

Abstract
Traditional drug screening models are often unable to faithfully recapitulate human physiology in health and disease, motivating the development of microfluidic organs-on-a-chip (OOC) platforms that can mimic many aspects of human physiology and in the process alleviate many of the discrepancies between preclinical studies and clinical trials outcomes. Linsitinib, a novel anti-cancer drug, showed promising results in pre-clinical models of Ewing Sarcoma (ES), where it suppressed tumor growth. However, a Phase II clinical trial in several European centers with patients showed relapsed and/or refractory ES. We report an integrated, open setting, imaging and sampling accessible, polysulfone-based platform, featuring minimal hydrophobic compound binding. Two bioengineered human tissues – bone ES tumor and heart muscle – were cultured either in isolation or in the integrated platform and subjected to a clinically used linsitinib dosage. The measured anti-tumor efficacy and cardiotoxicity were compared with the results observed in the clinical trial. Only the engineered tumor tissues, and not monolayers, recapitulated the bone microenvironment pathways targeted by linsitinib, and the clinically-relevant differences in drug responses between non-metastatic and metastatic ES tumors. The responses of non-metastatic ES tumor tissues and heart muscle to linsitinib were much closer to those observed in the clinical trial for tissues cultured in an integrated setting than for tissues cultured in isolation. Drug treatment of isolated tissues resulted in significant decreases in tumor viability and cardiac function. Meanwhile, drug treatment in an integrated setting showed poor tumor response and less cardiotoxicity, which matched the results of the clinical trial. Overall, the integration of engineered human tumor and cardiac tissues in the integrated platform improved the predictive accuracy for both the direct and off-target effects of linsitinib. The proposed approach could be readily extended to other drugs and tissue systems.
Introduction
The development of new cancer therapeutics has a lower success rate than most drugs, with only 1 in 15 new drug candidates from clinical trials receiving FDA approval.1 The current process of drug development is long, expensive, and inefficient, largely due to the lack of predictive preclinical testing models.2,3 Anti-cancer drugs, such as endostatin, have been notorious for yielding promising results in mice, such as full tumor elimination, and subsequently showing minimal results in humans.4,5 At the same time, many drugs pass preclinical trials only to be withdrawn due to the side effects detected during clinical trials or even after entering the market and being used in large numbers of patients. This is particularly true for drugs causing cardiac side effects. Rofecoxib, a COX-2 inhibitor used as an analgesic and anti-inflammatory drug, was approved by the FDA in 1999 but was removed from market in 2004 because of side effects not seen in preclinical and clinical trials. Unfortunately, by this time it had already caused an estimated 140 000 heart attacks.6
Recently, a multi-center Eurosarc Phase II clinical trial of linsitinib, a tyrosine kinase inhibitor of the insulin-like growth factor receptor (IGF-1R) and the insulin receptor (INSR), on patients with advanced Ewing Sarcoma (ES), found the drug largely ineffective.7 These clinical results contradict the previous patient-derived orthotopic xenograft models of ES and cancer cell monolayers that helped establish IGF-1R inhibitors like linsitinib to be safe and effective for inhibiting tumor growth.8-10 The discrepancies between the results of cancer cell monolayers, mouse models, and clinical studies suggest a need for testing IGF-1R inhibitors like linsitinib in human tissue models.
In addition, cardiotoxicity of linsitinib has been shown in clinical trials of other types of cancers, with patients presenting proarrhythmic events, like tachycardia and atrial fibrillation.11,12 IGF-1R signaling has an important role in normal cardiomyocyte function, with the IGF pathway being activated in the physiological hypertrophic response to exercise and hypertension.13,14 Animal studies with inactivation of the insulin and IGF-1 receptors showed the development of dilated cardiomyopathy and lethal heart failure, with the knock-out of this receptor further increasing mortality.15 The use of other tyrosine kinases inhibitors like herceptin and imatinib mesylate was also associated with heart failure.16,17
The need for preclinical models that could more accurately predict the efficacy and safety of new drugs has driven the development of human tissue models of cancer. Our group has previously established a tissue-engineered model of ES (TE-ES), by cultivation of ES tumor aggregates within bioengineered human bone.18-21 This model recapitulated the hypoxic, glycolytic tumor phenotype with a necrotic core surrounded by proliferative ES cells, as well as re-expression of genes related to focal adhesion, malignant deregulation, angiogenesis, and vasculogenic mimicry to levels similar to those observed in patient tumor samples.18
Human cardiac tissue, of high interest for testing the toxicity of anti-cancer drugs, has been studied by several research groups.22-27 Our approach involves the formation of cardiac tissues from human induced pluripotent stem (iPS) cells-derived cardiomyocytes and supporting fibroblasts encapsulated in hydrogel and electromechanical conditioning for tissue maturation. After four weeks in culture, engineered tissues displayed a number of molecular, ultrastructural and functional cardiac properties.23,28
Linking the tissues fluidically enables the crosstalk between tissues as well as more physiological drug delivery, distribution, and uptake. Several groups have developed multi-organ platforms to facilitate developmental drug testing.29-34 Notably, most organs-on-a-chip (OOC) devices currently in use are based on polydimethylsiloxane (PDMS), a material adopted for ease of fabrication, but known to absorb hydrophobic molecules, and most critically drugs and oxygen, thereby limiting the accuracy of testing.35-38
Here we describe a simple OOC with bioengineered human ES tumor and heart tissues and demonstrate its utility for testing the efficacy (using the ES tumor model) and cardiac safety (using the cardiac tissue model) of linsitinib, under the therapeutic regimen used in clinical studies. (Fig. 1, ESI† Fig. S1). Our goal was to recapitulate some of the clinical outcomes for metastatic and non-metastatic ES tumors.
Fig. 1.
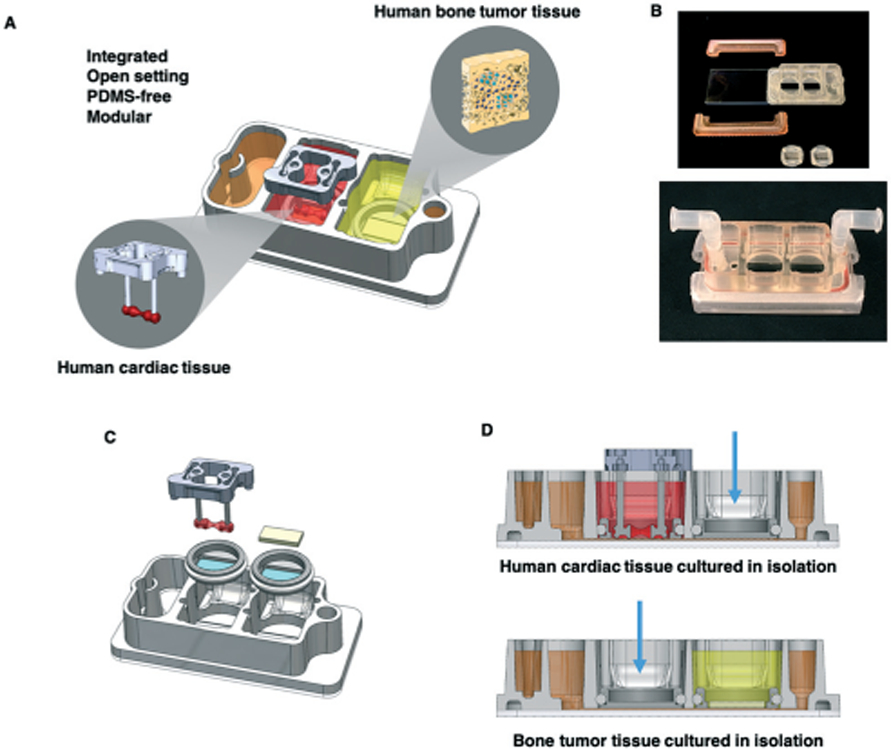
Experimental design. A. Schematic of the platform with two engineered human tissues: Ewing sarcoma (ES) tumor and cardiac tissues that were cultured either with microfluidic perfusion (integrated platform) or in isolation. Metastatic and non-metastatic ES tumors were studied at clinical dosages and treatment regimens of linsitinib. B. Photographs of the integrated platform and its components (top) and in its complete functional state (bottom). C. Platform assembly; note microfluidic connections for circulation at the left and right, and the reservoir for perfusate at the left. D. The platform setup for culturing tissues in isolation, as shown for the cardiac tissue (top) and the bone tumor tissue (bottom). Blue arrows indicate polypropylene plugs, allowing culture of one tissue at a time in isolation.
Results and discussion
Development of an integrated two-tissue platform
We developed a PDMS-free, modular and integrated two-tissue platform for studies of drug anti-tumor efficacy and cardiac safety (Fig. 1A). The platform has 4 main components: (i) the primary piece with tissue chambers and medium reservoir, (ii) 2 clamps, (iii) an O-ring, and (iv) a glass slide at the bottom (Fig. 1A and B, ESI† Video S1). The open setting of the central piece allows manual sampling, and the glass slide allows microscopic analysis. Each tissue is cultured in its own chamber, the bottom of which is a nylon mesh with 20 μm pores (Fig. 1C). These inserts can be replaced by polypropylene plugs when the tissues need to be cultured in isolation (Fig. 1D). Under the nylon mesh membrane, the tissues are linked by a channel that runs along the length of the platform, connecting the flow inlet, the individual tissue chambers, the reservoir where drugs can be introduced, and the flow outlet. The platform uses a single channel of a peristaltic pump to recirculate culture media at a desired flow rate and shear stress (Fig. 2A and B, ESI† Video S2, Fig. S2A-C), within the physiological range for human capillaries.39 Design details are summarized in ESI I† Table S1.
Fig. 2.
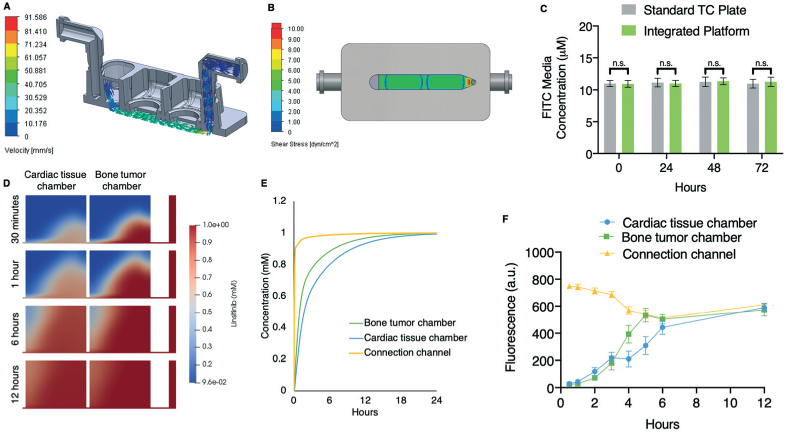
Concentration profiles of a hydrophobic small-molecule tracer and linsitinib circulation within the platform. A. Simulated fluid flow velocity of circulating medium in the platform. B. Simulated shear stress of circulating medium in the platform. C. Hydrophobic FITC (10 μM) was circulated in the platform and its concentration, relative to a control sample in a standard 12-well tissue culture plate, was assessed at 0, 24, 48, and 72 hours (mean ± s.e.m., n = 6). D. Simulated linsitinib concentration gradients within each tissue chamber at 30 minutes and 1, 6, and 12 hours after introduction of linsitinib to the media reservoir. E. Simulated linsitinib concentration in both tissue chambers and in the microfluidic channel over 24 hours. F. Empirical FITC concentrations across both individual tissue chambers and the microfluidic channel were measured every 2 hours for up to 12 hours (mean ± s.e.m., n = 4). *P < 0.05; **P < 0.01; ***P < 0.001 by unpaired, two-tailed Student's t test.
The platform sterility was confirmed by a 4-week incubation with soybean casein digest medium, which is specific for the growth of aerobic bacteria and fungi (ESI† Fig. S2D and E). The central piece of the platform is made of polysulfone, which is a tough, stable, and biocompatible thermoplastic polymer, that does not absorb hydrophobic molecules and is used for the fabrication of new OOC platforms.30,39,40 Fluorescein isothiocyanate (FITC), a low molecular weight, hydrophobic, fluorescent dye, with properties comparable to linsitinib, was circulated for 72 hours, without measurable absorption by the platform (Fig. 2C). The computational fluid dynamics software CoBi was used for simulations of linsitinib transport across the porous nylon mesh membranes separating the individual tissue chambers and flow channel. CoBi has been used previously to simulate drug analog transport in the eye and the lung airway.41,42
Linsitinib introduced into the circulation at a 3.3 mL min−1 flow rate reached uniform concentration between the connection channel and both tissue chambers within 12 hours, and diffused into the tissues within 6 hours (Fig. 2D and E). We also circulated fluorescent FITC, which has similar chemical properties as linsitinib including hydrophobicity and molecular weight, and measured its distribution in the platform (Fig. 2F).43,44 The simulated and experimental results agreed: FITC reached uniform concentration throughout the platform after reaching equilibrium across both models at approximately 6 hours. This is significant, as linsitinib is known to have a short halflife of approximately 5 hours.45 The delayed drug distribution by diffusion through tissues observed here has been documented as an issue for treating solid tumors in patients, with chemotherapeutic concentrations decreasing exponentially with distance from tumor blood vessels and often being limited to the tumor periphery even 12 hours after injection.46-48
To assess molecular diffusion in the platform, we added fluorescent FITC into the bone tumor chamber and showed that it reached uniform distribution across the entire platform after 6 hours of perfusion (ESI† Fig. S2F). To document the inter-chamber communication, we also measured the concentration of osteopontin (OPN), an established marker of osteoblast function, and showed that it distributed from the bone tumor chamber throughout the platform (ESI† Fig. S2G). Moreover, the platform modularity allows serial connections for tissue scaling (ESI† Fig. S3). In the platform, the tissues are cultured with a transwell located at the bottom of the chamber. Because of the location of the transwell, it was difficult to visualize the tissue with the inverted microscope we had available. Thus, we adapted an in-house microscope with an upright objective (Mitutoyo Inc., magnification: 2×) and a working distance of 34 mm to allow visualization of the tissue (ESI† Fig. S4). By incorporating additional optical filters and light sources, this system also enables fluorescent imaging of the tissue.
Validation of engineered Ewing sarcoma and cardiac tissue models
Two types of primary ES tumor cells were used in our bone tumor models: metastatic (SK-N-MC cell line) and non-metastatic (RD-ES cell line). Briefly, both cell lines were used to generate tumor aggregates that were introduced into and co-cultured with primary osteoblasts within a mineralized bone scaffold, where they maintained their native-like tumor morphology and expression of the ES cell marker CD99 (Fig. 3A). While monolayer cultures of ES cells failed to recapitulate tumor morphology and heterogeneity, in our bone tissues we observed heterogeneity in tumor size, morphology, and staining for the proliferation marker Ki67 (ESI† Fig. S5A).
Fig. 3.
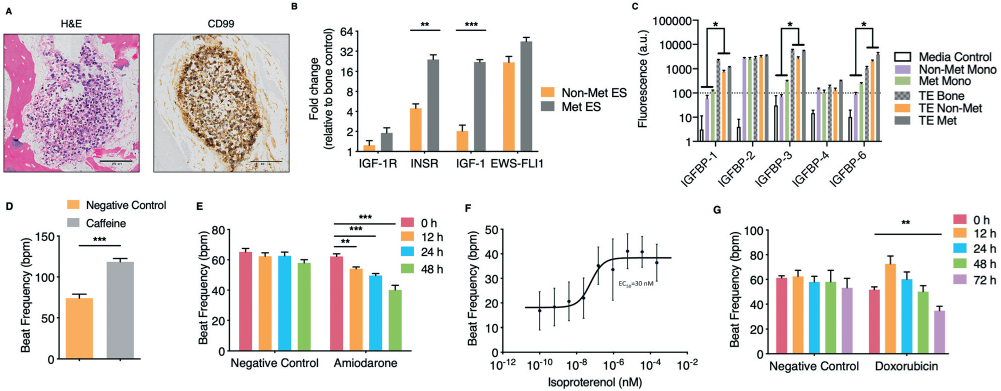
Development and validation of the engineered human Ewing sarcoma (ES) bone tumor and human cardiac tissue. A. Immunohistochemistry analysis of the engineered tumor tissues. H&E staining demonstrates tumor morphology within the tissue engineered bone, and positivity for ES marker CD99. Scale bars: 100 μm. B. Gene expression of ES translocation marker EWS-FLI1 and linsitinib targets in non-metastatic and metastatic ES engineered tissues. Levels were normalized first to β actin and subsequently to the tissue engineered bone control (mean ± s.d., n = 3). C. Proteomic analysis of IGF-1 binding proteins secreted by tumor cells grown in monolayer as compared to our engineered bone (control) and bone tumor tissues (mean ± s.d., n = 3). D. Human engineered cardiac tissue response to caffeine (50 mM) (mean ± s.e.m., n = 5). E. Human engineered cardiac tissue response to amiodarone (2.418 μM) over 48 hours (mean ± s.e.m., n = 6 for negative control; n = 7 for amiodarone). F. Isoproterenol dose-response study of engineered cardiac tissues (mean ± s.e.m., n = 63). G. Response of cardiac tissues to doxorubicin (1 μM) over 72 hours (mean ± s.e.m., n = 7). *P < 0.05; **P < 0.01; ***P < 0.001, by two-way ANOVA with Bonferroni post-test or unpaired, two-tailed Student's t test.
We selected linsitinib because it was a promising chemotherapeutic in a well-documented, ongoing Phase II clinical trial, and because we previously observed similarly upregulated IGF-1 ligand gene expression in native and bioengineered ES tumors relative to the monolayers of ES cells.18 Gene expression (by qRT-PCR) of linsitinib target IGF-1R in our TE-ES models revealed levels similar to those in engineered bone controls (Fig. 3B). Unlike our tissue engineered tumor models, tumor cell monolayers do not allow predictive testing of the drug target expression in the surrounding cells, in this case IGF-1R. Significantly higher expression of the insulin receptor (INSR) and the receptor ligand IGF-1 were observed in the metastatic than non-metastatic TE-ES models (Fig. 3B). This result is important because of the known roles of the INSR and IGF-1 ligand in activating resistance to inhibitors of this tumorigenic pathway, and is consistent with the clinically observed low responsiveness of metastatic ES.49,50
Both in the bloodstream and in the tissues, the IGF binding protein (IGFBP) family has high affinity for the IGF-1 ligand, thus being a critical regulator of the IGF-1R signaling pathway.51 For this reason, any predictive drug studies of IGF-1R inhibitors would need to be conducted at native-like concentrations of these binding proteins. Proteomic analysis of secreted IGFBPs showed significantly higher expression of IGFBP-1, 3, and 6 in both the TE-ES models and engineered bone tissue as compared to the corresponding tumor cell monolayers, which showed only traces of these IGFBPs (Fig. 3C). These transcriptional and proteomic results are also consistent with our previous studies that showed the importance of the tissue milieu in tumor models, including the upregulation of IGF-1 tumorigenic and anti-apoptotic pathways.18
The cardiac tissue model was generated from iPS cell-derived cardiomyocytes and the supporting fibroblasts that were encapsulated in fibrin hydrogel, as in our previous studies.23,28 The cell-loaded hydrogel was stretched between two elastic pillars inducing cell elongation and alignment, and was subjected to electrical stimulation to synchronously contract and work against the pillars. The tissues were matured over 4 weeks of culture and their functionality was validated by responses to drugs with known cardiac effects.
When exposed to caffeine, an inducer of ryanodine receptor-mediated calcium release with tachycardic effects, cardiac tissues displayed physiologic increases in beat frequency (Fig. 3D).52 Amiodarone, an antiarrhythmic therapeutic agent used to treat irregular heartbeats by blocking the potassium channel and increasing the effective refractory period, induced the expected decreases in the beat frequency (Fig. 3E).53 When exposed to isoproterenol, a non-selective beta-adrenergic agonist and the gold standard for assessing the ability of a model to recapitulate beta-adrenergic responses, the beat frequency increased, with expected values of EC50 (Fig. 3F). When exposed to doxorubicin, a chemotherapeutic with well documented cardiotoxic side effects (initial sinus tachycardia, supraventricular tachycardia, chronic dilated cardiomyopathy), the beat frequency initially increased, and then decreased during prolonged exposure to the drug (Fig. 3G).54 The cardiac model recapitulated the physiological effects observed clinically in patients for all four drugs, including doxorubicin.
Responses to linsitinib of engineered tumors cultured in isolation
The Phase II clinical trial of linsitinib that was administered for 3 weeks at the blood plasma concentration of 12 μM to patients with refractory or relapsed ES served as a basis for this study.7 To assess the drug efficacy and safety, we studied the engineered tissues under the same drug regimen used in this clinical study. We first confirmed the maintenance of the engineered bone tissue environment over the entire duration of tumor maturation and drug treatment (5 weeks). Immunohistochemical (IHC) staining of TE-ES samples showed sustained expression of functional osteoblast markers osteocalcin (OCN) and bone sialoprotein (BSP) (ESI† Fig. S5B). In order to track drug responses of ES cancer cell populations within the engineered bone niche, we labeled the metastatic and non-metastatic ES cells using an HIV-based lentiviral system, with a CMV-promoter combined GFP-luciferase vector. Cancer cell titrations demonstrated that the GFP-luciferase expression-dependent luminescence signal served as a reliable readout of viable cancer cells (ESI† Fig. S5C).55 We also monitored the tumor aggregates within the bone tissue by live imaging (ESI† Fig. S5D).
In ES cell monolayers, an MTT viability assay resulted in the IC50 for linsitinib that was two orders of magnitude lower than the effective plasma concentration observed in patients (ESI† Fig. S6A). However, when luminescence was used as a proxy for cell viability, the IC50 concentrations for linsitinib were in line with the 12 μM Cmax clinical concentration, suggesting the validity of this assay for evaluating tumor cell drug responses (ESI† Fig. S6B). Notably, treatment of the cancer cell monolayers with 12 μM linsitinib over 72 hours showed drug efficacy for both the non-metastatic and metastatic ES cells, an observation at odds with clinical data (ESI† Fig. S6C).49,50 These samples were also analyzed using an ELISA to verify linsitinib's mechanism of action—decreased levels of phosphorylated IGF-1R (ESI† Fig. S6D). Having determined that luminescence of the transduced cancer cells could serve as a reliable indicator of ES cell viability in monolayers, we next verified that this method could be used for the TE-ES models by exposing the non-metastatic TE-ES to 1 μM of doxorubicin for 72 hours (ESI† Fig. S7A and B).
The effects of linsitinib were studied in experiments recapitulating the 3 week treatment cycle used in the clinical trial (3 days of drug administration followed by 4 days without the drug, in 3 cycles), with luminescence serving as an indicator of cancer cell viability within the TE-ES. A dose-dependent response was observed for the non-metastatic TEES model, with significant reduction in cell viability at linsitinib concentration of 12 μM (ESI† Fig. S7C). TUNEL assays showed increases in apoptosis, corroborating the luminescence viability findings (ESI† Fig. S7D and E).
The linsitinib responses of metastatic and non-metastatic tumors were evaluated from luminescence signals measured following 3, 7, and 21 days of treatment. Already after 3 days, significant drug responses were observed in both TE-ES tumor models, just as in cancer cell monolayers (Fig. 4A, ESI† Fig. S6C). However, there was a difference between the non-metastatic and metastatic TE-ES model responses across the entire 21-day clinical drug treatment regimen, which was not observed in monolayers due to extensive cell proliferation. Linsitinib caused an initial decrease in cancer cell population in the non-metastatic model and the suppression of subsequent cell proliferation. In contrast, after an initial response to the drug after 3 days, the metastatic model displayed a decrease in drug efficacy, as the cancer cell population continued to expand over the 21-day treatment (Fig. 4A). Unlike the corresponding monolayer results, this observation is in line with the clinical results for metastatic ES—poor outcomes despite aggressive chemotherapy.56
Fig. 4.
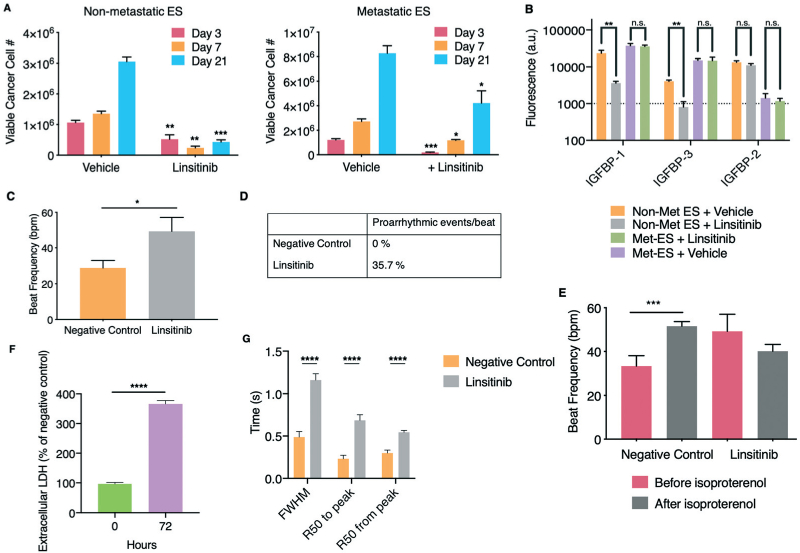
Responses of human engineered bone ES tumors and cardiac tissues to linsitinib in isolated platform chambers. A. Non-metastatic (left) and metastatic (right) ES tumors were exposed to linsitinib (12 μM) according to the 3 week drug treatment regimen used in a phase II clinical study. Luminescence as a function of cancer cell number and viability was measured (mean ± s.e.m., n = 6 for day 3, and n = 3 for day 7 and 21). B. At the culmination of the drug treatment regimen, sample protein lysates were collected for both linsitinib and vehicle treated non-metastatic and metastatic engineered ES bone tumors and comparative proteomic analysis of IGF-1 binding proteins was performed (mean ± s.d., n = 3 per group). C. Beat frequency of cardiac tissues after exposure to linsitinib (12 μM) (mean ± s.e.m., n = 11). D. Occurrence of proarrhythmic events/beat after exposure to linsitinib. E. Beat frequency of human cardiac tissues exposed to linsitinib after isoproterenol exposure (mean ± s.e.m., n = 6–9). F. Extracellular LDH before and after linsitinib exposure, as percentage of negative control (mean ± s.e.m., n = 3). G. Calcium transients of cardiac tissues characterized by the full-width half-maximum (FWHM), R50 to and from peak times (50% of the time to and from the maximal peak of the calcium transient) (mean ± s.e.m., n = 17–18). *P < 0.05; **P < 0.01; ***P < 0.001; ****P <0.0001 by two-way ANOVA with Bonferroni post-test or unpaired, two-tailed Student's t test.
Protein lysates from both metastatic and non-metastatic TE-ES samples at the end of the 21-day linsitinib treatment regimen were analyzed for IGF pathway binding proteins (Fig. 4B). In agreement with the luminescence cancer cell viability results, the metastatic model showed no difference in secretion of IGF binding proteins between the linsitinib treated and control samples, while the non-metastatic samples demonstrated significant decreases in both IGFBP-1 and −3.
Supernatants collected at regular intervals and analyzed for cytotoxicity and secreted proteins suggested the role of osteoblasts in responses to the linsitinib treatment (ESI† Fig. S8A). Lactic acid dehydrogenase (LDH) secretion indicated that cytotoxicity spiked in both models immediately following drug administration, but significantly more so in the responsive, non-metastatic ES model (ESI† Fig. S8A). OCN secretion decreased after drug treatment in both models, suggesting suppressed osteoblast function (ESI† Fig. S8A). Interestingly, the expression of osteopontin (OPN), known to play a stabilizing role for cancer ES cells, significantly increased over 21 days of treatment in the non-metastatic, linsitinib-responsive ES model, and decreased in the metastatic, non-responsive ES model (ESI† Fig. S8A).57
Given the responses to linsitinib observed in the non-metastatic ES tumor model, we isolated the drug-resistant cells by sorting, expanded this subpopulation, and used it to generate new tumor models. These tumors were subjected to another 21-day treatment regimen, to try to further assess the lack of their response to linsitinib. Interestingly, these ES resistant-cell derived tumors again showed a significant initial drug response (ESI† Fig. S8B), in line with the hypothesized transient insulin receptor dependent resistance, as opposed to the “inherited” pathway for IGF1-R inhibitor resistance.58
Responses to linsitinib of engineered cardiac tissues cultured in isolation
After documenting the capability of TE-ES tumors to model drug efficacy, we evaluated the capability of cardiac tissues to determine the cardiotoxicity of the same therapeutic concentration of linsitinib. The cardiac model responded with increased beating frequency after 3 days of drug exposure. Cardiotoxicity of linsitinib has been observed in clinical trials of other types of cancer, with patients presenting proarrhythmic events, like tachycardia (3.75–5% of patients) and atrial fibrillation (3.75–5%).11,12 We observed higher beat frequency and a higher rate of proarrhythmic events per beat (around 36%) than in clinical studies (Fig. 4C and D). Representative videos of a tissue before and after linsitinib treatment can be observed in ESI† Videos S3 and S4, respectively. When the cardiac tissues exposed to linsitinib were subsequently exposed to isoproterenol, the expected chronotropic response was not observed, suggesting lasting effects (Fig. 4E).
Cardiac tissues cultured in isolation responded to linsitinib with high levels of extracellular LDH (Fig. 4F). Because calcium is a key regulator of cardiac function and contraction, we studied calcium handling in cardiac tissues after drug treatment (ESI† Fig. S9A). The duration of calcium transients increased, along with increases in FWHM, R50 time from and to peak (Fig. 4G, ESI† Fig. S9B). Overall, when bioengineered cardiac tissues were exposed to linsitinib in an isolated setting, we observed induction of tachycardia, proarrhythmic events, altered physiological responses to isoproterenol, calcium mishandling, and high levels of LDH. The occurrence of proarrhythmic events at a rate higher than seen clinically and the increased sensitivity observed for beat frequency, isoproterenol response, and calcium handling suggest that this model on its own fails to accurately predict clinical responses. The same can be said for the non-metastatic TE-ES model, which showed significant drug response for the duration of the 3-week drug treatment regimen despite the lack of success in the Phase II clinical trial.
Responses to linsitinib of the ES tumor and cardiac tissues in an integrated setting
Tissue–tissue communication would further increase the physiological relevance of the tumor and cardiac models. In order to demonstrate that an integrated model (with the tumor and cardiac tissues connected by microfluidic perfusion) is more physiologically relevant for predictive drug screening, we studied the effects of linsitinib on the cardiac and tumor tissues simultaneously cultured and exposed to the drug in the integrated platform.
First, we determined the effects of the combined culture medium (1 : 1 mixture of bone tumor and cardiac media in the platform) on each engineered tissue. Importantly, the base media for both tissues are identical, except for one supplement (fetal bovine serum or B-27™). To this end, we cultured the non-metastatic TE-ES tumor (which responded to linsitinib treatment and therefore deviated from the clinically relevant observations) in bone tumor media (isolated culture), 1 : 1 mixed media (integrated platform), and in cardiac media (as a control) for the duration of the clinical drug treatment regimen (3 weeks). No significant differences were observed in the bone niche, and the OCN levels were also similar for the bone tumor media and the mixed media (ESI† Fig. S10A). Longitudinal luminescence readouts used to track ES cells showed faster growth in the 1 : 1 mixed media and cardiac media, suggesting that the B-27™ supplement could be contributing to increased proliferation (ESI† Fig. S10B).
The TE-ES models with mixed media were subjected to the same 12 μM linsitinib treatment regimen as the isolated cultures. Luminescence readings of cancer cell viability within the engineered tissues showed that despite significant increases in cancer cell proliferation in the mixed media, the drug was still effective at killing cancer cells and maintaining their population at a significantly lower level (~30% of their starting population) (ESI† Fig. S10B). Meanwhile OCN secretion increased only slightly, while peaks in LDH secretion (indicating cytotoxicity) were noted immediately following drug exposure at days 3, 11, and 17, similar to those observed with the bone tumor media (ESI† Fig. S10C and D). While some differences in cancer cell proliferation were noted in the mixed media, the responses to linsitinib were comparable. Engineered cardiac tissues in mixed media showed no change in beat frequency (ESI† Fig. S10E) or proarrhythmic events (ESI† Fig. S10F) relative to tissues in cardiac media.
The TE-ES and cardiac tissues were then cultured in the integrated platform with a perfusion of mixed media. Linsitinib was introduced into the reservoir and delivered to tissues via circulation of perfusate and diffusion into the tissues. Following 3 days of treatment, luminescence signals from the engineered non-metastatic ES bone tumor tissues revealed insignificant drug response, as observed in clinical studies, and in contrast to both the monolayer cell cultures and isolated TE-ES culture (Fig. 5A). Secretion of LDH showed no significant difference between the vehicle- and linsitinib-treated samples (Fig. 5B), in agreement with the luminescence viability data.
Fig. 5.
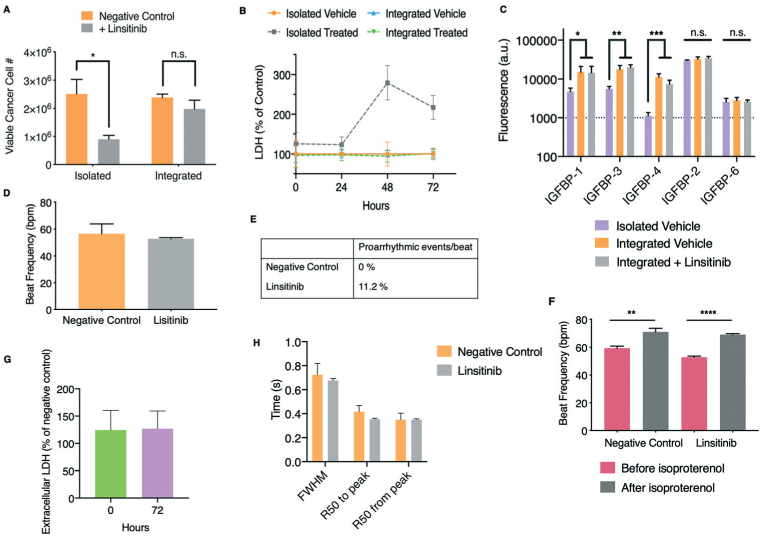
Responses of human engineered bone ES tumors and cardiac tissues to linsitinib in the integrated platform with microfluidic perfusion. A. and B. Non-metastatic ES bone tumors and cardiac tissues were exposed to linsitinib (12 μM) over a period of 72 hours in either isolated culture or within the perfused integrated platform. Luminescence (A) and LDH secretion (B) as functions of cancer cell number and viability as well as cytotoxicity, respectively, were measured (mean ± s.e.m., n = 3). C. Protein lysates were collected from non-metastatic ES bone tumors either grown in isolation, or exposed to perfusion and circulating linsitinib (12 μM) over a period of 72 hours in the integrated platform. Subsequently, comparative proteomic analysis of IGF-1 binding proteins was performed (mean ± s.d., n = 3 per group). D. Beat frequency of cardiac tissues after exposure to linsitinib (12 μM) within the perfused integrated platform (mean ± s.e.m., n = 9). E. Occurrence of proarrhythmic events/beat after exposure to linsitinib within the platform. F. Beat frequency of cardiac tissues that had been exposed to linsitinib in the platform after isoproterenol exposure (mean ± s.e.m., n = 9). G. Extracellular LDH before and after linsitinib exposure, as percentage of negative control (mean ± s.e.m. n = 3). H. Calcium transients of cardiac tissues characterized by the full-width half-maximum (FWHM), R50 to and from peak times (50% of the time to and from the maximal peak of the calcium transient) (mean ± s.e.m. n = 8–9). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 by two-way ANOVA with Bonferroni post-test or unpaired, two-tailed Student's t test.
ES cells, when co-cultured with mesenchymal stem cells and exposed to physiological shear stress in the platform, can become resistant to IGF-1R inhibitors.59 Therefore, we evaluated the role of flow shear in this newly found resistance of non-metastatic TE-ES bone tumor tissues to the IGF-1R inhibitor linsitinib. Initially, we observed increased secretion of OPN by bone tumors in the integrated, perfused culture as compared to isolated culture (ESI† Fig. S10G). This is interesting given the role of osteopontin in the drug resistance of cancer cells growing in bone, as described earlier.57 Proteomic analysis of the IGF pathway performed on TE-ES lysates cultured for 72 hours either in isolation (static culture) or in the integrated platform (perfusion culture), revealed significantly higher production of IGFBP-1, −3, and −4 in response to fluid flow (Fig. 5C). These proteins remained unaffected by linsitinib in the integrated platform, in contrast to isolated cultures discussed above, further demonstrating the loss of responsiveness (Fig. 4B and 5C).
Genomic analysis of IGFBP-3 expression in native ES tumors obtained from patients showed elevated levels over those in healthy individuals and 2D monolayer cultures of ES cell lines, including those used in our model (RD-ES and SK-N-MC) (ESI† Fig. S11A). Furthermore, high expression of IGFBP-3 correlated with poor survival of ES patients (ESI† Fig. S11B). High levels of IGFBP-3 in non-metastatic tumors cultured in the integrated platform agree with the genomic clinical data, and support the physiological relevance of perfusion for the tumor models.
Linsitinib was then introduced into the platform for 3 days (12 μM), either via perfusion or directly into the TE-ES tissue chamber, to distinguish the effects of flow-derived stimuli from drug diffusion into the tissues (ESI† Fig. S11C). Immediate exposure to the platform volume's amount of the drug resulted in a response to the drug akin to that observed in isolated cultures. In contrast, introduction of linsitinib into the circulation again showed no response. Taken together, all of these results were in agreement with the observations from the clinical trial, since linsitinib was unable to stop progression of ES, with none of the patients completing the trial. Culture of the TE-ES model in the integrated platform activated increased expression of both OPN and the IGF pathway binding proteins, both correlated with poor survival in patients, and treatment with drug via perfusion allowed for a more physiologically relevant distribution of the drug throughout the tumor tissue. We propose that the integrated model provides a better mimic of the clinical scenario than the isolated cultures, which did not match the clinical data.7
In the cardiac tissue model, we did not observe linsitinib-mediated changes in beat frequency, suggesting that the occurrence of false responses was reduced (Fig. 5D). Representative videos of a tissue before and after linsitinib treatment can be observed in ESI† Videos S5 and S6, respectively. Similarly, the rate of proarrhythmic events in the integrated model (~ 11%) was much closer to the rates observed clinically (Fig. 5E).11,12 When the cardiac tissues exposed to linsitinib were subsequently exposed to isoproterenol, we observed the expected chronotropic response (Fig. 5F). In the integrated platform, the cardiac tissues showed no major differences in extracellular LDH (Fig. 5G) and calcium handling (Fig. 5H, ESI† Fig. S12) between the drug-exposed and control tissues. Overall, in the integrated platform, linsitinib caused the incidence of proarrhythmic events similar to clinical data, while maintaining physiological response to isoproterenol and calcium handling, suggesting mild cardiotoxicity.
Conclusion
The platform design allowed real-time in situ monitoring of cancer cell growth and simultaneous assessment of the drug efficacy and cardiotoxicity. The platform's flexibility and ease of use allow the design to be tailored to the questions being asked. Also, the use of polysulfone as the main device fabrication material, instead of the widely utilized PDMS, avoids uncontrollable absorption of hydrophobic compounds, which most chemotherapeutics are. The open setting also allows for imaging and sampling of tissues and culture media. Because of the nature of linsitinib, we focused on cardiac function (contractile behavior and calcium handling) and cell viability, rather than on structure. In future experiments, if the drug being studied is suspected to induce structural changes, it should be looked into.
The integrated platform reported here contained the ES tumor (formed by introducing primary cancer cells into the engineered human bone) and the engineered human cardiac muscle (formed by electromechanical conditioning of iPS-derived cardiomyocytes and supporting fibroblasts in fibrin gel), connected by microfluidic circulation. The biological fidelity of the engineered tumor and heart tissues was documented by known responses to standard drugs. We also demonstrated advantages of engineered tissues over monolayer culture.
Tissues connected by a microfluidic circulation platform recapitulated the unfortunate results of a Phase II clinical trial of linsitinib. The integrated platform mimicked clinical results, while the isolated tissues mimicked preclinical results, a paradigm that can lead to expensive late stage drug failures. To overcome this, more predictive models, like the integrated platform developed herein, could be used preclinically to better predict clinical outcomes at an earlier stage. Future studies should demonstrate applicability of this system to patient-specific studies of other cancer drugs, in particular in the rapidly emerging field of cardio-oncology.
Methods
Integrated platform
The main manifold of the platform was machined using a 3-axis computer numerical control (CNC) milling machine from polysulfone and incorporated reservoirs for individual tissues and an additional reservoir and fluidic ports for circulating media. The connection channel was defined by a recessed slot within the main manifold and was sealed against a glass slide with machined polycarbonate clamps and an O-ring gasket. Each tissue reservoir was separated from the recirculation channel by a polypropylene insert over-molded onto a nylon mesh porous membrane. The membrane insert created a seal with the main manifold through the use of an elastomer O-ring. The plugs used to isolate tissue chambers (for culture in isolation) were machined from polycarbonate to create a seal via a fluoroelastomer O-ring.
The platform was connected to a peristaltic pump with a luer taper connector, with media flowing underneath through the connection channel. The media exited the channel into a reservoir, which also functions as a bubble trap. The reservoir was connected to the pump with a luer taper connector. PharmaMed pump tubing (Cole Parmer) routed the media back to the peristaltic pump (Cole Parmer) for recirculation.
The platform was contained within a 100 mm polystyrene dish that incorporated a secondary spacer between the dish and the lid to pass tubing in and out of the assembly without introducing gaps that would compromise sterility.
Software and equipment used for machined components include SolidWorks for 3D design, Mastercam for toolpath generation, and a Haas OM2 3 axis milling machine for physical manufacturing. Polycarbonate and polysulfone materials were sourced from McMaster-Carr. For injection molding of porous membrane inserts, nylon meshes were sourced from Millipore, polypropylene pellets (Flint Hills Resources P9M7R-056) sourced from PolyOne Distribution, and molds were machined in aluminum using the above fabrication equipment. Nylon mesh inserts were cut using a 40 W CO2 laser cutter and inserted into the mold. Injection molding was performed on an AB-200 semi-automatic plastic injector (AB Machinery).
Customized microscope system
The customized microscope was assembled on an optical breadboard (12″ × 12″). The system includes a 2× plan apochromat objective lens that allows a lager field of view, a CMOS monochromatic camera, and exchangeable LED light sources. The camera is mounted vertically on a motorized optical rail that enables focus of different horizontal plains of the tissues with enhanced precision. The LED light source provides either a white light or a light with a specific wavelength when coupled with an optical filter allowing bright-field or fluorescent imaging. All optomechanical components were obtained from Thorlabs, while the objective lens was purchased from Edmund Optics.
Sterility assay
The platform was incubated for 4 weeks, at 25 °C, with Soybean casein digest medium (SCDM), an aerobic bacteria and fungi specific medium. After the incubation period, any changes in the medium turbidity and the presence of microorganisms were assessed.
Cell culture
Human iPS cells were obtained through material transfer agreements from B. Conklin, Gladstone Institute (WTC11 line), maintained in mTeSR™1 medium (STEMCELL Technologies), supplemented with 1% penicillin/streptomycin, changed on a daily basis, on 1 : 60 growth-factor-reduced Matrigel (Corning) and passaged when 85–90% confluent using 0.5 mM EDTA (Invitrogen). For the first 24 hours after passaging, the culture medium was supplemented with 5 mM Y-27632 dihydrochloride (Tocris).
Human mesenchymal stem cells (MSCs) were isolated from commercially obtained fresh bone marrow aspirates (Cambrex) by attachment to the plastic surface, as previously described.20 Cells were expanded to the fourth passage in mesenchymal stem cell medium consisting of high glucose Dulbecco's modified Eagle's medium (DMEM; Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific), 1% penicillin–streptomycin (Life Technologies), and 0.1 ng mL−1 bFGF (Life Technologies).
The metastatic SK-N-MC (HTB-10) and non-metastatic RD-ES (HTB-166) ES cell lines were purchased from the American Type Culture Collection (ATCC). SK-N-MC cells were cultured in Eagle's minimum essential medium (EMEM; ATCC) and RD-ES cells were cultured in RPMI-1640 medium (ATCC), according to the manufacturer's specifications. Both culture media were supplemented with 10% FBS and 1% penicillin/streptomycin.
All cells were maintained at 37 °C and 5% CO2 in Heracell 150 incubators (Thermo Fisher Scientific). The cultures were maintained with 2 ml of medium per 10 cm2 of surface area and were routinely checked for mycoplasma contamination using a MycoAlert Plus Kit (Lonza). Pluripotent cells were routinely checked for expression of pluripotent markers.
GFP-luciferase transduction and cell sorting
A LentiSuite for HIV-based system (System Biosciences) was used according to the manufacturer's instructions to generate stable CMV-GFP-T2A-Luciferase vector expressing ES (SK-N-MC and RD-ES). Briefly, HEK-293 T (CRL-3216) cells were transfected with lentiviral and the GFP-Luciferase vector of interest, viral particles were purified and concentrated using a PEG-it Virus Precipitation Solution (System Biosciences). Cancer cell lines were transduced with the virus at MOI = 10 using Lipofectamine 3000 reagent (Thermo Fisher Scientific), according to the manufacturer's protocols. GFP+ transduced cancer cells were selected and sorted for using an Influx cell sorter (BD Biosciences) in collaboration with the Columbia Center for Translational Immunology (CCTI) Flow Cytometry Core at Columbia University Irving Medical Center.
Bone matrix scaffolds
Decellularized bone scaffolds were generated using a previously established protocol and cut into 2 mm thick axial sections.55 Sections to fabricate scaffolds were cleaned under high-pressure water beam, dried, and machined using a standard two-flute endmill to the final geometry of 6 mm × 3 mm × 1 mm (length x depth × thickness). To remove cellular material, the scaffolds were subjected to serial washes in 0.1% EDTA in phosphate-buffered saline (PBS; Santa Cruz Biotechnology), 0.1% EDTA in 10 mM Tris, and 0.5% SDS in 10 mM Tris, and a solution of 100 U mL−1 DNase and 1 U mL−1 RNase in 10 mM Tris buffer. Scaffolds were thoroughly rinsed in deionized water and freeze-dried. The scaffolds were selected within the density range of 0.37–0.45 mg mm−3, were sterilized overnight in 70% ethanol, and conditioned in mesenchymal stem cell medium overnight before seeding with cells. To monitor the effectiveness of the decellularization protocol, DNA content of the bone before and after decellularization was quantified using Quant-iT™ PicoGreen™ dsDNA Assay Kit (Thermo Fisher Scientific), following the manufacturer's protocol.
Tissue engineered ES tumors
Using an established protocol, expanded MSCs were seeded into the bone matrix scaffolds at a concentration of 106 cells per scaffold, using 40 μL of medium.60 The cells were allowed to attach for 2 hours, and then supplemented with additional mesenchymal stem cell medium overnight. After 24 hours, osteogenic differentiation was initiated by addition of low glucose DMEM supplemented with 1 mM dexamethasone (Sigma-Aldrich), 10 mM β-glycerophosphate (SigmaAldrich), and 50 mM L-ascorbic acid-2-phosphate (Sigma Aldrich). Each scaffold was incubated in 4 mL of osteogenic media with media changes 3 times a week for 3 weeks, allowing MSCs to differentiate into functional, maturing osteoblasts.
Two weeks following the initiation of osteogenic differentiation, aggregates of ES tumor cells were prepared as described previously, using 0.3 ×106 cells per aggregate.18 After 1 week of culture, corresponding to the end of bone tissue culture (3 weeks), the primary ES cell aggregates were placed into the engineered bone constructs (3 aggregates per construct, placed apart from each other). Tumor models were established for 2 different types of primary ES cells: non-metastatic (RD-ES) and metastatic (SK-N-MC). Tissue engineered RD-ES and SK-N-MC tumors were cultured in the RPMI and EMEM media, respectively, supplemented with 10% FBS and 1% penicillin/streptomycin. Bone constructs cultured without tumor cell aggregates (TE-bone) in RPMI and EMEM media were used as controls.
Upon maturation, bone tumors were transferred into the platform chambers and were cultured either in an isolated setting (stopping communication between the tissue chambers by inserting polypropylene plugs in the bottom of the chamber (Fig. 1D)), or in an integrated setting (tissue chambers connected by microfluidic perfusion).
Cardiac differentiation of human iPS cells
Using a previously established protocol, cardiac differentiation of human iPS cells was initiated in 90% confluent cell monolayers by replacing the mTeSR™1 medium with CDM3 (chemically defined medium) with 3 components: RPMI Medium 1640 (1×, Gibco), 500 μg mL−1 of recombinant human albumin (Sigma-Aldrich) and 213 μg mL−1 of l-ascorbic acid 2-phosphate (Sigma-Aldrich), supplemented with 1% penicillin/streptomycin.61 Medium was changed every 48 hours. For the first 48 hours, the medium was supplemented with 3 mM of glycogen synthase kinase 3 inhibitor CHIR99021 (Tocris). On day 2, the culture was switched to CDM3 medium supplemented with 2 mM of the Wnt inhibitor Wnt-C59 (Tocris). After day 4 of differentiation, the medium was changed to CDM3 with no supplements. Contracting cells were noted around day 10, when medium was changed to RPMI 1640 supplemented with B-27™ (50×; Gibco), and were used in experiments without selection for cardiomyocytes.
Tissue engineered cardiac muscle
Using a methodology established in our previous studies, cardiac tissues were formed between two elastic pillars (1mm in diameter, 9mm in length, 6mm in axis-to-axis distance) that were over-molded onto a polycarbonate support frame.23,28 The pillars were formed using Delrin (polyoxymethylene) molds fabricated by CNC machining. PDMS was centrifugal casted at 400 relative centrifugal force (RCF) for 5 minutes through the polycarbonate support structures inserted into the molds. After centrifugation, PDMS was cured in an oven at 60 °C for 1 hour and used at a 10 : 1 ratio of silicone elastomer base/curing agent. The resulting component pair of pillars to support the formation of one tissue, was inserted into the molds for cardiac tissues by press-fitting. An array of 6 reservoirs accommodates formation of 6 individual tissues.
Human iPS cell-derived cardiomyocytes at day 13 of differentiation were combined with normal human dermal fibroblasts (NHDF; Lonza) at a ratio of 75% human iPS cell-derived cardiomyocytes and 25% NHDF, for a total of 1 million cells per tissue. The hydrogel was formed by mixing 33 mg mL−1 of human fibrinogen (Sigma-Aldrich) with 25 U mL−1 of human thrombin (Sigma-Aldrich), at an 84 : 16 ratio. The cell suspension in hydrogel was dispensed into each well and allowed to polymerize around the pillars at 37 °C for 15 minutes before adding RPMI Medium 1640 supplemented with B-27™ containing 0.2mg mL−1 aprotinin (Sigma-Aldrich).
Tissues were formed by inserting the pillars into a mold for cardiac tissues (9 mm length × 3.2 mm width × 4.3 mm depth) that can be filled with 100 μL of cell suspension in hydrogel. Hydrogel compaction caused passive tension of the tissues stretched between the two pillars, inducing elongation and alignment. The medium was changed every other day and supplemented with 0.2 mg mL−1 aprotinin for the first 7 days. Cardiac tissues were transferred into the platform chambers and cultured in either isolation or integrated by perfusion, as previously explained at the end of the “Tissue engineered ES tumors” section.
Mathematical model of linsitinib transport in the platform
To evaluate drug transport in the blank platform, we performed computational fluid dynamics using a simultaneous finite volume solver (CoBi) that solves complex mass (continuity), momentum, energy, and drug conservation equations in two-dimensional discretization with heterogeneous properties (eqn (1)-(3)). The transport equations account for convection, diffusion, fluid–solid interaction, electrostatic drift and interfacial friction.
| (1) |
| (2) |
| (3) |
where P is the pressure, t is time, ρ is the fluid density, is the bulk fluid velocity, μ is the fluid viscosity, is the additional body force per unit mass, C is linsitinib concentration, D is the linsitinib diffusivity, and S is the source term. CoBi also has built-in modules to assign hydrodynamics (pressure, volumetric flux, and porous medium) and diffusion (partition coefficients, permeability, and diffusivity) properties.
Transwell membrane porosity was calculated by definition:
| (4) |
where Vvoid is the void volume, and VTotal is the total membrane volume. Using manufacturer's information for the total surface area, pore density, and pore size in the membrane, its porosity was calculated to be 5%.
The Polson equation (eqn (5)) was used to predict the diffusion coefficient:
| (5) |
where the parameters are dynamic viscosity (μ) at absolute temperature (T), and molecular weight (MW).62 Linsitinib diffusion in media was calculated to be 4.4 × 10−10 m2 s−1.
Estimation of linsitinib absorption and diffusive transport
Fluorescein isothiocyanate (FITC, 10 mM in DMSO; Sigma Aldrich) was circulated in the integrated platform to determine potential hydrophobic small molecule absorption, given its physical and chemical properties. FITC was added at a concentration of 10 μM to 1 : 1 bone tumor/cardiac mixed media and introduced into the platform. The control was the FITC-containing media in a 12-well tissue culture plate. Aliquots from the reservoir, bone tumor, and cardiac tissue chambers were taken at 0, 24, 48 and 72 hours and measured for fluorescent signal using a spectrophotometer (Biotek). A standard curve for FITC was generated to calculate the FITC concentrations from the measured fluorescence signals.
FITC concentrations were used to estimate the distribution of linsitinib within the platform; in the medium reservoir and each of the tissue chambers. Platforms were filled with 8 mL of 1 : 1 mixed media each, after which 10 μM of FITC was injected into one of the tissue chambers. The platforms were connected to the peristaltic pumps run at a flowrate of 3.3 mL min−1 to generate physiologically relevant fluid shear stress. Aliquots were taken from different locations in the platform and assayed for fluorescence on a spectrophotometer (Biotek).
Drug treatments
Cardiac tissues were studied using caffeine (50 mM in water; Sigma-Aldrich), amiodarone hydrochloride (2.418 μM in DMSO; Sigma-Aldrich), isoproterenol hydrochloride (a series of drug concentrations in water; Sigma-Aldrich), or doxorubicin hydrochloride (1 μM in DMSO; Sigma-Aldrich), all diluted in RPMI Medium 1640 supplemented with B-27™. Response to isoproterenol was analyzed 10 minutes after exposure to 1 mM isoproterenol hydrochloride, diluted in RPMI Medium 1630 supplemented with B27™.
ES bone tumor cell lines and tissues were studied using either doxorubicin hydrochloride (10 mM in water; Sigma-Aldrich), linsitinib (OSI-906) (various concentrations in DMSO; Santa Cruz Biotechnology), all diluted in either non-metastastic media (RPMI Medium 1640, 10% FBS, 1% PenStrep) or metastatic media (EMEM, 10% FBS, 1% PenStrep).
Both tissues were treated with linsitinib, dissolved at a 10 mM concentration in DMSO (Corning) and mixed in with the respective cell medium at a 12 mM concentration unless otherwise noted. Vehicle treatments involved just the addition of DMSO at identical volumes as a control. Tissues were randomly assigned to experimental groups. Medium was changed every day.
Histology, immunofluorescence, and microscopy
Bone tissue samples were washed in PBS, fixed in 10% formalin at room temperature for 24 hours, and decalcified for 24 hours with Immunocal solution (Decal Chemical Corp.). Samples were dehydrated in graded ethanol solutions, paraffin embedded, and sectioned to 5 μm thick. For immunohistochemistry, tissue sections were deparaffinized with CitriSolv (Thermo Fisher Scientific) and rehydrated with graded ethanol washes. Antigen retrieval was performed by incubation in citrate buffer (pH 6) at 90 °C for 30 minutes, while endogenous peroxidase activity was blocked with 3% H2O2. After washing with PBS, sections were blocked with horse serum (Vector Labs) and stained with primary antibodies overnight in a humidified environment. The primary antibodies used were polyclonal rabbit IgG to CD99 (1 : 500; ab108297), polyclonal rabbit IgG to Ki67 (1:100; ab15580), polyclonal rabbit IgG to osteopontin (1 : 500; ab1870), and polyclonal rabbit IgG to bone sialoprotein 2 (1 : 500, ab1854). After washing with PBS, samples were incubated with anti-rabbit secondary antibodies for 1 hour at 25 °C, developed as described previously (Vector Laboratories) and counterstained with Hematoxylin QS (Vector Labs).60 The images of histological sections were obtained by digitizing the tissue sections using the Olympus dotSlide 2.4 digital virtual microscopy system (Olympus) at a resolution of 0.32 μm.
To assess apoptosis, paraffin embedded tissue sections were first deparaffinized with CitriSolv, rehydrated with a graded series of ethanol washes, and stained with a Click-iT® TUNEL Alexa Fluor® imaging assay (Thermo Fisher Scientific). Following nuclear counterstaining with DAPI (Life Technologies), the TUNEL labelled slides were imaged with an IX81 inverted fluorescent microscope (Olympus) and a Pike F032B camera (ALLIED Vision), using NIS-Elements AR software, and processed using ImageJ (NIH). Four representative images per condition were then analyzed using the previously developed automatic TUNEL cell counter plugin for ImageJ to quantify DAPI+ cells and TUNEL+ cells.63 To view the transduced fluorescent bone tumor aggregates in situ, the TE-ES samples were captured using a Nikon A1 scanning confocal microscope on an Eclipse Ti microscope stand (Nikon Instruments, Melville, NY) using a 10×/0.3 Plan Fluor (Nikon) objective. The confocal pinhole was set at 1 airy unit to produce an optical section of approximately 17 μm. GFP was excited at 488 nm and emission was collected from 500–550 nm. Z series were collected through the depth of the tissue section and maximum projection renderings were generated using NIS Elements software (Nikon). Images were collected in the Confocal and Specialized Microscopy Shared Resource of the Herbert Irving Comprehensive Cancer Center.
Quantitative real-time PCR
Total RNA was isolated using Trizol (Life Technologies), following the manufacturer's instructions. RNA preparations (2 μg) were treated with a high-capacity cDNA reverse transcription kit (Applied Biosystems) to generate cDNA. Quantitative real-time PCR was performed using Fast SYBR™ Green Master Mix (Applied Biosystems). mRNA expression levels were quantified applying the ΔCt method, ΔCt = (Ct of gene of interest – Ct of β-Actin). Primer sequences were those that have been previously reported.18
Contractility videos
To measure the cardiac contractility online, we took contractility videos of the tissues that were analyzed using the native MATLAB code we previously developed.23,28 Tissue contractility was measured by tracking the change in tissue area as a function of time. Acquired video frames were inverted and an automated intensity threshold was used to identify cell location in the video frame. First, a baseline time point in the video corresponding to a relaxed tissue state was selected. Absolute differences in cell area from the baseline frame were then calculated to create a time course of cell area changes over time. The resulting time courses were analyzed using a native MATLAB (MATHWorks) automated peak finding algorithm to determine locations of maximum cell contractions in the time profiles. Beat period lengths were determined from the length of time between the pairs of local maxima, and the beat frequencies were determined by inverting beat periods. The rate of proarrhythmic events was calculated by the ratio of the number of proarrhythmic events over the total number of beats.
Calcium handling
After treatment with linsitinib, cardiac tissues were incubated with Fluo-4 (Invitrogen) in RPMI Medium 1640, supplemented with B-27™ and 10 μM blebbistatin (Sigma) for 30 minutes at 37 °C. Videos were acquired and analysed in MATLAB using a custom script that calculated the temporal changes in calcium fluorescence intensity. Each frame was normalized to a baseline background region to give baseline-corrected changes in minimum and maximum fluorescence values for each frame. The temporal changes in fluorescence intensity were presented by calcium transient traces. Full-width half max (FWHM) corresponds to the time between the calcium concentration transient value halfway through the contraction and the value halfway through the relaxation period. R50 values correspond to the time it takes for the cardiac tissue to contract from or relax to 50% of contracted state. Interbeat variability is the standard deviation of the time between beats, time to peak is the time it takes for the cardiac tissue to fully contract, and decay time reflects the time it takes for the tissue to fully relax.
Cell viability
Cell viability was analyzed using a previously established protocol.64 Cancer cell viability was measured for GFP-Luciferase labelled cancer cells using ONE-Glo luciferase substrate that was prepared according to the manufacturer's protocol (Promega). Samples were collected following 3, 7, and 21-day cycles of linsitinib treatment. Where noted, longitudinal cell viability was also assessed using luminescence, though at the cost of signal strength. Briefly, in vivo grade VivoGlo™ Luciferin (Promega) was made at a 200× stock concentration (30 mg mL−1) in water, added to the sample culture media at a 1 : 200 dilution, and scanned using a spectrophotometer (Biotek). Some of the IC50 values (as noted in the text) were determined using cell viability data generated using an MTT assay (RealTime-Glo™ MT cell viability assay, Promega) which were analyzed according to manufacturer's protocol. Cardiac cell viability was assessed by the Pierce LDH cytotoxicity assay kit (Thermo Fisher Scientific) in supernatant collected at 0 and 72 hours.
IGF pathway protein quantification
Proteomic analysis of secreted IGFBPs was performed using supernatants isolated from RD-ES and SK-N-MC monolayers as well as both non-metastatic and metastatic TE-ES samples. Where indicated, protein lysates were obtained from engineered ES tumor tissues using a cell lysis buffer to control for differences in media volume in the isolated setting versus that used in the integrated platform (RayBiotech). A Pierce™ BCA Protein Assay Kit (ThermoFisher) was used to quantify protein amounts across the samples, after which equivalent amounts were loaded and processed onto a Human IGF Signaling Array (RayBiotech) according to the manufacturer's instructions. The samples were shipped to RayBiotech for quantification.
In order to confirm linsitinib's mechanism of action in ES cells, both RD-ES and SK-N-MC monolayers were treated with 12 μM linsitinib for 6 hours, lysed, measured for protein quantity using a Pierce™ BCA Protein Assay Kit (ThermoFisher), and loaded equally onto a Human Phospho- and Total IGF1R ELISA (RayBiotech) to semi-quantitatively determine phosphorylated levels of the IGF-1 receptor, according to the manufacturer's instructions. Osteocalcin (OCN), osteopontin (OPN), and lactic acid dehydrogenase (LDH) secreted levels were all measured using a similar approach. Supernatants were isolated from controls and drug treated TE-ES (collected from isolated or integrated culture as indicated) and equal amounts were used in each assay according to the manufacturer's instructions. For OCN a Human Osteocalcin Quantikine ELISA (R&D Systems) was used, while for OPN it was a Human Osteopontin Quantikine ELISA (R&D Systems). LDH secretion was determined using a lactate dehydrogenase assay kit (Colorimetric; Abcam).
Genomic analysis of IGFBP-3 expression
The web-based genomics analysis and visualization application R2 Genomics Analysis and Visualization Platform (http://r2.amc.nl.) was used to determine IGFBP-3 average mRNA expression across multiple open access public ES data sets, described below. For consistency, we conducted comparative genomic analysis using the same microarray chips and normalization methods across studies.
Tumor Ewing's Sarcom–Savola (73 samples) source: GEO ID: gse17679 Dataset Date: 2000-01-01. Inflammatory gene profiling of Ewing sarcoma family of tumors.
Tumor Ewing's Sarcoma–Francesconi (37 samples) source: GEO ID: gse12102 Dataset Date: 2000-01-01. A genome-wide association study of at least 401 French ES patients compared to either 684 French or 3668 US self-described Caucasian controls consistently revealed candidate loci at chromosomes 1 and 10 (p < 10–6).
Tumor Ewing's Sarcoma–Delattre (117 samples) source: GEO ID: gse34620 Dataset Date: 2008-06-15. Expression profiling of Ewing sarcoma samples in the frame of the CIT program from the French Ligue Nationale Contre le Cancer.
Kaplan Meier survivability curve – IGFBP-3
Kaplan scanning was performed within the R2 Genomics Analysis and visualization platform (http://r2.amc.nl.). Briefly, the Kaplan scanner separates the samples of a dataset into 2 groups based on the gene expression of one gene, in this case IGFBP-3. In the order of expression, it uses every increasing expression value as a cut-off to create 2 groups and test the p-value in a log-rank test. Minimum group size was set to 8. The highest value is reported, accompanied by a Kaplan–Meier graph that shows the most significant expression cut-off for survival analysis by separating sample groups into high and low expression values. The best possible Kaplan–Meier curve that is based on the log-rank test is only possible for datasets where survival data is present (in our study the Savola dataset). Patients were enrolled in the Italian Cooperative Study (SE 91-CNR Protocol; started November 1991; ended November 1997) organized by the Italian Association for Pediatric Hematology–Oncology and the National Council of Research (CNR).
Statistical methods
Data were analyzed in Excel (Microsoft) and graphed in Prism (GraphPad). Data are presented as mean ± s.e.m., unless otherwise noted. Differences between experimental groups were analyzed by unpaired, two-tailed Student's t-test or two-way ANOVA with Bonferroni post-test. Significant differences defined by P < 0.05 for all statistical methods, unless otherwise noted. No blinding or randomization was used.
Supplementary Material
Acknowledgements
The authors gratefully acknowledge funding support of the NIH (grants EB025765, EB027062, HL076485 and CA249799 to G. V.-N.; grant S10OD020056 to the Columbia Center for Translational Immunology Flow Cytometry Core at Columbia University Irving Medical Center; grant P30CA013696 to the Confocal and Specialized Microscopy Shared Resource of the Herbert Irving Comprehensive Cancer Center at Columbia University), NSF (grant 16478 to G. V.-N.), and FCT (grant PD/BD/105819/2014 to D. T.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Authors thank Manuel Tamargo for assistance in editing the figures and the Yazawa Lab for assistance with the luminescence readings.
Footnotes
Conflicts of interest
G. V.-N. and K. R.-B. are co-founders of TARA Biosystems, a Columbia University spin-out that is commercializing the use of bioengineered human cardiac tissues for drug development.
Electronic supplementary information (ESI) available. See DOI: 10.1039/d0lc00424c
References
- 1.Hay M and Thomas DW, Nat. Biotechnol, 2014, 32, 40–51. [DOI] [PubMed] [Google Scholar]
- 2.Dickson M and Gangnon JP, Nat. Rev. Drug Discovery, 2004, 3, 417–429. [DOI] [PubMed] [Google Scholar]
- 3.DiMasi JA, Grabowski HG and Hansen RW, J. Health Econ, 2016, 47, 20–33. [DOI] [PubMed] [Google Scholar]
- 4.O'Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR and Folkman J, Cell, 1997, 88, 277–285. [DOI] [PubMed] [Google Scholar]
- 5.Kulke MH, Bergsland EK, Ryan DP, Enzinger PC, Lynch TJ, Zhu AX, Meyerhardt JA, Heymach JV, Fogler WE, Sidor C, Michelini A, Kinsella K, Venook AP and Fuchs CS, J. Clin. Oncol, 2006, 24, 3555–3561. [DOI] [PubMed] [Google Scholar]
- 6.Flowers RJ, Nat. Rev. Drug Discovery, 2003, 3, 179–191. [DOI] [PubMed] [Google Scholar]
- 7.ClinicalTrials.gov, Eurosarc Trial of Linsitinib in Advanced Ewing Sarcoma (LINES) [Internet], Bethesda: National Library of Medicine, 2015. [updated 2019 June 3; cited 2019 Nov 17], Available from: https:///clinicaltrials.gov/ct2/show/NCT02546544. [Google Scholar]
- 8.Murakami T, Singh AS, Kiyuna T, Dry SM, Li Y, James AW, Igarashi K, Kawaguchi K, DeLong JC, Zhang Y, Hiroshima Y, Tussel R, Eckardt MA, Yanagawa J, Federman N, Matsuyama R, Chishima T, Tanaka K, Bouvet M, Endo I, Eilber FC and Hoffman RM, Oncotarget, 2016, 7, 47556–47564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun H, Lin D-C, Cao Q, Guo X, Marijon H, Zhao Z, Gery S, Xu L, Yang H, Pang B, Lee VKM, Lim HJ, Doan N, Said JW, Chu P, Mayakonda A, Thomas T, Forscher C, Baloglu E, Shacham S, Rajalingam R and Koeffler HP, Cancer Res., 2016, 76, 2687–2697. [DOI] [PubMed] [Google Scholar]
- 10.Amaral AT, Garofalo C, Frapolli R, Manara MC, Mancarella C, Uboldi S, Giandomenico SD, Ordóñez JL, Sevillano V, Malaguarnera R, Picci P, Hassan AB, Alava ED, D'Incalci M and Scotlandi K, Clin. Cancer Res, 2015, 21, 1373–1382. [DOI] [PubMed] [Google Scholar]
- 11.ClinicalTrials.gov, Linsitinib in Treating Patients with Gastrointestinal StromaI Tumors, Bethesda: National Library of Medicine, 2012. [updated 2018 Sept 21; cited 2019 Nov 17], Available from: https:///clinicaltrials.gov/ct2/show/NCT01560260. [Google Scholar]
- 12.ClinicalTrials.gov, Linsitinib or Topotecan Hydrochloride in Treating Patients with Relapsed Small Cell Lung Cancer, Bethesda: National Library of Medicine, 2012. [updated 2016 Jan 14; cited 2019 Nov 17], Available from: https:///clinicaltrials.gov/ct2/show/NCT01533181. [Google Scholar]
- 13.Mcmullen JR, Shioi T, Huang W, Zhang L, Tarnavski O, Bisping E, Schinke M, Konh S, Sherwood MC, Brown J, Riggi L, Kang PM and Izumo S, J. Biol. Chem, 2004, 279, 4782–4793. [DOI] [PubMed] [Google Scholar]
- 14.Kuo WW, Chu C-Y, Wu CH, Lin JA, Liu J-Y, Hsieh Y-H, Ueng KC, Lee SD, Hsieh DJ, Hsu HH, Chen LM and Huang CY, Cell Biochem. Funct, 2005, 23, 325–331. [DOI] [PubMed] [Google Scholar]
- 15.Laustsen PG, Russell SJ, Cui L, Entingh-Pearsall A, Holzenberger M, Liao R and Kahn CR, Mol. Cell. Biol, 2007, 27, 1649–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crone SA, Zhao YY, Fan L, Gu Y, Minamisawa S, Liu Y, Peterson KL, Chen J, Kahn R, Condorelli G, Ross J Jr, Chein KR and Lee KF, Nat. Med, 2002, 8, 459–465. [DOI] [PubMed] [Google Scholar]
- 17.Kerkelä R, Grazette L, Yacobi R, Iliescu C, Patten R, Beahm C, WaIters B, Shevtsov S, Pesant S, Clubb FJ, Rosenzweig A, Salomon RN, Van Etten RA, Alroy J, Durand JB and Force T, Nat. Med, 2006, 12, 908–916. [DOI] [PubMed] [Google Scholar]
- 18.Villasante A, Marturano-Kruik A and Vunjak-Novakovic G, Biomaterials, 2014, 35, 5785–5794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marturano-Kruik A, Yeager K, Bach D, Villasante A, Cimetta E and Vunjak-Novakovic G, Conf. Proc. IEEE Eng. Med. Biol. Soc, 2015, 3561–3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marturano-Kruik A, Villasante A, Yaeger K, Ambati SR, Chramiec A, Raimondi MT and Vunjak-Novakovic G, Biomaterials, 2018, 150, 150–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chramiec A and Vunjak-Novakovic G, Adv. Drug Delivery Rev, 2019, 140, 78–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boudou T, Legant WR, Mu A, Borochin MA, Thavandiran N, Radisic M, Zandstra PW, Epstein JA, Margulies KB and Chen CS, Tissue Eng., Part A, 2012, 18, 910–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ronaldson-Bouchard K, Ma SP, Yeager K, Chen T, Song LJ, Sirabella D, Morikawa K, Teles D, Yazawa M and Vunjak-Novakovic G, Nature, 2018, 556, 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Conant G, Ahadian S, Zhao Y and Radisic M, Sci. Rep, 2017, 7, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hansen A, Eder A, Bönstrup M, Flato M, Mewe M, Schaaf S, Aksehirlioglu B, Schwoerer AP, Uebeler J and Eschenhagen T, Circ. Res, 2010, 107, 35–44. [DOI] [PubMed] [Google Scholar]
- 26.Ramade A, Legant WR, Picart C, Chen CS and Boudou T, Methods Cell Biol., 2014, 121, 191–211. [DOI] [PubMed] [Google Scholar]
- 27.Schaaf S, Shibamiya A, Mewe M, Eder A, Stöhr A, Hirt MN, Raun T, Zimmermann WH, Conradi K, Eschenhagen T and Hansen A, PLoS One, 2011, 6, e26397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ronaldson-Bouchard K, Yeager K, Teles D, Chen T, Ma S, Song L, Morikawa K, Wobma HM, Vasciaveo A, Ruiz EC, Yazawa M and Vunjak-Novakovic G, Nat. Protoc, 2019, 14, 2781–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oleaga C, Bernabini C, Smith AS, Srinivasan B, Jackson M, McLamb W, Platt V, Bridges R, Cai Y, Santhanam N, Berry B, Najjar S, Akanda N, Guo X, Martin C, Ekman G, Esch MB, Langer J, Ouedraogo G, Cotovio J, Breton K, Shuler ML and Jickman JJ, Sci. Rep, 2016, 6, 20030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Edington CD, Chen WLK, Geishecker E, Kassis T, Soenksen LR, Bhushan BM, Freake D, Kirschner J, Maass C, Tsamandouras N, Valdez J, Cook CD, Parent T, Snyder S, Yu J, Suter E, Shockley M, Velazquez J, Velazquez JJ, Stockdale L, Papps JP, Lee I, Vann N, Gamboa M, LaBarge ME, Zhong Z, Wang X, Boyer LA, Lauffenburger DA, Carrier RL, Communal C, Tannenbaum SR, Stokes CL, Hughes DJ, Rohatgi G, Trumper DL, Cirit M and Griffith LG, Sci. Rep, 2018, 8, 4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maschmeyer I, Lorenz AK, Schimek K, Hasenberg T, Ramme AP, Hubner J, Lindner M, Drewell C, Bauer S, Thomas S, Sambo NS, Sonntag F, Lauster R and Marx U, Lab Chip, 2015, 15, 2688–2699. [DOI] [PubMed] [Google Scholar]
- 32.Loskill P, Marcus SG, Mathur A, Reese WM and Healy KE, PLoS One, 2015, 10, e0139587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Skardal A, Murphy SV, Devarasetty M, Mead I, Kang H-W, Seol Y-J, Zhang YS, Shin S-R, Zhao L, Aleman J, Hall AR, Shupe TD, Kleensang A, Dokmeci MR, Lee SJ, Jackson JD, Yoo JJ, Hartung T, Khademhosseini A, Soker S, Bishop CE and Atala A, Sci. Rep, 2017, 7, 8837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen WLK, Edington C, Suter E, Yu J, Velazquez JJ, Velazquez JG, Shockley M, Large EM, Venkataramanan R, Hughes DJ, Stokes CL, Trumper DL, Carrier RL, Cirit M, Griffith LG and Lauffenburger DA, Biotechnol. Bioeng, 2017, 114, 2648–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Meer BJ, de Vries H, Firth KSA, van Weerd J, Tertoolen LGJ, Karperien HBJ, Jonkheijm P, Denning C, IJzerman AP and Mummery CL, Biochem. Biophys. Res. Commun, 2017, 482, 323–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gomez-Sjoberg R, Leyrat AA, Houseman BT, Shokat K and Quake SR, Anal. Chem, 2010, 82, 8954–8960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Toepke MW and Beebe DJ, Lab Chip, 2006, 6, 1484–1486. [DOI] [PubMed] [Google Scholar]
- 38.Wang JD, Douville NJ, Takayama S and Elsayed M, Ann. Biomed. Eng, 2012, 40, 1862–1873. [DOI] [PubMed] [Google Scholar]
- 39.Koutsiaris AG, Tachmitzi SV, Batis N, Kotoula MG, Karabatsas CH, Tsironi E and Chatzoulis DZ, Biorheology, 2007, 44, 375–386. [PubMed] [Google Scholar]
- 40.Xiao S, Coppeta JR, Rogers HB, Isenberg BC, Zhu J, Olalekan SA, McKinnon KE, Dokic D, Rashedi AS, Haisenleder DJ, Malpani SS, Arnold-Murray CA, Chen K, Jiang M, Bail L, Nguyen CT, Zhang J, Laronda MM, Hope TJ, Maniar KP, Pavone ME, Avram MJ, Sefton EC, Getsios S, Burdette JE, Kim JJ, Borenstein JT and Woodruff TK, Nat. Commun, 2017, 8, 14584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pak J, Chen ZJ, Sun K, Przekwas A, Walenga R and Fan J, Comput. Biol. Med, 2018, 92, 139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kannan R, Chen ZJ, Singh N, Przekwas A, Delvadia R, Tian G and Walenga R, Int. J. Numer. Method Biomed. Eng, 2017, 33, e2838. [DOI] [PubMed] [Google Scholar]
- 43.PubChem, Linsitinib [Internet], Bethesda: National Institutes of Health; ), 2006. [updated 2020 Jan 04; cited 2020 Jan 07], Available from: https://pubchem.ncbi.nlm.nih.gov/compound/Linsitinib#sectise=Chemical-and-Physical-Properties. [Google Scholar]
- 44.PubChem, Fluorescein-5-isothiocyanate [Internet], Bethesda: National Insitutes of Health, 2005. [updated 2020 Jan 04, cited 2020 Jan 07] Available from: https://pubchem.ncbi.nlm.nih.gov/compound/Fluorescein-5-isothiocyanate#section=Computed-Properties. [Google Scholar]
- 45.Macaulay VM, Middleton MR, Eckhardt SG, Rudin CM, Juergens RA, Gedrich R, Gogov S, McCarthy S, Poondru S, Stephens AW and Gadgeel SM, Clin. Cancer Res, 2016, 22, 2897–2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Primeau AJ, Rendon A, Hedley D, Lilge L and Tannock IF, Clin. Cancer Res, 2005, 11, 8782–8788. [DOI] [PubMed] [Google Scholar]
- 47.Lankelma J, Dekker H, Luque FR, Luykx S, Hoekman K, van der Valk P, van Diest PJ and Pinedo HM, Clin. Cancer Res, 1999, 5, 1703–1707. [PubMed] [Google Scholar]
- 48.Zheng JH, Chen CT, Au JL and Wientjes MG, AAPS PharmSci, 2000, 3, E15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Buck E, Gokhale PC, Koujak S, Brown E, Eyzaguirre A, Tao N, Rosenfeld-Franklin M, Lerner L, Chiu MI, Wild R, Epstein D, Pachter JA and Miglarese MR, Mol. Cancer Ther, 2010, 9, 2652–2664. [DOI] [PubMed] [Google Scholar]
- 50.Pishas K and Lessnick S, F1000Research, 2016, 5, 2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Allard JB and Duan C, Front. Endocrinol, 2018, 9,117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kong H, Jones PP, Koop A, Zhang L, Duff HJ and Chen SR, Biochem. J, 2008, 414, 441–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van Erven L and Schalij MJ, Heart, 2010, 96, 1593–1600. [DOI] [PubMed] [Google Scholar]
- 54.Chatterjee K, Zhang J, Honbo N and Karliner JS, Cardiology, 2010, 115, 155–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Logeart-Avramoglou D, Oudina K, Bourguignon M, Delpierre D, Nicola MA, Bensidhoum M, Arnaud E and Petite H, Tissue Eng., Part C, 2010, 16, 447–458. [DOI] [PubMed] [Google Scholar]
- 56.Cotterill SJ, Ahrens S, Paulussen M, Jürgens HF, Voûte PA, Gadner H and Craft AW, J. Clin. Oncol, 2000, 18, 3108–3114. [DOI] [PubMed] [Google Scholar]
- 57.Redini F and Heymann D, Front. Oncol, 2015, 5, 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Belfiore A, Frasca F, Pandini G, Sciacca L and Vigneri R, Endocr. Rev, 2009, 30, 586–623. [DOI] [PubMed] [Google Scholar]
- 59.Santoro M, Menegaz BA, Lamhamedi-Cherradi SE, Molina ER, Wu D, Priebe W, Ludwig JA and Mikos AG, Tissue Eng., Part A, 2017, 23, 80–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marcos-Campos I, Marolt D, Petridis P, Bhumiratana S, Schmidt D and Vunjak-Novakovic G, Biomaterials, 2012, 33, 8329–8342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Burridge PW, Matsa E, Shukla P, Lin ZC, Churko JM, Ebert AD, Lan F, Diecke S, Huber B, Mordwinkin NM, Plews JR, Abilez OJ, Cui B, Gold JD and Wu JC, Nat. Methods, 2014, 11, 855–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Polson A, J. Phys. Chem, 1950, 54, 649–652. [Google Scholar]
- 63.Maidana DE, Tsoka P, Tian B, Dib B, Matsumoto H, Kataoka K, Lin H, Miller JW and Vavvas DG, Invest. Ophthalmol. Visual Sci, 2015, 56, 6701–6708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bhumiratana S, Bernhard JC, Alfi DM, Yeager K, Eton RE, Bova J, Shah F, Gimble JM, Lopez MJ, Eisig SB and Vunjak-Novakovic G, Sci. Transl. Med, 2016, 8, 343ra83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.