

Abstract
For humans, companion animals, and food producing animals, vaccination has been touted as the most successful medical intervention for the prevention of disease in the twentieth century. However, vaccination is not without problems. With the development of new and less reactogenic vaccine antigens, which take advantage of molecular recombinant technologies, also comes the need for more effective adjuvants that will facilitate the induction of adaptive immune responses. Furthermore, current vaccine adjuvants are successful at generating humoral or antibody mediated protection but many diseases currently plaguing humans and animals, such as tuberculosis and malaria, require cell mediated immunity for adequate protection. A comprehensive discussion is presented of current vaccine adjuvants, their effects on the induction of immune responses, and vaccine adjuvants that have shown promise in recent literature.
Keywords: vaccine adjuvants, vaccines, immunology, mucosal immunization, biodegradable polymers, alum, liposomes, TLR ligands, polymeric biomaterials, controlled release/delivery
INTRODUCTION
Over the last 200 years, the use of vaccines has proven to be one of the most successful medical interventions in the reduction of disease caused by infectious agents.1 For example, through vaccination, disease caused by the human smallpox virus was eradicated worldwide. Europe, the Western Pacific, and the United States have been declared polio-disease free and have stopped using the Sabin (oral-live) vaccine, now including the killed version (Salk vaccine) as part of the childhood vaccination schedule.2 In veterinary medicine, control and eradication of diseases such as swine cholera, parvovirus-induced enteritis, distemper virus, and pseudorabies virus have all been achieved through intervention strategies employing vaccination programs.3 Indeed, vaccination has been touted as the greatest medical achievement in the 20th century.
Despite advancements and improvements in vaccine efficacy and implementation over the past several decades, infectious disease still remains the largest cause of death worldwide; unfortunately, many of these deaths occur in children and infants caused by diseases that are preventable by vaccination.4,5 According to the World Health Organization (WHO), 14% of the global childhood mortality is caused by vaccine preventable diseases including measles, Haemophilus influenzae type b (Hib), Bordetella pertussis (whooping cough), and neonatal tetanus.6 Many challenges still remain with regard to fully realizing the health benefits of active immunization programs. Some of these obstacles include the development of single dose vaccines, methods to overcome the poor immunogenicity of recombinant and subunit immunogens, and the ability to rapidly and rationally develop vaccines against emerging pathogens. One promising strategy for addressing these challenges is the development of new vaccine adjuvants, or carriers that enhance the effectiveness of vaccines.
Current immunization practices often require multiple doses to achieve protective immunity. Health care workers have observed that dropout rates in vaccination programs can reach as high as 70% in some developing countries.7 Recent failures of the human chicken pox vaccine demonstrated that the current recommended single dose is not protective in an outbreak situation.8 Many of the patients recently contracting mumps in Canada could not document more than a single immunization.9 The WHO listed the development of single dose vaccines as number one in their “Grand Challenges” for human health in 2005.10 While not receiving the full regimen of a vaccine may significantly impact the development of protective immunity for humans, in most livestock systems, it is often impractical in terms of cost, labor and stress on the animal to immunize more than once.11 Vaccination still remains a cost effective way to combat disease.12 Prophylactic administration of an efficacious vaccine can be more cost effective than therapeutic treatment, more ecologically friendly than the use of anti-microbial agents (i.e., less chance of antibiotic resistant bacteria in the environment) and offers greater flexibility in management options. It is estimated that for each $1 spent on vaccines, $5–10 are saved in what would have been lost to disease.13 It is estimated that 30–50% of the antibiotics produced are used in agriculture, many at subtherapeutic levels in feeds to promote growth by suppressing bacterial growth.14 Emerging antibiotic resistance, changes in consumer acceptance of anti-microbial use in food producing animals, and high cost of treatment as compared to prevention dictates that novel biologics for preventing disease must be developed.15 Vaccination against infectious agents has greatly improved the health of humans, companion animals, and livestock species worldwide. A single dose vaccine, whether for humans or animals, would greatly increase patient compliance, thus improving the efficacy of many vaccines (i.e., a full dosing regimen received at once), and reduce the costs associated with vaccination programs.
Recent developments in both synthetic and naturally derived adjuvants suggest that single dose vaccines for a variety of pathogens may be realized in the near future. However, no single adjuvant will be effective for all vaccine applications. Developing new adjuvants for improved immunotherapy requires the development of complementary strategies that address all the complex variables involved in immune surveillance.16 Thus, before discussing recent developments in vaccine adjuvants, we briefly discuss innate and adaptive immunity and the various types of vaccines currently used to confer protective immunity.
INNATE AND ADAPTIVE IMMUNITY
Innate and adaptive immune systems work together as a complex integrated system.17 When cells from innate defenses recognize foreign structures or pathogens, a cascade of events ensues which functions to eliminate or contain the threat. The innate immune system is involved in surveillance and detection of foreign invaders and as such is a key target for activation by vaccine adjuvants. Innate immunity comprises of a variety of hematopoietic and cellular factors including the complement system, phagocytic cells, NK cells, naturally occurring antibodies, γδ T cells, and anti-microbial peptides.18,19 The innate immune system uses relatively few molecules to recognize components of foreign invaders. These bacterial associated components were described by Janeway and Medzhitov as pathogen-associated molecular patterns (PAMPs).18 Depending on the vigor of the innate immune response, the adaptive immune response may or may not be actively engaged. In contrast to innate immunity, adaptive immunity recognizes antigen-specific epitopes via specialized cell surface receptors (antibody or T cell receptor) resulting in an antigen-specific and more directed immune response.18 It has been shown that a combination of innate immunity and prolonged presence of the pathogen-derived immunogens significantly influences the induction of a robust immune response.20 To enhance immune activation, adjuvants can be tailored to specifically activate the type of immune response needed against a particular disease (antibody, cell-mediated, or mucosal immunity) without the need to suffer the consequences of an active infection.21
A critical innate immune cell that is involved with induction of immune responses is the dendritic cell (DC). DCs are found in all body tissues and, as such, are effectively distributed to play a central role in stimulation and regulation of adaptive immunity (cell mediated and humoral immunity).22 In the blood and tissues, DCs are in an “immature” state, capable of phagocytosis, and express low levels of costimulatory molecules as well as molecules associated with cellular migration (CCR7, DC-SIGN, and DEC-205).23 In the basal and suprabasal epidermis, resident DCs or Langerhans’ cells are the first cells to encounter microbes or injected immunogens. These cells provide innate immune surveillance and are continually replenished form special progenitor cells that reside in the dermis.24 Dendritic-like cells are also resident in the lungs where they discriminate between pathogenic and harmless inhaled particles.24 In fact, pulmonary DCs are key producers of IL-10 and, as such, are suppressors of airway inflammation. Within the gut mucosa, DCs extend their pseudopodia between epithelial barriers to sample luminal contents.25 Among the many different pattern recognition receptors (PRRs) on DCs, Toll-like receptors (TLRs) allow DCs to recognize specific microbial ligands (e.g., CpG DNA, lipoteichoic acid (LTA), lipopolysaccharide (LPS), flagellin).17 TLRs are type I transmembrane proteins that mediate the initial recognition of microbial components and as such are likely targets for stimulation by vaccine adjuvants.26,27 Stimulation of TLR and other PRRs result in the activation of specific intracellular signaling pathways (e.g., MyD88-dependent and -independent) leading to activation of transcription factors (NFκB and/or AP-1) necessary for cellular migration, maturation, and antigen presentation. DCs acquire antigen by three main mechanisms: (1) phagocytosis or energy-dependent engulfment of bacteria, particulate matter or cellular debris; (2) macropinocytosis uptake of soluble antigens; (3) receptor mediated uptake triggered by mannose receptors, complement receptors, or Fc receptors. Upon activation via TLRs and/or other environmental cues, such as IL-8, DCs undergo maturation and migrate to the draining lymph node. Following maturation, DCs lose much of their phagocytic capacity while increasing surface expression of migratory and costimulatory molecules, such as MHC I/II, CD80, CD86, and CD40. This process is accompanied by migration to the draining lymph node(s). Within the lymph node, DCs continue maturation and serve as potent antigen presenting cells (APC) to naïve CD4+ and CD8+ T cells.
There are two antigenic processing pathways within DCs that lead to the major histocompatibility complex (MHC) molecules, whose function is to bind peptide fragments derived from pathogens and display them on the cell surface for T cell recognition.28,29 Antigens taken up by DCs via phagocytosis are contained within a phagosome or early endosome. The phagosome fuses with a lysosome generating a phagolysosome. Following changes in the pH of the phagolysosome, proteolytic enzymes are activated and the antigen is degraded into small peptide fragments (9–13 amino acids in length) in order to facilitate their presentation to T cells and B cells. Antigens contained within phagolysosomes representing exogenous antigens are loaded into MHC II and then presented on the cellular surface for stimulation of CD4+ T cells. A diagrammatic representation of a mature DC presenting antigen via MHC II, the exogenous pathway, is shown in Figure 1a.
Figure 1.
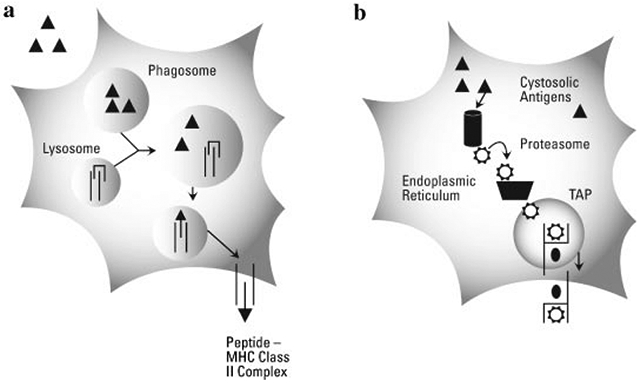
Exogenous and endogenous antigen presentation. (a) Following engulfment, a pathogen or immunogenic protein is contained within a phagosome or endosome. Fusion of the phagosome with the lysosome creates a phagolysosome bringing together the engulfed antigens and degradative enzymes and MHC II molecules. Following proteolytic cleavage, MHC II chaperone protein (CLIP) is displaced by the peptide (9–13 amino acids), which binds within the MHC II cleft. The vesicle containing the peptide-MHC II complex (pMHC II) traffics through the cytosol, eventually fusing with the cell membrane and the pMHC II is now displayed on the cell surface. (b) For antigens gaining access to the cytosol of the cell (self-antigens, viruses, or cytosolic bacteria) proteins are degraded by cytosolic proteosomes or immune proteosomes. Degraded peptides are guided to TAP (transporter protein associated with antigen processing) and enter the endoplasmic reticulum. Subsequently, the peptides are loaded into MHC I molecules and following intracellular trafficking, are presented on the surface of the cell.
Antigens generated within the cytosol of the cell, including viral antigens, antigen from bacteria that escape into the cytosol, and many cancer antigens are presented by the endogenous pathway. Cytostolic proteins are degraded by proteosomes in the cytosol, chaperone proteins (TAP) translocate the peptide fragments into the endoplasmic reticulum where it is loaded into MHC I molecules that are subsequently transported to the cell surface for presentation to CD8+ T cells as shown in Figure 1b. While all nucleated cells in the body express MHC I molecules, only DCs are able to efficiently stimulate naïve CD8+ cells.30 Antigen specific CD8+ T cells properly activated by DCs can directly kill infected cells, a powerful component of cell-mediated immunity. What also makes DCs excellent activators of adaptive immunity is that DCs regularly present antigen from the same source by both MHC I and MHC II pathways by phagocytosing necrotic or apoptotic cells, thus, allowing cytosolically derived antigens access to MHC II loading compartments.30,31 Thus, DCs are not only involved in immune surveillance, but also act as a bridge between innate and adaptive immunity.
Both the effector and regulatory aspects of CMI and humoral immunity are directly affected by the induction or activation of CD4+ T helper cells. These CD4+ T cells can be further classified as Th1, Th17, Th2, or Treg.32-34 A Th2-type immune response is characterized by the production of IL-4, IL-5, IL-10, and IL-13 and the secretion of IgG1 and IgE antibody isotypes. Th1-type responses are characterized by the production of IFN-γ and TNF-β, IgG2a antibodies and are usually associated with cell-mediated immunity including activated macrophages and delayed-type hypersensitivity.35 Immune responses of the Th1-type are directed more towards intracellular pathogens and are necessary for clearance of many viruses, some bacteria (e.g., Mycobacterium tuberculosis) and anti-tumor effects, whereas a Th2-type response is generally associated with the induction of antibodies that effectively neutralize toxins, viruses, and bacterial adhesion.36,37 Th17 responses are considered inflammatory in nature and are characterized by production of IL-17.32 These responses appear to provide protection during acute inflammatory reactions but have been associated with chronic inflammatory diseases. The role of Th17 cells in vaccinology or infectious disease has yet to be elucidated.
Induction of the appropriate immune response (humoral vs. CMI vs. regulatory) is essential for vaccine efficacy.37,38 For example, in the BALB/c model of leishmaniasis, an immune response dominated by IL-4 and IgG1 (i.e., Th2-biased response), in comparison to a protective Th1-biased response (IFN-γ and IgG2a), does not protect nor allow these mice to clear the infection.39-41 Furthermore, in regions of the world where tuberculosis is endemic, a large portion of the population is infected and presents with a preexisting immune response to Mycobacterium species that is usually Th2 dominant.42 It its hypothesized that the current tuberculosis vaccine (Bacillus Calmette-Guerin or BCG vaccine) is ineffective in preventing disease because the current BCG vaccine is unable to redirect the preexisting immune response (Treg and/or Th2) in to a protective, Th1 dominant immune response.42,43 In veterinary medicine, the current vaccines used against feline infectious peritonitis viruses enhances humoral immunity which has been shown to exacerbate the disease, whereas a CMI response would be protective.44
In addition to presentation of antigen to T cells, mature DCs help to shape the adaptive immune response by secretion of cytokines. Activated DCs produce the cytokines tumor necrosis factor alpha (TNF-α), which mediates acute inflammation, and a variety of interleukins, such as IL-1β, IL-6, IL-8, IL-12, and IL-10. The specific combination of cytokines released by activated DCs can influence the ensuing CD4+ T cell response. The bias of the immune response generated after antigen presentation can be characterized by measurement of the cytokine profiles upon induction of antigen-specific recall responses (Fig. 2). The production of cytokines by DCs is also a critical feature of efficacious immune induction. For example, DC secretion of IL-1β induces secretion of IL-2, which facilitates the maturation and proliferation of naïve antigen-specific T lymphocytes. Conversely, antigen presentation in the absence of effective costimulation (CD80/86, CD40) or cytokine secretion by DCs induces ineffective T cell activation that can result in either tolerance or anergy.30
Figure 2.
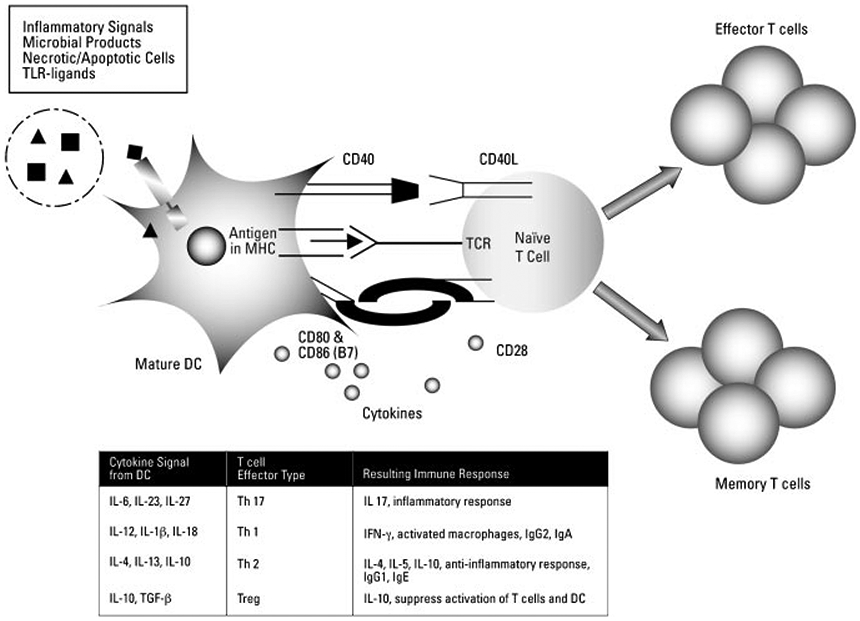
Signals from DCs can influence the differentiation of naïve T cells. Stimulated, mature DCs present not only antigen in the context of MHC but also costimulatory surface molecules necessary for T cell activation. Furthermore, the type and quantity of cytokines secreted by DCs in conjunction with these costimulatory molecules can direct the naïve T cell into different effectors phenotypes. IL-12 secretion from the DC initiates a Th1 type response characterized by secretion of IFNγ. IL-4 secretion from the DC results in a Th2 type response characterized by the secretion of IL-4, IL-5, and IL-10. The cytokines secreted by DCs are induced following ligation of cellular receptors (PRRs or TLRs) and signals from the surrounding tissues (i.e., IL-8). New evidence is emerging regarding the role of DCs in activating Th17 and Treg cells.
VACCINES
The most potent (i.e., protective) and lasting immune response in a host is induced following a natural infection with the pathogenic organism. However, for many individuals, the clinical outcome of a naturally occurring infection may not be favorable because of a lack of an effective treatment for the given disease, untoward morbidity or sequelae, or high mortality. For these reasons, vaccines have been designed to mimic the immune response that would otherwise be induced by an active infection, thereby avoiding the undesirable effects of disease. To be effective, a vaccine must contain some portion of the disease-causing agent (e.g., bacteria, virus, or toxin) and may include an immune-enhancer or adjuvant. Vaccine regimens generally employ an initial dose or priming dose followed by two to three booster doses. This prime-boost strategy allows for the presentation of high quantities of immunogen in the draining lymph node at several time points. The first dose initiates immune responses that particularly involve DCs and naïve immune cells. Repeated administration of this same immunogen induces activation of not only effector cells (e.g., immunoglobulin-committed B cells and T cells) but also memory immune cells.29 Upon subsequent exposure to the same immunogen, memory T and B cells provide for a secondary immune response characterized by a greater magnitude (e.g., high antibody titer) and one that occurs at a faster rate than the induction of a primary immune response.29,45 Regardless of the type of immunogen administered in currently licensed vaccines (e.g., killed organism, subunit), the primary mechanism of protection is mediated by the generation of neutralizing antibodies as opposed to the induction of cell-mediated immunity.46
Vaccines can be classified into three general categories: modified live, killed/inactivated, or subunit. Each has its advantages and disadvantages. A list of the current licensed vaccines for use in humans within the United States, is available on multiple websites managed by both the Department of Health and Human Services (DHHS) and the Centers for Disease Control and Prevention (CDC).12,47-49 The information provided includes the type of immunogen used, the age at which the vaccine should be administered, and the immunization schedules as recommended/required for the United States as issued by the DHHS.
Live Vaccines
Other than a natural infection, vaccines containing modified live organisms, relative to other vaccine formulations, induce the most potent and lasting immune response in the host. Modified live vaccines generally require the fewest number of inoculations, require no adjuvants, often confer lifelong immunity, and can be delivered through the same route as the natural infection would occur.50 The organism is able to replicate in the host, causing a mild, limited infection that stimulates the host immune response in a very similar fashion to that induced by a natural infection. Furthermore, these vaccines retain many of the natural microbial compounds that enhance immunity by activating the innate immune system.
Safe use of live vaccines requires that the organism first be attenuated, that is, the virulence capacity of the organism must be reduced. This can be achieved through repetitive passages (10–1,000 times) in a nonhuman host or in vitro. Alternatively, attenuated organisms can be developed by inducing genetic changes so that critical virulence attributes have been deleted or inactivated in the target organism. The Sabin oral polio vaccine and Flu-mist are two examples of modified-attenuated, live vaccines that are delivered along the same routes as the natural infection.51 A closely related but nonpathogenic organism can also be used if the nonpathogen and pathogen share immunoprotective epitopes. For example, Jenner observed that cowpox infection prevented smallpox, and an attenuated Ankara strain of vaccinia virus was used to vaccinate against smallpox.4 Likewise, attenuated Mycobacterium bovis used in the BCG vaccine is protective against disease caused by virulent M. tuberculosis.50
The largest drawback of modified live vaccines is that they are able to replicate in the host and, thus, are capable of persistent infection, recombination and reversion to the virulent wild-type. If the host is immunocompromised, the organism may be able to persist, and an otherwise nonpathogenic strain may be able to induce disease in the absence of a competent immune system. The live organism may also be able to spread and induce disease in other nonvaccinated individuals. Through horizontal gene transfer and natural random mutation, attenuated organism may acquire or reacquire virulence genes and become capable of disease induction. The attenuated strain of poliovirus used in oral vaccines has been shown to circulate throughout a given population and occasionally revert to virulence.51 While modified live vaccines are very effective at inducing both cellular and humoral immunity, they can cause severe reactions, ranging from inflammation at the site of inoculation to systemic disease. Furthermore, many current diagnostic tests cannot distinguish between an individual who is naturally infected and an individual that received a modified live vaccine.52-54 Effectiveness of live vaccines also requires that they be properly handled before administration. Keeping attenuated vaccines viable (i.e., proper storage) has been problematic in worldwide efforts to eradicate polio.51
Killed Vaccines
Killed or inactivated vaccines are comprised of the whole organism that has been treated with either heat or chemicals. In this way, the organism is not able to replicate in the host, yet cellular integrity of the pathogen is preserved. Dependent upon in vitro growth conditions, killed vaccines are also potent inducers of humoral immunity because most of the virulence factors and epitopes are present.50 Killed vaccines do not carry the same risks as live vaccines; the organism cannot replicate and, therefore, cannot establish persistent infection, spread to other individuals, or revert to a virulent form.50 These types of vaccines are generally cost effective to produce, possess a longer shelf life and are less sensitive to changes in temperature and handling when compared to modified live vaccines.55 Some killed vaccines can be administered orally (e.g., typhoid and cholera) more closely mimicking natural infection.35 Many injectable vaccines that contain killed/inactivated organisms include: polio virus (Salk injectable polio vaccine), whole-cell B. pertussis, hepatitis A virus, Yersinia pestis (causative agent of plague), and encephalitis viruses.56
The use of killed vaccines often requires multiple doses for the induction of protective immunity. The degree of CMI induced following immunization with killed vaccines can be weak. Like modified live vaccines, killed vaccines are highly reactogenic and are associated with adverse side effects. For example, the whole cell killed pertussis vaccine can induce a high fever accompanied by severe pain, redness and swelling at the injection site due to the presence of LPS and other TLR ligands in the vaccine.57,58
Subunit Vaccines
Subunit vaccines contain only a portion of the organism. Toxoids, inactivated bacterial toxins, were the first subunit vaccine to be employed for human use. Diphtheria (DT) and tetanus toxoids (TT) are formaldehyde-inactivated forms of the bacterial toxin that induce immune protection against the native toxin (i.e., neutralizing antibody). Other subunit vaccines currently in use include hemaglutinin-binding proteins of influenza virus and polysaccharide capsules of bacteria such as the vaccines that include conjugated forms of HiB (H. influenzae type B), pneumococcal (Streptococcus pneumoniae), and meningococcal (Neisseria meningitides) polysaccharides.56,59 Because of the poor immunogenicity of carbohydrate immunogens, these compounds are generally conjugated to a protein in order to enhance the immunogenicity; this strategy has been specifically used when developing vaccines for infants or the elderly, HiB and pneumococcal vaccines, respectively.
Another type of subunit vaccine being developed does not include protein or other structural components of the pathogen but utilizes the DNA of the pathogen. By injecting the DNA sequence encoding a protective epitope, immunity can be induced against a specific pathogen that bears the target epitope.56 DNA can be delivered using a viral vector with the epitope encoded on a plasmid or DNA-containing particulates to DCs.60 Host cells then express the epitope, it is presented in the context of MHCI or II molecules, subsequently inducing strong cellular immunity.29 While many DNA vaccines are still experimental, there are currently several DNA-based human vaccines in phase I, II, or III human trials, including vaccines against cytomegalovirus, Dengue virus, human immunodeficiency virus, herpes simplex virus-2, hepatitis B, and melanoma (skin cancer).61
Subunit vaccines offer several advantages including targeting the immune response to protective epitopes but retaining or deleting epitopes that can be used to differentiate “vaccinated” individuals from naturally exposed/infected individuals.62 Subunit vaccines may also eliminate many of the side effects and reactivity associated with modified live or killed whole organisms as they lack many of the microbial components that trigger innate immune recognition. The purified protein or other subunit components can be prepared free of LPS, CpG-DNA, or other TLR ligands that can induce an overt inflammatory response. Thus, subunit vaccines are very safe, and using new technologies, can be very cost effective to produce.
However, subunit vaccines still have many weaknesses. In general, subunit vaccines lack strong immunogenicity and require multiple doses for protection.63 Poor immunogenicity also generally requires that subunit vaccines be delivered with an adjuvant or immunoenhancer (e.g., monophosphoryl lipid A—MPLA). Many of the bacterial components that trigger a more robust immune response also enhance the protective response by inducing affinity maturation of the antibody response, increasing serum antibody titers, and immunoglobulin class switching.64 While current subunit vaccines can be formulated to induce high titer antibody responses, the induction of protective T cell responses (CD4+ or CD8+ cell-mediated immunity) are generally lacking.
Adjuvants enhance the immunogenicity of vaccine components where a live attenuated vaccine may not be desirable. Increasing numbers of immunocompromised patients, elderly populations, and infants represent a special problem to health officials as live-attenuated vaccines are not recommended in these groups. Subunit and recombinant protein vaccines are easier to produce and are generally considered safer than live vaccines, but require adjuvants to be efficacious.65
ADJUVANTS
An adjuvant is an agent that stimulates the immune system, increasing the response to a vaccine, while not having any specific antigenic effect. Adjuvants are immunoenhancing materials that perform three major functions: (i) provide a “depot” for the antigen, creating an antigenic reservoir for slow release, (ii) facilitate targeting of the antigen to immune cells (APCs) and enhance phagocytosis, and (iii) modulate and enhance the type of immune response induced by the antigen alone (e.g., isotype switching induces Th1 vs. Th2 bias).66-69 Adjuvants may also provide the danger signal that the immune system needs in order to respond to the antigen as it would during an active infection.29
The first function, providing a depot for the immunogen, is accomplished by entrapping the antigen in a poorly metabolized, nondegrading or slowly degrading substance, or otherwise sequestering the antigen to allow for the slow clearance of the antigen from the body. Some of these types of adjuvants are discussed in more depth in other sections of the review. Table 1 shows the adjuvants currently being used in licensed vaccines. Aluminum phosphate and aluminum hydroxide, commonly referred to as alum, are the adjuvants most often used in human vaccines and the resulting gel-like matrix that alum creates a slow-release environment for the immunogen. Oil-water emulsions also work by sequestering the antigen and slowly releasing it. The classic water-in-oil emulsion, incomplete Freund’s adjuvant, is widely used in livestock vaccines, even though it has a tendency to induce granulomas at the injection sites.65 It is not used in vaccine formulations for human use because of this tendency. Other mineral oil emulsions, such as Drakeol, Marcol, ISA-206, and ISA-25 are also used in various livestock vaccines.65 Recently, MF59, a variation of the biodegradable oil squalene, has proven to be a potent adjuvant with a satisfactory safety record and thus, is suitable for human use.65,70 Virosomes, virus-like particles, immunostimulatory complexes (ISCOMs), and liposomes all allow for the slow clearance of antigen by incorporating the antigen into small particles composed of stabilized lipids, phospholipids, or proteins. GlaxoSmithKline’s new class of adjuvants (AS02A, AS01B, AS04, and AS15-SB) combines stable mineral oil liposomes containing a squalene derivative, and immunostimulating MPLA.61,71 Furthermore, antigen sequestering can be achieved by incorporating the antigen into microspheres composed of polymeric units of a biodegradable material. As the microsphere degrades, the antigen is released. Thus, many different carrier formulations provide antigen depots once injected.
Table 1.
| Humans, US | Humans, United Kingdom, and European Union |
Livestock, Worldwide (General Categories)a |
|---|---|---|
| Aluminum hydroxide, aluminum phosphate, potassium aluminum sulfate (alum) | Aluminum hydroxide, aluminum phosphate, potassium aluminum sulfate (alum) | Aluminum hydroxide, aluminum phosphate, potassium aluminum sulfate (alum) |
| Calcium phosphate | Saponin (QS-21) | |
| MF-59 (Squalene, in Fluad) | Oil emulsions paraffin, mineral oil, lanolin, squalene, ISA-70, Montanide (IMS) | |
| AS04 (liposome formulation containing MPLA and QS-21) (FENDrix, Cervaix) | Glycerin |
Many livestock adjuvant-vaccine formulations are proprietary and their compositions have not been disclosed.
The second function of adjuvants is to enhance the immune response by targeting the antigen to immune cells, enhancing phagocytosis, and/or activating the APC. This can be accomplished by properties of the antigen, by a property of the carrier, or by inclusion of immunostimulatory molecules. Pertussis toxin binds with high affinity to epithelial cells, enhancing uptake of the vaccine.29 Other toxins, cholera toxin (CT) and Escherichia coli heat-labile toxin (LT), bind selectively to M cells of the intestinal tract.35 M cells efficiently translocate vaccine particles across the epithelial barrier into a region rich in APCs and lymphocytes.29,65,72 While bacterial toxins such as CT and E. coli LT augment a strong humoral immune response, the response to the anti-toxin may overshadow the response to the conjugate antigen.29 LPS, another bacterial-derived immunostimulant, is derived from the outer membrane of gram-negative bacteria such as B. pertussis. These bacterial products directly interact with the innate immune system via LPS receptors CD14 and TLR-4.72 Human TLRs, when triggered by LPS, stimulate the activation of NF-κB, a transcriptional activator for the production of pro-inflammatory cytokines.65 Because humans are very sensitive to endotoxins, LPS is toxic for inclusion in many human vaccine preparations, and the majority of injectable solutions for medical use are pyrogen- or LPS-free.
These first two mechanisms of immunity are illustrated in Figure 3. Some adjuvants may interact directly with TLRs on APC (Fig. 3b), and can be derived from pathogens that display highly conserved structures (e.g., PAMPs).73 As illustrated in Figure 3, an adjuvant can interact with the PAMP directly or release antigen as in the more traditional depot effect. Many adjuvants exhibit a combination of these characteristics.
Figure 3.
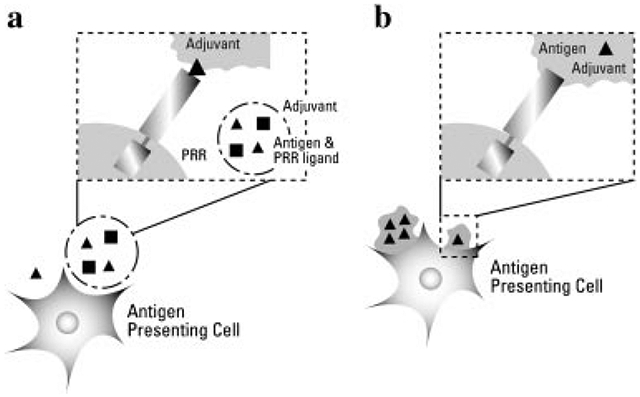
Recognition of antigen and PRR ligand by immature DC. An adjuvant may act as a depot, releasing both vaccine antigen and stimulatory PRR ligand over time (a) as in many a.u. or mineral oil formulations containing MDP, MPLA, or CpG. Conversely, the adjuvant may be directly recognized by the PRR (such as mannose receptor or TLRs) (b), as may be used in whole cell, killed bacterin vaccines or some polymer adjuvants.
Many biologically derived materials exhibit the third mechanism of adjuvanticity, modulation of the immune response mechanism. MPLA is a nontoxic LPS derivative obtained from Salmonella and has been shown to enhance IFN-γ production and induction of CD4+ T cell-mediated immunity.35,74 MPLA has been shown to interact through TLR-4, however it is not fully dependant upon TLR-4 for its effect.75,76 Ligation of TLR-4 and activation of TRIF transcription factors is responsible for activating both DCs and intraperitoneal macrophages resulting in T cell stimulation without induction of IL-6, IFN-γ, or other inflammatory molecules responsible for the toxic side-effects associated with LPS.76
Cytokines, when included in a vaccine mixture, can enhance the immune response and/or induce immune deviation. In theory, the inclusion of recombinant cytokines can enhance the activation of the APC and also selectively direct the immune response. Delivery of IL-6 or IL-12 along with antigen induces elevated serum antibody titers of both IgG1 and IgG2a isotypes, including increased production of mucosally secreted IgA.35,77 Inclusion of a plasmid encoding IL-2 in intranasal vaccines shifted the immune response to TT and CT (both dominant Th2-type antigens) to a Th1-type immune response.35 The antibody response to antigen delivered by osmotic pump was greatly enhanced by the inclusion of IL-1β with the antigen.78 The immune response to intramuscular plasmid DNA vaccination is enhanced by the inclusion of the gene sequence for GM-CSF.29 Inclusion of exogenous cytokines in a vaccine mixture acts directly on the APC or T cell providing the secondary signal needed to induce immune activation. Many of these properties have led to inclusion of cytokine adjuvants in experimental vaccines that are currently in phase I and phase II clinical trials (Tab. 2).
Table 2.
Adjuvants Currently Being Tested in U.S. Human Clinical Trials*
| Adjuvant | Phase | Disease |
|---|---|---|
| Aluminum hydroxide | I | Influenza, SARS |
| III | Leishmania | |
| Alhydrogel | II | Anthrax, plague, Leishmania |
| Montanide ISA | I/II | Melanoma, solid tumors, malaria |
| QS-21 | I | Cancer: breast, prostate, lung, HIV |
| II | Melanoma (skin) | |
| MF59 | I/II | Influenza A (H9N2), avian flu (H5N1), HIV |
| ISCOMATRIX | II | Melanoma |
| MLPA, MPL and other TLR-4 ligands | I | HIV, visceral Leishmaniasis |
| II | Allergy (tree pollen) | |
| MDP and other TLR-2 ligands | I | HIV |
| AS15-SB, (liposomes with MPLA and QS-21) | III | Lung cancer |
| AS02A, AS01B | I/II | Malaria, HIV, melanoma |
| CpG (TLR-9 ligand) | I | Malaria, cancer: breast, melanoma |
| II | Allergy: ragweed | |
| Imiquimod (TLR-7 ligand) | I/II | Influenza, melanoma |
| Heat Liable Toxin (LTK63 and LT-R192G) | I | HIV, tuberculosis, E. coli (ETEC) |
| Diphtheria Toxin | II | Hepatitis B |
| IMP321 (Th1 activating peptide) | I | Hepatitis B |
| IL-12 | I | HIV, Leishmania, melanoma |
| II | Prostate cancer | |
| IL-15 | I | HIV |
| IL-2 | I | HIV, melanoma |
| GM-CSF | I/II | HIV, cancer: melanoma, lung, ovarian, B cell lymphoma, Hepatitis B |
| Type I interferon | I | Influenza |
| Virus carrier: fowlpox, vaccina virus, canarypox | I/II | Cancer: solid tumors, breast, prostate |
| Bacterial carrier: Salmonella typhi CVD | I | HIV |
| PLG microparticles | I | HIV |
Data from http:clinicaltrials.gov.
Immune modulation can be influenced by other characteristics of the adjuvant/delivery system.79 As mentioned above, an immune response has been historically categorized as either Th1- or Th2-like. With the discovery of Th17 cells and the increasing role of antigen derived Treg cells in controlling disease, the relative simplicity of the Th1/Th2 paradigm will likely need modification. For the sake of this discussion, the Th1/Th2 paradigm provides a model and reference for understanding disease pathogenesis and host immunity. Many different factors can contribute to induction of an immunologically biased immune response including route of antigen delivery (intramuscular, subcutaneous, intranasal, oral), antigen dose, duration of antigen presentation, number or frequency of immunizations and inclusion of costimulatory molecules (e.g., LPS, exogenous cytokines) with the antigen.80 Adjuvants can affect all of these factors in different ways, and hence the role of the vaccinologist is to use the correct adjuvant to induce a protective immune response.80 In the mouse model of leishmaniasis, induction of Th2-biased immune responses by vaccination does not protect the mouse from infection nor does the mouse clear the parasite (i.e., cutaneous lesions develop and persist). On the other hand, induction of Th1-biased immunity was shown to prevent subsequent infection and lesion development illustrating that the Th1/Th2 bias of the immune response is important in the ability to induce protective immunity.39-41 Furthermore, in examining the efficacy of BCG vaccination on the clinical outcome of tuberculosis, preexisting immune responses (usually Th2 dominant) need to be overcome and appropriately redirected in order for vaccines to be efficacious.42,43 In laboratory animals, Ova-peptide (derived from hen egg ovalbumin, Ova) delivered in alum did not induce a T cell response that could be restimulated in vitro.81 Delivery of the same peptide within PLGA microspheres induced a significant in vitro proliferative response and production of IFN-γ when lymphocytes were restimulated in vitro with Ova.81 Cunningham et al.82 showed that they could alter the Th1/Th2 bias of the immune response to FliC flagellar antigen of Salmonella by changing the antigen delivery system. Antigens naturally delivered, on the surface of whole bacteria, induced predominantly IgG2a antibodies (Th1 response) whereas recombinant soluble or polymerized FliC induced primarily IgG1 and Th2 cytokines (IL-4).82
Th1/Th2 Immune Modulation
Induction of the appropriate type of immune response is essential for development of protective immunity. Once naïve T cells have been primed and a Th1 or Th2 type of immune response has been initiated, further immunizations to that antigen using different adjuvants cannot shift the initial immune bias.83,84 New or novel antigens are not affected by this previous vaccine induced bias.83 However, it is believed that repeated immunizations that favor a Th2 immune bias create a situation of immunological memory that affects the ability of the immune system as a whole to initiate Th1 immune responses to subsequently encountered immunogens.85
Table 3 summarizes the dominant antibody isotypes induced by some adjuvants, a reflection of Th1/Th2 biasing of an adjuvant. As illustrated by these examples, the form (e.g., particulate or soluble) of the antigen, delivery system, and route of delivery can all affect the Th1/Th2 bias of a subsequent immune response to a vaccine, and the type of immune response (cell-mediated or humoral) that will be protective varies with the disease in question. Antigen, adjuvants, and delivery systems need to be chosen with care to obtain the most protective response. Current licensed vaccines for the most part are lacking in their ability to induce Th1 type immune responses without also generating undesirable toxic side-effects such as the severe inflammation associated with whole-cell pertussis vaccines.36 While traditional alum-based vaccines initiate the Th2 response,65,86 a Th1 response may be more effective for preventing some diseases.87 Alum is still widely used in veterinary vaccines, but is frequently associated with the induction of tissue granulomas and subsequent carcass losses.88 Oil-based liposomes are capable of inducing a strong Th1 response, but are also associated with adverse tissue reactivity, granuloma formation, and subsequent carcass loss.89,90
Table 3.
Antibody Isotype Bias Induced in Laboratory Animals by Administration of Immunogens in Various Adjuvants
| Adjuvant | Examples | Basic Characteristics | Dominant Antibody Isotype |
Refs. |
|---|---|---|---|---|
| Inorganic | Aluminum hydroxide, aluminum phosphate, and calcium phosphate | Hydrogel emulsion—creates depot effect, enhance macrophages maturation | IgG1, IgE | 36,96,97,99 |
| Oil emulsions | Mineral oil (i.e., Freunds incomplete) | Basic water-in-oil emulsion with long standing record as research gold standard | IgG1 | 36,63,108 |
| MF59 | Blend of muramyl tripeptide, squalene, polyoxyethylene, sorbitan monooelate, and sorbitan trioleate | IgG2a | 36,74 | |
| QS-21 | Purified saponin from Quillaja saponica, used to stabilized lipid emulsions | IgG2a | 35,110 | |
| Montamide ISA-51 and ISA-720 | ‘Ready to use’ oil for water-in-oil emulsion | IgG1 | 62,66,202 | |
| Isocoms | ISCOMATRIX | Complex of saponins and lipids | IgG1 | 66,110 |
| Microbial derived | Monophosphoryl lipid A (MPLA) | Detoxified TLR-4 ligand | IgG1 and IgG2a/c | 75,76,107 |
| Macrophage activating protein-2 | TLR-2 ligand from Mycoplasma spp, and purified derivatives | IgG2a | 123,124 | |
| Virosomes | Stabilized lipid complexes containing viral proteins such as influenza hemaglutinin | IgG2a, IgA | 36,103,116 | |
| LT/CT | Modified bacterial toxins for mucosal adherence heat-liable enterotoxin and cholera toxin | LT: IgG1, IgG2a, and IgA | 36,141 | |
| CT: IgG1 | ||||
| CpG | Nonmethylated bacterial DNA, a TLR-9 ligand | IgG2a | 36,64,129 | |
| Cytokines as adjuvants | IL-1 | Pro-inflammatory cytokine | IgG2a, IgA | 35,66 |
| IL-2 | Lymphoproliferative cytokine | IgG2a | 35 | |
| IL-12 | Pro-inflammatory cytokine | IgG2a, IgA | 144 | |
| IL-6 | Anti-inflammatory cytokine | IgG1, IgA | 145 | |
| Natural polymers | Polysaccharides | Coating or emulsified with solid antigen | IgG1 or IgG2a depending on route | 36 |
| Synthetic polymers | Polyanhydrides | Antigen and immunostimulators emulsified into biodegradable particles ranging from 50 μm to 20 nm | Variable depending on inclusion of immunostimulants and polymer chemistry | 36,148,153 |
| Polyesters | Antigen and immunostimulators emulsified into biodegradable particles ranging from 50 μm to 20 nm | Variable depending on immunostimulants and antigen incorporated | 36,68,189,197 |
In the United States, the only adjuvant currently approved for use in humans is alum. However, in England and other European Union countries, MF59 is also used. MF59 is based on a biodegradable plant oil emulsion containing muramyl tripeptide (MTP).91 Highly purified MTP is a synthetic component similar to that found in mycobacterial cell walls and MTP retains immunostimulatory properties while eliminating much of the toxic effects associated with the whole bacterium.74 MF59 is used in the H5N1 bird flu vaccine developed by Novartis. MF59 was chosen for dose-sparing benefits (i.e., less immunogen needed) and is recommended in elderly (65 and older) including those with underlying chronic conditions such as diabetes.92,93
Vaccine adjuvants straddle a fine line between tissue toxicity and efficacy. Multiple studies in livestock species have shown that greater immunogenicity is achieved when adjuvants causing severe tissue reactivity were used. Greater antibody titers were observed in swine vaccinated with bacterins prepared with a paraffin oil or lecithin (>20%) adjuvant; however, these adjuvants are highly irritating leading to severe diffuse granulomatous tissue at the injection site with multiple foci of necrosis.94 While adjuvants containing lower amounts (5–10%) of lecithin-based oil or alum induce less tissue irritation, the corresponding antibody titers were also much lower.94 Vaccine adjuvants for veterinary medicine have many of the same concerns as adjuvants used for human medicine. Tissue irritation, granuloma formation, and abscess formation at the injection site are undesirable from an animal welfare viewpoint, but also can be costly to the producer due to carcass losses at time of slaughter.94
Another consideration for the development of new adjuvants is for the induction of mucosal immunity. With few exceptions (C. tetani, rabies virus, and other insect vector borne pathogens), most pathogens enter the host via mucosal surfaces (e.g., upper respiratory, gastrointestinal, vaginal, or urinary tracts). Induction of mucosal antibody (i.e., secretory IgA) by appropriate delivery of the antigen to the mucosal associated lymphoid tissue (MALT) is the most effective way to neutralize these pathogens or their secreted toxins.72
ALUM ADJUVANTS
Salts of aluminum hydroxide or aluminum phosphate, commonly referred to as alum, have long been used in vaccines and have an extensive safety record. Alum was first used as an adjuvant in 1926.95 Until recently, it was the only adjuvant approved for use in humans.95,96 Gels of aluminum phosphate are commercially available for clinical use and generate consistent, predictable results.97 Alum-based vaccines are prepared by suspending the antigen in a phosphate buffered solution and allowing the antigen to adsorb to the aluminum hydrogel.97 The amount of antigen that adsorbs onto alum depends upon the forces within the antigen, and between the antigen and the alum, including hydrophobic interactions, van der Waals forces, ionic charges, and hydrogen bonding. The typical quantity of alum in a human vaccine dose is 0.5 mg, while the upper allowable limit established by the U.S. Food and Drug Administration (FDA) and WHO is 1.25 mg per injection.97 Alum has proven safe for routine use in children, and enhances the production of antibody to protein toxoids and polysaccharide vaccines.97 Alum has a synergistic effect when combined with other adjuvants and can enhance the adjuvant properties of liposomes, QS-21, MPLA, and CpG.97 However, alum is not ideal for small peptide vaccines or for use with recombinant proteins due to their inherent low immunogenicity.35,80,98
Recently, the use of alum in vaccines has come under scrutiny. Alum has been occasionally associated with severe tissue reactions such as erythema, subcutaneous nodules, granulomas, and has been thought to induce hypersensitivity and macrophagic myofasciitis.96,99,100 It is well established that alum-based vaccines induce IgE and IL-4, which are associated with allergy and type IV immediate hypersensitivity.96 While alum is effective at inducing strong humoral immunity, alum-based vaccines generally fail to induce cell-mediated immune responses, such as cytotoxic T cells or delayed type hypersensitivity.36 Alum enhances a strongly biased Th2 immune response in animal models.99 Alum-based vaccines have other drawbacks besides the immune bias. Alum, because it is a semi-particulate hydrogel, cannot be lyophilized or frozen,101 thus limiting shelf life and storage conditions. Because the mode of action of alum includes the formation of antigenic deposits at the site of injection, alum is not suitable for oral or intranasal immunization.35,80,102 Finally, alum proved to be ineffective when used in conjunction with DNA-based vaccines.103
The mechanism of adjuvanticity for alum has been traditionally thought of as providing an antigenic depot in the tissue. The evidence of the depot effect, or delayed antigen release, of alum adjuvants was established by White in 1967 and Harris in 1935, by inducing immunity in a second animal by implanting granulomatous tissue that had developed as a result of immunizing the donor animal with an alum-based vaccine.97 Alum particles have been observed at the site of injection up to a year after immunization.97 Alum-precipitated antigens are somewhat particulate, and therefore, more readily ingested by phagocytes.104 Macrophages recovered from muscle tissue following injection of an alum-based vaccine and macrophages cultured in vitro in the presence of alum show persistence of crystalline inclusions.99 Alhydrogel and Adju-phos, commercially available prepared alum gels, produce particles roughly 3–4.5 μm in size.97
Excess alum in a vaccine mixture enhances the adjuvant effect, however alum is slightly cytotoxic to macrophages.97 Recent studies with cultured macrophages showed that aluminum hydroxide induces a distinct maturation pattern characterized by the expression of surface markers that resemble those found on mature myeloid DCs (HLA-DRhish/CD86high/CD83+/CD1a−/CD14−) endowing them with the ability to enhance activation of CD4+ T cells.99 Other recent studies have shown that alum may facilitate this DC maturation by inducing the release of uric acid crystals.105 Uric acid crystals are an endogenous ligand for TLR-2.43,106 Further evidence for TLR activation was shown by a diminished response to antigens in alum injected into MyD88-deficient mice.105
ADJUVANT ACTIVITY OF CALCIUM PHOSPHATE
Calcium phosphate has been used for many years as the adjuvant in childhood diphtheria–tetanus–pertussis (DTP) vaccine formulations in France.96,97 Furthermore, calcium phosphate is a normal body constituent and is readily absorbed.96,97 In contrast to aluminum phosphate, calcium phosphate does not induce IgE production in animals or humans.96,97 Because of this property, the most common use of calcium phosphate is the delivery of allergens in desensitization therapy for allergic patients.96,97 In laboratory animals (e.g., mice and guinea pigs), calcium phosphate elicits a lower antibody response than alum-based preparations, however, the opposite is true in humans.96 Using calcium phosphate-based vaccines, children and pregnant women developed higher neutralizing antibodies than those receiving an aluminum phosphate-based vaccine.97 The mode of action is thought to be the same as for alum compounds, functioning to create a depot for the immunogen and facilitating the uptake of the particulate antigen by APCs.97
FREUND′S COMPLETE ADJUVANT AND FREUND′S INCOMPLETE ADJUVANT
Freund’s complete and incomplete adjuvants (CFA and IFA, respectively) are the standard classical adjuvants to which all other adjuvants are compared.80 This very potent adjuvant system is comprised of a water-in-mineral oil emulsion with the emulsifier mannide monooleate.107 Freund’s complete adjuvant also contains heat-killed M. tuberculosis whereas IFA contains only the mineral oil emulsion and emulsifier.29,63,107 Classically, proteinaceous antigens administered in CFA induce a very strong immune response, including cell-mediated responses, whereas immunogenic proteins administered intraperitoneally in IFA were thought to induce tolerance.83,108 Advances in both knowledge of the immune system (induction of tolerance and Th2 responses) and methodology in measuring immune responses have shown that administration of antigens in IFA actually induces a Th2 response. This response is characterized by the induction of memory T cells that traffic to the spleen, rather than the draining lymph nodes.108 In addition, the cytokine response produced by these cells is small in quantity (as compared to Th1 cytokines in a lymph node) and may be below the limits of detection.83,109 The presence of the mycobacterial products in CFA provide a potent danger signal and induces costimulatory signals necessary for induction of Th1-type cytokines. Thus, the resultant immune responses induced by CFA and IFA provide the basis for the differential Th1/Th2 skewing of the immune response (i.e., immune deviation) observed when these two similar adjuvants are employed in a vaccine.108 Complete Freund’s adjuvant is capable of inducing high antibody titers and long lasting T cell responses, but is so reactogenic that its use even in laboratory animals is discouraged.107 The immune enhancing mechanisms of these adjuvants, the delayed release of antigen, slower antigen clearance, and targeting of the antigen to APCs is due to the mineral oil emulsion.29 Variations on mineral oil emulsion vaccine adjuvants are marketed by Chiron and Norvarits as Montamide ISA-51 and ISA-720.92
MF59 OIL-EMULSION ADJUVANTS
Introduced in Europe in 1997, MF59 is an oil-in-water microemulsion that includes squalene (derived from biodegradable plant oil), Polysor-bate 80, and Span 85 (stabilizers) and small amount of MTP, a novel synthetic component derived from mycobacterial cell walls.63,74 In clinical trials, the MTPs proved to be too toxic and have been excluded from current formulations.63,70,74 MF59 has been shown to stimulate a strong Th2 biased immune response to a large number of antigens and may be more suitable for subunit vaccines than alum.36 MF59-based vaccines that have incorporated recombinant antigens induce high titer antibody responses and T cell proliferative responses.74 Combination of MF59 with influenza subunits enhanced the immune response of elderly patients over that obtained using other adjuvants and is being evaluated for use in children.36 MF59 does not induce Th1-type immunity (e.g., IFN-γ) and, therefore, may not be suitable for vaccines where cell-mediated immunity is needed for protection.74 The mechanism of adjuvanticity for MF59 appears to be in directing delivery of the immunogen to APCs.74 Studies with MF59 have shown that macrophages, but not DCs, are the main cell type involved in clearing the oil depot from tissue, and DCs are the key APCs within the T cell zones of the lymph node.36 It was proposed that following uptake, adjuvant-induced cell death allowed for the transfer of the antigen from the macrophage to the DC for T cell induction (i.e., cross-presentation).36 Another observation that arose during the development of MF59 is that there is a difference in emulsion particle size and the resulting immune response in different animal species. Small laboratory animals (mice, guinea pigs, and rabbits) develop high antibody titers following immunization with oil emulsion formulas regardless of particle size. However, nonhuman primates (baboons, chimpanzees) and goats require stable, small droplet emulsions for optimal antibody induction.70 The key lesson here is that not all animal species respond equally to an adjuvant and testing in both large and small animals may be necessary to ensure applicability of a novel adjuvant. Mineral oil emulsions of various compositions are widely used in veterinary adjuvants, and as their safety record is improved, they are also being developed for human use.61,62,65
IMMUNOSTIMULATING COMPLEXES (ISCOMs)
ISCOMs were first described in 1984 by Morein et al.110 Cholesterol mixed with plant-derived saponins under controlled conditions creates 40 nm cage-like particles referred to as immunostimulating complexes. These synthetic adjuvants are based on the concept of packaging the antigen into micro/nanoparticles or micelles, where the particle size is a crucial determinant of efficient uptake. Many different plant-derived saponins have been investigated for adjuvant activity including saponins derived from Bupleurum chinense, Glycyrrhiza uralensis, Quillaja brasiliensis, and Quillaja saponaria.110-114 These heterogeneous compounds stabilize the lipid–cholesterol structure while adding immunostimulatory properties. However, these compounds are also generally hemolytic and their tissue-reactive toxic nature has plagued development. While saponins have been used in veterinary vaccines for many years, a balance between potency and adverse reactions will need to be achieved for widespread acceptance in human vaccines.64,107 A detoxified saponin derivative, QS-21, has exhibited marked decrease in toxicity while maintaining the strong immunoenhancing properties.35,110 This adjuvant has been shown to induce a strong Th1 immune response (CTL, IL-2, IFN-γ, and IgG2a) because of the lipid–cholesterol makeup. Like virosomes, ISCOMs have the ability to fuse with cellular membranes and to deliver the immunogen into the cytosol of the target cell. This results in the endogenous processing and presentation of the immunogenic peptide via MHC I.35,64,110 This property also makes ISCOMs good vehicles for intracellular delivery of DNA-based vaccines.110 To increase antigenic loading of ISCOMs, affinity tags or aliphatic regions can be incorporated into recombinant proteins for higher efficiencies of incorporation into ISCOM membranes; alternatively, chelating agents (e.g., Cu++) can be used to increase antigen binding.107
VIROSOMES AND VIRUS-LIKE PARTICLES
Virosomes are particles of stabilized membrane lipids and functional viral fusion proteins that can be used to deliver vaccine antigens.103,115 While theoretically a wide number of virus fusion proteins could be used, the majority of virosomes utilize the hemaglutinin (HA) and neuraminidase (NA) from influenza virus.103 Virus-like particles are the spontaneous assembly of viral coat proteins lacking in viral genetic material.116 Virosomes and virus-like particles can be generated by either inserting the viral fusion proteins and antigen into preformed small phospholipid vesicles (liposomes) or by separation and reconstitution of viral envelopes with the vaccine antigen.103 These particles retain the receptor binding capacity and mimic infectivity of native viruses without the risks associated with attenuated viruses and are capable of delivering vaccine antigens directly into the cytosol of the target cell.103 This allows for induction of both humoral and cell mediated immunity because some of the virosome-delivered antigens have the potential to be presented via MHC II following endosomal processing, and virosomes that escape into the cytosol will allow for antigenic presentation via the MHC I pathway.103 This type of delivery system has been shown to greatly enhance production of serum IgG and IgA at mucosal surfaces.35 A synergistic effect is observed when other adjuvants or immunomodulators are included, such as heat-labile toxin of E. coli.35 Virosomes and other virus-like particles are proving efficient for intranasal or mucosal delivery of many types of proteinaceous antigens (i.e., viral coat proteins) or DNA-based vaccines.35
LIPOPOLYSACCHARIDE (LPS)
Many antigenic preparations, particularly recombinantly derived antigens, contain residual amounts of bacterial LPS and other TLR ligands that may provide adjuvant activity.117 LPS is known to stimulate a variety of cells to produce cytokines and chemokines that control DC trafficking and maturation.118 An unusual feature of its adjuvanticity is that LPS can be delivered at a different site and a different time than the antigen and still enhance the immune response to the given antigen. But despite its potency, LPS has been used only as an experimental adjuvant in animal studies due to its toxicity and pyrogenicity in humans. Chemically modified forms of its active component such as MPLA, have been shown to possess many of the adjuvant properties of LPS but without the associated toxicity.
MONOPHOSPHORYL LIPID A (MPLA)
Gram-negative bacterial extracts have strong immunopotentiating effects, however they are too toxic for routine use in human vaccines. Most of the immunostimulatory or toxic effects are derived from the lipid A portion of LPS, which is located in the outer-membrane of gram-negative bacteria.107 Further analysis showed that by removing a phosphate group, sugar moiety, and an ester-linked fatty acid group the toxicity could be reduced 100- to 1000-fold, while still retaining the immunostimulatory function.107 MPLA, the resulting molecule, was derived from Salmonella minnesota.35 Similar to LPS, MPLA interacts with TLR-4 on APCs, although immune enhancement is observed in the absence of TLR-4.75,76 MPLA initiates signaling through TRIF transcriptional activation rather than NF-κB, which induces many pro-inflammatory cytokines associated with the toxic effects of LPS.76 Equivalent T-cell mediated responses were observed in mice immunized with Ova adjuvanted with LPS or MPLA indicating that the mechanism of TLR-4 signaling (TRIF vs. NF-κB), and not the magnitude of the response, was responsible for the reduction in toxicity.76 Binding of MPLA to TLR-4 initiates the synthesis of IL-1β, IL-12, and IFN-γ, all of which are necessary for DC maturation, migration, and initiation of the T cell response.35,119 In animal studies, MPLA induced a strong systemic Th1 type immune response, including cytotoxic T lymphocytes (CTLs).35 Furthermore, MPLA was shown to enhance the production of complement fixing antibodies and increased production of secretory IgA.35 While MPLA enhanced the resulting immune response to a given antigen in comparison to the immune response to the soluble antigen alone, MPLA is more effective when combined with other adjuvants or delivery systems such as alum, QS-21 (Quil A) and polymeric microspheres, or other adjuvants that provide a depot effect.35 Several vaccine formulations using MPLA as an adjuvant are in clinical trials for humans and livestock species.61,65
TLR-2 LIGANDS
Since the discovery of TLRs as a key sensing and signaling mechanism for APCs, efforts have been made to exploit TLRs as receptors for vaccine adjuvants.120,121 Many different derivatives of gram positive cell wall components have all been found to trigger immune activation through TLR-2. OspA of Borrelia burgdorferi was used in the vaccine against Lyme disease.122 Muramyl dipeptide (MDP) has been synthesized from several gram positive bacteria including several Mycobacterium species, Corynebacterium granulosum, B. pertussis, and N. meningitides. MDP derivatives have been shown to induce dichotomous effects on the immune system. When delivered in soluble delivery systems, MDP enhances humoral immunity; when delivered in liposomes, MDP enhances CMI.116 Addition of MDP to a vaccine formulation acts synergistically with mineral oil and alum carriers, enhancing the CMI response.107 Macrophage activating lipopeptide-2 (MALP-2) is another TLR-2 targeted ligand showing promise as a vaccine adjuvant. MALP-2 is an agonist of the TLR-2–TLR-6 heterodimer from Mycoplasma fermentans and has been shown to activate APCs via MyD88 signaling and activation of NFκB transcription factor.123 TLR-2 and TLR-6 are also present on B cells.123 Studies in mice lacking either B or T cells showed that MALP-2 activated B cells in a T cell-independent manner but enhanced T cell function via a B-cell dependant mechanism.123 Pam2Cys is a synthetic compound with structural similarity to MALP-2 and has been shown to enhance the CMI and humoral response in an experimental vaccine for Listeria monocytogenes and an intranasal administration of an influenza vaccine in mice.124 ESAT-6, a protein derived from the cell wall of M. tuberculosis, can also be recombinantly produced.125,126 ESAT-6 can act both as a protective antigen against tuberculosis or can non-specifically enhance CMI to coadministered antigens.52,125-127
CpG ADJUVANTS
Prokaryotic DNA contains unmethylated CpG dinucleotides within specific nucleic acid motifs that are recognized by the innate immune system of vertebrates.128 These immunostimulatory motifs are the ligand for TLR-9 which is found primarily in intracellular vesicles of phagocytic cells.128 Signaling through TLR-9 CpG ligands induces the production of reactive oxygen species and activation of NF-κB.129 These immunostimulatory sequences are species specific and unique sequences have been described for laboratory animals (mice, rats, and rabbits), humans, and nonhuman primates, as well as companion and farm animals.65 For humans, there have been two types of CpG motifs described, type K (also known as B-type) and type D (or A-type).128 The type K CpG motifs primarily stimulate B cell and monocyte proliferation, IgM, IL-10, and IL-6 secretion. Type D CpG motifs primarily activate DCs, a response which is characterized by upregulation of CD80, CD86, MHC II, and TNF-α and IL-8 secretion.128 Regardless, CpG motifs are capable of stimulating enhanced secretion of immunoglobulins, and may be capable of modulating preexisting immune responses.64,129 Addition of CpG motifs to vaccine formulations has been shown to induce both cellular and humoral response to immunogens, inducing a Th1 bias. CpG motifs have been shown to induce demonstrable immune responses to weak immunogens such as malarial antigens, anti-H. influenzae glycoconjugates and melanoma antigens.129 When both alum and CpG motifs were included in vaccine formulations, the resulting immune response was Th1-biased, with no IgE production or eosinphilia.84 Furthermore, addition of CpG motifs to intranasal vaccine formulations enhanced the total serum titer to TT and influenza (viral) antigens in mice indicating that they may be useful as immune enhancers for mucosal delivery of antigens.130 CpG motifs are also used to enhance the response to antigens encapsulated in biodegradable polymeric microspheres described in this review.129,131,132 CpG motifs have been included in many experimental vaccines demonstrating enhanced protection against a variety of pathogens including Ebola virus, Bacillus anthracis, Francisella tularensis, L. monocytogenes, and Cryptococcus neoformans and in models of polymicrobial intraabdominal sepsis.122,133-136
BACTERIAL TOXINS
Bacterial toxins have a high degree of immunogenicity and immune enhancing capabilities along with highly specific cellular receptors. These properties have led researchers to study the potential of bacterial toxins as vaccine adjuvants. Pertussigen, a complex mixture derived from B. pertussis, including pertussis toxin, has been used experimentally as an adjuvant.137 Pertussigen enhances levels of IgE and hypersensitivity reactions to codelivered antigens and may help adjuvant the response to TT and diphtheria toxoid which are part of the trivalent childhood DPT vaccine.6,137 Heat-labile enterotoxin (LT) from E. coli has also been shown to enhance mucosal immunity to coadministered antigens.35 LT exhibits adjuvant efficacy for induction of mucosal and parenteral immunity in mice. LT was also used as an oral adjuvant for Campylobacter killed whole-cell vaccines. In rhesus monkeys, LT was shown to be safe and provided superior performance over the Campylobacter killed whole-cell vaccines alone.138 Cholera enterotoxin (CT) is another bacterially derived protein that shows high immunogenic potential when delivered to mucosal surfaces.139,140 LT is highly homologous to CT, but CT stimulates predominantly Th2 responses to conjugated antigens while LT stimulates mixed Th1/Th2 response.141 However, cholera-like toxin adjuvants delivered by the nasal route have been found to be taken up by the olfactory nerve and the central nervous system, leading to potential unwanted side effects142 and CT can induce diarrhea in humans. Not much is known about the cell-mediated immunity or delayed hypersensitivity response to CT. The ability of CT to act as a mucosal adjuvant has been confirmed by many investigators with a variety of antigens, and administering CT by a route different from the antigen is not immune enhancing.139,140
CYTOKINES
The cytokine network controlling immunity and T cell development is complex and much research remains to be done to elucidate these pathways.143 The effect of a few cytokines and their relevance to immune activation has been well studied and these cytokines have been explored as adjuvants to provide potentially less toxic approaches to enhancing vaccine efficacy. For example, granulocyte macrophage-colony stimulating factor (GM-CSF) has been included in experimental vaccines due to its ability to enhance APC recruitment and activation.116 In attempts to improve the pneumococcal polysaccharide vaccine against S. pneumoniae, IL-12 was included as a mucosal adjuvant.144 The inclusion of IL-12 enhanced mucosal and systemic IgG2a and IgA following intranasal vaccination and showed a marked reduction in bacterial nasal carriage and prevention of bacterial systemic invasion.144 Inflammatory cytokines in the IL-1 family have been shown to enhance the production of serum and mucosal IgG and IgA antibodies and cell-mediated responses to codelivered Ova and tetanus toxoid.35 The choice of cytokine included in a vaccine formulation must be chosen with care. In a recent study, polylactide microspheres were investigated as intranasal delivery of recombinant V antigen (rV) of Y. pestis coencapsulated with IL-6, IFN-γ, or IL-4.145 While all formulations induced mucosal IgG1 and IgA antibodies, only formulations including IL-6 with the rV induced protection from systemic bacterial challenge.145 The challenge of cytokine delivery is the rapid utilization of cytokines and their pluripotent biological effects. One mechanism to reduce these effects is to deliver a plasmid including the sequence of the cytokine.66,116 With DNA based vaccine technologies, this has proven very effective for enhancing the immune response to the DNA-encoded antigen. Inclusion of the sequence for IL-2 or IL-12 with the sequence with HIV antigen enhanced the production of a strong Th1 immune response.35
POLYMER VACCINES
Biodegradable polymers have been studied for many years because they show promise for the development of single dose vaccines.146,147 Polymers have the ability to sustain the release of the vaccine antigen over an extended period of time, thus eliminating the need of subsequent doses of vaccines. Other potential advantages of these materials are that immunomodulatory properties (i.e., adjuvanticity) can also be achieved with the proper tailoring of the polymer chemistry.148 Studies evaluating the use of controlled-release, single dose polymeric vaccines in both laboratory animals and livestock species (i.e., sheep, mini-pigs, cattle, and horses) have shown promise when encapsulating protein antigens.149-153
Biodegradable polymers also offer the advantage that MPLA, CpG DNA motifs or other immunomodulatory molecules can be incorporated to create a pathogen-mimicking solid particle.154 Polymeric vaccine particles have been shown to induce demonstrable immune responses when administered by several routes including parenteral (e.g., intramuscularly or subcutaneously), intranasal, or oral.35
These materials also have the added advantage over stable (nondegradable) devices (e.g., pumps) in that after administration, there is no need to remove them, therefore eliminating another surgical procedure. Furthermore, most are manufactured from synthetic parent compounds, eliminating many potential reactive antigenic or allergenic epitopes that can accompany the use of animal or plant derived materials.
The two most widely studied polymer classes for controlled release vaccines are polyesters155-164 and polyanhydrides.87,165-176 Other classes of polymeric compounds have been evaluated and shown to successfully deliver antigen to laboratory animals.177-190 Key findings of research done with these polymeric systems as vaccines carriers are discussed below and the chemistries are shown in Table 4.
Table 4.
Structure of Biodegradable Polymers Studied for Use as Vaccine Adjuvants
| Polymer | Structure | Refs. |
|---|---|---|
| Polysaccharides | ||
| Dextran | 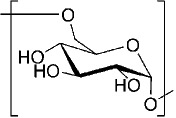 |
229 |
| Chitosan | 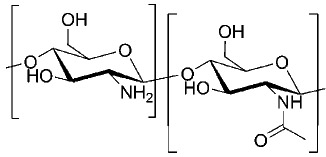 |
35,230 |
| N-trimethyl chitosan | 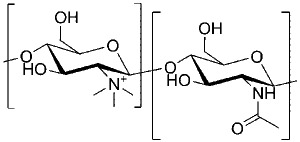 |
230 |
| Polyanhydrides | ||
| Poly(sebacic acid) SA | 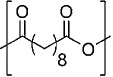 |
148,214,219,226 |
| 1,3-bis(p-carboxyphenoxy)propane CPP | 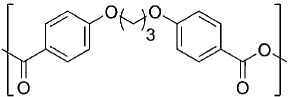 |
174,226 |
| 1,6-bis(p-carboxyphenoxy)hexane CPH | 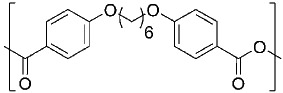 |
148,214,219 |
| l,8-bis(p-carboxyphenoxy)-3,6-dioxaoctane CPTEG | 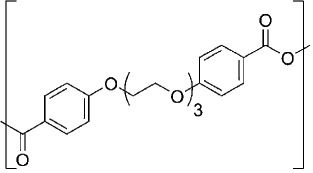 |
217,221,222 |
| Poly(trimellitylimido-l-tyrosine) | 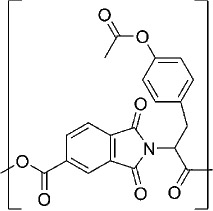 |
226 |
| Poly(ortho ester)s | 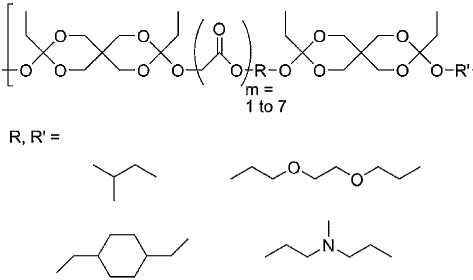 |
60 |
| Poly(ester-amide)s | ||
| Phenylalanine-based PEA | 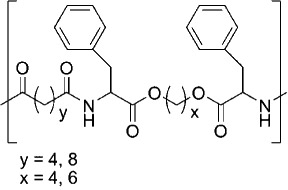 |
235,236 |
| Leucine-based PEA | 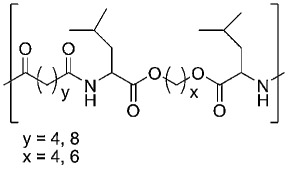 |
235,236 |
| Polyesters | ||
| Poly(lactic acid) LA | 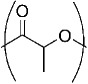 |
157,159,203 |
| Poly(glycolic acid) GA | 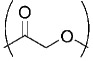 |
157,159,203 |
| Poly-ε-caprolactone | 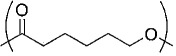 |
225 |
| Poloxamers | ||
| Poly(ethylene glycol) PEG | 237,238 | |
| Other | ||
| Poly(vinyl methyl ether-alt-maleicanhydride) PVM/MA | 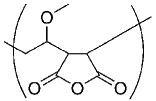 |
224 |
POLYESTERS
Microspheres composed of polyesters have been the most widely studied. Polymers of lactic acid and glycolic acid (e.g., poly(lactide-co-glycolide), PLGA) have been utilized in biomedical applications such as bone pins and dissolvable sutures for many years and recently have proven effective as vaccine delivery vehicles for the induction of protective immunity in laboratory animals.155-159 The greatest benefits of PLGA is that its degradation products, lactic acid, and glycolic acid, are naturally occurring metabolites and are readily absorbed by neighboring cells.160,161 However, as the polyester degrades and the acidic monomers are released, an acidic microenvironment is created. Prolonged exposure to aqueous or acidic environments has been shown to be detrimental to the stability and immunogenicity of proteins, especially the proteins used in recombinant and subunit vaccines, for example, TT and DT.162,163 Some attempts to minimize this acidity have been recently evaluated by incorporating a basic compound like magnesium carbonate (MgCO3) into PLGA microspheres.164 However, subsequent analysis indicated that while MgCO3 did not significantly improve peptide stability, it did enhance the antibody production, acting as a potential adjuvant.
Antigen-loaded PLGA microspheres function as an adjuvant by at least two mechanisms: (1) creating a depot for the antigen in vivo, and (2) enhancing phagocytic uptake of the antigen-loaded particle by APCs.159 The uptake of PLGA microspheres by macrophages or DCs has been demonstrated following administration by intraperitoneal or intradermal routes, respectively.191 Other immunostimulatory properties of PLGA were observed in studies showing an enhanced cytokine production and proliferation when cells were incubated in vitro with blank PLGA microspheres.161 Similarly, oral administration of PLGA nanoparticles containing type II collagen promoted the induction of tolerogenic immune responses that ameliorated arthritis.192 The prolonged presence of the nanoparticles in the Peyer’s patches and the induction of elevated TGF-β suggested the differential activation of DCs that modulated the subsequent immune response.
Vaccine formulations based on PLGA, PLA, or PGA variants have been successful in inducing immune responses in laboratory rodents to a large number of antigens including: Y. pestis antigens, HIV gp140, B. pertussis antigens, measles virus antigen, OVA antigen, TT, diphtheria toxin, type II collagen, malarial antigens, cancer cell antigens, E. coli adhesion proteins, Vibrio cholerae antigens, influenza virus antigens, hepatitis B viral antigens, and ricin toxoid.35,161,192,193 These vaccines have been delivered by a variety of routes including intradermally, intravaginally, intranasally, orally, or parenterally into laboratory animals to induce both serum antibodies, mucosal IgA, cell-mediated responses and facilitated the induction of secondary immune responses (e.g., isotype switching) as determined when individuals were analyzed up to a year after single immunization.161,194 Many groups have reported the successful induction of immunity following use of a single dose vaccine formulation composed of PLGA microspheres of various compositions.157,195-200 Furthermore, encapsulation of antigens in PLGA microspheres was shown to enhance antigen presentation via MHC I leading to increased activation of antigen specific cytotoxic T cells.147,193,195 However, most of these studies were conducted in vitro, and some investigations included MPLA, a known Th1 immune response activator, in the microsphere while others used multiple injection regimens in vivo. There is no consensus opinion, however, as to whether PLGA-based vaccines are more efficacious than current adjuvant systems such as alum. Antibody responses induced in mice and guinea pigs following vaccination with TT-loaded PLGA were greater than those induced by single injection of soluble TT alone or two doses of alum absorbed TT. Additionally, a stronger anamnestic response (higher titer) was observed when individuals that had received the TT-loaded PLGA microparticles were boosted 1 year later.194 On the other hand, Walker et al.199 observed that encapsulation of TT in PLGA microspheres did not induce serum antibody titers higher than alum-based TT vaccines. Only small amounts of antigenically active TT were released in the first 2 days from PLGA microspheres, even though protein continued to be released for up to 11 weeks.194 Collectively, evaluation of PLGA studies does not provide strong correlation between release of antigenic peptides, length of in vitro release of peptides, and immune response to those peptides in vivo.
Some studies have suggested that immunization with PLGA microspheres effects immune deviation. Moore et al.201 showed the ability of HIV gp120 protein loaded PLGA microspheres to shift the T cell response from a dominant Th2 or mixed Th1/Th2 to a more dominant Th1 immune response as indicated by the presence of IFN-γ producing CD4+ T cells. In other studies, the Th2-biased hepatitis B core antigen has been formulated with the Th1 immune stimulator MPLA in PLGA nanoparticles to develop a stronger Th1 response.68 More recently, a vaccine formulation prepared against malaria and composed of PLGA microspheres and Montanide ISA-720 was shown to induce an antibody response (IgG isotype class switching) characteristic of Th1 response.202
Variations in reported efficacy of PLGA microspheres may be due to dose of antigen, method of encapsulation (e.g., spray drying vs. solvent evaporation), route of immunization, and/or the size of the microspheres.193,203 Following primary immunization with small microspheres (10–20 μm), a greater anamnestic response was generated 1 year later following a low dose booster than that observed in animals initially receiving larger microspheres (>60 μm);194 however, nanoparticles (200–600 nm) were less effective at inducing cell-mediated immune response than microspheres.193 This may be because microspheres <10 μm in diameter are readily phagocytosed by macrophages and DCs that would enhance antigen processing and presentation.204-209 On the other hand, the route of immunization with PLGA microparticles influenced the type of immune response generated. The intraperitoneal route induced Th1 cell-mediated response while the intramuscular route induced a Th2 humoral response.193 Despite all the extensive research done with PLGA as antigen carriers, some with success in animal models, no formulation has been reported to induce a protective immunity in humans.210
POLYANHYDRIDES
Polyanhydrides are a class of surface erodible, biocompatible polymers that have been extensively used as carriers for controlled drug delivery.87,165-176 These biodegradable polymers are currently approved by the FDA for use in a variety of biomedical applications and can also be fabricated into protein-loaded microspheres.211 Biocompatibility studies have shown that these biomaterials degrade into carboxylic acids, which are nonmutagenic and noncytotoxic products.212,213 The surface erosion mechanism leads to a controlled release profile with predictable degradation profiles, which can range from days to months, depending on the copolymer composition.214,215 In addition, studies involving polyanhydride delivery systems for vaccines have shown attractive features such as improved adjuvanticity, antigen stabilization, and enhanced immune responses.165,175,176,216
The main advantage of polyanhydrides over polyesters as antigen carriers is associated with the enhanced protein stability following encapsulation. Studies have shown that polyanhydrides are capable of stabilizing polypeptides and sustaining their release without the inclusion of potentially reactive excipients or stabilizers.217-220 The hydrophobicity and surface erosion characteristics of polyanhydrides prevent water from penetrating to the interior of the microsphere thus preserving the encapsulated antigen in its native state (i.e., increased stability). Furthermore, the degradation products of polyanhydrides are less acidic than those of polyesters, which may further enhance the stability of encapsulated antigens and reduce tissue reactions to the polymer.217,219 Despite these beneficial characteristics, the use of polyanhydrides for vaccine delivery has not been extensively evaluated.
Recently, Kipper et al.148 performed in vivo studies to evaluate the induction of immune responses following immunization with antigen-loaded microspheres based on the anhydride monomers, sebacic acid (SA) and 1,6-bis(p-carboxyphenoxy)hexane (CPH). Microspheres encapsulating TT antigen were injected in C3H/HeOuJ mice. These studies demonstrated that TT maintained its immunogenicity and antigenicity following encapsulation. The type of immune response generated, Th1 versus Th2, was evaluated by antibody isotypes. It was observed that TT loaded 20:80 CPH/SA microspheres enhanced the immune response after a single dose and induced a dominant Th2-like immune response. However, the 50:50 CPH/SA produced a balanced Th1/Th2 response. Total TT-specific IgG titer remained high regardless of dominant isotype. The preferential enhancement of the Th1 immune response resulting in more balanced immune response (i.e., immune deviation) is a unique and valuable feature of this delivery vehicle that makes it a promising adjuvant candidate for vaccines. Currently, the groups led by Narasimhan and Wannemuehler are corroborating the immunomodulatory properties of the CPH/SA system with other antigens as well as investigating the adjuvant properties of novel amphiphilic polyanhydride chemistries. Copolymers of CPH and 1,8-bis(p-carboxyphenoxy)-3,6-dioxaoctane (CPTEG), which contains ethylene glycol moieties in the polymer backbone, are promising candidates for the development of vaccines as they have been shown to provide a conducive environment for protein stabilization.217,221-223
Anhydride monomers have been copolymerized with other chemistries and their potential as adjuvants have been evaluated. An immunogenic subcellular extract obtained from heat-killed Salmonella enteritidis cells (HE) has been encapsulated in nanoparticles of a copolymer comprised of methyl vinyl ether and maleic anhydride (PVM/MA), best known as Gantrez® polymer.224 In this study, 80% of the Gantrez®-HE immunized mice survived even when the nanoparticle formulation was administered 49 days prior to a lethal challenge. As early as 10 days after immunization, a Th1 immune response was demonstrable in these mice as determined by the IgG2a antibody titer in the serum. On the other hand, a dominant Th2 immune response was present at 49 days after immunization (IgG1 > IgG2a). Since it is known that a Th1/Th2 balance is required to protect against S. enteritidis infection, this copolymer is a promising candidate for the development of future vaccines. In this regard, blank nanoparticles of Gantrez® administered subcutaneously four weeks prior to challenge induced a level of protection similar to that induced by antigen-loaded nanoparticles or the Rv6 commercially available vaccine against S. enteritidis serovar abortusuis.225 While the authors did not demonstrate the presence of antigen-specific immunity, these data suggest that the blank nanoparticles were able to induce and sustain sufficient innate immunity to provide nonspecific protection against subsequent Salmonella infection. In the same study, abortusovis antigen-loaded poly(ε-caprolactone) microparticles did not induce protection.
In another attempt to design suitable carriers specifically intended for vaccine delivery, Hanes et al.226 synthesized poly(anhydrides-co-imides) with the adjuvant l-tyrosine incorporated in the polymer backbone. In these studies, a predictable and controlled protein release was observed from microspheres of poly[trimellitylimido-l-tyrosine-co-sebacic acid-co-1,3-bis(p-carboxyphenoxy)propane] and polymeric implants were well tolerated after subcutaneous implantation in rats. More recent studies demonstrating the suitability of polyanhydrides for use in single dose vaccines involved the design of a core-shelled cylindrical device composed of a biodegradable hydrophobic coating and laminated core of polyanhydrides and polyphosphazenes.227 Polyanhydrides based on SA were used as isolating layers of the cylinder in order to produce a pulsatile drug release, a mechanism which would minimize doses of vaccines. Even though these polyanhydride systems showed promising characteristics for vaccines design, no further in vivo studies evaluating the characteristics of the proposed adjuvant were validated.
A comparative study between polyanhydrides and polyesters has demonstrated the potential capabilities of polyanhydrides for oral vaccination.228 Microspheres (0.1–10 μm) composed of fumaric acid (FA) and SA proved to have strong adhesive interactions with the mucosal gastrointestinal lining of rats, as opposed to poly(lactic acid) (LA), which showed minimal uptake. The adhesive interactions are ideal to prolong the biological activity of the delivered antigen or bioavailability of encapsulated drugs. Not surprisingly, plasmid DNA- and anti-coagulant drug dicumarol-encapsulated FA/SA microspheres enhanced gene activity and plasma drug levels, respectively, when compared to the controls. In the same studies, blends of FA and LA were used for insulin delivery and groups that received the formulation were able to regulate glucose levels as opposed to the groups that received insulin only. Even though the biological activity of insulin was preserved, it was the adhesive characteristic of FA the responsible for the efficient delivery.
OTHER POLYMERS
Naturally Derived
Several naturally derived polymeric materials, such as dextran, chitosan, starch, and alginate have been evaluated in laboratory models for use as vaccine adjuvants. In the case of dextran, it has been chemically modified or use in conjunction with other adjuvants in order to improve its immunogenicity. Immunization of cattle with dextran in combination with mineral oil against Streptococcus bovis and Lactobacillus spp. induced the highest serum IgG responses when compared with other adjuvants (i.e., FCA, Quil A, alum), presumably due to the combined effect of both substances.177 In studies involving vaccination of cattle against M. tuberculosis, diethylaminoethyl (DEAE)-dextran induced high levels of IL-2 and low levels of IFN-γ, indicating a strong humoral response not desirable for this particular disease.178 Interesting results were obtained when a dietary supplementation of Lactobacillus casei with dextran enhances humoral immune responses, and chickens were able to maintain the growth of the bacteria in their intestines and prevent possible infections.179 Vaccines that have been evaluated utilizing cross-linked dextran microparticles, containing conjugated TT induced serum antibody to TT for long periods, eliminating the need of additional booster doses.229
Chitosan, a cationic polysaccharide derived from chitin in the exoskeleton of crustaceans, can also be formulated into microparticles capable of encapsulating antigen.230 Studies with chitosan showed that the immune bias induced by vaccination with antigen containing chitosan microparticles was more dependent on the route of delivery (e.g., intranasal vs. parenteral) than the nature of this adjuvant.35,180 An intranasal delivery of N-trimethyl chitosan chloride (TMC) containing diphtheria toxoid enhanced the immune response when compared with the conventional alum adsorbed vaccine.230 This enhancement of nasal vaccination is likely a result of the mucoadhesive properties of chitosan, which enhances penetration across nasal mucosae.181,182 More recent studies with chitosan and TMC establish that chemical variables, such as molecular weight in chitosan and degree of quaternization in TMC influence the magnitude of the immune response after nasal administration.183
Another natural polymer with potential in vaccines is starch, which also has been assessed in mucosal vaccines. Some advantages of starch include its inert properties, proven safety, and commercial availability.184 Heritage et al.185 found that human serum albumin delivered on starch microparticles grafted with polydimethyl-siloxane stimulated systemic and mucosal immune responses. Similarly to studies done with chitosan, the route of administration of starch influences the immune response.186 Among oral, subcutaneous, and intramuscular administrations, vaccines delivered by the subcutaneous routes induced stronger humoral responses. However, when comparing oral and intramuscular routes, stronger humoral response was induced after oral primary administration and a stronger cell-mediated response after oral booster doses. Although the adjuvant capabilities of starch were proved with success in mice studies, a human vaccine trial was not successful.231
Alginate microparticles offer several advantages for vaccine applications, including good biocompatibility, ease of preparation, and antigen protection during fabrication and administration.184,232 Alginate microparticles have been administered to several animal species (i.e., mice, rabbits, cattle, and chicken).232 The enhancement of the immune response induced in the animals after oral administration with antigen-loaded alginate microparticles shows promise for the development of veterinary vaccines. Nevertheless, in vitro studies show that alginate is not the optimum chemistry to activate human-derived DCs, as it decreases the expression of costimulatory molecules and antigen presenting complexes when compared to nontreated cells.233 Other in vitro studies that simulated gastric fluid environment showed that alginate microparticles were not able to stabilize live rotavirus vaccines.234
Synthetic
Other novel polymer chemistries have been researched to overcome the limitations of available polymers as vaccine carriers. The novel poly(ester-amide) (PEA) copolymers, composed of amino acid residues, diols, and dicarboxylic acids, have been shown to enhance cellular immunity.187 Polyamide gives PEA its superior mechanical and thermal properties, while the polyester portion is responsible for its flexibility and hydrolytic susceptibility, allowing PEA to degrade within a reasonable period of time. It is biodegradable, however, in contrast to polyester and polyanhydrides, PEA degrades by enzymatic cleavage within the body.235,236 Thus, shelf life and handling does not affect its degradation rate and the polymer remains intact until needed for therapy. PEA has been conjugated with several therapeutics peptides, including human melanoma antigen-derived peptides (MART), a synthetic peptide based on the gp120 protein of HIV, and a MHC II-restricted T-cell epitope from the influenza A virus hemaglutinin (HA) protein.187 In general, the studies evaluating PEA-peptide conjugates demonstrated that cellular immunity, encompassing both MHC I- and MHC II-restricted T-cell responses, was enhanced.
More recently, in vivo studies in mice have shown that poly(ethylene glycol)-stabilized poly (propylene sulfide) nanoparticles target the APCs directly in the lymph nodes.237,238 In these studies it was found that particles in the size range of 20–45 nm enter lymphatic vessels and subsequently target DCs in the lymph nodes. The cross-linked polymer system used here degrades into a water soluble polymer under oxidative conditions.
Polymers in Plasmid DNA Vaccines
Plasmid DNA vaccines represent a promising alternative against intracellular pathogens. Even though plasmid DNA immunogens have elicited strong cell-mediated responses in small laboratory animals, these have not had success in limited human clinical trials.188 Ideal adjuvants will improve the magnitude of plasmid DNA expression, must protect DNA from enzymatic degradation, and must facilitate the DNA plasmid uptake into cells. Several polymer chemistries have been evaluated in conjunction with DNA vaccines and a thorough discussion of this topic is beyond the scope of this review. In short, microspheres of polyesters, polycarbonates, polystyrene, and poly(orthoesters) have been used in DNA vaccination and their administration resulted in enhanced immune responses when compared to naked DNA administrations.60,189,190 Table 5 summarizes the pros and cons of polymeric adjuvants.
Table 5.
Advantages and Disadvantages of Polymers as Vaccine Adjuvants
| Polymer | Antigens | Advantages | Disadvantages | Refs. |
|---|---|---|---|---|
| Polysaccharides | ||||
| Dextran |
Streptococcus bovis Lactobacillus spp Mycobacterium tuberculosis Tetanus toxoid (TT) |
Induces strong humoral responses | Not desirable for some diseases (e.g., tuberculosis) | 101 |
| Chitosan N-trimethyl chitosan |
Diphtheria toxoid | Enhanced immune response compared to alum Mucoadhesive properties |
230 35 |
|
| Polyanhydrides | TT Salmonella enteritidis (HE) Plasmid DNA | Biocompatible nonmutagenic, noncytotoxic degradation products Degradation products have low acidity Enhanced protein stability Immunomodulatory Mucoadhesive properties |
Processing and storage | 90,113,214,219,217,221,222,226 |
| Poly(orthoester)s | Plasmid DNA | Enhanced immune response when compared to naked DNA | Plasmid DNA unsuccessful in human clinical trials | 60 |
| Poly (ester-amide)s | Melanoma antigen derived peptides (MART) HIV gp120 MHCII restricted T-cell epitope from influenza A virus hemaglutinin (HA) |
Degrades by enzymatic cleavage Enhanced cell-mediated) immunity |
235 | |
| Polyesters | TT Diphteria toxoid Yersinia pestis HIVgp140 Bordetella pertussis Measles virus antigen Ovalbumin Type II collagen Malarial antigens Cancer cell antigens Eschericia coli Ricin toxoid Vibrio cholerae Influenza virus antigens Hepatitis B viral antigens Plasmid DNA |
Degradation products are biocompatible and easily metabolizable Antigen-loaded microspheres enhance uptake by APCs Experiments have shown increase in both humoral and cellular immune responses |
Acidic microenvironments detrimental to antigens No protective immunity in humans have been reported yet Poor mucoadhesive properties |
57,99,101,159,161 |
| Poly(ethylene glycol) | Can target APCs in LNs | 237,238 | ||
| Poly(vinyl methyl ether-alt-maleic anhydride) | Salmonella enteritidis (HE) | Th1/Th2 balance Nonspecific protection against Salmonella | 224 |
THE IDEAL VACCINE ADJUVANT
Vaccines and their adjuvants interact with the patient’s immune system in a variety of ways. Thus, there is no single set of characteristics that would describe an ideal vaccine adjuvant for all situations. An adjuvant must be appropriate to the particular delivery route (e.g., intramuscular, mucosal, intraperitoneal, etc.), desired immune response (cell-mediated vs. humoral), pathogen, and stage of a disease. Additionally, biological traits of the patient may also be important including species, race, age, medical history, and genetic makeup. All of these factors may influence the effectiveness of a vaccine adjuvant, and the effects of these factors may be unknown. Nonetheless, there are certain characteristics that a good vaccine adjuvant must possess. These characteristics can be broadly grouped into two categories: biological characteristics and practical or economical characteristics.
Because vaccine adjuvants may enhance the immune response through different modes of action, the particular mechanism of adjuvanticity is of paramount importance. The mechanisms of adjuvant activity have been classified in different ways by different authors.66,119,239 The broadest classification distinguishes among two types of mechanisms: immune stimulation and targeting antigens to particular cells or tissue types.65 Adjuvants which act through the later mechanism target vaccines to DCs, through interactions with transmembrane TLR proteins or other cell surface receptors,97 or by virtue of their size.240,241 Polymer microspheres and liposomes <10 μm in diameter may be readily phagocytosed by macrophages and DCs.97 This specific targeting can reduce the quantity of antigen required to induce protective immunity. A good immunostimulatory vaccine adjuvant must stimulate the desired immune response without toxicity or inducing excessive inflammation. While some immunostimulatory adjuvants of bacterial origin have potent adjuvanticity (e.g., LPS), they can also be extremely toxic (e.g., induction of tumor necrosis factor).242 Less toxic adjuvants, such as alum, may also be less potent or ineffective at eliciting cell mediated immunity.97,243,244
Good immunostimulatory vaccine adjuvants activate DCs to mature into APC and migrate to the draining lymph node, coincident with induction of the cytokine profile appropriate to the desired immune response mechanism (i.e., IFN-γ, IL-2, and IL-12 for the Th1 response and IL-4, IL-5, and IL-6 for the Th2 response). Like adjuvants that target DCs, some immunostimulatory vaccine adjuvants also interact with TLR proteins. Though these proteins have affinity for a variety of ligands, different subpopulations of DCs express different TLR profiles and, thus, have different degrees of sensitivity to different antigens and adjuvants.31,245,246 Furthermore, the same TLR may activate different intracellular signaling cascades leading to different activated phenotypes in different DC subpopulations. Regardless of the mechanism of adjuvanticity, vaccine adjuvants must activate this desired adaptive immune response without over stimulating innate immune function.
Economical and practical considerations must also be taken into account when selecting an ideal vaccine adjuvant. Singh and O’Hagan64,74 list biodegradability, ease of manufacture, and low cost among important characteristics of vaccines. Other practical aspects to be considered include stability over time, ability to provide immunity with a single dose, and suitability for mucosal delivery. Such characteristics would enable more practical and economical strategies to fight infectious disease in remote areas that lack developed public health infrastructure and in communities that do not have access to modern medical care.247 Finally, while the “depot” effect (long thought to be the primary mechanism of adjuvanticity for alum) is no longer regarded as the essential mechanism behind adjuvant effectiveness,97,248 formulations such as degradable polymer microspheres may provide sustained exposure to antigens, obviating the need for multiple administrations. Practical considerations such as stability and cost may preclude the widespread use of some otherwise potent protein adjuvants such as cytokines.64,244
Thus, we can summarize the ideal vaccine adjuvant as one which selectively targets the antigen to the desired population of APCs, minimizes the amount of antigen required, induces the desired adaptive immune response while minimizing the innate immune response, is minimally toxic, low-cost, stable for long-term storage, and provides protective immunity in a single dose via a convenient delivery route.
NEW RESEARCH TOOLS TO STUDY DISEASE PREVENTION
New adjuvants are also needed that can be used to precisely tune the nature or outcome of the immune response to more effectively protect against particular diseases such as cancers and HIV. This may be done by controlling the induction of particular cytokine profiles and by more effectively targeting antigens to specific tissues, cells, or intracellular compartments (e.g., DNA vaccines to the nucleus of a cell). These new adjuvants could also be used as research tools to study the induction or regulation of different immune response mechanisms that are associated with autoimmune diseases, allergies, or tolerance. Excitedly so, many of these new adjuvants are being developed experimentally and much more research is needed to bring them to an application. As shown in Table 1, very few adjuvants are being used in licensed vaccines. Even the materials being tested in current clinical trials represent relatively few new immunostimulating adjuvants or chemistries, especially against infectious diseases (Tab. 2). Furthermore, there may be a need for a considerable shift in thinking about how vaccines are tested for efficacy. Antibody titer is almost universally used as the test for vaccine efficacy but often high antibody titers do not translate into the best protection.249 Many times a highly immunogenic antigen does not correlate to a protective immune response. This “deceptive imprinting” is a common evasion mechanism by pathogens and partially responsible for the slow development of HIV vaccines.250 Also there is the caveat that laboratory mice are not humans (or other livestock species) and what works in a mouse may not translate to other species. Numerous studies have highlighted differences in mouse and human immune systems including differences in complement reactivity,251 induction of Th17 cells,252 or response to a vaccine based on particle size.70
In 2006, the National Research Council convened a Workshop on Immunomodulation253 which made several recommendations to improve vaccine design including (1) an improved molecular level understanding of the innate immune system, (2) the need for effective delivery mechanisms, (3) the identification of potential molecular targets to modulate innate immunity without undesirable side effects, and (4) new strategies to target DCs and optimize antigen presentation. A key need that was identified by this panel was that in order to solve these important problems, it is critical for researchers from multiple disciplines to work together. These fields may include biochemistry, immunology, materials science, cell biology, computational biology/materials science, pathology, oncology, microbiology, and combinatorial science. It is important to combine expertise from antigen biochemistry, cell biology, and immunology to understand the mechanism of immunogenicity and how the preservation of various epitopes contributes to immunogenicity. As these antigens are combined with adjuvants, it is important for materials scientists to work closely with immunologists to understand how protein antigens can be stabilized during encapsulation and delivery and how adjuvants interact with APCs. As these adjuvanted systems enter the body, they encounter plasma proteins that may adsorb on to the surface of the adjuvant. How this affects the release of the antigen and how this influences APC activation or antigen processing is of great significance to the initiation of the desired immune response. Finally, the use of the appropriate animal models to study these phenomena is critical and immunohistochemical methods are needed to study how these adjuvants affect the local tissue response.
In this regard, the authors, who belong to chemical engineering and veterinary microbiology departments have worked towards providing a highly cross-disciplinary research environment for students and postdoctoral researchers in their respective groups. The chemical engineering graduate students have the opportunity to take courses on immunology and molecular biology techniques, participate in journal clubs, and several of them have completed an immunobiology certificate program on their way to a Ph.D. Likewise, the microbiology students have the opportunity to take courses on polymeric biomaterials and nanotechnology. Such an approach has immensely benefited students from both disciplinary groups and has prepared them to address diverse research problems with new and innovative perspectives. Similar examples of cross-disciplinary research groups exist and are much needed as scientists embark on new therapies for diseases such as cancer, HIV, and respiratory infections.
Over the last 200 years, the use of vaccines has proven to be one of the most successful medical interventions in the reduction of disease caused by infectious agents.1 However, many challenges still remain with regard to fully realizing the health benefits of active immunization programs. Some of these obstacles include the implementation of improved adjuvants, development of single dose vaccines, methods to overcome the poor immunogenicity of recombinant and subunit immunogens, and the ability to rapidly and rationally develop vaccines against emerging pathogens. In this regard, the mechanisms underpinning the effective modulation of cellular and molecular events associated with adjuvant enhancement of immune responses are still largely unknown. There is growing interest in the development of vaccine delivery systems based on micro- and nano-scale devices composed of biodegradable polymers, because they have the potential to act as effective adjuvants by encompassing all three of the classical adjuvant properties: providing an antigenic depot with a tailored and pulsatile release of the antigen over time, directing particulate antigens to the APCs and modulating the activation of innate immunity by altering polymer chemistry.95 However, the mechanism of adjuvanticity and the ability of adjuvant chemistry to selectively modulate the immune response are still largely unknown. In order to address these challenges, it is important to perform fundamental and systematic studies of the role of polymer chemistry in regulating activation of APCs (e.g., DCs), antigen uptake, processing, and presentation, migration to the draining lymph node, and modulation of the immune response.
The mechanisms by which adjuvants enhance and/or redirect the immune response (e.g., formation of high titer antibodies, CD4+ helper T lymphocytes and/or CD8+ T lymphocytes) in order to establish long-term immunologic memory are poorly understood. Upon antigen stimulation, T cells differentiate into two distinct populations described as Th1 and Th2 type immune responses.254 Furthermore, Th1- and Th2-related cytokines (IFNγ or IL-4/IL-13 respectively) can impact both the quality and magnitude of humoral and cell-mediated immunity. Humoral immunity, characterized by the activation of B cells that differentiate into antibody secreting plasma cells, is effective at neutralizing toxins, viruses, complement fixation, and opsonization of extra-cellular pathogens whereas the cell-mediated immunity (i.e., activation of cytotoxic T cells and macrophages) are crucial for protection against intracellular pathogens.255 The balance of humoral and cell-mediated immune responses has been shown to be important in the favorable outcome of many disease states. In this regard, vigorous and inappropriate cell-mediated immune responses have been implicated in the induction of autoimmune and chronically inflammatory diseases (multiple sclerosis and Crohn’s disease) while robust humoral immune responses are associated with allergic reactions.120 In order to control the induction of appropriate immune responses and reduce the risk of autoimmunity or allergic responses, there is an urgent need to develop new, well-characterized adjuvants that allow for tailored immune activation and deviation. In spite of these implications of immune deviation, the mechanisms by which adjuvants influence whether Th1 or Th2 cells dominate an immune response are not well understood. Additionally, it is also important to consider the use of adjuvants to induce regulatory T cell responses and to avoid the aberrant induction of Th17 cells that have been associated with chronic inflammatory diseases.
Both in vitro and in vivo studies with the adjuvants discussed above indicate that adjuvant chemistry and particle size may play an important role in regulating the cellular and molecular mechanisms responsible for modulating host immune responses. Additionally, in order to understand intracellular trafficking at a molecular level, it is important to employ the use of reporter molecules (e.g., quantum dots (QDs)) embedded within the adjuvant. Encapsulating QDs within adjuvants will provide adequate intracellular stability to effectively track the transport of nanospheres through intracellular compartments. The luminescence properties of QDs are expected to persist so long as the integrity of their nanocrystal structure is maintained, providing superior performance compared to conventional fluorescent dyes such as FITC, whose fluorescence is sensitive to pH.256,257 QDs can also dramatically enhance in vivo imaging of APC migration by using red and near-infrared emitting QDs as an alternative to Cy5, Cy5.5, or other traditional organic dyes. QDs have substantially larger absorption cross-sections than even the best commercial dyes developed specifically for such imaging applications.258 This improves the effective brightness of the fluorescence emission signal considerably. Additionally, QDs have unrivaled photostability that allows continuous long-term excitation without a substantial loss in fluorescence.256
Finally, in order to understand the cellular and molecular mechanisms that establish immunologic memory, it is very important to correctly choose appropriate in vitro/in vivo models that will promote the induction of cell-mediated as well as humoral (i.e., antibody) immune responses. Because there are likely to be subtle immunogenetic differences between mouse strains (and eventually individual human subjects), the use of combinatorial approaches evaluating cell–adjuvant interactions may provide a robust and versatile approach to the development of vaccines that will effectively stimulate immunity for different conditions and/or applications. These approaches may be used to rapidly screen a large number of adjuvant chemistries for their ability to differentially activate APCs, which will aid in the rational use of cocktails of micro- or nanoparticles in vaccine formulations. These formulations will possess the ability to stimulate the appropriate immune response depending upon the disease. The availability of transgenic models (e.g., OTI and OTII transgenic mice) provides for the capability to critically evaluate the activation of CD4+ and CD8+ pathways while other molecular biology tools enable researchers to evaluate the effect of new adjuvants on antigen processing and presentation both in vitro and in vivo.
In summary, an integrated and cross-disciplinary approach is needed that combines the development of novel adjuvants with: (i) molecular level studies that will elucidate the mechanisms of chemistry-mediated cellular activation by adjuvants; (ii) cellular level studies that will elucidate the uptake mechanisms of antigen-loaded adjuvants by immune cells and the activation and migration of these cells; and (iii) in vivo studies that highlight the underlying mechanisms governing immune modulation. Such an integrated approach is essential to solve the important challenge of rationally designing vaccine delivery systems that will effectively stimulate the immune system. It can provide new insights into the mechanisms of adjuvanticity and on the complex relationships between adjuvant chemistry, molecular mechanisms of APC activation, antigen uptake, processing/presentation by APCs, migration to the draining lymph node, and modulation of the immune response. To carry out such an approach, it is important to assemble highly cross-disciplinary teams of researchers with expertise in the areas of molecular and cellular immunology, intracellular trafficking, biomaterials chemistry, toxicology, nanotechnology, and pathology.
ACKNOWLEDGMENTS
B.N. and M.J.W. gratefully acknowledge financial support from the U.S. Department of Defense—Office of Naval Research (ONR Award # N00014-06-1-1176). B.N. also acknowledges financial support from the Whitaker Foundation. M.P.T. acknowledges financial support from NIH-NCI via the Ruth L. Kirschstein Fellowship.
REFERENCES
- 1.Pashine A, Valiante NM, Ulmer JB. 2005. Targeting the innate immune response with improved vaccine adjuvants. Nat Med 11:S63–68. [DOI] [PubMed] [Google Scholar]
- 2.http://news.bbc.co.uk/2/hi/health/4433507.stm. Polio not eradicated 50 years on: BBC. [Google Scholar]
- 3.Spickler AR, Roth JA. 2003. Adjuvants in veterinary vaccines: Modes of action and adverse effects. J Vet Intern Med 17:273–281. [DOI] [PubMed] [Google Scholar]
- 4.Birmingham M, Stein C. 2003. The burden of vaccine preventable diseases. In: Bloom BR, Lambert P-H, editors. The vaccine book, 1st edition. San Diego, CA: Academic Press. pp 1–22. [Google Scholar]
- 5.Bruggemann H, Baumer S, Fricke WF, Wiezer A, Leiesegang H, Decker I, Herzberg C, Martinez-Arias R, Merkl R, Henne A, Gottschalk G. 2003. The genome sequence of Clostridium tetani, the causative agent of tetanus disease. Proc Natl Acad Sci 100:1316–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.http://www.who.int/immunization/en/. Vaccines, immunizations and biologicals: World Health Organization. [Google Scholar]
- 7.Aguado MT, Lambert P-H. 1992. Controlled-release vaccines—Biodegradable polylactide/polyglycolide(PL/PG) microspheres as antigen vehicles. Immunobiology 184:113–125. [DOI] [PubMed] [Google Scholar]
- 8.Lopez AS, Guris D, Zimmerman L, Gladden L, Moore T, Haselow DT, Loparev VN, Schmid DS, Jumaan AO, Snow SL. 2006. One dose of varicella vaccine does not prevent school outbreaks: Is it time for a second dose? Pediatrics 117:e1070–e1077. [DOI] [PubMed] [Google Scholar]
- 9.Watson-Creed G, Saunders A, Scott J, Lowe L, Pettipas J, Hatchette TF. 2006. Two successive outbreaks of mumps in Nova Scotia among vaccinated adolescents and young adults. Can Med Assoc J 175:483–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.http://www.fnih.org/news/17october2003.shtml. Grand Challenges in Global Health: Foundation for the National Institutes of Health. [Google Scholar]
- 11.Bowersock TL. 2002. Evolving importance of biologics and novel delivery systems in the face of microbial resistance. AAPS PharmSci 4:E33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.http://who.int/vaccine_research/documents/index. Vaccine Research: World Health Organization. [Google Scholar]
- 13.Kaufmann SH. 2007. The contribution of immunology to the rational design of novel antibacterial vaccines. Nat Rev Microbiol 5:491–504. [DOI] [PubMed] [Google Scholar]
- 14.Bowersock TL, Martin S. 1999. Vaccine delivery to animals. Adv Drug Deliv Rev 38:167–194. [DOI] [PubMed] [Google Scholar]
- 15.Szucs TD. 2005. Health economic research on vaccinations and immunisation practices—An introductory primer. Vaccine 23:2095–2103. [DOI] [PubMed] [Google Scholar]
- 16.Gilboa E 2004. The promise of cancer vaccines. Nat Rev Cancer 4:401–411. [DOI] [PubMed] [Google Scholar]
- 17.Ehlers S 2004. Commentary: Adaptive immunity in the absence of innate immune responses? The un-Tolled truth of the silent invaders. Eur J Immunol 34:1783–1788. [DOI] [PubMed] [Google Scholar]
- 18.Harlan DM, Karp CL, Matzinger P, Munn DH, Ransohoff RM, Metzger DW. 2002. Immunological concerns with bioengineering approaches. Ann NY Acad Sci 961:323–330. [DOI] [PubMed] [Google Scholar]
- 19.Mayer G 2006. http://pathmicro.med.sc.edu/ghaf-far/innate.htm. Innate (non-Specific) Immunity: University of South Carolina. [Google Scholar]
- 20.Zinkernagel RM. 2000. Localization dose and time of antigens determine immune reactivity. Semin Immunol 12:163–171; discussion 257–344. [DOI] [PubMed] [Google Scholar]
- 21.Vogel FR. 2000. Improving vaccine performance with adjuvants. Clin Infect Dis 30:S266–S270. [DOI] [PubMed] [Google Scholar]
- 22.Iwasaki A 2007. Mucosal dendritic cells. Annu Rev Immunol 25:381–418. [DOI] [PubMed] [Google Scholar]
- 23.Gogolak P, Rethi B, Hajas G, Rajnavolgyi E. 2003. Targeting dendritic cells for priming cellular immune responses. J Mol Recognit 16:299–317. [DOI] [PubMed] [Google Scholar]
- 24.Granucci F, Feau S, Zanoni I, Raimondi N, Pavelka N, Vizzardelli C, Ricciardi-Castagnoli P. 2004. The regulatory role of dendritic cells in the innate immune response. In: Kaufmann SHE, Medzhitov R, Gordon S, editors. The innate immune response to infection, 1st edition. Washington, DC: ASM Press. pp 95–109. [Google Scholar]
- 25.Niess JH, Reinecker HC. 2006. Dendritic cells in the recognition of intestinal microbiota. Cell Microbiol 8:558–564. [DOI] [PubMed] [Google Scholar]
- 26.Akira S 2001. Toll-like receptors and innate immunity. Adv Immunol 78:1–56. [DOI] [PubMed] [Google Scholar]
- 27.Flo TH, Ryan L, Latz E, Takeuchi O, Monks BG, Lien E, Halaas O, Akira S, Skjak-Braek G, Golenbock DT, Espevik T. 2002. Involvement of toll-like receptor (TLR) 2 and TLR4 in cell activation by mannuronic acid polymers. J Biol Chem 277:35489–35495. [DOI] [PubMed] [Google Scholar]
- 28.Santana MA, Rosenstein Y. 2003. What it takes to become an effector T cell: The process, the cells involved, and the mechanisms. J Cell Physiol 195:392–401. [DOI] [PubMed] [Google Scholar]
- 29.Janeway CA, Travers P, Walport M, Shlomchik MJ, editors. 2001. Immunobiology the immune system in health and disease, 5th edition. New York, NY: Garland Publishing. [Google Scholar]
- 30.Heath WR, Belz GT, Behrens GM, Smith CM, Forehan SP, Parish IA, Davey GM, Wilson NS, Carbone FR, Villadangos JA. 2004. Cross-presentation, dendritic cell subsets, and the generation of immunity to cellular antigens. Immunol Rev 199:9–26. [DOI] [PubMed] [Google Scholar]
- 31.Pulendran B, Ahmed R. 2006. Translating innate immunity into immunological memory: Implications for vaccine development. Cell 124:849–863. [DOI] [PubMed] [Google Scholar]
- 32.Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, Parente E, Fili L, Ferri S, Frosali F, Giudici F, Romagnani P, Parronchi P, Tonelli F, Maggi E, Romagnani S. 2007. Phenotypic and functional features of human Th17 cells. J Exp Med 204:1849–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Villadangos JA, Schnorrer P. 2007. Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo. Nat Rev Immunol 7:543–555. [DOI] [PubMed] [Google Scholar]
- 34.Rogers PR, Croft M. 2000. CD28, Ox-40, LFA-1, and CD4 modulation of Th1/Th2 differentiation is directly dependent on the dose of antigen. J Immunol 164:2955–2963. [DOI] [PubMed] [Google Scholar]
- 35.McNeela EA, Mills KH. 2001. Manipulating the immune system: Humoral versus cell-mediated immunity. Adv Drug Deliv Rev 51:43–54. [DOI] [PubMed] [Google Scholar]
- 36.Brewer JM, Pollock KGJ. 2004. Adjuvant-induced Th2 and Th-1 dominated immune responses. In: Kaufmann SHE, editor. Novel vaccination strategies, 1st edition. Weinheim: WILEY-VCH Verlag GmbH & Co. KGaA. pp 147–163. [Google Scholar]
- 37.Finkelman FD, Urban JFJ. 1992. Cytokines: Making the right choice. Parasitol Today 8:311–314. [DOI] [PubMed] [Google Scholar]
- 38.Woodland DL. 2004. Jump-starting the immune system: Prime-boosting comes of age. Trends Immunol 25:98–104. [DOI] [PubMed] [Google Scholar]
- 39.Sedlik C, Dériaud E, Leclerc C. 1997. Lack of Th1 or Th2 polarization of CD4+ T cell response induced by particulate antigen targeted to phagocytic cells. Int Immunol 9:91–103. [DOI] [PubMed] [Google Scholar]
- 40.Chatelain R, Mauze S, Coffman RL. 1999. Experimental Leishmania major infection in mice: Role of IL-10. Parasite Immunol 21:211–218. [DOI] [PubMed] [Google Scholar]
- 41.Brady MT, Mahon BP, Mills KHG. 1998. Pertussis infection and vaccination induces Th1 cells. Immunol Today 19:534. [DOI] [PubMed] [Google Scholar]
- 42.Rook GAW, Dheda K, Zumla A. 2005. Do successful tuberculosis vaccines need to be immunoregulatory rather than merely Th1-boosting? Vaccine 23:2115–2120. [DOI] [PubMed] [Google Scholar]
- 43.Heath WR, Carbone FR. 2003. Immunology: Dangerous liaisons. Nature 425:460–461. [DOI] [PubMed] [Google Scholar]
- 44.Hebben M, Duquesne V, Cronier J, Rossi B, Aubert A. 2004. Modified vaccinia virus Ankara as a vaccine against feline coronavirus: Immunogenicity and efficacy. J Feline Med Surg 6:111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zinkernagel RM. 2003. Immunological memory and vaccines against acute cytopathic and noncytopathic infections. In: Bloom BR, Lambert P-H, editors. The vaccine book, 1st edition. San Diego, CA: Academic Press. pp 149–164. [Google Scholar]
- 46.Robbins JB, Schneeson R, Szu SC. 1996. Hypothesis: How licensed vaccines confer protective immunity. In: Cohen S, Shafferman A, editors. Novel strategies in design and production of vaccines, Series volume: 2 v.397, pp 169–182. [DOI] [PubMed] [Google Scholar]
- 47.http://www.cdc.gov/nip. The childhood and adolescent immunization schedule: Department of Health and Human Services. [Google Scholar]
- 48.http://www.fda.gov/cber/vaccine/licvacc.html. Vaccines Licensed for Immunization and Distributed in the U.S: U.S. FDA. [Google Scholar]
- 49.http://www.cdc.gov/nip. The Childhood and Adolescent Immunization Schedule: Department of Health and Human Services. [Google Scholar]
- 50.Ebensen T, Link C, Guzman CA. 2004. Classical bacterial vaccines. In: Kaufmann SH, editor. Novel vaccination strategies, Weinheim: Wiley-VCH. pp 221–242. [Google Scholar]
- 51.Roberts L 2004. Polio: The final assault? Science 303:1960–1971. [DOI] [PubMed] [Google Scholar]
- 52.Collins DM, Kawakami RP, Wards BJ, Campbell S, de Lisle GW. 2003. Vaccine and skin testing properties of two avirulent Mycobacterium bovis mutants with and without an additional esat-6 mutation. Tuberculosis (Edinb) 83:361–366. [DOI] [PubMed] [Google Scholar]
- 53.Breard E, Sailleau C, Coupier H, Mure-Ravaud K, Hammoumi S, Gicquel B, Hamblin C, Dubourget P, Zientara S. 2003. Comparison of genome segments 2, 7 and 10 of bluetongue viruses serotype 2 for differentiation between field isolates and the vaccine strain. Vet Res 34:777–789. [DOI] [PubMed] [Google Scholar]
- 54.Bramwell VW, Perrie Y. 2006. Particulate delivery systems for vaccines: What can we expect? J Pharm Pharmacol 58:717–728. [DOI] [PubMed] [Google Scholar]
- 55.De Quadros CA. 2003. Polio. In: Bloom BR, Lambert P-H, editors. The vaccine book. San Diego: Academic Press. pp 189–196. [Google Scholar]
- 56.Plotkin SA. 2003. Disease states and vaccination: Selected cases—Introduction. In: Bloom BR, Lambert P-H, editors. The vaccine book, 1st edition. San Diego, CA: Academic Press. pp 179–188. [Google Scholar]
- 57.Sato Y, Sato H. 1999. Development of acellular pertussis vaccines. Biologicals 27:61–69. [DOI] [PubMed] [Google Scholar]
- 58.Sidey FM, Furman BL, Wardlaw AC. 1989. Effect of hyperreactivity to endotoxin on the toxicity of pertussis vaccine and pertussis toxin in mice. Vaccine 7:237–241. [DOI] [PubMed] [Google Scholar]
- 59.Coenen F, Tolboom JTBM, Frijlink HW. 2006. Stability of influenza sub-unit vaccine: Does a couple of days outside the refrigerator matter? Vaccine 24:525–531. [DOI] [PubMed] [Google Scholar]
- 60.Wang C, Ge Q, Ting D, Nguyen D, Shen HR, Chen J, Eisen HN, Heller J, Langer R, Putnam D. 2004. Molecularly engineered poly(ortho ester) microspheres for enhanced delivery of DNA vaccines. Nat Mater 3:190–196. [DOI] [PubMed] [Google Scholar]
- 61.http://clinicaltrials.gov/ct/action/GetStudy. Clinical Trials: US National Institutes of Health. [Google Scholar]
- 62.Pastoret PP, Jones P. 2004. Veterinary vaccines for animal and public health. Dev Biol (Basel) 119:15–29. [PubMed] [Google Scholar]
- 63.Vogel FR, Powell MF. 1995. A compendium of vaccine adjuvants and excipients. In: Powell MF, Newman MJ, editors. Vaccine design—The subunit and adjuvant approach, 1st edition. New York: Plenum Press. pp 141–227. [DOI] [PubMed] [Google Scholar]
- 64.Singh M, O’Hagan D. 1999. Advances in vaccine adjuvants. Nat Biotechnol 17:1075–1081. [DOI] [PubMed] [Google Scholar]
- 65.Singh M, O’Hagan DT. 2003. Recent advances in veterinary vaccine adjuvants. Int J Parasitol 33:469–478. [DOI] [PubMed] [Google Scholar]
- 66.Cox E, Verdonck F, Vanrompay D, Goddeeris B. 2006. Adjuvants modulating mucosal immune responses or directing systemic responses towards the mucosa. Vet Res 37:511–539. [DOI] [PubMed] [Google Scholar]
- 67.Trujillo-Vargas CM, Mayer KD, Bickert T, Palmet-shofer A, Grunewald S, Ramirez-Pineda JR, Polte T, Hansen G, Wohlleben G, Erb KJ. 2005. Vaccinations with T-helper type 1 directing adjuvants have different suppressive effects on the development of allergen-induced T-helper type 2 responses. Clin Exp Allergy 35:1003–1013. [DOI] [PubMed] [Google Scholar]
- 68.Lutsiak ME, Kwon GS, Samuel J. 2006. Biodegradable nanoparticle delivery of a Th2-biased peptide for induction of Th1 immune responses. J Pharm Pharmacol 58:739–747. [DOI] [PubMed] [Google Scholar]
- 69.Petrovsky N 2006. Novel human polysaccharide adjuvants with dual Th1 and Th2 potentiating activity. Vaccine 24:S26–S29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ott G, Barchfeld GL, Chernoff D, Radhakrishnan R, van Hoogevest P, van Nest G. 1995. M F95: Design and evaluation of a safe and potent adjuvant for human vaccines. In: Powell MF, Newman MJ, editors. Vaccine design—The subunit and adjuvant approach, 1st edition. New York: Plenum. pp 277–294. [DOI] [PubMed] [Google Scholar]
- 71.http://www.gsk.com/reportsandpublications-other.htm. Reports and publications: GlaxoSmithKline. [Google Scholar]
- 72.Foster N, Hirst BH. 2005. Exploiting receptor biology for oral vaccination with biodegradable particulates. Adv Drug Deliv Rev 57:431–450. [DOI] [PubMed] [Google Scholar]
- 73.O’Hagan DT, Rappuoli R. 2004. Novel approaches to vaccine delivery. Pharm Res 21:1519–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.O’Hagan DT, Valiante NM. 2003. Recent advances in the discovery and delivery of vaccine adjuvants. Nat Rev Drug Discov 2:727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gavin AL, Hoebe K, Duong B, Ota T, Martin C, Beutler B, Nemazee D. 2006. Adjuvant-enhanced antibody responses in the absence of toll-like receptor signaling. Science 314:1936–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mata-Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TC. 2007. The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science 316:1628–1632. [DOI] [PubMed] [Google Scholar]
- 77.Albu DI, Jones-Trower A, Woron AM, Stellrecht K, Broder CC, Metzger DW. 2003. Intranasal vaccination using interleukin-12 and cholera toxin subunit B as adjuvants to enhance mucosal and systemic immunity to human immunodeficiency virus type 1 glycoproteins. J Virol 77:5589–5597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kemp JM, Kajihara S, Nagahara S, Sano A, Brandon M, Lofthouse S. 2002. Continous antigen delivery from controlled release implants induces significant and anamnestic immune responses. Vaccine 20:1089–1098. [DOI] [PubMed] [Google Scholar]
- 79.Newman KD, Sosnowski DL, Kwon GS, Samuel J. 1998. Delivery of MUC1 mucin peptide by Poly(d,l-lactic-co-glycolic acid) microspheres induces type 1 T helper immune responses. J Pharm Sci 87:1421–1427. [DOI] [PubMed] [Google Scholar]
- 80.Hunter RL. 2002. Overview of vaccine adjuvants: Present and future. Vaccine 20:s7–s12. [DOI] [PubMed] [Google Scholar]
- 81.Newman KD, Samuel J, Kwon G. 1998. Ovalbumin peptide encapsulated in Poly(d,l lactic-co-glycolic acid) microspheres is capable of inducing a T helper type 1 immune response. J Control Release 54:49–59. [DOI] [PubMed] [Google Scholar]
- 82.Cunningham AF, Khan M, Ball J, Toellner KM, Serre K, Mohr E, MacLennan IC. 2004. Responses to the soluble flagellar protein FliC are Th2, while those to FliC on Salmonella are Th1. Eur J Immunol 34:2986–2995. [DOI] [PubMed] [Google Scholar]
- 83.Yip HC, Karulin AY, Tary-Lehmann M, Hesse MD, Heinfried R, Heeger PS, Trezza RP, Heinzel FP, Forsthuber T, Lehmann PV. 1999. Adjuvant-guided type-1 and type-2 immunity: Infectious/noninfectious dichotomy defines the class of response. J Immunol 162:3942–3949. [PubMed] [Google Scholar]
- 84.Vasilakos JP, Smith RMA, Gibson SJ, Lindh JM, Pederson LK, Reiter MJ, Smith MH, Tomai MA. 2000. Adjuvant activities of immune response modifier R-848: Comparison with CpG ODN. Cell Immunol 204:64–74. [DOI] [PubMed] [Google Scholar]
- 85.Sacks T, Klinman DM. 1997. Long-term effect of primary immunization on subsequent immune responsiveness. Cell Immunol 177:162–168. [DOI] [PubMed] [Google Scholar]
- 86.Barth H, Berg PA, Klein R. 2003. Methods for the in vitro determination of an individual disposition towards Th1 or Th2-reactivity by the application of appropriate stimulatory antigens. Clin Exp Immunol 134:78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hanes J, Chiba M, Langer R. 1998. Degradation of porous poly(anhydride-co-imide) microspheres and implications for controlled macromolecule delivery. Biomaterials 19:163–172. [DOI] [PubMed] [Google Scholar]
- 88.Valtulini S, Macchi C, Ballanti P, Cherel Y, Laval A, Theaker JM, Bak M, Ferretti E, Morvan H. 2005. Aluminium hydroxide-induced granulomas in pigs. Vaccine 23:3999–4004. [DOI] [PubMed] [Google Scholar]
- 89.Aucouturier J, Deville S, Perret C, Vallee I, Boireau P. 2001. Assessment of efficacy and safety of various adjuvant formulations with a total soluble extract of Trichinella spiralis. Parasite 8:S126–S132. [DOI] [PubMed] [Google Scholar]
- 90.Straw BE, MacLachlan NJ, Corbett WT, Carter PB, Schey HM. 1985. Comparison of tissue reactions produced by Haemophilus pleuropneumoniae vaccines made with six different adjuvants in swine. Can J Comp Med 49:149–151. [PMC free article] [PubMed] [Google Scholar]
- 91.Hsu T, Hutto DL, Minion FC, Zuerner RL, Wannemuehler MJ. 2001. Cloning of a beta-hemolysin gene of Brachyspira (Serpulina) hyodysenteriae and its expression in Escherichia coli. Infect Immun 69:706–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.www.novartis-vaccines.com/press-room/news/20061128.shtml. Novartis submits first regulatory file for H5N1 adjuvanted influenza vaccine with European regulators for pre-pandemic avian influenza prevention: Novartis. [Google Scholar]
- 93.Podda A 2001. The adjuvanted influenza vaccines with novel adjuvants: Experience with the MF59-adjuvanted vaccine. Vaccine 19:2673–2680. [DOI] [PubMed] [Google Scholar]
- 94.Straw BE, Shin S, Callihan D, Petersen M. 1990. Antibody production and tissue irritation in swine vaccinated with Actinobacillus bacterins containing various adjuvants. J Am Vet Med Assoc 196:600–604. [PubMed] [Google Scholar]
- 95.Chiba M, Hanes J, Langer R. 1997. Controlled protein delivery from biodegradable tyrosine-containing poly(anhydride-co-imide) microspheres. Biomaterials 18:893–901. [DOI] [PubMed] [Google Scholar]
- 96.Gupta RK, Siber GR. 1994. Comparison of adjuvant activities of aluminum phosphate, calcium phosphate and stearyl tyrosine for tetanus toxoid. Biologicals 22:53–63. [DOI] [PubMed] [Google Scholar]
- 97.Gupta RK, Rost BE, Relyveld E, Siber GR. 1995. Adjuvant properties of aluminum and calcium compounds. In: Powell MF, Newman MJ, editors. Vaccine design—The subunit and adjuvant approach, 1st edition. New York: Plenum. pp 229–242. [DOI] [PubMed] [Google Scholar]
- 98.Lavigne MV, Castro M, Andino J, Manghi M. 2004. Alternative diphtheria, tetanus and whooping cough immunization schedule to evoke a Th2 tetanus and a Th1 pertussis immune response. Microbes Infect 6:481–484. [DOI] [PubMed] [Google Scholar]
- 99.Rimaniol AC, Gras G, Verdier F, Capel F, Grigoriev VB, Porcheray F, Sauzeat E, Fournier JG, Clayette P, Siegrist CA, Dormont D. 2004. Aluminum hydroxide adjuvant induces macrophage differentiation towards a specialized antigen-presenting cell type. Vaccine 22:3127–3135. [DOI] [PubMed] [Google Scholar]
- 100.Goldman M, Lambert P-H. 2004. Immunological safety of vaccines: Facts, hypotheses and allegations. In: Kaufmann SH, editor. Novel vaccination strategies. Weinhem: Wiley-VCH. pp 595–612. [Google Scholar]
- 101.Diwan M, Khar RK, Talwar GP. 2001. Tetanus toxoid loaded ‘preformed microspheres’ of cross-linked dextran. Vaccine 19:3853–3859. [DOI] [PubMed] [Google Scholar]
- 102.Kumar N, Langer R, Domb AJ. 2002. Polyanhydrides: An overview. Adv Drug Deliv Rev 54:889–910. [DOI] [PubMed] [Google Scholar]
- 103.Daemen T, de Mare A, Bungener L, de Jonge J, Huckriede A, Wilschut J. 2005. Virosomes for antigen and DNA delivery. Adv Drug Deliv Rev 57:451–463. [DOI] [PubMed] [Google Scholar]
- 104.Hilf N, Radsak M, Schild H. 2004. Host-derived adjuvants. In: Kaufmann SH, editor. Novel vaccination strategies. Weinheim: Wiley-VCH. pp 129–146. [Google Scholar]
- 105.Kool M, Soullie T, van Nimwegen M, Willart MA, Muskens F, Jung S, Hoogsteden HC, Hammad H, Lambrecht BN. 2008. Alum adjuvant boosts adaptive immunity by inducing uric acid and activating inflammatory dendritic cells. J Exp Med 205:869–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Seong SY, Matzinger P. 2004. Hydrophobicity: An ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol 4:469–478. [DOI] [PubMed] [Google Scholar]
- 107.Johansson J, Ledin A, Vernesson M, Lovgren-Bengtsson K, Hellman L. 2004. Identification of adjuvants that enhance the therapeutic antibody response to host IgE. Vaccine 22:2873–2880. [DOI] [PubMed] [Google Scholar]
- 108.Shibaki A, Katz SI. 2002. Induction of skewed Th1/Th2 T-cell differentiation via subcutaneous immunization with Freund’s adjuvant. Exp Dermatol 11:126–134. [DOI] [PubMed] [Google Scholar]
- 109.Guery JC, Galbiati F, Smiroldo S, Adorini L. 1996. Selective development of T helper (Th)2 cells induced by continuous administration of low dose soluble proteins to normal and beta(2)-microglobulin-deficient BALB/c mice. J Exp Med 183:485–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pearse MJ, Drane D. 2005. ISCOMATRIX adjuvant for antigen delivery. Adv Drug Deliv Rev 57:465–474. [DOI] [PubMed] [Google Scholar]
- 111.Fleck JD, Kauffmann C, Spilki F, Lencina CL, Roehe PM, Gosmann G. 2006. Adjuvant activity of Quillaja brasiliensis saponins on the immune responses to bovine herpesvirus type 1 in mice. Vaccine 24:7129–7134. [DOI] [PubMed] [Google Scholar]
- 112.Palatnik deSousa CB, Santos WR, Casas CP, Paraguai de Souza E, Tinoco LW, da Silva BP, Palatnik M, Parente JP. 2004. Protective vaccination against murine visceral leishmaniasis using aldehyde-containing Quillaja saponaria sapogenins. Vaccine 22:2470–2479. [DOI] [PubMed] [Google Scholar]
- 113.Sun HX. 2006. Haemolytic activities and adjuvant effect of Bupleurum chinense saponins on the immune responses to ovalbumin in mice. Vaccine 24:1324–1331. [DOI] [PubMed] [Google Scholar]
- 114.Sun HX, Pan HJ. 2006. Immunological adjuvant effect of Glycyrrhiza uralensis saponins on the immune responses to ovalbumin in mice. Vaccine 24:1914–1920. [DOI] [PubMed] [Google Scholar]
- 115.Kammer AR, Amacker M, Rasi S, Westerfeld N, Gremion C, Neuhaus D, Zurbriggen R. 2007. A new and versatile virosomal antigen delivery system to induce cellular and humoral immune responses. Vaccine 25:7065–7074. [DOI] [PubMed] [Google Scholar]
- 116.Aguilar JC, Rodriguez EG. 2007. Vaccine adjuvants revisited. Vaccine 25:3752–3762. [DOI] [PubMed] [Google Scholar]
- 117.Wakelin SJ, Sabroe I, Gregory CD, Poxton IR, Forsythe JL, Garden OJ, Howie SE. 2006. “Dirty little secrets”—Endotoxin contamination of recombinant proteins. Immunol Lett 106:1–7. [DOI] [PubMed] [Google Scholar]
- 118.Banchereau J, Steinman RM. 1998. Dendritic cells and the control of immunity. Nature 392:245–252. [DOI] [PubMed] [Google Scholar]
- 119.Singh M, Srivastava I. 2003. Advances in vaccine adjuvants for infectious diseases. Curr HIV Res 1:309–320. [DOI] [PubMed] [Google Scholar]
- 120.Akira S, Takeda K, Kaisho T. 2001. Toll-like receptors: Critical proteins linking innate and acquired immunity. Nat Immunol 2:675–680. [DOI] [PubMed] [Google Scholar]
- 121.van Duin D, Medzhitov R, Shaw AC. 2006. Triggering TLR signaling in vaccination. Trends Immunol 27:49–55. [DOI] [PubMed] [Google Scholar]
- 122.Ishii KJ, Akira S. 2007. Toll or toll-free adjuvant path toward the optimal vaccine development. J Clin Immunol 27:363–371. [DOI] [PubMed] [Google Scholar]
- 123.Borsutzky S, Kretschmer K, Becker PD, Muhlradt PF, Kirschning CJ, Weiss S, Guzman CA. 2005. The mucosal adjuvant macrophage-activating lipopeptide-2 directly stimulates B lymphocytes via the TLR2 without the need of accessory cells. J Immunol 174:6308–6313. [DOI] [PubMed] [Google Scholar]
- 124.Jackson DC, Lau YF, Le T, Suhrbier A, Deliyannis G, Cheers C, Smith C, Zeng W, Brown LE. 2004. A totally synthetic vaccine of generic structure that targets Toll-like receptor 2 on dendritic cells and promotes antibody or cytotoxic T cell responses. Proc Natl Acad Sci USA 101:15440–15445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wang B, Henao-Tamayo M, Harton M, Ordway D, Shanley C, Basaraba RJ, Orme IM. 2007. A Toll-like receptor-2-directed fusion protein vaccine against tuberculosis. Clin Vaccine Immunol 14:902–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Menon SA, Wannemuehler MJ, Mahairas GG, Minion FC. 2002. Mycobacterial ESAT-6 protein enhances mouse IFN-gamma responses to Mycoplasma hyopneumoniae P71 protein. J Interferon Cytokine Res 22:807–813. [DOI] [PubMed] [Google Scholar]
- 127.McMurray DN. 2003. Recent progress in the development and testing of vaccines against human tuberculosis. Int J Parasitol 33:547–554. [DOI] [PubMed] [Google Scholar]
- 128.Verthelyi D, Zeuner RA. 2003. Differential signaling by CpG DNA in DCs and B cells: Not just TLR9. Trends Immunol 24:519–522. [DOI] [PubMed] [Google Scholar]
- 129.Diwan M, Tafaghodi M, Samuel J. 2002. Enhancement of immune responses by co-delivery of a CpG oligodeoxynucleotide and tetanus toxoid in biodegradable nanospheres. J Control Release 85:247–262. [DOI] [PubMed] [Google Scholar]
- 130.Tafaghodi M, Sajadi Tabassi SA, Jaafari MR. 2006. Induction of systemic and mucosal immune responses by intranasal administration of alginate microspheres encapsulated with tetanus toxoid and CpG-ODN. Int J Pharm 319:37–43. [DOI] [PubMed] [Google Scholar]
- 131.Kazzaz J, Singh M, Ugozzoli M, Chesko J, Soenawan E, O’Hagan DT. 2006. Encapsulation of the immune potentiators MPL and RC529 in PLG microparticles enhances their potency. J Control Release 110:566–573. [DOI] [PubMed] [Google Scholar]
- 132.Diwan M, Elamanchili P, Lane H, Gainer A, Samuel J. 2003. Biodegradable nanoparticle mediated antigen delivery to human cord blood derived dendritic cells for induction of primary T cell responses. J Drug Target 11:495–507. [DOI] [PubMed] [Google Scholar]
- 133.Elkins KL, Rhinehart-Jones TR, Stibitz S, Conover JS, Klinman DM. 1999. Bacterial DNA containing CpG motifs stimulates lymphocyte-dependent protection of mice against lethal infection with intracellular bacteria. J Immunol 162:2291–2298. [PubMed] [Google Scholar]
- 134.Ishii KJ, Ito S, Tamura T, Hemmi H, Conover J, Ozato K, Akira S, Klinman DM. 2005. CpG-activated Thy1.2+ dendritic cells protect against lethal Listeria monocytogenes infection. Eur J Immunol 35:2397–2405. [DOI] [PubMed] [Google Scholar]
- 135.Miyagi K, Kawakami K, Kinjo Y, Uezu K, Kinjo T, Nakamura K, Saito A. 2005. CpG oligodeoxynucleotides promote the host protective response against infection with Cryptococcus neoformans through induction of interferon-gamma production by CD4+ T cells. Clin Exp Immunol 140:220–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Williamson ED, Bennett AM, Perkins SD, Beedham RJ, Miller J, Baillie LW. 2002. Co-immunisation with a plasmid DNA cocktail primes mice against anthrax and plague. Vaccine 20:2933–2941. [DOI] [PubMed] [Google Scholar]
- 137.Munoz JJ, Arai H, Bergman RK, Sadowski PL. 1981. Biological activities of crystalline pertussigen from Bordetella pertussis. Infect Immun 33:820–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Baqar S, Applebee LA, Bourgeois AL. 1995. Immunogenicity and protective efficacy of a prototype Campylobacter killed whole-cell vaccine in mice. Infect Immun 63:3731–3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Jackson RJ, Fujihashi K, Xu-Amano J, Kiyono H, Elson CO, McGhee JR. 1993. Optimizing oral vaccines: Induction of systemic and mucosal B-cell and antibody responses to tetanus toxoid by use of cholera toxin as an adjuvant. Infect Immun 61:4272–4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Elson CO. 1989. Cholera toxin and its subunits as potential oral adjuvants. Curr Top Microbiol Immunol 146:29–33. [DOI] [PubMed] [Google Scholar]
- 141.Freytag LC, Clements JD. 2005. Mucosal adjuvants. Vaccine 23:1804–1813. [DOI] [PubMed] [Google Scholar]
- 142.van Ginkel FW, Jackson RJ, Yoshino N, Hagiwara Y, Metzger DJ, Connell TD, Vu HL, Martin M, Fujihashi K, McGhee JR. 2005. Enterotoxin-based mucosal adjuvants alter antigen trafficking and induce inflammatory responses in the nasal tract. Infect Immun 73:6892–6902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chabalgoity JA, Baz A, Rial A, Grille S. 2007. The relevance of cytokines for development of protective immunity and rational design of vaccines. Cytokine Growth Factor Rev 18:195–207. [DOI] [PubMed] [Google Scholar]
- 144.Lynch JM, Briles DE, Metzger DW. 2003. Increased protection against pneumococcal disease by mucosal administration of conjugate vaccine plus interleukin-12. Infect Immun 71:4780–4788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Griffin KF, Eyles JE, Spiers ID, Alpar HO, Williamson ED. 2002. Protection against plague following immunisation with microencapsulated V antigen is reduced by co-encapsulation with IFN-gamma or IL-4, but not IL-6. Vaccine 20:3650–3657. [DOI] [PubMed] [Google Scholar]
- 146.Cleland JL. 1999. Single-administration vaccines: Controlled-release technology to mimic repeated immunizations. Trends Biotechnol 17:25–29. [DOI] [PubMed] [Google Scholar]
- 147.Audran R, Peter K, Dannull J, Men Y, Scandella E, Groettrup M, Gander B, Corradin G. 2003. Encapsulation of peptides in biodegradable microspheres prolongs their MHC class-I presentation by dendritic cells and macrophages in vitro. Vaccine 21:1250–1255. [DOI] [PubMed] [Google Scholar]
- 148.Kipper MJ, Wilson JH, Wannemuehler MJ, Narasimhan B. 2006. Single dose vaccine based on biodegradable polyanhydride microspheres can modulate immune response mechanism. J Biomed Mater Res 76A:798–810. [DOI] [PubMed] [Google Scholar]
- 149.O’Brien CN, Guidry AJ, Douglass LW, Westhoff DC. 2001. Immunization with Staphylococcus aureus lysate incorporated into microspheres. J Dairy Sci 84:1791–1799. [DOI] [PubMed] [Google Scholar]
- 150.Singh M, Vajdy M, Gardner J, Briones M, O’Hagan D. 2001. Mucosal immunization with HIV-1 gag DNA on cationic microparticles prolongs gene expression and enhances local and systemic immunity. Vaccine 20:594–602. [DOI] [PubMed] [Google Scholar]
- 151.Singh M, Briones M, O’Hagan DT. 2001. A novel bioadhesive intranasal delivery system for inactivated influenza vaccines. J Control Release 70:267–276. [DOI] [PubMed] [Google Scholar]
- 152.Munoz PM, Estevan M, Marin CM, Jesus De Miguel M, Jesus Grillo M, Barberan M, Irache JM, Blasco JM, Gamazo C. 2006. Brucella outer membrane complex-loaded microparticles as a vaccine against Brucella ovis in rams. Vaccine 24:1897–1905. [DOI] [PubMed] [Google Scholar]
- 153.Hanes J, Cleland JL, Langer R. 1997. New advances in microsphere-based single-dose vaccines. Adv Drug Deliv Rev 28:97–119. [DOI] [PubMed] [Google Scholar]
- 154.Diwan M, Elamanchili P, Cao M, Samuel J. 2004. Dose sparing of CpG oligodeoxynucleotide vaccine adjuvants by nanoparticle delivery. Curr Drug Deliv 1:405–412. [DOI] [PubMed] [Google Scholar]
- 155.Chong CS, Cao M, Wong WW, Fischer KP, Addison WR, Kwon GS, Tyrrell DL, Samuel J. 2005. Enhancement of T helper type 1 immune responses against hepatitis B virus core antigen by PLGA nanoparticle vaccine delivery. J Control Release 102:85–99. [DOI] [PubMed] [Google Scholar]
- 156.Hamdy S, Elamanchili P, Alshamsan A, Molavi O, Satou T, Samuel J. 2007. Enhanced antigen-specific primary CD4+ and CD8+ responses by codelivery of ovalbumin and toll-like receptor ligand monophosphoryl lipid A in poly(D,L-lactic-co-glycolic acid) nanoparticles. J Biomed Mater Res A 81:652–662. [DOI] [PubMed] [Google Scholar]
- 157.Alonso MJ, Gupta RK, Min C, Siber GR, Langer R. 1994. Biodegradable microspheres as controlled-release tetanus toxoid delivery systems. Vaccine 12:299–306. [DOI] [PubMed] [Google Scholar]
- 158.O’Brien CN, Guidry AJ. 1996. Formulation of poly(DL-lactide-co-glycolide) microspheres and their ingestion by bovine leukocytes. J Dairy Sci 79:1954–1959. [DOI] [PubMed] [Google Scholar]
- 159.Jiang W, Gupta RK, Deshpande MC, Schwendeman SP. 2005. Biodegradable poly(lactic-co-glycolic acid) microparticles for injectable delivery of vaccine antigens. Adv Drug Deliv Rev 57:391–410. [DOI] [PubMed] [Google Scholar]
- 160.Gunatillake PA, Adhikari R. 2003. Biodegradable synthetic polymers for tissue engineering. Eur Cells Mater 5:1–16; discussion 16. [DOI] [PubMed] [Google Scholar]
- 161.Gupta RK, Chang AC, Siber GR. 1998. Biodegradable polymer microspheres as vaccine adjuvants and delivery systems. Dev Biol Stand 92:63–78. [PubMed] [Google Scholar]
- 162.Jiang W, Schwendeman SP. 2001. Stabilization of a model formalinized protein antigen encapsulated in poly(lactide-co-glycolide)-based microspheres. J Pharm Sci 90:1558–1569. [DOI] [PubMed] [Google Scholar]
- 163.Xing DK, Crane DT, Bolgiano B, Corbel MJ, Jones C, Sesardic D. 1996. Physicochemical and immunological studies on the stability of free and microsphere-encapsulated tetanus toxoid in vitro. Vaccine 14:1205–1213. [DOI] [PubMed] [Google Scholar]
- 164.Cui C, Stevens VC, Schwendeman SP. 2007. Injectable polymer microspheres enhance immunogenicity of a contraceptive peptide vaccine. Vaccine 25:500–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Leong KW, D’Amore P, Marletta M, Langer R. 1986. Bioerodible polyanhydrides as drug-carrier matrices. II. Biocompatibility and chemical reactivity. J Biomed Mater Res 20:51–64. [DOI] [PubMed] [Google Scholar]
- 166.Rosen HB, Chang J, Wnek GE, Linhardt RJ, Langer R. 1983. Bioerodible polyanhydrides for controlled drug delivery. Biomaterials 4:131–133. [DOI] [PubMed] [Google Scholar]
- 167.Domb AJ, Amselem S, Shah J, Maniar M. 1993. Polyanhydrides: Synthesis and characterization. Adv Polym Sci 107:94–141. [Google Scholar]
- 168.Seidel JO, Uhrich KE, Laurencin CT, Langer R. 1996. Erosion of poly(anhydride-co-imides): A preliminary mechanistic study. J Appl Polym Sci 62:1277–1283. [Google Scholar]
- 169.Shen E, Patel P, Uhrich K, Narasimhan B. 1999. Morphological characterization of erodible polymer carriers for drug release. Proc Int Symp Control Rel Bioact Mater 26:717–718. [Google Scholar]
- 170.Shen E, Pizsczek R, Dziadul B, Narasimhan B. 2001. Microphase separation in bioerodible copolymers for drug delivery. Biomaterials 22:201–210. [DOI] [PubMed] [Google Scholar]
- 171.Erdmann L, Macedo B, Uhrich KE. 2000. Degradable poly(anhydride ester) implants: Effects of localized salicylic acid release on bone. Biomaterals 21:2507–2512. [DOI] [PubMed] [Google Scholar]
- 172.Fu J, Fiegel J, Krauland E, Hanes J. 2002. New polymeric carriers for controlled drug delivery following inhalation or injection. Biomaterials 23:4425–4433. [DOI] [PubMed] [Google Scholar]
- 173.Mathiowitz E, Ron E, Mathiowitz G, Amato C, Langer R. 1990. Morphological characterization of bioerodible polymers. 1. crystallinity of polyanhydride copolymers. Macromolecules 23:3212–3218. [Google Scholar]
- 174.Torres MP, Determan A, Mallapragada SK, Narasimhan B. 2006. Polyanhydrides. In: Lee S, editor. Encyclopedia of chemical processing. New York: Marcel Dekker. pp 2247–2257. [Google Scholar]
- 175.Tamada J, Langer R. 1992. The development of polyanhydrides for drug delivery applications. J Biomater Sci Polym Ed 3:315–353. [DOI] [PubMed] [Google Scholar]
- 176.Determan AS, Trewyn BG, Lin VS, Nilsen-Hamilton M, Narasimhan B. 2004. Encapsulation, stabilization, and release of BSA-FITC from polyanhydride microspheres. J Control Release 100:97–109. [DOI] [PubMed] [Google Scholar]
- 177.Shu Q, Hillard MA, Bindon BM, Duan E, Xu Y, Bird SH, Rowe JB, Oddy VH, Gill HS. 2000. Effects of various adjuvants on efficacy of a vaccine against Streptococcus bovis and Lactobacillus spp in cattle. Am J Vet Res 61:839–843. [DOI] [PubMed] [Google Scholar]
- 178.Wedlock DN, Keen DL, McCarthy AR, Andersen P, Buddle BM. 2002. Effect of different adjuvants on the immune responses of cattle vaccinated with Mycobacterium tuberculosis culture filtrate proteins. Vet Immunol Immunopathol 86:79–88. [DOI] [PubMed] [Google Scholar]
- 179.Ogawa T, Asai Y, Sakamoto H, Yasuda K. 2006. Oral immunoadjuvant activity of Lactobacillus casei subsp. casei in dextran-fed layer chickens. Br J Nutr 95:430–434. [DOI] [PubMed] [Google Scholar]
- 180.Baudner BC, Giuliani MM, Verhoef JC, Rappuoli R, Junginger HE, Giudice GD. 2003. The concomitant use of the LTK63 mucosal adjuvant and of chitosan-based delivery system enhances the immunogenicity and efficacy of intranasally administered vaccines. Vaccine 21:3837–3844. [DOI] [PubMed] [Google Scholar]
- 181.Illum L, Farraj NF, Davis SS. 1994. Chitosan as a novel nasal delivery system for peptide drugs. Pharm Res 11:1186–1189. [DOI] [PubMed] [Google Scholar]
- 182.Alpar HO, Somavarapu S, Atuah KN, Bramwell VW. 2005. Biodegradable mucoadhesive particulates for nasal and pulmonary antigen and DNA delivery. Adv Drug Deliv Rev 57:411–430. [DOI] [PubMed] [Google Scholar]
- 183.Boonyo W, Junginger HE, Waranuch N, Polnok A, Pitaksuteepong T. 2007. Chitosan and trimethyl chitosan chloride (TMC) as adjuvants for inducing immune responses to ovalbumin in mice following nasal administration. J Control Release 121:168–175. [DOI] [PubMed] [Google Scholar]
- 184.Singh M, O’Hagan D. 1998. The preparation and characterization of polymeric antigen delivery systems for oral administration. Adv Drug Deliv Rev 34:285–304. [DOI] [PubMed] [Google Scholar]
- 185.Heritage PL, Loomes LM, Jianxiong J, Brook MA, Underdown BJ, McDermott MB. 1996. Novel polymer-grafted starch microparticles for mucosal delivery of vaccines. Immunology 88:162–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Stertman L, Strindelius L, Sjoholm I. 2004. Starch microparticles as an adjuvant in immunisation: Effect of route of administration on the immune response in mice. Vaccine 22:2863–2872. [DOI] [PubMed] [Google Scholar]
- 187.Vassilev VP, DeFife KM, Landis GC, Mendy J, Minev BR, Cantwell MJ, Carpenter KW, Turnell WG. 2005. Inducing an immune response using a novel poly(ester-amide) co-polymer delivery platform. Drug Deliv Technol 5:54–59. [Google Scholar]
- 188.Greenland JR, Letvin NL. 2007. Chemical adjuvants for plasmid DNA vaccines. Vaccine 25:3731–3741. [DOI] [PubMed] [Google Scholar]
- 189.Alpar HO, Ozsoy Y, Bowen J, Eyles JE, Conway BR, Williamson ED. 1997. Potential of particulate carriers for the mucosal delivery of DNA vaccines. Biochem Soc Trans 25:337S. [DOI] [PubMed] [Google Scholar]
- 190.Petrovsky N, Aguilar JC. 2004. Vaccine adjuvants: Current state and future trends. Immunol Cell Biol 82:488–496. [DOI] [PubMed] [Google Scholar]
- 191.Newman KD, Elamanchili P, Kwon GS, Samuel J. 2002. Uptake of poly(D,L-lactic-co-glycolic acid) microspheres by antigen-presenting cells in vivo. J Biomed Mater Res 60:480–486. [DOI] [PubMed] [Google Scholar]
- 192.Kim W, Lee W, Ryoo J, Kim S, Kim J, Youn J, Min S, Bae E, Hwang S, Park S, Cho C, Park J, Kim H. 2002. Suppression of collagen-induced arthritis by single administration of poly(lactic-co-glycolic acid) nanoparticles entrapping type II collagen: A novel treatment strategy for induction of oral tolerance. Arthritis Rheum 46:1109–1120. [DOI] [PubMed] [Google Scholar]
- 193.Conway MA, Madrigal-Estebas L, McClean S, Brayden DJ, Mills KH. 2001. Protection against Bordetella pertussis infection following parenteral or oral immunization with antigens entrapped in biodegradable particles: Effect of formulation and route of immunization on induction of Th1 and Th2 cells. Vaccine 19:1940–1950. [DOI] [PubMed] [Google Scholar]
- 194.Gupta RK, Alroy J, Alonso MJ, Langer R, Siber GR. 1997. Chronic local tissue reactions, long-term immunogenicity and immunologic priming of mice and guinea pigs to tetanus toxoid encapsulated in biodegradable polymer microspheres composed of poly lactide-co-glycolide polymers. Vaccine 15:1716–1723. [DOI] [PubMed] [Google Scholar]
- 195.Evans JT, Ward JR, Kern J, Johnson ME. 2004. A single vaccination with protein-microspheres elicits a strong CD8 T-cell-mediated immune response against Mycobacterium tuberculosis antigen Mtb8.4. Vaccine 22:1964–1972. [DOI] [PubMed] [Google Scholar]
- 196.Men Y, Thomasin C, Merkle HP, Gander B, Corradin G. 1995. A single administration of tetanus toxoid in biodegradable microspheres elicits T cell and antibody responses similar or superior to those obtained with aluminum hydroxide. Vaccine 13:683–689. [DOI] [PubMed] [Google Scholar]
- 197.Raghuvanshi RS, Katare YK, Lalwani K, Ali MM, Singh O, Panda AK. 2002. Improved immune response from biodegradable polymer particles entrapping tetanus toxoid by use of different immunization protocol and adjuvants. Int J Pharm 245:109–121. [DOI] [PubMed] [Google Scholar]
- 198.Peyre M, Sesardic D, Merkle HP, Gander B, Johansen P. 2003. An experimental divalent vaccine based on biodegradable microspheres induces protective immunity against tetanus and diphtheria. J Pharm Sci 92:957–966. [DOI] [PubMed] [Google Scholar]
- 199.Walker KB, Xing DK, Sesardic D, Corbel MJ. 1998. Modulation of the immune response to tetanus toxoid by polylactide-polyglycolide microspheres. Dev Biol Stand 92:259–267. [PubMed] [Google Scholar]
- 200.Brannon-Peppas L, Vert M. 2000. Polylactic and polyglycolic acids as drug delivery carriers. In: Brannon-Peppas L, Klibanov AM, Langer R, Mikos AG, Peppas NA, Trantolo DJ, Yaszemski MJ, Wnek GE, editors. Handbook of pharmaceutical controlled release technology. New York: Marcel Dekker. pp 99–130. [Google Scholar]
- 201.Moore A, McGuirk P, Adams S, Jones WC, Paul McGee J, O’Hagan DT, Mills KHG. 1995. Immunization with a soluble recombinant HIV protein entrapped in biodegradable microparticles induces HIV-specific CD8+ cytotoxic T lymphocytes and CD4+ Th1 cells. Vaccine 13:1741–1749. [DOI] [PubMed] [Google Scholar]
- 202.Mata E, Carcaboso AM, Hernandez RM, Igartua M, Corradin G, Pedraz JL. 2007. Adjuvant activity of polymer microparticles and Montanide ISA 720 on immune responses to Plasmodium falciparum MSP2 long synthetic peptides in mice. Vaccine 25:877–885. [DOI] [PubMed] [Google Scholar]
- 203.Gupta RK, Singh M, O’Hagan DT. 1998. Poly(lactide-co-glycolide) microparticles for the development of single-dose controlled-release vaccines. Adv Drug Deliv Rev 32:225–246. [PubMed] [Google Scholar]
- 204.O’Brien CN, Guidry AJ. 1996. Formulation of poly(DL-lactide-co-glycolide) microspheres and their ingestion by bovine leukocytes. J Dairy Sci 79:1954–1959. [DOI] [PubMed] [Google Scholar]
- 205.Waeckerle-Men Y, Scandella E, Allmen EU-v, Ludewig B, Gillessen S, Merkle HP, Gander B, Groettrup M. 2004. Phenotype and functional analysis of human monocyte-derived dendritic cells loaded with biodegradable poly(lactide-co-glycolide) microspheres for immunotherapy. J Immunol Methods 287:109–124. [DOI] [PubMed] [Google Scholar]
- 206.Lacasse FX, Filion MC, Phillips NC, Escher E, McMullen JN, Hildgen P. 1998. Influence of surface properties at biodegradable microsphere surfaces: Effects on plasma protein adsorption and phagocytosis. Pharm Res 15:312–317. [DOI] [PubMed] [Google Scholar]
- 207.Pitaksuteepong T, Davies N, Baird M, Rades T. 2004. Uptake of antigen encapsulated in polyethylcyanoacrylate nanoparticles by D1-dendritic cells. Pharmazie 59:134–142. [PubMed] [Google Scholar]
- 208.Peyre M, Fleck R, Hockley D, Gander B, Sesardic D. 2004. In vivo uptake of an experimental microencapsulated diptheria vaccine following subcutaneous immunization. Vaccine 22:2430–2437. [DOI] [PubMed] [Google Scholar]
- 209.Raghuvanshi RS, Katare YK, Lalwani K, Ali MM, Singh O, Panda AK. 2002. Improved immune response from biodegradable polymer particles entrapping tetanus toxoid by use of different immunization protocol and adjuvants. Int J Pharm 245:109–121. [DOI] [PubMed] [Google Scholar]
- 210.des Rieux A, Fievez V, Garinot M, Schneider Y-J, Preat V. 2006. Nanoparticles as potential oral delivery systems of proteins and vaccines: A mechanistic approach. J Control Release 116:1–27. [DOI] [PubMed] [Google Scholar]
- 211.Leong KW, D’Amore P, Marletta M, Langer R. 1986. Bioerodible polyanhydrides as drug-carrier matrices. II. Biocompatibility and chemical reactivity. J Biomed Mater Res 20:51–64. [DOI] [PubMed] [Google Scholar]
- 212.Katti DS, Lakshmi S, Langer R, Laurencin CT. 2002. Toxicity, biodegradation and elimination of polyanhydrides. Adv Drug Deliv Rev 54:933–961. [DOI] [PubMed] [Google Scholar]
- 213.Kumar N, Langer RS, Domb AJ. 2002. Polyanhyrides: An overview. Adv Drug Deliv Rev 54:889–910. [DOI] [PubMed] [Google Scholar]
- 214.Kipper MJ, Shen E, Determan A, Narasimhan B. 2002. Design of an injectable system based on bioerodible polyanhydride microspheres for sustained drug delivery. Biomaterials 23:4405–4412. [DOI] [PubMed] [Google Scholar]
- 215.Shen E, Kipper MJ, Dziadul B, Lim M-K, Narasimhan B. 2002. Mechanistic relationships between polymer microstructure and drug release kinetics in bioerodible polyanhydrides. J Control Release 82:115–125. [DOI] [PubMed] [Google Scholar]
- 216.Schwendeman SP, Costantino HR, Gupta RK, Langer R. 1997. Peptide, protein, and vaccine delivery from implantable polymeric systems. In: Park K, editor. Controlled drug delivery: Challenges and strategies. Washington, DC: ACS. pp 229–267. [Google Scholar]
- 217.Torres MP, Determan AS, Anderson GL, Mallapragada SK, Narasimhan B. 2007. Amphiphilic polyanhydrides for protein stabilization and release. Biomaterials 28:108–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Tabata Y, Gutta S, Langer R. 1993. Controlled delivery systems for proteins using polyanhydride microspheres. Pharm Res 10:487–496. [DOI] [PubMed] [Google Scholar]
- 219.Determan AS, Wilson JH, Kipper MJ, Wannemuehler MJ, Narasimhan B. 2006. Protein stability in the presence of polymer degradation products: Consequences for controlled release formulations. Biomaterials 27:3312–3320. [DOI] [PubMed] [Google Scholar]
- 220.Ron E, Turek T, Mathiowitz E, Chasin M, Hageman M, Langer R. 1993. Controlled release of polypeptides from polyanhydrides. Proc Natl Acad Sci USA 90:4176–4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Vogel BM, Mallapragada SK. 2005. Synthesis of novel biodegradable polyanhydrides containing aromatic and glycol functionality for tailoring of hydrophilicity in controlled drug delivery devices. Biomaterials 26:721–728. [DOI] [PubMed] [Google Scholar]
- 222.Torres MP, Vogel BM, Narasimhan B, Mallapragada SK. 2006. Synthesis and Characterization of Novel Polyanhydrides with Tailored Erosion Mechanisms. J Biomed Mater Res 76A:102–110. [DOI] [PubMed] [Google Scholar]
- 223.Lopac SK, Torres MP, Wilson-Welder JH, Wannemuehler MJ, Narasimhan B. 2008. Effect of polymer chemistry and fabrication method on protein release and stability from polyanhydride microspheres. J Biomed Mater Res Part B Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Ochoa J, Irache JM, Tamayo I, Walz A, DelVecchio VG, Gamazo C. 2007. Protective immunity of biodegradable nanoparticle-based vaccine against an experimental challenge with Salmonella enteritidis in mice. Vaccine 25:4410–4419. [DOI] [PubMed] [Google Scholar]
- 225.Estevan M, Irache JM, Grillo MJ, Blasco JM, Gamazo C. 2006. Encapsulation of antigenic extracts of Salmonella enterica serovar: Abortusovis into polymeric systems and efficacy as vaccines in mice. Vet Microbiol 118:124–132. [DOI] [PubMed] [Google Scholar]
- 226.Hanes J, Chiba M, Langer R. 1998. Degradation of porous poly(anhydride-co-imide) microspheres and implications for controlled macromolecule delivery. Biomaterials 19:163–172. [DOI] [PubMed] [Google Scholar]
- 227.Qiu LY, Zhu KJ. 2001. Design of a core-shelled polymer cylinder for potential programmable drug delivery. Int J Pharm 219:151–160. [DOI] [PubMed] [Google Scholar]
- 228.Mathiowitz E, Jacob JS, Jong YS, Carino GP, Chickering DE, Chaturvedi P, Santos CA, Vijayaraghavan K, Montgomery S, Bassett M, Morrell C. 1997. Biologically erodable microspheres as potential oral drug delivery systems. Nature 386:410–414. [DOI] [PubMed] [Google Scholar]
- 229.Diwan M, Khar RK, Talwar GP. 2001. Tetanus toxoid loaded ‘preformed microspheres’ of cross-linked dextran. Vaccine 19:3853–3859. [DOI] [PubMed] [Google Scholar]
- 230.Amidi M, Pellikaan HC, Hirschberg H, de Boer AH, Crommelin DJA, Hennink WE, Kersten G, Jiskoot W. 2007. Diphtheria toxoid-containing microparticulate powder formulations for pulmonary vaccination: Preparation, characterization and evaluation in guinea pigs. Vaccine 25:6818–6829. [DOI] [PubMed] [Google Scholar]
- 231.Rydell N, Stertman L, Stalenheim G, Sjoholm I. 2006. Use of an oral diphtheria vaccine in human. Vaccine 24:5928–5930. [DOI] [PubMed] [Google Scholar]
- 232.Bowersock TL, Hogenesch H, Suckow M, Porter RE, Jackson R, Park H, Park K. 1996. Oral vaccination with alginate microsphere systems. J Control Release 39:209–220. [Google Scholar]
- 233.Babensee JE, Paranjpe A. 2005. Differential levels of dendritic cell maturation on different biomaterials used in combination products. J Biomed Mater Res 74:503–510. [DOI] [PubMed] [Google Scholar]
- 234.Duncan JD, Wang PX, Harrington CM, Schafer DP, Matsuoka Y, Mestecky JF, Compans RW, Novak MJ. 1996. Comparative analysis of oral delivery systems for live rotavirus vaccines. J Control Release 41:237–247. [Google Scholar]
- 235.Tsitlanadze G, Kviria T, Katsarava R. 2004. In vitro enzymatic biodegradation of amino acid based poly(ester amide)s biomaterials. J Mater Sci: Mat Med 15:185–190. [DOI] [PubMed] [Google Scholar]
- 236.Tsitlanadze G, Machaidze M, Kviria T, Djavakhishvili N, Chu CC, Katsarava R. 2004. Biodegradation of amino-acid-based poly(ester amide)s: In vitro weight loss and preliminary in vivo studies. J Biomater Sci Polym Ed 15:1–24. [DOI] [PubMed] [Google Scholar]
- 237.Reddy ST, Rehor A, Schmoekel HG, Hubbell JA, Swartz MA. 2006. In vivo targeting of dendritic cells in lymph nodes with poly(propylene sulfide) nanoparticles. J Control Release 112:26–34. [DOI] [PubMed] [Google Scholar]
- 238.Reddy ST, Swartz MA, Hubbell JA. 2006. Targeting dendritic cells with biomaterials: Developing the next generation of vaccines. Trends Immunol 27:573–579. [DOI] [PubMed] [Google Scholar]
- 239.Edelman R, Tacket CO. 1990. Adjuvants. Int Rev Immunol 7:51–66. [DOI] [PubMed] [Google Scholar]
- 240.O’Hagan DT, Singh M, Gupta RK. 1998. Poly (lactide-co-glycolide) microparticles for the development of single-dose controlled-release vaccines. Adv Drug Deliv Rev 32:225–246. [PubMed] [Google Scholar]
- 241.Schijns VE. 2002. Antigen delivery systems and immunostimulation. Vet Immunol Immunopathol 87:195–198. [DOI] [PubMed] [Google Scholar]
- 242.Salkowski CA, Detore GR, Vogel SN. 1997. Lipopolysaccharide and monophosphoryl lipid A differentially regulate interleukin-12, gamma interferon, and interleukin-10 mRNA production in murine macrophages. Infect Immun 65:3239–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.O’Hagan DT, MacKichan ML, Singh M. 2001. Recent developments in adjuvants for vaccines against infectious diseases. Biomol Eng 18:69–85. [DOI] [PubMed] [Google Scholar]
- 244.Nohria A, Rubin RH. 1994. Cytokines as potential vaccine adjuvants. Biotherapy 7:261–269. [DOI] [PubMed] [Google Scholar]
- 245.Jarrossay D, Napolitani G, Colonna M, Sallusto F, Lanzavecchia A. 2001. Specialization and complementarity in microbial molecule recognition by human myeloid and plasmacytoid dendritic cells. Eur J Immunol 31:3388–3393. [DOI] [PubMed] [Google Scholar]
- 246.Shortman K, Liu YJ. 2002. Mouse and human dendritic cell subtypes. Nat Rev Immunol 2:151–161. [DOI] [PubMed] [Google Scholar]
- 247.Brown F 1993. Review of accidents caused by incomplete inactivation of viruses. Dev Biol Stand 81:103–107. [PubMed] [Google Scholar]
- 248.Brewer JM. 2006. (How) do aluminium adjuvants work? Immunol Lett 102:10–15. [DOI] [PubMed] [Google Scholar]
- 249.Magyar T, Donko T, Kovacs F. 2008. Atrophic rhinitis vaccine composition triggers different serological profiles that do not correlate with protection. Acta Vet Hung 56:27–40. [DOI] [PubMed] [Google Scholar]
- 250.Nara PL, Lin G. 2005. HIV-1: The confounding variables of virus neutralization. Current drug targets. Infect Disord 5:157–170. [DOI] [PubMed] [Google Scholar]
- 251.Kemper C, Atkinson JP. 2007. T-cell regulation: With complements from innate immunity. Nat Rev Immunol 7:9–18. [DOI] [PubMed] [Google Scholar]
- 252.Stetson DB, Medzhitov R. 2007. T helper 17 cells get the NOD. Immunity 27:546–548. [DOI] [PubMed] [Google Scholar]
- 253.http://www.nap.edu/catalog/11471.html. Treating infectious diseases in a microbial world: Report of two workshops on novel antimicrobial therapeutics: National Academies Press. [PubMed] [Google Scholar]
- 254.Mosmann TR, Coffman RL. 1989. TH1 and TH2 cells: Different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol 7:145–173. [DOI] [PubMed] [Google Scholar]
- 255.Finkelman FD, Holmes J, Katona IM, Urban JF, Beckman MP, Park LS, Schooley KA, Coffman RL, Mosmann TR, Paul WE. 1990. Lymphokine control of in vivo immunoglobulin isotype selection. Annu Rev Immunol 8:303–333. [DOI] [PubMed] [Google Scholar]
- 256.Han M, Gao X, Su JZ, Nie S. 2001. Quantum-dot-tagged microbeads for multiplexed optical coding of biomolecules. Nat Biotechnol 19:631–635. [DOI] [PubMed] [Google Scholar]
- 257.Kim S, Lim YT, Soltesz EG, De Grand AM, Lee J, Nakayama A, Parker JA, Mihaljevic T, Laurence RG, Dor DM, Cohn LH, Bawendi MG, Frangioni JV. 2004. Near-infrared fluorescent type II quantum dots for sentinel lymph node mapping. Nat Biotechnol 22:93–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Delehanty JB, Medintz IL, Pons T, Brunel FM, Dawson PE, Mattoussi H. 2006. Self-assembled quantum dot-peptide bioconjugates for selective intracellular delivery. Bioconjug Chem 17:920–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.http://www.fda.gov/cber/vaccine/licvacc.html. Vaccines Licensed for Immunization in the US: US FDA. [Google Scholar]
- 260.http://apps.cfsph.iastate.edu/Vaccines/. Transboundary Veterinary Vaccine Directory: Iowa State University. [Google Scholar]
- 261.Beran J 2008. The importance of the second generation adjuvanted systems in “new” vaccines. Klin Mikrobiol Infekc Lek 14:5–12. [PubMed] [Google Scholar]