

Candida auris is a recently discovered human fungal pathogen and has shown an alarming potential for developing multi- and pan-resistance toward all classes of antifungals most commonly used in the clinic. Currently, C. auris has been globally recognized as a nosocomial pathogen of high concern due to this evolutionary potential.
KEYWORDS: Candida auris, multidrug resistance, microevolution, whole-genome sequencing, amphotericin B, antifungal agents, caspofungin, drug resistance evolution, experimental evolution, fluconazole, genome analysis
ABSTRACT
Candida auris is globally recognized as an opportunistic fungal pathogen of high concern, due to its extensive multidrug resistance (MDR). Still, molecular mechanisms of MDR are largely unexplored. This is the first account of genome-wide evolution of MDR in C. auris obtained through serial in vitro exposure to azoles, polyenes, and echinocandins. We show the stepwise accumulation of copy number variations and novel mutations in genes both known and unknown in antifungal drug resistance. Echinocandin resistance was accompanied by a codon deletion in FKS1 hot spot 1 and a substitution in FKS1 “novel” hot spot 3. Mutations in ERG3 and CIS2 further increased the echinocandin MIC. Decreased azole susceptibility was linked to a mutation in transcription factor TAC1b and overexpression of the drug efflux pump Cdr1, a segmental duplication of chromosome 1 containing ERG11, and a whole chromosome 5 duplication, which contains TAC1b. The latter was associated with increased expression of ERG11, TAC1b, and CDR2 but not CDR1. The simultaneous emergence of nonsense mutations in ERG3 and ERG11 was shown to decrease amphotericin B susceptibility, accompanied with fluconazole cross-resistance. A mutation in MEC3, a gene mainly known for its role in DNA damage homeostasis, further increased the polyene MIC. Overall, this study shows the alarming potential for and diversity of MDR development in C. auris, even in a clade until now not associated with MDR (clade II), stressing its clinical importance and the urge for future research.
INTRODUCTION
Over the course of a decade since its discovery (1), Candida auris has emerged in at least 39 countries in every inhabited continent (2), occasionally causing health care-associated outbreaks of lethal candidiasis (3). C. auris is substantially different from any other Candida species studied so far, as it behaves like a true multidrug-resistant (MDR) nosocomial pathogen (cf. methicillin-resistant Staphylococcus aureus, MRSA) (3). This was illustrated by the U.S. Centers for Disease Control and Prevention (CDC) in 2019 when they listed C. auris as the first fungus among urgent antimicrobial resistance threats (4). C. auris can become resistant to each drug and each combination of drugs from the three major antifungal drug classes, the azoles (e.g., fluconazole), echinocandins (e.g., caspofungin), and polyenes (e.g., amphotericin B).
Various clinical isolate screening reports indicate fluconazole resistance in over 80% (5–9) and amphotericin B resistance in up to 30% of the isolates tested (6, 7). Echinocandin resistance is less common, found in 2 to 10% in some screenings (6–8, 10), but it is alarmingly on the rise (11, 12). Overall, about 90% of the C. auris isolates are estimated to have acquired resistance to at least one drug, while 30 to 41% are resistant to two drugs, and roughly 4% are pan-resistant (resistance to the three major antifungal drug classes) (4, 7). These numbers show an unprecedented potential to acquire MDR, unlike any other pathogenic Candida species (3, 12, 13). The molecular mechanisms of antifungal drug resistance in C. auris, especially for amphotericin B resistance and MDR, are still poorly understood. Hundreds of resistant C. auris strains have been sequenced, and their decreased drug susceptibility for azoles and echinocandins has been associated with a few mutations in genes known to be involved in drug resistance. Still, the high levels of resistance and extensive MDR in some strains cannot be explained through the limited number of resistance-conferring mutations described so far (3, 7).
Azole resistance has been linked to three single nucleotide polymorphisms (SNPs) (7–9, 14) and an increased copy number (9, 15) of ERG11, the gene encoding the fluconazole target lanosterol 14-α-demethylase. The ATP binding cassette (ABC) transporter Cdr1 was proven to act as an efflux pump of azoles in C. auris (16–18), and another study suggests that gain of function (GOF) mutations in TAC1b can underly this mode of action (16). A recent study suggests that azole resistance can be the result of a duplication of chromosome V, which contains several genes involved in drug resistance and ergosterol biosynthesis (19). Reduced echinocandin susceptibility in C. auris was previously only linked to SNPs substituting amino acids S639 (9, 12, 20) and F635 (21) and a deletion of F635 (22) in Fks1, which is the echinocandin target, β(1,3) d-glucan synthase. The polyene amphotericin B works by sequestering ergosterol and induction of oxidative stress (23), rather than inhibiting a specific enzyme, and therefore, amphotericin B resistance is among the least understood drug resistance mechanisms in C. auris and Candida spp. in general (12, 23). So far, only an increased expression of genes involved in ergosterol biosynthesis (i.e., ERG1, ERG2, ERG6, and ERG16) (15) and SNPs in ERG2 (24), FLO8, and an unnamed membrane transporter-encoding gene (25) have been linked to amphotericin B resistance in C. auris (12, 20).
Overall, few studies have actually been able to validate the proposed drug resistance mechanisms in C. auris (16, 18, 26, 27), presumably because of the lack of an optimized gene-editing system. Here, we apply a strategy of serial transfer-based experimental evolution with the ability to trace back the emergence—and validate the cumulative effect—of single mutations or copy number changes throughout the evolutionary process. By designing allele-specific PCR primers, the presence or absence of specific mutations could easily be screened for by PCR on multiple single clones in the daily evolving populations. Doing so, we tracked down the emergence of 10 nonsynonymous mutations in 8 genes, evolved in 5 separate evolutionary lineages. In this study, we investigated MDR evolution in a clade II C. auris strain, which is understudied compared to other clades (28) and has been suggested to be less prone to drug resistance development (28, 29). Previously, five different clades (clades I to V, i.e., the South Asian, East Asian, African, South American, and Iranian clades, respectively) of C. auris were identified, each phylogenetically separated by thousands of SNPs (7, 30) and often associated with clade-specific virulence and/or drug resistance tendencies (9, 28, 29). This study shows that clade II C. auris can rapidly acquire MDR in vitro, and its mechanisms of resistance provide fundamental new insights on how resistance can be acquired by C. auris. Finally, our study presents both the power and challenges of using in vitro experimental evolution to discover molecular mechanisms of (multi)drug resistance.
RESULTS
C. auris clade II can acquire multidrug resistance rapidly in vitro.
A single colony of C. auris strain B11220, the original type strain described by Satoh et al. (1) from Japan in 2009, was subjected to an in vitro experimental microevolution assay as depicted and described in Fig. 1A and Materials and Methods, respectively. This parental progenitor (further referred to as the wild type [wt]) proved to be pan-susceptible (determined by MIC or MIC50, see Materials and Methods) to fluconazole (MIC50, 1 μg/ml), caspofungin (MIC50, 0.125 μg/ml), and amphotericin B (MIC50, 0.5 μg/ml). Based on these MIC50 values, the wild type strain was exposed (in triplicate) to three concentrations of each drug as follows: 2× MIC50, 1× MIC50, and 0.5× MIC50, or no drug, representing three selective pressures and a control, respectively. Serial transfer with conditional drug treatment (Fig. 1A) was maintained for 30 days or until drug resistance became evident from regular MIC testing. An overview of the ancestry of the evolved strains is depicted in Fig. 1B.
FIG 1.

Schematic overview of the in vitro experimental evolution assay. (A) A single colony is cultured in RPMI-MOPS medium (2% glucose) for 24 h at 37°C after which a standardized inoculum (106 cells) is resuspended in medium containing no drug (control), the drug at a concentration of 2× MIC50, 1× MIC50, and 0.5× MIC50 (shown here) of the particular starting strain. Daily, the culture is rediluted (1/10) in fresh RPMI-MOPS medium (2% glucose) with a concentration of drug based on the OD600 of the control culture (see Materials and Methods). All strains were evolved in triplicate. Daily aliquots of evolving populations were stored in RPMI-MOPS medium containing 25% glycerol at −80°C for later analysis. (B) Ancestry of the five evolved strains that were sequenced. WGS was performed on a single colony. The name of each strain represents the experimental treatment (letter) and day of isolation (number), respectively.
Five strains were evolved and sequenced as follows: F30, C20, A29, FC17, and CF16. Strain B11220 (wild type) was exposed to fluconazole (F-lineage), caspofungin (C-lineage), and amphotericin B (A-lineage). Next, these single resistant strains were exposed to a second drug to acquire multidrug resistance, yielding the FC-lineage for the F (fluconazole-resistant) strain that was given caspofungin, and the CF-lineages for the C (caspofungin-resistant) strain that was given fluconazole, respectively. The name of each strain represents the experimental lineage (letter which refers to the treatment) and day of isolation (number), respectively. Figure 2 and Fig. S1 (supplemental material) show the MIC50 values for each drug of each endpoint evolved strain (F30, C20, A29, FC17, and CF16). The length of the evolution experiment ranged from 16 (CF-lineage) to 30 days (F-lineage), although later it was shown that resistant clones emerged quite early (e.g., after 3 days in C-lineage; see Fig. 2).
FIG 2.
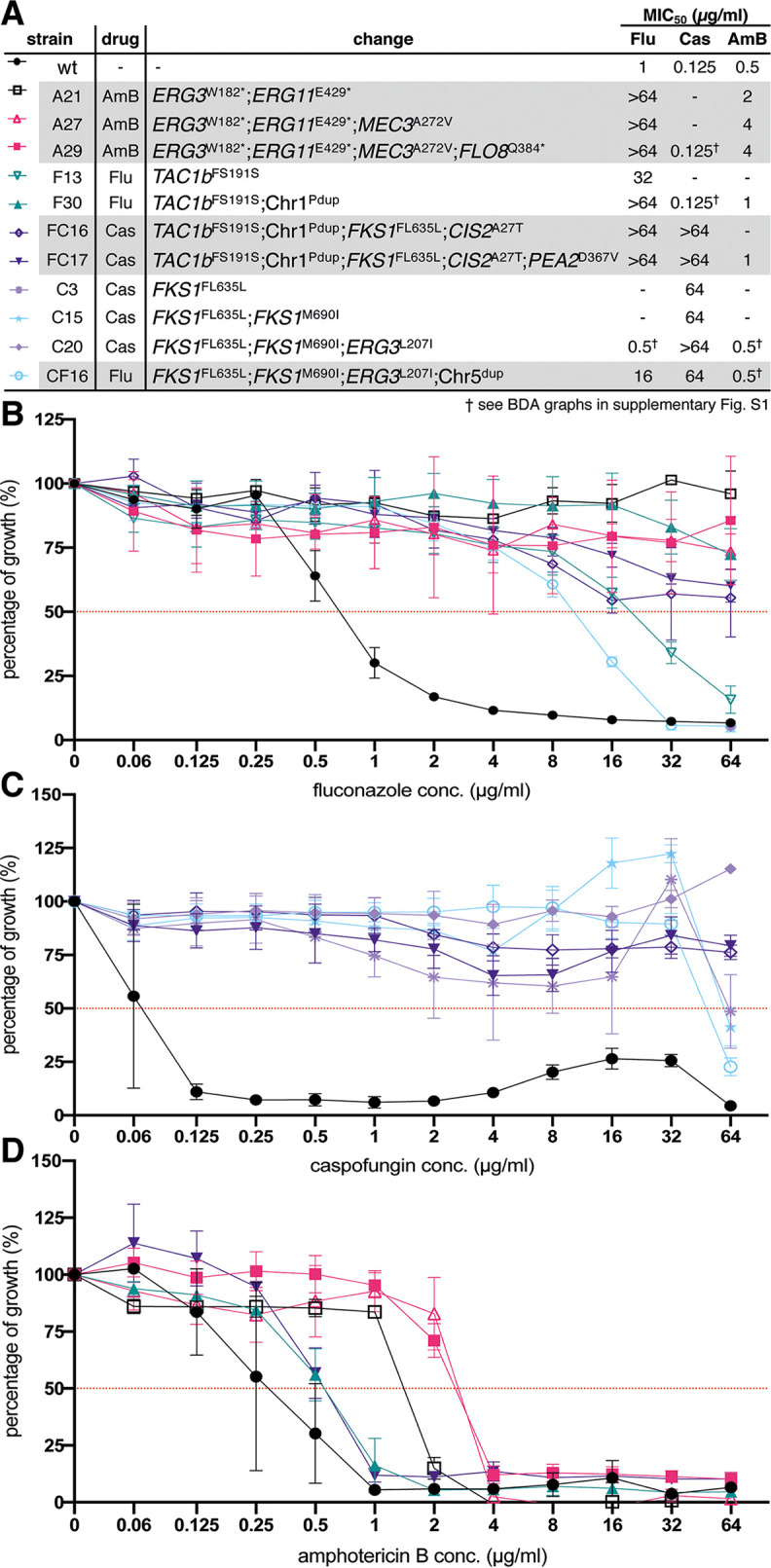
Resistance profiles of endpoint and intermediate evolved strains. (A) Summary of MIC50 values and associated mutations/CNVs for each endpoint strain and divergent intermediate strain. (B to D) Growth profiles of evolved strains relative to the wild-type strain (wt) in a broth dilution assay (BDA) of fluconazole (B), caspofungin (C), and amphotericin B (D). The percentage of growth was calculated from growth without drug and based on OD600 measurements after 48 h of incubation at 37°C. Each data point and its standard deviation is calculated from 3 biological repeats, each represented by the mean of 2 technical repeats. Pdup: partial duplication, dup: duplication. Resistance profiles of endpoint evolved strains for all drugs are found in supplemental material Fig. S1.
Allele-specific PCR is an effective method for tracing back the emergence of mutations during evolution in serial isolates.
After microevolution, whole-genome sequencing of the wild type, F30, C20, A29, FC17, and CF16 strains showed the acquisition of 10 nonsynonymous mutations (listed in Table 1) and two different aneuploidies (shown in Fig. 3 and Fig. S3). All mutations except the deletion of F635 in FKS1 (see below) were novel to C. auris based on literature review and comparison with a set of 304 globally distributed C. auris isolate sequences representing clades I, II, III, and IV (9). The impact and/or cumulative effect of individual mutations and copy number variations (CNVs) will be discussed in the following paragraphs. To validate the effect of single mutations or aneuploidies in strains that harbored more than one mutation, we applied a screening strategy on evolving serial isolates using allele-specific PCR (AS-PCR). AS-PCR primers were designed as described by Liu et al. (31), implementing a specific mismatch at the third position of the 3′ end of the allele-specific primer to increase specificity. An overview of the universal and allele-specific primers used to perform AS-PCR, is given in Table S1. The specificity and sensitivity of all AS-PCR primers was assessed by performing temperature gradient PCRs on serial dilutions of the reference DNA template (for one example, see Fig. S2). Cells were recultured from the −80°C collection of daily stored aliquots (serial isolates or “populations”), and AS-PCR was performed on genomic DNA (gDNA) extracted from a maximum of 30 single clones of each population. After confirmation of the emergence of a mutation of interest, alleles were verified by sequencing a ±1,000-bp region spanning the allele of interest. Primers used for PCR and sequencing are given in Table S1. Next, the effect of single mutations on the drug susceptibility was analyzed by comparing MIC values of the serial isolates through broth dilution assays (BDA; see “Antifungal Susceptibility Testing” in Materials and Methods). Figure 2 shows the impact of each individual mutation on the MIC for the drug of interest for each lineage evolved, except for the A-lineage (i.e., amphotericin B resistance evolution), in which the mutations in ERG3 and ERG11 (Table 1) were either present or absent simultaneously in all colonies that were checked.
TABLE 1.
All nonsynonymous mutations identified in the endpoint evolved strainsa
| Strain | Change | Type of change | Gene ID (B11220) | Gene ID (B8441) | Ortholog |
|---|---|---|---|---|---|
| A29 | tgG/tgA|W182* | Nonsense | CJI96_002270 | B9J08_003737 | ERG3 |
| Gag/Tag|E429* | Nonsense | CJI96_001197 | B9J08_001448 | ERG11 | |
| Cag/Tag|Q384* | Nonsense | CJI96_001121 | B9J08_000401 | FLO8 | |
| gCg/gTg|A272V | Missense | CJI96_001637 | B9J08_003102 | MEC3 | |
| F30 | ttcagt/agt|FS191S | Codon deletion | CJI96_004335 | B9J08_004820 | TAC1b |
| FC17 | ttcagt/agt|FS191S | Codon deletion | CJI96_004335 | B9J08_004820 | TAC1b |
| ttcttg/ttg|FL635L | Codon deletion | CJI96_001351 | B9J08_000964 | FKS1 | |
| gAt/gTt|D367V | Missense | CJI96_001286 | B9J08_001356 | PEA2 | |
| Gca/Aca|A27T | Missense | CJI96_001769 | B9J08_003232 | CIS2 | |
| C20 | atG/atA|M690I | Missense | CJI96_001351 | B9J08_000964 | FKS1 |
| ttcttg/ttg|FL635L | Codon deletion | CJI96_001351 | B9J08_000964 | FKS1 | |
| Cta/Ata|L207I | Missense | CJI96_002270 | B9J08_003737 | ERG3 | |
| CF16 | atG/atA|M690I | Missense | CJI96_001351 | B9J08_000964 | FKS1 |
| ttcttg/ttg|FL635L | Codon deletion | CJI96_001351 | B9J08_000964 | FKS1 | |
| Cta/Ata|L207I | Missense | CJI96_002270 | B9J08_003737 | ERG3 |
Nucleotide and amino acid changes compared to the reference parent strain genome (wild type) are displayed. Genes were identified based on orthologues annotated in the C. auris B8441 genome sequence as provided at http://www.candidagenome.org/.
FIG 3.

Coverage plot of whole-genome sequencing of endpoint evolved strains. The coverage displayed is calculated by normalizing the average coverage depth per 5-kb window. Each color represents 1 chromosome (from left to right, chromosomes 1 to 7). Indicated are the significant duplication in chromosome 1 (Chr1) in strain F30 and FC17 and chromosome 5 (Chr5) in strain CF16.
Resistance profiles of endpoint evolved strains. Growth profiles in a broth dilution assay (BDA) of the endpoint evolved strains (A29, F30, FC17, C20, CF16) and control strain (wt) for fluconazole (A), caspofungin (B), and amphotericin B (C). The percentage of growth was calculated from growth without drug and based on OD600 measurements after 48 h of incubation at 37°C. Each data point and its standard deviation is calculated from 3 biological repeats, each represented by the mean of 2 technical repeats. (D) Summary of MIC50 values for each strain and indication of (relatively low [↑] or high [↑↑]) increase or decrease (↓) of MIC in respect to the parental strain (wt) MIC. Download FIG S1, TIF file, 0.2 MB (156.7KB, tif) .
Copyright © 2021 Carolus et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Example of the temperature specificity range of allele-specific primers. Here, a temperature gradient PCR was performed using the MEC3 allele-specific primers on gDNA of the wild-type strain (wt) and strain A29, containing the gCg/gTg|A272V mutation in MEC3 (see Table 1). Primer pairs “WT: and “Δ” indicate the primers targeting the wild-type allele (CauMEC3_B11220_PCR/Seq1_F and CauMEC3_SNP_A272A_R) and mutated allele (CauMEC3_B11220_PCR/Seq1_F and CauMEC3_SNP_A272V_R), respectively, and are given in Table S1. The green grid indicates a primer-specific temperature range (63 to 64°C). Download FIG S2, TIF file, 0.1 MB (110.8KB, tif) .
Copyright © 2021 Carolus et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Copy number variation quantification of TAC1b (marker on the Chr5 duplication in the CF-lineage; see Fig. 3), ERG11 (marker on the segmental duplication of Chr1 in F- and FC-lineage; see Fig. 3), and ACT1 (reference) by qPCR on gDNA. CNVs were determined for 1 biological repeat represented by 3 technical repeats. Download FIG S3, TIF file, 0.03 MB (26.6KB, tif) .
Copyright © 2021 Carolus et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
All primers used in this study. Primers are arranged per marker. “Purpose” indicates whether the primer was used for PCR and sequencing (PCR/seq), CNV or expression analysis (qPCR), or allele-specific PCR (AS). Primer pairs for AS-PCR consist of a universal PCR-sequencing primer (indicated by “PCR/seq/AS”) or universal AS-primer (indicated by “AS”) and one allele-specific primer (indicated by “AS-wt” for the wild-type allele and “AS-mt” for the mutant allele). Download Table S1, DOCX file, 0.02 MB (18.4KB, docx) .
Copyright © 2021 Carolus et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
High-level echinocandin resistance without fitness discrepancies evolved through mutations in FKS1 and ERG3.
Caspofungin resistance was evolved twice in this study, once as monoresistance in the C-lineage and once in the context of multidrug resistance in the FC-lineage, derived from the fluconazole-resistant strain F30 (Fig. 2). The susceptibility to caspofungin decreased drastically in both strain C20 and FC17 (MIC50, >64 μg/ml) (Fig. 2). Whole-genome sequencing revealed three mutations in the C20 strain, a missense mutation (atG/atA|M690I) and codon deletion (ttcttg/ttg|FL635L) in FKS1 (B9J08_000964; Table 1), the gene encoding the catalytic subunit of the echinocandin drug target β(1,3) d-glucan synthase, and one missense mutation (Cta/Ata|L207I) in ERG3, encoding sterol Δ5,6-desaturase (B9J08_003737; Table 1). The exact same codon deletion (ttcttg/ttg|FL635L) in FKS1 emerged independently during caspofungin resistance evolution in the FC-lineage (Table 1). Two additional mutations emerged during the evolution of strain FC17, namely, a missense mutation (gAt/gTt|D367V) in the PEA2 gene, encoding a subunit of the polarisome (B9J08_001356; Table 1), and a missense mutation (Gca/Aca|A27T) in the CIS2 gene, encoding a γ-glutamylcysteine synthetase (B9J08_003232; Table 1). Tracing back the emergence of these mutations shows that the FKS1 mutation FL635L was accompanied by a 500-fold increase in MIC50, from 0.125 μg/ml to 64 μg/ml (Fig. 2). The emergence of mutations in CIS2 (emerged in FC16) and ERG3 (present in C20) is associated with the further increase of the caspofungin MIC50, exceeding 64 μg/ml (Fig. 2). Acquired echinocandin resistance in fungi has been associated with several specific mutations in three defined “hot spot” regions (HS) in the FKS1 gene (32). Figure 4 shows an amino acid sequence alignment of the FKS1 gene HS1, HS2, and HS3 regions, constructed to compare the mutations found in this study to those known to confer echinocandin resistance in C. auris and other fungi as described in the literature. This literature review shows that the codon deletion at position F635 as found in this study also has been reported to confer decreased echinocandin susceptibility in Candida glabrata (32) and recently in a C. auris clade I strain (22). The same amino acid was substituted (not deleted as in the C-lineage here) in echinocandin-resistant C. auris strains reported prior (21). The FKS1 mutation M690I is located in hot spot region 3 without comparable mutations in pathogenic fungi (Fig. 4) and seems to have no direct impact on the drug susceptibility to caspofungin as measured in the C-lineage (Fig. 2).
FIG 4.

Hot spot (HS) region mutations of the FKS genes that confer echinocandin resistance. Amino acid sequence of hot spots 1, 2, and 3 (HS1 to -3, respectively) of C. auris and other fungi are aligned along with all mutations found to decrease echinocandin susceptibility as described in the literature (references are given between brackets). Species-specific polymorphisms of HS are indicated in gray, and the mutations found to confer echinocandin resistance in this study are indicated by a grid. Δ, deletion; *, nonsense mutation; a, mutations R647G and P649L were exclusively heterozygous; b, FKS2 and FKS1 are functionally redundant in C. glabrata and both mutated in echinocandin-resistant isolates; c, the naturally occurring alanine at position 660 allows intrinsic reduced echinocandin susceptibility in C. parapsilosis.
The mutations in the essential FKS1 gene do not seem to have a significant effect on the fitness of the strains as shown in growth (Fig. 5), stress tolerance (Fig. S4), and cytotoxicity assays (Fig. S5). Interestingly, strain FC17 (mutations in FKS1 and CIS2, see Fig. 2) shows an increased cytotoxicity toward HeLa cells compared to all other strains (Fig. S5).
FIG 5.

Growth curves of endpoint evolved strains. Growth curves were plotted based on culture density (spectrophotometric quantification of OD600; see Materials and Methods) over 72 h of incubation in RPMI-MOPS medium containing (A) 0.2% glucose and (B) 2% glucose at 37°C. Data points are average values of three biological repeats, each represented by the average of two technical repeats.
Stress tolerance of evolved strains. To assess fitness, survival/growth of evolved strains was assessed in different conditions of temperature (A), oxidative stress (B), membrane stress (C), osmotic stress (D), and pH stress (E), as described in Materials and Methods. Only 1 of 3 biological repeats and 9 technical repeats is shown, as all repeats showed similar results. Stress conditions without any growth or with no differential growth between the strains are not shown. Download FIG S4, TIF file, 1.1 MB (1.1MB, tif) .
Copyright © 2021 Carolus et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Cytotoxicity of evolved strains. The relative cytotoxicity of the endpoint evolved strains toward HeLa cells after 24 h (A) and 72 h (B) was evaluated using an LDH cytotoxicity assay as described in Materials and Methods. Bars represent means with standard deviation accounting for data obtained from 3 biological repeats, each represented by the mean of 3 technical repeats. Asterisks indicate a significant difference in relative cytotoxicity; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001. Download FIG S5, TIF file, 0.03 MB (36KB, tif) .
Copyright © 2021 Carolus et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The high febrile temperature mimicking condition (growth at 42°C, see Fig. S4A) is the only condition for which strain C20 seems to slightly differ from the wt strain. This phenotype is also present in strain CF16, of which strain C20 is the progenitor (Fig. 1B). Remarkably, the growth curve of strain C20 shows higher optical density (OD600) values measured after 36 h in RPMI-MOPS medium supplemented with 2% glucose compared to the wt strain (Fig. 5B). Superior growth of C20 is not observed under the physiological condition of RPMI-MOPS medium supplemented with 0.2% glucose (Fig. 5A). Overall, the fitness assessment shows no significance trade-offs associated with the mutations in FKS1 and ERG3 of strain C20.
Cross-resistance to amphotericin B and fluconazole was established after mutagenesis of ERG3, ERG11, FLO8, and MEC3 and seems to be accompanied with fitness trade-offs.
During microevolution, the MIC of amphotericin B increased 8-fold in the A-lineage, from an MIC50 of 0.5 μg/ml (wt strain) to an MIC50 of 4 μg/ml (strain A29; Fig. 2). Simultaneously, cross-resistance to fluconazole emerged, with an MIC increase from 1 μg/ml to over 64 μg/ml (Fig. 2). Two nonsense mutations in genes involved in the ergosterol biosynthesis pathway were discovered (Table 1), namely, the tgG/tgA|W182* mutation in the ERG3 gene and the Gag/Tag|E429* mutation in the ERG11 gene, encoding lanosterol 14-alpha-demethylase (B9J08_001448; Table 1). The ERG11 mutation of strain A29 lies within a region of ERG11 that corresponds to a frequently mutated (“hot spot”) region of ERG11 in azole-resistant C. albicans (33, 34). It is, however, distinct from the three SNPs of ERG11 (namely, Y132F, K143R, and F126L) that have been linked to drug (azole) resistance in C. auris so far (7–9, 14) and are situated in another hot spot region of ERG11 (33, 34).
Additionally, a nonsense mutation (Cag/Tag|Q384*) was found in the transcription factor encoding the FLO8 gene (B9J08_000401; Table 1), and a missense mutation (gCg/gTg|A272V) emerged in the MEC3 gene which encodes a subunit of the Rad17p-Mec3p-Ddc1p sliding clamp (B9J08_003102; Table 1). Remarkably, the mutation in MEC3 is accompanied by a 2-fold decrease in amphotericin B susceptibility (from an MIC50 of 2 μg/ml in strain A21 to an MIC50 of 4 μg/ml in strain A27; see Fig. 2). The mutation in FLO8 did not seem to alter the drug susceptibility for fluconazole or amphotericin B for the concentrations tested. Additionally, strain A29 was found to significantly overexpress TAC1b and ERG11, as shown by reverse transcriptase quantitative PCR (RT-qPCR), depicted in Fig. 6. Characterization of the growth (Fig. 5), stress tolerance (Fig. S4), and cytotoxicity (Fig. S5) of strain A29 and the wt strain shows, however, that fitness trade-offs accompany the accumulation of the four above-mentioned mutations. Strain A29 shows a lower growth rate in exponential phase and a lower density at stationary phase than the wt strain (Fig. 5), especially in glucose-limiting conditions (Fig. 5A). This growth defect might explain the significantly reduced cytotoxicity towards HeLa cells in this strain (Fig. S5). The spot assays displayed in Fig. S4 show that this strain shows a reduced tolerance to high temperature (Fig. S4.A), membrane stress (Fig. S4.C), osmotic stress (Fig. S4.D), and low pH (Fig. S4.E).
FIG 6.

Relative expression of various genes of interest among evolved strains. Fold change of expression levels for CDR1, CDR2, ERG11, and TAC1b for the wild type (wt), endpoint evolved strains (A29, F30, FC17, C20, CF16) and intermediate strains F12 and F13 (for CDR1 and ERG11). Bars represent log2-transformed means with standard deviation accounting for data obtained from 3 biological repeats, each represented by the mean of 2 technical repeats. Asterisks indicate significant overexpression; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001.
Fluconazole resistance is linked to two aneuploidies and a mutation in transcription factor TAC1b.
In strain F13, a codon deletion (ttc/|F15) in the TAC1b gene was identified (B9J08_004820; Table 1) that corresponds to a 32-fold increase in the MIC50 of fluconazole (Fig. 2). Tac1b is a transcription factor that positively regulates the expression of the ATP binding cassette (ABC) transporter Cdr1, known to be involved in azole efflux and azole resistance in C. auris (16, 26). Gene expression analysis of strain F12 (no TAC1b mutation) and strain F13 (TAC1b mutation obtained) confirms that the acquisition of this mutation corresponds to a significantly increased expression of CDR1 (B9J08_000164), as shown in Fig. 6. The overexpression of CDR1 is maintained in strain F30 and in the multidrug-resistant strain FC17, as shown in Fig. 6. Although the mutation in strain F13 is novel to C. auris, it is located in a region of TAC1b that is known to harbor gain-of-function mutations in fluconazole-resistant clinical isolates of C. auris, as shown by Rybak et al. (16).
Two aneuploidies independently emerged during fluconazole resistance evolution. Read coverage of whole-genome sequencing was used to analyze copy number variation (CNV) by calculating the normalized depth read coverage per 5 kb window (see Materials and Methods). A visual representation of this normalized coverage for each chromosome in all endpoint sequenced strains is displayed in Fig. 3. This reveals that a segmental and whole-chromosome duplication emerged in the F- and CF-lineages, respectively.
The 191-kb segmental duplication of Chr1 in the F30 strain contained 75 protein-encoding genes (based on the B11220 reference genome annotation; GenBank accession numbers CP043531 to CP043537), including ERG11. During further evolution to caspofungin resistance in the FC-lineage (see Fig. 1B), this segmental duplication was maintained but decreased in size to 161 kb containing 67 protein-encoding genes (still including ERG11). The segmental Chr1 duplication is the only genetic difference between strain F13 and strain F30 that can be attributed to the increase in MIC50 of 32 μg/ml (in strain F13) to a MIC50 of >64 μg/ml (in strain F30), as shown in Fig. 2. Expression analysis showed that the duplication in strain F30 is accompanied by an increased expression of ERG11, not present in strain F13 (Fig. 6). Strain F30 was also marked by a slight decrease in amphotericin B susceptibility, retained in the FC17 strain (Fig. 2). This is possibly due to ERG11 overexpression.
The whole chromosome 5 (Chr5) duplication in the CF16 strain contained a region of 933 kb encompassing 405 protein-encoding genes, including TAC1b. This aneuploidy marks the only genetic difference between strain C20 and strain CF16 and is therefore suggested to confer the 32-fold decrease in fluconazole susceptibility between those strains (Fig. 2). Expression analysis showed that the duplication of Chr5 correlates with a significant overexpression of TAC1b and CDR2 (B9J08_002451), but not CDR1 in strain CF16 (Fig. 6).
The mutation in TAC1b and segmental duplication of Chr1 did not seem to have any major effect on the growth (Fig. 5), stress tolerance (Fig. S4), and cytotoxicity (Fig. S5) of strain F30 compared to the wt strain. The chromosome 5 duplication might, however, drive the slight reduction in growth at stationary phase (Fig. 5) and tolerance toward certain stressors in strain CF16 compared to its progenitor (C20) and the wt strain (Fig. S4).
DISCUSSION
This study shows the evolution of multiple mechanisms known to be involved in drug resistance in fungi, albeit new to C. auris.
In the largest screening of C. auris clade II strains, 62.3% of a total of 61 isolates proved to be fluconazole resistant, although only 3 isolates harbored a known azole-conferring mutation in ERG11 (K143R) (35). This indicates that other mechanisms of azole resistance play a role in C. auris clade II (35). Here, we suggest that at least four molecular mechanisms, none of which include the most common mutations in ERG11, might underlie the decreased fluconazole susceptibility in clade II C. auris. Previous reports show that many GOF mutations in TAC1 or homologues of this transcription factor can confer azole resistance in Candida species (36–38), including C. auris (16), through an overexpression of the drug efflux pump Cdr1. Many GOF mutations are found in the region encoding the putative transcriptional activation domain of TAC1, situated in the C-terminal portion of the protein in Candida (39). Rybak et al. report one mutation in this region (codon deletion at position F862) to be associated with fluconazole resistance in C. auris, although all other reported resistance-associated mutations lie between the DNA binding, transcription factor, and activation domain of TAC1b (16). One such mutation (F214S), discovered in an experimentally evolved strain of C. auris (16), lies in the proximity of the codon deletion at position 191 as we discover here. Based on these reports and our findings, we therefore hypothesize that F191Δ is a new potential gain of function mutation of C. auris TAC1b, conferring azole resistance through CDR1 overexpression. Nevertheless, previous reports have shown that Tac1b might function in other, Cdr1-independent ways to decrease azole susceptibility in C. auris (16, 26). Another genetic adaptation linked to reduced azole susceptibility in this study involves aneuploidies. In C. albicans, both TAC1 and ERG11 are located on Chr5, and the duplication of this region by forming an isochromosome [i(5L)] has been reported to confer azole resistance in vitro and in vivo (40). Based on this, and other reports on azole resistance in C. auris due to CNVs and/or overexpression of ERG11 (9, 15, 41), we hypothesize that a similar mode of action is at play in strain F30. Moreover, comparing drug susceptibility between strain F13 and F30, the overexpression of ERG11 in strain F30 more than doubles the fluconazole MIC50 compared to—the already resistant—strain F13, while it decreases the susceptibly for amphotericin B (Fig. 2). Recently, azole resistance in an experimentally evolved C. auris clade I strain was linked to the duplication of chromosome 5 and consequent upregulation of ERG11 (Chr1) and TAC1b but not CDR1 (Chr5) (19). In this study, the duplication of C. auris Chr5 is the only genomic alteration that distinguishes strain CF16 from strain C20, and therefore, we propose that this duplication is, here too, responsible for azole resistance. Expression analysis shows that the duplication and subsequent overexpression of TAC1b (Fig. 6) does not correspond to an increased expression of CDR1, making our findings consistent with the report of Bing et al. (19). TAC1b may play a CDR1-independent role in azole resistance of C. auris, as suggested by Mayr et al. (26), and since CDR2 is significantly upregulated in strain CF16, its function in azole resistance should be further investigated.
The acquisition of resistance to polyenes is among the least understood of all antifungal drugs and has been linked to mutations in genes involved in the ergosterol biosynthesis pathway in Candida spp., including alterations in ERG2 (42), ERG6 (43), ERG11 (44), and ERG3 (45). Cross-resistance to azoles and amphotericin B has often been associated with the abrogation of two ERG genes simultaneously (46). One such example is the combination of the loss of function (LOF) of ERG11 and ERG3 in C. tropicalis (47, 48). Upon the abrogation of ERG11, due to a LOF mutation or the action of azoles, a toxic 3,6-diol derivative is produced through the action of the sterol Δ5,6 desaturase, encoded by ERG3 (49). Simultaneous disruption of the function of both ERG3 and ERG11 can prevent this detrimental effect (46). Here, we hypothesize that such a mechanism of cross-resistance might be established also in C. auris. The fact that all clones tested (over 60) between generation A20 and A21 had either both or no nonsense mutations can be explained by simultaneous emergence or an epistatic effect (i.e., the effect of a mutation in one gene depends on a mutation in another gene) of ERG3 and ERG11 LOF mutations in strain A21 (Fig. 2). In the latter scenario, the AmB conferring nonsense mutation in ERG11 was only possible in an ERG3-LOF background and both emerged in a quick and dependent fashion.
Target alteration is the most commonly observed and most studied mechanism of echinocandin resistance in Candida species (50). In C. auris, most echinocandin resistance-conferring FKS1 mutations occur at position S639 (12, 20). Most recently, however, a SNP (21) and codon deletion (22) of FKS1 F635, the same codon deleted in strain C3 in this study, was linked to reduced echinocandin susceptibility of C. auris in the clinic. In general, mutations in echinocandin-resistant Candida species lie within two small, strictly defined hot spot regions of FKS1 (50). However, the codon substitution at position 690 that emerged in strain C15, occurs in the elusive hot spot 3, a third potent hot spot region discovered by site-directed mutagenesis of FKS1 in Saccharomyces cerevisiae (32). This mutation occurred after the codon deletion at position 635 (in HS1) in the C-lineage but did not affect the echinocandin MIC50, possibly indicating functional compensation of the altered Fks1 protein. A third mutation of strain C20 occurred in ERG3. One report shows that a mutation in ERG3 in a clinical Candida parapsilosis strain conferred resistance to both azoles and echinocandins (17). Here, we observe a slight increase, rather than a decrease, in fluconazole susceptibility upon the emergence of the ERG3 mutation in strain C20 (Fig. 2). Most interestingly this mutation further increases the MIC50 for caspofungin in strain C20 compared to strain C15, which only harbored FKS1 mutations (Fig. 2). Overall, the caspofungin-resistant strains (FC17, C20) obtained in this study show echinocandin MIC values (>64 μg/ml) that significantly exceed any previously reported MIC values for echinocandins in C. auris (8, 51–53). As these mutations evolved rapidly (Fig. 2) and do not seem to greatly impact the overall fitness of these strains (see Fig. 5 and Fig. S4 and S5), they might impose a significant clinical threat. We therefore suggest that the clinical significance of the reported mutations, including the role of ERG3, should be further investigated.
Four genes were mutated that were previously not or vaguely associated with drug resistance in fungi.
FLO8, mutated in the amphotericin B-resistant strain A29, encodes a transcription factor known to be essential for filamentation in C. albicans (54). This filamentation was shown to decrease the rate of programmed cell death in C. albicans when exposed to amphotericin B (55). Flo8 has multiple downstream effects, one of which is the positive regulation of ERG11 expression, shown in S. cerevisiae (56), and thus potentially playing a role in azole and amphotericin B resistance. In a recent study of clinical C. auris isolates from South America, a nonsynonymous mutation in the FLO8 gene significantly correlated with amphotericin B resistance (25). In a follow-up study on the structure of Flo8, the authors suggest a potential role of Flo8 in C. auris virulence and drug resistance, arguing that the FLO8 mutation found before (25) could be a gain of function mutation (57). In our study, however, we see a nonsense mutation, abrogating Flo8 at amino acid 100 and thus assumed to be disruptive to its function. Earlier, a LOF of FLO8 was found to play a role in azole resistance, with a FLO8 deletion correlated to increased TAC1, CDR1, and CDR2 expression, while FLO8 overexpression led to decreased CDR1 expression (58). Although these reports strengthen the suspicion of a role of Flo8 in drug resistance, we cannot further explain the influence of the FLO8 mutation on the resistance phenotype observed here. Further research on Flo8 in drug resistance is therefore highly desirable.
The fourth gene mutated during amphotericin B resistance evolution is an ortholog of MEC3, encoding a DNA damage checkpoint protein as part of the Rad17p-Mec3p-Ddc1p sliding clamp, primarily involved in DNA damage recognition and repair in S. cerevisiae (59). No clear reports of a function for MEC3 in antifungal drug resistance were found, although two studies mention the upregulation of MEC3 upon the acquisition of azole resistance in an experimentally evolved C. glabrata strain (38, 60). Our results show that the mutation in MEC3 has a significant influence on susceptibility to amphotericin B (Fig. 2). The mechanism behind increased amphotericin B resistance upon acquiring a mutation in MEC3 remains unclear, although an altered DNA damage recognition response might prevent apoptosis upon exposure to amphotericin B. Multiple reports concerning the mode of action of polyenes suggest that, besides a pore-forming and sterol adsorption effect, polyenes such as amphotericin B are cytotoxic through oxidative damage, which includes protein carbonylation, lipid peroxidation, and DNA damage (23, 61–65). The latter is recognized by DNA damage checkpoint mechanisms and can lead to apoptosis (66), possibly explaining the phenomenon we observe here. Other reports show that defects in DNA damage recognition might drive the emergence of drug resistance in fungi, as they can increase mutation rates (67–69). Notably, a recent report of resistant C. auris in a hospital in India, shows that all (n = 9) sequenced isolates harbored a nonsense mutation in MSH2, a DNA mismatch repair gene (24).
Strain FC17 harbored a mutation in CIS2. The S. cerevisiae ortholog (ECM38) of CIS2 encodes a γ-glutamyltranspeptidase, involved in glutathione degradation (70), detoxification of xenobiotics (71), and cell wall biogenesis (72). The role that CIS2 plays in the latter, regarding echinocandin resistance, remains unclear, but a study from Maras and colleagues (73) illustrated that fluconazole and micafungin resistance were accompanied by altered levels of glutathione in C. albicans, hypothesized to counteract oxidative stress caused by these antifungal drugs. Interestingly, strain FC17 shows an oxidative stress tolerance similar to the wt and mono-resistant strains and higher than the other MDR strains (strain A29 and CF16; see Fig. S4B). Additionally, the cytotoxicity assessment shows that strain FC17 has a significantly higher cytotoxic effect than the wt and other strains after 72 h (Fig. S5B). This might be linked to the mutation in CIS2 and an increased oxidative stress tolerance. Nevertheless, in the study by Maras and colleagues (73), the increased levels of glutathione were accompanied by the overexpression of γ-glutamylcysteine synthetase (73). A role for CIS2 and glutathione catabolism in drug resistance, mediated by an altered redox metabolism remains to be elucidated.
The fourth mutation in the FC-lineage lies within a gene predicted to encode PEA2, a subunit of the polarisome, involved in polarized growth and morphogenesis in S. cerevisiae (74). This mutation has, however, no significant effect on the drug resistance profile and might thus be the result of random genetic drift.
Experimental evolution can be a powerful tool to research resistance, although it has limitations.
Due to recent advances in next-generation sequencing technology, genome-wide studies of drug resistance have become more common (75, 76). The classic approach of sequencing drug-resistant clinical isolates directly from patients (75) has many limitations, including the frequent unavailability of the original drug-susceptible genotype and the difficulty in resolving mutations associated with drug resistance from those that have accumulated due to host-pathogen interactions and random genetic drift. In vitro experimental evolution copes with most of these problems (75, 77), is highly repeatable, and allows controlled long-term monitoring of different strains and conditions. Moreover, the ability to isolate and investigate each generation separately allows monitoring of both the speed and the stepwise progression of drug resistance development. Nevertheless, in vitro experimental evolution has its own limitations, such as the homogeneity of the selective pressure in the absence of metabolization of the drug, tissue-specific exposure, and host-pathogen interactions. However, studies of drug resistance by in vitro evolution often resemble acquired resistance found in clinical isolates (13, 16, 75, 77, 78). In regard to our results, a comparative analysis of mutations reported in the literature and reanalysis of variants predicted in 304 sequenced clinical isolates of C. auris (9) show that most mutations reported here are novel to this species. Although the in vitro context in which these mutations evolved might explain this, one must be careful by regarding these findings to be nonrelevant to the in vivo setting or clinical environment. Reports on resistance mutations (providing whole-genome analysis) are still scarce and the database of 304 sequenced clinical isolates of C. auris (9) is limited, with only 23% of isolates reported being multidrug-resistant and only 7 clinical isolates belonging to clade II (6 isolates pan-susceptible, 1 isolate fluconazole-resistant) (9). This study and other studies of bacteria (79) and fungi (13, 77, 80) show that in vitro experimental evolution can be a powerful tool, especially if combined with an effective approach to trace the full evolutionary history of mutation events, as we did here using allele-specific PCR. Nevertheless, we need to point out that the preferred way of validating molecular mechanisms for resistance should rely on gene-editing the evolved mutations in the parental (wild type) background and/or editing mutant alleles to the wild type form in evolved strains. For this, a CRISPR-based gene editing system would be ideal. So far, no scarless allele-specific CRISPR gene editing system in C. auris has been optimized or reported, although alleles have been deleted, replaced, and complemented with an inducible promoter (16, 18, 27). The CRISPR system reported by Kim et al. (18), based on a system for C. albicans (81), could be used for allele editing in C. auris, but it makes use of the stable integration of the CRISPR cassette into the ENO1 locus (81). Disruption of the ENO1 locus in C. albicans has been reported to alter growth, virulence, and drug susceptibility (82). If the same phenotypes would apply to C. auris, it would complicate the interpretation of the effect that introduced mutations have in the GMO strain. Moreover, constitutive cas9 expression can have off-target effects (83), including an effect on the fitness of the transformed cells, as reported for C. glabrata (84). In conclusion, neutral gene editing, which preferably relies on transient expression of cas9, is a highly desired tool for future molecular research in C. auris and should be regarded as a priority in the field.
MATERIALS AND METHODS
Strains and growth conditions.
All experiments were performed with C. auris strain B11220 (CBS10913) from the Westerdijk Fungal Biodiversity Center (wi.knaw.nl/). Strains were grown on yeast extract-peptone-dextrose (YPD) agar (2% glucose) at 37°C and enriched in RPMI 1640 (Thermo Fisher Scientific)-MOPS (morpholinepropanesulfonic acid) liquid medium containing 2% glucose at 37°C in a shaking incubator overnight. All strains, including daily aliquots of serially transferred populations in the microevolution assay, were stored at −80°C in RPMI-MOPS medium containing 25% glycerol.
Antifungal susceptibility testing.
The MIC was determined using a broth dilution assay (BDA) based on Clinical and Laboratory Standards Institute (CLSI) guidelines (85). In short, a dilution of 64 μg/ml to 0.06 μg/ml of each drug was prepared in RPMI-MOPS medium in a 96-well polystyrene microtiter plate. A standardized amount of 100 to 500 cells was dissolved in a final volume of 200 μl per well, and plates were incubated at 37°C. Growth was measured after 48 h of incubation through spectrophotometric quantification of the OD600 in a SPECTRAmax Plus 384 microplate reader (Molecular Devices). Resistance was determined through tentative breakpoints provided by the CDC (6).
In vitro experimental evolution assay.
An overview of the design of the experimental evolution assay is given in Fig. 1A. At the start of the evolution experiment, 106 cells are diluted in a 5 ml volume of RPMI-MOPS medium (2% glucose) containing no drug (control) or a drug at a concentration of 0.5× MIC50, 1× MIC50, or 2× MIC50. All conditions were performed in triplicate (3 evolving populations per condition). After 24 h of incubation at 37°C in a shaking incubator, growth of each population was compared to the average growth of 3 controls (no drug) by spectrophotometric quantification (OD600). Next, 500 μl of each population was transferred to 4,500 μl of fresh medium with a concentration of drug equal to the previous culture when OD600 (evolving population) ≤ OD600 (average control) or double compared to the previous culture when OD600 (evolving population) > OD600 (average control). The experiment was terminated after 30 days or if the MIC50 exceeded the resistance breakpoint value, as evaluated by intermediate MIC testing (using BDA). At the end of each evolution experiment, a single colony was picked as the progenitor for a consecutive evolution experiment.
Analysis of growth.
Growth was assessed by spectrophotometric observation (OD600) over time in a Multiskan GO (Thermo Scientific) automated plate reader using flat-bottom 96-well plates and intermittent (30 min interval) pulsed shaking (medium strength, 5 min). Cultures were diluted in RPMI-MOPS medium containing 0.2% or 2% glucose, to a final volume of 106 cells per well. Growth was measured for 72 h at 37°C. Growth curves were plotted as an average value of 3 biological repeats with 3 technical repeats per biological repeat.
Analysis of stress tolerance.
The survival of C. auris cells in various degrees of temperature-, oxidative-, membrane-, osmotic- and pH-induced stress was assessed using spot assays. C. auris cells were resuspended in phosphate-buffered saline (PBS) at concentrations of 107, 106, 105, and 104 cells/ml and 1 μl of each dilution was spotted on YPD agar containing 2% glucose. Plates were incubated for 48 h at 37°C or at 30°C, 37°C, and 42°C for the evaluation of temperature stress. Oxidative stress was evaluated on agar containing 6, 8, or 10 mM H2O2 (Sigma-Aldrich). Membrane stress was evaluated on agar containing 0.005, 0.05, or 0.2% sodium dodecyl sulphate (SDS; Sigma-Aldrich). Osmotic stress was evaluated on agar containing 1, 1.75, or 2.5 M NaCl (Sigma-Aldrich). pH stress was evaluated on agar adjusted to pH 4, 5, 6, or 7 by supplementing the agar with HCl or KOH. Agar adjusted to pH 4 and 5 was buffered with 50 mM citrate buffer, while agar adjusted to pH 6 was buffered with 50 mM morpholineethanesulfonic acid (MES) buffer, and agar adjusted to pH 7 was buffered with 100 mM MOPS buffer. All spot assays were performed on 3 biological repeats with 3 technical repeats per biological repeat, of which one example is shown.
Analysis of relative cytotoxicity.
HeLa cells (Thermo Fisher Scientific) were cultured in complete Dulbecco’s modified Eagle medium (DMEM) at 37°C and 5% CO2, using 25-cm2 polystyrene culture flasks. Upon reaching 80% confluence, cells were washed twice with PBS, trypsinized using 0.5% trypsin EDTA (Gibco), and replenished to allow cell adhesion in polystyrene 96-well plates as a 100-μl suspension of 105 cells/ml fresh DMEM culture medium for 24 h, at 37°C and 5% CO2. A 10 μl volume of 105 C. auris cells or PBS (negative control) was added to the HeLa cells, followed by incubation at 37°C and 5% CO2 for 24 h or 72 h. Cytotoxicity of C. auris toward HeLa cells was assessed using a lactate dehydrogenase (LDH) cytotoxicity assay as follows: 10 μl of cell lysis buffer (Invitrogen) was added to cultures of HeLa cells with C. auris or pure HeLa cells (positive control), followed by incubation for 45 min at 37°C and 5% CO2. Then, 50 μl of supernatant (lysate) was mixed with 50 μl of substrate mix (Invitrogen) in a separate 96-well plate and incubated for 30 min at room temperature in darkness. Next, 50 μl of stop solution (Invitrogen) was added, followed by spectrophotometric quantification of the OD490 and OD680 in a Synergy H1 hybrid plate reader (BioTek). LDH activity was calculated by subtracting the OD680 from the OD490, and relative toxicity was calculated as . Relative cytotoxicity for each strain was measured for 3 biological repeats, each represented by the average of 3 technical repeats, and compared statistically with GraphPad Prism using a one-way analysis of variance (ANOVA) with multiple comparisons in respect to the wt strain.
DNA extraction.
Genomic DNA for whole-genome sequencing was extracted using the MasterPure yeast DNA purification kit (Lucigen, USA) following the manufacturer’s protocol. For (AS-)PCR and Sanger sequencing, DNA was isolated from the cells through phenol chloroform isoamyl alcohol (PCI) extraction. Cells were dissolved in 300 μl Tris EDTA (TE) buffer with 300 μl PCI solution (phenol pH 6.7, chloroform, and isoamylalcohol at 25:24:1) and lysed by microbead shearing in a FastPrep-24TM Classic lysis system (20 sec, 6m/sec) (MP Biomedicals). After cell lysis, DNA was isolated and purified using ethanol precipitation. The resulting DNA was diluted to a concentration of 200 ng/μl in milliQ H2O, based on the DNA concentration measured through absorbance at 260 nm with a NanoDrop spectrophotometer (Isogen).
Whole-genome sequencing and analysis.
Genomic libraries were created using the NEBNext Ultra DNA library prep kit for Illumina sequencing (New England Biolabs, USA), and genomes were sequenced on an Illumina MiSeq v2 500 instrument (Illumina, USA), obtaining a coverage of at least 50×. Standard quality control was performed using FastQC v0.11.7 (86). Paired-end reads were aligned using the Burrows-Wheeler Aligner MEM algorithm BWA-MEM v0.7.17 (87) to the annotated genome assemblies of strain B8441 (clade I; GenBank accession number GCA_002759435.2 [15]) and B11220 (clade II; GenBank accession number CP043531 to CP043537 [29]). For SNP and indel identification, the assembly alignment to the annotated genome of strain B8441 (clade I) was used, while CNV analysis was performed using the assembly alignment to reference genome B11220 (clade II), respectively. The genome sequences of all endpoint experimentally evolved strains were deposited in the NCBI Sequence Read Archive (SRA) under BioProject PRJNA664007. Variants were identified and filtered using GATK v4.1.2.1 (88, 89), with the haploid mode, including GATK tools HaplotypeCaller and variant filtration using “QD < 2.0 ‖ FS > 60.0 ‖ MQ < 40.0”. In addition, variants were filtered if they had a minimum genotype (GT) quality of <50, alternate allele frequency of <0.8, or allelic depth (DP) of <10. The final variant call format (VCF) was annotated using SnpEff v4.3T (90). CNVs were identified using CNVnator v0.3 (91), selecting for 1 kb genomic windows of significant (P < 0.01) variation in normalized coverage. The average depth per 5 kb window was normalized to the coverage of the whole-genome sequence for each isolate and plotted in R (92). Candidate variants were compared with a set of 304 globally distributed C. auris isolates representing clades I, II, III, and IV (9).
PCR and Sanger sequencing.
Primers for PCR and Sanger sequencing were designed in silico using CLC Genomics Workbench v20.0.3. Primer design was based on B11220 whole-genome sequencing (WGS) consensus sequences (GenBank accession number CP043531 to CP043537 [29]) of the regions of interest and sequences of the genes of interest in reference genome C. auris B8441 downloaded from the Candida genome database (candidagenome.org). Sequencing primers were designed to include a ±1,000-bp region of interest (spanning the region with the mutation of interest). All primers are given in Table S1.
Amplification of regions of interest was achieved through PCR using Q5 high-fidelity DNA polymerase (New England Biolabs, Inc.). The total reaction volume of 50 μl consisted of 200 ng/μl DNA extract, 5 μl deoxynucleoside triphosphate (dNTP) (0.2 mM), 10 μl Q5 buffer, 0.5 μl Q5 polymerase (2 units), milliQ, and 0.4 μl of both forward and reverse primer (1 μM). The PCR program consisted of initial denaturation at 98°C for 3 sec, 30 cycles of 98°C for 15 sec, 56°C for 25 sec, 72°C for 2 min, and a final elongation step at 72°C for 2 min in a Labcycler Basic thermocycler (Bioké). Correct amplification was verified by performing electrophoresis on a 1% agarose gel at 135 V for 25 min. After verification, the sequencing primers (10 μM) were added to PCR amplicons, and the DNA was sequenced using Sanger sequencing by Eurofins (GATC, Germany).
Allele-specific PCR (AS-PCR).
The emergence of SNPs and indels was traced back in whole populations and a maximum of 30 single clones (colonies) per population using a rapid sequencing-free method: allele-specific SNP-PCR. Two primer pairs per gene of interest were designed according to Liu et al. (31), consisting of one universal primer and/or one mutant-allele primer or wild-type allele primer, respectively. Primers consist of an allele-specific region at the 3′ terminal nucleotide of the mutant or wild-type allele-specific primer. Additionally, a mismatch at the 3rd nucleotide from the 3′ terminal was included to increase annealing specificity at a wider temperature range (31). All primers used for AS-PCR are listed in Table S1.
To validate primer specificity, a temperature gradient PCR was performed in which the annealing temperature varied between 60°C and 70°C. AS-PCR sensitivity was assessed by performing PCR on serial dilution of the reference DNA template. All PCRs were performed in a total reaction volume of 20 μl consisting of 1 μl of 1/20 dilution of the pure PCI DNA extract, 5 μl dNTP (0.2 mM), 10 μl TaqE buffer, 0.5 μl TaqE polymerase (2 units), milliQ, and 0.4 μl of both forward and reverse primer (1 μM). The PCR program consisted of initial denaturation at 98°C for 3 sec, 30 cycles of 98°C for 15 sec, 25 sec annealing at 61°C (for primers of CIS2, PEA2, TAC1b, FKS1FL635L, ERG3W182*, and ERG11) or at 63°C (for primers of ERG3L207I, FKS1M690I, FLO8, and MEC3), 72°C for 2 min, and a final elongation step at 72°C for 2 min in a Labcycler Basic thermocycler (Bioké). Amplification and thus the presence or absence of a mutation were verified by performing electrophoresis on a 1% agarose gel at 135 V for 25 min.
Gene expression and copy number variation analysis.
C. auris cells from a single colony grown overnight on YPD agar (2% glucose) were enriched in RPMI-MOPS (2% glucose) medium for 16 h. These cultures were diluted to 108 cells in a volume of 50 ml fresh RPMI-MOPS (2% glucose) medium and incubated for 8 h at 37°C in a shaking incubator to ensure the harvested cells were growing in the exponential growth phase. Next, cells were harvested by centrifugation, washing in ice-cold PBS, and snap-freezing in liquid nitrogen to store at −80°C.
For gene expression analysis (RNA extraction and RT-qPCR), cells were resuspended in 1 ml TRIzol (Thermo Fisher Scientific) and lysed by microbead shearing in FastPrep-24TM Classic lysis system (20 sec, 6m/sec) (MP Biomedicals). Nucleotides were extracted by washing the lysate supernatant with chloroform (360 μl) and isopropanol (350 μl) and precipitated by washing three times with 70% ethanol. Nucleotide concentrations and purity were measured spectrophotometrically using a NanoDrop ND-1000 instrument (Isogen Life Science). Extracts were diluted to 1 μg pure nucleotide concentration and purified by DNase treatment (New England Biolabs). cDNA was synthesized from RNA by using the iScript cDNA synthesis kit (Bio-Rad) according to the manufacturer’s recommendations. Real-time qPCR was performed using GoTaq polymerase (Promega) and the StepOnePlus real-time PCR thermocycler (Thermo Fisher) as follows: activation at 95°C for 2 min, 40 cycles of denaturation at 95°C for 3 sec, and annealing/extension at 60°C for 30 sec. Primers used for qPCR were designed with the PrimerQuest tool of IDT and are listed in Table S1. A total of 8 housekeeping genes involved in various cellular processes were assessed, of which the 3 most stable candidates were used in the analysis (i.e., ACT1, LSC2, UBC4). Gene expression analysis was performed using qBasePlus software. Fold change (with standard deviation [SD]) was plotted from log2(Y) transformed data and compared statistically (using a one-way ANOVA with multiple comparisons in respect to wt) with GraphPad Prism. Expression analysis in each strain was performed using 3 biological repeats each represented by the average of 2 technical repeats.
For copy number variation analysis, gDNA was extracted as described in “DNA Extraction,” above, and standardized concentrations of 0.5 ng/μl of gDNA were used to quantify target markers (TAC1b and ERG11) by qPCR using the same protocol and analysis as described above.
ACKNOWLEDGMENTS
This work was supported by FWO personal research grant no. 11D7620N awarded to H.C. A travel grant awarded by FWO, grant no. K204920N, allowed a fruitful research stay of H.C. at Broad Institute (Cambridge, MA, USA). C.A.C. and J.F.M. were supported by the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under award U19AI110818 to the Broad Institute.
We thank the Steenackers group (KU Leuven) for their advice on experimental evolution.
Footnotes
Citation Carolus H, Pierson S, Muñoz JF, Subotić A, Cruz RB, Cuomo CA, Van Dijck P. 2021. Genome-wide analysis of experimentally evolved Candida auris reveals multiple novel mechanisms of multidrug resistance. mBio 12:e03333-20. https://doi.org/10.1128/mBio.03333-20.
REFERENCES
- 1.Satoh K, Makimura K, Hasumi Y, Nishiyama Y, Uchida K, Yamaguchi H. 2009. Candida auris sp. nov., a novel ascomycetous yeast isolated from the external ear canal of an inpatient in a Japanese hospital. Microbiol Immunol 53:41–44. doi: 10.1111/j.1348-0421.2008.00083.x. [DOI] [PubMed] [Google Scholar]
- 2.Kean R, Brown J, Gulmez D, Ware A, Ramage G. 2020. Candida auris: a decade of understanding of an enigmatic pathogenic yeast. J Fungi (Basel) 6:30. doi: 10.3390/jof6010030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Forsberg K, Woodworth K, Walters M, Berkow EL, Jackson B, Chiller T, Vallabhaneni S. 2019. Candida auris: the recent emergence of a multidrug-resistant fungal pathogen. Med Mycol 57:1–12. doi: 10.1093/mmy/myy054. [DOI] [PubMed] [Google Scholar]
- 4.CDC. 2019. Antibiotic Resistance Threats in the United States, 2019. www.cdc.gov/DrugResistance/Biggest-Threats.html. Accessed 26 March 2020.
- 5.Magobo RE, Corcoran C, Seetharam S, Govender NP. 2014. Candida auris-associated candidemia, South Africa. Emerg Infect Dis 20:1250–1251. doi: 10.3201/eid2007.131765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.CDC. 2020. Candida auris: antifungal susceptibility testing and interpretation. https://www.cdc.gov/fungal/candida-auris/c-auris-antifungal.html. Accessed 26 March 2020.
- 7.Lockhart SR, Etienne KA, Vallabhaneni S, Farooqi J, Chowdhary A, Govender NP, Colombo AL, Calvo B, Cuomo CA, Desjardins CA, Berkow EL, Castanheira M, Magobo RE, Jabeen K, Asghar RJ, Meis JF, Jackson B, Chiller T, Litvintseva AP. 2017. Simultaneous emergence of multidrug-resistant Candida auris on 3 continents confirmed by whole-genome sequencing and epidemiological analyses. Clin Infect Dis 64:134–140. doi: 10.1093/cid/ciw691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chowdhary A, Prakash A, Sharma C, Kordalewska M, Kumar A, Sarma S, Tarai B, Singh A, Upadhyaya G, Upadhyay S, Yadav P, Singh PK, Khillan V, Sachdeva N, Perlin DS, Meis JF. 2018. A multicentre study of antifungal susceptibility patterns among 350 Candida auris isolates (2009–17) in India: role of the ERG11 and FKS1 genes in azole and echinocandin resistance. J Antimicrob Chemother 73:891–899. doi: 10.1093/jac/dkx480. [DOI] [PubMed] [Google Scholar]
- 9.Chow NA, Muñoz JF, Gade L, Berkow E, Li X, Welsh RM, Forsberg K, Lockhart SR, Adam R, Alanio A, Alastruey-Izquierdo A, Althawadi S, Belén Araúz A, Ben-Ami R, Bharat A, Calvo B, Desnos-Ollivier M, Escandón P, Gardam D, Gunturu R, Heath CH, Kurzai O, Martin R, Litvintseva AP, Cuomo CA. 2020. Tracing the evolutionary history and global expansion of Candida auris using population genomic analyses. mBio 11:e03364-19. doi: 10.1128/mBio.03364-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bishop L, Cummins M, Guy R, Hoffman P, Jeffery K, Jeffery-Smith A, Brown C. 2017. Guidance for the laboratory investigation, management and infection prevention and control for cases of Candida auris. Public Health England. Updated 11 August 2017.
- 11.Kathuria S, Singh PK, Sharma C, Prakash A, Masih A, Kumar A, Meis JF, Chowdhary A. 2015. Multidrug-resistant Candida auris misidentified as Candida haemulonii: characterization by matrix-assisted laser desorption ionization-time of flight mass spectrometry and DNA sequencing and its antifungal susceptibility profile variability by Vitek 2, CLSI broth microdilution, and Etest method. J Clin Microbiol 53:1823–1830. doi: 10.1128/JCM.00367-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lockhart SR. 2019. Candida auris and multidrug resistance: defining the new normal. Fungal Genet Biol 131:103243. doi: 10.1016/j.fgb.2019.103243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ksiezopolska E, Gabaldon T. 2018. Evolutionary emergence of drug resistance in Candida opportunistic pathogens. Genes (Basel) 9:461. doi: 10.3390/genes9090461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Healey KR, Kordalewska M, Jimenez Ortigosa C, Singh A, Berrio I, Chowdhary A, Perlin DS. 2018. Limited ERG11 mutations identified in isolates of Candida auris directly contribute to reduced azole susceptibility. Antimicrob Agents Chemother 62:e01427-18. doi: 10.1128/AAC.01427-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muñoz JF, Gade L, Chow NA, Loparev VN, Juieng P, Berkow EL, Farrer RA, Litvintseva AP, Cuomo CA. 2018. Genomic insights into multidrug-resistance, mating and virulence in Candida auris and related emerging species. Nat Commun 9:5346. doi: 10.1038/s41467-018-07779-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rybak JM, Muñoz JF, Barker KS, Parker JE, Esquivel BD, Berkow EL, Lockhart SR, Gade L, Palmer GE, White TC, Kelly SL, Cuomo CA, Rogers PD. 2020. Mutations in TAC1B: a novel genetic determinant of clinical fluconazole resistance in Candida auris. mBio 11:e00365-20. doi: 10.1128/mBio.00365-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rybak JM, Dickens CM, Parker JE, Caudle KE, Manigaba K, Whaley SG, Nishimoto AT, Luna-Tapia A, Roy S, Zhang Q, Barker KS, Palmer GE, Sutter TR, Homayouni R, Wiederhold NP, Kelly SL, Rogers PD. 2017. Loss of C-5 sterol desaturase activity results in increased resistance to azole and echinocandin antifungals in a clinical isolate of Candida parapsilosis. Antimicrob Agents Chemother 61:e00651-17. doi: 10.1128/AAC.00651-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim SH, Iyer KR, Pardeshi L, Munoz JF, Robbins N, Cuomo CA, Wong KH, Cowen LE. 2019. Genetic analysis of Candida auris implicates Hsp90 in morphogenesis and azole tolerance and Cdr1 in azole resistance. mBio 10:e02529-18. doi: 10.1128/mBio.02529-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bing J, Hu T, Zheng Q, Muñoz JF, Cuomo CA, Huang G. 2020. Experimental evolution identifies adaptive aneuploidy as a mechanism of fluconazole resistance in Candida auris. Antimicrob Agents Chemother 65:e01466-20. doi: 10.1128/AAC.01466-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chaabane F, Graf A, Jequier L, Coste AT. 2019. Review on antifungal resistance mechanisms in the emerging pathogen Candida auris. Front Microbiol 10:2788. doi: 10.3389/fmicb.2019.02788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharma D, Paul RA, Chakrabarti A, Bhattacharya S, Soman R, Shankarnarayan SA, Chavan D, Das P, Kaur H, Ghosh A. 2020. Caspofungin resistance in Candia auris due to mutations in Fks1 with adjunctive role of chitin and key cell wall stress response pathway genes. BioRxiv doi: 10.1101/2020.07.09.196600. [DOI]
- 22.Alfouzan W, Ahmad S, Dhar R, Asadzadeh M, Almerdasi N, Abdo NM, Joseph L, de Groot T, Alali WQ, Khan Z, Meis JF, Al-Rashidi MR. 2020. Molecular epidemiology of Candida auris outbreak in a major secondary-care hospital in Kuwait. J Fungi (Basel) 6:307. doi: 10.3390/jof6040307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carolus H, Pierson S, Lagrou K, Van Dijck P. 2020. Amphotericin B and other polyenes: discovery, clinical use, mode of action and drug resistance. J Fungi (Basel) 6:321. doi: 10.3390/jof6040321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yadav A, Singh A, Wang Y, van Haren MH, Singh A, de Groot T, Meis JF, Xu J, Chowdhary A. 2021. Colonisation and transmission dynamics of Candida auris among chronic respiratory diseases patients hospitalised in a chest hospital, Delhi, India: a comparative analysis of whole genome sequencing and microsatellite typing. J Fungi (Basel) 7:81. doi: 10.3390/jof7020081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Escandon P, Chow NA, Caceres DH, Gade L, Berkow EL, Armstrong P, Rivera S, Misas E, Duarte C, Moulton-Meissner H, Welsh RM, Parra C, Pescador LA, Villalobos N, Salcedo S, Berrio I, Varon C, Espinosa-Bode A, Lockhart SR, Jackson BR, Litvintseva AP, Beltran M, Chiller TM. 2019. Molecular epidemiology of Candida auris in Colombia reveals a highly related, countrywide colonization with regional patterns in amphotericin B resistance. Clin Infect Dis 68:15–21. doi: 10.1093/cid/ciy411. [DOI] [PubMed] [Google Scholar]
- 26.Mayr EM, Ramirez-Zavala B, Kruger I, Morschhauser J. 2020. A zinc cluster transcription factor contributes to the intrinsic fluconazole resistance of Candida auris. mSphere 5:e00279-20. doi: 10.1128/mSphere.00279-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rybak JM, Doorley LA, Nishimoto AT, Barker KS, Palmer GE, Rogers PD. 2019. Abrogation of triazole resistance upon deletion of CDR1 in a clinical isolate of Candida auris. Antimicrob Agents Chemother 63:e00057-19. doi: 10.1128/AAC.00057-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Welsh RM, Sexton DJ, Forsberg K, Vallabhaneni S, Litvintseva A. 2019. Insights into the unique nature of the East Asian clade of the emerging pathogenic yeast Candida auris. J Clin Microbiol 57:e00007-19. doi: 10.1128/JCM.00007-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muñoz JF, Welsh RM, Shea T, Batra D, Gade L, Howard D, Rowe LA, Meis JF, Litvintseva AP, Cuomo CA. 2021. Clade-specific chromosomal rearrangements and loss of subtelomeric adhesins in Candida auris. Genetics iyab029. doi: 10.1093/genetics/iyab029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chow NA, de Groot T, Badali H, Abastabar M, Chiller TM, Meis JF. 2019. Potential fifth clade of Candida auris, Iran, 2018. Emerg Infect Dis 25:1780–1781. doi: 10.3201/eid2509.190686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu J, Huang S, Sun M, Liu S, Liu Y, Wang W, Zhang X, Wang H, Hua W. 2012. An improved allele-specific PCR primer design method for SNP marker analysis and its application. Plant Methods 8:34. doi: 10.1186/1746-4811-8-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson ME, Katiyar SK, Edlind TD. 2011. New Fks hot spot for acquired echinocandin resistance in Saccharomyces cerevisiae and its contribution to intrinsic resistance of Scedosporium species. Antimicrob Agents Chemother 55:3774–3781. doi: 10.1128/AAC.01811-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marichal P, Koymans L, Willemsens S, Bellens D, Verhasselt P, Luyten W, Borgers M, Ramaekers F, Odds FC, Bossche Vande H. 1999. Contribution of mutations in the cytochrome P450 14α-demethylase (Erg11p, Cyp51p) to azole resistance in Candida albicans. Microbiology 145:2701–2713. doi: 10.1099/00221287-145-10-2701. [DOI] [PubMed] [Google Scholar]
- 34.Wang H, Kong F, Sorrell TC, Wang B, McNicholas P, Pantarat N, Ellis D, Xiao M, Widmer F, Chen SCA. 2009. Rapid detection of ERG11 gene mutations in clinical Candida albicans isolates with reduced susceptibility to fluconazole by rolling circle amplification and DNA sequencing. BMC Microbiol 9:167. doi: 10.1186/1471-2180-9-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kwon YJ, Shin JH, Byun SA, Choi MJ, Won EJ, Lee D, Lee SY, Chun S, Lee JH, Choi HJ, Kee SJ, Kim SH, Shin MG. 2019. Candida auris clinical isolates from South Korea: identification, antifungal susceptibility, and genotyping. J Clin Microbiol 57:e01624-18. doi: 10.1128/JCM.01624-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coste A, Selmecki A, Forche A, Diogo D, Bougnoux ME, d'Enfert C, Berman J, Sanglard D. 2007. Genotypic evolution of azole resistance mechanisms in sequential Candida albicans isolates. Eukaryot Cell 6:1889–1904. doi: 10.1128/EC.00151-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coste A, Karababa M, Ischer F, Bille J, Sanglard D. 2004. TAC1, transcriptional activator of CDR genes, is a new transcription factor involved in the regulation of Candida albicans ABC transporters CDR1 and CDR2. Eukaryot Cell 3:1639–1652. doi: 10.1128/EC.3.6.1639-1652.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vermitsky JP, Earhart KD, Smith WL, Homayouni R, Edlind TD, Rogers PD. 2006. Pdr1 regulates multidrug resistance in Candida glabrata: gene disruption and genome-wide expression studies. Mol Microbiol 61:704–722. doi: 10.1111/j.1365-2958.2006.05235.x. [DOI] [PubMed] [Google Scholar]
- 39.Sanglard D, Coste A, Ferrari S. 2009. Antifungal drug resistance mechanisms in fungal pathogens from the perspective of transcriptional gene regulation. FEMS Yeast Res 9:1029–1050. doi: 10.1111/j.1567-1364.2009.00578.x. [DOI] [PubMed] [Google Scholar]
- 40.Selmecki A, Gerami-Nejad M, Paulson C, Forche A, Berman J. 2008. An isochromosome confers drug resistance in vivo by amplification of two genes, ERG11 and TAC1. Mol Microbiol 68:624–641. doi: 10.1111/j.1365-2958.2008.06176.x. [DOI] [PubMed] [Google Scholar]
- 41.Bhattacharya S, Holowka T, Orner EP, Fries BC. 2019. Gene duplication associated with increased fluconazole tolerance in Candida auris cells of advanced generational age. Sci Rep 9:1–13. doi: 10.1038/s41598-019-41513-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hull CM, Bader O, Parker JE, Weig M, Gross U, Warrilow AG, Kelly DE, Kelly SL. 2012. Two clinical isolates of Candida glabrata exhibiting reduced sensitivity to amphotericin B both harbor mutations in ERG2. Antimicrob Agents Chemother 56:6417–6421. doi: 10.1128/AAC.01145-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vandeputte P, Tronchin G, Berges T, Hennequin C, Chabasse D, Bouchara JP. 2007. Reduced susceptibility to polyenes associated with a missense mutation in the ERG6 gene in a clinical isolate of Candida glabrata with pseudohyphal growth. Antimicrob Agents Chemother 51:982–990. doi: 10.1128/AAC.01510-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hull CM, Parker JE, Bader O, Weig M, Gross U, Warrilow AG, Kelly DE, Kelly SL. 2012. Facultative sterol uptake in an ergosterol-deficient clinical isolate of Candida glabrata harboring a missense mutation in ERG11 and exhibiting cross-resistance to azoles and amphotericin B. Antimicrob Agents Chemother 56:4223–4232. doi: 10.1128/AAC.06253-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sanglard D, Ischer F, Parkinson T, Falconer D, Bille J. 2003. Candida albicans mutations in the ergosterol biosynthetic pathway and resistance to several antifungal agents. Antimicrob Agents Chemother 47:2404–2412. doi: 10.1128/aac.47.8.2404-2412.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sanglard D. 2016. Emerging threats in antifungal-resistant fungal pathogens. Front Med (Lausanne) 3:11. doi: 10.3389/fmed.2016.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eddouzi J, Parker JE, Vale-Silva LA, Coste A, Ischer F, Kelly S, Manai M, Sanglard D. 2013. Molecular mechanisms of drug resistance in clinical Candida species isolated from Tunisian hospitals. Antimicrob Agents Chemother 57:3182–3193. doi: 10.1128/AAC.00555-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vincent BM, Lancaster AK, Scherz-Shouval R, Whitesell L, Lindquist S. 2013. Fitness trade-offs restrict the evolution of resistance to amphotericin B. PLoS Biol 11:e1001692. doi: 10.1371/journal.pbio.1001692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kelly SL, Lamb DC, Corran AJ, Baldwin BC, Kelly DE. 1995. Mode of action and resistance to azole antifungals associated with the formation of 14 alpha-methylergosta-8,24(28)-dien-3 beta,6 alpha-diol. Biochem Biophys Res Commun 207:910–915. doi: 10.1006/bbrc.1995.1272. [DOI] [PubMed] [Google Scholar]
- 50.Perlin DS. 2015. Mechanisms of echinocandin antifungal drug resistance. Ann N Y Acad Sci 1354:1–11. doi: 10.1111/nyas.12831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Biagi MJ, Wiederhold NP, Gibas C, Wickes BL, Lozano V, Bleasdale SC, Danziger L. 2019. Development of high-level echinocandin resistance in a patient with recurrent Candida auris candidemia secondary to chronic candiduria. Open Forum Infect Dis 6:ofz262. doi: 10.1093/ofid/ofz262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kordalewska M, Lee A, Park S, Berrio I, Chowdhary A, Zhao Y, Perlin DS. 2018. Understanding echinocandin resistance in the emerging pathogen Candida auris. Antimicrob Agents Chemother 62:e00238-18. doi: 10.1128/AAC.00238-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.O’Brien B, Liang J, Chaturvedi S, Jacobs JL, Chaturvedi V. 2020. Pan-resistant Candida auris: New York subcluster susceptible to antifungal combinations. Lancet Microbe 1(5):e193–e194. doi: 10.1016/S2666-5247(20)30090-2. [DOI] [PubMed] [Google Scholar]
- 54.Cao F, Lane S, Raniga PP, Lu Y, Zhou Z, Ramon K, Chen J, Liu H. 2006. The Flo8 transcription factor is essential for hyphal development and virulence in Candida albicans. Mol Biol Cell 17:295–307. doi: 10.1091/mbc.e05-06-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Laprade DJ, Brown MS, McCarthy ML, Ritch JJ, Austriaco N. 2016. Filamentation protects Candida albicans from amphotericin B-induced programmed cell death via a mechanism involving the yeast metacaspase, MCA1. Microb Cell 3:285–292. doi: 10.15698/mic2016.07.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Woods K, Hofken T. 2016. The zinc cluster proteins Upc2 and Ecm22 promote filamentation in Saccharomyces cerevisiae by sterol biosynthesis-dependent and -independent pathways. Mol Microbiol 99:512–527. doi: 10.1111/mmi.13244. [DOI] [PubMed] [Google Scholar]
- 57.Misas E, Escandon P, McEwen JG, Clay OK. 2019. The LUFS domain, its transcriptional regulator proteins, and drug resistance in the fungal pathogen Candida auris. Protein Sci 28:2024–2029. doi: 10.1002/pro.3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li WJ, Liu JY, Shi C, Zhao Y, Meng LN, Wu F, Xiang MJ. 2019. FLO8 deletion leads to azole resistance by upregulating CDR1 and CDR2 in Candida albicans. Res Microbiol 170:272–279. doi: 10.1016/j.resmic.2019.08.005. [DOI] [PubMed] [Google Scholar]
- 59.Majka J, Burgers PM. 2003. Yeast Rad17/Mec3/Ddc1: a sliding clamp for the DNA damage checkpoint. Proc Natl Acad Sci U S A 100:2249–2254. doi: 10.1073/pnas.0437148100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tsai HF, Sammons LR, Zhang X, Suffis SD, Su Q, Myers TG, Marr KA, Bennett JE. 2010. Microarray and molecular analyses of the azole resistance mechanism in Candida glabrata oropharyngeal isolates. Antimicrob Agents Chemother 54:3308–3317. doi: 10.1128/AAC.00535-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cao Y, Zhu Z, Chen X, Yao X, Zhao L, Wang H, Yan L, Wu H, Chai Y, Jiang Y. 2013. Effect of amphotericin B on the metabolic profiles of Candida albicans. J Proteome Res 12:2921–2932. doi: 10.1021/pr4002178. [DOI] [PubMed] [Google Scholar]
- 62.Liu TT, Lee RE, Barker KS, Lee RE, Wei L, Homayouni R, Rogers PD. 2005. Genome-wide expression profiling of the response to azole, polyene, echinocandin, and pyrimidine antifungal agents in Candida albicans. Antimicrob Agents Chemother 49:2226–2236. doi: 10.1128/AAC.49.6.2226-2236.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sangalli-Leite F, Scorzoni L, Mesa-Arango AC, Casas C, Herrero E, Gianinni MJSM, Rodríguez-Tudela JL, Cuenca-Estrella M, Zaragoza O. 2011. Amphotericin B mediates killing in Cryptococcus neoformans through the induction of a strong oxidative burst. Microbes Infect 13:457–467. doi: 10.1016/j.micinf.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 64.Sokol-Anderson M, Sligh JE, Jr, Elberg S, Brajtburg J, Kobayashi GS, Medoff G. 1988. Role of cell defense against oxidative damage in the resistance of Candida albicans to the killing effect of amphotericin B. Antimicrob Agents Chemother 32:702–705. doi: 10.1128/aac.32.5.702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sokol-Anderson ML, Brajtburg J, Medoff G. 1986. Amphotericin B-induced oxidative damage and killing of Candida albicans. J Infect Dis 154:76–83. doi: 10.1093/infdis/154.1.76. [DOI] [PubMed] [Google Scholar]
- 66.Burhans WC, Weinberger M, Marchetti MA, Ramachandran L, D’Urso G, Huberman JA. 2003. Apoptosis-like yeast cell death in response to DNA damage and replication defects. Mutat Res 532:227–243. doi: 10.1016/j.mrfmmm.2003.08.019. [DOI] [PubMed] [Google Scholar]
- 67.Legrand M, Chan CL, Jauert PA, Kirkpatrick DT. 2007. Role of DNA mismatch repair and double-strand break repair in genome stability and antifungal drug resistance in Candida albicans. Eukaryot Cell 6:2194–2205. doi: 10.1128/EC.00299-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Healey KR, Jimenez Ortigosa C, Shor E, Perlin DS. 2016. Genetic drivers of multidrug resistance in Candida glabrata. Front Microbiol 7:1995–1995. doi: 10.3389/fmicb.2016.01995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Boyce KJ, Wang Y, Verma S, Shakya VP, Xue C, Idnurm A. 2017. Mismatch repair of DNA replication errors contributes to microevolution in the pathogenic fungus Cryptococcus neoformans. mBio 8:e00595-17. doi: 10.1128/mBio.00595-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kumar C, Sharma R, Bachhawat AK. 2003. Utilization of glutathione as an exogenous sulfur source is independent of gamma-glutamyl transpeptidase in the yeast Saccharomyces cerevisiae: evidence for an alternative gluathione degradation pathway. FEMS Microbiol Lett 219:187–194. doi: 10.1016/S0378-1097(03)00059-4. [DOI] [PubMed] [Google Scholar]
- 71.Ubiyvovk VM, Blazhenko OV, Gigot D, Penninckx M, Sibirny AA. 2006. Role of gamma-glutamyltranspeptidase in detoxification of xenobiotics in the yeasts Hansenula polymorpha and Saccharomyces cerevisiae. Cell Biol Int 30:665–671. doi: 10.1016/j.cellbi.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 72.Lussier M, White AM, Sheraton J, di Paolo T, Treadwell J, Southard SB, Horenstein CI, Chen-Weiner J, Ram AF, Kapteyn JC, Roemer TW, Vo DH, Bondoc DC, Hall J, Zhong WW, Sdicu AM, Davies J, Klis FM, Robbins PW, Bussey H. 1997. Large scale identification of genes involved in cell surface biosynthesis and architecture in Saccharomyces cerevisiae. Genetics 147:435–450. doi: 10.1093/genetics/147.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Maras B, Angiolella L, Mignogna G, Vavala E, Macone A, Colone M, Pitari G, Stringaro A, Dupre S, Palamara AT. 2014. Glutathione metabolism in Candida albicans resistant strains to fluconazole and micafungin. PLoS One 9:e98387. doi: 10.1371/journal.pone.0098387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Song Q, Johnson C, Wilson TE, Kumar A. 2014. Pooled segregant sequencing reveals genetic determinants of yeast pseudohyphal growth. PLoS Genet 10:e1004570. doi: 10.1371/journal.pgen.1004570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Robbins N, Caplan T, Cowen LE. 2017. Molecular evolution of antifungal drug resistance. Annu Rev Microbiol 71:753–775. doi: 10.1146/annurev-micro-030117-020345. [DOI] [PubMed] [Google Scholar]
- 76.Sanglard D. 2019. Finding the needle in a haystack: mapping antifungal drug resistance in fungal pathogen by genomic approaches. PLoS Pathog 15:e1007478. doi: 10.1371/journal.ppat.1007478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pais P, Galocha M, Viana R, Cavalheiro M, Pereira D, Teixeira MC. 2019. Microevolution of the pathogenic yeasts Candida albicans and Candida glabrata during antifungal therapy and host infection. Microb Cell 6:142–159. doi: 10.15698/mic2019.03.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Maclean RC, Hall AR, Perron GG, Buckling A. 2010. The evolution of antibiotic resistance: insight into the roles of molecular mechanisms of resistance and treatment context. Discov Med 10:112–118. [PubMed] [Google Scholar]
- 79.Lukacisinova M, Bollenbach T. 2017. Toward a quantitative understanding of antibiotic resistance evolution. Curr Opin Biotechnol 46:90–97. doi: 10.1016/j.copbio.2017.02.013. [DOI] [PubMed] [Google Scholar]
- 80.Fisher KJ, Lang GI. 2016. Experimental evolution in fungi: an untapped resource. Fungal Genet Biol 94:88–94. doi: 10.1016/j.fgb.2016.06.007. [DOI] [PubMed] [Google Scholar]
- 81.Vyas VK, Barrasa MI, Fink GR. 2015. A Candida albicans CRISPR system permits genetic engineering of essential genes and gene families. Sci Adv 1:e1500248-e1500248. doi: 10.1126/sciadv.1500248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ko H-C, Hsiao T-Y, Chen C-T, Yang Y-L. 2013. Candida albicans ENO1 null mutants exhibit altered drug susceptibility, hyphal formation, and virulence. J Microbiol 51:345–351. doi: 10.1007/s12275-013-2577-z. [DOI] [PubMed] [Google Scholar]
- 83.Zhang X-H, Tee LY, Wang X-G, Huang Q-S, Yang S-H. 2015. Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol Ther Nucleic Acids 4:e264. doi: 10.1038/mtna.2015.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Enkler L, Richer D, Marchand AL, Ferrandon D, Jossinet F. 2016. Genome engineering in the yeast pathogen Candida glabrata using the CRISPR-Cas9 system. Sci Rep 6:35766. doi: 10.1038/srep35766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.CLSI. 2008. Reference method for broth dilution antifungal susceptibility testing of yeasts; Approved standard , 3rd ed. CLSI document M27-A3. 28. CLSI, Wayne, PA. [Google Scholar]
- 86.Andrews S. 2010. FastQC, Babraham Bioinformatics. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- 87.Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 1303.3997. https://arxiv.org/abs/1303.3997.
- 88.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cingolani P. 2017. SnpEff v4.3T: Genomic variant annotations and functional effect prediction toolbox. http://snpeff.sourceforge.net/.
- 91.Abyzov A, Urban AE, Snyder M, Gerstein M. 2011. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res 21:974–984. doi: 10.1101/gr.114876.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Team RDC. 2008. The R project for statistical computing. http://www.R-project.org.
- 93.Garcia-Effron G, Park S, Perlin DS. 2009. Correlating echinocandin MIC and kinetic inhibition of fks1 mutant glucan synthases for Candida albicans: implications for interpretive breakpoints. AAC 53:112–122. doi: 10.1128/AAC.01162-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Castanheira M, Woosley LN, Diekema DJ, Messer SA, Jones RN, Pfaller MA. 2010. Low prevalence of fks1 hot spot 1 mutations in a worldwide collection of Candida strains. AAC 54:2655–2659. doi: 10.1128/AAC.01711-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lackner M, Tscherner M, Schaller M, Kuchler K, Mair C, Sartori B, Istel F, Arendrup MC, Lass-Flörl C. 2014. Positions and numbers of FKS mutations in Candida albicans selectively influence in vitro and in vivo susceptibilities to echinocandin treatment. Antimicrob Agents Chemother 58:3626–3635. doi: 10.1128/AAC.00123-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kritikos A, Neofytos D, Khanna N, Schreiber PW, Boggian K, Bille J, Schrenzel J, Mühlethaler K, Zbinden R, Bruderer T, Goldenberger D, Pfyffer G, Conen A, Van Delden C, Zimmerli S, Sanglard D, Bachmann D, Marchetti O, Lamoth F, Bregenzer T, Conen A, Flückiger U, Khanna N, Orasch C, Heininger U, Franciolli M, Damonti L, Zimmerli S, Rothen M, Zellweger C, Tarr P, Fleisch F, Chuard C, Erard V, Emonet S, Garbino J, van Delden C, Genne D, Bochud P, Calandra T, Lamoth F, Marchetti O, Chave J, Graber P, Monotti R, Regionale O, Bernasconi E, Civico O, Rossi M, Krause M, et al. 2018. Accuracy of Sensititre YeastOne echinocandins epidemiological cut-off values for identification of FKS mutant Candida albicans and Candida glabrata: a ten year national survey of the Fungal Infection Network of Switzerland (FUNGINOS). Clin Microbiol Infect 24:1214.e1–1214.e4. doi: 10.1016/j.cmi.2018.05.012. [DOI] [PubMed] [Google Scholar]
- 97.Garcia-Effron G, Lee S, Park S, Cleary JD, Perlin DS. 2009. Effect of Candida glabrata FKS1 and FKS2 mutations on echinocandin sensitivity and kinetics of 1,3-beta-d-glucan synthase: implication for the existing susceptibility breakpoint. Antimicrob Agents Chemother 53:3690–3699. doi: 10.1128/AAC.00443-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zimbeck AJ, Iqbal N, Ahlquist AM, Farley MM, Harrison LH, Chiller T, Lockhart SR. 2010. FKS mutations and elevated echinocandin MIC values among Candida glabrata isolates from U.S. population-based surveillance. Antimicrob Agents Chemother 54:5042–5047. doi: 10.1128/AAC.00836-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Prigent G, Aït-Ammar N, Levesque E, Fekkar A, Costa J-M, El Anbassi S, Foulet F, Duvoux C, Merle J-C, Dannaoui E, Botterel F. 2017. Echinocandin resistance in Candida species isolates from liver transplant recipients. Antimicrob Agents Chemother 61:e01229-16. doi: 10.1128/AAC.01229-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Park S, Kelly R, Kahn JN, Robles J, Hsu M-J, Register E, Li W, Vyas V, Fan H, Abruzzo G, Flattery A, Gill C, Chrebet G, Parent SA, Kurtz M, Teppler H, Douglas CM, Perlin DS. 2005. Specific substitutions in the echinocandin target Fks1p account for reduced susceptibility of rare laboratory and clinical Candida sp. isolates. Antimicrob Agents Chemother 49:3264–3273. doi: 10.1128/AAC.49.8.3264-3273.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Martí-Carrizosa M, Sánchez-Reus F, March F, Cantón E, Coll P. 2015. Implication of Candida parapsilosis FKS1 and FKS2 mutations in reduced echinocandin susceptibility. Antimicrob Agents Chemother 59:3570–3573. doi: 10.1128/AAC.04922-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jiménez-Ortigosa C, Moore C, Denning DW, Perlin DS. 2017. Emergence of echinocandin resistance due to a point mutation in the FKS1 gene of Aspergillus fumigatus in a patient with chronic pulmonary aspergillosis. Antimicrob Agents Chemother 61:e01277-17. doi: 10.1128/AAC.01277-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Resistance profiles of endpoint evolved strains. Growth profiles in a broth dilution assay (BDA) of the endpoint evolved strains (A29, F30, FC17, C20, CF16) and control strain (wt) for fluconazole (A), caspofungin (B), and amphotericin B (C). The percentage of growth was calculated from growth without drug and based on OD600 measurements after 48 h of incubation at 37°C. Each data point and its standard deviation is calculated from 3 biological repeats, each represented by the mean of 2 technical repeats. (D) Summary of MIC50 values for each strain and indication of (relatively low [↑] or high [↑↑]) increase or decrease (↓) of MIC in respect to the parental strain (wt) MIC. Download FIG S1, TIF file, 0.2 MB (156.7KB, tif) .
Copyright © 2021 Carolus et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Example of the temperature specificity range of allele-specific primers. Here, a temperature gradient PCR was performed using the MEC3 allele-specific primers on gDNA of the wild-type strain (wt) and strain A29, containing the gCg/gTg|A272V mutation in MEC3 (see Table 1). Primer pairs “WT: and “Δ” indicate the primers targeting the wild-type allele (CauMEC3_B11220_PCR/Seq1_F and CauMEC3_SNP_A272A_R) and mutated allele (CauMEC3_B11220_PCR/Seq1_F and CauMEC3_SNP_A272V_R), respectively, and are given in Table S1. The green grid indicates a primer-specific temperature range (63 to 64°C). Download FIG S2, TIF file, 0.1 MB (110.8KB, tif) .
Copyright © 2021 Carolus et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Copy number variation quantification of TAC1b (marker on the Chr5 duplication in the CF-lineage; see Fig. 3), ERG11 (marker on the segmental duplication of Chr1 in F- and FC-lineage; see Fig. 3), and ACT1 (reference) by qPCR on gDNA. CNVs were determined for 1 biological repeat represented by 3 technical repeats. Download FIG S3, TIF file, 0.03 MB (26.6KB, tif) .
Copyright © 2021 Carolus et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
All primers used in this study. Primers are arranged per marker. “Purpose” indicates whether the primer was used for PCR and sequencing (PCR/seq), CNV or expression analysis (qPCR), or allele-specific PCR (AS). Primer pairs for AS-PCR consist of a universal PCR-sequencing primer (indicated by “PCR/seq/AS”) or universal AS-primer (indicated by “AS”) and one allele-specific primer (indicated by “AS-wt” for the wild-type allele and “AS-mt” for the mutant allele). Download Table S1, DOCX file, 0.02 MB (18.4KB, docx) .
Copyright © 2021 Carolus et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Stress tolerance of evolved strains. To assess fitness, survival/growth of evolved strains was assessed in different conditions of temperature (A), oxidative stress (B), membrane stress (C), osmotic stress (D), and pH stress (E), as described in Materials and Methods. Only 1 of 3 biological repeats and 9 technical repeats is shown, as all repeats showed similar results. Stress conditions without any growth or with no differential growth between the strains are not shown. Download FIG S4, TIF file, 1.1 MB (1.1MB, tif) .
Copyright © 2021 Carolus et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Cytotoxicity of evolved strains. The relative cytotoxicity of the endpoint evolved strains toward HeLa cells after 24 h (A) and 72 h (B) was evaluated using an LDH cytotoxicity assay as described in Materials and Methods. Bars represent means with standard deviation accounting for data obtained from 3 biological repeats, each represented by the mean of 3 technical repeats. Asterisks indicate a significant difference in relative cytotoxicity; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001. Download FIG S5, TIF file, 0.03 MB (36KB, tif) .
Copyright © 2021 Carolus et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.