

Abstract
We interrogated at nucleotide resolution the spatiotemporal order of chromatin changes that occur immediately following a site-specific double-strand break (DSB) upstream of the PHO5 locus and its subsequent repair by nonhomologous end joining (NHEJ). We observed the immediate eviction of a nucleosome flanking the break and the repositioning of adjacent nucleosomes away from the break. These early chromatin events were independent of the end-processing Mre11-Rad50-Xrs2 (MRX) complex and preceded the MRX-dependent broad eviction of histones and DNA end-resectioning that extends up to ∼8 kb away from the break. We also examined the temporal dynamics of NHEJ-mediated repair in a G1-arrested population. Concomitant with DSB repair by NHEJ, we observed the redeposition and precise repositioning of nucleosomes at their originally occupied positions. This re-establishment of the prelesion chromatin landscape suggests that a DNA replication-independent mechanism exists to preserve epigenome organization following DSB repair.
Failure to repair DNA double-strand breaks (DSBs) results in cell cycle arrest and ultimately programmed cell death, whereas improper repair can lead to profound alterations or loss of genomic information through translocations, inversions, deletions, and other genomic aberrations (Hanahan and Weinberg 2000; Halazonetis et al. 2008). Accurate and timely recognition and repair of DSBs are critical to maintaining genomic integrity and must be accomplished within the context of chromatin (Hauer and Gasser 2017). The canonical access-repair-restore model (Soria et al. 2012; Polo and Almouzni 2015) for DNA repair in eukaryotic genomes recognizes that chromatin presents a significant obstacle for DNA double-strand break recognition and repair processes. Chromatin must thus undergo obligatory remodeling to evict existing nucleosomes and allow break-recognition factors to access and signal for repair complexes to assemble and mend the broken DNA. Although repair of the broken DNA ensures that genome integrity is preserved, chromatin architecture must also be restored to preserve the integrity of the epigenome.
Much of our understanding of DNA DSBs in the context of chromatin structure has been informed by experiments at the yeast MAT locus (Haber 2012). This locus is specifically and efficiently cut by the HO endonuclease in vivo and has served as a model system for many genetic and molecular investigations of DNA damage and repair (Sugawara and Haber 2006; Haber 2012). Immediately following a DSB, the MRX complex recognizes the broken ends of DNA and recruits the downstream kinase Tel1 which, in conjunction with Mec1, phosphorylates histone H2A (analogous to H2AX in mammalian cells) at up to 50 kb on either side of the break (Shroff et al. 2004; Kim et al. 2007; Lee et al. 2014). Moreover, nucleosomes surrounding the break site are evicted in an Mre11-Rad50-Xrs2 (MRX)-dependent manner up to 8 kb surrounding the break, and this process serves to facilitate the access and activity of subsequent repair factors (Tsukuda et al. 2005; Shim et al. 2007). This nucleosome eviction that occurs surrounding a DSB appears to be a conserved phenomenon, as similar results are also observed at specific breaks induced in the genomes of mammalian systems (Tsukuda et al. 2005; Berkovich et al. 2007; Goldstein et al. 2013). The MRX and Yku70/Yku80 (KU) complexes each associate with the broken ends of chromatin both prior to and during the nucleosome remodeling/eviction steps, and this association is critical to their roles in the repair of the break via nonhomologous end joining (NHEJ) (Boulton and Jackson 1996; Moore and Haber 1996). Although it is clear that the KU complex retards 5′-3′ end-resectioning and that MRX is critical in the recognition and processing of the end of the DSB (Lee et al. 1998; Cannavo and Cejka 2014; Symington 2014), these roles do not describe the function of MRX or KU complexes relative to nucleosome dynamics and histone octamer eviction immediately following a break.
The two primary pathways to repair DSBs are homologous recombination (HR) and nonhomologous end joining. HR involves the generation of single-stranded DNA (ssDNA) around the DSB via end-resection machinery and subsequently requires the presence of a homologous sequence of DNA (most commonly a sister chromatid) to serve as a template that can be used to resynthesize DNA sequences. However, except in special circumstances such as MAT switching, where the donor is heterochromatic with highly positioned nucleosomes that are not found at the recipient locus (Weiss and Simpson 1998; Ravindra et al. 1999), it is difficult to distinguish the chromatin structures of homologous regions undergoing recombination (Hicks et al. 2011; Tsabar et al. 2016). NHEJ, however, is a simpler process and requires no template for repair of the DNA strand. Instead, the Yku70-Yku80 heterodimer along with MRX recognizes both ends of a DSB and, in conjunction with Lif1 and Nej1, the enzyme Dnl4 catalyzes their religation (Boulton and Jackson 1996; Teo and Jackson 1997; Wilson et al. 1997; Lieber 2010). Though the process of reconstituting the DNA backbone is straightforward at the molecular level, it remains unclear how the local chromatin environment is perturbed and restored following a break and how similar this chromatin architecture is to the prelesion state.
In both yeast and higher eukaryotic systems, it has been demonstrated that specific chromatin assembly factors and chromatin remodelers are associated with and required for proper repair and restoration of the chromatin state following a DSB (Chai et al. 2005; Tsukuda et al. 2005; Liang et al. 2007; Shim et al. 2007; Kim and Haber 2009; Neumann et al. 2012; Horigome et al. 2014; Kwon et al. 2015; Polo 2015). However, chromatin remodelers may exhibit pleiotropic activities such as regulation of cell-cycle dependent gene expression and may not directly influence the chromatin environment surrounding a DSB. Thus, despite a critical role for ATP-dependent chromatin remodeling complexes in DSB repair, the precise structural outcomes of nucleosome eviction and subsequent reassembly and positioning of these nucleosomes to their final prelesion state has yet to be elucidated. Further, it has been challenging to discriminate between replication-independent chromatin re-establishment and the replication-coupled chromatin reassembly that occurs following S-phase (Tsabar et al. 2016). Although DNA replication-dependent mechanisms of chromatin reassembly following break repair are expected to restore the original parental chromatin organization (Li and Tyler 2016), the fidelity and accuracy of replication-independent mechanisms for histone octamer deposition have not been fully explored. Specifically, it is unknown if an independent mechanism is competent to restore the complex regulatory landscape, including transcription factor binding and nucleosome positioning, to its predamage state. We sought to interrogate, at nucleotide resolution, the spatiotemporal kinetics of chromatin remodeling and eviction in response to a single DSB and the subsequent restoration of chromatin organization following replication-independent NHEJ-mediated repair.
Results
Site-specific inducible DSB at the PHO5 locus
The eviction of histone octamers from the broken chromatin and subsequent remodeling of nucleosomes are required steps in the recognition and processing of double-strand breaks (Polo and Almouzni 2015; Hauer and Gasser 2017). To understand the relationship between the spatial and temporal kinetics of the chromatin remodeling events that accompany a DSB, we inserted a 117-bp HO endonuclease recognition sequence upstream of the PHO5 locus (Fig. 1A), which is marked by a well-positioned +1 nucleosome (nucleosome 4L in Fig. 1A) and has been frequently used as a model for well-defined chromatin organization (Almer and Hörz 1986; Almer et al. 1986; Schmid et al. 1992; Gaudreau et al. 1997; Hertel et al. 2005; Tsabar et al. 2016). Insertion of the HO recognition site did not impact the positioning of the +1 nucleosome at PHO5 but did have a modest effect on local chromatin structure flanking the insertion site (Supplemental Fig. 1). Specifically, we noticed that the nucleosomes flanking the inserted HO recognition site were more organized than in the native locus. Using a galactose-inducible HO endonuclease, we found that the PHO5 117-bp HO sequence cuts just as efficiently as the endogenous MAT locus (Lee et al. 1998; Tsukuda et al. 2005) with 70% of the target locus being cut within 15 min and >90% within 1 h of galactose induction as measured by Southern blot quantification (Fig. 1B; Supplemental Fig. 2). We also observe similar cutting kinetics in strains lacking MRE11 or YKU70. If HO expression is turned off, NHEJ can readily repair DSB ends; however, when HO is continually expressed, nearly all DSBs persist (Moore and Haber 1996; Aylon et al. 2004).
Figure 1.

Inducible DSB at PHO5. (A) Schematic illustrating the ectopic 117-bp HO cut site (HOcs) that was engineered 579 bp upstream of the endogenous PHO5 locus on Chr II (marked in red). The +1 nucleosome of PHO5 is annotated 4L in this illustration. A predicted Sum1 protein binding site is illustrated with a small yellow square. Expression of the HO endonuclease is regulated by an inducible promoter. Nucleosomes are numbered and annotated based on their distance and orientation (left/right) relative to the HO cut site. (B) Southern blot analysis of HO endonuclease-induced cutting near PHO5 with fraction of cut DNA (y-axis) plotted as a function of time (x-axis) for the WT, yku70Δ, and mre11Δ strains. Dotted red line depicts cleavage of 90% of the DNA. (C) Chromatin accessibility surrounding the break greatly increases for WT, yku70Δ, but is limited in mre11Δ strains. Greater time postinduction (min) is plotted in darker shades of blue indicated in the legend (right). (D) Direct comparison between the WT and two mutant strains for the terminal (120 min) MNase sensitivity (in C) which has been smoothed.
In response to a persistent DSB, histone octamers are broadly evicted from the DNA at a rate of ∼4 kb/h, symmetrically from the DSB (Tsukuda et al. 2005). The DSB is then subject to NHEJ-mediated repair or 5′ to 3′ end-resectioning for subsequent homology-driven repair (Frank-Vaillant and Marcand 2002; Tran et al. 2004; Tsukuda et al. 2005; Mimitou and Symington 2008; Zhu et al. 2008; Symington and Gautier 2011; Jasin and Rothstein 2013). We first profiled the temporal changes in chromatin organization surrounding the DSB using sensitivity to micrococcal nuclease (MNase) as a proxy for chromatin accessibility. We found a time-dependent increase in chromatin accessibility (loss of sequence fragment coverage) following induction of the break in wild-type cells (Fig. 1C, top panel). A similar increase in chromatin accessibility surrounding the break was observed in the absence of YKU70 (Fig. 1C, middle panel); however, we observed only a minimal degree of increased accessibility (less than twofold relative to pre-induction) at the break site in mre11Δ cells (Fig. 1C, bottom panel) as has been previously reported at lower resolution (Tsukuda et al. 2005). By 120 min, chromatin accessibility surrounding the break had increased nearly fourfold relative to pre-induction and spread ∼16 kb in both wild-type and yku70Δ cells (with ∼8 kb on either side of the break) (Fig. 1D). As histone eviction and DNA end-resectioning are closely coupled events, we also profiled total DNA coverage to estimate the extent of end-resectioning in each sample (Supplemental Fig. 3). Similar to the MNase chromatin accessibility data, we observed a decrease in genomic DNA coverage for wild-type cells (∼1.9-fold), which was further attenuated in the mre11Δ cells (∼1.5-fold). DNA resectioning was more evident in yku70Δ cells (∼3.7-fold) at the terminal time point; however, resectioning lagged chromatin accessibility at all time points. These results establish our PHO5 GAL::HO- inducible system as a robust model to interrogate chromatin surrounding DSBs.
MNase-seq epigenome mapping resolves chromatin dynamics at base-pair resolution
We sought to precisely define the spatiotemporal dynamics of octamer eviction and chromatin remodeling at nucleotide resolution following induction of a DSB at the PHO5 locus. In order to assess changes in chromatin occupancy, we generated genome-wide chromatin occupancy profiles (GCOPs) (Henikoff et al. 2011; Belsky et al. 2015; Gutiérrez et al. 2019) by coupling paired-end sequencing with limited MNase digestion to reveal the factor-agnostic location of both nucleosomes and smaller DNA-binding factors. DNA protected by the histone octamer will yield fragments of ∼150 bp, whereas smaller DNA-binding proteins such as transcription factors will protect fragments between 20 and 80 bp. GCOPs are visualized by plotting the fragment length as a function of chromosomal coordinate for the midpoint of each recovered fragment (Fig. 2A); thus, well-positioned and phased nucleosomes appear as tight and evenly arrayed clouds of data points with a fragment size of ∼150 bp. Similarly, DNA protected by smaller sequence-specific DNA-binding factors is represented by a focus of smaller sized fragments. We chose an unrelated locus on Chromosome IV containing two genes, CTH1 and GIR2, with well-defined chromatin organization, to serve as an internal control representing static chromatin for these studies. This locus contains nine well-positioned nucleosomes and a clearly defined Aft2 footprint upstream of the CTH1 gene body (Fig. 2B) that do not change over the time course. In contrast, the GAL7 locus exhibits a change in chromatin organization in response to the addition of galactose (Fig. 2C). These changes include the rapid eviction and dephasing of the nucleosomes within the gene body due to high levels of transcription (Yarger and Hopper 1979; Lohr et al. 1995; Platt and Reece 1998). To further quantify our results, we employed a two-dimensional cross correlation analysis with an idealized nucleosome (Supplemental Fig. 4) to define the position, occupancy, and fuzziness (correlation score with an idealized nucleosome; lower scores being more fuzzy) for each nucleosome. The idealized nucleosome was derived from MNase fragment length data centered on the nucleosome dyads of ∼8000 nucleosomes mapped by an orthogonal chemical cleavage-based approach (Brogaard et al. 2012). These metrics are summarized in pictograph form for both the CTH1/GIR2 and GAL7 loci (Fig. 2D,E). Specifically, the location, size, and color opacity of the circles represent position, occupancy, and fuzziness. The occupancy, positioning, and fuzziness of the nucleosomes at CTH1/GIR2 remain stable and unchanged throughout the time course (Fig. 2D); in contrast, we observe profound chromatin architectural changes at GAL7 following induction (Fig. 2E). These findings are consistent across all strains profiled in this study (Supplemental Figs. 5, 6) and demonstrate the specificity and sensitivity of GCOPs for describing the spatiotemporal dynamics of chromatin occupancy.
Figure 2.

Genome-wide chromatin occupancy profiles (GCOPs) to detect chromatin changes at base-pair resolution. (A) Schematic of the MNase-seq based assay to generate GCOPs. MNase digests unprotected DNA, the subsequent nucleosome and TF protected fragments of DNA are subjected to paired-end sequencing, and the fragment size is plotted as a function of the position of its midpoint along the chromosome. (B) GCOP for the CTH1 and GIR2 loci which serve as a control for unaltered chromatin. Gray boxes depict gene bodies, with arrows indicating the direction of transcription. The Aft2p binding site is annotated between these two genes. A two-dimensional (2D) cross correlation with an idealized nucleosome (see Methods) is calculated at every base pair and depicted as a continuous black trace. The peaks of this cross-correlation analysis represent the most likely position of a locally detected nucleosome, and the shaded regions represent 0.5 standard deviations of nucleosome positions from the center of each detected peak. The intensity of the shaded color is proportional to the nucleosome's peak cross-correlation score. (C) GCOP for the GAL7 locus following galactose induction. (D) and (E) serve as pictographic summaries of the chromatin changes observed, where each blue circle represents a corresponding nucleosome from the plots in B and C. The size of a given circle is proportional to its occupancy; its x position is the location of the peak of the 2D cross-correlation score, and the intensity of color is proportional to the peak cross-correlation value (fuzziness).
High-resolution analysis of DSB-induced chromatin changes at PHO5
To interrogate chromatin structure in response to a DSB at the PHO5 locus, GCOPs were generated for wild-type, yku70Δ, and mre11Δ mutant strains (Fig. 3). Prior to DSB induction (Fig. 3, pre-induction, top panels), all strains exhibit a similar chromatin organization, with five nucleosomes positioned both left and right of the HO cut site (HOcs) (illustrated in Fig. 1A). In wild-type cells, we observe the rapid loss of the 1L nucleosome upon galactose induction. The 1R and 2R nucleosomes shift away from the break and toward each other and are positioned so closely together after 30 min that they are difficult to discern as individual nucleosomes. The crowding of the 1R and 2R nucleosomes appears to be limited by a small DNA-binding factor site (presumably Sum1) because the three nucleosomes downstream from this footprint do not alter their positioning following break induction in wild-type or mutant strains. We also observe a time-dependent loss of reads within the HO cut site as well as an increase in smaller length fragments on either side of the cut site. We interpret the increase in protected small fragments to represent the occupancy of a DNA-binding factor smaller than a nucleosome. This small occupancy footprint is not symmetric however; it is larger on the left side of the break and resides at the sequence previously occupied by the evicted 1L nucleosome (Fig. 3A, blue arrowhead; Supplemental Fig. 7). The presence of this footprint is also seen in both yku70Δ and mre11Δ mutants, suggesting that it is not dependent on these DSB end binding factors (Fig. 3B,C; Supplemental Fig. 7). Additionally, we observe a gain in nucleosomal-sized fragments in the linker region between the 1L and 2L nucleosomes (denoted by the red arrowhead), suggesting the sliding of histone octamers away from the break (Fig. 3). Immediately downstream from the Sum1 binding site, we observe a relatively static nucleosome (3R) (gold arrowhead) whose positioning is not impacted by the break.
Figure 3.

Local chromatin changes in response to a DSB at the PHO5 locus. GCOPs of WT (A), yku70Δ (B), and mre11Δ (C) strains over the experimental time course. Gene bodies are annotated above the plots in gray, with arrows indicating the direction of transcription. Dotted blue lines denote the 117-bp HO recognition site, and a predicted Sum1 protein binding site is also depicted. Similar to Figure 2, B and C, the black trace represents the 2D cross-correlation score and the blue shaded regions represent 0.5 standard deviations of nucleosome positions from the center of each identified nucleosome. The regions quantified in Figure 4 are marked by colored arrowheads for the 1L (blue) and 3R (gold) nucleosomes and the 1L-2L linker (red) region.
To summarize our observations in Figure 3, we generated pictograms for nucleosome positioning, occupancy, and fuzziness over the experimental time course (Fig. 4A). The pictogram captures the rapid eviction of the 1L nucleosome, a leftward shift for nucleosomes 2L, 3L, and 4L, as well as the rightward shift and crowding of nucleosomes 1R and 2R (Fig. 4A). Over the course of 120 min following induction, we observe a time-dependent decrease in the overall occupancy which may also, in part, be due to end-resectioning. Despite this reduction in nucleosome occupancy, nucleosome positioning does not change appreciably when compared to early postinduction time points (<30 min), indicating that the nucleosomes which remain following induction of the break retain their early postbreak positioning (Fig. 4A).
Figure 4.

MRE11-independent and -dependent nucleosome dynamics at a DSB. (A) Pictographs depicting the chromatin changes near PHO5 following DSB induction. Increasing time is plotted downward on the y-axis and the boundaries of the 117-bp HO recognition site are denoted by dotted blue lines. (B) The FPKM corrected relative occupancy of the 1L nucleosome (Fig. 3, blue arrowhead) for the WT, yku70Δ, and mre11Δ mutant strains (*) P < 0.05, (**) P < 0.01, Student's t-test. n = 2. (C) Occupancy of the 1L-2L linker region was quantified as in B (Fig. 3; red arrowhead). (D) The same relative occupancy analysis performed in B is plotted for the 3R nucleosome (Fig. 3, gold arrowhead). This nucleosome is positioned immediately to the right of a predicted Sum1 protein binding site (Fig. 1A).
Using 2D density kernel cross-correlation analysis, we are unable to algorithmically detect the 1L nucleosome later than 15 min following break induction in wild-type and mutant strains (Fig. 3). However, this cross-correlation metric assumes a homogeneous single-state system (i.e., an idealized nucleosome precisely localized in the bulk of the population), and it fails to detect more complex state mixtures (i.e., partial nucleosomes, poorly positioned nucleosomes, occupancy by either a nucleosome or a transcription factor, etc.). Thus, significant changes in nucleosome occupancy between wild-type and mutant strains may become obscured by low 2D cross-correlation values. Given the apparent competition between the 1L nucleosome being evicted, the accumulation of smaller protected fragments over the same sequence, as well as the gain in nucleosomal-sized fragments in the linker region between the 1L and 2L nucleosomes, we sought to employ a more sensitive and direct method to analyze the data. We directly inferred the kinetics of 1L nucleosome eviction illustrated in Figure 3 by measuring fragment loss at the pre-induction 1L nucleosome position (Fig. 4B) and similarly assessed the occupancy of the 1L-2L nucleosomal linker region by measuring fragment gain at this position (Fig. 4C). As a local control, we also analyzed the 3R nucleosomal fragments (Fig. 4D). These metrics demonstrated that the nucleosomes immediately around the break were dynamic, whereas the 3R nucleosome behaved consistently across wild-type and mutant strains. Although the loss of the 1L nucleosome is similar in all strains up to 30 min, at later time points (≥60 min), we detected delayed 1L nucleosome loss in the mre11Δ strain relative to the loss observed in the wild-type and yku70Δ strains (Fig. 4B). The kinetics of nucleosome appearance in the 1L-2L linker region—either by histone deposition or nucleosome sliding—were consistent between wild type and the mutant strains (Fig. 4C). The relative fragment loss observed at the 1L nucleosome position parallels the relative gain in nucleosome fragments in the 1L-2L linker region in all strains, suggesting that this 1L nucleosome is initially shifted left of the break. Together, these results indicate that the 1L nucleosome and the 1L-2L linker region experience early occupancy changes independent of the MRX and Yku70-Yku80 binding proteins and that these alterations precede the eventual nucleosome loss and broad changes in chromatin accessibility we observe at later time points.
Genome-wide changes in response to a single DSB
Despite the early perturbations in nucleosome positioning that were localized to the immediate vicinity of the break, the broad increase in chromatin accessibility surrounding the break at terminal time points led us to question if any other distant chromatin changes had occurred. Given that our data provide nucleosome occupancy and position throughout the entire genome at base-pair resolution, we sought to interrogate the postinduction nucleosome occupancy changes genome-wide in response to a single, persistent DSB. We determined the relative ratio between the 2D cross-correlation score (similarity to an idealized nucleosome) for genic nucleosomes in the pre-induction state versus the 120-min postinduction state and discovered that the most significant changes were confined to the genes within ∼8 kb of the DSB and the GAL loci on Chromosome II (Supplemental Fig. 8). The subtle changes in nucleosome occupancy that were detected on the remaining 15 unbroken chromosomes were likely associated with perturbations in gene expression elicited by a DSB (Lee et al. 2000). In contrast to the changes in nucleosome occupancy that spread 8 kb from the break, changes in nucleosome positioning were largely constrained to the sequences immediately surrounding the break (Supplemental Fig. 9). Taken together, these findings suggest that the structure of the chromatin landscape (except for the region immediately around the DSB) is largely preserved across the genome in response to a DSB.
DNA replication-independent restoration of chromatin following NHEJ
How is the local eviction and nucleosome displacement restored following repair of a DSB? Specifically, how rapidly is the chromatin structure restored and does it return to the pre-induction state? It remains unclear whether chromatin organization is precisely restored in an independent manner following NEHJ (Chiruvella et al. 2013; Geuting et al. 2013; Emerson and Bertuch 2016; Gao et al. 2016; Li and Tyler 2016). To profile the changes that occur following repair via NHEJ, we arrested cells in the G1 phase of the cell cycle by the addition of α-factor for 3.5 h and then induced HO expression for 1 h with galactose to ensure complete cutting and chromatin disruption at the target locus. Repair and potential chromatin reassembly at PHO5 were allowed to occur by adding 2% dextrose to repress GAL::HO expression (Fig. 5A). In the absence of continued transcription, the HO endonuclease is rapidly degraded, allowing NHEJ to rejoin the 4-nt 3′ overhanging ends (Moore and Haber 1996; Kaplun et al. 2000). In G1-arrested cells, 5′ to 3′ resectioning is hindered in wild-type cells and replication is blocked (Aylon et al. 2004; Ira et al. 2004; Clerici et al. 2008; Zierhut and Diffley 2008). This condition allowed us to specifically interrogate replication-independent chromatin reassembly following NHEJ repair. Following repression of GAL::HO, we observed a progressive increase in the fraction of DNA being repaired, with rejoining of ∼50% occurring within 6 h. We confirmed that this repair was DNL4-dependent and thus represented classical NHEJ (Fig. 5A; Supplemental Fig. 10; Teo and Jackson 1997; Wilson et al. 1997).
Figure 5.

NHEJ-mediated DSB repair and chromatin dynamics at the PHO5 locus. (A) Cutting/rejoining kinetics of the ectopic 117-bp HO recognition site that was engineered upstream of the PHO5 gene. Suppression of HO (starts at the time denoted by the dotted red line) facilitates repair by NHEJ, a Dnl4-dependent repair process, in this donorless system. (B,C). Bulk MNase coverage over the cut/repair time course in the WT strain (B) and the dnl4Δ mutant (C). The red line denotes the HO cut site. Time pre-induction (−1 h after HO induction [0 h]), and after repression of HO expression (1 h, 2 h, etc.) are shown in darker shades of blue. (D,E) Chromatin occupancy profiles of the WT (D) and dnl4Δ (E) strains. As in Figures 2 and 3, the black trace represents the 2D cross-correlation score and the blue shaded regions represent 0.5 standard deviations of nucleosome positions from the center of each identified nucleosome.
We first examined broad chromatin accessibility via sensitivity to MNase digestion throughout the process of NHEJ and noted only a modest twofold increase in chromatin accessibility at the sequences surrounding the break. These changes were consistent with the reconstitution of chromatin on the 50% of remaining DNA that underwent repair (Fig. 5B). In contrast, we observed substantially more chromatin accessibility and loss of DNA surrounding the break in a dnl4Δ strain (Fig. 5C), consistent with the broken DNA ends being slowly degraded in the absence of a competent repair pathway (Aylon et al. 2004). We were able to verify transcriptional repression of GAL::HO by generating GCOPs for the HO gene body, where we observed the restoration and maintenance of pre-induction nucleosome structure within the HO gene body following dextrose addition (Supplemental Fig. 11). Lastly, we confirmed the G1 arrest of the experimental cell population for the duration of the experiment via FACS (Supplemental Fig. 12).
We also examined the spatiotemporal dynamics of chromatin structure in high resolution at the sequences immediately surrounding the DSB in both wild type and the dnl4Δ strain. The chromatin structure was similar in the pre-induction state and following HO-induced cutting for 1 h. Following repression of HO, we began to detect NHEJ repair within 1 h and the reappearance of the 1L nucleosome in the wild-type strain (Fig. 5D); this nucleosome reappearance was not observed in the dnl4Δ strain, suggesting that repair of the DNA was necessary and sufficient for restoring the 1L nucleosome (Fig. 5E). We also observed that the 1R and 2R nucleosomes assume their pre-induction positioning, and the overall chromatin state at 6 h postdextrose recapitulates that of the pre-induction state in the wild-type strain (Fig. 6A, top panel), whereas in the dnl4Δ strain, we observed a profound loss of recovered fragments, suggesting much of the DNA surrounding the broken locus had been degraded. Despite the extensive histone eviction and resectioning that occurred in the dnl4Δ strain by the terminal time point, at earlier time points (up to 2 h postdextrose addition), the chromatin organization and structure around the break was similar to the chromatin structure in the wild-type strain. This result indicates that the break-induced changes are stable until degradation or repair of the broken chromosome and not dependent on the continued expression of HO endonuclease (Figs. 5E, 6A, bottom panel). We again quantified the occupancy kinetics of the 1L and 3R nucleosomes in these G1-arrested cells (Fig. 6B) and observed a close association between the loss of the 1L nucleosome and near complete cutting of the PHO5 locus. The eviction and redeposition of the 1L nucleosome closely parallels the break and subsequent repair of the DNA in the wild-type strain but is never restored in the dnl4Δ strain. In contrast, the 3R nucleosome exhibits a decrease in occupancy reflective of the proportion of DNA that remains unrepaired by NHEJ. Furthermore, the kinetics of reappearance or deposition of the 1L nucleosome along the chromatin parallels the fraction of DNA repaired (Fig. 5A,D). These observations suggest that the processes intrinsic to NHEJ-mediated DSB repair are competent to facilitate the re-establishment and positioning of nucleosomes to their prelesion arrangement along the chromatin in a replication-independent but repair-coupled manner (Fig. 6C).
Figure 6.

NHEJ-mediated DSB repair restores chromatin structure independent of replication. (A) Pictographs depicting local nucleosome changes over the experimental time course in the WT and dnl4Δ strains. Similar to the analysis performed in Figure 4, we precisely quantified the alterations in chromatin structure following break induction and NHEJ-mediated repair. Increasing time is plotted downward on the y-axis and the boundaries of the HO cut site are denoted by dotted blue lines. (B) The log2-relative (to pre-induction) occupancy of the 1L (WT squares/solid line; dnl4Δ triangles/dashed line) and 3R (WT circles/solid line; dnl4Δ diamonds/dashed line) nucleosomes (denoted 1L and 3R, respectively, in Fig. 6C) are plotted over time for this experiment. (C) Model for the replication-independent nucleosome reassembly and chromatin restoration following NHEJ.
Discussion
The structural changes that chromatin undergoes following a double-strand break and its ensuing repair have only been investigated at low-resolution (Shim et al. 2005; Tsukuda et al. 2005; Li and Tyler 2016; Tsabar et al. 2016). To interrogate the kinetics of chromatin dynamics following a DSB and throughout NHEJ-mediated repair, we developed a system that would allow us to survey the genome-wide chromatin occupancy changes at nucleotide resolution with an inducible site-specific break upstream of the PHO5 locus. We identified an early cascade of discrete chromatin changes that occurred immediately following break induction which were independent of MRE11; specifically, the rapid eviction of the 1L nucleosome which appeared to proceed through an intermediate remodeling event of the 1L nucleosome into the adjacent linker region. These immediate changes in chromatin structure at the break were followed by the MRE11-dependent broader eviction of histone octamers. The early and local changes to chromatin were reversible through repair of the genetic lesion by NHEJ and occurred in a replication-independent manner.
Previous investigations of chromatin structure surrounding a DSB have relied on ChIP or PCR/probe-based methods to query the presence of specific sequences/loci (Tsukuda et al. 2005; Shim et al. 2007; Goldstein et al. 2013; Li and Tyler 2016; Tsabar et al. 2016). These methods have established a low-resolution model of broad nucleosome eviction dependent on MRX activity and facilitated by Sgs1, Exo1, Dna2, Fun30, and Ino80 (Dubrana et al. 2007; Mimitou and Symington 2008; Eapen et al. 2012; Westmoreland and Resnick 2016). This combination of chromatin remodeling complexes, helicases, and nucleases suggests that these broad chromatin changes are related to and coupled with DNA end-resectioning (Symington and Gautier 2011). At 120 min postinduction, we observe approximately a twofold reduction in the occupancy of nucleosomes to the left and right of the DSB in wild-type and yku70Δ strains, but this occupancy loss is abrogated in the mre11Δ strain. This degree and extent of MNase sensitivity we observed at our terminal time point (120 min)—which, in wild type strains, extends ∼8 kb from the break—is consistent with prior reports (Tsukuda et al. 2005). During these eviction and resection phenomena, we do not observe any shifts or positioning changes in the remaining nucleosomes despite their loss in occupancy. Similarly, when we examined the remaining 15 unbroken chromosomes, we were also unable to detect any significant changes in nucleosome occupancy or positioning. These data suggest that the structure of the chromatin landscape may be largely preserved up until the moment of eviction and resectioning. Although changes in global transcript levels, elevated rates of histone turnover, higher order changes in nuclear organization, and changes in chromatin fiber condensation following double-strand breaks have been described (Lee et al. 2000; Amitai et al. 2017; Hauer and Gasser 2017; Hauer et al. 2017; Herbert et al. 2017; Seeber and Gasser 2017), our work suggests that the fundamental organization of chromatin (at the level of the individual nucleosome) does not undergo radical remodeling and repositioning immediately following a single induced DSB break. This stability is in contrast to the widespread loss of chromatin occupancy observed following acute treatment with the radiomimetic drug zeocin (Hauer et al. 2017) and likely reflects a difference in the extent of DNA damage. Preservation of pre-existing chromatin structure surrounding the break and throughout the genome may be critical for maintaining regulated gene expression and preventing potential spurious or cryptic transcription.
Prior to these broad and late changes to chromatin structure, rapid (within 15–30 min) local alterations to nucleosome positioning and occupancy were observed concomitant with break induction. Specifically, we documented the repositioning of a single nucleosome to the left of the break, the gain of small-factor footprints immediately bordering both sides of the break, and the local dephasing of nucleosomes around the break. The ATP-dependent chromatin remodeling complexes INO80, SWR1, and RSC have been implicated in facilitating repair/recognition factor access to DSB ends (Morrison et al. 2004; Tsukuda et al. 2005; Liang et al. 2007; Shim et al. 2007; Horigome et al. 2014). However, the immediate and early changes (<60 min) were independent of MRE11 and implicate another biochemical mechanism or chromatin remodeling process in the early phase of a DSB, or, alternately, the chromatin changes at the ends of the break are entropically driven.
In concert with these early local chromatin changes, we were able to detect the appearance of a small-factor footprint on both sides of the DSB within 15 min following induction of the DSB. We initially hypothesized that these small-factor occupancy footprints were the result of canonically associated DSB recognition and repair factors. The MRX complex and YKU70-80 heterodimer are known to rapidly (∼20 min) associate with the ends of broken DNA (Palmbos et al. 2008; Wu et al. 2008). However, the asymmetry and appearance of this footprint was independent of MRE11 or YKU70, suggesting that this footprint is the result of another DNA end binding factor. We investigated the sequences bordering the DSB and found no well-described motifs that would explain the observed footprint or account for differential digestion of the recovered up-/downstream sequence following MNase digestion. The creation of two new free DNA ends at a DSB serves as a substrate for chromatin remodeling complexes such as RSC and INO80—which have previously been described to be associated with DSBs with rapid kinetics (Liang et al. 2007; Shim et al. 2007). Whereas the recruitment of INO80 is thought to be MRX-dependent, the RSC complex may interact with the broken DNA to create space for recognition and repair factors by sliding nucleosomes away from the DSB and also prevent histone octamers from sliding towards the free ends and off the DNA. The displacement of the H2A/H2B dimer immediately around a DSB (Shroff et al. 2004) may also suggest the presence of H3-H4 tetrasomes at a DSB and potentially explain the smaller sized footprint we observe. Finally, we also considered the possibility that the HO endonuclease itself associates with the ends of the 117-bp HO recognition sequence following break induction, and HO itself could be responsible for this footprint; however, the asymmetric small-factor footprint remains long after dextrose repression and degradation of HO in a dnl4Δ strain (Kaplun et al. 2000).
Following recognition and repair of a DSB, the concomitant re-establishment of chromatin structure surrounding the repaired locus is critical for maintaining epigenome integrity. Previous studies with HO-induced DSBs have demonstrated the re-establishment of chromatin structure following recombinatorial repair of the break, a process which necessitates template driven new DNA synthesis and requires specific chromatin remodeling machinery and histone chaperones (Tsabar et al. 2016; Mehta et al. 2017). The process of NHEJ-mediated repair, however, intrinsically lacks a DNA synthesis-coupled chromatin assembly step, and it is unclear if the prelesion chromatin structure and organization is capable of being re-established prior to the next S-phase. Chromatin remodeling complexes (HIRA, RSC) and histone chaperones (CAF-1 and ASF-1) have been implicated in NHEJ-mediated repair and in the re-establishment of H3 histone occupancy at the repaired break (Linger and Tyler 2005; Shim et al. 2005; Kim and Haber 2009; Li and Tyler 2016). These studies suggest an active mechanism to re-establish chromatin occupancy following NHEJ-mediated DSB repair, but the structure of this repaired chromatin specifically at the level of individual nucleosome occupancy and positioning has yet to be elucidated. Furthermore, the implication of a replication-associated histone chaperone, specifically CAF-1, in re-establishing histone occupancy following NHEJ-mediated repair in cycling cells suggests that this process might yet be coupled to DNA synthesis or necessitate a replication-dependent re-assembly of the chromatin. The notion of a replication-dependent reset of chromatin following repair of a DSB is augmented by work which has demonstrated that the dephosphorylation of H2A, a DSB-coupled modification which extends up to 50 kb away from the DSB, is most efficient on free histone dimers and not in the DNA packaged nucleosome (Keogh et al. 2006; Nakada et al. 2008). Taken together, these observations postulate a model that re-establishment or resetting of the chromatin state following repair of a DSB by NHEJ is replication-coupled.
By arresting donorless haploid cells in G1 with ɑ-factor throughout the entire experiment, we were able to prevent replication and eliminate the possibility of repair via sister chromatid recombination. The break-induced eviction, dephasing, and small-factor footprints that we previously described as a consequence of a DSB are all reversible within 1 h of NHEJ-mediated repair and are most clearly evident at 6 h postrepair, when a majority of the broken DNA has been religated. This result suggests that repair-coupled but replication-independent chromatin machinery is competent to reset chromatin structure following a DSB and occurs at a rate limited by repair of the genetic lesion. It remains to be determined whether there will be locus-specific differences in this replication-independent re-establishment of chromatin structure. For example, the PHO5 locus is known for its well-positioned +1 nucleosome and may have sequence features that facilitate and contribute to the chromatin organization. In the broader context of DNA repair, particularly in higher eukaryotes, it is important to consider that NHEJ (and related/derivative nontemplate-mediated end-joining processes) are the predominant and rapid repair pathways used to mend a DSB (Mao et al. 2008). Given that cells experiencing genotoxic stress, and DSBs in particular, may be postmitotic, our work suggests that a concerted and rapid chromatin reassembly mechanism exists coupled to NHEJ which serves to maintain the structure of the epigenome in the absence of DNA replication or synthesis.
Methods
Strains
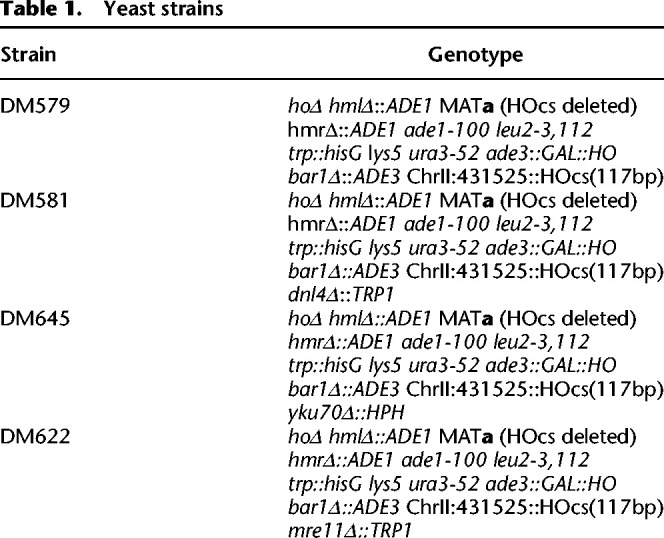
We built our model system in strain JKM139 lacking HML, HMR (Sandell and Zakian 1993; Lee et al. 1998). By selecting survivors on a YEP-GAL plate, we recovered alterations of the MATa cleavage site (designated HOcs deleted) that could no longer be cut by the endonuclease (Lee et al. 1998). Strains used in this study have the genotypes listed in Table 1.
Table 1.
Yeast strains
Single gene deletions were carried out using single-step-PCR-mediated transformation (Sikorski and Hieter 1989). Markerless insertion of the 117-bp MATa HO cut site (Kostriken and Heffron 1984) was accomplished through the use of CRISPR-Cas9 editing, by using the plasmid pGM031, which contains Pho5-gRNA-f (DM1806) and Pho5-gRNA-r (DM1807) cloned into the BplI cut site in bRA90 plasmid (Anand et al. 2017). Primer and gBlock sequences are provided in Supplemental Table 1.
Galactose inductions
Cells were grown to an OD600 of ∼0.4–0.5 in YEP with 2% raffinose and 0.1% dextrose. Galactose was added to a final concentration of 2% to induce HO expression. Cells were harvested at specified time intervals for subsequent Southern blotting and chromatin preparation.
Southern blotting
Southern blots were performed with DNA probes labeled with αP32-ATP (PerkinElmer) as previously described (Southern 2006) using Church & Gilbert hybridization buffer (Church and Gilbert 1984). To assess the fraction of DNA cut in the wild-type, yku70Δ, and mre11Δ strains, we performed band densitometry analysis (with ImageJ) of each lane and computed the fraction of cut DNA relative to the sum of the cut and uncut DNA bands (Supplemental Fig. 11). To interrogate the fraction of DNA rejoined via NHEJ, we computed the fraction of each postinduction uncut band to the pre-induction sample (−1 lane in Supplemental Fig. 11) and then normalized this value to an unrelated control fragment which was probed on Chr IX.
Cell cycle arrest and HO repression
Wild-type cells were grown to an OD600 of ∼0.2–0.3 in YEP with 2% raffinose and 0.1% dextrose, at which point alpha factor was added at a final concentration of 50 ng/mL for 3.5–4 h (GenWay). Galactose was added to a final concentration of 2% to induce HO expression. To repress HO, cells were pelleted, washed twice in YEP + 2% dextrose + 50 ng/mL alpha factor, and resuspended in YEP + 2% dextrose + 50 ng/mL alpha factor. Cells were harvested at specified time intervals for subsequent Southern blotting and chromatin preparation.
Flow cytometry
Flow cytometry was performed as previously described (Gutiérrez et al. 2019).
Chromatin preparation
Chromatin preparation was performed as previously described (Henikoff et al. 2011; Belsky et al. 2015). Briefly, cells were cross-linked by adding formaldehyde to a final concentration of 1% for 30 min at room temperature with stirring. Formaldehyde was quenched by adding glycine to a final concentration of 125 mM for 5 min at room temperature with stirring. Cells were centrifuged at 2000 rpm for 5 min, washed with sterile water, and resuspended with 20 mL of buffer Z (0.56 M sorbitol, 50 mM Tris at pH 7.4, autoclaved). Next, 14 μL of β-ME (United States Biological) and 0.5 mL of a 10 mg/mL solution of zymolyase prepared in buffer Z were added. Samples were incubated for 30 min at 24°C with shaking. Cells were centrifuged at 1500 rpm for 6 min at 4°C and then resuspended in 2.5 mL of NP buffer (1 M sorbitol, 50 mM NaCl, 10 mM Tris at pH 7.4, 5 mM MgCl2, 1 mM CaCl2, autoclaved) supplemented with 0.5 mM spermidine, 0.007% β-ME, and 0.075% NP-40. Aliquots of 15 U/μL MNase (Worthington) were prepared to determine the best digestion conditions: 1.5-mL tubes contained 4 μL, 2μL, 1 μL, 0.5 μL, 0.25 μL, and 0 μL of MNase, to which 400 μL of the resuspended cells were added. Samples were inverted to mix and digested on the benchtop for 20 min. The reaction was halted by adding 100 μL of stop buffer (5% SDS, 50 mM EDTA). Next, Proteinase K was added to a 0.2 mg/mL final concentration, and the samples were inverted and incubated overnight at 65°C. DNA was recovered by phenol extraction and isopropanol precipitation. To ensure that samples were not overdigested, we selected samples from the MNase titration where 4–5 rungs of the nucleosomal ladder are visible.
Sequencing library preparation
Sequencing libraries were prepared as previously described (Belsky et al. 2015) with the following modifications: NEBNext multiplex oligos for Illumina kit (New England Biolabs) were used in adapter ligation, followed by 12 cycles of PCR. PCR reactions were cleaned using Agencourt AMPure XP beads (Beckman).
Data analysis
All figures and plots were generated using R version 3.2.0 (R Core Team 2015).
Alignment
Sequencing reads were aligned with Bowtie (Langmead et al. 2009) in paired-end mode to the sacCer3/R64 version of the Saccharomyces cerevisiae genome and an edited version of Chr II derived from this same genome with the 117-bp HO cut site inserted upstream of PHO5.
Analysis of chromatin structure and occupancy
Sequencing reads from biological replicates were sampled to equivalent read depth and then merged for downstream data analysis, with the exception of the nucleosome occupancy figures (Figs. 4B–D, 6B). Sequencing depth and concordance between replicates is described in Supplemental Table S2. Two independent metrics were used to determine the concordance or correlation between the replicates. The first is the coverage of reads in 1000-bp windows across the genome (mean correlation of coverage for all experiments is R = 0.9633). To ensure we are recovering similar chromatin occupancy patterns in the data (and not just similar levels of coverage), we used information theory to calculate the entropy (well-organized chromatin = low entropy; disorganized chromatin = high entropy) for each 1000-bp window, and then these entropy scores were compared between replicates for concordance (mean correlation of entropy for all experiments is R = 0.9480) (Tran et al. 2020).
Chromatin accessibility was determined for each sample by calculating the total number of reads in a 500-bp window stepping every 10 bases for the 50 kb surrounding the HO recognition site. The log2-ratio of each sample relative to the pre-induction sample was then calculated to interrogate MNase sensitivity of the region. The boundaries of MNase sensitivity were determined by smoothing the MNase coverage curves and determining the widths of the sensitivity for all strains.
Nucleosomes were called based on a sliding 2D cross-correlation score at every base on an “idealized” nucleosome 2D density kernel. This kernel was derived by analyzing 8632 unique nucleosome positions on Chr IV that were mapped utilizing a highly sensitive chemical mapping methodology (Brogaard et al. 2012). This analysis provided us with the approximate size and distribution of reads in our data that corresponded to a canonical well-positioned nucleosome. The variance of the size (y) and position (x) of the fragments is how we derived the idealized nucleosome 2D kernel. We derived idealized nucleosome kernels for the pre-induction sample of each time course to control for slight MNase digestion variability across wild-type and mutant strains. Density plots of these idealized kernels are provided in Supplemental Figure 4 and demonstrate the consistency between data sets. Nucleosomes were defined as peaks of this 2D cross-correlation trace above the 10th percentile of all cross-correlation values on Chr IV. We then shaded each nucleosomal peak ±0.5 standard deviations of the x variance of the idealized nucleosome to highlight the area of interest. The intensity of this shading is proportional to the maximal nucleosome occupancy within a plotting window time course. Nucleosome occupancy is defined as the number of fragments that fall between 0.5 standard deviations of position (x) and 1 standard deviation of size (y). Nucleosome “fuzziness” is separate from the occupancy and positioning metrics and is defined as the value of the peak of the 2D cross-correlation score. This metric captures how similar or dissimilar a called nucleosome peak is to an idealized nucleosome.
Data access
The sequencing data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA597604. All code necessary to reproduce the analysis and figures presented in the manuscript is available at GitLab (https://gitlab.oit.duke.edu/dmm29/tripuraneni_dsb_2021) and as Supplemental Code.
Supplementary Material
Acknowledgments
We thank members of the MacAlpine and Haber laboratories for critical comments and suggestions. We also thank Yulong Li for advice on data analysis. This work was supported by the following grants from the National Institute of General Medical Sciences: R35 GM127062 (D.M.M.), R01 GM61766 (J.E.H.), R35 GM127029 (J.E.H.), R01 GM118551 (A.J.H.).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.271155.120.
Competing interest statement
The authors declare no competing interests.
References
- Almer A, Hörz W. 1986. Nuclease hypersensitive regions with adjacent positioned nucleosomes mark the gene boundaries of the PHO5/PHO3 locus in yeast. EMBO J 5: 2681–2687. 10.1002/j.1460-2075.1986.tb04551.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almer A, Rudolph H, Hinnen A, Hörz W. 1986. Removal of positioned nucleosomes from the yeast PHO5 promoter upon PHO5 induction releases additional upstream activating DNA elements. EMBO J 5: 2689–2696. 10.1002/j.1460-2075.1986.tb04552.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amitai A, Seeber A, Gasser SM, Holcman D. 2017. Visualization of chromatin decompaction and break site extrusion as predicted by statistical polymer modeling of single-locus trajectories. Cell Rep 18: 1200–1214. 10.1016/j.celrep.2017.01.018 [DOI] [PubMed] [Google Scholar]
- Anand R, Memisoglu G, Haber J. 2017. Cas9 -mediated gene editing in Saccharomyces cerevisiae. Protoc Exch 10.1038/protex.2017.021a [DOI] [Google Scholar]
- Aylon Y, Liefshitz B, Kupiec M. 2004. The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. EMBO J 23: 4868–4875. 10.1038/sj.emboj.7600469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belsky JA, MacAlpine HK, Lubelsky Y, Hartemink AJ, MacAlpine DM. 2015. Genome-wide chromatin footprinting reveals changes in replication origin architecture induced by pre-RC assembly. Genes Dev 29: 212–224. 10.1101/gad.247924.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkovich E, Monnat RJ, Kastan MB. 2007. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat Cell Biol 9: 683–690. 10.1038/ncb1599 [DOI] [PubMed] [Google Scholar]
- Boulton SJ, Jackson SP. 1996. Saccharomyces cerevisiae Ku70 potentiates illegitimate DNA double-strand break repair and serves as a barrier to error-prone DNA repair pathways. EMBO J 15: 5093–5103. 10.1002/j.1460-2075.1996.tb00890.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brogaard K, Xi L, Wang J-P, Widom J. 2012. A map of nucleosome positions in yeast at base-pair resolution. Nature 486: 496–501. 10.1038/nature11142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavo E, Cejka P. 2014. Sae2 promotes dsDNA endonuclease activity within Mre11–Rad50–Xrs2 to resect DNA breaks. Nature 514: 122–125. 10.1038/nature13771 [DOI] [PubMed] [Google Scholar]
- Chai B, Huang J, Cairns BR, Laurent BC. 2005. Distinct roles for the RSC and Swi/Snf ATP-dependent chromatin remodelers in DNA double-strand break repair. Genes Dev 19: 1656–1661. 10.1101/gad.1273105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiruvella KK, Liang Z, Wilson TE. 2013. Repair of double-strand breaks by end joining. Cold Spring Harb Perspect Biol 5: a012757. 10.1101/cshperspect.a012757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church GM, Gilbert W. 1984. Genomic sequencing. Proc Natl Acad Sci 81: 1991–1995. 10.1073/pnas.81.7.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerici M, Mantiero D, Guerini I, Lucchini G, Longhese MP. 2008. The Yku70–Yku80 complex contributes to regulate double-strand break processing and checkpoint activation during the cell cycle. EMBO Rep 9: 810–818. 10.1038/embor.2008.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubrana K, van Attikum H, Hediger F, Gasser SM. 2007. The processing of double-strand breaks and binding of single-strand-binding proteins RPA and Rad51 modulate the formation of ATR-kinase foci in yeast. J Cell Sci 120: 4209–4220. 10.1242/jcs.018366 [DOI] [PubMed] [Google Scholar]
- Eapen VV, Sugawara N, Tsabar M, Wu W-H, Haber JE. 2012. The Saccharomyces cerevisiae chromatin remodeler Fun30 regulates DNA end resection and checkpoint deactivation. Mol Cell Biol 32: 4727–4740. 10.1128/MCB.00566-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson CH, Bertuch AA. 2016. Consider the workhorse: nonhomologous end-joining in budding yeast. Biochem Cell Biol 94: 396–406. 10.1139/bcb-2016-0001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank-Vaillant M, Marcand S. 2002. Transient stability of DNA ends allows nonhomologous end joining to precede homologous recombination. Mol Cell 10: 1189–1199. 10.1016/S1097-2765(02)00705-0 [DOI] [PubMed] [Google Scholar]
- Gao S, Honey S, Futcher B, Grollman AP. 2016. The non-homologous end-joining pathway of S. cerevisiae works effectively in G1-phase cells, and religates cognate ends correctly and non-randomly. DNA Repair 42: 1–10. 10.1016/j.dnarep.2016.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudreau L, Schmid A, Blaschke D, Ptashne M, Hörz W. 1997. RNA polymerase II holoenzyme recruitment is sufficient to remodel chromatin at the yeast PHO5 promoter. Cell 89: 55–62. 10.1016/S0092-8674(00)80182-8 [DOI] [PubMed] [Google Scholar]
- Geuting V, Reul C, Löbrich M. 2013. ATM release at resected double-strand breaks provides heterochromatin reconstitution to facilitate homologous recombination. PLoS Genet 9: e1003667. 10.1371/journal.pgen.1003667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein M, Derheimer FA, Tait-Mulder J, Kastan MB. 2013. Nucleolin mediates nucleosome disruption critical for DNA double-strand break repair. Proc Natl Acad Sci 110: 16874–16879. 10.1073/pnas.1306160110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez MP, MacAlpine HK, MacAlpine DM. 2019. Nascent chromatin occupancy profiling reveals locus- and factor-specific chromatin maturation dynamics behind the DNA replication fork. Genome Res 29: 1123–1133. 10.1101/gr.243386.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber JE. 2012. Mating-type genes and MAT switching in Saccharomyces cerevisiae. Genetics 191: 33–64. 10.1534/genetics.111.134577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halazonetis TD, Gorgoulis VG, Bartek J. 2008. An oncogene-induced DNA damage model for cancer development. Science 319: 1352–1355. 10.1126/science.1140735 [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. 2000. The hallmarks of cancer. Cell 100: 57–70. 10.1016/S0092-8674(00)81683-9 [DOI] [PubMed] [Google Scholar]
- Hauer MH, Gasser SM. 2017. Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev 31: 2204–2221. 10.1101/gad.307702.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauer MH, Seeber A, Singh V, Thierry R, Sack R, Amitai A, Kryzhanovska M, Eglinger J, Holcman D, Owen-Hughes T, et al. 2017. Histone degradation in response to DNA damage enhances chromatin dynamics and recombination rates. Nat Struct Mol Biol 24: 99–107. 10.1038/nsmb.3347 [DOI] [PubMed] [Google Scholar]
- Henikoff JG, Belsky JA, Krassovsky K, MacAlpine DM, Henikoff S. 2011. Epigenome characterization at single base-pair resolution. Proc Natl Acad Sci 108: 18318–18323. 10.1073/pnas.1110731108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert S, Brion A, Arbona J-M, Lelek M, Veillet A, Lelandais B, Parmar J, Fernández FG, Almayrac E, Khalil Y, et al. 2017. Chromatin stiffening underlies enhanced locus mobility after DNA damage in budding yeast. EMBO J 36: 2595–2608. 10.15252/embj.201695842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertel CB, Längst G, Hörz W, Korber P. 2005. Nucleosome stability at the yeast PHO5 and PHO8 promoters correlates with differential cofactor requirements for chromatin opening. Mol Cell Biol 25: 10755–10767. 10.1128/MCB.25.24.10755-10767.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks WM, Yamaguchi M, Haber JE. 2011. Real-time analysis of double-strand DNA break repair by homologous recombination. Proc Natl Acad Sci 108: 3108–3115. 10.1073/pnas.1019660108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horigome C, Oma Y, Konishi T, Schmid R, Marcomini I, Hauer MH, Dion V, Harata M, Gasser SM. 2014. SWR1 and INO80 chromatin remodelers contribute to DNA double-strand break perinuclear anchorage site choice. Mol Cell 55: 626–639. 10.1016/j.molcel.2014.06.027 [DOI] [PubMed] [Google Scholar]
- Ira G, Pellicioli A, Balijja A, Wang X, Fiorani S, Carotenuto W, Liberi G, Bressan D, Wan L, Hollingsworth NM, et al. 2004. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 431: 1011–1017. 10.1038/nature02964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasin M, Rothstein R. 2013. Repair of strand breaks by homologous recombination. Cold Spring Harb Perspect Biol 5: a012740. 10.1101/cshperspect.a012740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplun L, Ivantsiv Y, Kornitzer D, Raveh D. 2000. Functions of the DNA damage response pathway target Ho endonuclease of yeast for degradation via the ubiquitin-26S proteasome system. Proc Natl Acad Sci 97: 10077–10082. 10.1073/pnas.97.18.10077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keogh M-C, Kim J-A, Downey M, Fillingham J, Chowdhury D, Harrison JC, Onishi M, Datta N, Galicia S, Emili A, et al. 2006. A phosphatase complex that dephosphorylates γH2AX regulates DNA damage checkpoint recovery. Nature 439: 497–501. 10.1038/nature04384 [DOI] [PubMed] [Google Scholar]
- Kim J-A, Haber JE. 2009. Chromatin assembly factors Asf1 and CAF-1 have overlapping roles in deactivating the DNA damage checkpoint when DNA repair is complete. Proc Natl Acad Sci 106: 1151–1156. 10.1073/pnas.0812578106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J-A, Kruhlak M, Dotiwala F, Nussenzweig A, Haber JE. 2007. Heterochromatin is refractory to γ-H2AX modification in yeast and mammals. J Cell Biol 178: 209–218. 10.1083/jcb.200612031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostriken R, Heffron F. 1984. The product of the HO gene is a nuclease: purification and characterization of the enzyme. Cold Spring Harb Symp Quant Biol 49: 89–96. 10.1101/SQB.1984.049.01.012 [DOI] [PubMed] [Google Scholar]
- Kwon S-J, Park J-H, Park E-J, Lee S-A, Lee H-S, Kang SW, Kwon J. 2015. ATM-mediated phosphorylation of the chromatin remodeling enzyme BRG1 modulates DNA double-strand break repair. Oncogene 34: 303–313. 10.1038/onc.2013.556 [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SE, Moore JK, Holmes A, Umezu K, Kolodner RD, Haber JE. 1998. Saccharomyces Ku70, mre11/rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell 94: 399–409. 10.1016/S0092-8674(00)81482-8 [DOI] [PubMed] [Google Scholar]
- Lee SE, Pellicioli A, Demeter J, Vaze MP, Gasch AP, Malkova A, Brown PO, Botstein D, Stearns T, Foiani M, et al. 2000. Arrest, adaptation, and recovery following a chromosome double-strand break in Saccharomyces cerevisiae. Cold Spring Harb Symp Quant Biol 65: 303–314. 10.1101/sqb.2000.65.303 [DOI] [PubMed] [Google Scholar]
- Lee C-S, Lee K, Legube G, Haber JE. 2014. Dynamics of yeast histone H2A and H2B phosphorylation in response to a double-strand break. Nat Struct Mol Biol 21: 103–109. 10.1038/nsmb.2737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Tyler JK. 2016. Nucleosome disassembly during human non-homologous end joining followed by concerted HIRA- and CAF-1-dependent reassembly. eLife 5: e15129. 10.7554/eLife.15129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang B, Qiu J, Ratnakumar K, Laurent BC. 2007. RSC functions as an early double-strand-break sensor in the cell's response to DNA damage. Curr Biol 17: 1432–1437. 10.1016/j.cub.2007.07.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber MR. 2010. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem 79: 181–211. 10.1146/annurev.biochem.052308.093131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linger J, Tyler JK. 2005. The yeast histone chaperone chromatin assembly factor 1 protects against double-strand DNA-damaging agents. Genetics 171: 1513–1522. 10.1534/genetics.105.043000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr D, Venkov P, Zlatanova J. 1995. Transcriptional regulation in the yeast GAL gene family: a complex genetic network. FASEB J 9: 777–787. 10.1096/fasebj.9.9.7601342 [DOI] [PubMed] [Google Scholar]
- Mao Z, Bozzella M, Seluanov A, Gorbunova V. 2008. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair 7: 1765–1771. 10.1016/j.dnarep.2008.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta A, Beach A, Haber JE. 2017. Homology requirements and competition between gene conversion and break-induced replication during double-strand break repair. Mol Cell 65: 515–526.e3. 10.1016/j.molcel.2016.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou EP, Symington LS. 2008. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455: 770–774. 10.1038/nature07312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JK, Haber JE. 1996. Capture of retrotransposon DNA at the sites of chromosomal double-strand breaks. Nature 383: 644–646. 10.1038/383644a0 [DOI] [PubMed] [Google Scholar]
- Morrison AJ, Highland J, Krogan NJ, Arbel-Eden A, Greenblatt JF, Haber JE, Shen X. 2004. INO80 and γ-H2AX interaction links ATP-dependent chromatin remodeling to DNA damage repair. Cell 119: 767–775. 10.1016/j.cell.2004.11.037 [DOI] [PubMed] [Google Scholar]
- Nakada S, Chen GI, Gingras A-C, Durocher D. 2008. PP4 is a γH2AX phosphatase required for recovery from the DNA damage checkpoint. EMBO Rep 9: 1019–1026. 10.1038/embor.2008.162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann FR, Dion V, Gehlen LR, Tsai-Pflugfelder M, Schmid R, Taddei A, Gasser SM. 2012. Targeted INO80 enhances subnuclear chromatin movement and ectopic homologous recombination. Genes Dev 26: 369–383. 10.1101/gad.176156.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmbos PL, Wu D, Daley JM, Wilson TE. 2008. Recruitment of Saccharomyces cerevisiae Dnl4-Lif1 complex to a double-strand break requires interactions with Yku80 and the Xrs2 FHA domain. Genetics 180: 1809–1819. 10.1534/genetics.108.095539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt A, Reece RJ. 1998. The yeast galactose genetic switch is mediated by the formation of a Gal4p-Gal80p-Gal3p complex. EMBO J 17: 4086–4091. 10.1093/emboj/17.14.4086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo SE. 2015. Reshaping chromatin after DNA damage: the choreography of histone proteins. J Mol Biol 427: 626–636. 10.1016/j.jmb.2014.05.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo SE, Almouzni G. 2015. Chromatin dynamics after DNA damage: the legacy of the access-repair-restore model. DNA Repair 36: 114–121. 10.1016/j.dnarep.2015.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindra A, Weiss K, Simpson RT. 1999. High-resolution structural analysis of chromatin at specific loci: Saccharomyces cerevisiae silent mating-type locus HMRa. Mol Cell Biol 19: 7944–7950. 10.1128/MCB.19.12.7944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2015. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. https://www.R-project.org/. [Google Scholar]
- Sandell LL, Zakian VA. 1993. Loss of a yeast telomere: arrest, recovery, and chromosome loss. Cell 75: 729–739. 10.1016/0092-8674(93)90493-A [DOI] [PubMed] [Google Scholar]
- Schmid A, Fascher KD, Hörz W. 1992. Nucleosome disruption at the yeast PHO5 promoter upon PHO5 induction occurs in the absence of DNA replication. Cell 71: 853–864. 10.1016/0092-8674(92)90560-Y [DOI] [PubMed] [Google Scholar]
- Seeber A, Gasser SM. 2017. Chromatin organization and dynamics in double-strand break repair. Curr Opin Genet Dev 43: 9–16. 10.1016/j.gde.2016.10.005 [DOI] [PubMed] [Google Scholar]
- Shim EY, Ma J-L, Oum J-H, Yanez Y, Lee SE. 2005. The yeast chromatin remodeler RSC complex facilitates end joining repair of DNA double-strand breaks. Mol Cell Biol 25: 3934–3944. 10.1128/MCB.25.10.3934-3944.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim EY, Hong SJ, Oum J-H, Yanez Y, Zhang Y, Lee SE. 2007. RSC mobilizes nucleosomes to improve accessibility of repair machinery to the damaged chromatin. Mol Cell Biol 27: 1602–1613. 10.1128/MCB.01956-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shroff R, Arbel-Eden A, Pilch D, Ira G, Bonner WM, Petrini JH, Haber JE, Lichten M. 2004. Distribution and dynamics of chromatin modification induced by a defined DNA double-strand break. Curr Biol 14: 1703–1711. 10.1016/j.cub.2004.09.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122: 19–27. https://www.genetics.org/content/122/1/19.short. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soria G, Polo SE, Almouzni G. 2012. Prime, repair, restore: the active role of chromatin in the DNA damage response. Mol Cell 46: 722–734. 10.1016/j.molcel.2012.06.002 [DOI] [PubMed] [Google Scholar]
- Southern E. 2006. Southern blotting. Nat Protoc 1: 518–525. 10.1038/nprot.2006.73 [DOI] [PubMed] [Google Scholar]
- Sugawara N, Haber JE. 2006. Repair of DNA double strand breaks: in vivo biochemistry. Methods Enzymol 408: 416–429. 10.1016/S0076-6879(06)08026-8 [DOI] [PubMed] [Google Scholar]
- Symington LS. 2014. End resection at double-strand breaks: mechanism and regulation. Cold Spring Harb Perspect Biol 6: a016436. 10.1101/cshperspect.a016436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS, Gautier J. 2011. Double-strand break end resection and repair pathway choice. Annu Rev Genet 45: 247–271. 10.1146/annurev-genet-110410-132435 [DOI] [PubMed] [Google Scholar]
- Teo SH, Jackson SP. 1997. Identification of Saccharomyces cerevisiae DNA ligase IV: involvement in DNA double-strand break repair. EMBO J 16: 4788–4795. 10.1093/emboj/16.15.4788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran PT, Erdeniz N, Symington LS, Liskay RM. 2004. EXO1-A multi-tasking eukaryotic nuclease. DNA Repair 3: 1549–1559. 10.1016/j.dnarep.2004.05.015 [DOI] [PubMed] [Google Scholar]
- Tran TQ, MacAlpine HK, Tripuraneni V, Mitra S, MacAlpine DM, Hartemink AJ. 2020. Linking the dynamics of chromatin occupancy and transcription with predictive models. bioRxiv 10.1101/2020.06.28.176545 [DOI] [PMC free article] [PubMed]
- Tsabar M, Hicks WM, Tsaponina O, Haber JE. 2016. Re-establishment of nucleosome occupancy during double-strand break repair in budding yeast. DNA Repair 47: 21–29. 10.1016/j.dnarep.2016.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukuda T, Fleming AB, Nickoloff Ja, Osley MA. 2005. Chromatin remodelling at a DNA double-strand break site in Saccharomyces cerevisiae. Nature 438: 379–383. 10.1038/nature04148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss K, Simpson RT. 1998. High-resolution structural analysis of chromatin at specific loci: Saccharomyces cerevisiae silent mating type locus HMLα. Mol Cell Biol 18: 5392–5403. 10.1128/MCB.18.9.5392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westmoreland JW, Resnick MA. 2016. Recombinational repair of radiation-induced double-strand breaks occurs in the absence of extensive resection. Nucleic Acids Res 44: 695–704. 10.1093/nar/gkv1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson TE, Grawunder U, Lieber MR. 1997. Yeast DNA ligase IV mediates non-homologous DNA end joining. Nature 388: 495–498. 10.1038/41365 [DOI] [PubMed] [Google Scholar]
- Wu D, Topper LM, Wilson TE. 2008. Recruitment and dissociation of nonhomologous end joining proteins at a DNA double-strand break in Saccharomyces cerevisiae. Genetics 178: 1237–1249. 10.1534/genetics.107.083535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarger JG, Hopper JE. 1979. Positive transcriptional control of inducible galactose pathway enzymes in yeast. In From gene to protein: information transfer in normal and abnormal cells (ed. Russell TR, et al. ), p. 642. Academic Press, New York. [Google Scholar]
- Zhu Z, Chung W-H, Shim EY, Lee SE, Ira G. 2008. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 134: 981–994. 10.1016/j.cell.2008.08.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zierhut C, Diffley JFX. 2008. Break dosage, cell cycle stage and DNA replication influence DNA double strand break response. EMBO J 27: 1875–1885. 10.1038/emboj.2008.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.