII, possibly including Spt5 (Schwer et al. 2015). Therefore, although FCP1 clearly has a conserved function in removing pSer2 and facilitating recycling of RNAPII after rounds of productive elongation, there is abundant evidence—both genetic and biochemical—hinting at a more expansive portfolio for this phosphatase during the transcription cycle.
The SCPs (encoded in human by the genes CTDSP1, CTDSP2, CTDSPL, and CTDSPL2) were identified on the basis of homology with FCP1 and shown to have CTD phosphatase activity in vitro, but with a preference for pSer5 over pSer2 (Yeo et al. 2003). Human SCP1/CTDSP1 is similar in structure to FCP1 but lacks a BRCT domain, allowing a different mode of phospho-CTD substrate binding and likely accounting for the different specificities of the two enzymes for pSer2 versus pSer5 (Kamenski et al. 2004; Ghosh et al. 2008). SCPs do not appear to play a major role in transcription, as suggested by early loss-of-function studies (Yeo et al. 2005). Interestingly, SCP expression is restricted to nonneuronal lineages, where they promote the silencing of a specific subset of genes involved in neuronal development, presumably by locally maintaining RNAPII in unphosphorylated form (Yeo et al. 2005; Xue et al. 2013; Nesti et al. 2014).
Rtr1 and RPAP2: founding members of a new phosphatase family
Wild-type FCP1 has only weak activity toward pSer5 in vitro, and in most studies, inactivation or depletion of FCP1 preferentially affected pSer2 (and pThr4) levels in vivo, leaving open a role for a pSer5 phosphatase. Such an activity might be needed to (1) remove phosphates from the CTD of RNAPII after escape from the promoter region, where pSer5 generally peaks (Fig. 4; Kim et al. 2010; Mayer et al. 2010; Tietjen et al. 2010; Bataille et al. 2012), or (2) promote reinitiation by dephosphorylating pSer5 of soluble RNAPII, possibly in conjunction with a pSer2-specific enzyme such as FCP1 (see above). One candidate emerged with the identification and characterization of Rtr1, the budding yeast ortholog of the human RNAPII-associated protein 2 (RPAP2), which had been detected, but not functionally characterized, in an earlier proteomic analysis (Jeronimo et al. 2007). The RTR1 gene of S. cerevisiae was implicated in transcription and stress tolerance and shown to interact genetically with components of the RNAPII machinery including Mediator, Spt4/Spt5, and the PAF complex (Gibney et al. 2008). Its protein product bound both RNAPII and pSer5-containing phosphopeptides, and cross-linked to chromatin of active genes between the peaks of pSer5 and pSer2 (Fig. 4), suggesting a role in cotranscriptional pSer5 removal (Mosley et al. 2009). In rtr1 deletion strains, pSer5 levels increased both in extracts and on chromatin, and there was evidenceof transcriptional impairment—reductions in specific mRNAs coinciding with decreased occupancy by RNAPII, and readthrough transcription of certain genes suggestive of a termination defect. This study detected a pSer5-specific phosphatase activity associated with Rtr1 purified from yeast extracts, but a structural and biochemical analysis of the Kluyveromyces lactis enzyme raised doubts about whether it possessed an identifiable active site or intrinsic phosphatase activity (Xiang et al. 2012); another study suggested that K. lactis Rtr1 might be especially prone to inactivation during purification (Hsu et al. 2014). A structure of the S. cerevisiae Rtr1 resolved this uncertainty by revealing a novel active site for phosphoryl transfer, definitively demonstrating intrinsic activity in vitro and firmly establishing Rtr1 and RPAP2 as founding members of yet another class of non-PPPs involved in transcription (Fig. 2; Irani et al. 2016).
In vitro, activity of the purified Rtr1 monomer toward model substrates was weak (Irani et al. 2016), suggesting that additional protein–protein interactions or accessory subunits might facilitate its function in vivo. Additional complexity was also suggested by studies implicating RPAP2 in 3′ end processing of snRNA genes via its association with the Integrator complex; RPAP2 specifically binds pSer7, previously known to promote Integrator recruitment, and depletion of RPAP2 mimicked effects of Ser7 → Ala substitutions in the CTD by increasing levels of pSer5, both in extracts and on chromatin at the gene encoding U2 snRNA (Egloff et al. 2012). Importantly, pSer7 was not required for RPAP2 occupancy of the ACTB gene (where pSer5 also increased upon RPAP2 knockdown), indicating that there are multiple mechanisms for recruitment of the phosphatase to RNAPII transcribed genes. Beyond pSer5, pTyr1 might be a target of Rtr1 (Hsu et al. 2014). Moreover, two CTD-interacting domain (CID)-containing proteins, RPRD1A and RPRD1B, were shown to form both heterodimers and homodimers that bind preferentially to pSer2- or pSer7-containing repeats in vitro, erecting a “scaffold” to recruit RPAP2 and thereby stimulate it to dephosphorylate pSer5 on an adjacent repeat; depletion by RNAi indicated that the RPRDs also mediate RPAP2 interaction with RNAPII in vivo (Ni et al. 2014).
Even harder to pin down than their enzymatic activity have been the precise functions of Rtr1 and RPAP2 in gene expression and cellular physiology. A recent study combining proteomics, genomics, and bioinformatics suggested roles of Rtr1 and pSer5 in determining termination pathway choice at genes transcribed by RNAPII in budding yeast (Victorino et al. 2020). In rtr1 deletion strains with elevated levels of pSer5, interactions of the CTD with cleavage factor Ia (CFIa) were diminished, while those with another termination factor—Nrd1, which preferentially binds pSer5-containing repeats—were increased. Nrd1 is required, together with Nab3, Sen1, and additional factors, in the NNS pathway, an alternative termination mechanism in yeast. Loss of Rtr1 altered the distribution of RNAPII on protein-coding genes, shifting it toward the TSS, consistent with premature termination, possibly via engagement of NNS factors. Interestingly, loss of Ssu72, another pSer5-specific phosphatase residing in the cleavage and polyadenylation factor (CPF) required for termination of polyadenylated transcripts (discussed in the next section), has roughly the opposite effect: impaired, rather than premature, termination. A clear message that emerges from this cloudy picture is that the function of transcription-associated phosphatases is not dictated solely by their specificities for positions within the CTD repeat.
SSU72 links the beginning and end of transcription
Ssu72 is a pSer5-specific phosphatase, which was affinity-purified as a stoichiometric subunit of the CPF in budding yeast (Dichtl et al. 2002) and fission yeast (Vanoosthuyse et al. 2014). Interestingly, the SSU72 gene was first characterized on the basis of a genetic interaction with a GTF required for initiation, TFIIB, encoded by the SUA7 gene in budding yeast; an ssu72 mutation enhanced a defect in TSS specification of a sua7 mutant, presaging a linkage between the initiation and termination steps of transcription (Sun and Hampsey 1996). Ssu72 is essential for viability in S. cerevisiae (but not in S. pombe), and temperature-sensitive or inducibly degraded mutant variants elicit a constellation of transcription-associated phenotypes: extensive readthrough transcription indicative of a general termination defect (Dichtl et al. 2002), a specific defect in snRNA termination similar to that seen in NNS pathway mutants (Ganem et al. 2003), increased pSer5 (but not pSer2) (Krishnamurthy et al. 2004; Reyes-Reyes and Hampsey 2007), and a loss of the transcription-dependent physical interaction between promoter and terminator regions, or “gene looping,” detected by chromosome conformation capture (3C) analysis (Ansari and Hampsey 2005; Tan-Wong et al. 2012).
Ssu72 can also remove pSer7, another phosphate mark on the RNAPII CTD that is placed by both Cdk7 and Cdk9 orthologs (Glover-Cutter et al. 2009; St. Amour et al. 2012; Zhang et al. 2012). In budding yeast, both pSer5 and pSer7 decrease ∼350 bp upstream of the polyadenylation signal (PAS) (Fig. 4) but persist beyond the PAS in ssu72 mutants (Bataille et al. 2012). Failure of pSer7 dephosphorylation in ssu72 mutants was suggested to contribute to their associated defects in transcription termination (Zhang et al. 2012).
Ssu72 contains a CX5R motif typically found in protein tyrosine phosphatases (PTPases); mutation of the catalytic Cys residue abolished activity toward p-nitrophenylphosphate and CTD-derived substrates in vitro (Meinhart et al. 2003; Krishnamurthy et al. 2004). A Cys → Ser substitution at this position was lethal in S. cerevisiae (Sun and Hampsey 1996). A catalytically dead C15S variant also failed to restore normal levels of pSer5 after depletion of an Ssu72-degron fusion protein but was able to rescue 3′ end processing of transcripts in an Ssu72-depleted extract, possibly reflecting a structural role of the protein in maintaining CPF integrity. In contrast, Ssu72 depletion impaired promoter-dependent transcription in yeast extracts, with a concomitant increase in pSer5 and depletion of RNAPIIA, and these defects were rescued by wild-type but not catalytically inactive Ssu72 (Krishnamurthy et al. 2004). Similarly, gene looping depends on Ssu72 catalytic activity and on the core CPF subunit Pta1, both of which associate with promoter and terminator regions but not with intervening gene bodies (Ansari and Hampsey 2005). In yeast, gene looping—and the cis and trans regulators that enforce it, which include the promoter, PAS, TFIIB, and CPF—is proposed to enforce directionality of transcription from intrinsically bidirectional promoters and thereby prevent transcriptional interference in tandem gene pairs (Tan-Wong et al. 2012). More generally, juxtaposing terminators and promoters would be an efficient means to recycle RNAPII for new rounds of transcription—a role that might require CTD phosphatases such as Ssu72 to erase phosphate marks placed in the previous cycle. Gene looping per se has not been observed in metazoans, but it seems likely that phosphatases such as SSU72 will play prominent roles in RNAPII recycling mechanisms yet to be uncovered.
PPPs emerge as key regulators of RNAPII transcription
Ssu72 is only one of the two phosphatases resident in the CPF; both budding and fission yeast CPF complexes also contain a PP1 holoenzyme (Dichtl et al. 2002; Vanoosthuyse et al. 2014). This was the first demonstration of a PPP family member associating with the core transcriptional machinery and was a harbinger of the central roles of these enzymes now emerging throughout the RNAPII cycle. The budding yeast genome encodes a single, essential PP1 catalytic subunit, Glc7, which, together with the regulatory subunit Ref2 and other factors, forms a subcomplex within the CPF (Fig. 2; Nedea et al. 2003). Glc7 has been implicated in transcription termination, and putative targets of the phosphatase include Sen1 in the NNS termination pathway (Nedea et al. 2008) and pTyr1 of the Rpb1-CTD, removal of which is important for recruitment of the cleavage factors Rtt103 and Pcf11 (Schreieck et al. 2014). Depletion of the PP1 nuclear targeting subunit (PNUTS) led to RNAPII readthrough of termination signals in mouse cells, also consistent with a role for a PP1 holoenzyme in 3′ end formation (Austenaa et al. 2015). S. pombe has two genes encoding PP1 catalytic isoforms, dis2+ and sds21+, neither of which is essential for viability (Fig. 2; Ohkura et al. 1989). Only Dis2, however, is found, together with its regulatory subunits Ppn1 (ortholog of PNUTS) and Swd2.2, in the CPF, where it plays nonredundant roles in chromosome condensation and transcription termination (Vanoosthuyse et al. 2014; Parua et al. 2018). Inactivation of Dis2 in a cold-sensitive lethal dis2-11 mutant did not affect levels of Rpb1-pTyr1 on chromatin but increased levels of phosphorylation on the C-terminal repeats (CTRs) of Spt5 and led to readthrough transcription extending beyond normal termination zones, genome-wide, detected by precision run-on transcription and sequencing (PRO-seq) (Parua et al. 2018). The Spt5 CTRs are phosphorylated by Cdk9 to accelerate elongation by RNAPII (Booth et al. 2018); their PP1-dependent dephosphorylation downstream from the PAS (Fig. 4) appears to apply a brake, leading to RNAPII accumulation in a 3′ paused complex prone to termination—a mechanism conserved between S. pombe (Kecman et al. 2018; Parua et al. 2018) and human cells (Fig. 5; Cortazar et al. 2019; Eaton et al. 2020; Parua et al. 2020).
Figure 5.

The PP1-PNUTS holoenzyme acts at multiple steps of transcription. (Top) PP1-PNUTS activity couples transcription to splicing. PNUTS is recruited to RNAPII paused at the first exon–intron boundary but lacks the PP1 catalytic subunit. Concomitantly, PP1 joins another holoenzyme (likely with the NIPP1 splicing factor) and inhibits the U2 spliceosome. The NUAK1 kinase phosphorylates PNUTS and triggers a PP1 subunit switch to bind PNUTS, thereby promoting both pause release and splicing activation (Cossa et al. 2020). (Bottom) PP1-PNUTS is required for efficient termination. PP1-PNUTS dephosphorylates SPT5, slowing RNAPII elongation and ensuring proper, spatially precise termination (Kecman et al. 2018; Parua et al. 2018; Cortazar et al. 2019; Cossa et al. 2020; Eaton et al. 2020; Parua et al. 2020). (Yellow circles) Phosphorylation.
PP1 regulates the RNAPII transcription cycle at multiple steps
Whereas the defined functions of yeast PP1 are executed near the end of transcription, critical roles have been ascribed to metazoan PP1 throughout the cycle. All of these functions involve the same regulatory subunit, PNUTS, encoded by PPP1R10 (Fig. 5). A role of PP1-PNUTS during early elongation was first suggested by the apparent influence of mouse PNUTS on RNAPII transcriptional polarity; the depletion of PNUTS and of an interactor, Wdr82, induced antisense transcription from bidirectional promoters (Austenaa et al. 2015). This study did not identify the relevant PP1-PNUTS substrate, but antisense transcription induction might involve the known target Spt5, which in its unphosphorylated form restrains movement of RNAPII both upstream of and downstream from the TSS on human genes (Brannan et al. 2012) and has been implicated in suppression of antisense transcription initiating at intragenic sites in S. pombe (Shetty et al. 2017; Sansó et al. 2020). A role for PP1 in the promoter-proximal region is also consistent with recent ChIP-seq analysis of PNUTS, which revealed a distribution similar to that of RNAPII between the TSS and pause site (Cossa et al. 2020).
In ChIP analysis, PNUTS cross-links between the RNAPII pause site and the first exon–intron boundary. PNUTS becomes phosphorylated dependent on the kinase NUAK1 and thus more prone to bind and recruit PP1 catalytic subunits. The incoming PP1 is mainly complexed to the nuclear inhibitor of PP1 (NIPP1), a PP1 regulatory subunit involved in splicing (Fig. 5). Therefore, a NIPP1-PNUTS switch might couple splicing to RNAPII pause release in a temporally and spatially coordinated manner (Cossa et al. 2020). The NUAK1-PP1 interaction also affects later events in transcription; NUAK1 inhibition led to a loss of PNUTS binding to chromatin at both upstream sites and the 3′ ends of genes, where nascent RNA-seq analysis indicated transcriptional readthrough, in keeping with results of PNUTS depletion in mice and with a role of PP1 in termination (Fig. 5; Austenaa et al. 2015; Cortazar et al. 2019; Eaton et al. 2020; Parua et al. 2020).
Other reports have suggested a role of PP1-PNUTS as a CTD phosphatase. Embryos lacking Drosophila PNUTS (dPNUTS), which colocalizes and interacts with RNAPII, had increased pSer5 and impaired gene transcription (Ciurciu et al. 2013). In two recent studies, PNUTS pull-down from human cell extracts coprecipitated RNAPII, and phosphatase assays indicated selectivity for pSer5 in vitro (Wu et al. 2018; Landsverk et al. 2020). The fission yeast Dis2-Ppn1 holoenzyme has also been suggested to dephosphorylate the Rpb1-CTD, albeit on a different residue, Thr4 (Fig. 4; Kecman et al. 2018). It is unclear in either organism whether these phosphorylations increased as direct or indirect results of PP1 inactivation in vivo, and thus, the status and specificity of PP1-PNUTS as an RNAPII-CTD phosphatase remain uncertain.
RNAPII pausing and elongation control: a job opening for PPP family phosphatases
After initiation and promoter clearance on a large fraction of human genes, RNAPII transcribes ∼50–100 nt and becomes stably paused, awaiting additional signals to trigger resumption of processive elongation. This promoter-proximal pausing underlies a distinctive feature of RNAPII ChIP profiles in metazoans: a sharp peak in occupancy just downstream from the TSS on pause-regulated genes. Different organisms and cell types have different proportions of paused versus nonpaused genes (at which RNAPII recruitment or initiation is limiting for expression), typically differentiated by the ratio of RNAPII in the TSS region versus the gene body, known as a pause index (Core and Adelman 2019). Promoter-proximal pausing is nonetheless a pervasive feature of gene regulation in metazoans and also occurs in fission yeast (but not budding yeast), albeit at a smaller fraction of genes (Booth et al. 2016). Importantly, the machinery that governs pausing—pause-inducing and -releasing factors, and the kinases and phosphatases that modify them—is conserved in all eukaryotes, including fungi that lack detectable pausing (with the exception of the negative elongation factor NELF, which is absent in yeast, plants, and worms). Moreover, that machinery appears to be engaged—and Cdk9 activity is required for normal elongation—at virtually all genes transcribed by RNAPII, whether or not release from the pause is the rate-limiting step in their expression (Jonkers et al. 2014; Booth et al. 2018; Luse et al. 2020). The significance of pausing in regulating gene expression has been inferred mainly from the classes of genes most likely to harbor a paused RNAPII elongation complex; highly paused gene subsets are typically enriched for ones involved in stimulus responses or cell cycle regulation, possibly because the release of paused RNAPII, rather than recruitment and initiation de novo, allows a faster or more switch-like (“all or none”) transcriptional response (Core and Adelman 2019).
The mechanisms underlying pausing and pause release have been extensively studied. Both steps—establishment and release—are phosphorylation-dependent and thus regulated by kinases and phosphatases in vivo. A paused RNAPII ternary complex was reconstituted in vitro by addition of DSIF and NELF (Vos et al. 2018b). This complex was severely impaired for transcript elongation, relative to the one containing RNAPII alone, but could be activated by the addition of P-TEFb together with the elongation-promoting factors SPT6 and the PAF complex. This activation, which is likely to mimic pause release in vivo, was accompanied by CDK9-mediated phosphorylation of RNAPII (on both the CTD and a linker region connecting the CTD to the rest of RPB1), DSIF, NELF, SPT6, and PAF complex subunits. Phosphorylated DSIF, SPT6, and PAF were all retained in the active elongation complex, whereas NELF was released (Vos et al. 2018a).
After pause release and during elongation in vivo, Ser5 becomes progressively dephosphorylated (Fig. 4), making pSer5 phosphatases candidates for a role in pause release. No added phosphatase was needed, however, to convert a paused RNAPII elongation complex to an actively elongating one in a defined system comprising purified components (Vos et al. 2018a). Moreover, in ChIP analysis, pSer5 undergoes a gradual decrease rather than a sharp drop in occupancy downstream from the pause site, suggesting that pSer5 removal is not a strict prerequisite for efficient elongation (Bataille et al. 2012). To date, studies of the dedicated pSer5 phosphatases SSU72, Rtr1/RPAP2, and the SCPs have left uncertain their functional relevance to RNAPII pausing and elongation control. It is now clear, however, that other phosphatases—and other targets—play decisive roles in these events. In particular, PPPs have emerged as the major phosphatases that oppose kinase-driven elongation by RNAPII. These studies specifically implicate PP1, PP4, and PP2A, acting through the CTD, SPT5, and possibly other targets, as gatekeepers of RNAPII pause release and brakes on RNAPII elongation (Fig. 4).
PP4 and PP2A keep RNAPII in the starting blocks
Beyond PP1, two other PPP family phosphatases have recently emerged as regulators of early steps in the RNAPII transcription cycle: PP4 and PP2A. By orthogonal approaches in different organisms, two groups independently identified PP4 as a phospho-SPT5 phosphatase. In the worm Caenorhabditis elegans, expression of certain stress-resistance and longevity-promoting genes driven by the transcription factor DAF-16 is dependent on SMK-1, which is a regulatory subunit of a PP4 holoenzyme (ortholog of the human PPP4R3B gene product). Depletion of PP4 subunits by RNAi led to increased phosphorylation on multiple sites in SPT-5 and impaired transcription of specific DAF-16 target genes, although the block appeared to be at initiation rather than during elongation (Sen et al. 2020). Independently, a human PP4 complex was shown to be a target of negative regulation by CDK9, through an inhibitory phosphorylation on another regulatory subunit, PPP4R2, first detected in a chemical-genetic screen for CDK9 substrates (Sansó et al. 2016). Both PPP4R2 and -3 were previously shown to be phosphorylated by a cell cycle CDK to inhibit PP4 and regulate microtubule nucleation at centrosomes (Voss et al. 2013). Treatment of human cells with a CDK9 inhibitor diminished PPP4R2-Thr173 phosphorylation on chromatin, and increased phosphatase activity recovered in anti-PP4 immunoprecipitates, relative to mock-treated controls. Conversely, treatment of PP4 complexes with purified P-TEFb inhibited their dephosphorylating activity toward phosphopeptide substrates. In vitro, PP4 dephosphorylated a site on SPT5 that was refractory to PP1 (Parua et al. 2020). Both that site—pSer666 (Fig. 4), within a linker connecting conserved KOW4 and KOW5 motifs—and sites within CTR1 were phosphorylated when paused complexes were converted to actively elongating ones by P-TEFb in vitro (Figs. 4, 6; Vos et al. 2018a). PP4 complex subunits cross-linked to the promoter-proximal regions of human genes, and their depletion by RNAi increased SPT5 phosphorylation on Ser666 (and CTR1) and caused changes in RNAPII distribution, suggesting attenuation of promoter-proximal pausing. This pattern, detected on three highly paused genes, suggests that human PP4 is a negative regulator of RNAPII entry into processive elongation and a potential enforcer of promoter-proximal pausing (Parua et al. 2020). Taken together, the results in worms and humans indicate a conserved function of a PP4 holoenzyme in dephosphorylating SPT5 and possibly other components of the paused complex to regulate early steps in RNAPII transcription.
Figure 6.
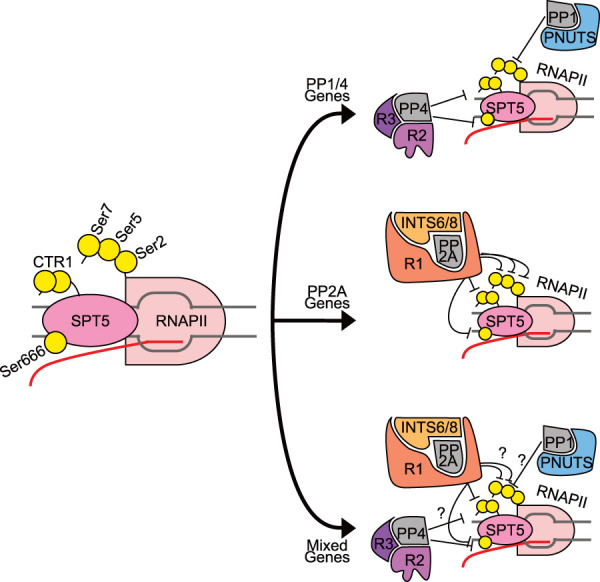
Different PPP holoenzymes cooperate during RNAPII pausing. (Left) Relevant phosphorylation sites on SPT5 and RNAPII. SPT5 C-terminal repeat region 1 (CTR1) typically contains Thr-Pro repeats, where Thr can be phosphorylated by CDK9 (e.g., Thr806 in human SPT5). SPT5 contains another region—the KOW4–KOW5 linker—with multiple Ser residues phosphorylated by CDK9 (including Ser666 in human SPT5). The RNAPII CTD contains multiple heptad repeats, which include phosphorylated residues Ser2, Ser5, and Ser7 (Tyr1 and Thr4 are omitted for clarity). Different PPP family phosphatases have been proposed to act at the promoter-proximal pause, as indicated at right. We speculate that these enzymes might act at different, hypothetical gene classes: “PP1/PP4 genes,” at which PP4 targets SPT5 (Parua et al. 2020) and PP1-PNUTS dephosphorylates pSer5 (Wu et al. 2018; Landsverk et al. 2020); “PP2A genes,” at which PP2A-PPP2R1A, associated with the Integrator, targets both SPT5 and RNAPII residues (Huang et al. 2020; Zheng et al. 2020; Vervoort et al. 2021), and “mixed genes,” where RNAPII and SPT5 are targeted by PP1, PP2A, and PP4 holoenzymes.
Now, three independent and mutually reinforcing studies have implicated PP2A in key aspects of RNAPII transcription, including pause regulation, elongation, and termination (Huang et al. 2020; Zheng et al. 2020; Vervoort et al. 2021). One analysis, in Drosophila cells in culture, uncovered a large number of protein-coding genes that were derepressed by depletion of INTS8, a subunit of the Integrator complex (Huang et al. 2020). Proteomic analysis revealed an interaction of INTS8 with both the A (PR65) and C subunits of PP2A (as well as other Integrator subunits) but no identifiable, regulatory B subunit. Instead, INTS8 appears to provide surrogate B subunit function within the Integrator–PP2A complex; mutating four residues in a short PP2A-recruiting motif of INTS8 (WFEF) to Ala led to up-regulation of Integrator target genes, mimicking knockdown of INTS8, PP2A-C, or PP2A-A or inhibition of PP2A with small molecules. The mechanism of increased transcription appeared to be enhanced release from the promoter-proximal pause; of the most highly paused genes (top decile of genes ranked by pause index), over half were induced by shRNA targeting INTS8, and PRO-seq analysis revealed redistribution of RNAPII away from the pause site when PP2A recruitment was prevented. Similar results were obtained in human embryonic kidney (HEK) 293T cells. Interestingly, perturbation of Integrator–PP2A activity or integrity in cells caused selective increases in SPT5 phosphorylation on Ser666 (or its equivalent residue in Drosophila SPT5), but not on C-terminal repeat region 1 (CTR1), and on both Ser5 and Ser7 (but not Ser2) of the RNAPII CTD (Fig. 4; Huang et al. 2020).
In a parallel study, a proteomic search for binding partners of nuclear PP2A in human cells yielded subunits of the Integrator (Zheng et al. 2020). In agreement with the previous work, this Integrator–PP2A complex (termed “INTAC”), recovered from human cell extracts, contained A and C but not B subunits. Moreover, in a cryo-electron microscopy (cryo-EM) structure of Integrator bound to PP2A-A and -C subunits, the possibility of a “missing” B subunit was effectively excluded due to steric constraints. Instead, the canonical functions of the B subunit in substrate selection are likely to be performed by the Integrator subunits that directly contact PP2A: INTS8, which provides a PP2A-docking site on Integrator, and INTS6, which is even more intimately associated with the PP2A core in a phosphatase module that occupies one side of a cruciform structure comprising backbone and (INTS8-containing) shoulder subcomplexes. The endonuclease subunit INTS11, implicated in premature termination of promoter-proximally engaged RNAPII and in snRNA 3′ end formation (Elrod et al. 2019; Tatomer et al. 2019), binds to the other side of the cruciform. In ChIP-seq analysis, PP2A recruitment to chromatin was dependent on the Integrator subunits but not vice versa. Specific loss of PP2A recruitment led to up-regulation of Integrator target genes, but a preferential effect on pausing or elongation was not detected in this study. In vitro, the purified INTAC holoenzyme dephosphorylated pSer2, pSer5, and pSer7 of RNAPII phosphorylated by TFIIH, whereas in vivo, depletion of PP2A or INTS8 subunits increased pSer2 and pSer5 ChIP signals at target genes. SPT5 was not evaluated as a PP2A target in this study, either in vivo or in vitro.
A third study reinforces the central observation of an Integrator–PP2A holoenzyme with key roles in the RNAPII transcription cycle while providing strong evidence that this complex works in specific opposition to P-TEFb (Vervoort et al. 2021). Approaching the question from yet a third angle, the investigators performed screens of CRISPR-mediated gene knockouts in leukemia-derived cell lines to identify mechanisms of resistance to CDK9 inhibitors. Among the top hits were both INTS6 and INTS8. After identifying, validating, and characterizing an Integrator–PP2A complex in human-cell extracts (also lacking any evidence of a B subunit), the investigators implicated this complex in regulation of RNAPII transcription genome-wide. In ChIP-seq analysis, RNAPII, Integrator, and PP2A colocalized at all active genes, and PP2A was recruited de novo to stimulus-responsive genes concomitant with their transcriptional activation by lipopolysaccharide or epidermal growth factor treatments. These results suggested a specific role of PP2A in antagonizing CDK9; impairing PP2A function, through knockout or knockdown of INTS6 or INTS8 or through pharmacological phosphatase inhibition, overcame effects of CDK9 inhibitors on target protein phosphorylation—including Ser2, Ser5, and Ser7 of the CTD and CTR1 of SPT5—and on transcription by RNAPII. Preventing PP2A recruitment rescued the decreased levels of nascent transcription and attenuated the enhancement of promoter-proximal pausing elicited by CDK9 inhibition, likely explaining the CDK9 inhibitor resistance conferred by INTS6 or INTS8 knockouts. Conversely, treating cells with a small-molecule activator of PP2A (SMAP) further enhanced pausing due to CDK9 inhibition and synergized with CDK9 inhibitors in killing human tumor cells both in culture and in mouse xenograft models.
The pause-enforcing functions and phosphoprotein substrates assigned to the Integrator–PP2A complex overlap with those inferred for PP4 complexes in human cells (Parua et al. 2020). The simplest way to rationalize the existence of two pause-enhancing phosphatases is that each acts at a distinct subset of genes, possibly to promote transcriptional responses to different environmental stimuli (Fig. 6, top and middle). Different analyses have led to subtly different conclusions about the ubiquity of PP2A on protein-coding genes, and genome-wide analyses of PP4 occupancy have yet to be reported. Another, more interesting (and maybe more likely) possibility is that PP2A and PP4 might cohabitate on many or even most genes, either to respond to different stimuli or to work together (Fig. 6, bottom). In this light, it is intriguing that immunoprecipitates of SMK-1 from C. elegans extracts contained, in addition to PP4 subunits, the PP2A scaffold subunit PAA-1 (Sen et al. 2020). Future investigations should aim to reveal the division of labor among different PPP family members in restraining elongation at the promoter-proximal pause and, possibly, other transitions in the RNAPII cycle.
Taken together, these recent studies implicate both PP4 and PP2A in promoter-proximal pausing, but it has yet to be determined whether dephosphorylating enzymes are needed in vivo to establish or maintain the stably paused complex (or both). Whereas the P-TEFb-dependent activation of RNAPII elongation complexes reconstituted in vitro is likely to resemble physiologic pause release, the paused complex on which CDK9 acted was assembled with the constituent proteins purified in their unphosphorylated form (Vos et al. 2018a,b). It seems highly likely that phosphatases of the PPP family will prove to be instrumental, either individually or in combination, in opposing kinase functions in both promoter-proximal pausing and pause release in vivo, but definitive tests of these requirements might only come from more complex systems that can recapitulate phosphoregulation of both steps in vitro.
Next steps: challenges met and unmet in the study of PPPs
The unique strategies by which PPPs achieve functional specificity and their ubiquitous roles in cell physiology render them particularly challenging to study, in contrast to non-PPPs such as FCP1, Rtr1/RPAP2, and SSU72, with more restricted or even dedicated roles in the RNAPII transcription cycle. These phosphatases are not as abundant as PP1 or PP2A (Fig. 2), but their manipulation led to phenotypes more readily interpreted as owing to transcriptional perturbation.
A more surgical approach to dissection of PPP function might be achieved through targeting individual regulatory subunits, but here too, there are challenges. First, the sheer number of PIPs that associate with PP1 appears to be huge and may be underestimated; lack of sequence conservation makes their identification difficult (Verbinnen et al. 2017). Contrast this situation with that of kinases that depend on binding to a regulatory subunit for biological activity: a typical CDK partners with only one or a few cyclins, which in most cases are readily identifiable as such by structural homology. In addition, the depletion of one phosphatase regulatory subunit affects the balance of others, such that the liberated catalytic subunit is redistributed among other partners, potentially influencing unrelated cellular events. One recent advance on this front is the development of expression systems comprising a catalytic subunit covalently linked to a regulatory one (Wu et al. 2018). This approach allows the study of individual regulatory subunits without unbalancing endogenous holoenzymes and was recently used to address the role of human PP1-PNUTS in termination (Cortazar et al. 2019).
Another key difference between kinases and phosphatases is in their “druggability”; i.e., their susceptibility to selective inhibition by small molecules. Compounds such as okadaic acid and calyculin A have been used to dissect the functions of different PPPs, but their specificity is limited: Whereas both compounds preferentially target PP2A over PP1, they are likely to inhibit both enzymes at the concentrations typically used in cell-based experiments. Moreover, active site-directed compounds cannot distinguish among different holoenzymes containing the same catalytic subunit. Attempts to use PPP inhibitors therapeutically are also problematic, as targeting of the PP1 or PP2A catalytic center can have pleiotropic, potentially toxic effects. A more promising approach, with molecules targeting specific holoenzymes, has been pioneered by several laboratories in recent years and is explored below.
Can phosphatases in transcription be targeted therapeutically?
The RNAPII machinery is an attractive target for cancer therapeutics, because cancer cells can become dependent on high-level transcription driven, for example, by oncogenic transcription factors (Bradner et al. 2017). That CDKs acting in the transcription cycle might be targeted therapeutically was first indicated by the cancer-specific cytotoxic effects of the covalent kinase inhibitor THZ1, which is specific for CDK7, CDK12, and CDK13 (Chipumuro et al. 2014; Christensen et al. 2014; Kwiatkowski et al. 2014; Wang et al. 2015). Small molecules more selective for CDK7 were less effective on their own but could be combined with compounds targeting other CDKs, or other pathways, to achieve synergistic effects, suggesting that multiple enzymatic activities might need to be ablated for the best therapeutic effects (Kalan et al. 2017; Minzel et al. 2018; Olson et al. 2019). Similarly, new insights into transcriptional phosphatase function might be harnessed to target cancer, for example, with inhibitors of NUAK1, the kinase that regulates activity of PP1 in splicing (Cossa et al. 2020).
Although compounds directly targeting the catalytic centers of PPPs are likely to have wide-ranging effects in cells and toxicity in clinical settings, alternative strategies have recently been developed to target PP2A, and promising approaches have been proposed for PP1 inhibition. PP2A dysregulation in multiple diseases is well established. For example, levels of PP2A are commonly decreased in cancer or neurodegenerative diseases, and compounds have been developed that boost its activity (Sangodkar et al. 2016). PP2A is inactivated by interacting proteins such as SET and CIP2A. Several compounds have been identified on the basis of their ability to disrupt SET/PP2A or CIP2A/PP2A complexes. This disruption is typically an indirect effect of molecules targeting other activities; for example, the proteasome inhibitor bortezomib or the EGFR inhibitor erlotinib, both of which induce CIP2A down-regulation. An additional approach is the targeting of PME1, an esterase that counteracts a carboxymethylation reaction required for PP2A activation (O'Connor et al. 2018).
An exciting recent advance was the development of SMAPs (see above)—derivatives of phenothiazines (PPZs), compounds historically used for neurological or psychiatric illnesses, which displayed the off-target effect of activating PP2A. PPZs were therefore optimized to remove their psychotropic activity and increase selectivity for PP2A (Sangodkar et al. 2017). A recent report investigated the mechanism of SMAP action, taking advantage of a cryo-EM structure of a PP2A trimer containing the prototypical SMAP DT-061 (Leonard et al. 2020). Interestingly, DT-061 bound a pocket composed of residues in all three subunits, suggesting that SMAPs can act as “molecular glue,” promoting the allosteric activation of PP2A holoenzymes (Leonard et al. 2020; Westermarck and Neel 2020). A concurrent report independently described iHAP1, a SMAP mechanistically distinct from DT-061 with promising efficacy in T-cell lymphoblastic leukemia models (Morita et al. 2020). These studies show the potential of SMAPs for therapeutic PP2A reactivation. Indeed, as described above, the reactivation of Integrator–PP2A complexes with a SMAP was recently shown to synergize with CDK9 inhibitors to trigger apoptosis in cancer cells (Vervoort et al. 2021).
The development of drugs targeting PP1 holoenzymes lagged behind similar efforts aimed at PP2A. Attempts to target PP1 catalytic subunits with derivatives of natural compounds such as calyculin A were largely unsuccessful, indicating that selective PP1 inhibition might not be feasible (Vagnarelli and Alessi 2018). PP1 holoenzymes are structurally and functionally more diverse than PP2A complexes, which have a more constant quaternary structure (although the Integrator–PP2A complex is a clear exception). Moreover, the pervasive role of PP2A in restraining proliferation provided a stronger impetus to PP2A targeted drug discovery. There is no such unity of function among different PP1 holoenzymes, which play disparate roles in diverse cellular pathways; it remains unclear whether drug development campaigns should aim for inhibitors or activators. Nevertheless, PP1 holoenzymes are clearly relevant in human disease, as a recent review of genetically engineered mouse models (GEMMs) of PP1 isoforms or PIPs revealed (Ferreira et al. 2019). Dozens of the GEMMs had disease-associated phenotypes. Different diseases were associated with PP1 activation or inactivation, and there was specificity in both the tissues and cellular compartments involved (Ferreira et al. 2019). Moreover, the diversity of PP1 holoenzymes augurs accessibility to pharmacophores that target less-frequented patches or pockets that constitute protein–protein interaction surfaces, rather than the catalytic centers. A recent success story that illustrates this point is the development of Raphin1, a compound specifically targeting the holoenzyme PP1-PPP1R15B (but not the highly similar PP1-PPP1R15A), which controls the phosphorylation status of the translation initiation factor eIF2α (Krzyzosiak et al. 2018).
Conclusions and perspective: kinase and phosphatase circuits and cycles in transcription
Work over nearly three decades has produced a detailed timeline of kinase functions needed in transcription, and an extensive but still growing inventory of the phosphatases with which kinases collaborate to drive the RNAPII cycle. The cell cycle is also driven by coordinated action of CDKs and opposing phosphatases and provides precedents and paradigms for studying transcription; deeper understanding will emerge as we learn how the key regulatory enzymes signal to the core machinery and to each other. Examples of CDK-CDK cross talk in transcription include a function of the Mediator-associated CDK8 in recruiting CDK7 and CDK9 to initiation or elongation complexes (Donner et al. 2010), the direct activation of CDK9 by CDK7 (Larochelle et al. 2012), and substrate “priming”—the preference of certain CDKs for the CTD previously phosphorylated by a different CDK (Viladevall et al. 2009; Czudnochowski et al. 2012; St. Amour et al. 2012; Bösken et al. 2014; Greifenberg et al. 2016; Mayfield et al. 2019).
Connections have also been uncovered between CDKs and phosphatases. Phosphorylation of CDK9 within its activation loop (T-loop) is required for full activity but also for binding to an inhibitory 7SK-HEXIM1/2 ribonucleoprotein (RNP) complex; PP1, PP2A, and PP2B have been implicated, directly or indirectly, in regulating CDK9 T-loop phosphorylation and thereby the distribution of CDK9 between RNP-bound and RNP-free forms (Ammosova et al. 2005; Chen et al. 2008). Conversely, CDK9 places inhibitory phosphorylations on PP1 catalytic subunits in fission yeast and human cells, thereby preventing dephosphorylation of another CDK9 target, the SPT5 CTRs, and maintaining a rapid elongation rate (Sansó et al. 2016; Parua et al. 2018, 2020). A nearly identical signaling module, comprising PP1 and CDK1, controls the switch-like entry to and exit from mitosis in both fission yeast and metazoans (Grallert et al. 2015). Similarly, a CDK9–PP4 circuit duplicates one previously found to contain a cell cycle CDK and PP4 (Voss et al. 2013). Moreover, just as kinases are often arranged in linear cascades, phosphatase relays play decisive roles in cell signaling pathways; a relevant example occurs during mitotic exit, when PP1 autodephosphorylates to become active and, in turn, dephosphorylates and activates PP2A (Grallert et al. 2015). Similar phosphatase cascades are likely to emerge in the RNAPII transcription cycle, given the recently documented involvement of both PP1 (Kecman et al. 2018; Parua et al. 2018, 2020; Cortazar et al. 2019; Eaton et al. 2020) and PP2A (Huang et al. 2020; Zheng et al. 2020; Vervoort et al. 2021) and the reported interaction of PP4 and PP2A subunits in C. elegans (Sen et al. 2020).
Both the CDK9–PP4 and CDK9–PP1 interactions are likely to make transcription cycle transitions—between promoter-proximally paused and elongating RNAPII and between elongating and 3′ paused RNAPII, respectively—more switch-like and spatially precise (Parua et al. 2020). Moreover, a network in which different phosphatases (PP1, PP2A, and PP4) oppose the actions of a single kinase (CDK9) can achieve spatial and temporal complexity in the modification patterns it creates on multisite phosphorylation substrates such as RNAPII and SPT5, another regulatory strategy previously uncovered in the cell cycle (Swaffer et al. 2016).
An important question still to be addressed is whether phosphatase activity is truly regulatory under physiologic conditions. Transcriptional CDKs have attracted intense interest not only because of their inherent druggability, but also because of their roles in transducing extrinsic signals to the RNAPII machinery to promote or repress gene expression. For example, CDK9 can be induced by genotoxic stress or co-opted by viral or oncogenic trans-activators to drive specific transcription programs (Bacon and D'Orso 2019). We are not aware of a clear example of phosphatases acting in such a decisive way, but the involvement of PP1 holoenzymes in the control of RNAPII elongation rate (Parua et al. 2018, 2020; Cortazar et al. 2019; Eaton et al. 2020) suggests where we might look to find one. In yeast and metazoans, nutrient-sensing and other signaling pathways use transcriptional interference between neighboring genes as a regulatory switch (Kaikkonen and Adelman 2018; Shuman 2020). In fission yeast, key genes encoding proteins needed for organic phosphate acquisition (pho genes) are arranged in tandem with lncRNA genes; under phosphate-replete conditions, readthrough transcription driven by the upstream lncRNA promoters disrupts DNA binding by an activator of the downstream protein-coding genes, limiting their expression. Upon phosphate starvation, the lncRNA transcripts terminate farther upstream, and expression of the pho genes—pho1+, pho84+, and tgp1+—is induced (Shuman 2020). This control can be disrupted by a mutation in rpb1 that decreases the intrinsic rate of elongation by RNAPII, favoring termination of lncRNA transcripts near a more upstream PAS and derepressing tgp1+ under phosphate-replete conditions (Yague-Sanz et al. 2020). Conversely, hyperrepression of pho genes was elicited by mutations that impair 3′ end processing or termination: a CTD-T4A substitution in the rpb1 gene, and mutations in subunits of CPF, including ssu72 and ppn1 (Sanchez et al. 2018). In contrast to the slow rpb1 mutant described above, loss of CPF-associated PP1 is likely to attenuate or delay the deceleration of RNAPII that promotes termination downstream from the PAS (Kecman et al. 2018; Parua et al. 2018), possibly contributing to pho gene hyperrepression. The choice of a lncRNA 3′ end processing site is responsive to cellular levels of specific inositol pyrophosphates (IPPs) (Sanchez et al. 2019; Garg et al. 2020); it will be interesting to map the downstream targets of this signaling, which might include transcriptional kinases, phosphatases, or both.
It is a safe bet that more regulatory circuits, involving other kinases and phosphatases, will emerge in the next few years. We predict the traffic will flow both ways, with phosphatases also signaling to kinases, and include all permutations of negative and positive regulation. An intriguing question for the future is how phosphatases coordinate with the CDKs to regulate liquid–liquid phase separation as RNAPII transits in and out of condensates with different proteins (Cramer 2019b). Phosphorylation by CDKs can dissolve transcription “hubs” containing RNAPII and PIC components and facilitate formation of distinct condensates containing RNAPII and splicing factors (Boehning et al. 2018; Lu et al. 2018; Guo et al. 2019). It seems nearly inevitable that phosphatases will play complementary roles as “cogatekeepers” controlling ingress and egress at these assemblies. Given the rapid recent progress in the field, moreover, a final, seemingly safe prediction is that there will be more surprises, and new players enlisted—including other, “famous” phosphatases such as the tumor suppressor PTEN (Steinbach et al. 2019)—in a truly comprehensive model of the RNAPII transcription cycle.
Acknowledgments
We thank Rui Sun for critical review of the manuscript. This work was supported by grants from the European Research Council (AuroMYC) and the European Fonds for Regional Development (to M.E.) and National Institutes of Health grant R35 GM127289 (to R.P.F.).
Footnotes
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.348315.121.
Competing interest statement
The authors declare no competing interests.
References
- Adelman K, Lis JT. 2012. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet 13: 720–731. 10.1038/nrg3293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammosova T, Washington K, Debebe Z, Brady J, Nekhai S. 2005. Dephosphorylation of CDK9 by protein phosphatase 2A and protein phosphatase-1 in Tat-activated HIV-1 transcription. Retrovirology 2: 47. 10.1186/1742-4690-2-47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari A, Hampsey M. 2005. A role for the CPF 3′-end processing machinery in RNAP II-dependent gene looping. Genes Dev 19: 2969–2978. 10.1101/gad.1362305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archambault J, Chambers RS, Kobor MS, Ho Y, Cartier M, Bolotin D, Andrews B, Kane CM, Greenblatt J. 1997. An essential component of a C-terminal domain phosphatase that interacts with transcription factor IIF in Saccharomyces cerevisiae. Proc Natl Acad Sci 94: 14300–14305. 10.1073/pnas.94.26.14300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archambault J, Pan G, Dahmus GK, Cartier M, Marshall N, Zhang S, Dahmus ME, Greenblatt J. 1998. FCP1, the RAP74-interacting subunit of a human protein phosphatase that dephosphorylates the carboxyl-terminal domain of RNA polymerase IIO. J Biol Chem 273: 27593–27601. 10.1074/jbc.273.42.27593 [DOI] [PubMed] [Google Scholar]
- Austenaa LM, Barozzi I, Simonatto M, Masella S, Della Chiara G, Ghisletti S, Curina A, de Wit E, Bouwman BA, de Pretis S, et al. 2015. Transcription of mammalian cis-regulatory elements is restrained by actively enforced early termination. Mol Cell 60: 460–474. 10.1016/j.molcel.2015.09.018 [DOI] [PubMed] [Google Scholar]
- Bacon CW, D'Orso I. 2019. CDK9: a signaling hub for transcriptional control. Transcription 10: 57–75. 10.1080/21541264.2018.1523668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bataille AR, Jeronimo C, Jacques PE, Laramée L, Fortin ME, Forest A, Bergeron M, Hanes SD, Robert F. 2012. A universal RNA polymerase II CTD cycle is orchestrated by complex interplays between kinase, phosphatase, and isomerase enzymes along genes. Mol Cell 45: 158–170. 10.1016/j.molcel.2011.11.024 [DOI] [PubMed] [Google Scholar]
- Beltrao P, Bork P, Krogan NJ, van Noort V. 2013. Evolution and functional cross-talk of protein post-translational modifications. Mol Syst Biol 9: 714. 10.1002/msb.201304521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolotti A. 2018. The split protein phosphatase system. Biochem J 475: 3707–3723. 10.1042/BCJ20170726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehning M, Dugast-Darzacq C, Rankovic M, Hansen AS, Yu T, Marie-Nelly H, McSwiggen DT, Kokic G, Dailey GM, Cramer P, et al. 2018. RNA polymerase II clustering through carboxy-terminal domain phase separation. Nat Struct Mol Biol 25: 833–840. 10.1038/s41594-018-0112-y [DOI] [PubMed] [Google Scholar]
- Booth GT, Wang IX, Cheung VG, Lis JT. 2016. Divergence of a conserved elongation factor and transcription regulation in budding and fission yeast. Genome Res 26: 799–811. 10.1101/gr.204578.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth GT, Parua PK, Sansó M, Fisher RP, Lis JT. 2018. Cdk9 regulates a promoter-proximal checkpoint to modulate RNA polymerase II elongation rate in fission yeast. Nat Commun 9: 543. 10.1038/s41467-018-03006-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bösken CA, Farnung L, Hintermair C, Merzel Schachter M, Vogel-Bachmayr K, Blazek D, Anand K, Fisher RP, Eick D, Geyer M. 2014. The structure and substrate specificity of human Cdk12/Cyclin K. Nat Commun 5: 3505. 10.1038/ncomms4505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradner JE, Hnisz D, Young RA. 2017. Transcriptional addiction in cancer. Cell 168: 629–643. 10.1016/j.cell.2016.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brannan K, Kim H, Erickson B, Glover-Cutter K, Kim S, Fong N, Kiemele L, Hansen K, Davis R, Lykke-Andersen J, et al. 2012. mRNA decapping factors and the exonuclease Xrn2 function in widespread premature termination of RNA polymerase II transcription. Mol Cell 46: 311–324. 10.1016/j.molcel.2012.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitkreutz A, Choi H, Sharom JR, Boucher L, Neduva V, Larsen B, Lin ZY, Breitkreutz BJ, Stark C, Liu G, et al. 2010. A global protein kinase and phosphatase interaction network in yeast. Science 328: 1043–1046. 10.1126/science.1176495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratowski S. 2003. The CTD code. Nat Struct Biol 10: 679–680. 10.1038/nsb0903-679 [DOI] [PubMed] [Google Scholar]
- Buratowski S. 2009. Progression through the RNA polymerase II CTD cycle. Mol Cell 36: 541–546. 10.1016/j.molcel.2009.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceulemans H, Bollen M. 2004. Functional diversity of protein phosphatase-1, a cellular economizer and reset button. Physiol Rev 84: 1–39. 10.1152/physrev.00013.2003 [DOI] [PubMed] [Google Scholar]
- Chambers RS, Dahmus ME. 1994. Purification and characterization of a phosphatase from HeLa cells which dephosphorylates the C-terminal domain of RNA polymerase II. J Biol Chem 269: 26243–26248. 10.1016/S0021-9258(18)47186-4 [DOI] [PubMed] [Google Scholar]
- Chambers RS, Kane CM. 1996. Purification and characterization of an RNA polymerase II phosphatase from yeast. J Biol Chem 271: 24498–24504. 10.1074/jbc.271.40.24498 [DOI] [PubMed] [Google Scholar]
- Chambers RS, Wang BQ, Burton ZF, Dahmus ME. 1995. The activity of COOH-terminal domain phosphatase is regulated by a docking site on RNA polymerase II and by the general transcription factors IIF and IIB. J Biol Chem 270: 14962–14969. 10.1074/jbc.270.25.14962 [DOI] [PubMed] [Google Scholar]
- Chen R, Liu M, Li H, Xue Y, Ramey WN, He N, Ai N, Luo H, Zhu Y, Zhou N, et al. 2008. PP2B and PP1α cooperatively disrupt 7SK snRNP to release P-TEFb for transcription in response to Ca2+ signaling. Genes Dev 22: 1356–1368. 10.1101/gad.1636008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MJ, Dixon JE, Manning G. 2017. Genomics and evolution of protein phosphatases. Sci Signal 10: eaag1796. 10.1126/scisignal.aag1796 [DOI] [PubMed] [Google Scholar]
- Chipumuro E, Marco E, Christensen CL, Kwiatkowski N, Zhang T, Hatheway CM, Abraham BJ, Sharma B, Yeung C, Altabef A, et al. 2014. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 159: 1126–1139. 10.1016/j.cell.2014.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Kim TK, Mancebo H, Lane WS, Flores O, Reinberg D. 1999. A protein phosphatase functions to recycle RNA polymerase II. Genes Dev 13: 1540–1552. 10.1101/gad.13.12.1540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho EJ, Kobor MS, Kim M, Greenblatt J, Buratowski S. 2001. Opposing effects of Ctk1 kinase and Fcp1 phosphatase at Ser 2 of the RNA polymerase II C-terminal domain. Genes Dev 15: 3319–3329. 10.1101/gad.935901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen CL, Kwiatkowski N, Abraham BJ, Carretero J, Al-Shahrour F, Zhang T, Chipumuro E, Herter-Sprie GS, Akbay EA, Altabef A, et al. 2014. Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell 26: 909–922. 10.1016/j.ccell.2014.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciurciu A, Duncalf L, Jonchere V, Lansdale N, Vasieva O, Glenday P, Rudenko A, Vissi E, Cobbe N, Alphey L, et al. 2013. PNUTS/PP1 regulates RNAPII-mediated gene expression and is necessary for developmental growth. PLoS Genet 9: e1003885. 10.1371/journal.pgen.1003885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corden JL. 1990. Tails of RNA polymerase II. Trends Biochem Sci 15: 383–387. 10.1016/0968-0004(90)90236-5 [DOI] [PubMed] [Google Scholar]
- Core L, Adelman K. 2019. Promoter-proximal pausing of RNA polymerase II: a nexus of gene regulation. Genes Dev 33: 960–982. 10.1101/gad.325142.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortazar MA, Sheridan RM, Erickson B, Fong N, Glover-Cutter K, Brannan K, Bentley DL. 2019. Control of RNA Pol II speed by PNUTS-PP1 and Spt5 dephosphorylation facilitates termination by a ‘sitting duck torpedo’ mechanism. Mol Cell 76: 896–908.e4. 10.1016/j.molcel.2019.09.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossa G, Roeschert I, Prinz F, Baluapuri A, Silveira Vidal R, Schülein-Völk C, Chang YC, Ade CP, Mastrobuoni G, Girard C, et al. 2020. Localized inhibition of protein phosphatase 1 by NUAK1 promotes spliceosome activity and reveals a MYC-sensitive feedback control of transcription. Mol Cell 77: 1322–1339.e11. 10.1016/j.molcel.2020.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer P. 2019a. Eukaryotic transcription turns 50. Cell 179: 808–812. 10.1016/j.cell.2019.09.018 [DOI] [PubMed] [Google Scholar]
- Cramer P. 2019b. Organization and regulation of gene transcription. Nature 573: 45–54. 10.1038/s41586-019-1517-4 [DOI] [PubMed] [Google Scholar]
- Czudnochowski N, Bösken CA, Geyer M. 2012. Serine-7 but not serine-5 phosphorylation primes RNA polymerase II CTD for P-TEFb recognition. Nat Commun 3: 842. 10.1038/ncomms1846 [DOI] [PubMed] [Google Scholar]
- Descostes N, Heidemann M, Spinelli L, Schüller R, Maqbool MA, Fenouil R, Koch F, Innocenti C, Gut M, Gut I, et al. 2014. Tyrosine phosphorylation of RNA polymerase II CTD is associated with antisense promoter transcription and active enhancers in mammalian cells. Elife 3: e02105. 10.7554/eLife.02105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dichtl B, Blank D, Ohnacker M, Friedlein A, Roeder D, Langen H, Keller W. 2002. A role for SSU72 in balancing RNA polymerase II transcription elongation and termination. Mol Cell 10: 1139–1150. 10.1016/S1097-2765(02)00707-4 [DOI] [PubMed] [Google Scholar]
- Dingar D, Tu WB, Resetca D, Lourenco C, Tamachi A, De Melo J, Houlahan KE, Kalkat M, Chan PK, Boutros PC, et al. 2018. MYC dephosphorylation by the PP1/PNUTS phosphatase complex regulates chromatin binding and protein stability. Nat Commun 9: 3502. 10.1038/s41467-018-05660-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donner AJ, Ebmeier CC, Taatjes DJ, Espinosa JM. 2010. CDK8 is a positive regulator of transcriptional elongation within the serum response network. Nat Struct Mol Biol 17: 194–201. 10.1038/nsmb.1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton JD, Francis L, Davidson L, West S. 2020. A unified allosteric/torpedo mechanism for transcriptional termination on human protein-coding genes. Genes Dev 34: 132–145. 10.1101/gad.332833.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egloff S, Murphy S. 2008. Cracking the RNA polymerase II CTD code. Trends Genet 24: 280–288. 10.1016/j.tig.2008.03.008 [DOI] [PubMed] [Google Scholar]
- Egloff S, Zaborowska J, Laitem C, Kiss T, Murphy S. 2012. Ser7 phosphorylation of the CTD recruits the RPAP2 Ser5 phosphatase to snRNA genes. Mol Cell 45: 111–122. 10.1016/j.molcel.2011.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elrod ND, Henriques T, Huang KL, Tatomer DC, Wilusz JE, Wagner EJ, Adelman K. 2019. The integrator complex attenuates promoter-proximal transcription at protein-coding genes. Mol Cell 76: 738–752.e7. 10.1016/j.molcel.2019.10.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira M, Beullens M, Bollen M, Van Eynde A. 2019. Functions and therapeutic potential of protein phosphatase 1: insights from mouse genetics. Biochim Biophys Acta Mol Cell Res 1866: 16–30. 10.1016/j.bbamcr.2018.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuda NJ, Buckley MS, Wei W, Core LJ, Waters CT, Reinberg D, Lis JT. 2012. Fcp1 dephosphorylation of the RNA polymerase II C-terminal domain is required for efficient transcription of heat shock genes. Mol Cell Biol 32: 3428–3437. 10.1128/MCB.00247-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem C, Devaux F, Torchet C, Jacq C, Quevillon-Cheruel S, Labesse G, Facca C, Faye G. 2003. Ssu72 is a phosphatase essential for transcription termination of snoRNAs and specific mRNAs in yeast. EMBO J 22: 1588–1598. 10.1093/emboj/cdg141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg A, Shuman S, Schwer B. 2020. A genetic screen for suppressors of hyper-repression of the fission yeast PHO regulon by Pol2 CTD mutation T4A implicates inositol 1-pyrophosphates as agonists of precocious lncRNA transcription termination. Nucleic Acids Res 48: 10739–10752. 10.1093/nar/gkaa776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelens L, Qian J, Bollen M, Saurin AT. 2018. The importance of kinase–phosphatase integration: lessons from mitosis. Trends Cell Biol 28: 6–21. 10.1016/j.tcb.2017.09.005 [DOI] [PubMed] [Google Scholar]
- Ghosh A, Shuman S, Lima CD. 2008. The structure of Fcp1, an essential RNA polymerase II CTD phosphatase. Mol Cell 32: 478–490. 10.1016/j.molcel.2008.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibney PA, Fries T, Bailer SM, Morano KA. 2008. Rtr1 is the Saccharomyces cerevisiae homolog of a novel family of RNA polymerase II-binding proteins. Eukaryot Cell 7: 938–948. 10.1128/EC.00042-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover-Cutter K, Larochelle S, Erickson B, Zhang C, Shokat K, Fisher RP, Bentley DL. 2009. TFIIH-associated Cdk7 kinase functions in phosphorylation of C-terminal domain Ser7 residues, promoter-proximal pausing, and termination by RNA polymerase II. Mol Cell Biol 29: 5455–5464. 10.1128/MCB.00637-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grallert A, Boke E, Hagting A, Hodgson B, Connolly Y, Griffiths JR, Smith DL, Pines J, Hagan IM. 2015. A PP1-PP2A phosphatase relay controls mitotic progression. Nature 517: 94–98. 10.1038/nature14019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greifenberg AK, Hönig D, Pilarova K, Düster R, Bartholomeeusen K, Bösken CA, Anand K, Blazek D, Geyer M. 2016. Structural and functional analysis of the Cdk13/Cyclin K complex. Cell Rep 14: 320–331. 10.1016/j.celrep.2015.12.025 [DOI] [PubMed] [Google Scholar]
- Guo YE, Manteiga JC, Henninger JE, Sabari BR, Dall'Agnese A, Hannett NM, Spille JH, Afeyan LK, Zamudio AV, Shrinivas K, et al. 2019. Pol II phosphorylation regulates a switch between transcriptional and splicing condensates. Nature 572: 543–548. 10.1038/s41586-019-1464-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heroes E, Lesage B, Görnemann J, Beullens M, Van Meervelt L, Bollen M. 2013. The PP1 binding code: a molecular-lego strategy that governs specificity. FEBS J 280: 584–595. 10.1111/j.1742-4658.2012.08547.x [DOI] [PubMed] [Google Scholar]
- Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E. 2015. Phosphositeplus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 43: D512–D520. 10.1093/nar/gku1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsin JP, Manley JL. 2012. The RNA polymerase II CTD coordinates transcription and RNA processing. Genes Dev 26: 2119–2137. 10.1101/gad.200303.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsin JP, Xiang K, Manley JL. 2014. Function and control of RNA polymerase II C-terminal domain phosphorylation in vertebrate transcription and RNA processing. Mol Cell Biol 34: 2488–2498. 10.1128/MCB.00181-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PL, Yang F, Smith-Kinnaman W, Yang W, Song JE, Mosley AL, Varani G. 2014. Rtr1 is a dual specificity phosphatase that dephosphorylates Tyr1 and Ser5 on the RNA polymerase II CTD. J Mol Biol 426: 2970–2981. 10.1016/j.jmb.2014.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang KL, Jee D, Stein CB, Elrod ND, Henriques T, Mascibroda LG, Baillat D, Russell WK, Adelman K, Wagner EJ. 2020. Integrator recruits protein phosphatase 2A to prevent pause release and facilitate transcription termination. Mol Cell 80: 345–358.e9. 10.1016/j.molcel.2020.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irani S, Yogesha SD, Mayfield J, Zhang M, Zhang Y, Matthews WL, Nie G, Prescott NA, Zhang YJ. 2016. Structure of Saccharomyces cerevisiae Rtr1 reveals an active site for an atypical phosphatase. Sci Signal 9: ra24. 10.1126/scisignal.aad4805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeronimo C, Forget D, Bouchard A, Li Q, Chua G, Poitras C, Thérien C, Bergeron D, Bourassa S, Greenblatt J, et al. 2007. Systematic analysis of the protein interaction network for the human transcription machinery reveals the identity of the 7SK capping enzyme. Mol Cell 27: 262–274. 10.1016/j.molcel.2007.06.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers I, Kwak H, Lis JT. 2014. Genome-wide dynamics of Pol II elongation and its interplay with promoter proximal pausing, chromatin, and exons. Elife 3: e02407. 10.7554/eLife.02407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaikkonen MU, Adelman K. 2018. Emerging roles of non-coding RNA transcription. Trends Biochem Sci 43: 654–667. 10.1016/j.tibs.2018.06.002 [DOI] [PubMed] [Google Scholar]
- Kalan S, Amat R, Schachter MM, Kwiatkowski N, Abraham BJ, Liang Y, Zhang T, Olson CM, Larochelle S, Young RA, et al. 2017. Activation of the p53 transcriptional program sensitizes cancer cells to Cdk7 inhibitors. Cell Rep 21: 467–481. 10.1016/j.celrep.2017.09.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenski T, Heilmeier S, Meinhart A, Cramer P. 2004. Structure and mechanism of RNA polymerase II CTD phosphatases. Mol Cell 15: 399–407. 10.1016/j.molcel.2004.06.035 [DOI] [PubMed] [Google Scholar]
- Kauko O, Imanishi SY, Kulesskiy E, Yetukuri L, Laajala TD, Sharma M, Pavic K, Aakula A, Rupp C, Jumppanen M, et al. 2020. Phosphoproteome and drug-response effects mediated by the three protein phosphatase 2A inhibitor proteins CIP2A, SET, and PME-1. J Biol Chem 295: 4194–4211. 10.1074/jbc.RA119.011265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kecman T, Kuś K, Heo DH, Duckett K, Birot A, Liberatori S, Mohammed S, Geis-Asteggiante L, Robinson CV, Vasiljeva L. 2018. Elongation/termination factor exchange mediated by PP1 phosphatase orchestrates transcription termination. Cell Rep 25: 259–269.e5. 10.1016/j.celrep.2018.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Erickson B, Luo W, Seward D, Graber JH, Pollock DD, Megee PC, Bentley DL. 2010. Gene-specific RNA polymerase II phosphorylation and the CTD code. Nat Struct Mol Biol 17: 1279–1286. 10.1038/nsmb.1913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobor MS, Archambault J, Lester W, Holstege FC, Gileadi O, Jansma DB, Jennings EG, Kouyoumdjian F, Davidson AR, Young RA, et al. 1999. An unusual eukaryotic protein phosphatase required for transcription by RNA polymerase II and CTD dephosphorylation in S. cerevisiae. Mol Cell 4: 55–62. 10.1016/S1097-2765(00)80187-2 [DOI] [PubMed] [Google Scholar]
- Kobor MS, Simon LD, Omichinski J, Zhong G, Archambault J, Greenblatt J. 2000. A motif shared by TFIIF and TFIIB mediates their interaction with the RNA polymerase II carboxy-terminal domain phosphatase Fcp1p in Saccharomyces cerevisiae. Mol Cell Biol 20: 7438–7449. 10.1128/MCB.20.20.7438-7449.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy S, He X, Reyes-Reyes M, Moore C, Hampsey M. 2004. Ssu72 Is an RNA polymerase II CTD phosphatase. Mol Cell 14: 387–394. 10.1016/S1097-2765(04)00235-7 [DOI] [PubMed] [Google Scholar]
- Krzyzosiak A, Sigurdardottir A, Luh L, Carrara M, Das I, Schneider K, Bertolotti A. 2018. Target-based discovery of an inhibitor of the regulatory phosphatase PPP1R15B. Cell 174: 1216–1228.e19. 10.1016/j.cell.2018.06.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, Dastur A, Amzallag A, Ramaswamy S, Tesar B, et al. 2014. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 511: 616–620. 10.1038/nature13393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landsverk HB, Sandquist LE, Bay LTE, Steurer B, Campsteijn C, Landsverk OJB, Marteijn JA, Petermann E, Trinkle-Mulcahy L, Syljuåsen RG. 2020. WDR82/PNUTS-PP1 prevents transcription-replication conflicts by promoting RNA polymerase II degradation on chromatin. Cell Rep 33: 108469. 10.1016/j.celrep.2020.108469 [DOI] [PubMed] [Google Scholar]
- Larochelle S, Batliner J, Gamble MJ, Barboza NM, Kraybill BC, Blethrow JD, Shokat KM, Fisher RP. 2006. Dichotomous but stringent substrate selection by the dual-function Cdk7 complex revealed by chemical genetics. Nat Struct Mol Biol 13: 55–62. 10.1038/nsmb1028 [DOI] [PubMed] [Google Scholar]
- Larochelle S, Amat R, Glover-Cutter K, Sansó M, Zhang C, Allen JJ, Shokat KM, Bentley DL, Fisher RP. 2012. Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat Struct Mol Biol 19: 1108–1115. 10.1038/nsmb.2399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard D, Huang W, Izadmehr S, O'Connor CM, Wiredja DD, Wang Z, Zaware N, Chen Y, Schlatzer DM, Kiselar J, et al. 2020. Selective PP2A enhancement through biased heterotrimer stabilization. Cell 181: 688–701.e16. 10.1016/j.cell.2020.03.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Yu D, Hansen AS, Ganguly S, Liu R, Heckert A, Darzacq X, Zhou Q. 2018. Phase-separation mechanism for C-terminal hyperphosphorylation of RNA polymerase II. Nature 558: 318–323. 10.1038/s41586-018-0174-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luse DS, Parida M, Spector BM, Nilson KA, Price DH. 2020. A unified view of the sequence and functional organization of the human RNA polymerase II promoter. Nucleic Acids Res 48: 7767–7785. 10.1093/nar/gkaa531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M. 2014. Cyclin-dependent kinases. Genome Biol 15: 122. 10.1186/gb4184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal SS, Cho H, Kim S, Cabane K, Reinberg D. 2002. FCP1, a phosphatase specific for the heptapeptide repeat of the largest subunit of RNA polymerase II, stimulates transcription elongation. Mol Cell Biol 22: 7543–7552. 10.1128/MCB.22.21.7543-7552.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manke IA, Lowery DM, Nguyen A, Yaffe MB. 2003. BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science 302: 636–639. 10.1126/science.1088877 [DOI] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. 2002. The protein kinase complement of the human genome. Science 298: 1912–1934. 10.1126/science.1075762 [DOI] [PubMed] [Google Scholar]
- Mayer A, Lidschreiber M, Siebert M, Leike K, Söding J, Cramer P. 2010. Uniform transitions of the general RNA polymerase II transcription complex. Nat Struct Mol Biol 17: 1272–1278. 10.1038/nsmb.1903 [DOI] [PubMed] [Google Scholar]
- Mayfield JE, Burkholder NT, Zhang YJ. 2016. Dephosphorylating eukaryotic RNA polymerase II. Biochim Biophys Acta 1864: 372–387. 10.1016/j.bbapap.2016.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayfield JE, Irani S, Escobar EE, Zhang Z, Burkholder NT, Robinson MR, Mehaffey MR, Sipe SN, Yang W, Prescott NA, et al. 2019. Tyr1 phosphorylation promotes phosphorylation of Ser2 on the C-terminal domain of eukaryotic RNA polymerase II by P-TEFb. Elife 8: e48725. 10.7554/eLife.48725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinhart A, Silberzahn T, Cramer P. 2003. The mRNA transcription/processing factor Ssu72 is a potential tyrosine phosphatase. J Biol Chem 278: 15917–15921. 10.1074/jbc.M301643200 [DOI] [PubMed] [Google Scholar]
- Minzel W, Venkatachalam A, Fink A, Hung E, Brachya G, Burstain I, Shaham M, Rivlin A, Omer I, Zinger A, et al. 2018. Small molecules co-targeting CKIα and the transcriptional kinases CDK7/9 control AML in preclinical models. Cell 175: 171–185.e25. 10.1016/j.cell.2018.07.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorhead GB, Trinkle-Mulcahy L, Nimick M, De Wever V, Campbell DG, Gourlay R, Lam YW, Lamond AI. 2008. Displacement affinity chromatography of protein phosphatase one (PP1) complexes. BMC Biochem 9: 28. 10.1186/1471-2091-9-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita K, He S, Nowak RP, Wang J, Zimmerman MW, Fu C, Durbin AD, Martel MW, Prutsch N, Gray NS, et al. 2020. Allosteric activators of protein phosphatase 2A display broad antitumor activity mediated by dephosphorylation of MYBL2. Cell 181: 702–715.e20. 10.1016/j.cell.2020.03.051 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Morrison DK, Murakami MS, Cleghon V. 2000. Protein kinases and phosphatases in the Drosophila genome. J Cell Biol 150: F57–F62. 10.1083/jcb.150.2.F57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosley AL, Pattenden SG, Carey M, Venkatesh S, Gilmore JM, Florens L, Workman JL, Washburn MP. 2009. Rtr1 is a CTD phosphatase that regulates RNA polymerase II during the transition from serine 5 to serine 2 phosphorylation. Mol Cell 34: 168–178. 10.1016/j.molcel.2009.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedea E, He X, Kim M, Pootoolal J, Zhong G, Canadien V, Hughes T, Buratowski S, Moore CL, Greenblatt J. 2003. Organization and function of APT, a subcomplex of the yeast cleavage and polyadenylation factor involved in the formation of mRNA and small nucleolar RNA 3′-ends. J Biol Chem 278: 33000–33010. 10.1074/jbc.M304454200 [DOI] [PubMed] [Google Scholar]
- Nedea E, Nalbant D, Xia D, Theoharis NT, Suter B, Richardson CJ, Tatchell K, Kislinger T, Greenblatt JF, Nagy PL. 2008. The Glc7 phosphatase subunit of the cleavage and polyadenylation factor is essential for transcription termination on snoRNA genes. Mol Cell 29: 577–587. 10.1016/j.molcel.2007.12.031 [DOI] [PubMed] [Google Scholar]
- Nesti E, Corson GM, McCleskey M, Oyer JA, Mandel G. 2014. C-terminal domain small phosphatase 1 and MAP kinase reciprocally control REST stability and neuronal differentiation. Proc Natl Acad Sci 111: E3929–E3936. 10.1073/pnas.1414770111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Z, Xu C, Guo X, Hunter GO, Kuznetsova OV, Tempel W, Marcon E, Zhong G, Guo H, Kuo WW, et al. 2014. RPRD1A and RPRD1B are human RNA polymerase II C-terminal domain scaffolds for Ser5 dephosphorylation. Nat Struct Mol Biol 21: 686–695. 10.1038/nsmb.2853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor CM, Perl A, Leonard D, Sangodkar J, Narla G. 2018. Therapeutic targeting of PP2A. Int J Biochem Cell Biol 96: 182–193. 10.1016/j.biocel.2017.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkura H, Kinoshita N, Miyatani S, Toda T, Yanagida M. 1989. The fission yeast dis2+ gene required for chromosome disjoining encodes one of two putative type 1 protein phosphatases. Cell 57: 997–1007. 10.1016/0092-8674(89)90338-3 [DOI] [PubMed] [Google Scholar]
- Olson CM, Liang Y, Leggett A, Park WD, Li L, Mills CE, Elsarrag SZ, Ficarro SB, Zhang T, Düster R, et al. 2019. Development of a selective CDK7 covalent inhibitor reveals predominant cell-cycle phenotype. Cell Chem Biol 26: 792–803.e10. 10.1016/j.chembiol.2019.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Örd M, Loog M. 2019. How the cell cycle clock ticks. Mol Biol Cell 30: 169–172. 10.1091/mbc.E18-05-0272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parua PK, Fisher RP. 2020. Dissecting the Pol II transcription cycle and derailing cancer with CDK inhibitors. Nat Chem Biol 16: 716–724. 10.1038/s41589-020-0563-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parua PK, Booth GT, Sansó M, Benjamin B, Tanny JC, Lis JT, Fisher RP. 2018. A Cdk9–PP1 switch regulates the elongation–termination transition of RNA polymerase II. Nature 558: 460–464. 10.1038/s41586-018-0214-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parua PK, Kalan S, Benjamin B, Sansó M, Fisher RP. 2020. Distinct Cdk9-phosphatase switches act at the beginning and end of elongation by RNA polymerase II. Nat Commun 11: 4338. 10.1038/s41467-020-18173-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pons T, Paramonov I, Boullosa C, Ibáñez K, Rojas AM, Valencia A. 2014. A common structural scaffold in CTD phosphatases that supports distinct catalytic mechanisms. Proteins 82: 103–118. 10.1002/prot.24376 [DOI] [PubMed] [Google Scholar]
- Poss ZC, Ebmeier CC, Odell AT, Tangpeerachaikul A, Lee T, Pelish HE, Shair MD, Dowell RD, Old WM, Taatjes DJ. 2016. Identification of mediator kinase substrates in human cells using cortistatin A and quantitative phosphoproteomics. Cell Rep 15: 436–450. 10.1016/j.celrep.2016.03.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Reyes M, Hampsey M. 2007. Role for the Ssu72 C-terminal domain phosphatase in RNA polymerase II transcription elongation. Mol Cell Biol 27: 926–936. 10.1128/MCB.01361-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimel JK, Poss ZC, Erickson B, Maas ZL, Ebmeier CC, Johnson JL, Decker TM, Yaron TM, Bradley MJ, Hamman KB, et al. 2020. Selective inhibition of CDK7 reveals high-confidence targets and new models for TFIIH function in transcription. Genes Dev 34: 1452–1473. 10.1101/gad.341545.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez AM, Shuman S, Schwer B. 2018. RNA polymerase II CTD interactome with 3′ processing and termination factors in fission yeast and its impact on phosphate homeostasis. Proc Natl Acad Sci 115: E10652–E10661. 10.1073/pnas.1810711115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez AM, Garg A, Shuman S, Schwer B. 2019. Inositol pyrophosphates impact phosphate homeostasis via modulation of RNA 3′ processing and transcription termination. Nucleic Acids Res 47: 8452–8469. 10.1093/nar/gkz567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangodkar J, Farrington CC, McClinch K, Galsky MD, Kastrinsky DB, Narla G. 2016. All roads lead to PP2A: exploiting the therapeutic potential of this phosphatase. FEBS J 283: 1004–1024. 10.1111/febs.13573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangodkar J, Perl A, Tohme R, Kiselar J, Kastrinsky DB, Zaware N, Izadmehr S, Mazhar S, Wiredja DD, O'Connor CM, et al. 2017. Activation of tumor suppressor protein PP2A inhibits KRAS-driven tumor growth. J Clin Invest 127: 2081–2090. 10.1172/JCI89548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansó M, Fisher RP. 2013. Pause, play, repeat: CDKs push RNAP II's buttons. Transcription 4: 146–152. 10.4161/trns.25146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansó M, Levin RS, Lipp JJ, Wang VY, Greifenberg AK, Quezada EM, Ali A, Ghosh A, Larochelle S, Rana TM, et al. 2016. P-TEFb regulation of transcription termination factor Xrn2 revealed by a chemical genetic screen for Cdk9 substrates. Genes Dev 30: 117–131. 10.1101/gad.269589.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansó M, Parua PK, Pinto D, Svensson JP, Page V, Bitton DA, MacKinnon S, Garcia P, Hidalgo E, Bahler J, et al. 2020. Cdk9 and H2Bub1 signal to Clr6-CII/Rpd3S to suppress aberrant antisense transcription. Nucleic Acids Res 48: 7154–7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schier AC, Taatjes DJ. 2020. Structure and mechanism of the RNA polymerase II transcription machinery. Genes Dev 34: 465–488. 10.1101/gad.335679.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreieck A, Easter AD, Etzold S, Wiederhold K, Lidschreiber M, Cramer P, Passmore LA. 2014. RNA polymerase II termination involves C-terminal-domain tyrosine dephosphorylation by CPF subunit Glc7. Nat Struct Mol Biol 21: 175–179. 10.1038/nsmb.2753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder SC, Schwer B, Shuman S, Bentley D. 2000. Dynamic association of capping enzymes with transcribing RNA polymerase II. Genes Dev 14: 2435–2440. 10.1101/gad.836300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweighofer A, Meskiene I. 2015. Phosphatases in plants. Methods Mol Biol 1306: 25–46. 10.1007/978-1-4939-2648-0_2 [DOI] [PubMed] [Google Scholar]
- Schwer B, Shuman S. 2011. Deciphering the RNA polymerase II CTD code in fission yeast. Mol Cell 43: 311–318. 10.1016/j.molcel.2011.05.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Ghosh A, Sanchez AM, Lima CD, Shuman S. 2015. Genetic and structural analysis of the essential fission yeast RNA polymerase II CTD phosphatase Fcp1. RNA 21: 1135–1146. 10.1261/rna.050286.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen I, Zhou X, Chernobrovkin A, Puerta-Cavanzo N, Kanno T, Salignon J, Stoehr A, Lin XX, Baskaner B, Brandenburg S, et al. 2020. DAF-16/FOXO requires protein phosphatase 4 to initiate transcription of stress resistance and longevity promoting genes. Nat Commun 11: 138. 10.1038/s41467-019-13931-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty A, Kallgren SP, Demel C, Maier KC, Spatt D, Alver BH, Cramer P, Park PJ, Winston F. 2017. Spt5 plays vital roles in the control of sense and antisense transcription elongation. Mol Cell 66: 77–88.e5. 10.1016/j.molcel.2017.02.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y. 2009. Serine/threonine phosphatases: mechanism through structure. Cell 139: 468–484. 10.1016/j.cell.2009.10.006 [DOI] [PubMed] [Google Scholar]
- Shuman S. 2020. Transcriptional interference at tandem lncRNA and protein-coding genes: an emerging theme in regulation of cellular nutrient homeostasis. Nucleic Acids Res 48: 8243–8254. 10.1093/nar/gkaa630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims RJ 3rd, Rojas LA, Beck DB, Bonasio R, Schuller R, Drury WJ 3rd, Eick D, Reinberg D. 2011. The C-terminal domain of RNA polymerase II is modified by site-specific methylation. Science 332: 99–103. 10.1126/science.1202663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoly I, Shemesh N, Ziv-Ukelson M, Ben-Zvi A, Yeger-Lotem E. 2017. An asymmetrically balanced organization of kinases versus phosphatases across eukaryotes determines their distinct impacts. PLoS Comput Biol 13: e1005221. 10.1371/journal.pcbi.1005221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- St. Amour CV, Sansó M, Bösken CA, Lee KM, Larochelle S, Zhang C, Shokat KM, Geyer M, Fisher RP. 2012. Separate domains of fission yeast Cdk9 (P-TEFb) are required for capping enzyme recruitment and primed (Ser7-phosphorylated) Rpb1 carboxyl-terminal domain substrate recognition. Mol Cell Biol 32: 2372–2383. 10.1128/MCB.06657-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbach N, Hasson D, Mathur D, Stratikopoulos EE, Sachidanandam R, Bernstein E, Parsons RE. 2019. PTEN interacts with the transcription machinery on chromatin and regulates RNA polymerase II-mediated transcription. Nucleic Acids Res 47: 5573–5586. 10.1093/nar/gkz272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun ZW, Hampsey M. 1996. Synthetic enhancement of a TFIIB defect by a mutation in SSU72, an essential yeast gene encoding a novel protein that affects transcription start site selection in vivo. Mol Cell Biol 16: 1557–1566. 10.1128/MCB.16.4.1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaffer MP, Jones AW, Flynn HR, Snijders AP, Nurse P. 2016. CDK substrate phosphorylation and ordering the cell cycle. Cell 167: 1750–1761 e1716. 10.1016/j.cell.2016.11.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan-Wong SM, Zaugg JB, Camblong J, Xu Z, Zhang DW, Mischo HE, Ansari AZ, Luscombe NM, Steinmetz LM, Proudfoot NJ. 2012. Gene loops enhance transcriptional directionality. Science 338: 671–675. 10.1126/science.1224350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatomer DC, Elrod ND, Liang D, Xiao MS, Jiang JZ, Jonathan M, Huang KL, Wagner EJ, Cherry S, Wilusz JE. 2019. The integrator complex cleaves nascent mRNAs to attenuate transcription. Genes Dev 33: 1525–1538. 10.1101/gad.330167.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tietjen JR, Zhang DW, Rodríguez-Molina JB, White BE, Akhtar MS, Heidemann M, Li X, Chapman RD, Shokat K, Keles S, et al. 2010. Chemical-genomic dissection of the CTD code. Nat Struct Mol Biol 17: 1154–1161. 10.1038/nsmb.1900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagnarelli P, Alessi DR. 2018. PP1 phosphatase complexes: undruggable no longer. Cell 174: 1049–1051. 10.1016/j.cell.2018.08.007 [DOI] [PubMed] [Google Scholar]
- Vanoosthuyse V, Legros P, van der Sar SJ, Yvert G, Toda K, Le Bihan T, Watanabe Y, Hardwick K, Bernard P. 2014. CPF-associated phosphatase activity opposes condensin-mediated chromosome condensation. PLoS Genet 10: e1004415. 10.1371/journal.pgen.1004415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbinnen I, Ferreira M, Bollen M. 2017. Biogenesis and activity regulation of protein phosphatase 1. Biochem Soc Trans 45: 89–99. 10.1042/BST20160154 [DOI] [PubMed] [Google Scholar]
- Vervoort SJ, Welsh SA, Devlin JR, Barbieri E, Knight DA, Offley S, Bjelosevic S, Costacurta M, Todorovski I, Kearney CJ, et al. 2021. The PP2A-Integrator-CDK9 axis fine-tunes transcription and can be targeted therapeutically in cancer. Cell (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victorino JF, Fox MJ, Smith-Kinnaman WR, Peck Justice SA, Burriss KH, Boyd AK, Zimmerly MA, Chan RR, Hunter GO, Liu Y, et al. 2020. RNA polymerase II CTD phosphatase Rtr1 fine-tunes transcription termination. PLoS Genet 16: e1008317. 10.1371/journal.pgen.1008317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viladevall L, St Amour CV, Rosebrock A, Schneider S, Zhang C, Allen JJ, Shokat KM, Schwer B, Leatherwood JK, Fisher RP. 2009. TFIIH and P-TEFb coordinate transcription with capping enzyme recruitment at specific genes in fission yeast. Mol Cell 33: 738–751. 10.1016/j.molcel.2009.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virshup DM, Shenolikar S. 2009. From promiscuity to precision: protein phosphatases get a makeover. Mol Cell 33: 537–545. 10.1016/j.molcel.2009.02.015 [DOI] [PubMed] [Google Scholar]
- Vos SM, Farnung L, Boehning M, Wigge C, Linden A, Urlaub H, Cramer P. 2018a. Structure of activated transcription complex Pol II-DSIF-PAF-SPT6. Nature 560: 607–612. 10.1038/s41586-018-0440-4 [DOI] [PubMed] [Google Scholar]
- Vos SM, Farnung L, Urlaub H, Cramer P. 2018b. Structure of paused transcription complex Pol II-DSIF-NELF. Nature 560: 601–606. 10.1038/s41586-018-0442-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss M, Campbell K, Saranzewa N, Campbell DG, Hastie CJ, Peggie MW, Martin-Granados C, Prescott AR, Cohen PT. 2013. Protein phosphatase 4 is phosphorylated and inactivated by Cdk in response to spindle toxins and interacts with γ-tubulin. Cell Cycle 12: 2876–2887. 10.4161/cc.25919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang T, Kwiatkowski N, Abraham BJ, Lee TI, Xie S, Yuzugullu H, Von T, Li H, Lin Z, et al. 2015. CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell 163: 174–186. 10.1016/j.cell.2015.08.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermarck J, Neel BG. 2020. Piecing together a broken tumor suppressor phosphatase for cancer therapy. Cell 181: 514–517. 10.1016/j.cell.2020.04.005 [DOI] [PubMed] [Google Scholar]
- Wu P, Nielsen TE, Clausen MH. 2015. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol Sci 36: 422–439. 10.1016/j.tips.2015.04.005 [DOI] [PubMed] [Google Scholar]
- Wu D, De Wever V, Derua R, Winkler C, Beullens M, Van Eynde A, Bollen M. 2018. A substrate-trapping strategy for protein phosphatase PP1 holoenzymes using hypoactive subunit fusions. J Biol Chem 293: 15152–15162. 10.1074/jbc.RA118.004132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang K, Manley JL, Tong L. 2012. The yeast regulator of transcription protein Rtr1 lacks an active site and phosphatase activity. Nat Commun 3: 946. 10.1038/ncomms1947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Ouyang K, Huang J, Zhou Y, Ouyang H, Li H, Wang G, Wu Q, Wei C, Bi Y, et al. 2013. Direct conversion of fibroblasts to neurons by reprogramming PTB-regulated microRNA circuits. Cell 152: 82–96. 10.1016/j.cell.2012.11.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yague-Sanz C, Vanrobaeys Y, Fernandez R, Duval M, Larochelle M, Beaudoin J, Berro J, Labbé S, Jacques PE, Bachand F. 2020. Nutrient-dependent control of RNA polymerase II elongation rate regulates specific gene expression programs by alternative polyadenylation. Genes Dev 34: 883–897. 10.1101/gad.337212.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Yamaguchi Y, Inukai N, Okamoto S, Mura T, Handa H. 2006. P-TEFb-mediated phosphorylation of hSpt5 C-terminal repeats is critical for processive transcription elongation. Mol Cell 21: 227–237. 10.1016/j.molcel.2005.11.024 [DOI] [PubMed] [Google Scholar]
- Yeo M, Lin PS, Dahmus ME, Gill GN. 2003. A novel RNA polymerase II C-terminal domain phosphatase that preferentially dephosphorylates serine 5. J Biol Chem 278: 26078–26085. 10.1074/jbc.M301791200 [DOI] [PubMed] [Google Scholar]
- Yeo M, Lee SK, Lee B, Ruiz EC, Pfaff SL, Gill GN. 2005. Small CTD phosphatases function in silencing neuronal gene expression. Science 307: 596–600. 10.1126/science.1100801 [DOI] [PubMed] [Google Scholar]
- Zhang DW, Mosley AL, Ramisetty SR, Rodríguez-Molina JB, Washburn MP, Ansari AZ. 2012. Ssu72 phosphatase-dependent erasure of phospho-Ser7 marks on the RNA polymerase II C-terminal domain is essential for viability and transcription termination. J Biol Chem 287: 8541–8551. 10.1074/jbc.M111.335687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Qi Y, Hu S, Cao X, Xu C, Yin Z, Chen X, Li Y, Liu W, Li J, et al. 2020. Identification of integrator-PP2A complex (INTAC), an RNA polymerase II phosphatase. Science 370: eabb5872. 10.1126/science.abb5872 [DOI] [PubMed] [Google Scholar]