

The cell wall is the interface between the fungal cell and its environment and disruption of cell wall assembly is an effective strategy for antifungal therapies. Therefore, a detailed understanding of how cell walls form is critical to identify potential drug targets and develop therapeutic strategies.
KEYWORDS: cell wall, chitin synthase, chitin deacetylase, lipid droplet, chlamydospore
ABSTRACT
The polysaccharide chitosan is found in the cell wall of specific cell types in a variety of fungal species where it contributes to stress resistance, or in pathogenic fungi, virulence. Under certain growth conditions, the pathogenic yeast Candida dubliniensis forms a cell type termed a chlamydospore, which has an additional internal layer in its cell wall compared to hyphal or yeast cell types. We report that this internal layer of the chlamydospore wall is rich in chitosan. The ascospore wall of Saccharomyces cerevisiae also has a distinct chitosan layer. As in S. cerevisiae, formation of the chitosan layer in the C. dubliniensis wall requires the chitin synthase CHS3 and the chitin deacetylase CDA2. In addition, three lipid droplet-localized proteins—Rrt8, Srt1, and Mum3—identified in S. cerevisiae as important for chitosan layer assembly in the ascospore wall are required for the formation of the chitosan layer of the chlamydospore wall in C. dubliniensis. These results reveal that a conserved machinery is required for the synthesis of a distinct chitosan layer in the walls of these two yeasts and may be generally important for incorporation of chitosan into fungal walls.
IMPORTANCE The cell wall is the interface between the fungal cell and its environment and disruption of cell wall assembly is an effective strategy for antifungal therapies. Therefore, a detailed understanding of how cell walls form is critical to identify potential drug targets and develop therapeutic strategies. This study shows that a set of genes required for the assembly of a chitosan layer in the cell wall of S. cerevisiae is also necessary for chitosan formation in a different cell type in a different yeast, C. dubliniensis. Because chitosan incorporation into the cell wall can be important for virulence, the conservation of this pathway suggests possible new targets for antifungals aimed at disrupting cell wall function.
INTRODUCTION
The cell wall is the interface between the fungal cell and the environment (1). In pathogenic fungi, the cell wall is critical for virulence as it mediates interactions with, and evasion of, the host immune system (2). Fungal cell walls are essential for viability and are a common target of antifungal drugs (3–6). Therefore, understanding the structure and assembly of the fungal wall is important for the development of antifungal therapies.
Fungal cell walls are composed primarily of heavily mannosylated proteins (referred to as mannan) and polysaccharides (1). In particular β-1,3 glucans and chitin, a β-1,4-N-acetylglucosamine polymer, are common structural components of fungal cell walls (1, 7, 8). Chitosan, a β-1,4-glucosamine polymer created by deacetylation of chitin, is also found in fungal cell walls but is often limited to specific cell types or developmental stages (9–12). The presence of chitosan in cell walls can be critical for the organism. For example, in the pathogen Cryptococcus neoformans chitosan in the wall dampens the host inflammatory response, and Cryptococcus strains unable to synthesize chitosan are avirulent (13–15). Chitosan is often found in conjunction with polyphenolic compounds, which has led to the proposal that chitosan-polyphenol complexes are a conserved architectural motif in fungal walls (16).
How chitosan is incorporated into the cell wall is not yet well understood. This process has been best studied in the budding yeast, Saccharomyces cerevisiae, where chitosan is found uniquely in the walls of ascospores, a dormant cell type produced after meiosis by a process termed sporulation (17, 18). The ascospore wall consists of four distinct layers, named for their primary constituents, which are deposited in a sequential manner: mannan, glucan, chitosan, and dityrosine (10, 19–22). The mannan and glucan layers form the inner layers of the ascospore wall and are similar in composition to layers in the vegetative cell wall (21). The outer ascospore wall, containing a layer of chitosan and a layer of the polyphenol dityrosine, is unique to ascospores and confers resistance against environmental insults (10, 23, 24).
The chitin in the vegetative cell wall of S. cerevisiae is produced by three different chitin synthases, Chs1, -2, and -3 (25–27). However, during sporulation chitin is produced exclusively by Chs3 (28). Chitosan is generated when acetyl groups on chitin are removed by the sporulation-specific deacetylases, Cda1 and Cda2 (11, 29). Deletion of both CDA1 and CDA2 results in spore walls that contain chitin but lack the chitosan layer. In addition, while the mannan and beta-glucan layers are present, the dityrosine layer is missing. Chitosan is therefore necessary for the formation of both layers of the outer cell wall (29). In contrast, formation of the chitosan layer is independent of the formation of dityrosine. Dityrosine is synthesized from l-tyrosine in the spore cytosol by the sequential action of the Dit1 and Dit2 enzymes (30), and mutants in either DIT1 or DIT2 result in loss of the dityrosine without any obvious effect on the chitosan layer (23).
In addition to the genes directly involved in chitosan or dityrosine synthesis, several other genes are required for the formation of one or more layers of the outer spore wall (31–35). Genes of unknown function such as MUM3 and OSW1, as well as the cis-prenyltransferase encoded by SRT1, lack both the chitosan and dityrosine layers (34). In an srt1Δ mutant, Chs3 activity is reduced, suggesting that Srt1 contributes to spore wall formation through regulation of Chs3 (34). Srt1 is localized to a class of lipid droplets that is physically associated with the developing spore wall (34, 36). Mutants in the paralogous genes LDS1, LDS2, and RRT8, which encode lipid droplet-localized proteins, are specifically defective in the dityrosine layer (35). Whether the genes required for chitosan layer formation in S. cerevisiae are functionally conserved in other fungi has not been reported.
The human fungal pathogen Candida albicans and its close relative, Candida dubliniensis, exhibit cell types with various morphologies (37, 38). Although these Candida species are not known to produce ascospores, under certain conditions they produce a distinct, thick-walled cell type at hyphal tips termed a chlamydospore (37, 39). Chlamydospores are large round cells that are the result of mitotic divisions, unlike ascospores, which package the haploid products of meiosis. The function of chlamydospores in the Candida life cycle is unknown. Nutrient limitation or low-oxygen conditions are often required to induce the appearance of chlamydospores, and C. dubliniensis appears to undergo chlamydosporulation more readily than C. albicans (40, 41).
Ultrastructural studies revealed that the chlamydospore wall is more extensive than the wall of budding or hyphal C. dubliniensis cells with an internal layer not found in those cell types (42). The structure and composition of this layer has not been well characterized. In the present study, we investigated the organization of the chlamydospore wall in C. dubliniensis. This study demonstrated that the unique internal layer of the chlamydospore wall is composed of chitosan. Moreover, genes encoding orthologs of S. cerevisiae proteins necessary for chitosan layer synthesis in ascospores are also required chlamydospore wall assembly. These results reveal that a conserved pathway underlies chitosan synthesis and incorporation in these two yeasts.
RESULTS
C. dubliniensis forms chlamydospores on solid medium containing nonfermentable carbon sources.
In examining the growth of clinical isolates of C. dubliniensis, we discovered that growth on certain carbon sources induced chlamydospore formation. While chlamydospores were not observed in cultures grown on synthetic medium containing glucose or galactose, growth on N-acetylglucosamine, glucosamine, glycerol, or acetate all led to hyphal growth and the appearance of chlamydospores (Fig. 1). Three different clinical isolates of C. dubliniensis as well as the established C. dubliniensis strain SN90 (43) displayed this behavior, whereas C. albicans did not form chlamydospores on any of these media (K. Min, unpublished data). Solid glycerol medium was particularly efficient at inducing chlamydospores (no chlamydospores were seen in liquid medium with any carbon source) (Fig. 1). We took advantage of these induction conditions to examine the properties of the chlamydospore wall in C. dubliniensis, using the clinical isolate that showed the most robust chlamydospore formation.
FIG 1.
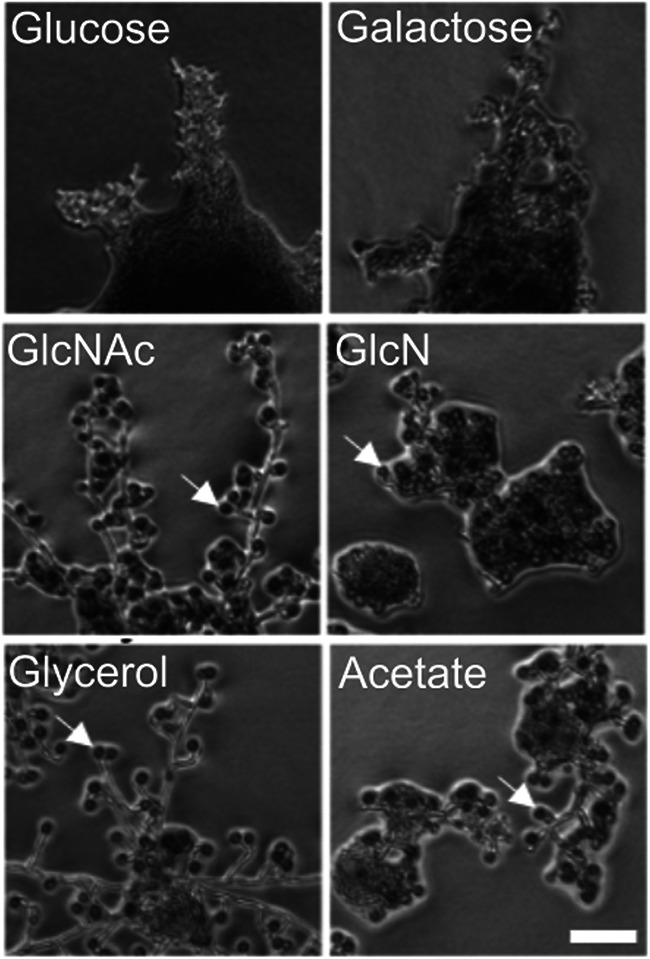
Effect of different carbon sources on the chlamydospore formation. A wild-type C. dubliniensis strain (Cd1465) was spotted on synthetic agar medium containing the indicated carbon sources and photographed on agar after 24 h of growth. Gal, galactose; GlcNAc, N-acetylglucosamine; GlcN, glucosamine. White arrows highlight examples of chlamydospores. Scale bar, 50 nm.
The chlamydospore wall of C. dubliniensis contains chitosan but not dityrosine.
In the S. cerevisiae ascospore wall, a layer of chitosan underlies the dityrosine layer and chitosan is found in association with polyphenol components in other fungal cell walls (9). The observation that chlamydospore walls of C. albicans contain dityrosine suggested that chlamydospore walls might contain chitosan as well (44). Chitosan can be specifically visualized using the stain Eosin Y, which has affinity for chitosan but not chitin (9, 35). When C. dubliniensis chlamydospores were stained with Eosin Y and examined by fluorescence microscopy, bright Eosin Y-dependent fluorescence was visible at the periphery of the chlamydospore (Fig. 2). The fluorescent signal was not observed on hyphal cells, consistent with the presence of chitosan specifically in the chlamydospore wall. Similar staining of C. albicans chlamydospores with Eosin Y has recently been reported (45).
FIG 2.
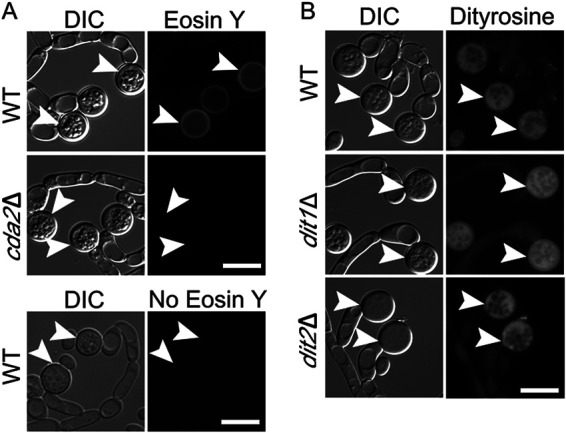
Fluorescence analysis of the chlamydospore wall of C. dubliniensis. (A) Chlamydospores of WT (Cd1465) and cda2Δ (BEM7) strains were stained with Eosin Y to visualize the chitosan layer and imaged using a GFP filter set. Wild-type (WT) chlamydospores with no Eosin Y staining are shown as control. (B) WT (Cd1465), dit1Δ (BEM9), or dit2Δ (BEM10) strains were grown on SG medium to induce chlamydospores and then visualized by differential interference contrast (DIC) or fluorescence microscopy using a dityrosine filter set (excitation [Ex.], 320 nm; emission [Em.], 410 nm). Arrowheads indicate examples of chlamydospores visible in the images. Scale bar, 10 μm.
To prove whether Eosin Y staining was specifically detecting chitosan, a genetic approach was used. The C. dubliniensis genome encodes one member of the chitin deacetylase enzyme family, Cda2 (Cd36_25340), required to convert chitin to chitosan. If Eosin Y staining is due to the presence of chitosan in the chlamydospore wall, then this staining should be reduced or absent in a cda2 deletion that lacks chitin deacetylase activity (9, 35).
C. dubliniensis is a diploid organism. To generate a cda2Δ/cda2Δ deletion strain in C. dubliniensis, we utilized a transient CRISPR-Cas9 system originally developed for C. albicans (46). Double-strand breaks in the two CDA2 alleles were generated by CRISPR-Cas9 and used to target integration of a SAT1 cassette (47), which confers resistance to the drug nourseothricin (NAT), into the CDA2 locus. By selecting for NAT-resistant transformants, diploids homozygous for cda2Δ were obtained. Chlamydospore formation was induced in the cda2Δ/cda2Δ diploid on glycerol medium and examined by Eosin Y staining. No Eosin Y staining was observed, confirming the presence of chitosan in the wild-type chlamydospore wall (Fig. 2A).
To test whether the chlamydospore wall of C. dubliniensis also contains dityrosine, chlamydospores were analyzed by fluorescence microscopy using a filter cube optimized for dityrosine (48). Unlike earlier reports in C. albicans, no fluorescence was seen specifically in the cell wall, though fluorescence was visible throughout the cytoplasm that was brighter than background fluorescence in the hyphal cells (Fig. 2B). This fluorescence is not due to dityrosine, however, since deletion of the C. dubliniensis DIT1 or DIT2 genes (which are required for making dityrosine in budding yeast) also exhibited the cytoplasmic fluorescence (Fig. 2B). Therefore, a common feature in chlamydospores from C. dubliniensis and C. albicans and the ascospores from budding yeast is the presence of a chitosan layer in the cell wall.
The chlamydospore wall of C. albicans also lacks dityrosine fluorescence.
The lack of dityrosine fluorescence in the C. dubliniensis chlamydospore wall led us to examine C. albicans chlamydospores under our microscopy conditions. Cells were spread on a corn meal agar plate and a glass cover slip placed on top of the cells. Chlamydospores were examined after 5 days incubation as described previously (44). Using a filter set similar to what was used in reference 44, we also see a distinct fluorescence signal from the chlamydospore wall (Fig. 3). However, using a filter set optimized for dityrosine excitation and emission, the C. albicans chlamydospores display a diffuse fluorescence throughout the cytoplasm, as in C. dubliniensis. Also similar to C. dubliniensis, the C. albicans chlamydospores stain brightly with Eosin Y. Thus, the C. albicans chlamydospore wall appears identical to C dubliniensis, suggesting that the wall fluorescence seen under UV illumination is not due to dityrosine.
FIG 3.
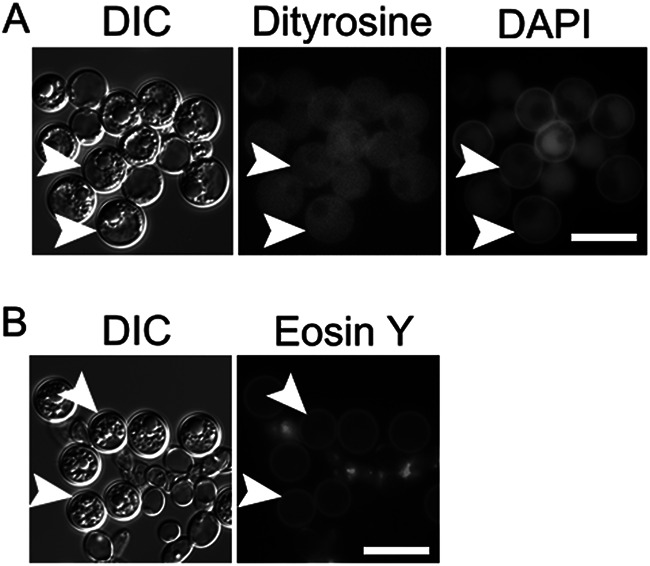
Fluorescence analysis of the C. albicans chlamydospore wall. (A) Unstained chlamydospores of C. albicans strain NLC1 were imaged using either a dityrosine filter set (Ex., 320 nm; Em., 410 nm) or a DAPI filter set (Ex., 375 nm; Em., 475 nm). (B) Chlamydospores of NLC1 stained with Eosin Y and imaged using a GFP filter set. Arrowheads indicate examples of chlamydospores visible in the images. Scale bar, 10 μm.
A chitosan synthesis pathway is conserved in C. dubliniensis.
S. cerevisiae encodes three different chitin synthases, but chitin synthase 3 (CHS3) is specifically used in the synthesis of the chitosan layer of the spore wall (28). C. dubliniensis encodes four different predicted chitin synthases, and the ORF Cd36_12160 encodes the ortholog of S. cerevisiae CHS3 (49, 50). To examine whether the use of the Chs3 ortholog for chitosan synthesis is conserved, a C. dubliniensis chs3Δ/chs3Δ mutant was constructed, and chlamydospores were stained with Eosin Y. Interestingly, as for the cda2Δ/cda2Δ mutant, greatly reduced fluorescence signal from the Eosin Y staining was seen in the chs3Δ/chs3Δ chlamydospore wall (Fig. 4A). In S. cerevisiae, the ascospore wall does not stain with the dye Calcofluor White (CFW), but deletion of CDA1 and CDA2 leads to the accumulation of chitin in the ascospore wall and bright CFW staining (29). In contrast, in C. dubliniensis, deletion of CDA2 or CHS3 leads to, at best, a modest increase in staining around the cell wall (Fig. 4A). In the mutant and wild-type strains, CFW predominantly stains the septa, consistent with earlier reports in C. albicans that chitin at the septum is deposited by chitin synthase 2 (51) (Fig. 4A). In sum, these results indicate that Chs3 and Cda2, the same enzymes that generate chitosan in ascospores, collaborate to generate chitosan in the chlamydospore wall.
FIG 4.

Effect of mutations in C. dubliniensis orthologs of S. cerevisiae spore wall genes on the chlamydospore wall. (A) Cells of strains of the indicated genotype were grown on SGlycerol medium and then stained with both Eosin Y to label chitosan and Calcofluor White (CFW) to label chitin or chitosan. Arrowheads indicate examples of chlamydospores visible in the images. Arrows indicate examples of CFW-stained septa. Scale bar, 10 μm. (B) The intensity of the Eosin Y fluorescence was categorized as bright, dim, or no fluorescence for each chlamydospore, and the number of chlamydospores in each category for each strain was quantified. For each strain, the value represents the average for 100 chlamydospores in each of three independent experiments. Error bars indicate one standard deviation. One asterisk (*) indicates significant difference at P < 0.05; two asterisks (**) indicates significant difference at P < 0.0005 (Student t test).
C. dubliniensis encodes uncharacterized orthologs for several of genes required for making ascospore outer cell walls. If the process of chitosan assembly in the wall is conserved, then these same genes may function in chitosan deposition into the chlamydospore wall as well. In particular, we focused on the orthologs of S. cerevisiae MUM3 (Cd36_82000), SRT1 (Cd36_11510), and RRT8 (Cd36_33980). Homozygous deletions for all three of the C. dubliniensis genes were constructed, and chlamydospores of the mutant strains were examined by Eosin Y and CFW staining. Relative to the wild type, the intensity of the Eosin Y fluorescence was reduced in all of the mutant strains, while the fluorescence from CFW staining was unaltered (Fig. 4A). These results are similar to the effects of chs3Δ and cda2Δ and suggest that these genes are important for chitosan formation in C. dubliniensis.
To more carefully assess the effect of the mutants, the fluorescence intensity of the Eosin Y staining of individual chlamydospores was categorized as bright, reduced, or absent and the number of chlamydospores in each category was scored for each strain (Fig. 4B). The cda2Δ/cda2Δ and chs3Δ/chs3Δ mutant strains displayed a sharp reduction in the fraction of chlamydospores with bright fluorescence intensity and a corresponding increase in chlamydospores displaying no Eosin Y fluorescence. As expected, mutation of DIT1 had no obvious effect on Eosin Y staining. In contrast, the mum3Δ/mum3Δ, srt1Δ/srt1Δ, and rrt8Δ/rrt8Δ diploids all showed phenotypes similar to chs3Δ and cda2Δ strains with a significant, though not quite as strong, reduction in brightly staining spores and an increase in unstained spores (Fig. 4B).
To confirm that the loss of Eosin Y staining was due to the deletion alleles and not an off-target effect from CRISPR/Cas9, the ability of the wild-type gene to complement each mutant was tested. Each wild-type gene was cloned into the integrating plasmid CIp10-SAT, which can be targeted to integrate into the RPS1 locus (52). This vector uses the same SAT1 selectable marker that was used to make the deletion alleles. Therefore, prior to transformation with the plasmids, the SAT1 genes at both copies of each deletion had to be removed. This removal was possible because the knockout cassette included not only the SAT1 gene but also a maltose-inducible FLP recombinase gene, both of which are flanked by flippase recognition target (FRT) sites (46). Induction of the FLP recombinase on maltose medium results in recombination between the FRT sites, thereby deleting the SAT1 and FLP genes. Recombinants that lost both copies of SAT1 were detected by identification of NAT-sensitive colonies. Introduction of CHS3, CDA2, MUM3, or SRT1 into the corresponding knockout strains restored Eosin Y staining to the chlamydospores, confirming that the phenotypes are caused by loss of the specific gene function (Fig. 5). We were unable to do the complementation experiment for rrt8Δ since the deletion strain failed to grow on the maltose medium used to induce the FLP recombinase. Whether the maltose phenotype is a property of the RRT8 knockout or due to some other change in the strain is unknown. In sum, these results demonstrate that CHS3, CDA2, MUM3, SRT1, and probably RRT8 all contribute to formation of a chitosan component of the chlamydospore wall, suggesting that they constitute a conserved machinery mediating chitosan synthesis for incorporation into yeast cell walls.
FIG 5.
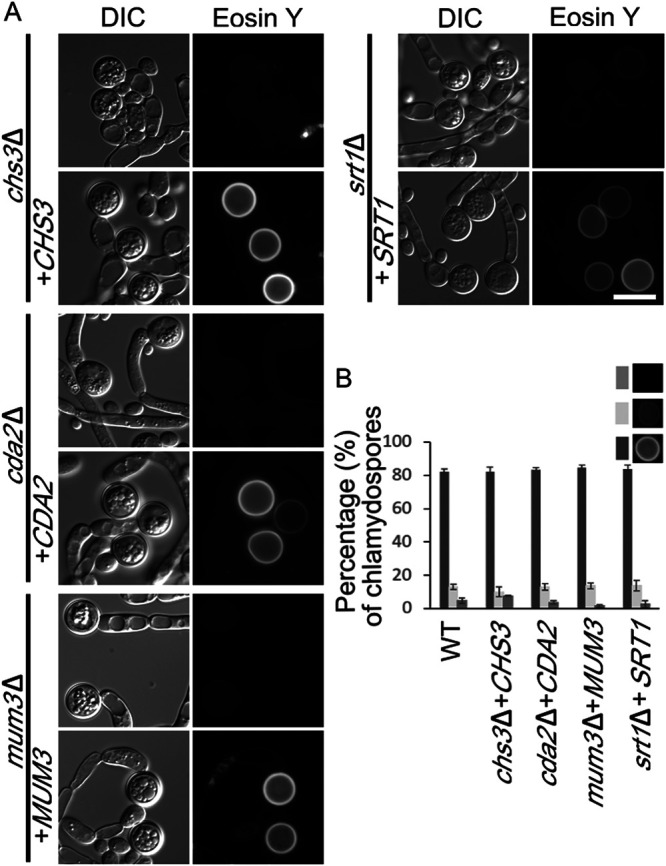
Complementation of the chitosan defect by the wild-type alleles. (A) A wild-type copy of CHS3, CDA2, MUM3, or SRT1 gene, respectively, was integrated into the corresponding deletion mutant (strains BEM15 to BEM18). Cells were grown on SGlycerol medium, and Eosin Y staining of chlamydospores with or without reintroduction of the wild-type allele was examined. DIC, differential interference contrast. Scale bar, 10 μm. (B) Rescue of Eosin Y staining by the wild-type alleles was quantified as in Fig. 4B..
Ultrastructural analysis identifies a chitosan layer in the chlamydospore wall.
The fluorescence images from the Eosin Y staining suggest that chitosan is missing or reduced in the chlamydospore wall of various mutants. Previous ultrastructural studies have revealed that the chlamydospore wall of C. albicans is distinct from the hyphal wall in having a darkly staining inner layer of unidentified material underneath what appear to be beta-glucan and mannan layers (42, 53). To examine the ultrastructure of the C. dubliniensis chlamydospore wall, cells were stained using osmium and thiocarbohydrazide and examined by electron microscopy (31). Similar to previous reports, the cell walls of wild-type chlamydospores displayed a layer of darkly staining material close to the plasma membrane with outer, lighter layers resembling the walls of the adjacent hyphal cells (Fig. 6).
FIG 6.

Electron microscopy of the chlamydospore wall of C. dubliniensis. (A) Chlamydospores were induced, and cells of different strains were stained with osmium-thiocarbohydrazide: WT (CD1465), cda2Δ (BEM7), chs3Δ (BEM8), mum3Δ (BEM11), srt1Δ (BEM14), and rrt8Δ (BEM13). For each strain, a pair of images is shown. The lower image is a higher magnification of the boxed region in upper image. Arrowheads indicate the inner cell wall layer. (B) Quantification of the thickness of the chitosan layer in each strain. Data represented are the means of measurements from 20 chlamydospores. The thickness of the chitosan layer was measured at 5 different positions on each chlamydospore. Error bars indicate one standard deviation. One asterisk (*) indicates a significant difference at P < 0.00005; two asterisks (**) in indicates P < 5E–10 (Student t test). Scale bar, 500 nm.
Given that the chitosan-containing outer ascospore wall of S. cerevisiae also stains darkly under these conditions (31), this inner, electron dense material in the chlamydospore wall may be chitosan. Consistent with this possibility and with the Eosin Y fluorescence results, this inner layer was dramatically reduced in both the chs3Δ/chs3Δ and the cda2Δ/cda2Δ strains (Fig. 6A). Thus, as in the ascospore wall, chitosan in the chlamydospore wall forms a discrete layer. Again, consistent with the Eosin Y fluorescence results, the chitosan layer appeared reduced or absent in chlamydospores of the mum3Δ, srt1Δ, and rrt8Δ mutants as well (Fig. 6A).
The reduction in the chitosan layer visible in the electron micrographs was somewhat variable between chlamydospores in individual strains. Therefore, to measure the effect of the mutants, the thickness of the chitosan layer in the micrographs was measured as an indicator of the amount of chitosan deposited. In each strain, the thickness of the chitosan layer was measured at five locations in 20 different chlamydospores (Fig. 6B). All of the mutants displayed significantly reduced chitosan layers, with the chs3Δ strain displaying the strongest phenotype. In sum, the ultrastructural analysis confirms that chitosan is present in a discrete layer of the chlamydospore wall and a conserved set of genes is required for proper formation of this layer.
C. dubliniensis Rrt8, Mum3, and Srt1 are all localized on lipid droplets.
In S. cerevisiae, the Srt1 and Rrt8/Lds1/Lds2 proteins are localized to lipid droplets, and lipid droplets are associated with the forming spore wall, suggesting some connection between lipid droplets and the assembly of the outer spore wall layers (34–36). C. albicans chlamydospores are reported to be rich in neutral lipids and lipid droplets based on both biochemical fractionation and staining with a lipid droplet dye (45, 54). To examine lipid droplets in C. dubliniensis chlamydospores, the cells were stained with the lipid droplet dye monodansylpentane (MDH) (55). This treatment revealed a very high density of lipid droplets within the chlamydospore compared to C. dubliniensis cells growing in yeast phase that was not changed in any of the mutant strains (Fig. 7).
FIG 7.
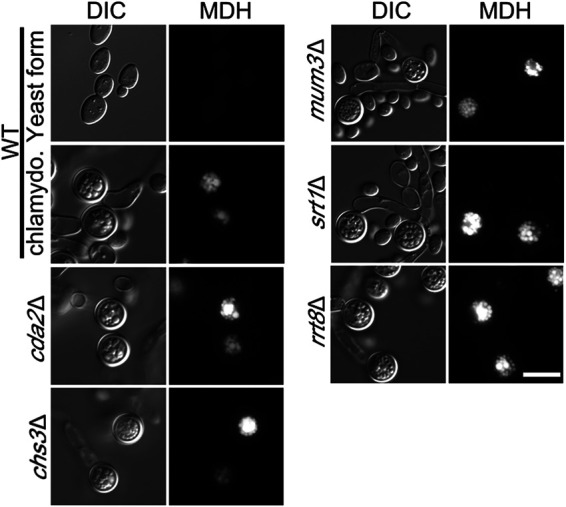
Lipid droplets in chlamydospores. WT cells (CD1465) growing on SGlucose or SGlycerol medium or the indicated cda2Δ (BEM7), chs3Δ (BEM8), mum3Δ (BEM11), srt1Δ (BEM14), and rrt8Δ (BEM13) mutant strains grown on SGlycerol were stained with MDH to label lipid droplets and visualized using a BFP filter. Scale bar, 10 μm.
The abundance of lipid droplets in the chlamydospore and the connection of the S. cerevisiae proteins to lipid droplets led us to examine the localization of the different C. dubliniensis proteins. Each gene, under the control of its native promoter, was fused at its 3′ end to a gene encoding a Candida codon-optimized red fluorescent protein (yEmRFP) (56). Plasmids containing the fusion genes were then integrated at the RPS1 locus in the appropriate deletion strains (except for rrt8Δ where we were unable to eliminate the SAT1 gene from the deletion, so a wild-type strain was used).
To confirm that the fusion proteins are functional, the appropriate deletion strains carrying MUM3::yEmRFP or SRT1::yEmRFP were examined for the ability of the fusion to rescue the mutant phenotype by staining of chlamydospores with Eosin Y (Fig. 8A). Both fusions restored bright Eosin Y staining indicating that the lipid droplet-localized fusion proteins are functional.
FIG 8.
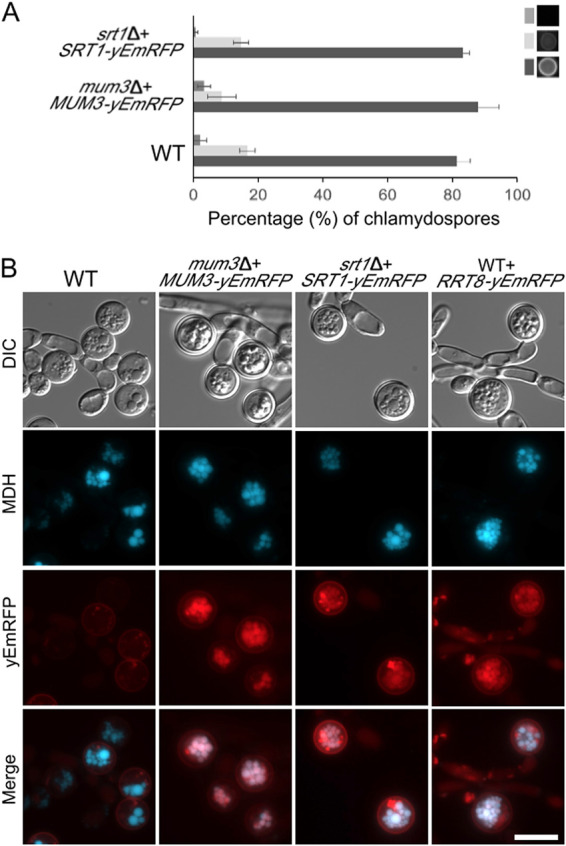
Localization of Cda2, Mum3, Rrt8, and Srt1 in chlamydospores. (A) Eosin Y staining of chlamydospores in WT (CD1465) mum3Δ MUM3-yEMRFP (BEM20) and srt1Δ SRT1-yEmRFP (BEM22) strains was quantified as in Fig. 4B. (B) WT (Cd1456) cells expressing no RFP fusion or strains expressing different MUM3-, SRT1-, or RRT8-yEmRFP fusions (BEM20, -21, or -22) were grown on SGlycerol medium, stained with MDH, and visualized through both BFP and RFP filters. Scale bar, 10 μm.
C. dubliniensis cells carrying the different yEmRFP fusions were then grown under chlamydospore-inducing conditions, stained with MDH to detect lipid droplets, and examined by fluorescence microscopy. For the MUM3, SRT1, and RRT8 fusions, red fluorescence colocalized with the lipid droplet marker in the chlamydospores (Fig. 8B). Red fluorescence at the cell periphery was also visible in the wild-type strain carrying no yEmRFP and so is background fluorescence visible due to the longer exposures necessary to visualize the yEmRFP fusions. Importantly, no background fluorescence was seen at the lipid droplets. The localization of all three proteins suggests that lipid droplets promote chitosan layer formation in C. dubliniensis.
DISCUSSION
We report that C. dubliniensis efficiently forms chlamydospores when incubated on synthetic medium containing different nonfermentable carbon sources. While the molecular signals that trigger chlamydosporulation are complex (40), nutritional signals are known to be involved and induction by changing carbon sources suggests that central carbon metabolism may play a role. Whether this induction mechanism is unique to C. dubliniensis remains to be seen. Though we did not observe chlamydospores with C. albicans under our conditions, growth on N-acetylglucosamine has been reported to induce chlamydospores in C. albicans (57, 58). Previous studies reported that C. dubliniensis can form chlamydospores in Staib medium (a seed extract) (59). Wild-type C. albicans does not form chlamydospores efficiently under these conditions, but deletion of the C. albicans NRG1 gene leads to chlamydosporulation in Staib medium similar to C. dubliniensis (41). The signals triggering chlamydosporulation may be different in SGlyerol and Staib medium, however, since no chlamydospores were seen on SGlycerol when a C. albicans nrg1 mutant was used (L. D. Bemena, unpublished data).
To create mutant strains in C. dubliniensis, we utilized a transient CRISPR-Cas9 system originally developed for C. albicans (46). Combining this transient system with the recyclable SAT1-FLP cassette allowed us to do multistep strain constructions directly in clinical isolates without the need for auxotrophic markers, greatly accelerating our analysis. That this system works well in both C. dubliniensis and C. albicans suggests that it will be useful for other Candida species as well.
Previously, the chlamydospore wall of C. albicans was reported to contain dityrosine based on fluorescence under UV illumination and the observation that deletion of the CYP56/DIT2 gene abolished chlamydospore formation (44). In contrast, using a dityrosine-optimized filter, we find only cytoplasmic fluorescence from chlamydospores of both C. albicans and C. dubliniensis, and this fluorescence is not altered in dit1 or dit2 mutants of C. dubliniensis, indicating that the cytoplasmic signal is not dityrosine. While it is possible that the Candida species produce a form of dityrosine polymer with different fluorescence characteristics from S. cerevisiae, the simplest interpretation of these results is that dityrosine is not a component of the chlamydospore wall in Candida.
We show here that chitosan is a major constituent of the previously described dark, inner layer of the Candida chlamydospore wall. Chitosan also forms a discrete layer in the S. cerevisiae ascospore wall, however, the position of the chitosan layer with respect to other cell wall components is distinct in the two cell walls (Fig. 9). In the ascospore, the chitosan is located toward the outside of the structure, while in the chlamydospore it is on the interior of the wall. In both cases, however, the chitosan is localized adjacent to the beta-glucan components of the wall, suggesting that the presence of the beta-glucan may also be important for organizing the chitosan into a distinct layer. In the ascospore wall, loss of the chitosan layer does not disrupt the shape or integrity of the ascospore but renders the spores sensitive to many environmental insults (29, 31). Similarly, in the chlamydospore wall the chitosan layer is not required for cellular integrity, though whether the chitosan layer also contributes stress resistance is not clear.
FIG 9.

Model for organization of the C. dubliniensis chlamydospore and S. cerevisiae ascospore walls. The organization of the different layers of the walls is shown with respect to the cell plasma membrane. The linkages between components are based on the known linkages in the vegetative cell wall of S. cerevisiae (25). The nature of the cross-links within and between the chitosan and dityrosine layers is unknown.
Our results reveal a conserved machinery required for chitosan layer synthesis. Multiple chitin synthases are present in both S. cerevisiae and C. dubliniensis, and yet in both yeasts CHS3 is uniquely required for synthesis of the chitosan layer of the ascospore and chlamydospore cell walls. Whether this reflects a specific association of this chitin synthase with the chitin deacetylase protein or with some other aspect of Chs3 activity remains to be determined. For example, the Chs3 enzyme might synthesize chitin strands of a chain length or organization that is more amenable to deacetylation. Indeed, C. albicans Chs3 has been reported to synthesize shorter chitin fibrils than Chs8 (50).
The lipid droplet-localized proteins Srt1, Rrt8, and Mum3 are required for proper chitosan layer formation in both yeasts. Since these proteins are localized on cytosolic lipid droplets, their effects on chitosan assembly must be somewhat indirect. MUM3 and SRT1 encode predicted lipid-synthesizing enzymes. The Mum3 protein is homologous to O-acyltransferase enzymes, and Srt1 is a subunit of a cis-prenyltransferase responsible for synthesizing a lipid droplet-localized pool of polyprenols (31, 34). In earlier work, we proposed a model in which Srt1-generated long-chain polyprenols in the lipid droplet that are transferred to the plasma membrane to enhance Chs3 activity (34). It is possible that a similar mechanism occurs during chlamydospore formation. An alternative possibility was recently suggested by nuclear magnetic resonance (NMR) studies of chitosan-containing cell wall preparations from both S. cerevisiae and Cryptococcus neoformans that revealed neutral lipids are directly incorporated into the cell wall (16, 60). Thus, the Rrt8, Srt1, and Mum3 proteins may be involved in the synthesis of some lipid component that is then transferred from the lipid droplet to play a structural role during chitosan layer assembly. Further biochemical work will be necessary to clarify how these proteins and their lipid products contribute to the formation of this cell wall structure.
NMR studies suggest that there is a common architecture for chitosan-containing elements in the fungal cell wall from ascomycetes to basidiomycetes (16). Orthologs of the genes described here that underlie formation of chitosan cell wall layers in Candida and Saccharomyces can be found throughout the fungi. Thus, the similar architecture may reflect a broadly conserved genetic network regulating the synthesis of chitosan-containing cell wall structures in fungi. Given the importance of chitosan to virulence of some pathogenic fungi, the genes described here may be useful targets for antifungal therapies (13).
MATERIALS AND METHODS
Strain and growth conditions.
Strains used are listed in Table 1. C. dubliniensis strain Cd1465 is derived from a clinical specimen isolated from a patient sample at the Stony Brook hospital. This strain was routinely cultured at 30°C on YPD medium (2% Bacto peptone, 2% dextrose, 1% yeast extract, and 2% agar). C. dubliniensis transformants were selected on YPD_NAT (2% Bacto peptone, 2% dextrose, 1% yeast extract, 2% agar, and 400 μg/ml nourseothricin (Werner BioAgents) for nourseothricin-resistant isolates. Synthetic glycerol (SGlycerol) solid medium (1.7% yeast nitrogen base without amino acids, 2% agar, and 0.1 M glycerol) was used to induce chlamydospores, as described below.
TABLE 1.
Strains used in this study
| Strain | Genotype | Source or reference |
|---|---|---|
| C. dubliniensis | ||
| Cd1465 | Wild type | This study |
| Cd1466 | Wild type | This study |
| Cd1467 | Wild type | This study |
| BEM7 | Cd1465, plus cda2Δ::FRT-SAT1::FLIP-FRT /cda2Δ:: FRT-SAT1::FLIP-FRT | This study |
| BEM8 | Cd1465, plus chs3Δ:: FRT-SAT1::FLIP-FRT /chs3Δ:: FRT-SAT1::FLIP-FRT | This study |
| BEM9 | Cd1465, plus dit1Δ:: FRT-SAT1::FLIP-FRT /dit1Δ:: FRT-SAT1::FLIP-FRT | This study |
| BEM10 | Cd1465, plus dit2Δ:: FRT-SAT1::FLIP-FRT /dit2Δ:: FRT-SAT1::FLIP-FRT | This study |
| BEM11 | Cd1465, plus mum3Δ:: FRT-SAT1::FLIP-FRT /mum3Δ::FRT-SAT1::FLIP-FRT | This study |
| BEM13 | Cd1465, plus rrt8Δ:: FRT-SAT1::FLIP-FRT /rrt8Δ:: FRT-SAT1::FLIP-FRT | This study |
| BEM14 | Cd1465, plus srt1Δ:: FRT-SAT1::FLIP-FRT /srt1Δ:: FRT-SAT1::FLIP-FRT | This study |
| BEM15 | Cd1465, plus cda2Δ::FRT/cda2Δ::FRT RPS1::PCDA2CDA2-CIp10-SAT1/RPS1 | This study |
| BEM16 | Cd1465, plus chs3Δ::FRT/chs3Δ::FRT RPS1::PCHS3CHS3-CIp10-SAT1/RPS1 | This study |
| BEM17 | Cd1465, plus mum3Δ::FRT/mum3Δ::FRT RPS1::PMUM3MUM3-CIp10-SAT1/RPS1 | This study |
| BEM18 | Cd1465, plus srt1Δ::FRT/srt1Δ::FRT RPS1::PSRT1SRT1-CIp10-SAT1/RPS1 | This study |
| BEM19 | Cd1465, plus cda2Δ::FRT/cda2Δ::FRT RPS1::PCDA2CDA2-yEmRFP-CIp10-SAT1/RPS1 | This study |
| BEM20 | Cd1465, plus mum3Δ::FRT/mum3Δ::FRT RPS1::PMUM3MUM3-yEmRFP-CIp10-SAT1/RPS1 | This study |
| BEM21 | Cd1465, plus RPS1::PRRT8RRT8-yEmRFP-CIp10-SAT1/RPS1 | This study |
| BEM22 | Cd1465, plus srt1Δ::FRT/srt1Δ::FRT RPS1::PSRT1SRT1-yEmRFP-CIp10-SAT1/RPS1 | This study |
| C. albicans | ||
| NLC1 | arg4/arg4 his1/his1 leu2/leu2 nrg1ΔCmLEU2/nrg1ΔCdHIS1 | 65 |
Induction of chlamydospores.
To induce chlamydospore formation in C. dubliniensis, wild-type and mutant strains were inoculated in 5 ml of YPD liquid and incubated at 30°C with shaking at 220 rpm for overnight. A suspension of 1× 107 cells/ml was prepared from the overnight culture. The cell suspension was then diluted 100 times and 1 ml was spread on a SGlycerol plate. Excess liquid was removed by pipetting, and the plates were left to dry at room temperature. All the plates were incubated in 30°C for 24 h. The chlamydospores were collected by adding 500 μl of distilled water to the plate and gently scraping the surface of the plate with a glass rod. To induce chlamydospores in C. albicans, a slice was made in a plate of corn meal agar with Tween 80 (Hardy Diagnostics, USA). 100 μl of an overnight culture in YPD medium was inoculated into the slice, and a cover slip placed over the line of inoculation. The plates were incubated at 25°C for 5 days.
CRISPR-Cas9 mutagenesis in C. dubliniensis.
To create knockout mutations in C. dubliniensis, we adapted a CRISPR/CAS9 system developed for C. albicans (61). The pV1093 vector carries both Cas9 and single-guide RNA (sgRNA) expression cassettes (61). Guide RNAs targeting specific genes were designed using the CCTop (CRISPR/Cas9 target online) program (62). The CAS9 gene expression cassette and the sgRNA scaffold were amplified separately from pV1093 using the primers BLD1 and BLD2. The sgRNA scaffold contains the SNR52 promoter was assembled by the single-joint PCR method (63). Briefly, three-DNA synthesis step was used to generate the sgRNA cassette. The first step consists to amplify by PCR the SNR52 promoter and sgRNA scaffold using gene-specific flanking primers (Table 2) and internal chimeric primers (BLD3 and BLD4). Twenty complementary bases overlapped and specified the sgRNA of each gene to be knocked out. For the second step, both components were fused by primer extension, relying upon annealing of the complementary chimeric primer extensions. The third step consists of amplifying the joined product with nested primers (BLD5 and BLD6) to yield the sgRNA cassette.
TABLE 2.
Oligonucleotide primers used in this study
| Primer | Key feature | Sequence (5′–3′) |
|---|---|---|
| BLD1 | CaCas9 forward | ATCTCATTAGATTTGGAACTTGTGGGTT |
| BLD2 | CaCas9 reverse | TTCGAGCGTCCCAAAACCTTCT |
| BLD3 | SNR52 forward | AAGAAAGAAAGAAAACCAGGAGTGAA |
| BLD4 | sgRNA reverse | ACAAATATTTAAACTCGGGACCTGG |
| BLD5 | SNR52 NGG | GCGGCCGCAAGTGATTAGACT |
| BLD6 | sgRNA NGG | GCAGCTCAGTGATTAAGAGTAAAGATGG |
| BLD17 | CDA2 FLP forward | CGGTTTAATAGTCATTTAATAAAAACTCTTGAAATTCTTATCAAATAAACTAATCATTCTTCAATTACCATAAAGGGAACAAAAGCTGGG |
| BLD18 | CDA2 FLP reverse | CAACACTAAATTCTTCTTTGTAACCACCTACCTACCTACATACATACATACATACAATACAAGAATTTTTGTATTGATCTCTAGAACTAGTGGATCTG |
| BLD19 | DIT1 SNR52 reverse | GATGATTTACATGGAAAGGCCAAATTAAAAATAGTTTACGCAAGTC |
| BLD20 | DIT1 sgRNA forward | GCCTTTCCATGTAAATCATCGTTTTAGAGCTAGAAATAGCAAGTTAAA |
| BLD23 | DIT1 FLP forward | CGTTGAATTCAAATACAAGTAGTAATACCACGGTTGATACAGATTCGTTTGAACAAAAGCAACAACAAATATTGAAGCTAAAGGGAACAAAAGCTGGG |
| BLD24 | DIT1 FLP reverse | CGTTTTCACTCTCGTCACAGTTGGCCACAACCTATCGTCAGAAGAAGAAACAATAATCCAACGGAACAAACCTCTAGAACTAGTGGATCTG |
| BLD25 | DIT1 upstream verification forward | GGCTGCAATTTCCCCAAAAG |
| BLD26 | DIT1 downstream verification reverse | GCCAGAGTAGCCAACAAGTTA |
| BLD27 | CDA2 upstream verification forward | TTCCGGTGGTAATTTTGATGAGA |
| BLD29 | DIT1 midgene verification reverse | GGTCCCATGATGATGACAGG |
| BLD30 | CDA2 midgene verification reverse | TTTGTTGAGAGCATCCCACC |
| BLD31 | CDA2 downstream verification forward | GACTCGGTGCAATCTTGTCA |
| BLD36 | MUM3 sgRNA forward | GTAGTCCAAATATTTACTTCGTTTTAGAGCTAGAAATAGCAAGTTAAA |
| BLD37 | MUM3 SNR52 reverse | GAAGTAAATATTTGGACTACCAAATTAAAAATAGTTTACGCAAGTC |
| BLD38 | MUM3 FLP forward | GGCGACACTACCGATGCCAATCCCGCTGTGGTAGTAAGTAACCATGCATCTTTAGCGGACTGCTTTGTTATTCTAAAGGGAACAAAAGCTGGG |
| BLD39 | MUM3 FLP reverse | CTACCGGATTCAAAGAGATGAAAGTAGTAAATCAAGAATTTATAGTTTACCTATAGGTAGGATTCAAGAGAACCTCTAGAACTAGTGGATCTG |
| BLD40 | MUM3 upstream verification forward | CAGCATTTGAATAAGGTAAA |
| BLD41 | MUM3 midgene verification reverse | TGTCCCTGTAACGTTGCTCC |
| BLD42 | MUM3 downstream verification reverse | GGGAGATAGGTTTACTGATC |
| BLD43 | RRT8 sgRNA forward | GGTACGGAGTCGTTGCACTTGTTTTAGAGCTAGAAATAGCAAGTTAAA |
| BLD44 | RRT8 SNR52 reverse | AAGTGCAACGACTCCGTACCCAAATTAAAAATAGTTTACGCAAGTC |
| BLD45 | RRT8 FLP forward | CCAATCTTCTAGACGTGGGCTAAAGGCACATGCAAGATACTTTAAGTTGAAAGGGTTTCTGCGTAGCGACTAAAGGGAACAAAAGCTGGG |
| BLD46 | RRT8 FLP reverse | GCAGGTTGGTTGTTGAGGTCTAAGTTTAGTAGCAGCAATGAAGGTGGAGTTGCTGCTGGGTTTGGATGTGCTCTAGAACTAGTGGATCTG |
| BLD47 | RRT8 upstream verification forward | GTGGGCCCAATCATTGTCTTG |
| BLD48 | RRT8 midgene verification reverse | TGATAAATGGGAACAGCTCG |
| BLD49 | RRT8 downstream verification reverse | CGGGTGAAATCTTGACCAAC |
| BLD50 | SRT1 sgRNA forward | TGGGAAAGAACCTCGTGTCCGTTTTAGAGCTAGAAATAGCAAGTTAAA |
| BLD51 | SRT1 SNR52 reverse | GGACACGAGGTTCTTTCCCACAAATTAAAAATAGTTTACGCAAGTC |
| BLD52 | SRT1 FLP forward | CAAAATAAGTTAACCAGAAAAGCAATACTTGTCTTGTAAGTCGGAAAGCTTTTTACAAGATCATAGTTCCAGTAAAGGGAACAAAAGCTGGG |
| BLD53 | SRT1 FLP reverse | GAATCTATTCATGACAACTTTGCATATTCTAGCTAAAATACAAAATACAATCGTAAAGCAAGGCTCTAGAACTAGTGGATCTG |
| BLD54 | SRT1 upstream verification forward | GGATTAATTGTCGAGTGGCA |
| BLD55 | SRT1 midgene verification reverse | GTAATACTGGTGGAATAAC |
| BLD56 | SRT1 downstream verification reverse | TAAATAACCAGGTAGACTTG |
| BLD64 | CHS3 sgRNA forward | AAGGTGGACGTGAAGTTTATGTTTTAGAGCTAGAAATAGCAAGTTAAA |
| BLD65 | CHS3 SNR52 reverse | ATAAACTTCACGTCCACCTTCAAATTAAAAATAGTTTACGCAAGTC |
| BLD66 | CHS3 FLP forward | CCCTTGCATTAACACCAAAACTTATAGACAACAGAAACATTAGTCTTTTTTGTTTTCTACATTTATTCCTCTAAAGGGAACAAAAGCTGGG |
| BLD67 | CHS3 FLP reverse | GTACAATGCATGCAATAAACAAGGCAGAAATTTGAAATATTCTGGAGCCTCTATGTTATAAAGCAGCGTTGCTCTAGAACTAGTGGATCTG |
| BLD68 | CHS3 upstream verification forward | GTTTTCAATTACAATTAATC |
| BLD69 | CHS3 midgene verification reverse | CATAATCGTTAATTTCATCG |
| BLD70 | CHS3 downstream verification reverse | TTTGTGTTTGTAAGAGATTC |
| BLD71 | CDA2 sgRNA forward | ATCCGATCCATTTATTATGGGTTTTAGAGCTAGAAATAGCAAGTTAAA |
| BLD72 | CDA2 SNR52 reverse | CCATAATAAATGGATCGGATCAAATTAAAAATAGTTTACGCAAGTC |
| BLD73 | CDA2 verification reverse | CATGAATTTAGATTGAAGTC |
| BLD74 | DIT2 sgRNA forward | TTAGTGCTCATGGAGAATTGGTTTTAGAGCTAGAAATAGCAAGTTAAA |
| BLD75 | DIT2 SNR52 reverse | CAATTCTCCATGAGCACTAACAAATTAAAAATAGTTTACGCAAGTC |
| BLD76 | DIT2 FLP forward | GCACAGATAACCCTTTTGCTATTTGAGAACCATCCGGGTGATACTAGCCTTGCTCTTTCCTCTTAAACAAGTAAAGGGAACAAAAGCTGGG |
| BLD77 | DIT2 FLP reverse | GTGAGTGTGGGGTGTTTTCTGTTAGCAAACGCAAGTTATATACTATATGGTATGTACTGCATTCTTCATTCCTCTAGAACTAGTGGATCTG |
| BLD78 | DIT2 upstream verification forward | GACAATGAAATTTCCAAGACTCC |
| BLD79 | DIT2 midgene verification reverse | GGGCAACAACATCTCGGTATAG |
| BLD80 | DIT2 downstream verification reverse | AAATGCTTAGCTTACAGGGG |
| BLD97 | CIp10_CHS3 forward | CGATACCGTCGACCTCGAGGACAGACAGAGAGAGAGATCAGAGATTGAA |
| BLD104 | CIp10_CDA2 forward | CACTATAGGGCGAATTGGGTACCCGAAATTTAAAGAGACAATTGAAAAAATTACAAGGAG |
| BLD105 | CIp10_CDA2 reverse | GGGAACAAAAGCTGGGTACCTCATTTTGGGAAAGTTTTAATATAATCAATACCACC |
| BLD111 | CIp10_CHS3 reverse | CAAAAGCTGGGTACCGGGCCCTCAACCAGACCCCGAAGATGATCC |
| BLD112 | CIp10_MUM3 forward | CTTATCGATACCGTCGACCTCGAGATGGAATTCATTGAGCATTTAGGAGTCAAGC |
| BLD113 | CIp10_MUM3 reverse | CAAAAGCTGGGTACCGGGCCCCTACAGAGCTACAGAAAAATCATCTTGCAATATACG |
| BLD116 | CIp10_SRT1 forward | TACCGTCGACCTCGAGACAATTATAAATGTTTTCATTAGTGTTGGTAGTGTATCATATGC |
| BLD117 | CIp10_SRT1 reverse | GGGAACAAAAGCTGGGTACCGGGCCCTTAAATAACTGATGTAGCAGGTGGAGGG |
| BLD118 | CIp10_SRT1 verification | GGACAATCTCTTGTTTTTACC |
| BLD121 | CIp10 first half forward | CCCGGTACCCAGCTTTTGTTCCCTTTAGTG |
| BLD123 | CIp10 second half reverse | CTCGAGGTCGACGGTATCG |
| BLD125 | CIp10_RRT8 forward | CGATACCGTCGACCTCGAGATTGTTAATGGGACCACTAGGGGTG |
| BLD126 | CIp10_RRT8 reverse | CAAAAGCTGGGTACCGGGCCCTCAGATGGTATTTGTAGCAGTCTTTGGG |
| BLD142 | yEmRFP forward | ATGGTTTCAAAAGGTGAAGAAGATAATATGGC |
| BLD143 | CIp10_CDA2_yEmRFP reverse | TCTTCACCTTTTGAAACCATTTTTGGGAAAGTTTTAATATAATCAATACCACCAACAC |
| BLD144 | CIp10_MUM3_yEmRFP reverse | CTTCTTCACCTTTTGAAACCATCAGAGCTACAGAAAAATCATCTTGCAATATACG |
| BLD145 | CIp10_RRT8_yEmRFP reverse | CTTCTTCACCTTTTGAAACCATGATGGTATTTGTAGCAGTCTTTGGGG |
| BLD146 | CIp10_SRT1_yEmRFP reverse | CTTCTTCACCTTTTGAAACCATAATAACTGATGTAGCAGGTGGAGGG |
| BLD148 | yEmRFP reverse | CGATACCGTCGACCTCGAGTTATTTATATAATTCATCCATACCACCAGTTGAATGTCT |
| BLD153 | CIp10_RRT8_yEmRFP verification | TGTTACGACAAAAGGCTCAA |
| BLD154 | CIp10_CDA2_SAT1 verification | TACATTTATATAAAACCAGT |
| BLD155 | CIp10_CDA2_yEmRFP verification | GATGAAAAATAATAAAGGTT |
| BLD156 | CIp10_MUM3_yEmRFP verification | ACCGGTAGATCTGTTGATCA |
| BLD157 | CIp10_SRT1_yEmRFP verification | GGAGTTATTATAGAACTATT |
| OKZ67 | CIp10 first half reverse | GTATTCAACATTTCCGTGTCG |
| OKZ68 | CIp10 second half forward | CGACACGGAAATGTTGAATAC |
The FLP recombination target sequence target (FRT) and the SAT1 cassette both encoded in pGR_NAT vector, were flanked by ∼20-bp homology to the 5′ and 3′ regions of the gene to be knocked out. This fragment was PCR amplified and used as the gene deletion construct (46). The oligonucleotides used in this study are listed in Table 2. PCR amplifications were conducted using Ex Taq in accordance with the manufacturer’s instructions (TaKaRa Bio, Inc.).
For the mutagenesis, PCR products for transformation were purified and concentrated with a commercial PCR purification kit (Qiagen, Germantown, MD). The deletion constructs (3 μg) were cotransformed with the CdCAS9 cassette (1 μg) and the sgRNA cassette (1 μg) using the lithium acetate transformation method (64). At least five independent homozygous deletion strains were tested for each mutant.
Rescue of mutant strains.
For each mutant, to confirm that the observed phenotypes were due to the deletion, an integrating plasmid carrying the wild-type gene was constructed. CIp10-SAT (a gift from N. Dean) was used as the vector. To construct the complementing plasmids, CIp10 was amplified as two separate fragments by PCR. The first fragment, amplified with BLD121 and OKZ67, contains the ApaI site at the one end and part of the Amp locus at the other end. The second fragment, amplified with BLD123 and OKZ68, harbors an overlapping fragment of the Amp locus at one end and an XhoI site at the other end. Each gene of interest was amplified by PCR from C. dubliniensis genomic DNA with 15-bp homologous sequence to the region of CIp10 carrying the ApaI or XhoI sites at the opposite ends. CDA2 was amplified with BLD104 and BLD105, CHS3 with BLD97 and BLD11, MUM3 with BLD112 and BLD113, and SRT1 with BLD116 and BLD117. The three fragments were fused by Gibson Assembly (BioLabs) and transformed into Escherichia coli. All of the plasmids used in this study are listed in Table 3.
TABLE 3.
Plasmids used in this study
| Plasmid | Name | Key feature | Source or reference |
|---|---|---|---|
| pNAT | pNAT | PURA3URA3 SAT1 | 46 |
| pV1093 | pV1093 | CaCas9/SAT1 flipper ENO1 | 61 |
| CIp10-SAT | CIp10-SAT | CaRPS1 SAT1 | N. Dean |
| yEpGAP_Cherry | yEpGAP_Cherry | URA3 yEmRFP | 56 |
| pLB1 | CIp10_CDA2 | CaRPS1 PCDA2CDA2 SAT1 | This study |
| pLB2 | CIp10_CHS3 | CaRPS1 PCHS3CHS3 SAT1 | This study |
| pLB3 | CIp10_MUM3 | CaRPS1 PMUM3MUM3 SAT1 | This study |
| pLB4 | CIp10_RRT8 | CaRPS1 PRRT8RRT8 SAT1 | This study |
| pLB5 | CIp10_SRT1 | CaRPS1 PSRT1SRT1 SAT1 | This study |
| pLB6 | CIp10_CDA2_yEmRFP | CaRPS1 PCDA2CDA2 yEmRFP SAT1 | This study |
| pLB7 | CIp10_MUM3_yEmRFP | CaRPS1 PMUM3MUM3 yEmRFP SAT1 | This study |
| pLB8 | CIp10_RRT8_yEmRFP | CaRPS1 PRRT8RRT8 yEmRFPSAT1 | This study |
| pLB9 | CIp10_SRT1_yEmRFP | CaRPS1 PSRT1SRT1 yEmRFP SAT1 | This study |
In order to rescue the mutant strains, we first recycled the selectable marker SAT1. To allow the recycling, the mutant strains were plated on YPM (2% Bacto peptone, 2% maltose, 1% yeast extract, 2% agar) to induce expression of the FLP recombinase (47) and then replica plated to YPD_NAT medium. Colonies that became sensitive to nourseothricin were selected for transformation with the integrating plasmid carrying the corresponding wild-type gene. The plasmids were linearized by digestion with NcoI before transformation into the mutant strains by lithium acetate transformation method (64) with modifications. Briefly, fresh overnight cultures (12 h to 16 h) were diluted 1:50 and incubated for ∼6 h (optical density at 600 nm of 5.0. The cells were harvested, washed once with H2O and once with 100 mM lithium acetate (LiOAc), and resuspended in 100 μl of LiOAc (100 mM). We used a transformation mixture composed of 240 μl of polyethylene glycol (50%), 32 μl of LiOAc (1 M), 33 μl of linearized plasmid (∼30 μg), and 5 μl of ssDNA, to which 100 μl of cell suspension was added. The mixture tube was incubated for overnight at 30°C. The next day, the tube was heat shocked at 44°C for 15 min. The cells were harvested and washed with YPD and then resuspended in 1 ml. The suspension was incubated at 30°C with shaking for 6 h. After the incubation period, the cells were harvested and spread on YPD_NAT plates. The plates were incubated at 30°C, and colonies were visible after 2 days.
Localization of Cda2, Mum3, Rrt8, and Srt1.
To localize the proteins of interest, plasmids were constructed by creating fusion genes that express C-terminal fusions to yEmRFP. First, the CIp10 vector was digested with KpnI and XhoI. Next, the gene of interest was amplified without the stop codon, using genomic DNA obtained from strain Cd1465. The yEmRFP fragment was amplified by PCR using yEpGAP-Cherry vector (56) as the template. As described above, the three fragments were fused by Gibson assembly. The plasmids were linearized by digestion with NcoI and transformed into the nourseothricin-sensitive mutants by the lithium acetate transformation method.
CFW/Eosin Y staining.
Chlamydospores were collected and washed with 1 ml of McIlvaine’s buffer (0.2 M Na2HPO4, 0.1 M citric acid [pH 6.0]), followed by staining with 30 μl of Eosin Y disodium salt (Sigma; 5 mg/ml) in 500 μl of McIlvaine’s buffer for 10 min at room temperature in the dark. Chlamydospores were then washed twice in McIlvaine’s buffer to remove residual dye and resuspended in 200 μl of McIlvaine’s buffer. One microliter of a 1-mg/ml Calcofluor White (CFW) solution (Sigma) was then added to the Eosin Y-stained cells before transfer to microscope slides. The fluorescence of CFW and Eosin Y stains was then examined using DAPI (4′,6′-diamidino-2-phenylindole) and fluorescein isothiocyanate filter sets, respectively.
MDH staining of lipid droplets.
To stain lipid droplets in chlamydospores with monodansylpentane (MDH; Abgent), chlamydospores collected as described above were washed once with 1× PBS, followed by incubation in 1 ml of PBS containing 100 mM MDH for 15 min in 37°C. Chlamydospores were then washed twice with 1× PBS and examined by fluorescence microscopy using a BFP optimized filter set to visualize MDH fluorescence.
Microscopy.
All images were collected on a Zeiss Axio-Imager microscope using a Hamamatsu ER-G camera and Zen 3.0 software. Different exposure times used for the different fluors as follows: Eosin Y, 200 ms; CFW, 5 ms; dityrosine, 2s; DAPI, 2s; yEmRFP, 2s; and MDH, 10 ms.
Transmission electron microscopy.
Chlamydospores were collected as described above and stained for electron microscopy using osmium and thiocarbohydrazide staining as described previously (31). Briefly, chlamydospores were fixed by resuspension in 3% glutaraldehyde in cacodylate buffer, for 1 h, washed once in 0.1 M cacodylate buffer (pH 7.4), and then resuspended in 1% osmium tetroxide and 1% potassium ferricyanide in cacodylate buffer for 30 min at room temperature. Chlamydospores were then washed four times in dH2O, resuspended in 1% thiocarbohydrazide in water, and incubated for 5 min at room temperature, followed by one wash in dH2O and an additional 5-min incubation in 1% osmium tetroxide and 1% potassium ferricyanide. The chlamydospores were then incubated in saturated uranyl acetate for 2 h and dehydrated through a graded series of acetone washes. The dehydrated samples were then treated with 100% propylene oxide for 10 min, embedded in Epon 812, and sectioned, and images were collected on an FEI BioTwin microscope at 80 kV.
Statistics.
Data are presented as means ± the standard errors of the indicated numbers of independent samples. Statistical significance was determined with Student t test (two tailed, heteroscedastic) using Microsoft Excel software. Differences between the analyzed samples were considered significant at P < 0.05.
ACKNOWLEDGMENTS
We thank Neta Dean for reagents and advice, members of the Konopka and Neiman labs for helpful discussions, and Nancy Hollingsworth for comments on the manuscript.
This study was supported by NIH grant GM072540 to A.M.N. and NIH grants R01GM116048 and R01AI047837 to J.B.K.
REFERENCES
- 1.Free SJ. 2013. Fungal cell wall organization and biosynthesis. Adv Genet 81:33–82. doi: 10.1016/B978-0-12-407677-8.00002-6. [DOI] [PubMed] [Google Scholar]
- 2.Garcia-Rubio R, de Oliveira HC, Rivera J, Trevijano-Contador N. 2019. The fungal cell wall: Candida, Cryptococcus, and Aspergillus species. Front Microbiol doi: 10.3389/fmicb.2019.02993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cortes JCG, Curto MA, Carvalho VSD, Perez P, Ribas JC. 2019. The fungal cell wall as a target for the development of new antifungal therapies. Biotechnol Adv 37:107352. doi: 10.1016/j.biotechadv.2019.02.008. [DOI] [PubMed] [Google Scholar]
- 4.Denning DW. 2003. Echinocandin antifungal drugs. Lancet 362:1142–1151. doi: 10.1016/S0140-6736(03)14472-8. [DOI] [PubMed] [Google Scholar]
- 5.Georgopapadakou NH, Tkacz JS. 1995. The fungal cell wall as a drug target. Trends Microbiol 3:98–104. doi: 10.1016/s0966-842x(00)88890-3. [DOI] [PubMed] [Google Scholar]
- 6.Selitrennikoff CP, Nakata M. 2003. New cell wall targets for antifungal drugs. Curr Opin Invest Drugs 4:200–205. [PubMed] [Google Scholar]
- 7.Brown HE, Esher SK, Alspaugh JA. 2020. Chitin: a “hidden figure” in the fungal cell wall. Curr Top Microbiol Immunol 425:83–111. doi: 10.1007/82_2019_184. [DOI] [PubMed] [Google Scholar]
- 8.Kollar R, Reinhold BB, Petrakova E, Yeh HJ, Ashwell G, Drgonova J, Kapteyn JC, Klis FM, Cabib E. 1997. Architecture of the yeast cell wall: β(1→6)-glucan interconnects mannoprotein, β(1→)3-glucan, and chitin. J Biol Chem 272:17762–17775. doi: 10.1074/jbc.272.28.17762. [DOI] [PubMed] [Google Scholar]
- 9.Baker LG, Specht CA, Donlin MJ, Lodge JK. 2007. Chitosan, the deacetylated form of chitin, is necessary for cell wall integrity in Cryptococcus neoformans. Eukaryot Cell 6:855–867. doi: 10.1128/EC.00399-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Briza P, Ellinger A, Winkler G, Breitenbach M. 1988. Chemical composition of the yeast ascospore wall: the second outer layer consists of chitosan. J Biol Chem 263:11569–11574. doi: 10.1016/S0021-9258(18)37997-3. [DOI] [PubMed] [Google Scholar]
- 11.Christodoulidou A, Bouriotis V, Thireos G. 1996. Two sporulation-specific chitin deacetylase-encoding genes are required for the ascospore wall rigidity of Saccharomyces cerevisiae. J Biol Chem 271:31420–31425. doi: 10.1074/jbc.271.49.31420. [DOI] [PubMed] [Google Scholar]
- 12.Matsuo Y, Tanaka K, Matsuda H, Kawamukai M. 2005. cda1+, encoding chitin deacetylase is required for proper spore formation in Schizosaccharomyces pombe. FEBS Lett 579:2737–2743. doi: 10.1016/j.febslet.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 13.Baker LG, Specht CA, Lodge JK. 2011. Cell wall chitosan is necessary for virulence in the opportunistic pathogen Cryptococcus neoformans. Eukaryot Cell 10:1264–1268. doi: 10.1128/EC.05138-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hole CR, Lam WC, Upadhya R, Lodge JK. 2020. Cryptococcus neoformans chitin synthase 3 plays a critical role in dampening host inflammatory responses. mBio 11:e03373-19. doi: 10.1128/mBio.03373-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lam WC, Upadhya R, Specht CA, Ragsdale AE, Hole CR, Levitz SM, Lodge JK. 2019. Chitosan biosynthesis and virulence in the human fungal pathogen Cryptococcus gattii. mSphere 4:e00644. doi: 10.1128/mSphere.00644-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chrissian C, Lin CP, Camacho E, Casadevall A, Neiman AM, Stark RE. 2020. Unconventional constituents and shared molecular architecture of the melanized cell wall of Cryptococcus neoformans and spore wall of Saccharomyces cerevisiae. J Fungi 6:329. doi: 10.3390/jof6040329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neiman AM. 2005. Ascospore formation in the yeast Saccharomyces cerevisiae. Microbiol Mol Biol Rev 69:565–584. doi: 10.1128/MMBR.69.4.565-584.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neiman AM. 2011. Sporulation in the budding yeast Saccharomyces cerevisiae. Genetics 189:737–765. doi: 10.1534/genetics.111.127126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Briza P, Ellinger A, Winkler G, Breitenbach M. 1990. Characterization of a dl-dityrosine-containing macromolecule from yeast ascospore walls. J Biol Chem 265:15118–15123. doi: 10.1016/S0021-9258(18)77231-1. [DOI] [PubMed] [Google Scholar]
- 20.Ishihara S, Hirata A, Nogami S, Beauvais A, Latge JP, Ohya Y. 2007. Homologous subunits of 1,3-β-glucan synthase are important for spore wall assembly in Saccharomyces cerevisiae. Eukaryot Cell 6:143–156. doi: 10.1128/EC.00200-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lynn RR, Magee PT. 1970. Development of the spore wall during ascospore formation in Saccharomyces cerevisiae. J Cell Biol 44:688–692. doi: 10.1083/jcb.44.3.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tachikawa H, Bloecher A, Tatchell K, Neiman AM. 2001. A Gip1p-Glc7p phosphatase complex regulates septin organization and spore wall formation. J Cell Biol 155:797–808. doi: 10.1083/jcb.200107008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Briza P, Breitenbach M, Ellinger A, Segall J. 1990. Isolation of two developmentally regulated genes involved in spore wall maturation in Saccharomyces cerevisiae. Genes Dev 4:1775–1789. doi: 10.1101/gad.4.10.1775. [DOI] [PubMed] [Google Scholar]
- 24.Whelan WL, Ballou CE. 1975. Sporulation in d-glucosamine auxotrophs of Saccharomyces cerevisiae: meiosis with defective ascospore wall formation. J Bacteriol 124:1545–1557. doi: 10.1128/JB.124.3.1545-1557.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Orlean P. 2012. Architecture and biosynthesis of the Saccharomyces cerevisiae cell wall. Genetics 192:775–818. doi: 10.1534/genetics.112.144485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shaw JA, Mol PC, Bowers B, Silverman SJ, Valdivieso MH, Duran A, Cabib E. 1991. The function of chitin synthases 2 and 3 in the Saccharomyces cerevisiae cell cycle. J Cell Biol 114:111–123. doi: 10.1083/jcb.114.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Silverman SJ. 1989. Similar and different domains of chitin synthases 1 and 2 of S. cerevisiae: two isozymes with distinct functions. Yeast 5:459–467. doi: 10.1002/yea.320050605. [DOI] [PubMed] [Google Scholar]
- 28.Pammer M, Briza P, Ellinger A, Schuster T, Stucka R, Feldmann H, Breitenbach M. 1992. DIT101 (CSD2, CAL1), a cell cycle-regulated yeast gene required for synthesis of chitin in cell walls and chitosan in spore walls. Yeast 8:1089–1099. doi: 10.1002/yea.320081211. [DOI] [PubMed] [Google Scholar]
- 29.Christodoulidou A, Briza P, Ellinger A, Bouriotis V. 1999. Yeast ascospore wall assembly requires two chitin deacetylase isozymes. FEBS Lett 460:275–279. doi: 10.1016/s0014-5793(99)01334-4. [DOI] [PubMed] [Google Scholar]
- 30.Briza P, Eckerstorfer M, Breitenbach M. 1994. The sporulation-specific enzymes encoded by the DIT1 and DIT2 genes catalyze a two-step reaction leading to a soluble ll-dityrosine-containing precursor of the yeast spore wall. Proc Natl Acad Sci U S A 91:4524–4528. doi: 10.1073/pnas.91.10.4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coluccio A, Bogengruber E, Conrad MN, Dresser ME, Briza P, Neiman AM. 2004. Morphogenetic pathway of spore wall assembly in Saccharomyces cerevisiae. Eukaryot Cell 3:1464–1475. doi: 10.1128/EC.3.6.1464-1475.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Felder T, Bogengruber E, Tenreiro S, Ellinger A, Sa-Correia I, Briza P. 2002. Dtrlp, a multidrug resistance transporter of the major facilitator superfamily, plays an essential role in spore wall maturation in Saccharomyces cerevisiae. Eukaryot Cell 1:799–810. doi: 10.1128/ec.1.5.799-810.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gomez-Esquer F, Rodriguez-Pena JM, Diaz G, Rodriguez E, Briza P, Nombela C, Arroyo J. 2004. CRR1, a gene encoding a putative transglycosidase, is required for proper spore wall assembly in Saccharomyces cerevisiae. Microbiology (Reading) 150:3269–3280. doi: 10.1099/mic.0.27314-0. [DOI] [PubMed] [Google Scholar]
- 34.Hoffmann R, Grabińska K, Guan Z, Sessa WC, Neiman AM. 2017. Long-chain polyprenols promote spore wall formation in Saccharomyces cerevisiae. Genetics 207:1371–1386. doi:10.1534/genetics.117.300322. doi: 10.1534/genetics.117.300322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin CP, Kim C, Smith SO, Neiman AM. 2013. A highly redundant gene network controls assembly of the outer spore wall in Saccharomyces cerevisiae. PLoS Genet 9:e1003700. doi: 10.1371/journal.pgen.1003700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ren J, Pei-Chen Lin C, Pathak MC, Temple BR, Nile AH, Mousley CJ, Duncan MC, Eckert DM, Leiker TJ, Ivanova PT, Myers DS, Murphy RC, Brown HA, Verdaasdonk J, Bloom KS, Ortlund EA, Neiman AM, Bankaitis VA. 2014. A phosphatidylinositol transfer protein integrates phosphoinositide signaling with lipid droplet metabolism to regulate a developmental program of nutrient stress-induced membrane biogenesis. Mol Biol Cell 25:712–727. doi: 10.1091/mbc.E13-11-0634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Staib P, Morschhauser J. 2007. Chlamydospore formation in Candida albicans and Candida dubliniensis: an enigmatic developmental programme. Mycoses 50:1–12. doi: 10.1111/j.1439-0507.2006.01308.x. [DOI] [PubMed] [Google Scholar]
- 38.Sudbery P, Gow N, Berman J. 2004. The distinct morphogenic states of Candida albicans. Trends Microbiol 12:317–324. doi: 10.1016/j.tim.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 39.Martin SW, Douglas LM, Konopka JB. 2005. Cell cycle dynamics and quorum sensing in Candida albicans chlamydospores are distinct from budding and hyphal growth. Eukaryot Cell 4:1191–1202. doi: 10.1128/EC.4.7.1191-1202.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bottcher B, Pollath C, Staib P, Hube B, Brunke S. 2016. Candida species rewired hyphae developmental programs for chlamydospore formation. Front Microbiol 7:1697. doi: 10.3389/fmicb.2016.01697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Staib P, Morschhauser J. 2005. Differential expression of the NRG1 repressor controls species-specific regulation of chlamydospore development in Candida albicans and Candida dubliniensis. Mol Microbiol 55:637–652. doi: 10.1111/j.1365-2958.2004.04414.x. [DOI] [PubMed] [Google Scholar]
- 42.Jansons VK, Nickerson WJ. 1970. Induction, morphogenesis, and germination of the chlamydospore of Candida albicans. J Bacteriol 104:910–921. doi: 10.1128/JB.104.2.910-921.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Staib P, Morschhauser J. 1999. Chlamydospore formation on Staib agar as a species-specific characteristic of Candida dubliniensis. Mycoses 42:521–524. doi: 10.1046/j.1439-0507.1999.00516.x. [DOI] [PubMed] [Google Scholar]
- 44.Melo NR, Moran GP, Warrilow AG, Dudley E, Smith SN, Sullivan DJ, Lamb DC, Kelly DE, Coleman DC, Kelly SL. 2008. CYP56 (Dit2p) in Candida albicans: characterization and investigation of its role in growth and antifungal drug susceptibility. Antimicrob Agents Chemother 52:3718–3724. doi: 10.1128/AAC.00446-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hernandez-Cervantes A, Znaidi S, van Wijlick L, Denega I, Basso V, Ropars J, Sertour N, Sullivan D, Moran G, Basmaciyan L, Bon F, Dalle F, Bougnoux ME, Boekhout T, Yang Y, Li Z, Bachellier-Bassi S, d’Enfert C. 2020. A conserved regulator controls asexual sporulation in the fungal pathogen Candida albicans. Nat Commun 11:6224. doi: 10.1038/s41467-020-20010-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Min K, Ichikawa Y, Woolford CA, Mitchell AP. 2016. Candida albicans gene deletion with a transient CRISPR-Cas9 system. mSphere 1:e00130-16. doi: 10.1128/mSphere.00130-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reuss O, Vik A, Kolter R, Morschhauser J. 2004. The SAT1 flipper, an optimized tool for gene disruption in Candida albicans. Gene 341:119–127. doi: 10.1016/j.gene.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 48.Suda Y, Rodriguez RK, Coluccio AE, Neiman AM. 2009. A screen for spore wall permeability mutants identifies a secreted protease required for proper spore wall assembly. PLoS One 4:e7184. doi: 10.1371/journal.pone.0007184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Henar Valdivieso M, Duran A, Roncero C. 1999. Chitin synthases in yeast and fungi. EXS 87:55–69. doi: 10.1007/978-3-0348-8757-1_4. [DOI] [PubMed] [Google Scholar]
- 50.Lenardon MD, Whitton RK, Munro CA, Marshall D, Gow NA. 2007. Individual chitin synthase enzymes synthesize microfibrils of differing structure at specific locations in the Candida albicans cell wall. Mol Microbiol 66:1164–1173. doi: 10.1111/j.1365-2958.2007.05990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mio T, Yabe T, Sudoh M, Satoh Y, Nakajima T, Arisawa M, Yamada-Okabe H. 1996. Role of three chitin synthase genes in the growth of Candida albicans. J Bacteriol 178:2416–2419. doi: 10.1128/jb.178.8.2416-2419.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murad AM, Lee PR, Broadbent ID, Barelle CJ, Brown AJ. 2000. CIp10, an efficient and convenient integrating vector for Candida albicans. Yeast 16:325–327. doi: 10.1002/1097-0061(20000315)16:4<325::AID-YEA538>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 53.Shannon JL. 1981. Scanning and transmission electron microscopy of Candida albicans chlamydospores. J Gen Microbiol 125:199–203. doi: 10.1099/00221287-125-1-199. [DOI] [PubMed] [Google Scholar]
- 54.Jansons VK, Nickerson WJ. 1970. Chemical composition of chlamydospores of Candida albicans. J Bacteriol 104:922–932. doi: 10.1128/JB.104.2.922-932.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Currie E, Guo X, Christiano R, Chitraju C, Kory N, Harrison K, Haas J, Walther TC, Farese RV, Jr.. 2014. High-confidence proteomic analysis of yeast LDs identifies additional droplet proteins and reveals connections to dolichol synthesis and sterol acetylation. J Lipid Res 55:1465–1477. doi: 10.1194/jlr.M050229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Keppler-Ross S, Noffz C, Dean N. 2008. A new purple fluorescent color marker for genetic studies in Saccharomyces cerevisiae and Candida albicans. Genetics 179:705–710. doi: 10.1534/genetics.108.087080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Strippoli V, Simonetti N. 1975. Specific induction of chlamydospore formation in Candida albicans by N-acetyl-d-glucosamine. Experientia 31:130–131. doi: 10.1007/BF01924719. [DOI] [PubMed] [Google Scholar]
- 58.Torosantucci A, Cassone A. 1983. Induction and morphogenesis of chlamydospores in an agerminative variant of Candida albicans. Sabouraudia 21:49–57. doi: 10.1080/00362178385380081. [DOI] [PubMed] [Google Scholar]
- 59.Palige K, Linde J, Martin R, Bottcher B, Citiulo F, Sullivan DJ, Weber J, Staib C, Rupp S, Hube B, Morschhauser J, Staib P. 2013. Global transcriptome sequencing identifies chlamydospore specific markers in Candida albicans and Candida dubliniensis. PLoS One 8:e61940. doi: 10.1371/journal.pone.0061940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chrissian C, Camacho E, Kelly JE, Wang H, Casadevall A, Stark RE. 2020. Solid-state NMR spectroscopy identifies three classes of lipids in Cryptococcus neoformans melanized cell walls and whole fungal cells. J Biol Chem 295:15083–15096. doi: 10.1074/jbc.RA120.015201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vyas VK, Barrasa MI, Fink GR. 2015. A Candida albicans CRISPR system permits genetic engineering of essential genes and gene families. Sci Adv 1:e1500248. doi: 10.1126/sciadv.1500248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stemmer M, Thumberger T, Del Sol Keyer M, Wittbrodt J, Mateo JL. 2015. CCTop: an intuitive, flexible, and reliable CRISPR/Cas9 target prediction tool. PLoS One 10:e0124633. doi: 10.1371/journal.pone.0124633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu JH, Hamari Z, Han KH, Seo JA, Reyes-Dominguez Y, Scazzocchio C. 2004. Double-joint PCR: a PCR-based molecular tool for gene manipulations in filamentous fungi. Fungal Genet Biol 41:973–981. doi: 10.1016/j.fgb.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 64.Walther A, Wendland J. 2003. An improved transformation protocol for the human fungal pathogen Candida albicans. Curr Genet 42:339–343. doi: 10.1007/s00294-002-0349-0. [DOI] [PubMed] [Google Scholar]
- 65.Noble SM, French S, Kohn LA, Chen V, Johnson AD. 2010. Systematic screens of a Candida albicans homozygous deletion library decouple morphogenetic switching and pathogenicity. Nat Genet 42:590–598. doi: 10.1038/ng.605. [DOI] [PMC free article] [PubMed] [Google Scholar]