

Abstract
Neospora caninum primarily infects cattle, causing abortions, with an estimated impact of a billion dollars on the worldwide economy annually. However, the study of its biology has been unheeded by the established paradigm that it is virtually identical to its close relative, the widely studied human pathogen Toxoplasma gondii. By revisiting the genome sequence, assembly, and annotation using third-generation sequencing technologies, here we show that the N. caninum genome was originally incorrectly assembled under the presumption of synteny with T. gondii. We show that major chromosomal rearrangements have occurred between these species. Importantly, we show that chromosomes originally named Chr VIIb and VIII are indeed fused, reducing the karyotype of both N. caninum and T. gondii to 13 chromosomes. We reannotate the N. caninum genome, revealing more than 500 new genes. We sequence and annotate the nonphotosynthetic plastid and mitochondrial genomes and show that although apicoplast genomes are virtually identical, high levels of gene fragmentation and reshuffling exist between species and strains. Our results correct assembly artifacts that are currently widely distributed in the genome database of N. caninum and T. gondii and, more importantly, highlight the mitochondria as a previously oversighted source of variability and pave the way for a change in the paradigm of synteny, encouraging rethinking the genome as basis of the comparative unique biology of these pathogens.
The Apicomplexa comprise a large phylum of parasitic alveolates of medical and veterinary importance, causing deadly diseases such as malaria, cryptosporidiosis, neosporosis, and toxoplasmosis, among others. With the exception of a few commonalities such as their obligatory intracellular lifestyle and the presence of specialized secretory organelles and of secondary endosymbionts, the apicomplexans differ greatly in morphology, host range specificity, pathogenicity, reproductive strategy, and transmission. Understanding the molecular basis of these differences has been the focus of much research. Comparative genomic analyses revealed that, albeit all small, apicomplexans genomes vary greatly in size, ranging from 9 to 130 Mb (Debarry and Kissinger 2011; Blazejewski et al. 2015). Having diverged from a common ancestor 350–824 myr ago (Escalante and Ayala 1995), shy of 900 genes are conserved among them, whereby major genomic rearrangements can be observed (Debarry and Kissinger 2011).
High synteny, defined as conserved content and order of a given genomic locus, is rarely observed (Debarry and Kissinger 2011). A seemingly stark exception to this is the genomes of Toxoplasma gondii and Neospora caninum. Morphologically, these parasites are virtually indistinguishable, so much so, that N. caninum was only recognized as a separate species in 1988 (Dubey 2003; Dubey et al. 2002). Moreover, both species show similar tropism within their hosts, where they can infect virtually any nucleated cell. They both show a fast-replicating form (tachyzoite), causing acute disease, that transitions into a slow-dividing form (bradyzoite), which persists in immune-privileged sites, such as the brain, establishing chronic infection. In line with this, initial comparative analysis concluded that these species have largely conserved genomic content and are largely syntenic (Reid et al. 2012). Despite their commonalities, however, the biology of these pathogens also differs significantly. T. gondii infects a wide range of intermediate hosts, including humans, causing deadly disease in immunocompromised individuals or by congenital transmission. In contrast, N. caninum infects primarily cattle, causing abortions with an estimated impact of a billion dollars on the worldwide economy annually (Reichel et al. 2013). Feline species act as definitive hosts of T. gondii, whereas sexual replication of N. caninum occurs only in canids (McAllister et al. 1998; Gondim et al. 2004; King et al. 2010). These biological differences have been largely ascribed to absence, point mutations, and pseudogenization of T. gondii virulence factors in N. caninum and the comparative amplification of surface protein-coding gene families in N. caninum (Khan et al. 2009; Reid et al. 2012).
Advancements in genome sequencing technologies have accompanied the fast-paced genomics era. Particularly, third-generation sequencing technologies, such as Pacific Biosciences (PacBio) and Oxford Nanopore Technologies sequencing, outperform prior technologies by providing very long reads that can span regions containing repetitive sequences. This has led to improvements in the assembly of previously unattainable genomes, such as those presenting high proportions of repetitive sequences, allowing whole new genomes to be assembled with high accuracy. Here, we set out to sequence and de novo assemble two N. caninum strain genomes and the T. gondii genome, using PacBio and Oxford Nanopore.
Results
N. caninum and T. gondii long-read assembly genomes
To assemble the N. caninum genome, we sequenced DNA from the reference strain N. caninum Liverpool (NcLiv), and a recent isolate from an experimental farm in Uruguay, named N. caninum Uru1 (NcUru1) (Cabrera et al. 2019). Sequencing was performed by PacBio technology, Oxford Nanopore, and Illumina. Reads derived from each sequencing were assembled independently and then combined into a single assembly per strain (Supplemental Fig. 1A). For NcLiv, PacBio and Nanopore assemblies matched completely, with the exception of a single conflict region, originally assembled in Chromosome VIIa, which was resolved by PCR and shown to belong to the newly assembled Chromosome X (Supplemental Fig. 1B,C). The assembled genomes were corrected with publicly available Illumina reads or those obtained in house. For NcLiv, the sequencing resulted in more than 100× depth, vastly improving the currently available genome coverage (Table 1). Genomes of the N. caninum strains were assembled separately.
Table 1.
Metrics of the de novo genome assembly of N. caninum Liverpool
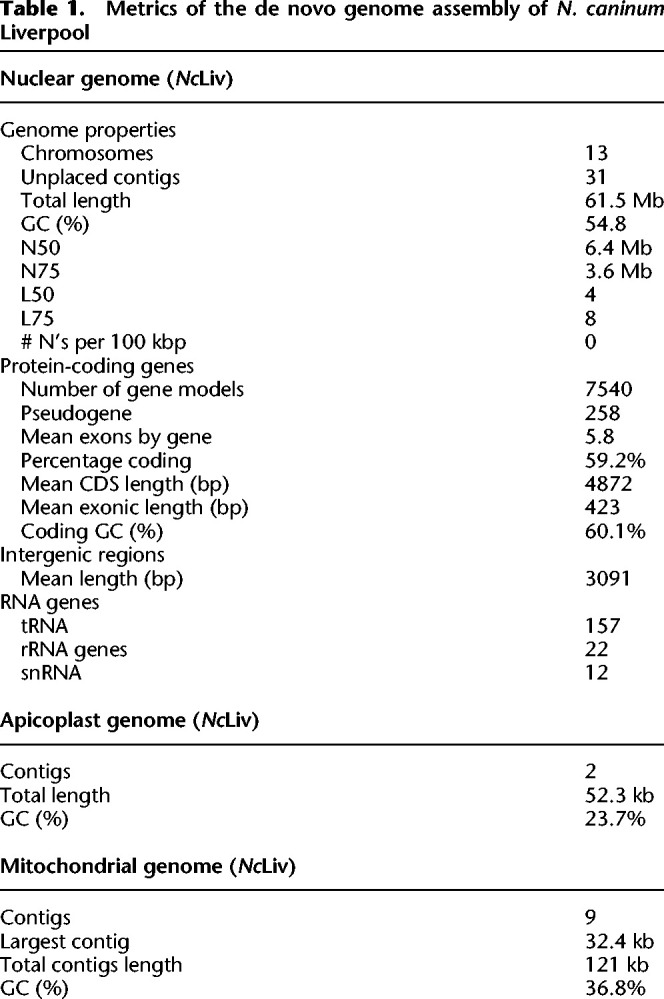
Assemblies for both Neospora strains were practically indistinguishable, indicating high genome similarity. The NcLiv genome consisted of 13 large contigs and 31 short (<122-kb) unplaced fragments. With an N50 of 6.4 Mb, 75% of the 61.6-Mb nuclear genome is in eight of the largest chromosomes (Table 1). The 13 largest contigs correspond to complete chromosomes; both 5′ and 3′ telomeres were mapped for 10, whereas we found one telomere at one end and subtelomeric regions at the other end for three (Supplemental Table 1). Putative centromeric sequences were identified in silico either by blasting the T. gondii centromeres or by identifying large regions devoid of gene-coding sequences flanked by syntenic genes flanking the centromeric regions of T. gondii (Fig. 1B–E;Supplemental Fig. 5; Supplemental Table 2). Our assembly revealed that, in contrast to what has been reported, the N. caninum genome is not organized in 14 chromosomes. Rather, previous Chr VIIb and Chr VIII are in fact a single chromosome (Fig. 1B).
Figure 1.

Comparative analysis of genome assemblies of Neospora caninum and Toxoplasma gondii using third-generation sequencing data reveals misassembly and karyotype differences. (A) Comparative analysis of the T. gondii type II (TgME49) genome assembly and the N. caninum Liverpool (NcLiv) strain genome assembly, obtained based on Sanger technology sequencing data. (B) Comparative alignment of the NcLiv genome assemblies using Sanger and third-generation (long-read) technology. (C) Comparative alignment of the T. gondii type II (TgME49) genome assemblies based on Sanger technology sequencing data or third-generation (long-read) technology of T. gondii type I (TgRH). (D) Comparative alignment of the T. gondii type I (TgRH) and the NcLiv genome assemblies based on third-generation (long-read) sequencing technology. (E) Chromosomal layout of N. caninum. Karyotype, chromosome length, telomeres, putative centromeres, and large repeats are shown.
Given that the previous N. caninum genome assembly had been constructed using the T. gondii genome as the reference, by assuming that these species conserve synteny and gene order, we next examined how our new N. caninum genome assembly compared with that of the T. gondii reference genome (Fig. 1A). Surprisingly, not only did the newly assembled N. caninum genome differ from that of T. gondii in the total number of chromosomes, but a number of large chromosomal rearrangements could be observed (Fig. 1B). Only half of the chromosomes’ structure, corresponding to N. caninum’s chromosomes originally named Ib, II, III, IX, X, and XII (now named according to their increasing size: XII, XI, IX, V, and III, respectively), is maintained between these species.
Next, we wondered whether the observable differences between the species were an artifact of the T. gondii genome assembly based on shorter reads. To this end, we sequenced the genome of a widely used T. gondii strain, known as the ΔKu80, both by PacBio and Oxford Nanopore and de novo assembled the genome. Our long-read sequencing resulted in a 108× coverage, and its assembly closely matched that reported by using Sanger and 454 sequencing (Fig. 1C), confirming that the differences observed with N. caninum are not artifactual. However, we were able to once again observe the fusion of Chromosomes VIIb and VIII in Toxoplasma, thereby indicating that the karyotype for both these apicomplexans is 13, and not 14 as previously reported (Fig. 1D). The sequenced strain was generated by directed insertion of a selectable marker in the Ku80 coding region using type I RH as the parental strain (Huynh and Carruthers 2009). Here, we were able to observe that the strain shows the expected replacement as per the original disruption strategy and that, despite lacking a major mechanism of DNA repair, it has not suffered any major chromosomal rearrangement (Supplemental Fig. 6A–C).
We next explored whether the breaks in synteny between these closely related species correlated with any distinguishable genomic features. We noticed that the level of sequence identity between the species varies along chromosomes. Coding regions seem to be highly conserved (82.4%). Nonetheless, we noticed that there is a detectable shift in codon usage between the two species, whereby N. caninum tends to use GC-richer codons than does T. gondii. The difference in codon usage conveys a detectable difference in composition whereby the mean GC content for N. caninum is 58.3% whereas it is 56.4% for T. gondii; this being a statistically significant difference (P-value 1.4 × 10−9). The same trend can be observed for GC3 (63.7% and 61.0%, respectively) and less markedly for GC1 (Supplemental Fig. 2). On the other hand, noncoding regions, including intronic and intergenic regions, are on average 80.7% identical. Noncoding regions throughout the genome of N. caninum are interspaced with short low complexity elements rich in A/G or A/T, with no identifiable conserved sequence motif. In contrast, three conserved sequence motifs, present in tandem with a defined periodicity in terms of nucleotide composition, could be identified in those regions of N. caninum chromosomes (I–X) where synteny with T. gondii is interrupted (Fig. 2A,B). The number of repeats present at each site and its specific sequence composition are detailed in Supplemental Table 3. Conspicuously, virtually identical domains are also present in regions where chromosomal rearrangements have occurred in the T. gondii genome (Supplemental Fig. 7A,B).
Figure 2.

Regions of synteny breaks between N. caninum and T. gondii are populated by three conserved domains. (A) Sequence identity of domains identified at regions where chromosomal rearrangements have occurred. (B) Graphical representation of Chromosome VIII of NcLiv. Comparative alignment to the T. gondii chromosomes. Percentages of sequence identity are shown. Regions examined for the presence of motifs are indicated (light green). The position of the putative centromere is indicated in orange. Note that large repetitive regions were not identified in this chromosome. 5′ (light purple) and 3′ (dark purple) telomeres are indicated. The identity and number of domains found per region, in Chromosome VII, are indicated.
Third-generation sequencing technologies allow better assembly and resolution of highly repetitive sequences. We assayed the presence of large repetitive elements in the genomes of NcLiv and NcUru1. We identified 119 large (>5000-bp) repetitive elements in the genome of NcLiv, of which 113 are shared between the two strains (Supplemental Table 7). We note, however, that in many cases, the strains show differences between them in the total length of the conserved repeats. Four repeats are found exclusively in NcLiv, whereas two are only found in NcUru1. Of the shared repetitive elements, five contain open reading frames (ORFs), coding for a total of 12 genes, 11 of which are annotated as coding for hypothetical proteins.
Gene annotation
Annotation of the newly assembled NcLiv genome, using homology-based searches and RNA-seq data sets, resulted in the annotation of 7540 genes and 254 pseudogenes. Of these, 7502 are distributed in 13 chromosomes, whereas 38 are in unplaced sequences. The products of 4553 genes have a putative assigned function, whereas 2987 code for hypothetical proteins. Comparatively, 554 new genes were identified in our annotation; nine are coded for in unassembled sequences, whereas 545 are distributed in the chromosomes. Albeit not originally annotated, 516 of these can be found in the original Sanger sequence reads of the NcLiv genome, with >95% identity in >95% of the gene's length. Of these, 494 mapped to the originally assembled chromosomes, whereas 22 belong to unplaced sequences.
No common theme was observed among the newly annotated genes. Of these, the most frequently found are SRS domain–containing proteins, followed by WD-domain, and zinc-finger containing proteins. This is consistent with previous findings showing that the SAG-related family of surface proteins is amplified in N. caninum (Reid et al. 2012). Previously, 227 members of this family had been identified. Here, we annotate a total of 231 SAG-related proteins. Finally, 38 novel genes were uniquely identified in the newly sequenced genome; 36 are distributed in 11 of the assembled chromosomes, whereas two remain in unplaced sequences (Fig. 3A). Of these, six have putative assigned functions or recognizable domains: a Syntaxin 6, N-terminal/SNARE domain–containing protein; an SPX domain/VTC domain–containing protein; a Kelch motif/Galactose oxidase, central domain/BTB/POZ domain–containing protein; a glutathione S-transferase, N-terminal domain–containing protein; a cyclophilin-type peptidyl-prolyl cis-trans isomerase (Fig. 3B; Supplemental Table 4); and a GYF domain-containing protein. The products of the remaining 32 genes are regarded as hypothetical. We note that many of these new genes resulted either from the annotation of sequencing gaps (Fig. 3A) or from sequencing that extended through repetitive regions where previous sequencing had failed (for a representative example, see Fig. 3C).
Figure 3.

Gene annotation reveals previously unknown genes in the genome of N. caninum. (A) Graphical representation of the position of novel genes in the newly assembled chromosomes. Red lines mark the position of novel genes along chromosomes. Alignment to the previously assembled NcLiv genome is partially shown for reference. Three insets are shown to highlight the annotation of three new genes in three newly assembled genomic regions. (B) Putative function of six out of the 38 newly identified genes. The remaining 32 genes are annotated as hypothetical. (C) Representative example of the improvement in annotation in regions that had been previously collapsed owing to the presence of tandem repeats. Several new genes were annotated, all of whose annotation is supported by RNA-seq data.
Next, we explored whether the new annotation revealed the presence of uncharacterized virulence factors. We surveyed the genome for homologs of major virulence factors characterized in T. gondii, the majority being kinases involved in protecting the parasitophorous vacuole from host-cell intrinsic defenses: ROP5, ROP16, ROP17, ROP18, and ROP38; dense granule secreted effectors such as GRA16, GRA24, GRA25, and GRA44; and IST. No major differences were found between the annotations regarding virulence factors with the exception of ROP38, for which four gene copies had been reported. We could resolve nine ROP38 copies arranged in tandem. Our annotation is consistent with previous reports describing the absence of GRA24 and IST homologs in N. caninum.
N. caninum apicoplast genome
The apicoplast is a relic nonphotosynthetic chloroplast-like organelle of bacterial origin present in most apicomplexan parasites, whereby essential lipid synthesis occurs (Fichera and Roos 1997; Waller and McFadden 2005). This organelle is of great importance as it has been validated as the target of antiparasitic drugs such as clindamycin (Fichera and Roos 1997). Most apicoplast proteins are encoded for in the nucleus and later imported (DeRocher et al. 2000; Waller 2000; Van Dooren et al. 2008; Lim et al. 2009; Sheiner et al. 2011; Glaser et al. 2012; Fellows et al. 2017). However, the apicoplast harbors its own genome, which has been traditionally regarded as coding for proteins needed for its maintenance (Reiff et al. 2012).
When assembling the core genome of NcLiv, we originally identified two contigs of 44 and 26 kb, with markedly lower GC content than that of the nuclear genome (21.5% vs. 59.2%). The assembled sequences bear significant homology at their ends, allowing their manual collapse into a single molecule of 56 kb. However, only a handful of long reads mapped to this pseudomolecule support its existence (Supplemental Fig. 3). Corrected long reads that mapped to those contigs were isolated and used to reassemble the apicoplast genome using both Canu and Unicycler. The removal of four incongruent reads in the case of NcLiv, as well as two incongruent reads in NcUru1, results in a circularized 35-kb apicoplast genome molecule, which is identical between the two strains with homogenous coverage (averaging 122×) along the length and bears a 10-kb inverted repeat sequence (Fig. 4A–D).
Figure 4.

N. caninum apicoplast genome structure and annotation. (A) Schematic representation of the circularized apicoplast genome and annotation. The presence of an inverted repeat sequence is highlighted by a shaded area. %GC is shown in gray. GC skew is represented in variable colors. Open reading frames present within the 35-kb circular apicoplast genome are shown (for details, see Supplemental Table 5). (B) Read coverage count along the length of the 35-kb apicoplast genome is shown. (C) The presence of an inverted repeat sequence is graphically represented in a YASS plot. (D) Alignment of long-read sequences to the apicoplast genome. A few reads spanning the end regions, supporting its circular topology, are highlighted.
Annotation of these contigs revealed 60 ORFs, most of which correspond to RNA polymerase subunits and ribosomal proteins. In addition, the coding regions for SulfB and Clp protease and a hypothetical protein were identified (Fig. 4A; Supplememtal Table 5). These have been shown to be resident proteins of the apicoplast, thereby confirming that this sequence corresponds to the apicoplast genome of N. caninum.
Mitochondrial genomes
A number of large contigs (up to 32.4 kb) amounting to a total of 121 kb were identified upon sequencing NcLiv (Table 1; Fig. 5A–G; Supplemental Fig. 4). These contain sequences with homology with classically known mitochondrial genes such as cox1 and cob1 and with ribosomal RNAs (Supplemental Fig. 4; Supplemental Table 6). In addition, these contigs display a GC content averaging 36.8%, markedly lower than that of the nuclear and apicoplast genomes (Table 1). We could not assemble these contigs together, and neither one of them circularizes on its own. All of these contigs contain fragments of cox1 and cob1 ORFs; however, the vast majority of them are interrupted by internal STOP codons. Three of the contigs feature replication origins of the heavy strand (OH) sequences, and only one of them features a potentially functional cox1 gene with no internal STOPs. One other contig lacks an origin of replication but features a potentially functional cob1 copy (Supplemental Table 6).
Figure 5.

Comparative analysis of mitochondrial genome structures and annotations of Neospora and Toxoplasma reveals gene fragmentation and reshuffling between species and strains. (A) The repetitive nature of the gene structure in a 32-kb mitochondrial DNA contig of NcLiv is graphically represented in a YASS plot. (B) The repetitive nature of the gene structure in a 16-kb mitochondrial DNA contig of NcLiv is graphically represented in a YASS plot. (C) Comparative alignment between two NcLiv mitochondrial contigs of 16 and 32 kb, respectively. (D) Comparative alignment between a NcLiv mitochondrial contig of 32 kb and a NcUru1 mitochondrial contig of 38 kb. (E) Comparative alignment between two NcUru1 mitochondrial contigs of 16 and 38 kb, respectively. (F) The repetitive nature of the gene structure in a 16-kb mitochondrial DNA contig of NcUru1 is graphically represented in a YASS plot. (G) Comparative alignment between a NcLiv mitochondrial contig of 32 kb and a T. gondii mitochondrial contigs of 39 kb.
The mitochondrial genome of NcUru1 is distributed in 20 contigs, which vary in size ranging from 1437 bp to 86 kb, with a medium of 12 kb in size and average GC content of 36.95%. The largest contig, of 86 kb, features three origins of replication (OH sequences), 36 fragments with homology with cytochrome b, 56 fragments with homology with cytochrome c oxidase subunits I and III, and several ribosomal RNA encoding genes. All genes encoding for cob, cox1, and cox3 contain internal STOP sequences, which would result in truncated proteins. Likewise, both genomes encode for the intronic endonuclease LAGLI.
Although apicoplast genomes are 95% identical between the two N. caninum strains sequenced, the contigs corresponding to the mitochondrial genomes of NcUru1 and NcLiv did not coincide with each other (Fig. 5C–G). They both showcase a similar structure consisting of multiple linear contigs, featuring pseudogenized copies of cox1 and cob1, whose order seems to reshuffle in every contig (Fig. 5A–G). The gene order, distribution, and length are unique to each strain.
Twenty-nine contigs ranging from 1.1 to 39 kb in size, with a median of 8.0 kb, were identified as the mitochondrial genome of T. gondii. The contigs’ GC content averages 36.6%. A similar structure of shuffled pseudogenes can be observed, whereby multiple copies of different size coding fragments of cob, cox1, and cox3 populate the mitochondrial chromosomes, together with rRNAs (Fig. 5C–G). Origins of replication of the heavy strand (OH sequences) can be identified in six contigs. Multiple coding sequences for the intronic endonucleases GIY and LAGLI can be identified in six contigs. Both between N. caninum strains or between N. caninum and T. gondii, no single contig bearing the same combination or order of gene coding fragments could be identified (Fig. 5E,G).
Discussion
Our ability to sequence genomes and annotate genes has greatly enhanced our understanding of the molecular basis of life, health, and disease. Comparative genomics has allowed us to establish evolutionary relationships among living organisms and aided in the development of specific molecular diagnosis and the rational prediction of selective drug targets. Widely used sequencing technologies such as Sanger, 454, and Illumina have played a pivotal part in these advancements. However, the limitations of these technologies, namely, their trouble reading through repetitive regions and their short-read outputs, have led to assembly artifacts that are currently widely distributed in genome and proteome databases (Tørresen et al. 2019). A number of protozoan parasite genomes have been recently revisited using third-generation sequencing technologies. One noteworthy example is the genome of Trypanosoma cruzi, the causative agent of Chagas disease. This genome was massively improved for several strains. Its assembly went from being highly fragmented in more than 4000 short contigs to less than 2000 contigs, doubling the overall genome size (Berná et al. 2018, 2019). Such improvements have allowed more accurate accounts of gene copy number and the identification of an underlying genome structure. Such advances are essential to profit from the advent of highly specific guided genome editing technologies, such as CRISPR-Cas9, to understand virulence traits linked to gene copy number and to inter- and intra-species differences.
Here, we disentangled the genomes of two closely related species; N. caninum and T. gondii, by de novo assembling them. The N. caninum genome had been previously assembled using T. gondii as a reference, under the presumption that they were largely syntenic, using short reads (Reid et al. 2012). We uncovered that even though half of the genome is indeed structured quite similarly between N. caninum and T. gondii, seven out of 13 chromosomes differ significantly from each other. Our unbiased assembly of both genomes revealed that the karyotype of these apicomplexans is of 13 chromosomes. Our results suggest that chromosomes previously mapped as Chr VIIb and VIII are, in fact, a single chromosome. A number of previous findings support this fusion. Several linkage analysis assays to uncover virulence factors showed that these chromosomes always segregate together (Su et al. 2002; Khan et al. 2005). Genome interactions mapped by Hi-C analysis revealed Chr VIIb and Chr VIII had a higher number of interactions with each other than any other combination of chromosomes did. The latter study also showed that the number of contacts between the right telomere of Chr VIIb and the left telomere of Chr VIII were the highest of all, suggesting that these could be physically linked (Bunnik et al. 2019). In addition, identification of the centromere of Chr VIIb was unattainable by ChIP-chip or ChIP-seq (Brooks et al. 2011; Gissot et al. 2012).
We detect a large inversion in the original Chr XII of N. caninum with respect to the T. gondii chromosome. This inversion is suggested in the 3D analysis of genome structure performed by Hi-C using the T. gondii type II strain ME49 (Bunnik et al. 2019). However, we do not detect such inversion in our T. gondii RH genome assembly, suggesting that the Chr XII inversion could be strain specific.
Genome structure differences and rearrangements are widely observed among apicomplexans (Debarry and Kissinger 2011). However, the driving forces of these differences are ill understood. It is well established that genomic divergence among Trypanosomatids, for example, can be partially ascribed to the presence of transposable elements within their genomes. Apicomplexans’ genomes, however, are devoid of such sequences (Debarry and Kissinger 2011). We identified low-complexity, repetitive regions, but their appearance did not correlate with recombination prone chromosomes but were rather evenly distributed throughout the genome. We did, however, identify a number of repetitive motifs frequently located in the vicinity of regions where synteny is lost. Experimental validation of these motifs as drivers for recombination would be needed to mechanistically link them to chromosomal rearrangements.
Unlike the situation for T. gondii for which multiple strains have been fully sequenced, whole genomes of N. caninum were so far limited to the reference strain NcLiv. Detailed population genomics studies based on whole-genome sequences from multiple strains worldwide are lacking and so is our current understanding of population genetic structure of Neospora. Very recently, however, a study analyzing 19 linked and unlinked genetic markers of 50 isolates collected worldwide resolved a single genotype of N. caninum (Khan et al. 2019). This is consistent with our results in which our whole-genome assembly of NcUru1 is practically indistinguishable from that of NcLiv, despite deriving from completely different geographical locations (Europe vs. South America). Nonetheless, it is well established that great genetic variability exists among N. caninum strains in the form of SNPs at particular loci and that these genetic differences underlie phenotypic variability (Al-Qassab et al. 2009; Regidor-Cerrillo et al. 2013; Cabrera et al. 2019). In this context, it is noteworthy that linkage assays, in which these minimal differences can be correlated to virulence phenotypes, rely on correct genome assemblies.
The apicoplast is a validated drug target to fight apicomplexan-caused diseases such as toxoplasmosis and malaria (Fichera and Roos 1997; Lell and Kremsner 2002). Despite its importance, however, very few complete apicomplexan apicoplast genome sequences have been reported. The N. caninum plastid genome sequence had not been identified before this study. However, plastid genome physical characterization suggested a size of ∼35 kb; whereby formation of oligomeric molecules, migrating as linear molecules in approximate multiples of the unit length, were detected (Gleeson and Johnson 1999). With ample coverage of an average of 122× and read lengths of up to 24 kb, we still needed to carefully analyze our sequencing data to find evidence that unequivocally supported a circularized plastid genome of 35 kb, as it has been previously reported.
On the other hand, the apicomplexan mitochondrial genome had been described before as consisting of repeated elements of 6–7 kb in length (Esseiva et al. 2004). Mitochondrial genomes commonly encode a number of proteins required for its maintenance; part of the translation apparatus, tRNAs, and large and small ribosomal RNAs rns and rnl; membrane-associated proteins that catalyze oxidative phosphorylation—cytochrome b (cob), subunits of cytochrome c oxidase (cox), ATP synthase subunits 6, 8, and 9, and subunits of the NADH dehydrogenase complex; and additional ORFs of unknown functions. Here, we found that the mitochondrial genomes of N. caninum code for fragments of cob, cox1, cox3, rrns, rrnl, Lagli, and Giy.
Although the nuclear and apicoplast genomes are virtually identical between the two N. caninum strains sequenced, mitochondria genomes are notably different. It is noteworthy that randomly shuffled partial ORF segments, in chromosomes of varying lengths, are observed between species (N. caninum and T. gondii) and between strains of N. caninum. This complex structure has been recently observed in T. gondii by others using Oxford Nanopore technology to sequence (Namasivayam et al. 2021). Mitochondria also differed greatly in size. Varying-size mtDNA fragments had been inadvertently observed before for Eimeria tenella, whereby Southern blot of cox3 yielded a smear pattern of 4 to 20 kb (Hikosaka et al. 2011). In addition, despite our ample sequencing coverage, we did not find any circular molecule, suggesting that the mtDNA is composed of linear fragments. This has been reported for Babesia, Theileria, and Plasmodium mtDNA (Preiser et al. 1996; Hikosaka et al. 2010). Gene structure organization differences are also observable among these closely related hematozoa apicomplexans.
No mitochondrial contig in N. caninum or T. gondii contained a fully functional copy of cox1 or cob. Likewise, Plasmodium mitochondrial ribosomal RNAs display a high degree of fragmentation. It is unclear how these small RNA fragments come together to form a ribosome (Hillebrand et al. 2018). A similar gene structure of fragmented gene pieces (divided in modules) has been described in the free living kinetoplastids. The mitochondrial gene modules of the unicellular flagellate Diplonemid are separately transcribed, followed by the joining of partial transcripts to contiguous RNAs (Moreira et al. 2016). Nonetheless, examination of available T. gondii PRU RNA sequence data from Oxford Nanopore has revealed numerous reads capable of encoding full-length cytochrome transcripts. In addition, recent RT-PCR experiments have detected nearly full-length cob and cox3 and a partial cox1 (Namasivayam et al. 2021). In addition, the investigators conclude that many ORFs are followed by variant polyadenylation signals that may be affecting the detection of full-length mitochondrial reads by Illumina-based RNA-seq as sequencing libraries are typically generated following oligo(dT) purification (Namasivayam et al. 2021).
Mitochondrial gene editing, in which transcripts are altered post-transcriptionally to render a functional product, has so far not described for apicomplexans. However, this process is quite common and mechanistically diverse in this organelle in a wide range of species ranging from protozoa to plants. On the other hand, functional copies of mitochondrial proteins could be encoded for in the nuclear genome and imported into the organelle. Protein import is largely used by the apicoplast, whereby proteins required for its metabolic functions are all imported from the nucleus (DeRocher et al. 2000; Waller 2000; Van Dooren et al. 2008; Glaser et al. 2012; Agrawal et al. 2013). Only those proteins required for the maintenance of its genome are fully transcribed and translated within the organelle (Reiff et al. 2012). Mitochondrial tRNAs and ATP synthase subunits are known to be imported in T. gondii (Esseiva et al. 2004; Huet et al. 2018; Salunke et al. 2018). It is noteworthy that a functional cox1 copy has been annotated in the T. gondii nuclear genome (TgME49_209260), and a homolog is present in the reference NcLiv genome (NCLIV_003650). TgME49_209260 has been proposed to localize to mitochondrial membranes by spatial proteomics (Crook et al. 2018; Barylyuk et al. 2020) and has been put forward as an essential gene in T. gondii by genome-wide CRISPR-Cas9-mediated deletion (Sidik et al. 2016), strongly suggesting that it codes for a functional protein. In addition, a number of other nuclear-encoded divergent cox-related proteins have been identified in the mitochondrial proteome of T. gondii (Seidi et al. 2018). Finally, recent profiling has shown that the respiratory chain complex IV in T. gondii, which in humans and yeast houses MT-COX1 and MT-COX3, contains peptides of these proteins and 17 other divergent Apicomplexa-specific proteins, all of which are encoded for in the nucleus (Maclean et al. 2021). Nonetheless, our results pose questions regarding the mechanisms of mitochondrial protein synthesis and transport that merit consideration.
The molecular mechanisms underpinning such high variability of genetic content among mitochondrial genomes are unknown. However, we identified the presence of LAGLI and GIY among the mitochondria encoded genes. Both these proteins are endonucleases encoded in invasive introns shown to be mobile elements. Intraspecific variability of fungal mitochondrial genomes has been mechanistically linked to the movement of these endonucleases (Juhász et al. 2008). Likewise, a GIY-type endonuclease present in the second intron of the mitochondrial cytochrome b gene in the fungus Podospora curvicolla was shown to autonomously transfer from an ORF-containing intron to an ORF-less allele (Saguez 2000). Our results pave the way for exploring the exact contribution of these endonucleases to genome variability. It remains to be determined, as well, whether the mitochondrial genome variability observed corresponds to a collection of multiple fragments, homogenously present within a population, or whether the contigs assembled represent a cohort of heteroplasmic mtDNA differentially distributed at the population level.
Overall, our results highlight distinct nuclear genomic structures, as well as a variable mitochondrial genome, as previously unexplored sources of genetic variability among apicomplexans. Variability is observed not only among closely related species but also between strains. Efforts to explore the mechanistic contributions of this variability could shed light onto the molecular underpinnings of virulence-related traits ranging from fitness to differences in drug susceptibility.
Methods
Cell culture
NcLiv was acquired from ATCC (50845). NcUru1 was isolated from a local congenitally infected calf as described by Cabrera et al. (2019). The T. gondii RH ΔKu80 (Huynh and Carruthers 2009) strain was kindly provided by Boris Striepen. All strains were maintained and grown as described by Cabrera et al. (2019).
High-molecular-weight DNA extraction and sequencing
For Oxford Nanopore and Illumina sequencing, DNA was extracted from NcLiv, NcUru1, and T. gondii RH ΔKu80 (Huynh and Carruthers 2009) strains, using a DNA purification kit from Zymo Research (D4074). Minion Oxford Nanopore sequencing was performed in house as described by Díaz-Viraqué et al. (2019). Sequencing libraries were prepared using a ligation sequencing kit and a native barcoding expansion kit (EXP-NBD103/SQK-LSK108, Oxford Nanopore Technologies) according to the method previously described (Quick et al. 2017), starting from 1 µg of total genomic DNA. Twelve-hour sequencing was performed in an R9.4 flow cell (FLO-MIN106, Oxford Nanopore Technologies). Base-calling and sequence retrieval was performed using Guppy basecaller version 3.0.3. For PacBio sequencing, DNA was extracted from NcLiv, NcUru1, and T. gondii RH ΔKu80 (Huynh and Carruthers 2009) strains by overnight incubation in lysis buffer (Tris-EDTA buffer supplemented with 1 µg/mL of RNase A, 10% SDS, 20% Proteinase K) at 55°C, phenol/chloroform extraction, ethanol precipitation, and resuspension in ultrapure water. PacBio sequencing was performed at the Integrative Genomics Core (Beckman Research Institute), using five SMRT cells per sample for the N. caninum and T. gondii genomes. Illumina sequencing of N. caninum DNA was also performed at the Integrative Genomics Core to 65× coverage.
Genome assembly and annotation
PacBio reads were assembled using HGAP Assembly software (Chin et al. 2013). Oxford Nanopore reads were assembled using Canu (Koren et al. 2017). Assemblies were merged using Quickmerge software (Chakraborty et al. 2016). A single conflict region in the assembly of Chromosome IV was solved by PCR using primers: M1_F, GAGGCGCTTACAATCAACCC; H37_F_M1_R, GAGACAGGACGGACTGAAGA; H37_R, CTGCTCTGTCTGAACAGGTT; M37_F, GCGAACAGCACGAAGTGAGA; M37_R, TCGTGCTTTGAGCATCCTCT. Short insertions and deletions, a common artifact produced by long-read technologies, were corrected using Illumina reads in Pilon (Walker et al. 2014). Reads used include our own, from DNA purified as described above, and raw reads obtained from the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) (accession IDs: PRJNA531306; WGS of N. caninum NC-Liverpool tachyzoites source: genomic, 2 × 100, ERR012899 and ERR012900; genomic, WGS of N. caninum NcLiv strain, 2 × 75). Gene annotation was performed using the automated annotation tool COMPANION (Steinbiss et al. 2016) using AUGUSTUS Threshold 0.2, taxon ID 5811, align reference proteins to target sequence as parameters (others as default), and supporting RNA-seq data to produce a transcript assembly. This was first aligned with Cufflinks (Trapnell et al. 2010) and assembled with TopHat (Trapnell et al. 2009) using SRAs with accession IDs ERR690607 and ERR690608 (organism: NcLiv, ssRNA-seq, 2 × 100, SRR4013168, SRR4013169, SRR4013170, SRR4013171, SRR4013172, and SRR4013173; RNA-seq of NcLiv, 2 × 100, as input sequences). Apicoplast and mitochondrial genomes were identified by manual GC filtering and confirmed by BLAST. Apicoplast genome was annotated using MFannot (https://github.com/BFL-lab/Mfannot). Mitochondrial genome annotation was performed in MITOS (http://mitos2.bioinf.uni-leipzig.de/index.py) using Opisthokont as reference.
Large repetitive sequences were identified using Tandem Repeats Finder (Benson 1999).
Comparative genomic analysis
Assembled genomes were compared using NUCmer (Delcher et al. 1999) to create the alignments between the assemblies being compared and Assemblytics (Nattestad and Schatz 2016) for visualization. Plots comparing the synteny between assemblies were obtained with the visualization tool Circos (Krzywinski et al. 2009), using the output from BLAST to create links between chromosomes. Repetitive regions were analyzed using YASS (Noé and Kucherov 2005). Corrected reads were obtained by Canu using corOutCoverage = 200. Blasr (Chaisson and Tesler 2012) and minimap2 (Li 2018) were used to align corrected long reads to the assemblies. BWA (Li 2013) and Bowtie 2 (Langmead and Salzberg 2012) were used to align Illumina read (wgs and RNA-seq) data to the assemblies.
SAMtools (Li et al. 2009) was used for postprocessing alignment and was used to obtain statistics and coverage information. Individual chromosome comparisons were performed with Artemis (Carver et al. 2012) and ACT. Integrative Genomics Viewer (IGV) (Robinson et al. 2017) was used for visual inspection of aligned reads (wgs and RNA-seq) on the assemblies.
Specific scripts generated in this study, available as Supplemental Code, were written in an R environment (RStudio Team 2020) and Bash to parse results and automate pipelines. Telomeres were identified by searching on chromosome ends for the typical TTTAGGG and AAACCCT heptameric repeats. Centromeric regions in Neospora were determined by BLAST against previously identified centromere sequences in T. gondii by ChIP-chip (Brooks et al. 2011).
Data access
The PacBio and MinION data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA597814.
Supplementary Material
Acknowledgments
We thank Dr. Fernando Alvarez, Natalia Rego, and Dr. Jon Boyle for helpful advice and critical reading of this manuscript. This work was funded by a grant from the National Agency for Research and Innovation (ANII) and the National Institute for Agricultural Research (INIA) number FSSA_X_2014_1_106026 awarded to C.R. and an installment grant from the Banco de Seguros del Estado (BSE) awarded to M.E.F.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.262832.120.
Competing interest statement
The authors declare no competing interests.
References
- Agrawal S, Chung DWD, Ponts N, van Dooren GG, Prudhomme J, Brooks CF, Rodrigues EM, Tan JC, Ferdig MT, Striepen B, et al. 2013. An apicoplast localized ubiquitylation system is required for the import of nuclear-encoded plastid proteins. PLoS Pathog 9: e1003426. 10.1371/journal.ppat.1003426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Qassab S, Reichel MP, Ivens A, Ellis JT. 2009. Genetic diversity amongst isolates of Neospora caninum, and the development of a multiplex assay for the detection of distinct strains. Mol Cell Probes 23: 132–139. 10.1016/j.mcp.2009.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barylyuk K, Koreny L, Ke H, Butterworth S, Crook OM, Lassadi I, Gupta V, Tromer E, Mourier T, Stevens TJ, et al. 2020. A comprehensive subcellular atlas of the Toxoplasma proteome via hyperLOPIT provides spatial context for protein functions. Cell Host Microbe 28: 752–766.e9. 10.1016/j.chom.2020.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson G. 1999. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27: 573–580. 10.1093/nar/27.2.573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berná L, Rodriguez M, Chiribao ML, Parodi-Talice A, Pita S, Rijo G, Alvarez-Valin F, Robello C. 2018. Expanding an expanded genome: long-read sequencing of Trypanosoma cruzi. Microb Genom 4: e000177.. 10.1099/mgen.0.000177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berná L, Pita S, Laura Chiribao M, Parodi-Talice A, Alvarez-Valin F, Robello C. 2019. Biology of the Trypanosoma cruzi genome. In Biology of Trypanosoma cruzi (ed. De Souza W). IntechOpen Ltd., London. 10.5772/intechopen.80373 [DOI] [Google Scholar]
- Blazejewski T, Nursimulu N, Pszenny V, Dangoudoubiyam S, Namasivayam S, Chiasson MA, Chessman K, Tonkin M, Swapna LS, Hung SS, et al. 2015. Systems-based analysis of the Sarcocystis neurona genome identifies pathways that contribute to a heteroxenous life cycle. mBio 6: e02445-14. 10.1128/mBio.02445-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks CF, Francia ME, Gissot M, Croken MM, Kim K, Striepen B. 2011. Toxoplasma gondii sequesters centromeres to a specific nuclear region throughout the cell cycle. Proc Natl Acad Sci 108: 3767–3772. 10.1073/pnas.1006741108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunnik EM, Venkat A, Shao J, McGovern KE, Batugedara G, Worth D, Prudhomme J, Lapp SA, Andolina C, Ross LS, et al. 2019. Comparative 3D genome organization in apicomplexan parasites. Proc Natl Acad Sci 116: 3183–3192. 10.1073/pnas.1810815116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera A, Fresia P, Berná L, Silveira C, Macías-Rioseco M, Arevalo AP, Crispo M, Pritsch O, Riet-Correa F, Giannitti F, et al. 2019. Isolation and molecular characterization of four novel Neospora caninum strains. Parasitol Res 118: 3535–3542. 10.1007/s00436-019-06474-9 [DOI] [PubMed] [Google Scholar]
- Carver T, Harris SR, Berriman M, Parkhill J, McQuillan JA. 2012. Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 28: 464–469. 10.1093/bioinformatics/btr703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJ, Tesler G. 2012. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics 13: 238. 10.1186/1471-2105-13-238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty M, Baldwin-Brown JG, Long AD, Emerson JJ. 2016. Contiguous and accurate de novo assembly of metazoan genomes with modest long read coverage. Nucleic Acids Res 44: e147. 10.1093/nar/gkw654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin C-SS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE, et al. 2013. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods 10: 563–569. 10.1038/nmeth.2474 [DOI] [PubMed] [Google Scholar]
- Crook OM, Mulvey CM, Kirk PDW, Lilley KS, Gatto L. 2018. A Bayesian mixture modelling approach for spatial proteomics. PLoS Comput Biol 14: e1006516. 10.1371/journal.pcbi.1006516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debarry JD, Kissinger JC. 2011. Jumbled genomes: missing apicomplexan synteny. Mol Biol Evol 28: 2855–2871. 10.1093/molbev/msr103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcher AL, Kasif S, Fleischmann RD, Peterson J, White O, Salzberg SL. 1999. Alignment of whole genomes. Nucleic Acids Res 27: 2369–2376. 10.1093/nar/27.11.2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRocher A, Hagen CB, Froehlich JE, Feagin JE, Parsons M. 2000. Analysis of targeting sequences demonstrates that trafficking to the Toxoplasma gondii plastid branches off the secretory system. J Cell Sci 113: 3969–3977. [DOI] [PubMed] [Google Scholar]
- Díaz-Viraqué F, Pita S, Greif G, de Souza R de CM, Iraola G, Robello C. 2019. Nanopore sequencing significantly improves genome assembly of the protozoan parasite Trypanosoma cruzi. Genome Biol Evol 11: 1952–1957. 10.1093/gbe/evz129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey JP. 2003. Review of Neospora caninum and neosporosis in animals. Korean J Parasitol 41: 1–16. 10.3347/kjp.2003.41.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey JP, Barr BC, Barta JR, Bjerkås I, Björkman C, Blagburn BL, Bowman DD, Buxton D, Ellis JT, Gottstein B, et al. 2002. Redescription of Neospora caninum and its differentiation from related coccidia. Int J Parasitol 32: 929–946. 10.1016/s0020-7519(02)00094-2 [DOI] [PubMed] [Google Scholar]
- Escalante AA, Ayala FJ. 1995. Evolutionary origin of Plasmodium and other Apicomplexa based on rRNA genes. Proc Natl Acad Sci 92: 5793–5797. 10.1073/pnas.92.13.5793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esseiva AC, Naguleswaran A, Hemphill A, Schneider A. 2004. Mitochondrial tRNA import in toxoplasma gondii. J Biol Chem 279: 42363–8. 10.1074/jbc.M404519200 [DOI] [PubMed] [Google Scholar]
- Fellows JD, Cipriano MJ, Agrawal S, Striepena B. 2017. A plastid protein that evolved from ubiquitin and is required for apicoplast protein import in Toxoplasma gondii. mBio 8: e00950-17. 10.1128/mBio.00950-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fichera ME, Roos DS. 1997. A plastid organelle as a drug target in apicomplexan parasites. Nature 390: 407–409. 10.1038/37132 [DOI] [PubMed] [Google Scholar]
- Gissot M, Walker R, Delhaye S, Huot L, Hot D, Tomavo S. 2012. Toxoplasma gondii chromodomain protein 1 binds to heterochromatin and colocalises with centromeres and telomeres at the nuclear periphery. PLoS One 7: e32671. 10.1371/journal.pone.0032671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser S, Van Dooren GG, Agrawal S, Brooks CF, McFadden GI, Striepen B, Higgins MK. 2012. Tic22 is an essential chaperone required for protein import into the apicoplast. J Biol Chem 287: 39505–39512. 10.1074/jbc.M112.405100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleeson MT, Johnson AM. 1999. Physical characterisation of the plastid DNA in Neospora caninum. Int J Parasitol 29: 1563–1573. 10.1016/S0020-7519(99)00117-4 [DOI] [PubMed] [Google Scholar]
- Gondim LFP, McAllister MM, Pitt WC, Zemlicka DE. 2004. Coyotes (Canis latrans) are definitive hosts of Neospora caninum. Int J Parasitol 34: 159–161. 10.1016/j.ijpara.2004.01.001 [DOI] [PubMed] [Google Scholar]
- Hikosaka K, Watanabe YI, Tsuji N, Kita K, Kishine H, Arisue N, Palacpac NMQ, Kawazu SI, Sawai H, Horii T, et al. 2010. Divergence of the mitochondrial genome structure in the apicomplexan parasites, babesia and theileria. Mol Biol Evol 27: 1107–1116. 10.1093/molbev/msp320 [DOI] [PubMed] [Google Scholar]
- Hikosaka K, Nakai Y, Watanabe Yi, Tachibana SI, Arisue N, Palacpac NMQ, Toyama T, Honma H, Horii T, Kita K, et al. 2011. Concatenated mitochondrial DNA of the coccidian parasite Eimeria tenella. Mitochondrion 11: 273–278. 10.1016/j.mito.2010.10.003 [DOI] [PubMed] [Google Scholar]
- Hillebrand A, Matz JM, Almendinger M, Müller K, Matuschewski K, Schmitz-Linneweber C. 2018. Identification of clustered organellar short (cos) RNAs and of a conserved family of organellar RNA-binding proteins, the heptatricopeptide repeat proteins, in the malaria parasite. Nucleic Acids Res 46: 10417–10431. 10.1093/nar/gky710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huet D, Rajendran E, van Dooren GG, Lourido S. 2018. Identification of cryptic subunits from an apicomplexan ATP synthase. eLife 7: e38097. 10.7554/eLife.38097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh M-H, Carruthers VB. 2009. Tagging of endogenous genes in a toxoplasma gondii strain lacking Ku80. Eukaryot Cell 8: 530–539. 10.1128/EC.00358-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhász Á, Pfeiffer I, Keszthelyi A, Kucsera J, Vágvölgyi C, Hamari Z. 2008. Comparative analysis of the complete mitochondrial genomes of Aspergillus niger mtDNA type 1a and Aspergillus tubingensis mtDNA type 2b. FEMS Microbiol Lett 281: 51.– . 10.1111/j.1574-6968.2008.01077.x [DOI] [PubMed] [Google Scholar]
- Khan A, Taylor S, Su C, Mackey AJ, Boyle J, Cole R, Glover D, Tang K, Paulsen IT, Berriman M, et al. 2005. Composite genome map and recombination parameters derived from three archetypal lineages of Toxoplasma gondii. Nucleic Acids Res 33: 2980–2992. 10.1093/nar/gki604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A, Taylor S, Ajioka JW, Rosenthal BM, Sibley LD. 2009. Selection at a single locus leads to widespread expansion of Toxoplasma gondii lineages that are virulent in mice. PLoS Genet 5: e1000404. 10.1371/journal.pgen.1000404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A, Fujita AW, Randle N, Regidor-Cerrillo J, Shaik JS, Shen K, Oler AJ, Quinones M, Latham SM, Akanmori BD, et al. 2019. Global selective sweep of a highly inbred genome of the cattle parasite Neospora caninum. Proc Natl Acad Sci 116: 22764.– . 10.1073/pnas.1913531116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King JS, Šlapeta J, Jenkins DJ, Al-Qassab SE, Ellis JT, Windsor PA. 2010. Australian dingoes are definitive hosts of Neospora caninum. Int J Parasitol 40: 945–950. 10.1016/j.ijpara.2010.01.008 [DOI] [PubMed] [Google Scholar]
- Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. 2017. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 27: 722–736. 10.1101/gr.215087.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. 2009. Circos: an information aesthetic for comparative genomics. Genome Res 19: 1639–1645. 10.1101/gr.092759.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lell B, Kremsner PG. 2002. Clindamycin as an antimalarial drug: review of clinical trials. Antimicrob Agents Chemother 9: 357.– . 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997 [q-bio.GN].
- Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34: 3094–3100. 10.1093/bioinformatics/bty191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim L, Kalanon M, McFadden GI. 2009. New proteins in the apicoplast membranes: time to rethink apicoplast protein targeting. Trends Parasitol 25: 197–200. 10.1016/j.pt.2009.02.001 [DOI] [PubMed] [Google Scholar]
- Maclean AE, Bridges HR, Silva MF, Ding S, Ovciarikova J, Hirst J, Sheiner L. 2021. Complexome profile of Toxoplasma gondii mitochondria identifies divergent subunits of respiratory chain complexes including new subunits of cytochrome bc1 complex. PLoS Pathog 17: e1009301. 10.1371/journal.ppat.1009301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister MM, Dubey JP, Lindsay DS, Jolley WR, Wills RA, McGuire AM. 1998. Dogs are definitive hosts of Neospora caninum. Int J Parasitol 28: 1473–1479. 10.1016/S0020-7519(98)00138-6 [DOI] [PubMed] [Google Scholar]
- Moreira S, Valach M, Aoulad-Aissa M, Otto C, Burger G. 2016. Novel modes of RNA editing in mitochondria. Nucleic Acids Res 44: 4907–4919. 10.1093/nar/gkw188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namasivayam S, Baptista RP, Xiao W, Hall EM, Doggett JS, Troell K, Kissinger JC. 2021. A novel fragmented mitochondrial genome in the protist pathogen Toxoplasma gondii and related tissue coccidia. Genome Res (this issue) 31: 852–865. 10.1101/gr.266403.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattestad M, Schatz MC. 2016. Assemblytics: a web analytics tool for the detection of variants from an assembly. Bioinformatics 32: 3021–3023. 10.1093/bioinformatics/btw369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noé L, Kucherov G. 2005. YASS: enhancing the sensitivity of DNA similarity search. Nucleic Acids Res 33: W540–W543. 10.1093/nar/gki478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preiser PR, Wilson RJ, Moore PW, McCready S, Hajibagheri MA, Blight KJ, Strath M, Williamson DH. 1996. Recombination associated with replication of malarial mitochondrial DNA. EMBO J 15: 684–693. 10.1002/j.1460-2075.1996.tb0040.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick J, Grubaugh ND, Pullan ST, Claro IM, Smith AD, Gangavarapu K, Oliveira G, Robles-Sikisaka R, Rogers TF, Beutler NA, et al. 2017. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat Protoc 12: 1261–1276. 10.1038/nprot.2017.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regidor-Cerrillo J, Díez-Fuertes F, García-Culebras A, Moore DP, González-Warleta M, Cuevas C, Schares G, Katzer F, Pedraza-Díaz S, Mezo M, et al. 2013. Genetic diversity and geographic population structure of bovine Neospora caninum determined by microsatellite genotyping analysis. PLoS One 8: e72678. 10.1371/journal.pone.0072678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichel MP, Alejandra Ayanegui-Alcérreca M, Gondim LFP, Ellis JT. 2013. What is the global economic impact of Neospora caninum in cattle: the billion dollar question. Int J Parasitol 43: 133–142. 10.1016/j.ijpara.2012.10.022 [DOI] [PubMed] [Google Scholar]
- Reid AJ, Vermont SJ, Cotton JA, Harris D, Hill-Cawthorne GA, Könen-Waisman S, Latham SM, Mourier T, Norton R, Quail MA, et al. 2012. Comparative genomics of the apicomplexan parasites toxoplasma gondii and Neospora caninum: coccidia differing in host range and transmission strategy. PLoS Pathog 8: e1002567. 10.1371/journal.ppat.1002567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiff SB, Vaishnava S, Striepena B. 2012. The HU protein is important for apicoplast genome maintenance and inheritance in toxoplasma gondii. Eukaryot Cell 11: 905–915. 10.1128/EC.00029-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Wenger AM, Zehir A, Mesirov JP. 2017. Variant review with the integrative genomics viewer. Cancer Res 77: e31–e34. 10.1158/0008-5472.CAN-17-0337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- RStudio Team. 2020. RStudio: integrated development for R. RStudio, PBC, Boston. http://www.rstudio.com/. [Google Scholar]
- Saguez C. 2000. Intronic GIY-YIG endonuclease gene in the mitochondrial genome of Podospora curvicolla: evidence for mobility. Nucleic Acids Res 28: 1299–1306. 10.1093/nar/28.6.1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salunke R, Mourier T, Banerjee M, Pain A, Shanmugam D. 2018. Highly diverged novel subunit composition of apicomplexan F-type ATP synthase identified from Toxoplasma gondii. PLoS Biol 16: e2006128. 10.1371/journal.pbio.2006128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidi A, Muellner-Wong LS, Rajendran E, Tjhin ET, Dagley LF, Aw VYT, Faou P, Webb AI, Tonkin CJ, van Dooren GG. 2018. Elucidating the mitochondrial proteome of Toxoplasma gondii reveals the presence of a divergent cytochrome c oxidase. eLife 7: e38131. 10.7554/eLife.38131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheiner L, Demerly JL, Poulsen N, Beatty WL, Lucas O, Behnke MS, White MW, Striepen B. 2011. A systematic screen to discover and analyze apicoplast proteins identifies a conserved and essential protein import factor. PLoS Pathog 7: e1002392. 10.1371/journal.ppat.1002392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidik SM, Huet D, Ganesan SM, Huynh MH, Wang T, Nasamu AS, Thiru P, Saeij JPJ, Carruthers VB, Niles JC, et al. 2016. A genome-wide CRISPR screen in toxoplasma identifies essential apicomplexan genes. Cell 166: 1423–1435.e12. 10.1016/j.cell.2016.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbiss S, Silva-Franco F, Brunk B, Foth B, Hertz-Fowler C, Berriman M, Otto TD. 2016. Companion: a web server for annotation and analysis of parasite genomes. Nucleic Acids Res 44: W29–W34. 10.1093/nar/gkw292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su C, Howe DK, Dubey JP, Ajioka JW, Sibley LD. 2002. Identification of quantitative trait loci controlling acute virulence in Toxoplasma gondii. Proc Natl Acad Sci 99: 10753–10758. 10.1073/pnas.172117099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tørresen OK, Star B, Mier P, Andrade-Navarro MA, Bateman A, Jarnot P, Gruca A, Grynberg M, Kajava AV, Promponas VJ, et al. 2019. Tandem repeats lead to sequence assembly errors and impose multi-level challenges for genome and protein databases. Nucleic Acids Res 47: 10994–11006. 10.1093/nar/gkz841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25: 1105–1111. 10.1093/bioinformatics/btp120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, Van Baren MJ, Salzberg SL, Wold BJ, Pachter L. 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515. 10.1038/nbt.1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dooren GG, Tomova C, Agrawal S, Humbel BM, Striepen B. 2008. Toxoplasma gondii Tic20 is essential for apicoplast protein import. Proc Natl Acad Sci 105: 13574–13579. 10.1073/pnas.0803862105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, et al. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9: e112963. 10.1371/journal.pone.0112963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waller RF. 2000. Protein trafficking to the plastid of Plasmodium falciparum is via the secretory pathway. EMBO J 19: 1794–1802. 10.1093/emboj/19.8.1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waller RF, McFadden GI. 2005. The apicoplast: a review of the derived plastid of apicomplexan parasites. Curr Issues Mol Biol 7: 57–79. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.