

Abstract
Toxoplasma gondii is a useful model for intracellular parasitism given its ease of culture in the laboratory and genomic resources. However, as for many other eukaryotes, the T. gondii genome contains hundreds of sequence gaps owing to repetitive and/or unclonable sequences that disrupt the assembly process. Here, we use the Oxford Nanopore Minion platform to generate near-complete de novo genome assemblies for multiple strains of T. gondii and its near relative, N. caninum. We significantly improved T. gondii genome contiguity (average N50 of ∼6.6 Mb) and added ∼2 Mb of newly assembled sequence. For all of the T. gondii strains that we sequenced (RH, ME49, CTG, II×III progeny clones CL13, S27, S21, S26, and D3X1), the largest contig ranged in size between 11.9 and 12.1 Mb in size, which is larger than any previously reported T. gondii chromosome, and found to be due to a consistent fusion of Chromosomes VIIb and VIII. These data were validated by mapping existing T. gondii ME49 Hi-C data to our assembly, providing parallel lines of evidence that the T. gondii karyotype consists of 13, rather than 14, chromosomes. By using this technology, we also resolved hundreds of tandem repeats of varying lengths, including in well-known host-targeting effector loci like rhoptry protein 5 (ROP5) and ROP38. Finally, when we compared T. gondii with N. caninum, we found that although the 13-chromosome karyotype was conserved, extensive, previously unappreciated chromosome-scale rearrangements had occurred in T. gondii and N. caninum since their most recent common ancestry.
Toxoplasma gondii and its Apicomplexan relatives are highly successful animal pathogens, infecting a wide variety of warm-blooded animals, including humans and domesticated animals. T. gondii infection can lead to severe toxoplasmosis in immunocompromised individuals and in congenitally infected fetuses (Joynson and Wreghitt 2005), and is a leading cause of blindness owing to its ability to infect the eye, causing ocular toxoplasmosis (Jones and Holland 2010). T. gondii belongs to the phylum Apicomplexa, a large group of animal and human pathogens including Neospora, Eimeria, Plasmodium, and Cryptosporidium. The ease of genetic manipulation, accessibility to cellular and biochemical experiments, and well-established animal model make T. gondii an important system for studying Apicomplexan biology (Kim and Weiss 2004). Genomic analysis tools for this organism have been under development for decades. Data housed at ToxoDB (https://toxodb.org), the primary genomic repository for T. gondii genome-wide data, presently include sequence, de novo assemblies, and annotation of multiple T. gondii genomes; next-generation sequence data for an additional 60 T. gondii genomes; and draft assemblies for both Hammondia hammondi and Neospora caninum (Lorenzi et al. 2016).
Availability of a complete reference genome that contains accurate representations of all small- or large-scale structural variants is essential to have a better understanding of gene content, genotype–phenotype relationships, and the evolution of unique traits in parasites of humans and other animals. However, like all eukaryotic genomes, a substantial part of the T. gondii genome consists of repetitive elements (Matrajt et al. 1999), making gap-free de novo assembly impossible using standard first- or second-generation sequencing approaches. Even with exceptionally high coverage, these approaches fail to resolve repetitive regions or complex structural variants with repeat units that are larger than the size of the individual reads. Three of the T. gondii reference genomes in ToxoDB (Gajria et al. 2008) were constructed by combining high-quality first-generation Sanger (Sanger et al. 1977) and second-generation 454 (Roche Applied Science) sequence data, yet these genomes still have hundreds of sequence gaps of unknown sequence content and length. Assembly gaps mask repetitive regions, which can contain previously unknown protein-coding genes and additional copies of genes found in tandem gene arrays, and in some cases, they may also lead to incorrect predictions of chromosomal structure. This problem is not unique to T. gondii and other apicomplexan genomes. For example, all versions of the human genome have thousands of gaps owing to incorrect assembly of repetitive sequence (Vollger et al. 2020), including assemblies generated recently using new sequencing technologies like those applied here.
In recent years, single-molecule sequencing approaches (developed by Oxford Nanopore Technologies and Pacific Biosciences [PacBio]) have revolutionized de novo sequence assembly by enabling high-throughput generation of kilobase-sized sequence reads. These approaches have allowed for resolution of many repeat-driven sequence assembly gaps and the detection and assembly of previously intractable structural variants within species, and when combined with second-generation sequencing, data can be used to generate near-complete de novo genome assemblies with high (>99%) nucleotide accuracy. Indeed, whole-genome assemblies of several organisms including bacteria (Madoui et al. 2015; Fournier et al. 2017; Díaz-Viraqué et al. 2019), parasites (Lapp et al. 2018), plants (Schmidt et al. 2017; Michael et al. 2018), and mammals (Jain et al. 2018) using a such a hybrid approach have been reported, generating assemblies of unprecedented contiguity. Here, we apply Oxford Nanopore sequencing and de novo assembly using the MinION Platform to multiple isolates of T. gondii, F1 progeny of a cross between two canonical T. gondii strains, and one of its nearest extant relatives, N. caninum. We used this approach to improve the overall contiguity of existing T. gondii and N. caninum genome assemblies, to resolve tandem gene arrays and determine how they change in size during sexual recombination, and to perform a robust synteny analysis between T. gondii and N. caninum.
Results
De novo assembly of TgRH88 genome using Nanopore reads revises the T. gondii karyotype
The majority of the T. gondii isolates collected from North America and Europe belong to three predominant clonal lineages, types I, II, and III (Sibley and Ajioka 2008), and RH strain is a representative strain of the type I lineage (Pfefferkorn and Pfefferkorn 1976). High-molecular-weight (HMW) genomic DNA of the TgRH88 strain was extracted using an optimized protocol that was originally designed for extraction of Gram-negative bacteria and mammalian cell DNA (authored by Josh Quick; https://www.protocols.io/view/ultra-long-read-sequencing-protocol-for-rad004-mrxc57n). A 48-h sequencing run on a single flow cell yielded 648,491 reads containing 7.40 Gb of sequences for the TgRH88 genome. Although more detailed sequence metrics are described below, the sequence reads we obtained robustly aligned to the TgGT1 reference genome found at ToxoDB (Fig. 1A,B). The Canu-corrected reads were subjected to de novo assembly using Canu v1.7.1, which yielded a TgRH88 primary assembly with a size of 64.40 Mb consisting of 23 contigs. By aligning the TgRH88 assembly sequences to the ToxoDB-48_TgGT1 reference genome, we noticed that the sequences annotated as Chr VIIb and Chr VIII were parts of a single contig in our TgRH88 assembly (TgRH88_tig00000001) (Fig. 1C). This contig, which was 11.93 Mb in length, was longer than any previously reported T. gondii chromosome and suggested to us that the published T. gondii karyotype of 14 chromosomes was incorrect. Given that prior work using Hi-C chromosome conformation capture sequencing suggested a fusion between Chromosomes VIIb and VIII (Bunnik et al. 2019), we mapped the Hi-C reads from that study onto our TgRH88 de novo assembly to determine if it has similar contact counts. As shown in Figure 1D, the Hi-C data identified the position of 13, rather than 14, inter-chromosomal contact points (representing centromeres) (Bunnik et al. 2019), and an intra-chromosomal contact map across TgRH88_tig00000001 indicated that this did indeed represent a single contiguous chromosome (Fig. 1E). These parallel findings provide assembly-based evidence that sequence fragments previously referred to as distinct chromosomes (VIIb and VIII) were in fact two parts of the same chromosome. We have named this contig TgRH88_tig00000001_ChrVIII.
Figure 1.

Primary de novo assembly of TgRH88 genome using Nanopore reads revises T. gondii karyotype. (A) Bivariate plot showing a comparison of the aligned read length with the sequenced read length. (B) Bivariate plot showing a comparison of the aligned–corrected read length (log10-transformed) with the percentage identity. In this case, corrected reads refer to the method deployed by Canu using read overlap. (C) Histogram showing comparison of chromosome size between the ToxoDB-48_TgGT1 genome and TgRH88 initial long-read assembly. (D) Inter-chromosomal Hi-C contact-count heat map plotted using the TgRH88 initial long-read assembly sequence showing 13 chromosomes in the assembly. (E) Intra-chromosomal Hi-C contact-count heat map plotted using the sequence of TgRH88_tig00000001 in TgRH88 initial long-read assembly showing no aberrant signal along the contig.
The RH strain is one of the most commonly used laboratory strains given its genetic tractability and robust in vitro growth characteristics (Saeij et al. 2005), but at the genomic level, this strain has been subject to much less formal annotation compared with the strain types GT1, ME49, and VEG. However, in addition to that generated here, there are two additional RH strain de novo assemblies in public databases, one generated using Illumina technology (NCBI BioProject [https://www.ncbi.nlm.nih.gov/bioproject/] accession number PRJNA294483) (Lau et al. 2016) and the other using a hybrid approach consisting of single-molecule long-read (PacBio RS technology) and short-read (Illumina) sequencing (BioProject accession number PRJNA279557). Our Nanopore and the PacBio RS assembly were more contiguous than the de novo Illumina assembly, having higher contig N50 values (6.7 Mb for both long-read assemblies compared to ∼65 kb for the Illumina assembly) (Fig. 2A). This increase in contiguity translated into more examples of complete or near-complete whole-chromosome assemblies. For example, both long-read technologies predicted the ∼12-Mb Chromosome VIII, whereas the Illumina assembly did not, and both also resulted in a near-complete assembly of Chromosome IV in a single contig (Fig. 2B). The total sizes of the predicted nuclear-encoded genomes were similar, differing by only 0.8% (a raw difference of 513,977 bp of additional sequence present in the Nanopore assembly). Based on NUCmer alignments, ∼362 kb of this unplaced sequence is shared between the two assemblies, whereas the remaining sequences (∼70 and 2700 kb for the Nanopore and PacBio RH assemblies, respectively) were unshared.
Figure 2.

Comparisons between the current Nanopore assembly of T. gondii strain RH88 and existing long-read (using PacBio RS and Illumina technology) and short-read (Illumina only) assemblies. (A) Assembly statistics for each. (B) Circos plot of NUCmer pairwise alignments across all three assemblies for T. gondii Chromosome IV. All alignments >10,000 bp and >90% identity are shown. (C) Pairwise alignments for Chromosomes IV (top) and VI (bottom) along with locations of select tandem repeats identified either de novo (orange or blue bars on the chromosome scaffolds) or known from prior studies (above or below chromosome scaffolds). For T. gondii Chromosome IV, comparisons to both the PacBio and Illumina assemblies are shown, whereas only the long-read comparison is shown for T. gondii Chromosome VI.
To explore differences in these assemblies further, we also mapped all tandem repeats with periods >500 bp as well as select known repeats in all three RH assemblies onto NUCmer-generated pairwise sequence alignments for Chromosomes IV and VI (Fig. 2C). For Chromosome IV, the most striking difference between the long-read assemblies and that generated using Illumina is the expanded assembly at the locus harboring the well-characterized 529-bp repeat (Fig. 2C, top). For T. gondii Chromosome VI (Fig. 2C) the subtelomeric repeat arrays for SAT350 and TGR4 were larger in the Nanopore assembly compared with the PacBio assembly, as was the case for the known tandem rhoptry gene array encoding ROP38. In contrast, gene TGME49_240310 was found in a larger array in the PacBio assembly. Overall, these data indicate the utility of long-read sequencing for resolving complex tandem repeats in T. gondii, especially those that are larger than typical first- or second-generation sequence reads. Moreover, with a few minor exceptions, both long-read sequencing technologies give similar results for the size and complexity of these repeats and give results in similar estimates for nuclear chromosome and plastid genome sizes.
We also used BLAST to identify contigs harboring what appeared to be sequence derived from either of the two organellar genomes of T. gondii, the apicoplast and the mitochondrion, and compared them to the other existing RH assemblies (Fig. 2A). One contig ∼35 kb in size from each assembly was derived from the plastid genome, which is the expected size for this organellar genome (Lorenzi et al. 2016). For the mitochondrial genome, we identified two nonchromosomal contigs of ∼15 kb in size in our assembly that likely harbored at least fragments of this genome (Fig. 2A), whereas there were no contigs in the other RH assemblies with any resemblance to a mitochondrial genome (likely due to being filtered out during sequence analysis or submission). Given the complexities of the mitochondrial genome and its assembly for T. gondii (as evidenced by recent work from the Kissinger group using Oxford Nanopore technology) (Namasivayam et al. 2021), we did not perform any extensive analyses of these sequences but include them in our assemblies submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA638608 with the annotation “location = mitochondria.”
De novo assembly of multiple T. gondii strains, their sexual recombinants, and N. caninum
To determine whether the Chr VIIb/VIII fusion was unique to TgRH88 or was also present in other isolates, we sequenced and assembled genomes of TgME49, TgCTG, TgME49×TgCTG F1 progeny (CL13, S27, S21, S26, and D3X1), and N. caninum Liverpool strain (Table 1). Aligning the reads against their most relevant “Reference” genome in ToxoDB (based on species and then closest genotype) revealed that 96% of the T. gondii reads and 85% of the N. caninum reads could be mapped (Supplemental Tables S2, S3). Assembly characteristics for all strains and species can be found in Supplemental Table S4, including chromosome and organellar genome sizes. The final polished and scaffolded assemblies (see Methods) of TgRH88, TgME49, or TgCTG consisted of 13 chromosome contigs/scaffolds and varying numbers of unplaced fragments, with an average total size of ∼64.8 Mb (Table 2). The polished N. caninum Liverpool assembly was composed of 58 contigs, showing a cumulative size of 62.1 Mb (Table 2). As reported in Table 2, with one exception (II×III F1 progeny S26), all the T. gondii final assemblies were composed of 23 to 59 contigs, representing a 43- to 109-fold reduction in the number of contigs in comparison to the ToxoDB-48_TgME49 assembly (2511 contigs) (Table 2). We performed one-to-one mappings between our Nanopore chromosome-sized contigs and all aligning contigs from the ToxoDB-48_TgME49 assembly (Supplemental Fig. S1A) using minimap2 and found that the most significant contribution to overall genome structure was the elimination of nearly all of the breaks within the ToxoDB-48_TgME49 scaffolds that are indicated within that assembly as strings of N's at least 100 bp long (Supplemental Fig. S1A). We also compared our de novo assembly sequences to their cognate reference sequences and identified 42 regions of at least 10,000 bp in size that were unique to our de novo assemblies (summing to 926 kb in total; yellow boxes in Supplemental Fig. S1A). To assess genome assembly completeness, we used BUSCO analysis on the polished TgRH88, TgME49, and TgCTG assemblies and compared the results to a similar analysis for the unpolished assemblies. This analysis, which counts the number of single-copy orthologs unambiguously identified in a genome assembly, found that for 215 such loci 88% of the complete genes were recovered from the polished, long-read-based assemblies, whereas 23.3%–54% were identified in the unpolished assemblies (Table 3).
Table 1.
Description of the Toxoplasma gondii and Neospora caninum strains sequenced in this study

Table 2.
Metrics of long-read assemblies and the reference genomes after polishing
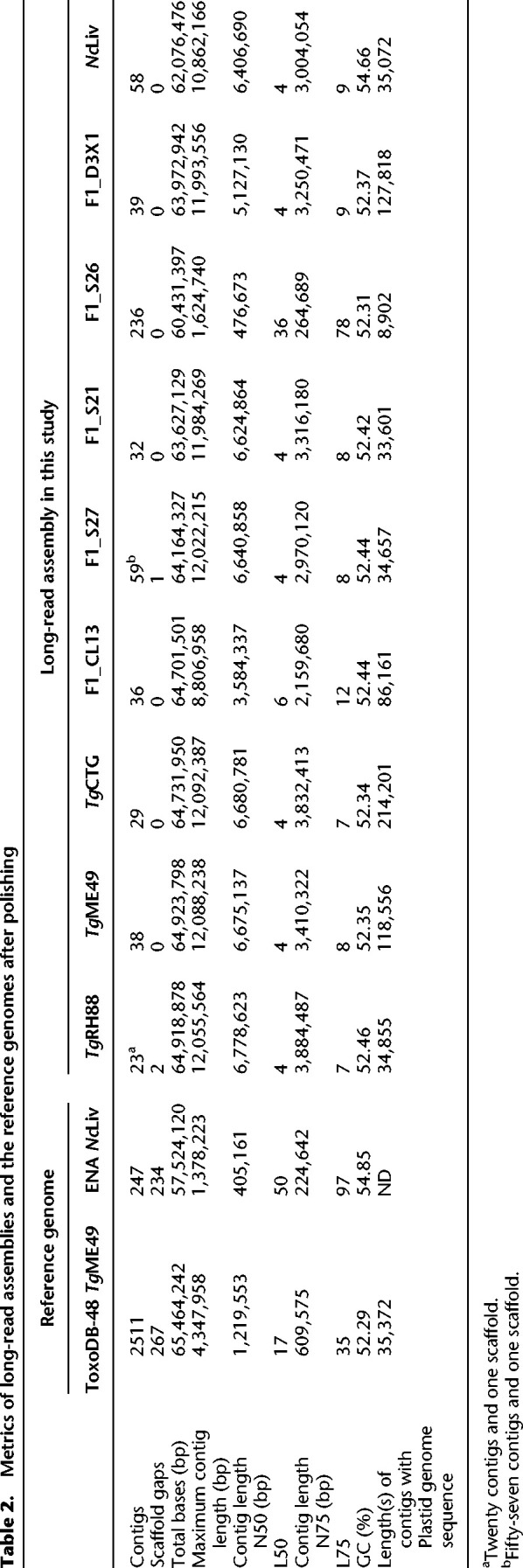
Table 3.
Metrics of the long-read assemblies before and after polishing

To assess the structural correctness of all of the long-read assemblies, we aligned our T. gondii assembly sequences to the reference genome using NUCmer. As can be seen in Supplemental Figure S1B, all of the T. gondii long-read assemblies showed strong collinearity with their corresponding reference genomes, barring a small number of putative inversions. Consistent with our finding for TgRH88 primary assembly, the Chr VIIb/VIII fusion was observed in each of our T. gondii assemblies (TgME49 is represented in Fig. 3A, red box; Supplemental Fig. S1A,B). To further confirm this observation, we aligned our TgME49 corrected reads back against the TgME49 long-read assembly and found an average read depth of 40× for the entirety of TgME49_tig00000001_ChrVIII, and 37× for the “breakpoint” (TgME49_tig00000001_ChrVIII: 5,090,422 bp) of Chr VIIb and Chr VIII, indicating that this was unlikely owing to an assembly error (Fig. 3B). We then mapped the corrected Nanopore reads again to the ToxoDB-48_TgME49 reference genome, and the alignments showed that all of the reads that were mapped either to the end of Chr VIIb or to the beginning of Chr VIII spanned the gap between the two chromosomes, with average coverage of 105× (Fig. 3C). Such a high-coverage link between reads aligning to chromosome ends was not present in any other chromosome pair (e.g., between Chromosomes IX and X) (Fig. 3D).
Figure 3.

Long-read assembly identifies 13 chromosomes in the T. gondii genome from multiple strains. (A) Dot plot showing the comparison of the TgME49 long-read assembly and the ToxoDB-48_TgME49 genome. Red box shows that the Chromosomes VIIb and VIII in the ToxoDB-48_TgME49 genome are fused in a single contig, TgME49_tig00000001_ChrVIII, in the TgME49 long-read assembly. (B) Coverage of the “breakpoint” (TgME49_tig00000001_ChrVIII: 5,090,422 bp, indicated by a vertical red line) of Chromosomes VIIb and VIII with 37 Nanopore reads in the TgME49 long-read assembly. (C) Coverage of the edges (indicated by a vertical red line) of Chromosomes VIIb and VIII with 105 Nanopore reads mapped to the ToxoDB-48_TgME49 genome. (D) Nanopore reads mapping to the end of Chromosomes IX and X in the ToxoDB-48_TgME49 genome assembly, showing that Nanopore reads only map to the end of each chromosome and do not span the junction between these chromosomes (indicated by a vertical red line). (E) Inter-chromosomal Hi-C contact-count heat map plotted using the TgME49 initial long-read assembly sequence showing 13 chromosomes in the assembly. (F) Intra-chromosomal Hi-C contact-count heat map plotted using the sequence of TgME49_tig00000001 in the TgME49 initial long-read assembly showing no aberrant signal along the contig.
This observation of a fusion between Chr VIIb and Chr VIII in T. gondii ME49 was in agreement with the observations in our TgRH88 assembly described above (Fig. 1C–E), and we also mapped Hi-C data (Bunnik et al. 2019) to our TgME49 long-read assembly to validate this finding in parallel. The resulting inter-chromosomal contact-count map revealed 13 chromosomes in the TgME49 long-read assembly (instead of 14) by showing that each chromosome showed a single centromeric interaction with each other chromosome (Fig. 3E). As we found for TgRH, we confirmed that TgME49_tig00000001_ChrVIII was a complete single chromosome (Fig. 3F; Supplemental Fig. S2). The intra-chromosomal contact-count map of TgME49_tig00000001_ChrVIII showed a strong and broad diagonal and no aberrant signal along the contig or at the “breakpoint” of Chr VIIb and Chr VIII (Supplemental Fig. S2B). Similar patterns were also observed in our TgCTG, S27, and S21 assemblies (Supplemental Fig. S2B). Collectively, these data show the T. gondii karyotype has been incorrectly calculated and contains 13, rather than 14, chromosomes. We refer to this fused chromosome as Chr VIII in our assembly and have eliminated Chr VIIb.
The TgCTG assembly had chromosome-scale resolution, in which 13 contiguous sequences (contigs) corresponded to the 13 chromosomes. However, Chromosome VIIa in the TgME49 assembly and Chromosome XI in the TgRH88 assembly were spread across two contigs. Hi-C data have historically been used to improve genome assemblies on the basis of contact frequency, depending strongly on one-dimensional distance (Dudchenko et al. 2017). That is, Hi-C alignment to contigs in the correct order and orientation would reveal the canonical intra-chromosomal pattern of enriched interactions along the diagonal (where one-dimensional genomic distance between bins is the smallest). By using pre-existing Hi-C reads from T. gondii strain ME49 (Bunnik et al. 2019) and mapping them to our de novo genome assemblies, we were able to determine the order and orientation of the contigs for Chromosomes Chr VIIa and XI in ME49 and RH88, respectively. These changes were incorporated before submission of the assemblies to GenBank (under BioProject accession number PRJNA638608).
Long-read assembly detects structural rearrangements in the T. gondii genome
We aligned the final assemblies of the eight T. gondii strains to the reference genomes and searched for large-scale structural variants using MUMmer (Supplemental Fig. S1B). Consistent with the data shown in Supplemental Figure S1B, most contigs in the long-read assemblies were collinear with the chromosomes in the ToxoDB-48 genomes. We did observe a 15.7-kb inversion on Chr III in our TgRH88 assembly, which was absent in TgME49, TgCTG, or any F1 progeny assembly (Fig. 4A). We also detected another ∼20-kb inversion on Chr XII, which was present in TgME49, TgCTG, and the F1 progeny assembly, but not in the TgRH88 assembly (TgME49 is represented in Fig. 4B).
Figure 4.

Long-read assembly reveals previously unknown inversions and the centromere location on Chr IV in T. gondii. (A) Inversion in the RH88 long-read assembly on Chromosome III relative to the ToxoDB-48_TgGT1 assembly. (B) Inversion in the ME49 long-read assembly on Chromosome XII relative to the ToxoDB-44_TgME49 genome. (C) Dot plot comparison of the TgRH88 long-read assembly and the ToxoDB-48_TgGT1 genome showing a 429.3-kb inversion at 2,096,529–2,525,795 bp on Chr IV. (D) Intra-chromosomal Hi-C contact-count heat map plotted using the sequence of tig00000014 in TgRH88 long-read assembly showing a clear centromere signal at position 2.2–2.3 Mb. (E) ChIP-on-chip signal of centromeric histone 3 variant (CenH3) (Brooks et al. 2011) plotted using the TgRH88 long-read assembly as coordinate.
Centromeres for 12 of the 13 T. gondii chromosomes have been identified using chromatin immunoprecipitation coupled with DNA microarrays (ChIP-on-chip) of centromeric and peri-centromeric proteins, but locations of centromere of Chr VIIb and Chr IV remain unknown (Brooks et al. 2011; Gissot et al. 2012). For Chr VIIb, no hybridization of the centromeric probe to the genomic chip was detected in the ChIP-on-chip assay (Brooks et al. 2011), which could be explained by our observation that Chr VIIb and Chr VIII are a single chromosome, and the centromere of this large chromosome appears to be in the center of this “fused” chromosome, in an area that was previously thought to be the beginning of Chromosome VIII (Fig. 1E). For Chromosome IV, two inconsecutive peaks of hybridization were detected at positions 2,501,171–2,527,417 bp on Chr IV and 1–9968 bp in the unplaced contig AAQM03000753 in the TgGT1 genome based on published ChIP-on-chip data (Brooks et al. 2011). Our TgRH88 assembly successfully relocated the sequences in AAQM03000753 into Chr IV and revealed a 430.9-kb inversion event at 2,096,529–2,527,423 bp on Chr IV relative to the ToxoDB GT1 reference genome (Fig. 4C). This inversion was unlikely to be owing to an assembly error because it was shown in all of our T. gondii assemblies and was supported by alignment of both Canu-corrected and raw reads spanning the boundaries of the inversion in the TgRH88 assembly. When we mapped the ChIP-on-chip data obtained from Brooks et al. (2011) to our TgRH88 assembly (after remapping the probe sequences to our TgRH88 assembly), we resolved the Chr IV centromere to a single significant signal peak at 2.20–2.23 Mb on Chr IV (Fig. 4E; Supplemental Fig. S3). This finding was also supported by published Hi-C data realigned to our Nanopore assemblies, because the intra-chromosomal contact count map showed a clear inter-chromosomal contact signal at 2.20–2.30 Mb on Chr IV in the TgRH88 assembly (Fig. 4D). Collectively, our data not only resolved the molecular karyotype of T. gondii but also resolved the precise location of the Chr IV centromere.
Long-read assembly adds new sequences to the T. gondii reference genome
As shown in Figure 1C and Supplemental Table S5, each chromosome-sized contig of our long-read de novo assemblies was longer than its cognate chromosome of the ToxoDB-48 T. gondii reference genome, and for TgRH88, TgME49, and TgCTG, we were able to assemble between 1.7 and 3.9 Mb of previously unlocated and/or unassembled sequence to the chromosomes of these assemblies. These new sequences were scattered across the genome and filled in nearly all of the sequence gaps found in the reference genome. The new sequences added by the long-read assemblies extended the subtelomeric regions of the chromosomes in the T. gondii reference genome. Although four chromosomes of the ToxoDB-48_TgME49 genome contained no telomeric repeat and seven were missing one of the telomeric repeats, all of the chromosome contigs in the TgME49 long-read assembly were assembled up until both telomeric caps (Supplemental Table S5). Both telomeres were found in 12 out of the 13 chromosomes in the TgCTG long-read assembly, and one chromosome contig lacked one of the telomeric repeats, whereas only five chromosomes in the ToxoDB-48_TgVEG genome contained one telomere, and no telomeric repeat was found in the rest of the chromosomes (Supplemental Table S5). Similarly, both telomeres in seven chromosomes and one telomere in six chromosomes were resolved in the TgRH88 long-read assembly, whereas there were only two chromosomes in ToxoDB-48_TgGT1 genome that contained one telomere (Supplemental Table S5).
The bulk of the remaining new sequence was owing to tandem arrays of sequence (both coding and noncoding). For example, two repetitive gene sequences are used for high-sensitivity detection of T. gondii in tissue and environmental samples, the B1 gene (Burg et al. 1989) and the so-called “529-bp repeat” (Reischl et al. 2003; Edvinsson et al. 2006). The precise copy number for these genes has been difficult to determine using first- and second-generation sequencing technologies and/or molecular biological experiments like Southern blotting. Therefore, we used a curated BLASTN approach to quantify copy number for each of these sequences across our respective Nanopore assemblies. As shown in Figure 5B, copy number for the B1 gene was significantly higher in our Nanopore assemblies compared with existing ToxoDB assemblies. However, the copy number for this gene was lower than that predicted in the literature, ranging between nine and 19 tandem copies depending on the strain (Fig. 5B), compared with quantitative blotting-based estimates of 35 (Burg et al. 1989). Copy number at this locus was stable, in that for all of the queried II×III F1 progeny, the copy number for each was identical to the parent from which it obtained that chromosomal segment. In contrast to the B1 locus, the “529-bp repeat” locus varied significantly between isolates and these same F1 progeny. The copy number ranged from 85 to 205, and the copy number at this locus for all F1 progeny varied independently of the underlying genotype for that region (Fig. 5A, white letters and green/blue). The size of this genome expansion is best illustrated by the whole-chromosome alignment shown in Figure 5E comparing the T. gondii ME49 529-bp repeat locus in the version 48 assembly on ToxoDB to our Nanopore assembly (Fig. 5E). It is likely not a coincidence that the 529-bp repeat locus occurs near a sequence assembly gap, and our long-read assembly closed this gap (see below) (Fig. 5E,F), giving the most accurate estimate of 529-bp repeat copy number in any T. gondii strain, which again varies compared with estimates in the literature (ranging from 200 to 300 copies) (e.g., Reischl et al. 2003; Edvinsson et al. 2006). Regardless, similar to tandem gene arrays discussed in Figure 6, it appears that even noncoding repeats like the B1 gene and the 529-bp repeat can also change in number irrespective of whether there is sexual recombination. Although the 529-bp repeat and B1 gene are found in a single locus, other tandem repeats like TgIRE and SAT350 are found across multiple subtelomeric genomic locations (Echeverria et al. 2000; Clemente et al. 2004). We mapped these sequences across the genome in all of our sequenced T. gondii strains and again found different results depending on the queried locus. For the TgIRE sequence, a 1919-bp repeat, chromosome-wide copy number in our sequenced strains was approximately two times that found in the reference sequences (Fig. 5C), but the overall copy number was similar across all of our Nanopore-derived sequences. The SAT350 sequence was also found at a much higher copy number in our Nanopore-assembled chromosomes compared with the reference sequences (Fig. 5D) but was more variable in the F1 progeny clones. This could be owing to changes during sexual recombination as for the 529-bp repeat above or to differences in sequence coverage for the F1 progeny. Regardless, our approach has provided better resolution of copy number for four well-known tandem repeat sequences and allowed us to determine which may be more susceptible to copy number change. Moreover, unappreciated strain differences in copy number at these loci may adversely affect interpretation of PCR-based detection assays, especially those using more quantitative methods.
Figure 5.

Long-read sequence assemblies precisely resolve canonical repeat sequences and identify additional expansions at gene-harboring loci. (A–D) Estimated copy number for Nanopore assemblies and existing genome assemblies on ToxoDB (“v48”) for T. gondii strain types I, II, and III and II×III F1 progeny. In all cases, Nanopore assemblies identified higher numbers of each repeat locus. In the F1 progeny, B1 gene copy number tracked directly with the genotype (type II or III) at that locus (A), whereas these same F1 progeny harbored unique numbers of 529-bp repeat copies, all of which were not only distinct from their respective genotypes of origin but distinct from one another (B). The TgIRE and SAT350 repeats also were better resolved in our Nanopore assemblies (C,D), although determining genotype of the corresponding region is not possible because these repeats are found at multiple locations throughout the genome. (E) Whole-chromosome alignment focused on the 529-bp repeat region for the v48 ToxoDB assembly (bottom) and our Nanopore-based assembly (top). Expansion of the known genome sequence at this locus in the Nanopore sequence compared with the ToxoDB assembly is clear, as well as consistent with our identification of approximately 140 previously unknown 529-bp repeats in the ME49 genome. (F) Alignment and annotation of repeat sequences of ME49 v48 ToxoDB Chromosome IV and that from our polished Nanopore assembly. Gray bars with red borders indicate mapping regions ≥10,000 bp determined using NUCmer, whereas orange boxes with blue borders indicate tandem repeats with period sizes ≥500 bp and at least two copies. Bars that appear orange are larger than those that are only blue. Incorrect inversion on the right arm of Chromosome IV in the ToxoDB assembly is evident, as is the more accurately resolved 529-bp repeat locus, which was likely a cause for the inversion in standard assemblies from multiple strains.
Figure 6.

Long-read assembly resolves duplicated locus structure in T. gondii genome. (A) Two unresolved scaffold gaps on Chr Ia in the ToxoDB-48_TgME49 genome span a 17.5-kb tandem repeat containing multiple copies of ROP4 and ROP7. The ROP4/7 gaps are closed by the TgME49 long-read assembly (TgME49_tig00000028), revealing a tandem array of five copies of this gene in the order shown. (B) BLASTN alignment of the ROP4/ROP7 coding sequence in the ToxoDB-48_TgME49 genome (upper panel) and the TgME49 long-read assembly (lower panel). (C) Copy number determination at six canonical tandem gene arrays across eight T. gondii strains and one N. caninum strain. Data from CL13, S27, S21, and S26 show that copy number can change during sexual recombination because the copy number in these F1 progeny clones does not match copy number in either parent. (D–F) Whole-chromosome alignments between ME49ToxoDB-48 and our Nanopore assemblies at loci harboring tandem gene arrays. Gray boxes with red borders indicate one-to-one mapping regions ≥10,000 bp determined by NUCmer, and orange/blue boxes are as described in Figure 5. Black bars indicate size of select tandem repeats in the ToxoDB and Nanopore assemblies.
With respect to Chromosome IV, mapping the 529-bp repeat to our Nanopore assemblies while comparing them to reference genomes in ToxoDB identified what appears to be an inversion in the right arm of Chromosome IV (Fig. 5F). This inversion was present in the T. gondii VEG and GT1 reference genomes, suggesting that it was owing to systematic assembly error. This inversion in the ToxoDB reference genome is flanked by multiple tandem repeats (identified using tandem repeats finder) (Fig. 5F, blue/orange boxes). In addition, the 529-bp repeat can be found at the end of each inversion (Fig. 5F, “529_rpt”) in the ToxoDB chromosome, but in our Nanopore assembly, the 529-bp repeat cluster is only found in one location (where it is greatly expanded compared with those in the ToxoDB reference). These data provide strong evidence that Chromosome IV is incorrectly assembled in multiple ToxoDB reference genomes owing to misassembly of the 529-bp repeat locus, and our Nanopore assembly has resolved this discrepancy in multiple T. gondii genomes.
Long-read assembly resolves tandem duplicated locus structures in the T. gondii genome
Our long-read assembly closed nearly all of the gaps in the T. gondii and N. caninum genomes (Table 2; Supplemental Table S6). Furthermore, many unplaced sequences in the T. gondii reference genome were assembled into contigs in our assembly. For instance, the unplaced contig KE140372 in the ToxoDB-48_TgME49 genome, which was 2194 bp in length and contained a sequence encoding a rhoptry protein 4 paralog, was assembled in TgME49_ tig00000028_ChrIa in our TgME49 assembly. Unplaced contigs in the ToxoDB-48_TgGT1 genome, AAQM03000823 and AAQM03000824, were assembled in TgRH88_tig00000013_ChrIII in our TgRH88 assembly.
We have had a long-standing interest in variation at tandemly expanded gene clusters and how this affects T. gondii virulence, which was first spurred by our identification of ROP5 gene cluster as being a critical determinant of virulence in the mouse (Reese et al. 2011). Our genome-wide analyses of copy number variation across multiple strains and species (Adomako-Ankomah et al. 2014) identified 53 putative tandemly expanded gene clusters in T. gondii (shown in Supplemental Table S6), some of which (such as MAF1) have been shown to be important in host–pathogen interactions (Adomako-Ankomah et al. 2016). In that initial study, we used sequence coverage to infer copy number, and we were eager to refine this analysis using the long-read de novo assemblies presented here.
The gap closure enabled us to determine the number, order, and orientation of T. gondii duplicated loci, including ROP7, ROP5, ROP38, MIC17, MAF1, and TSEL8. The ROP7 locus is represented in Figure 6A, where we identified two unresolved scaffold gaps on Chr Ia in the ToxoDB-48_TgME49 genome (black bars), and these gaps marked the site of ROP4/7 locus. This entire region was spanned by a single contig, TgME49_tig00000028_ChrIa, in our TgME49 assembly (Fig. 6A blue bar). Aligning the ROP7 genomic sequence (ToxoDB: TgME49_295110) against TgME49_ tig00000028_ChrIa using BLASTN revealed three copies of the ROP7 repeat, whereas only one was predicted in the ToxoDB-48_TgME49 genome (Fig. 6A,B). Although the ToxoDB-48_TgME49 genome identified one copy of the ROP4 gene (GenBank: EU047558.1), our TgME49 assembly showed that two copies of ROP4 exist in this locus, one of which was found between the first and the second copy of ROP7 (Fig. 6B). To validate this finding, we identified 12 individual Canu-corrected reads that spanned this entire tandem array, and each one that we examined provided evidence for three copies of ROP7 and two copies of ROP4 arranged in the order ROP4_1-ROP7_1-ROP4_2-ROP7_2-ROP7_3 (Supplemental Table S6). The copy number and copy order of other known tandem locus expansions (taken from the supplemental table by Adomako-Ankomah et al. 2014) in the strains we sequenced were identified and are listed in Supplemental Table S6. After conducting these analyses genome-wide and across multiple assemblies, we observed changes in copy number at some of these loci when we compared the parental (TgME49 or TgCTG) and progeny (CL13, S27, S21, S26, and D3X1) assemblies (Fig. 6C). For example, although the ROP5 locus had nine copies in our TgME49 assembly and four in our TgCTG assembly, it harbored six copies in the F1 progeny S27 and S21 and seven in S26 (Fig. 6C). There was an array of seven tandem copies of MIC17 in the TgME49 and TgCTG assemblies, whereas it was present in six copies in the S26 assembly (Fig. 6C). These data indicated that changes in copy number and order at tandem gene arrays can occur during sexual recombination. Moreover the ease with which a Nanopore assembly can be generated and assembled provides a new way to assess the occurrence and impact of acute changes in copy number that occur during asexual and sexual propagation of parasites like T. gondii. Whole-chromosome alignments are shown for Chromosomes Ia, VI, and XII to further illustrate the increase in sequence size at the chromosome level for loci like ROP7, ROP38, and ROP5, as well as other known tandem gene arrays (Fig. 6D–F).
We performed a similar analysis genome-wide for the remaining previously identified tandem expansions (Supplemental Table S7; Adomako-Ankomah et al. 2014) and were able to further curate this list of putative repetitive loci. Although in some cases our predictions from shotgun sequence read coverage was similar to that predicted using Nanopore assembly (as for those genes described in Fig. 6, as well as many others in Supplemental Table S7), for some, we were able to determine that there was in fact no evidence for the presence of a tandem gene array at that locus. For example, expanded locus 9 (EL9; TGME49_319090), EL19 (TGME49_204560), and EL27 (TGME49_264420) are all likely to be single-copy genes based on our Nanopore assemblies (Supplemental Table S7), even though based on sequence coverage alone they ranged in predicted copy number between 15 and 60 (Supplemental Table S7; Adomako-Ankomah et al. 2014). For these three genes, our prior overestimation of copy number using sequence coverage was owing to the presence of low complexity sequence (e.g., short tandem repeats) across the single-copy gene rather than being owing to actual tandem expansion (all three have stretches of low-complexity/repetitive sequence; ToxoDB). Overall, our current assemblies refine and further curate this locus list, providing the most accurate estimate to date of which loci encode tandem gene arrays and how their copy number varies across strain and species.
As described above, all of our sequence assemblies increased the size of the contiguous assemblies by 1–3 Mb. Although some of this new sequence is most certainly derived from gene-poor regions containing simple tandem repeats (and some of these regions are annotated as yellow boxes in Supplemental Fig. S2), the relatively high gene density of the T. gondii genome led us to hypothesize that some of this “new” sequence should be derived from gene sequences that were previously masked by assembly gaps. Therefore we used BLASTN to identify genome expansions in our Nanopore assembly relative to the v48 sequence on ToxoDB specifically using all available predicted genes as query sequences. Overall, we identified 62 gene-containing loci that were at least 10 kb larger in our Nanopore assembly compared with ToxoDB v48, representing 1.2 Mb of sequence. These expansions are represented in Supplemental Figure S4 as yellow blocks and are shown along with known tandem gene arrays (red blocks) and existing sequence gaps (black lines). Well-known tandem gene arrays that are collapsed in first- and second-generation sequence-based assemblies like MAF1 and ROP5 were identified in this analysis (Supplemental Fig. S4), confirming the accuracy of the approach. What was unexpected was the unequal distribution of these “expansions” in our genome-wide analysis across chromosomes (e.g., cf. Chromosomes XI and X). In addition to these gene-containing loci, we have identified all tandem repeats with a period size >500 bp in our de novo assembly of T. gondii ME49 and estimated their copy number in our de novo assemblies of T. gondii RH88 and CTG (Supplemental Table S8).
Standard error correction methods for tandem gene arrays fail to remove extensive homopolymeric repeats that lead to artifactual pseudogenization
Error correction using Pilon is a common practice after generation of single-molecule long-read assemblies because the overlap-based correction used by assemblers like Canu fails to correct systematic errors in the data. For Nanopore R9 flow cells, homopolymer runs as short as 3 bp are often truncated in the consensus (e.g., Bowden et al. 2019), leading to extensive artificial gene pseudogenization. As described above, a variety of error correction methods can eliminate many of these systematic errors leading to improved protein coding gene annotations (Table 3). To examine this in greater detail, we used TBLASTN to align T. gondii ME49 (version 48) protein sequences for all single-exon genes to the ToxoDB reference genome, the primary Canu assembly, and to our Pilon-corrected assembly. As shown in Figure 7A, the raw Canu assembly had only four full-length identical sequence hits to single-exon genes, whereas the Pilon-corrected assembly predicted nearly all of the query single-exon genes with 100% identity and 100% sequence coverage (1054/1121; 94%) (Fig. 7A). The effectiveness of Pilon error correction is shown for two single-exon, single-copy genes (ROP16 and ROP18) (Fig. 7B), in which the coding sequence had numerous frameshifts causing fragmented mapping in the raw assembly but resolved to a single gene with 100% identity and sequence coverage after Pilon correction. In contrast to these single-copy genes, we found that Pilon-based error correction performed much more poorly at multicopy loci like those encoding ROP5 and ROP38. As shown in Figure 7, C and D, many of the predicted ROP38 and ROP5 coding sequences are still highly fragmented after Pilon error correction, presumably owing to an inability of Pilon to assign enough reads to each copy to correct what are mostly homopolymeric repeat errors. We wrote a custom Perl script (provided in the Supplemental Code) to correct remaining homopolymer errors in tandem gene arrays (using Illumina sequence read alignments to either extend homopolymers up to 10 bp or truncate by a single base pair) and found that this eliminated many of the artifactual pseudogenes for the tandem gene expansions at the ROP5 and ROP38 loci (see Fig. 7C,D, bottom). Note that unlike single-copy genes where Canu plus Pilon correction was sufficient to correct them (Fig. 7B), we only eliminated these likely artifactual pseudogenes after running our supplemental correction scripts (for specific details about the correction script, see Methods).
Figure 7.

Error polishing with Pilon is effective for most single-copy genes, but resolution of tandem gene expansion errors requires supplemental correction. (A) We identified 1121 single-exon genes with predicted proteins that mapped with 100% identity and 100% coverage using TBLASTN against the ToxoDB-48 ME49 genome. Then, we mapped these against the raw assembly generated by Canu, the polished assembly generated by four rounds of Pilon, and after four rounds of supplemental error correction. Pilon error correction was sufficient for perfect mapping of 94% of the query single-exon genes (compared with only 0.4% for the raw Canu assembly), and supplemental error correction only increased this mapping percentage slightly. (B) Plots representing TBLASTN analysis of protein sequences from two single-copy genes showing the improved mapping achieved by Pilon-based error correction. Mapping identity is indicated by the color of the box representing the alignment. (C,D) Plots representing protein-coding sequences from the ROP5 (C) or ROP38 (D) gene mapped using TBLASTN against the raw Canu-only assembly, the Pilon-corrected assembly, and the region corrected using our supplemental approach tailored to tandem gene arrays. Both loci have multiple pseudogenes in the Canu-only and Canu-plus Pilon assemblies, but many of these errors are removed upon supplemental correction. The presence of a pseudogene in the ME49 ROP5 locus has been predicted before based on direct sequencing, suggesting that this may represent the most accurate version of the ME49 ROP5 locus sequenced to date.
Long-read assembly revises N. caninum karyotype and shows its lack of synteny with T. gondii
The comparison of TgRH88 long-read assembly and annotation with the ToxoDB-48_TgGT1 genome revealed a high level of collinearity between the two genomes with no large-scale rearrangement between chromosomes (except for the Chr VIIb/VIII fusion) (Fig. 8A), whereas a large number of chromosomal translocations and inversions were observed in the NcLiv long-read assembly with respect to ENA_NcLiv genome (Fig. 8B). For instance, the NcLiv_tig0000052 in NcLiv long-read assembly contained a portion of Chr IX and a portion of Chr X in the current N. caninum genome (Fig. 8B). A portion of NcLiv_tig00000006 in NcLiv long-read assembly was mapped to the current N. caninum Chr X, whereas the remainder of NcLiv_tig00000006 was mapped to Chr IX and a region of it was inverted (Fig. 8B). In addition to this, some chromosomal regions in the NcLiv long-read assembly did not show any synteny with the ENA_NcLiv genome (Fig. 8B). Similar chromosomal rearrangement patterns were observed in other N. caninum strains (as described in the cosubmitted paper [Berná et al. 2021]).
Figure 8.

Long-read assembly reveals N. caninum karyotype and its synteny with T. gondii. (A) Circos plot showing high synteny between the TgRH88 long-read assembly and the ToxoDB-48_TgGT1 genome. (B) Circos plot showing the chromosomal translocations and inversions in NcLiv long-read assembly compared with the ENA_NcLiv genome. (C) Circos plot showing the syntenic relationship between TgRH88 and NcLiv long-read assembly.
Although previous studies (Reid et al. 2012; Lorenzi et al. 2016) showed that the current T. gondii and N. caninum reference genomes were highly syntenic, the comparison of TgRH88 long-read assembly with the NcLiv long-read assembly showed smaller blocks of synteny in most regions between the two genomes (Fig. 8C). Many individual contigs of the NcLiv long-read assembly were shown to be mapped to multiple chromosome contigs in the TgRH88 long-read assembly. For example, a 3.26-Mb region and a 4.43-Mb region of TgRH88_tig00000006 of the NcLiv long-read assembly were in synteny with regions on TgRH88_tig00000001 and TgRH88_tig00000003 of TgRH88 long-read assembly, respectively (Fig. 8C). Moreover, some regions of NcLiv long-read assembly (e.g., NcLiv_tig00007783: 2,000,000-2,500,000 bp) show no synteny with the chromosomes of TgRH88 genome (Fig. 8C). Overall, our long-read assembly revealed a new and accurate N. caninum karyotype and revised the genomic synteny between the two closely related species, T. gondii and N. caninum.
Discussion
The first wave of genome sequencing using first- and second-generation sequencing technologies revolutionized our ability to link phenotype to genotype in diverse strains of T. gondii and its near relatives. Sequence from multiple isolates have been made publicly available and hosted on ToxoDB and were outlined in a recent publication (Lorenzi et al. 2016). These genomes, although of great use, were expectedly incomplete owing to the presence of hundreds to thousands of sequence assembly gaps, depending on sequencing depth and methodology. Recent years have experienced the impact of so-called “third-generation” technologies that have revolutionized the speed, cost, and efficacy of de novo sequence assembly and provide a new means to significantly improve existing sequence assemblies. This technology provides a unique opportunity to greatly improve the assembly of the T. gondii genome, particularly at repetitive loci, which are known to encode diversified secreted effectors (Adomako-Ankomah et al. 2014, 2016).
Our work revises the T. gondii and N. caninum karyotypes by identifying a previously unappreciated fusion between segments previously thought to be distinct chromosomes (VIIb and VIII). Throughout the history of the T. gondii genome, the number of linkage groups has been a moving target and has become more precise as new mapping and sequencing technologies have become available. Initial genetic mapping experiments and HMW Southern blotting identified 11 linkage groups (Sibley and Boothroyd 1992), whereas a denser map in later studies identified 13 linkage groups (Su et al. 2002). This particular map was still not fully representative of the T. gondii karyotype, as markers known to be on Chromosome VI were included in linkage group X, Chromosome XII was split into two linkage groups (“Unknown 1” and “Unknown 2”), and Chromosome XI was missing (Su et al. 2002). A clearer picture emerged when this linkage map was integrated with shotgun sequence assembly data using first-generation sequencing, leading to a consensus karyotype of 14 chromosomes. In all three of these assemblies (from strains GT1, ME49, and VEG), Chromosomes VIIb and VIII always assembled into separate contigs. However, in some studies significant genetic linkage between markers on former Chromosomes VIIb and VIII was observed (e.g., see the discussion by Khan et al. 2005 and supplemental Fig. S2 by Khan et al. 2014), but the lack of any contiguous assemblies for these two genome fragments (as found on ToxoDB as well as outlined by Lorenzi et al. 2016) led to continued acceptance of the 14 chromosome model. It was only until recently where reports using chromosome-capture technologies (“Hi-C”) (Bunnik et al. 2019) suggested a fusion between the VIIb and VIII genome fragments, and this was the first study to propose a 13-chromosome karyotype that is most consistent with our nuclear genome assembly. The reason for the consistent prediction that fragments VIIb and VIII were distinct in T. gondii was clearly owing to repetitive sequences near the breakpoint (hence the consistent and artificial fragmentation of this chromosome across multiple de novo sequenced strains using both first- and second-generation sequencing technology) (Lorenzi et al. 2016). This karyotype that is robustly supported by our assemblies are consistent with existing Hi-C data (Bunnik et al. 2019) and existing genetic linkage maps from F1 progeny derived from type IxII and IxIII crosses (Khan et al. 2005; Khan et al. 2014). Similar results were obtained in a cosubmitted manuscript appearing in this same issue (Berná et al. 2021).
Tandem gene expansion followed by selection-driven diversification provides a means for genome innovation and neofunctionalization, and this has occurred at multiple loci in the T. gondii genome (Adomako-Ankomah et al. 2014, 2016; Blank and Boyle 2018). These loci can differ in copy number between strains, including those belonging to the same clonal lineage (e.g., MAF1 and ROP5 copy number differs between “type I” strains GT1 and RH [Adomako-Ankomah et al. 2014, 2016], and MAF1 copy number differs between “type III” strains CTG and VEG [Adomako-Ankomah et al. 2016]). Although it is generally assumed that copy number changes can occur during errors in DNA replication, this could occur with different frequency during sexual versus asexual propagation. Here we show that some loci can change in gene number and content during sexual recombination by sequencing multiple F1 progeny from a well-defined cross between type II and type III T. gondii. Specific changes in copy number and/or content at specific loci could have a significant impact on the overall virulence phenotypes of individual F1 progeny that emerge from natural crosses. Although the above analysis was sufficient to accurately determine the number, order, and orientation of these genes in multiple strains of T. gondii, including the F1 progeny clones, the loci were still artifactually pseudogenized even after multiple rounds of polishing with Pilon. We are not aware a comprehensive attempt to solve this problem, possibly because single-copy genes tend to be very accurately corrected in Nanopore and PacBio data using tools like Pilon (Senol Cali et al. 2019). As a case in point, a hybrid PacBioRS/Hi-C assembly of the Plasmodium knowlesi genome (Lapp et al. 2018) required manual assembly correction to removed hundreds of incorrectly pseudogenized members of the SICAvar gene family, whereas other parts of the genome had much more accurate consensus sequences. Based on our analyses of these sequences, they appear to be the most precise version of these sequences to date, given that current versions of these genes deposited to GenBank from prior publications from our group and others for ROP5 (Reese et al. 2011) and MAF1 (Adomako-Ankomah et al. 2014, 2016) were obtained by PCR and subject to artifactual chimerism. This is particularly evident for the MAF1 locus, which is made up of tandem expansions of a two-gene cassette (which we have dubbed MAF1a and MAF1b), and these genes are functionally distinct (only MAF1b drives the unique T. gondii host mitochondrial association phenotype) (Adomako-Ankomah et al. 2016; Blank et al. 2018).
These data represent a new era in genome sequencing in T. gondii and its near relatives, allowing for near-complete telomere-to-telomere assemblies of T. gondii strains to be generated with minimal effort and cost. Moreover, with existing and supplementary correction methods that are targeted to the systematic error that can occur in single-molecule sequencing approaches (e.g., incorrect calls of homopolymer nucleotide runs), we have been able to generate the most accurate version of a key subset of virulence effector genes in this organism with such an enormous impact on human global health.
Methods
Parasite and cell culture
All T. gondii strains and the N. caninum Liverpool strain were maintained by serial passage of tachyzoites in human foreskin fibroblasts (HFFs). HFFs were cultured in Dulbecco's Modified Eagle's Medium (DMEM) containing 10% fetal bovine serum (FBS), 2 mM glutamine, and 50 mg/mL each of penicillin and streptomycin at 37°C in a 5% CO2 incubator.
HMW genomic DNA extraction
Before DNA purification, tachyzoites of T. gondii or N. caninum Liverpool strain were grown in 2 × 107 HFFs for ∼5–7 d until the monolayer was fully infected. The infected cells were then scraped and syringe-lysed to release the parasites, and the parasites were harvested by filtering (5.0-μm syringe filter, MilliporeSigma) and centrifugation. The pelleted parasites were resuspended and lysed in 10 mL TLB buffer (100 mM NaCl, 10 mM Tris-HCl at pH 8.0, 25 mM EDTA at pH 8.0, 0.5% [w/v] SDS) containing 20 μg/mL RNase A for 1 h at 37°C, followed by a 3-h Proteinase K (20 mg/mL) digestion at 50°C. The lysate was split into two tubes containing phase-lock gel (Quantabio), and 5 mL TE-saturated phenol (MilliporeSigma) was added to each tube, mixed by rotation for 10 min, and centrifuged for 10 min at 4750g. The DNA was isolated by removing the aqueous phase to two tubes containing phase-lock gel, followed by a 25:24:1 phenol-chloroform-isoamyl alcohol (MilliporeSigma) extraction. The DNA in the aqueous phase was further purified by ethanol precipitation by adding 4 mL 3 M NaOAc (pH 5.2) and then mixing 30 mL ice-cold 100% ethanol. The solution was mixed by gentle inversion and briefly centrifuged at 1000g for 2 min to pellet the DNA. The resulting pellet was washed three times with 70% ethanol, and all visible traces of ethanol were removed from the tube. The DNA was allowed to air dry for 5 min on a 40°C heat block and resuspended in 40 μL elution buffer (10 mM Tris-HCl at pH 8.5) without mixing on pipetting, followed by an overnight incubation at 4°C. The concentration and purity of the eluted DNA were measured using a NanoDrop spectrophotometer (Thermo Fisher Scientific), and ∼400 ng of DNA was used for sequencing library preparation.
MinION library preparation and sequencing
The MinION sequencing libraries were prepared using the SQK-RAD004 or SQK-RBK004 kit (Oxford Nanopore Technologies) protocol accompanying all pipetting steps performed using pipette tips with ∼1 cm cut off of the end. HMW DNA (7.5 μL corresponding to 400 ng of DNA) was mixed with 2.5 μL of fragmentation mix (SQK-RAD004 kit) or barcoded fragmentation mix (SQK-RBK004 kit), and then incubated for 1 min at 30°C, followed for 1 min at 80°C on a thermocycler. After incubation, 1 μL of rapid adapter mix was added and mixed gently by flicking the tube, and the library was incubated for 5 min at room temperature. Before the library loading, the flow cell (MinION R9.4.1 flow cell; FLO-MIN106, Oxford Nanopore Technologies) was primed by loading 800 μL of priming mix (flush tether and flush buffer mix, Oxford Nanopore Technologies) into the priming port on the flow cell and left for 5 min. After priming, 11 μL of DNA library was mixed with 34 μL of sequencing buffer (Oxford Nanopore Technologies), 25.5 μL of resuspended loading beads (Oxford Nanopore Technologies), and 4.5 μL of nuclease-free water. To initiate sequencing, 75 μL of the prepared library was loaded onto the flow cell through the SpotON sample port in a drop-by-drop manner. Sequencing was performed immediately after platform QC, which determined the number of active pores. The sequencing process was controlled using MinKNOW (Oxford Nanopore Technologies), and the resulting FAST5 files were base-called using Guppy v3.2.1 (Oxford Nanopore Technologies). The barcoded sequencing reads were demultiplexed using Deepbinner (https://github.com/rrwick/Deepbinner). Read statistics were computed and graphed using NanoPlot v1.0.0 (De Coster et al. 2018).
Read quality control and de novo genome assembly
To assess read quality, raw sequencing reads were aligned against the reference genomes (information on the reference genomes used in this study is shown in Supplemental Table S1) using minimap2 (Li 2018) with the following parameter: -ax map-ont. All reads >1000 bp in length were input into Canu v1.7.1 (Koren et al. 2017) for de novo assembly using the complete Canu pipeline (correction, trimming, and assembly) (Koren et al. 2017) with the following parameters: correctedErrorRate = 0.154, gnuplotTested = TRUE, minReadLength = 1000, and –nanopore-raw. Assembly was performed based on an estimated 65-Mb genome size for T. gondii strains, as well as 57 Mb for the N. caninum Liverpool strain, and was run using the Slurm management system on the high-throughput computing (HTC) cluster at University of Pittsburgh.
Error correction and assembly polishing
For the Canu-yielded TgRH88, TgME49, TgCTG, and NcLiv assemblies, assembly errors were corrected by Pilon v1.23 (Walker et al. 2014) with four iterations using the alignment of select whole-genome Illumina paired-end reads (NCBI Sequence Read Archive [SRA; https://www.ncbi.nlm.nih.gov/sra] accession numbers: SRR5123638, SRR2068653, SRR5643140, or ERR701181) to the assembly contigs generated by BWA-MEM (Li 2013). The resulting corrected contigs were reassembled using Flye v2.5 (Kolmogorov et al. 2019). For the II×III F1 progeny assemblies, CL13, S27, S21, S26, and D3X1, the assembly contigs were directly subjected to Flye for reassembly without Pilon correction. The final contigs/scaffolds in the TgRH88, TgME49, and TgCTG assemblies were assigned, ordered, and oriented to chromosomes using ToxoDB-48 genomes as references (Supplemental Table S1). These genome sequences were then deposited in GenBank and can be found under BioProject accession number PRJNA638608.
Supplemental assembly correction
Because tandem gene arrays were still artificially pseudogenized after Pilon-based error correction, we performed multiple rounds of error correction to eliminate any remaining homopolymer errors using a custom Perl script and modules, as well as a separate Perl module from James Tisdall (2001). First, we aligned the Illumina sequence reads used in Pilon to the polished assembly using Bowtie 2 (Langmead and Salzberg 2012) (using the -k 10 parameter to allow reads to map to up to 10 distinct locations). We then broke the genome into 250-kb fragments and counted all possible 30-bp k-mers in the raw reads aligning to that 250-kb region. Then, we walked through the assembly 1 bp at a time, extracting the 30-bp starting at that position and counting the number of times that 30 bp was found in the reads mapping to the 250-kb region. If the read count was less than five, we attempted to determine the length of the homopolymer by adding sequence iteratively until the number of reads harboring that sequence increased above 10. The nucleotide to be added was selected only if ≥90% of the reads had the same nucleotide at that position. If the read was not improved after addition of 10 nucleotides, no changes were made to the sequence, unless the truncation of 1 nucleotide from the end of the 30-bp assembly fragment increased coverage, in which case that change was made to the assembly. We repeated this correction four times iteratively and used the corrected assemblies to determine the sequence of individual paralogs at tandem gene arrays such as MAF1, ROP5, and ROP38.
Long-read assembly evaluation
Assembly statistics were computed using Canu v1.7.1 and QUAST v5.0.2 (Mikheenko et al. 2018). Genome assembly completeness assessment was performed using BUSCO v3.0.2 (Waterhouse et al. 2018) against the Protists Ensembl data set. Gene predictions were performed using AUGUSTUS v3.3 (Keller et al. 2011) with the T. gondii–specific training set.
Whole-genome alignment
Whole-genome alignments between the long-read assemblies and the reference genomes were performed using MUMmer v4.0.0 (Kurtz et al. 2004) and Mauve v2.4.0 (Darling et al. 2004). Dotplots were generated using D-Genies (Cabanettes and Klopp 2018). BWA-MEM was used for remapping the corrected reads to the reference genomes, and all SAM files were parsed to sorted BAM files using SAMtools v1.9 (Li et al. 2009). Alignments were visualized using a variety of tools including the Integrative Genomics Viewer (IGV) v2.4.15 (Thorvaldsdottir et al. 2013), Mauve (Darling et al. 2004), Circos (Krzywinski et al. 2009), and custom scripts implemented in R statistical software (R Core Team 2020).
Structural variant detection
Structural variant differences between the long-read assemblies and reference genomes (from ToxoDB or GenBank, depending on the strains analyzed) were identified by processing the delta file generated by the MUMmer alignment generator NUCmer with the parameter “show-diff.” In addition, the manual curation of structural variants was performed by visual inspection of chromosomal rearrangements based on the whole-genome alignments generated using Mauve and MUMmer and using BLASTN to identify and count repetitive loci. Select alignment plots were generated to integrate these data using R statistical software (R Core Team 2020).
Copy number variant detection
For all gene-coding tandem expansions in T. gondii or N. caninum identified previously (Adomako-Ankomah et al. 2014), all predicted gene sequences were downloaded from ToxoDB and aligned using BLASTN, and alignments showing >80% identity and covering at least 80% of the query gene were used to count tandem repeat numbers in the T. gondii strains RH88, ME49, and CTG. For a subset of these tandemly expanded loci (ROP5, ROP38, MIC17, MAF1, ROP4/7, and TSEL8), similar analyses were performed manually against all queried T. gondii strains (including F1 progeny clones), and in this case, only alignments that showed >95% identity and >98% coverage were considered as a match. Paralogs were grouped based on alignment identity, and the number of copies at these loci was estimated by alignment match counts. Only matches that were within a single assembled Nanopore-derived contig were considered for copy number estimates, and the length of the sequence between the upstream of the first match or the downstream from the last match on the genomic coordinate and the edge of the corresponding contig had to be longer than that of the sequence between two adjacent matches.
Identification and analysis of new sequences in the long-read assemblies
To identify new sequences that filled reference assembly gaps, we aligned the long-read chromosome contigs to the reference assembly chromosomes using NUCmer with the “show-diff -q” parameter. The coordinates of (1) sequence expansions and (2) unaligned sequences from our de novo assembly were determined, and the corresponding sequences were extracted using custom scripts. Repeats in all genome assemblies were detected using Tandem Repeat Finder (TRF) v4.09 (Benson 1999), and all repeats with a period size of ≥500 bp and having at least two copies were used to determine the impact of long-read assembly on resolution of repeats >500 bp in size (which are poorly resolved by second- or third-generation sequencing technologies.
Hi-C data analysis
Published Hi-C reads (Bunnik et al. 2019) were realigned to assemblies TgRH88, TgME49, TgCTG, S27, and S21 and then processed further by assigning fragments and removing invalid and duplicate pairs using the processing pipeline HiC-Pro (Servant et al. 2015). The resulting raw intra-chromosomal and inter-chromosomal contact maps were built at 10-kb resolution and corrected for experimental and technical biases using ICE normalization (Imakaev et al. 2012).
Data access
Raw sequence data (in FASTQ format) and assemblies generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA638608. Custom Perl scripts and their dependent modules are provided as Supplemental Code. Some modifications may be required to run the scripts on different systems. Contact the corresponding author for information if necessary.
Supplementary Material
Acknowledgments
We thank members of the Boyle laboratory for critical reading of the manuscript and Josh Quick for public sharing of protocols for DNA isolation that maximize read length. This work was supported by National Institutes of Health (NIH) grants R01AI116855, R01AI114655, and R21AI154386 to J.P.B., State Scholarship Fund from the China Scholar Council (201708440340) to J.X., and grants R21AI142506 (NIH) and NIFA-Hatch-225935 (University of California, Riverside) to K.G.L.R.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.262816.120.
Freely available online through the Genome Research Open Access option.
Competing interest statement
The authors declare no competing interests.
References
- Adomako-Ankomah Y, Wier GM, Borges AL, Wand HE, Boyle JP. 2014. Differential locus expansion distinguishes Toxoplasmatinae species and closely related strains of Toxoplasma gondii. mBio 5: e01003-13. 10.1128/mBio.01003-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adomako-Ankomah Y, English ED, Danielson JJ, Pernas LF, Parker ML, Boulanger MJ, Dubey JP, Boyle JP. 2016. Host mitochondrial association evolved in the human parasite Toxoplasma gondii via neofunctionalization of a gene duplicate. Genetics 203: 283–298. 10.1534/genetics.115.186270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson G. 1999. Tandem Repeats Finder: a program to analyze DNA sequences. Nucleic Acids Res 27: 573–580. 10.1093/nar/27.2.573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berná L, Marquez P, Cabrera A, Greif G, Francia ME, Robello C. 2021. Reevaluation of the Toxoplasma gondii and Neospora caninum genomes reveals misassembly, karyotype differences, and chromosomal rearrangements. Genome Res (this issue) 31: 823–833. 10.1101/gr.262832.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank ML, Boyle JP. 2018. Effector variation at tandem gene arrays in tissue-dwelling coccidia: Who needs antigenic variation anyway? Curr Opin Microbiol 46: 86–92. 10.1016/j.mib.2018.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank ML, Parker ML, Ramaswamy R, Powell CJ, English ED, Adomako-Ankomah Y, Pernas LF, Workman SD, Boothroyd JC, Boulanger MJ, et al. 2018. A Toxoplasma gondii locus required for the direct manipulation of host mitochondria has maintained multiple ancestral functions. Mol Microbiol 108: 519–535. 10.1111/mmi.13947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowden R, Davies RW, Heger A, Pagnamenta AT, de Cesare M, Oikkonen LE, Parkes D, Freeman C, Dhalla F, Patel SY, et al. 2019. Sequencing of human genomes with nanopore technology. Nat Commun 10: 1869. 10.1038/s41467-019-09637-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks CF, Francia ME, Gissot M, Croken MM, Kim K, Striepen B. 2011. Toxoplasma gondii sequesters centromeres to a specific nuclear region throughout the cell cycle. Proc Natl Acad Sci 108: 3767–3772. 10.1073/pnas.1006741108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunnik EM, Venkat A, Shao J, McGovern KE, Batugedara G, Worth D, Prudhomme J, Lapp SA, Andolina C, Ross LS, et al. 2019. Comparative 3D genome organization in apicomplexan parasites. Proc Natl Acad Sci 116: 3183–3192. 10.1073/pnas.1810815116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burg JL, Grover CM, Pouletty P, Boothroyd JC. 1989. Direct and sensitive detection of a pathogenic protozoan, Toxoplasma gondii, by polymerase chain reaction. J Clin Microbiol 27: 1787–1792. 10.1128/JCM.27.8.1787-1792.1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabanettes F, Klopp C. 2018. D-GENIES: dot plot large genomes in an interactive, efficient and simple way. PeerJ 6: e4958. 10.7717/peerj.4958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemente M, de Miguel N, Lia VV, Matrajt M, Angel SO. 2004. Structure analysis of two Toxoplasma gondii and Neospora caninum satellite DNA families and evolution of their common monomeric sequence. J Mol Evol 58: 557–567. 10.1007/s00239-003-2578-3 [DOI] [PubMed] [Google Scholar]
- Darling AC, Mau B, Blattner FR, Perna NT. 2004. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res 14: 1394–1403. 10.1101/gr.2289704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Coster W, D'Hert S, Schultz DT, Cruts M, Van Broeckhoven C. 2018. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34: 2666–2669. 10.1093/bioinformatics/bty149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz-Viraqué F, Pita S, Greif G, de Souza RCM, Iraola G, Robello C. 2019. Nanopore sequencing significantly improves genome assembly of the protozoan parasite Trypanosoma cruzi. Genome Biol Evol 11: 1952–1957. 10.1093/gbe/evz129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey JP, Hattel AL, Lindsay DS, Topper MJ. 1988. Neonatal Neospora caninum infection in dogs: isolation of the causative agent and experimental transmission. J Am Vet Med Assoc 193: 1259–1263. [PubMed] [Google Scholar]
- Dudchenko O, Batra SS, Omer AD, Nyquist SK, Hoeger M, Durand NC, Shamim MS, Machol I, Lander ES, Aiden AP, et al. 2017. De novo assembly of the aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356: 92–95. 10.1126/science.aal3327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echeverria PC, Rojas PA, Martin V, Guarnera EA, Pszenny V, Angel SO. 2000. Characterisation of a novel interspersed Toxoplasma gondii DNA repeat with potential uses for PCR diagnosis and PCR-RFLP analysis. FEMS Microbiol Lett 184: 23–27. 10.1111/j.1574-6968.2000.tb08984.x [DOI] [PubMed] [Google Scholar]
- Edvinsson B, Lappalainen M, Evengård B, ESCMID Study Group for Toxoplasmosis. 2006. Real-time PCR targeting a 529-bp repeat element for diagnosis of toxoplasmosis. Clin Microbiol Infect 12: 131–136. 10.1111/j.1469-0691.2005.01332.x [DOI] [PubMed] [Google Scholar]
- Fournier T, Gounot JS, Freel K, Cruaud C, Lemainque A, Aury JM, Wincker P, Schacherer J, Friedrich A. 2017. High-quality de novo genome assembly of the Dekkera bruxellensis yeast using nanopore MinION sequencing. G3 (Bethesda) 7: 3243–3250. 10.1534/g3.117.300128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajria B, Bahl A, Brestelli J, Dommer J, Fischer S, Gao X, Heiges M, Iodice J, Kissinger JC, Mackey AJ, et al. 2008. ToxoDB: an integrated Toxoplasma gondii database resource. Nucleic Acids Res 36: D553–D556. 10.1093/nar/gkm981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gissot M, Walker R, Delhaye S, Huot L, Hot D, Tomavo S. 2012. Toxoplasma gondii chromodomain protein 1 binds to heterochromatin and colocalises with centromeres and telomeres at the nuclear periphery. PLoS One 7: e32671. 10.1371/journal.pone.0032671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imakaev M, Fudenberg G, McCord RP, Naumova N, Goloborodko A, Lajoie BR, Dekker J, Mirny LA. 2012. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat Methods 9: 999–1003. 10.1038/nmeth.2148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Koren S, Miga KH, Quick J, Rand AC, Sasani TA, Tyson JR, Beggs AD, Dilthey AT, Fiddes IT, et al. 2018. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat Biotechnol 36: 338–345. 10.1038/nbt.4060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JL, Holland GN. 2010. Annual burden of ocular toxoplasmosis in the US. Am J Trop Med Hyg 82: 464–465. 10.4269/ajtmh.2010.09-0664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joynson DH, Wreghitt TG. 2005. Toxoplasmosis: a comprehensive clinical guide. Cambridge University Press, Cambridge. [Google Scholar]
- Kasper LH, Ware PL. 1985. Recognition and characterization of stage-specific oocyst/sporozoite antigens of Toxoplasma gondii by human antisera. J Clin Invest 75: 1570–1577. 10.1172/JCI111862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller O, Kollmar M, Stanke M, Waack S. 2011. A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics 27: 757–763. 10.1093/bioinformatics/btr010 [DOI] [PubMed] [Google Scholar]
- Khan A, Taylor S, Su C, Mackey AJ, Boyle J, Cole R, Glover D, Tang K, Paulsen IT, Berriman M, et al. 2005. Composite genome map and recombination parameters derived from three archetypal lineages of Toxoplasma gondii. Nucleic Acids Res 33: 2980–2992. 10.1093/nar/gki604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A, Shaik JS, Behnke M, Wang Q, Dubey JP, Lorenzi HA, Ajioka JW, Rosenthal BM, Sibley LD. 2014. NextGen sequencing reveals short double crossovers contribute disproportionately to genetic diversity in Toxoplasma gondii. BMC Genomics 15: 1168. 10.1186/1471-2164-15-1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Weiss LM. 2004. Toxoplasma gondii: the model apicomplexan. Int J Parasitol 34: 423–432. 10.1016/j.ijpara.2003.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolmogorov M, Yuan J, Lin Y, Pevzner PA. 2019. Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol 37: 540–546. 10.1038/s41587-019-0072-8 [DOI] [PubMed] [Google Scholar]
- Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. 2017. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 27: 722–736. 10.1101/gr.215087.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. 2009. Circos: an information aesthetic for comparative genomics. Genome Res 19: 1639–1645. 10.1101/gr.092759.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. 2004. Versatile and open software for comparing large genomes. Genome Biol 5: R12. 10.1186/gb-2004-5-2-r12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapp SA, Geraldo JA, Chien JT, Ay F, Pakala SB, Batugedara G, Humphrey J, MaHPIC consortium; De Barry J, Le Roch KG, et al. 2018. PacBio assembly of a Plasmodium knowlesi genome sequence with Hi-C correction and manual annotation of the SICAvar gene family. Parasitology 145: 71–84. 10.1017/S0031182017001329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau YL, Lee WC, Gudimella R, Zhang G, Ching XT, Razali R, Aziz F, Anwar A, Fong MY. 2016. Deciphering the draft genome of Toxoplasma gondii RH strain. PLoS One 11: e0157901. 10.1371/journal.pone.0157901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997v2 [q-bio.GN].
- Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34: 3094–3100. 10.1093/bioinformatics/bty191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzi H, Khan A, Behnke MS, Namasivayam S, Swapna LS, Hadjithomas M, Karamycheva S, Pinney D, Brunk BP, Ajioka JW, et al. 2016. Local admixture of amplified and diversified secreted pathogenesis determinants shapes mosaic Toxoplasma gondii genomes. Nat Commun 7: 10147. 10.1038/ncomms10147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madoui MA, Engelen S, Cruaud C, Belser C, Bertrand L, Alberti A, Lemainque A, Wincker P, Aury JM. 2015. Genome assembly using nanopore-guided long and error-free DNA reads. BMC Genomics 16: 327. 10.1186/s12864-015-1519-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matrajt M, Angel SO, Pszenny V, Guarnera E, Roos DS, Garberi JC. 1999. Arrays of repetitive DNA elements in the largest chromosomes of Toxoplasma gondii. Genome 42: 265–269. 10.1139/g98-120 [DOI] [PubMed] [Google Scholar]
- Michael TP, Jupe F, Bemm F, Motley ST, Sandoval JP, Lanz C, Loudet O, Weigel D, Ecker JR. 2018. High contiguity Arabidopsis thaliana genome assembly with a single nanopore flow cell. Nat Commun 9: 541. 10.1038/s41467-018-03016-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikheenko A, Prjibelski A, Saveliev V, Antipov D, Gurevich A. 2018. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 34: i142–i150. 10.1093/bioinformatics/bty266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namasivayam S, Baptista RP, Xiao W, Hall EM, Doggett JS, Troell K, Kissinger JC. 2021. A novel fragmented mitochondrial genome in the protist pathogen Toxoplasma gondii and related tissue coccidia. Genome Res (this issue) 31: 852–865. 10.1101/gr.266403.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfefferkorn ER, Pfefferkorn LC. 1976. Toxoplasma gondii: isolation and preliminary characterization of temperature-sensitive mutants. Exp Parasitol 39: 365–376. 10.1016/0014-4894(76)90040-0 [DOI] [PubMed] [Google Scholar]
- Pfefferkorn ER, Pfefferkorn LC, Colby ED. 1977. Development of gametes and oocysts in cats fed cysts derived from cloned trophozoites of Toxoplasma gondii. J Parasitol 63: 158–159. [PubMed] [Google Scholar]
- R Core Team. 2020. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. https://www.R-project.org/. [Google Scholar]
- Reese ML, Zeiner GM, Saeij JP, Boothroyd JC, Boyle JP. 2011. Polymorphic family of injected pseudokinases is paramount in Toxoplasma virulence. Proc Natl Acad Sci 108: 9625–9630. 10.1073/pnas.1015980108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid AJ, Vermont SJ, Cotton JA, Harris D, Hill-Cawthorne GA, Könen-Waisman S, Latham SM, Mourier T, Norton R, Quail MA, et al. 2012. Comparative genomics of the apicomplexan parasites Toxoplasma gondii and Neospora caninum: coccidia differing in host range and transmission strategy. PLoS Pathog 8: e1002567. 10.1371/journal.ppat.1002567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reischl U, Bretagne S, Krüger D, Ernault P, Costa JM. 2003. Comparison of two DNA targets for the diagnosis of Toxoplasmosis by real-time PCR using fluorescence resonance energy transfer hybridization probes. BMC Infect Dis 3: 7. 10.1186/1471-2334-3-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabin AB. 1941. Toxoplasmic encephalitis in children. JAMA 116: 801–807. 10.1001/jama.1941.02820090001001 [DOI] [Google Scholar]
- Saeij JP, Boyle JP, Boothroyd JC. 2005. Differences among the three major strains of Toxoplasma gondii and their specific interactions with the infected host. Trends Parasitol 21: 476–481. 10.1016/j.pt.2005.08.001 [DOI] [PubMed] [Google Scholar]
- Saeij JPJ, Coller S, Boyle JP, Jerome ME, White MW, Boothroyd JC. 2007. Toxoplasma co-opts host gene expression by injection of a polymorphic kinase homologue. Nature 445: 324–327. 10.1038/nature05395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger F, Nicklen S, Coulson AR. 1977. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci 74: 5463–5467. 10.1073/pnas.74.12.5463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt MH, Vogel A, Denton AK, Istace B, Wormit A, van de Geest H, Bolger ME, Alseekh S, Maß J, Pfaff C, et al. 2017. De novo assembly of a new Solanum pennellii accession using Nanopore sequencing. Plant Cell 29: 2336–2348. 10.1105/tpc.17.00521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senol Cali D, Kim JS, Ghose S, Alkan C, Mutlu O. 2019. Nanopore sequencing technology and tools for genome assembly: computational analysis of the current state, bottlenecks and future directions. Brief Bioinformatics 20: 1542–1559. 10.1093/bib/bby017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servant N, Varoquaux N, Lajoie BR, Viara E, Chen CJ, Vert JP, Heard E, Dekker J, Barillot E. 2015. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol 16: 259. 10.1186/s13059-015-0831-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibley LD, Ajioka JW. 2008. Population structure of Toxoplasma gondii: clonal expansion driven by infrequent recombination and selective sweeps. Annu Rev Microbiol 62: 329–351. 10.1146/annurev.micro.62.081307.162925 [DOI] [PubMed] [Google Scholar]
- Sibley LD, Boothroyd JC. 1992. Construction of a molecular karyotype for Toxoplasma gondii. Mol Biochem Parasitol 51: 291–300. 10.1016/0166-6851(92)90079-Y [DOI] [PubMed] [Google Scholar]
- Sibley LD, LeBlanc AJ, Pfefferkorn ER, Boothroyd JC. 1992. Generation of a restriction fragment length polymorphism linkage map for Toxoplasma gondii. Genetics 132: 1003–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su C, Howe DK, Dubey JP, Ajioka JW, Sibley LD. 2002. Identification of quantitative trait loci controlling acute virulence in Toxoplasma gondii. Proc Natl Acad Sci 99: 10753–10758. 10.1073/pnas.172117099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, Mesirov JP. 2013. Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinformatics 14: 178–192. 10.1093/bib/bbs017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisdall JD. 2001. Beginning Perl for bioinformatics. O'Reilly, Beijing. [Google Scholar]
- Vollger MR, Logsdon GA, Audano PA, Sulovari A, Porubsky D, Peluso P, Wenger AM, Concepcion GT, Kronenberg ZN, Munson KM, et al. 2020. Improved assembly and variant detection of a haploid human genome using single-molecule, high-fidelity long reads. Ann Hum Genet 84: 125–140. 10.1111/ahg.12364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, et al. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9: e112963. 10.1371/journal.pone.0112963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse RM, Seppey M, Simão FA, Manni M, Ioannidis P, Klioutchnikov G, Kriventseva EV, Zdobnov EM. 2018. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol Biol Evol 35: 543–548. 10.1093/molbev/msx319 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.