

Abstract
Breast cancer (BC) affects women globally; the majority of breast cancer-related mortalities are due to metastasis. Acquisition of a mesenchymal phenotype has been implicated in the progression of breast cancer cells to an invasive, metastatic state. Triple-negative breast cancer (TNBC) subtypes have high rates of metastases, recurrence, and have poorer prognoses compared to other BC types, partially due to lack of commonly targeted receptors. Kinases have diverse and pivotal functions in metastasis in TNBC, and discovery of new kinase targets for TNBC is warranted. We previously used a screening approach to identify intermediate-synthesis non-potent, non-selective small molecule inhibitors from the Published Kinase Inhibitor Set that reversed the mesenchymal phenotype in TNBC cells. Two of these inhibitors (GSK346294A, GSK448459A) are structurally similar, but have unique kinase activity profiles and exhibited differential biologic effects on TNBC cells, specifically on epithelial-to-mesenchymal transition (EMT). Here, we further interrogate these effects and compare activity of these inhibitors on transwell migration, gene (qRT-PCR) and protein (Western blot) expressions, and cancer stem cell-like behavior. We incorporated translational patient-derived xenograft models in these studies, and we focused on the lead inhibitor hit, GSK346294A, to demonstrate the utility of our comparative analysis as a screening modality to identify novel kinase targets and/or signaling pathways to pursue in TNBC. This study introduces a new method for discovering novel kinase targets that reverse the EMT phenotype; this screening approach can be applied to all cancer types and is not limited to breast cancer.
Keywords: triple negative breast cancer, metastasis, epithelial-to-mesenchymal transition, patient-derived xenograft, screen approach, small molecule inhibitor, therapeutic target
Introduction
Breast cancer, second only to lung cancer, remains a leading cause of cancer in women worldwide, despite profound advances in discovery of targeted therapeutics. Breast cancer alone accounts for 30% of all new cancer diagnoses in women [1]. Invasive and metastatic breast cancer is more difficult to manage and treat; metastatic tumors are genetically distinct from the primary tumors from which they were derived, emphasizing the need to discover novel therapeutic targets in invasive and metastatic breast cancer subtypes [2]. Breast cancers are categorized based on the expression (estrogen receptor (ER+), progesterone receptor (PR+)) or amplification (HER2/Neu amplified) of pharmacologically tractable receptors. Triple negative breast cancers (TNBCs) lack these targetable receptors, and the standard first-line therapy is systemic chemotherapy with potentially fatal side effects [3]. Discovery of novel therapeutic targets in TNBC is hindered by the diverse inter- and intratumoral cellular heterogeneity [4–6]. Furthermore, because TNBC is loosely defined as any breast cancer in which the cells lack ER, PR or HER2/Neu receptors, tumors in this class have a diverse variety of molecular signatures and protein expressions.
Aberrant protein phosphorylation is a key event in driving oncogenesis. Kinases regulate all aspects of cellular biology through phosphorylation and subsequent activation of diverse pathways including cell proliferation, apoptosis, cell migration and invasion in addition to other oncogenic signaling pathways [7]. The development of kinase-targeting inhibitors drastically improved the treatment of breast cancer; currently there are over 150 kinase-targeting drugs in clinical phase trials [7]. Although 37 small molecule kinase inhibitors have been clinically approved in other breast cancer subtypes, kinase inhibitors for TNBC are lacking [7, 8]. Despite promising therapeutic advances, small molecule inhibitors often fail in early clinical trial stages in TNBC subtypes [4, 9]. Discovery and characterization of novel targetable proteins and/or pathways in TNBCs is necessary.
Acquisition of mesenchymal characteristics in a process known as epithelial-mesenchymal transition (EMT) is a hallmark feature of TNBC cells and drives the metastatic transformation of this BC subtype [10–12]. One approach to therapeutic discovery research in TNBC is to identify metastatic drivers and proteins that regulate the EMT axis. Kinases that regulate specific metastatic processes, such as cytoskeletal rearrangement, increased expression of mesenchymal gene signatures, and increased expression and activity of proteins that modulate the tumor microenvironment, have been under investigation as novel therapeutic targets in TNBC [9, 13, 14]. The human kinome consists of 538 kinases; a significant number of these kinases have uncharacterized or understudied functions [12, 15, 16]. A program initiated by the NIH, Illuminating the Druggable Genome, further quantified this cohort: 31 kinases are understudied in that there is a lack of publications, small molecule inhibitors, and gene references to functionality in the literature. A larger portion of the human kinome [17, 18], 163 kinases, have functions characterized, but do not yet have small molecules or drugs that target them [18]. There are various approaches to identify novel kinase targets in cancer, including high-throughput phenotypic and proliferation screening, in silico modeling, and machine learning [8, 19]. In a preliminary screen, we employed intermediate synthesis, non-selective kinase inhibitors from the Published Kinase Inhibitor Set (PKIS) library and treated a panel of TNBC cell lines. Because our primary aim was to identify kinase targets that reverse the mesenchymal phenotype in TNBC, compound ‘hits’ were those that affected cell morphology, epithelial and mesenchymal gene expression changes, and transwell migration most significantly [20]. Two of the compound hits from this initial screen (GSK346294A, GSK448459A) were synthesized in the same medicinal campaign and are categorized as thiophene benzimidazole PLK1 inhibitors. However, because these intermediate synthesis inhibitors are non-selective and non-potent, they have affinity for a number of kinase targets beyond PLK1 [21]. While these compounds are structurally similar, they exerted different effects on TNBC biology, specifically with respect to reversal of the EMT phenotype. We proposed that the unique kinases inhibited by the pharmacologically similar kinases are responsible for the observed differential effects on EMT reversal. Identifying those kinases introduces new candidate targets or kinase-mediated signaling pathways that regulate the EMT axis to explore in TNBC.
For these studies, we used new preclinical models of TNBC patient-derived xenograft (PDX) tumors established by our laboratory that represent patients in the greater New Orleans community. The implementation of patient-derived xenograft models (PDXs) in therapeutic research has dramatically advanced preclinical therapeutic discovery research in oncology [22, 23]. While immortalized cell lines are limited to the study of therapies on a single cell system, PDX models facilitate the study of biological effects on the entire tumor system [24]. The diverse applications of PDX models include in vitro (both adherent and suspension culture systems), ex vivo (treatment of intact tumors) and in vivo experiments that are essential for preclinical therapeutic research. We have previously demonstrated the application of PDX models we established from local TNBC patients in therapeutic discovery [24]. In this study, we demonstrated the utility of this translational comparative analysis approach using non-selective inhibitors to identify novel signaling pathways and targets to pursue for TNBC.
Results
Lead PKIS compounds differentially affected cell proliferation and viability
Various biologic endpoints were used to evaluate effects of the non-selective inhibitor ‘hits’ from the medium-throughput screen on TNBC biology. First, cell proliferation effects of the PKIS compounds selected from the initial screen (GSK346294A, GSK448459A) were tested (Supplementary Figure 1). To evaluate proliferation, we utilized immunofluorescence staining for Ki-67, a transcription factor that is increased in response to protein synthesis and is a marker for proliferating cells [25]. The potent PLK inhibitor, BI2536, was included as a positive control [26], which selectively targets PLK1, PLK2 and PLK3. As expected, treatment of MDA-MB-231 cells with BI2536 significantly reduced the number of Ki-67 positive cells per high powered field, indicating suppression of cell proliferation. Treatment with the PKIS compounds did not suppress cell proliferation. In fact, treatment with GSK346294A increased the total number of cells expressing the Ki-67 marker, while GSK448459A decreased Ki-67 positive cells (Figure 1A–B). Next, to measure drug effects on TNBC cells we observed differential responses in total cells remaining in MDA-MB-231, BT-549 and our PDX-derived TU-BcX-4IC cells treated with the PKIS compounds using crystal violet staining. GSK448459A treatment resulted in increased number of cells remaining, while GSK346294A decreased the number of cells remaining compared to DMSO controls (Figure 1C). Finally, a dose-response study using crystal violet staining to evaluate number of cells remaining after treatment was performed. After 72 hours’ treatment, in MDA-MB-231 and BT-549 cells, GSK448459A (50 μM and 7.5 μM respectively) had higher IC-50 values compared to treatment with GSK346294A (50μM and 1mM respectively). In the TU-BcX-4IC cells, both compounds had similar dose-dependent profiles, with an IC-50 of 5mM (Figure 1D). Representative images from the dose-response experiments are shown in Supplementary Figure 2.
Figure 1. Differential effects of PKIS compounds on TNBC cell proliferation and viability.
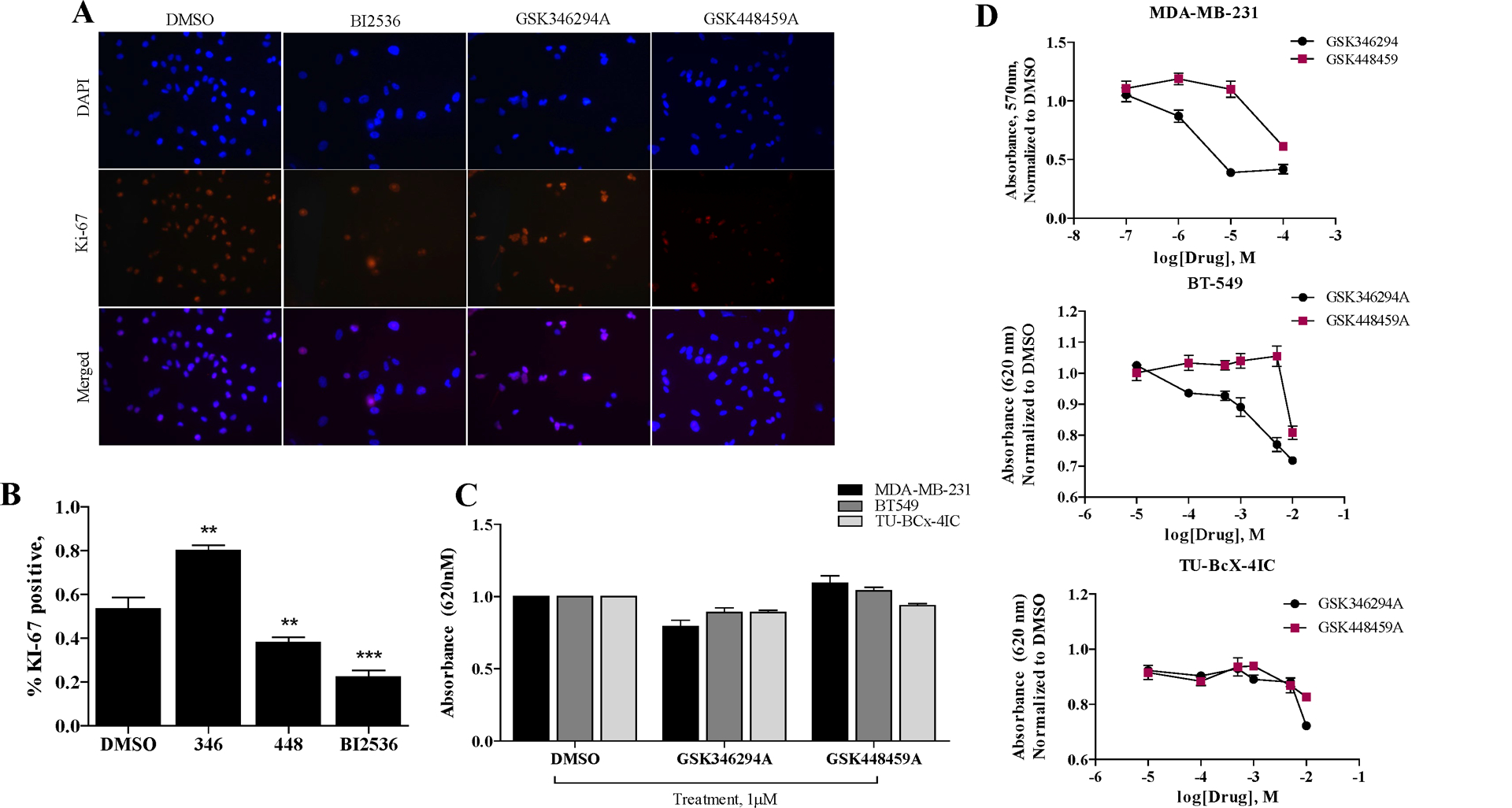
(A) Immunofluorescence staining of MDA-MB-231 cells with Ki-67 after 72 hours treatment with PKIS compounds (GSK346294A, GSK448459A) at 1μM or DMSO control. Red = Ki-67, Blue = DAPI nuclear stain. Images were taken at 200X magnification. (B) Quantification of cell proliferation data. Percent of Ki-67 positive cells compared to total DAPI positive cells per image is shown. Error bars represent S.E.M. and significance is defined as * p < 0.05, *** p < 0.001. Abbreviations for inhibitors are GSK346294A (346), GSK448459A (448)). (C) Cell viability of TNBC cells (MDA-MB-231, BT549, TU-BcX-4IC) in response to PKIS inhibitors treated at1 μM for 72 hours compared to DMSO control. Cells were fixed, stained with crystal violet, lysed, and absorbance was measured (620 nm excitation). (D) Cell viability dose-response of TNBC cells (MDA-MB-231, BT549, TU-BcX-4IC) in response to PKIS inhibitors normalized to DMSO control. Error bars represent S.E.M.
Lead PKIS compounds reverse mesenchymal morphology in TNBC cells
GSK346294A and GSK448459A were identified as lead compounds in our previously published phenotypic screen because they reversed the characteristic mesenchymal phenotype of TNBC cells. To further validate these observations, we repeated the cell morphology staining with crystal violet using additional TNBC cell lines BT549 and TU-BcX-4IC (Figure 2A). First, based on microscopic imaging of the stained cells, it was observed that the compounds selected from the PKIS screen reversed the mesenchymal morphology of TNBC cells to epithelial phenotypes. Mesenchymal cells were characterized as bipolar, spindly, fibroblast-like with minimal cell-cell contact; the epithelial phenotype had increased circularity and cell-cell contact with neighboring cells, as well as larger cytoplasm-to-nuclear ratios. Interestingly, we did not observe obvious morphologic changes in response to drug treatments in the BT-549 cell line, suggesting a cell-line specific effect. We observed a similar mesenchymal reversal after treatment with the PKIS compounds in another PDX-derived cell line, TU-BcX-49S (Supplementary Figure 3). These morphologic observations were quantified using immunofluorescence staining with phalloidin to highlight intermediate filaments (Figure 2B). Because it is difficult to evaluate overall morphology changes based on visual observation of a few select cells in representative images, we performed morphometric quantification of the cell shapes to quantify our observations. Based on various aspects of the imaged cells, we compared features that describe the morphologies of the cells including overall cell area, nuclear area fraction, and length-by-width aspect ratio. Larger cell areas, a smaller nuclear area fraction, and reduced length-by-width aspect ratios characterize an epithelial cell phenotype, and we have previously used these parameters to quantify morphology effects of drug treatments in vitro [14]. Although both PKIS compounds significantly increased the total areas of TU-BcX-4IC cells, out of the two inhibitors GSK346294A treatment resulted in the largest overall areas (Figure 2C). Neither inhibitor affected the aspect ratio of the cells (Figure 2D). GSK346294A most significantly reduced nuclear area fraction, or the ratio of the nuclear area to cell areas (Figure 2E). Overall, GSK346294A had the most dramatic effects on TNBC cells morphology and proliferation.
Figure 2. Cell morphology changes in TNBC cells induced by treatment with lead PKIS inhibitors.
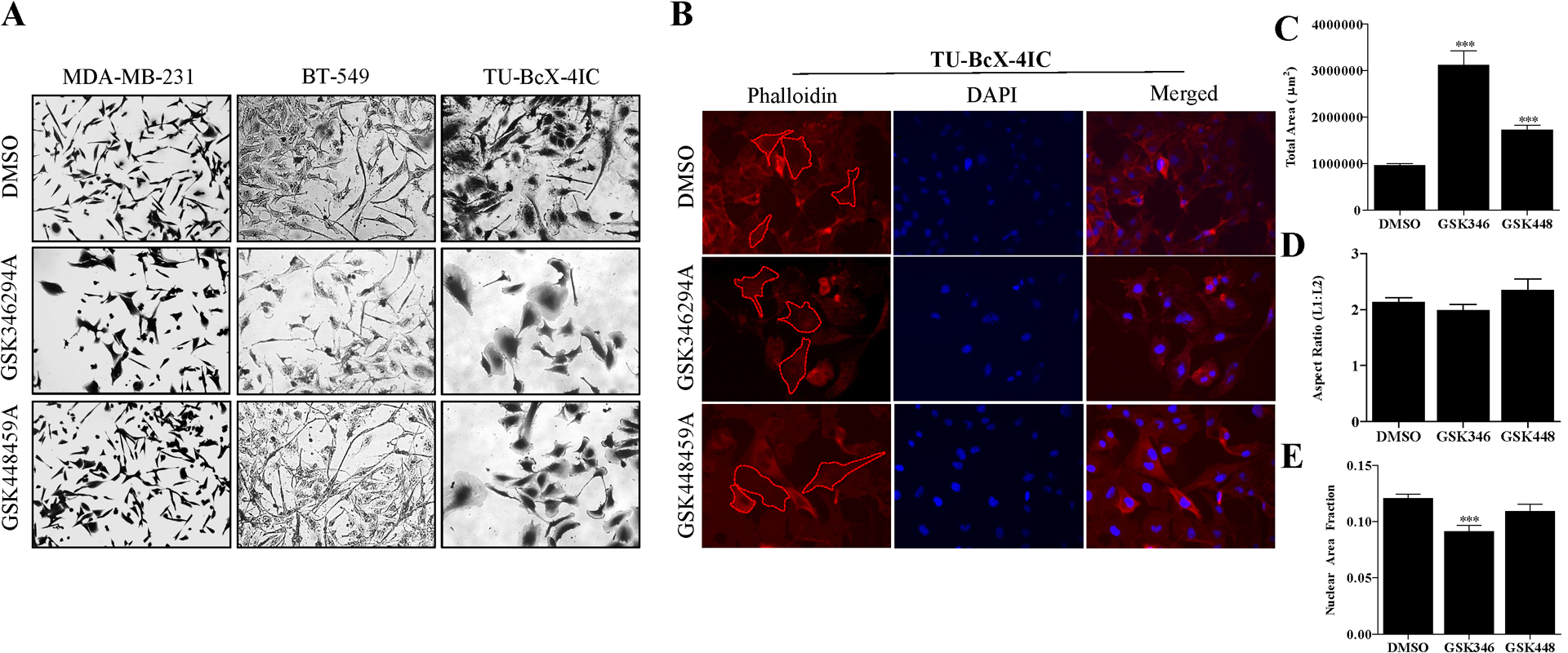
(A) Crystal violet staining of established TNBC cell lines (MDA-MB-231, BT-549) and the PDX-derived cell line TU-BcX-4IC with GSK346294A and GSK448459A compared to DMSO control (1 μM, 72 hours). Cells were fixed in glutaraldehyde before staining. Images were taken with brightfield microscopy and representative images are shown at 100x magnification. (B) Immunofluorescence staining with phalloidin of TU-BcX-4IC cells treated with DMSO, GSK346294A and GSK448459A (1 μM). After 72 hours, cells were fixed stained with phalloidin (red) or DAPI nuclear stain (blue). Red dashed shapes outline cells to demonstrate examples of individual cells that were selected for morphometric quantification. Quantification of post-treatment cell morphologies based on (C) total area, (D) aspect ratio and (E) nuclear area fraction. Area is quantified as length × width; aspect ratio is length divided by width, and nuclear area fraction is nuclear area:total cell area. * p < 0.05, *** p < 0.001.
Lead PKIS inhibitors suppress cellular migration and mesenchymal gene expression
Effects of the lead PKIS inhibitors on two additional aspects of the mesenchymal phenotype were then explored: mesenchymal gene expression and cell migration. BT549 and TU-BcX-4IC cells were pre-treated for 72 hours with PKIS compounds before viable cells were seeded on transwell migration chambers for 24 hours. Both PKIS compounds significantly suppressed migration of BT549 and TU-BcX-4IC cells, although GSK346294A and GSK448459A had the most suppressive effects on cell migration (Figure 3A, B). To compare expression changes of mesenchymal genes in TNBC cells after treatment with the PKIS compounds, we employed qRT-PCR. For these studies, drug effects were compared in MDA-MB-231, BT549 and TU-BcX-4IC cells, and normalized the data to both DMSO and actin controls. The mesenchymal genes analyzed included ZEB1, MMP9, cFOS, SNAI1, SLUG and TWIST, and the epithelial gene analyzed was CDH1. Consistent with our previously published findings, both PKIS compounds significantly increased CDH1 expression in MDA-MB-231 cells. In BT-549 cells, GSK346294A and GSK448459A significantly reduced CDH1 mRNA expression. CDH1 expression was not altered by the PKIS compounds in TU-BcX-4IC cells (Figure 3C). These findings contradict our hypothesis based on preliminary findings in a larger scale drug screen that the PKIS compounds promote an epithelial phenotype through CDH1 gene expression. These data demonstrate, however, that these findings are cell-line specific. When another TNBC PDX-derived cell line was analyzed, TU-BcX-49S, only GSK346294A increased CDH1 expression compared to DMSO controls (Supplementary Figure 4). Interestingly, GSK448459A significantly increased ZEB1 expression in MDA-MB-231 cells. Neither of the lead PKIS compounds affected ZEB1 expression in BT-549 cells, and GSK346294A and GSK448459A suppressed ZEB1 expression in TU-BcX-4IC cells albeit not significantly (Figure 3D). With respect to cFOS expression, both GSK346294A and GSK448459A had suppressive effects in all three TNBC cell lines, although not significantly (Supplementary Figure 5). Neither GSK346294A nor GSK448459A suppressed cFOS mRNA expression in TU-BcX-49S cells (Supplementary Figure 4). MMP9, a matrix gene upregulated in the mesenchymal phenotype, was also examined. GSK448459A significantly downregulated MMP9 expression in MDA-MB-231 cells and GSK346294A downregulated MMP9 expression, but not significantly. Neither GSK346294A nor GSK448459A affected MMP9 expression in BT-549 nor in TU-BcX-4IC (Figure 3E). Other transcriptions factors known to drive the mesenchymal phenotype, SNAI1, SLUG and TWIST, were also examined (Supplementary Figure 6). GSK448459A significantly decreased SNAI1 expression in MDA-MB-231 cells, while GSK346294A did not. In contrast, GSK448459A increased SNAI1 expression in BT-549 cells significantly, while GSK346294A had no significant effects on SNAI1 expression. None of the inhibitors significantly affected SLUG nor TWIST expression in TNBC cell lines, Together, these data demonstrate the pharmacologically similar PKIS compound had different effects on mesenchymal and epithelial gene expression changes in TNBC cells. The individual PKIS drugs affected different aspects of the EMT phenotype, and while one inhibitor can suppress some mesenchymal-associated genes, it had no effect on other genes that promote a mesenchymal phenotype through a different signaling pathway. These data indicate the unique kinases inhibited by the individual PKIS drugs affect the EMT phenotype through different signaling pathways.
Figure 3. Comparison of PKIS inhibitors effects on transwell cell migration and mesenchymal gene expression.
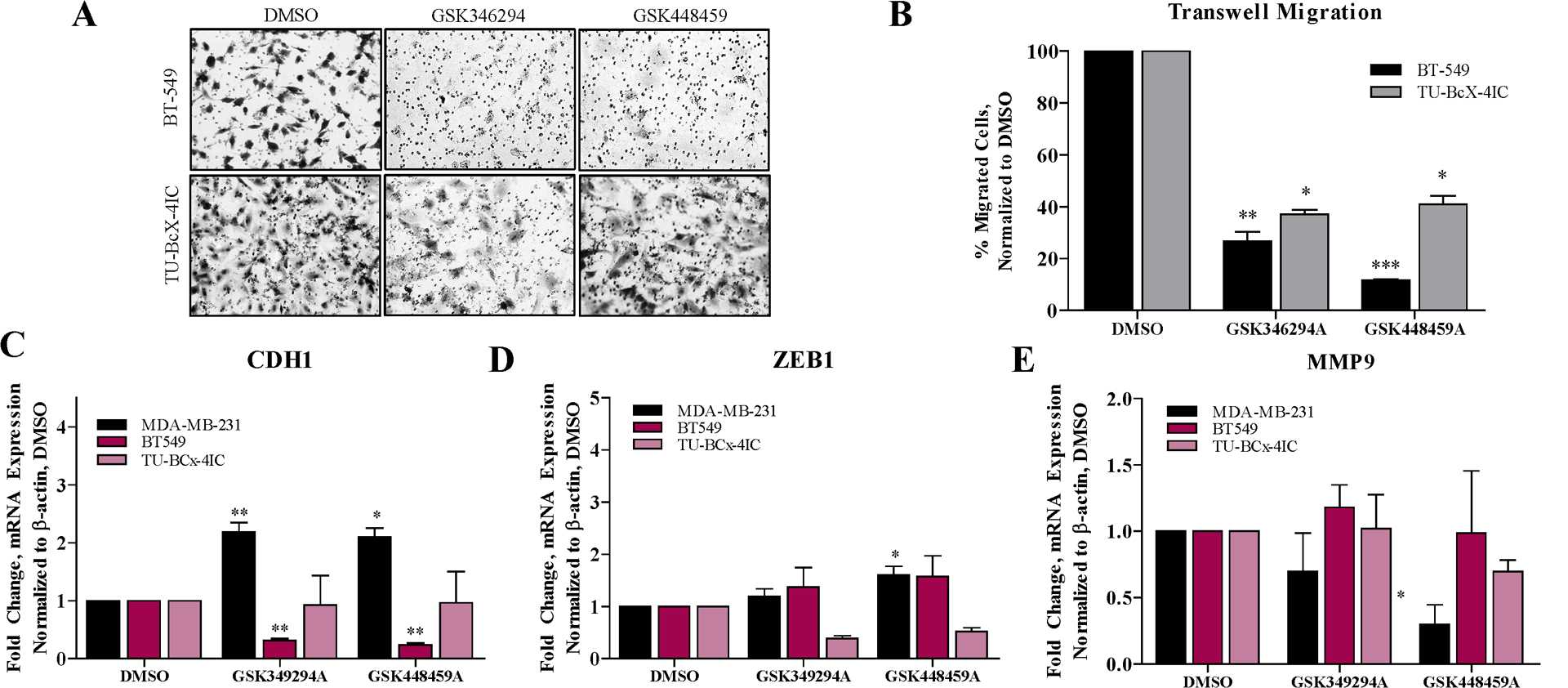
(A) Transwell migration assay of BT549 and TU-BcX-4IC cells pretreated with PKIS compounds (1 μM) or DMSO controls for 72 hours. Representative images of migrated cells fixed and stained with crystal violet were captured with brightfield microscopy. (B) Quantification of crystal violet stained membranes from transwell migration assays using Image J software. Counted cells in the treatment groups were normalized to DMSO controls for each cell line. (C) CDH1, (D) ZEB1 or(E) cFOS gene expression changes induced by 72 hours treatment with PKIS inhibitors (1 μM) or DMSO treatments in TNBC cells (MDA-MB-231, BT549, TU-BcX-4IC) were evaluated using qRT-PCR. MDA-MB-231 cells had very low endogenous expression of TWIST, and gene expression analyses could not be performed. Error bars represent S.E.M. * p < 0.05, ** p < 0.01, *** p < 0.001.
GSK346294A suppresses the mesenchymal phenotype in PDX models
Suppression of the mesenchymal phenotype was then confirmed on a protein level in treated TNBC PDX tumors. TU-BcX-4IC was established from the mastectomy specimen of a Caucasian woman in Louisiana and represents a high-grade metaplastic breast carcinoma and a TNBC subtype. After three passages in mice (T3), the TU-BcX-4IC PDX tumor was excised, tumor pieces were dissected (4 × 4 mm) and plated and treated for 24 hours with the lead PKIS compounds and PLK inhibitors BI2536 and BI6727 [26, 27]. GSK346294A increased CDH1 protein and suppressed the mesenchymal-associated proteins MMP9 and ZEB1 in TU-BcX-4IC treated explants (Figure 4A–B, Supplementary Figure 7). GSK448459 did not suppress MMP9 and ZEB1 protein expressions in TU-BcX-4IC explants. Because the selective PLK1 inhibitor BI6727 suppressed the mesenchymal markers ZEB1 and MMP9 expression while the non-selective PLK inhibitor BI2536 did not suggests that PLK1 promotes a mesenchymal phenotype. These data provide further evidence of the unique biological profiles of the pharmacologically similar PKIS compounds on TNBC PDX tumors.
Figure 4. Effects of GSK346294A on patient-derived tumors.
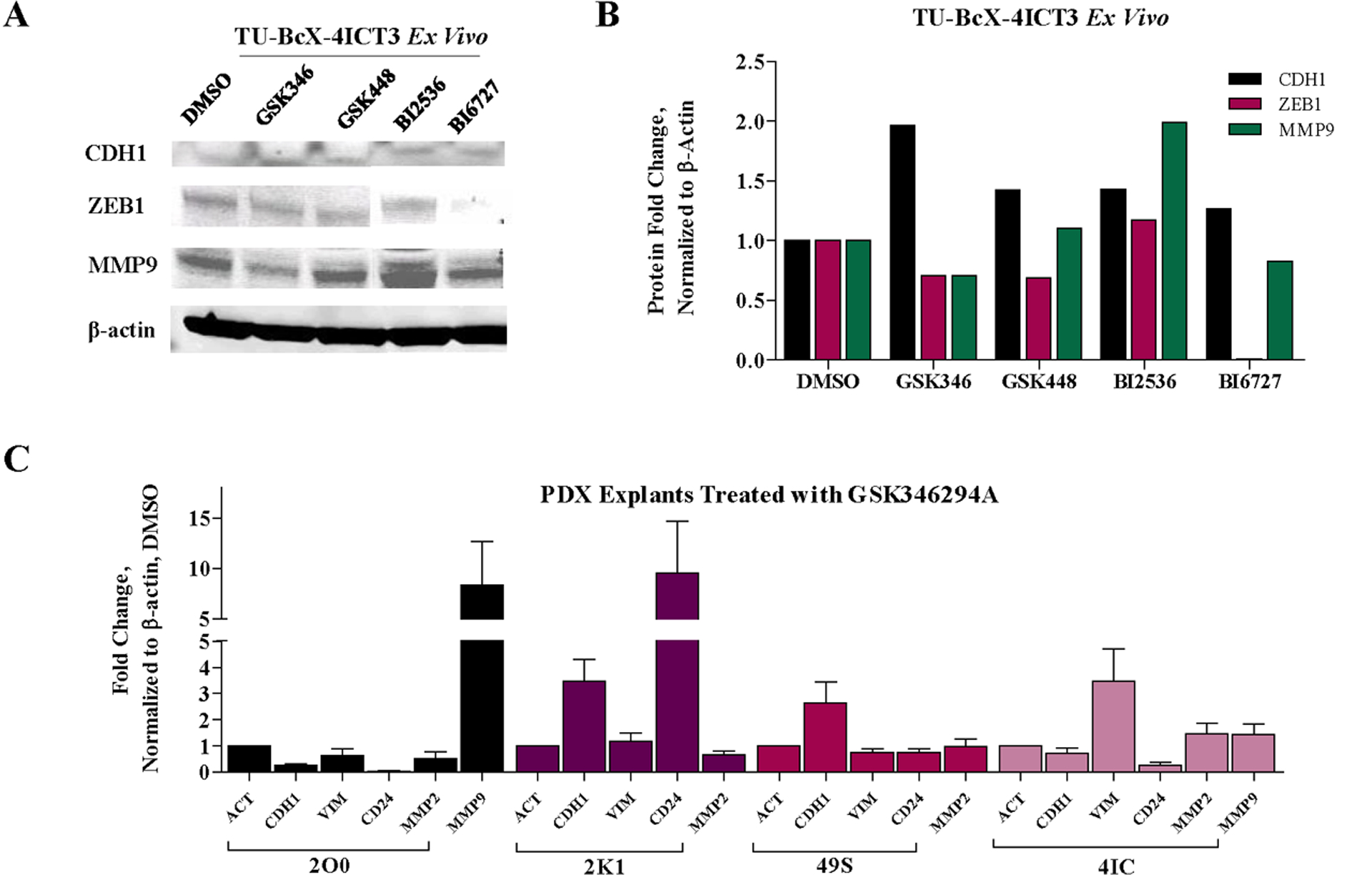
(A) Western blot of TU-BcX-4IC tumor pieces treated with PKIS inhibitor (GSK346294A, GSK448459A) and PLK-targeted compounds (BI2536, BI6727) compared to DMSO controls. Proteins analyzed included CDH1, ZEB1, and MMP9. All data was normalized to β-actin. (B) Quantification of Western blot analysis performed using Image Lab software (C) Epithelial (CDH1, CD24) and mesenchymal (VIM, MMP9) gene expression changes of tumor pieces from TNBC PDX models (TU-BcX-2O0, TU-BcX-2K1, TU-BcX-49S and TU-BcX-4IC) treated with GSK346294A for 72 hours analyzed with qRT-PCR.
GSK346294A was chosen as the lead compound to pursue for the next series of experiments because this PKIS compound had the most consistent and significant mesenchymal reversal effects based on cell morphology, biomass effect, migration, gene and protein expression changes compared to GSK448459A, although each compound had unique and distinct effect on the different endpoints. In the next experiment, tumor pieces from other TNBC PDX models established in our laboratory (TU-BcX-2K1, TU-BcX-2O0, TU-BcX-49S, TU-BcX-4IC) were treated and qRT-PCR was performed to observe epithelial (CDH1, CD24) and mesenchymal (VIM, MMP9) genes. These genes were chosen because endogenous levels of these genes were present in all four of the PDX models. We found that 72 hours treatment of these intact tumor pieces with GSK346294A increased epithelial markers CDH1 mRNA expression in TU-BcX-2K1 and TU-BcX-49S tumors, and increased CD24 expression in TU-BcX-2K1 tumors (Figure 4C). We also observed suppression of MMP9 in TU-BcX-2K1 tumors. GSK346294A did not reverse the mesenchymal phenotype in the TU-BcX-2O0 nor in the TU-BcX-4IC tumors on a transcript level. When MMP9 expression was examined, TU-BcX-2K1 nor TU-BcX-49S tumors had undetectable endogenous expression of MMP9 mRNA. Treatment of TU-BcX-2O0 and TU-BcX-4IC tumors with GSK346294A did not reduce MMP9 levels. Importantly, treatment with GSK346294A compared to DMSO control resulted in differential responses to individual patient-derived tumors, emphasizing the importance of utilizing PDX models in therapeutic discovery research. We then evaluated baseline tumor expression of the top kinases inhibited by the PKIS compounds in the four PDX models utilized in the previous study: PLK1, STK10, NEK2, TNK1, PDGFRA, PDGFRB, HIPK4, MST2, RET and FLT4 (Supplementary Figure 8). Endogenous PLK1 and NEK2 mRNA expression was higher in TU-BcX-2K1 and TU-BcX-49S tumors (Supplementary Figure 9). Endogenous PDGFRA expression was highest in TU-BcX-2O0; PDGFR and FLT4 endogenous expression was highest in TU-BcX-2O0 and TU-BcX-2K1 compared to the other models. Endogenous RET mRNA expression was highest in TU-BcX-2O0 and TU-BcX-4IC tumors (Supplementary Figure 9). Notably, all four PDX tumors tested had low endogenous mRNA expression of TNK1 and MST2, while all four models expressed HIPK4. Together these data show unique gene expression patterns amongst the individual patient-derived tumors utilized in this study.
GSK346294A does not significantly suppress metastasis but reduces the tumor initiating cell population in TNBC tumors
To evaluate in vivo effects of GSK346294A on tumor growth and metastasis, TU-BcX-4IC tumor pieces were implanted bilaterally in the mammary fat pads (mfp) of SCID/Beige mice and treatments with DMSO vehicle or GSK346294A (25 mg/kg bid) were injected intraperitoneally the following day. This model exhibits rapid tumorigenesis and extensive metastases to both the lungs and livers after implantation in SCID/Beige mice. After 15 days, tumors were resected, and mice were monitored for an additional 20 days for metastasis. Treatment with GSK346294A did not affect growth of the tumor at the site of implantation (i.e. the primary tumor) compared to DMSO vehicle control (Supplementary Figure 10). Lungs and livers were harvested, formalin-fixed paraffin embedded and stained with H & E to visualize and quantify metastatic lesions. Overall, GSK346294A treatment did not effectively suppress metastasis to both lungs and livers compared to mice treated with DMSO controls. Specifically, GSK346294A did not affect the area of lung metastases per stained lung section (Figure 5A) but did minimally reduce the overall number of metastases per lung section (Figure 5B). GSK346294A also minimally suppressed the area of liver metastases per section (Figure 5C) as well as the overall number of liver metastases per section (Figure 5D). Representative images of the lung and liver sections support our previous findings that TU-BcX-4IC is a highly metastatic PDX model (Figure 5E).
Figure 5. GSK346294A effects on tumorigenesis and metastasis of TU-BcX-4IC PDX model.
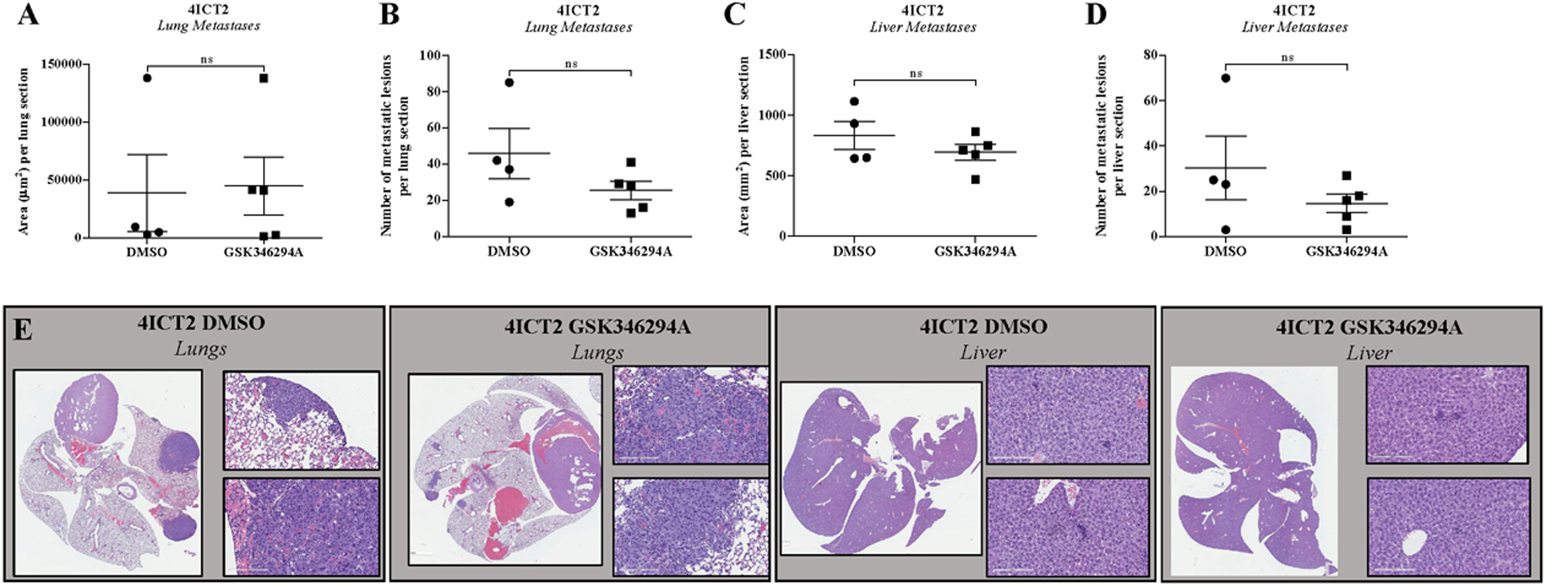
TU-BcX-4IC tumor pieces (3 × 3 mm) were implanted bilaterally in the mfp of SCID/Beige mice (5 mice/group). Mice were treated with intraperitoneal injections of DMSO vehicle or GSK346294A (25mg/kg bid) for 15 days. After 15 days, mice were sacrificed, and lungs and livers were harvested, fixed in formalin and embedded in paraffin, then sectioned and stained with H & E. Metastases were quantified. (A) Area of lung metastatic lesions per section and (B) overall number of metastases per section of DMSO treatment groups compared to GSK346294A. (C) Area of liver metastatic lesions per section and (D) overall number of metastases per section. (E) Representative images of the stained lung and liver sections of the DMSO control mice and GSK346294A treated mice. Error bars represent S.E.M.
Peripheral blood from the TU-BcX-4IC in vivo treatment experiment was harvested and analyzed using flow cytometry to evaluate differences in circulating tumor stem-like cell populations between GSK346294A-treated mice compared to DMSO vehicle-treated. Cancer stem cells (CSCs) were characterized as HLA+CD44+CD24−. Treatment with GSK346294A suppressed the CSC populations in the peripheral blood of mice implanted with TU-BcX-4IC tumors, although not significantly (Figure 6A). GSK346294A was not cytotoxic to PDX organoids (PDX-Os) created from TU-BcX-4IC, as was determined in a live/dead immunofluorescence stain (Figure 6B). To further evaluate the effects of GSK346294A on stem cell-like behavior, second generation (M2) spheres derived from TU-BcX-4IC and MDA-MB-231 cells were treated with DMSO vehicle or PKIS compounds. We compared effects of two lead PKIS compounds from the initial screen (GSK346294A, GSK448459A) on sphere formation. After 7 days treatment, spheres were imaged, and areas quantified (Figure 6C–E). GSK346294A and GSK448459A significantly suppressed mammosphere formation of TU-BcX-4IC cells. GSK346294A and GSK448459A significantly increased sphere area in MDA-MB-231 cells. CD44 and CD24 are two genes that define the cancer stem cell phenotype: CSCs are CD44 high and CD24 low. The PKIS inhibitors did not significantly suppress the CSC population in MDA-MB-231 nor in TU-BcX-4IC cells (Supplementary Figure 11). Together, these data suggest that the PKIS compounds do not significantly suppress the canonical CSC population in TNBC.
Figure 6. GSK346294A suppresses mammosphere formation and stem-cell like properties of TNBC cells.
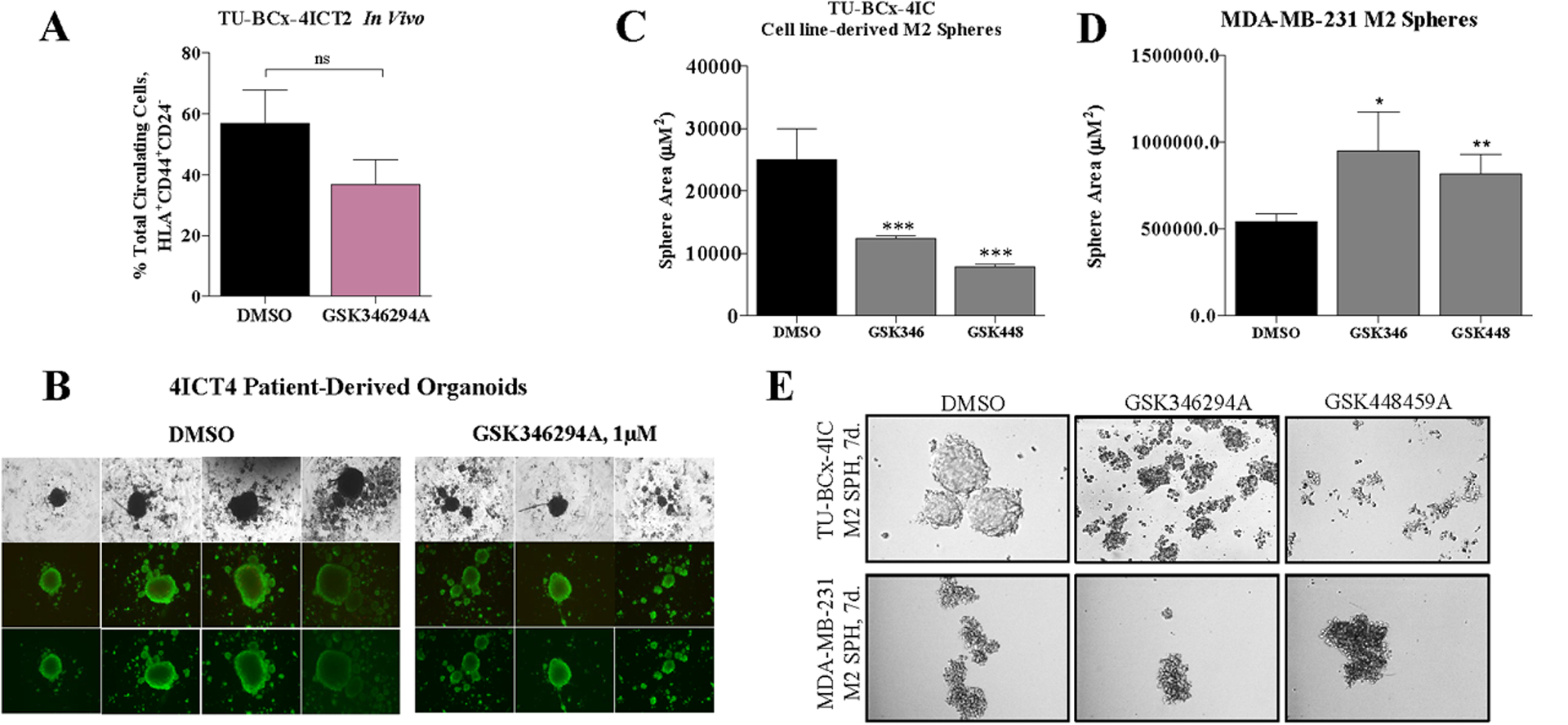
(A) Peripheral blood harvested from the TU-BcX-4IC in vivo treatment experiment (DMSO vehicle, GSK346294A treatments) was analyzed using flow cytometry for the presence of tumor circulating stem cells characterized as HLA+CD44+CD24-. n = 4 mice/treatment group. (B) Patient-derived organoids from the TU-BcX-4IC model were treated with GSK346294A (1μM) or DMSO control for 72 hours before staining with a Live/Dead kit (PromoKine). Calcein-AM (green) highlights viable cells and EthD-III (red) highlights dead cells when imaged with immunofluorescence. (C) TU-BcX-4IC and (D) MDA-MB-231 M2 mammospheres were treated with the PKIS compounds (GSK346294A, GSK448459A) as well as potent PLK inhibitors (BI2536, BI6727). After 7 days of treatment, the mammospheres were imaged and areas were quantified. (E) Representative images of TU-BcX-4IC and MDA-MB-231 mammospheres treated with PKIS compounds and selective PLK inhibitors.
Next, we evaluated how the inhibitors affected the tumor-initiating capacity of TNBC cells. We pre-treated MDA-MB-231 M2 spheres with DMSO or GSK346294A and then injected the cells into mice. We used various cell concentrations: 200,000 cells/tumor, 100,000 cells/tumor, 50,000 cells/tumor, 10,000 cells/tumor, 1,000 cells/tumor. Pre-treatment of MDA-MB-231 M2 spheres with GSK346294A inhibited tumorigenesis at all cell concentrations (Figure 7A, B; Supplementary Figure 12). Quantification of metastatic lesions in H & E stained lungs and livers harvested from this experiment showed that GSK346294A consistently suppressed both overall area and number of metastases in all cell concentrations, although not significantly (Supplementary Figure 13). TU-BcX-4IC M2 spheres also pre-treated with DMSO or GSK346294A, and then mice were inoculated with pre-treated spheres (100,000 cells per injection). The number of cells injected was based on the preliminary in vivo dose-response experiment with MDA-MB-231 cells. GSK346294A significantly suppressed tumor formation compared to DMSO pre-treatment in TU-BcX-4IC cell-derived xenografts (Figure 7C, D). Quantification of metastatic lesions in lungs and livers harvested from the TU-BcX-4IC pre-treatment experiment demonstrate GSK346294A pre-treatment suppressed area of lung metastasis (Figure 7E) and significantly suppresses total number of lesions (Figure 7F). GSK346294A did not suppress area (Figure 7G) or number (Figure 7H) of liver metastases. Together, these data show GSK346294A suppresses cancer stem cell-like behavior in vivo.
Figure 7. GSK346294A suppresses stem cell-like behavior of TNBC cells.
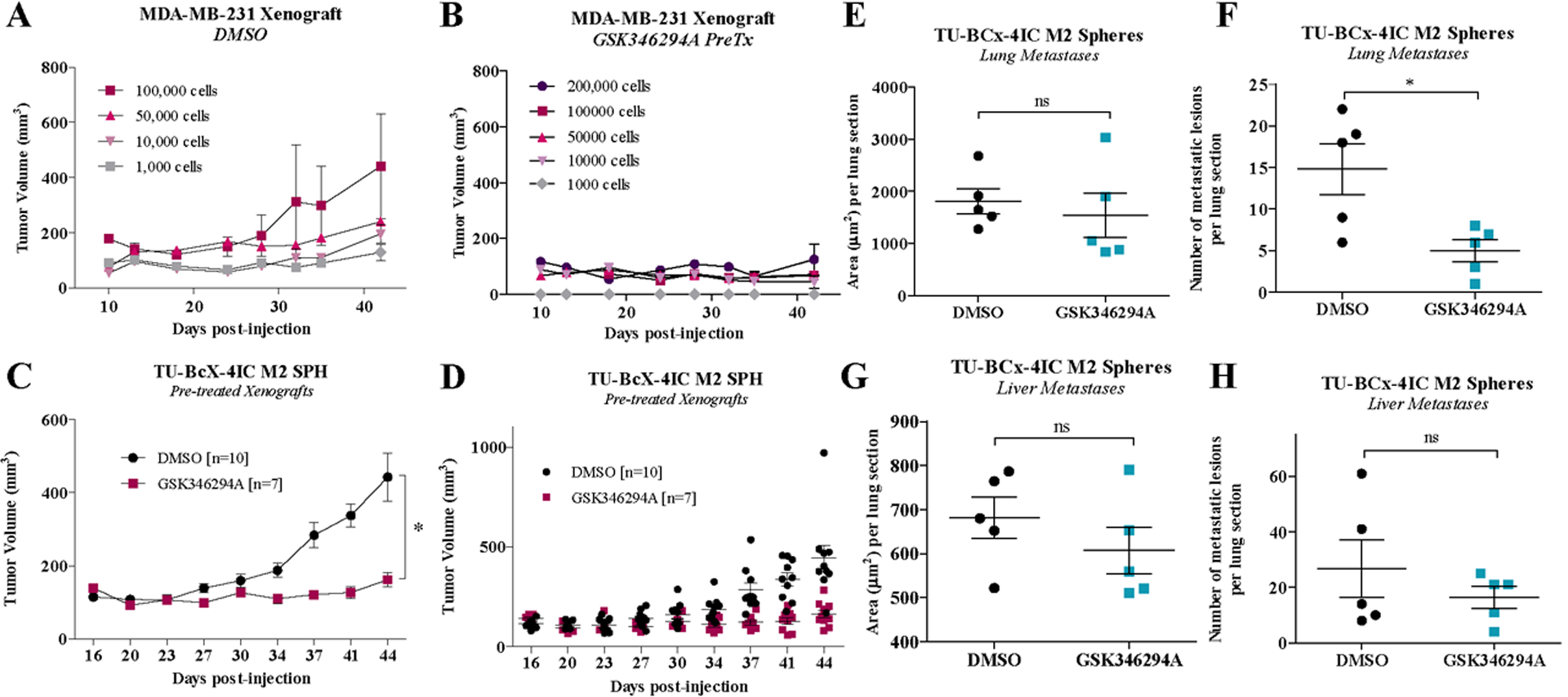
Pre-treated MDA-MB-231 M2 mammospheres (DMSO, GSK346294A (1μM)) were injected bilaterally into the mfp of SCID/Beige mice at different numbers of cell concentrations (200,000 cells, 100,000 cells, 50,000 cells, 10,000 cells, 1,000 cells). Notably, in the DMSO treatment group, the mouse injected with 200,000 cells/tumor died three days after injections. Tumorigenesis data of the pre-treated (A) DMSO and (B) GSK346294A groups are shown. Pre-treated TU-BcX-4IC M2 mammospheres (DMSO, GSK346294A (1μM)) were injected bilaterally into the mfp of SCID/Beige mice (100,000 cells/injection; n = 5 mice/group). (C, D) Tumorigenesis data of the pre-treated DMSO and GSK346294A groups are shown. Tumor areas were measured biweekly with calipers. Tumors were resected and after 20 additional days mice were sacrificed. Lungs and livers were harvested, formalin fixed, paraffin-embedded and H & E stained to visualize metastasis. Metastatic lesions were measured and quantified using the AperioImage program.
GSK346294A downregulates cell cycle-associated and cytoskeletal rearrangement pathways
To discover signaling pathways responsible for the observed reversal of the mesenchymal phenotype in TNBC cells, RNA sequencing was performed on TNBC cells (MDA-MB-231, TU-BcX-4IC) treated with GSK346294A or DMSO controls for 72 hours. In MDA-MB-231 cells, the mitotic spindle, apical junction and myogenesis signaling pathways were the most significantly downregulated pathways after GSK346294A treatment in MDA-MB-231 cells (Supplementary Figure 14A). Pathway analyses show pathways affected in MDA-MB-231 cells treated with GSK346294A compared to DMSO control included HER2-induced breast cancer cell invasion, the metaphase component of the cell cycle, development of EMT, and regulation of cytoskeletal arrangements (Supplementary Figure 14B). The common pathways between the sequencing analyses were regulation of cell cycle and regulation of cytoskeletal organization. We compared the most downregulated genes common in MDA-MB-231 and TU-BcX-4IC cells treated with GSK346294A normalized to DMSO control. NES, COL1A1, FASN, TNC, COL6A3, COL12A1 and COL8A1 were amongst the most downregulated genes common to the two treated TNBC cell lines compared to DMSO control (Supplementary Figure 15). Together, these data show that GSK346294A affects cytoskeletal organization and has potential functions in downregulation of extracellular matrix genes, confirming our findings that GSK346294A suppresses the mesenchymal and migratory phenotype in TNBC cells.
Discussion
Despite impressive advances in therapeutic development in oncology, triple negative breast cancer remains a difficult malignancy to therapeutically manage, due to its high rates of recurrence, metastasis and acquisition of drug resistance. Systemic chemotherapy continues to be the first-line treatment option for TNBC; while some small molecule inhibitors are in pre-clinical studies for TNBC, discovery of novel targets is warranted. There are various methods for discovering novel targets; we previously described a screening approach using a broad range of kinase inhibitors and lead compound ‘hits’ in TNBC cell lines based on changes to cell morphology, suppression of migratory behavior and suppression of canonical mesenchymal genes [20]. After lifting the blind on that study, we found that many of the compound hits were from the same original medicinal chemistry campaign and were pharmacologically and structurally similar but had differential effects on EMT in TNBC [20]. This implies unique and/or common kinase targets were responsible for the observed reversal of the EMT phenotype. Here, we further interrogate these findings and compare the biologic effects of pharmacologically similar compounds on TNBC cell behavior. Two of the selected compound hits that were compounds from a PLK1-targeting project [21] were selected for further investigation: GSK346294A and GSK448459A. In these studies, we utilized novel TNBC patient-derived xenografts established in our laboratory.
The two selected inhibitors were originally made to establish structure activity relationships for a thiophene benzimidazole series designed to target PLK1 [21]. However, as non-optimized compounds, they also have activity against kinases besides PLK1, which we demonstrated with the kinase inhibition assay. As expected, both compounds inhibited PLK1. With respect to other kinases, both compounds exhibited binding activity to LOK, but to varying degrees: GSK448459A at 99.1% and GSK346294A at 36% binding activity. There were other kinases in the screens that were uniquely bound by one or two PKIS drugs. For example, GSK448459A uniquely bound to NEK2, with a 97.1% binding activity compared to GSK346294A (5%). This was a proof-of-principle screen to demonstrate that unique and shared kinase targets exist amongst the pharmacologically similar PKIS compounds. We proposed using these unique and shared kinase profiles could lead to the discovery of novel kinase targets to pursue that regulate the EMT axis in TNBC.
The role of PLK1 in cell cycle progression in cancer has been well-documented [28]. Because PLK1 is a common target of the two PKIS compounds, we first evaluated the proliferative effects of the compounds on MDA-MB-231 cells. Because our kinome profile was performed at the same dose (1μM), we consistently utilized this dose for the proliferation experiments. Importantly, the PKIS compound that suppressed cell proliferation (GSK448459A) exhibited significant binding activity against kinases that were not inhibited by, or less inhibited by, PKIS drugs that increased proliferation (GSK346294A). These kinases included NEK2, HIPK4, MST2 and MINK, suggesting inhibition of these kinase(s) resulted in suppression of TNBC cell proliferation. Additionally, cell biomass remaining was dose-dependent, and the responses of the PKIS compounds were similar in three TNBC cell lines tested, including our PDX-derived cell line, TU-BcX-4IC, MDA-MB-231 and BT-549 cells had higher IC-50 values for GSK448459A compared to GSK346294A, which had similar IC-50 values. With respect to quantification of cell proliferation, GSK448459A suppressed Ki-67 expression most significantly in TNBC cells. Because Ki-67 accumulates in S-phase entry, if drug inhibitors affect the S-phase this can result in paradoxically increased Ki-67 expression, which is what we speculate occurred with GSK346294A [29]. While GSK346294A increased number of Ki-67 positive cells, it decreased cell abundance in a dose-dependent manner. A possible reason for this observation is that the drug inhibited cell adherence mechanisms, which resulted in fewer cells remaining on the plate for quantification.
TNBC cells are characterized as mesenchymal in appearance, with elongated, fibroblast-like cells with protrusions and minimal cell-cell contact. In our initial kinase inhibitor screen, we previously reported that the PKIS compounds characterized in this study reversed the mesenchymal phenotype of MDA-MB-231 cells to represent a more epithelial phenotype. This observation was quantified using morphometric measurements such as total area of the cells, aspect ratio and nuclear area fraction. These parameters have been shown to describe the mesenchymal cell phenotype. Nuclear area fraction is the ratio of the nuclear area to the total cellular area. Because a larger nucleus is associated with phenotypically mesenchymal cells, a smaller nuclear area fraction is associated with the epithelial phenotype while a larger fraction describes a mesenchymal phenotype [30, 31]. Here, using the same parameters, we observed that GSK346294A most significantly altered cell morphology compared to GSK448459A in our PDX-derived TU-BcX-4IC cells. Notably, we did not observe obvious morphological changes in response to PKIS inhibitor treatment in BT-549 cell, suggesting a cell line-specific effect.
Acquisition of cell migratory capabilities and expression of mesenchymal-associated genes drive the mesenchymal phenotype in TNBC. We confirm our previous findings that both PKIS compounds suppressed transwell migration of TNBC cells, including our PDX-derived cell line. One compound in particular, GSK346294A, consistently and most significantly suppressed transwell migration of BT-549 and TU-BcX-4IC cells compared to the other compounds and the DMSO controls. Comparing epithelial (CDH1) and mesenchymal-associated (ZEB1, MMP9, cFOS, SNAI1, SLUG, TWIST) gene expressions between the two pharmacologically similar compounds led us to identify common and unique genes that were affected by the inhibitors. Overall, the PKIS drugs had minimal effect on EMT-associated gene expression changes in the TU-BcX-4IC cell line. A possible reason for this observation is that the inhibitors were not sufficiently potent at the dose utilized to exert a response. Another possible explanation is because the cell line was derived directly from human tumors, the drugs were not able to reverse the EMT phenotype overall due to the heterogeneity of the cancer cells. We found the compounds had differential effects on EMT gene expressions, and these data were cell-line specific. For example, although the compounds increased CDH1 expression in MDA-MB-231 cells, this was not translated to the other TNBC cell lines tested. These data demonstrate differential activity of these pharmacologically similar compounds; since these compounds have different off-target kinase activity it was inferred that there are unique kinase-mediated mechanisms that were responsible for the observed EMT reversal.
The application of patient-derived tumor models in drug discovery research is more translational than immortalized cell lines, because PDX models recapitulate molecular signatures and tumor characteristics that were present in the original patient’s tumor. Furthermore, intact tumor pieces can be treated with PDX models in an ex vivo approach to evaluate drug effects on the intact tumor system, and not just isolated components. Throughout this manuscript we utilized a TNBC PDX model established in our laboratory, TU-BcX-4IC. Protein analysis of EMT-related genes in treated TU-BcX-4IC explants confirms that out of the two compounds, GSK346294A treatment most consistently reversed the EMT phenotype, demonstrated by increased CDH1 expression and suppressed ZEB1 and MMP9 expressions. Because GSK346294A consistently has the most overall effects on reversal of the mesenchymal phenotype compared to the other PKIS compound GSK448459A, we focused on further describing the activity of GSK346294A treatment in TNBC for the remainder of this study. Other TNBC PDX models established by our group were used to for ex vivo treatment studies: TU-BcX-2O0 (a claudin-low TNBC subtype) [32] and TU-BcX-2K1 are minimally metastatic models while TU-BcX-4IC and TU-BcX-49S are highly metastatic. GSK346294A had different effects on EMT-related gene expressions (CDH1, CD24, ZEB1, MMP2, MMP9) in the various models. Individual patient tumors have unique cellular compositions and baseline endogenous expression of genes. Because relative drug response on individual patients’ tumors varies depending on endogenous expression of the drug’s targets, baseline expression of the kinases inhibited, or not inhibited by, GSK346294A expression was assessed. TU-BcX-2K1 and TU-BcX-49S tumors had elevated endogenous PLK1 mRNA expression compared to TU-BcX-2O0 and TU-BcX-4IC. The next challenge was to narrow down which kinases from the kinase inhibition screen were more likely to affect the EMT phenotype. GSK346294A exhibited strong activity against PLK1 and moderate activity against LOK, PDGFRB, RET and BRSK2. Because GSK346294A inhibited PLK1 activity, and treatment of TU-BcX-2K1 and −49S tumors resulted in a more significant reversal of the EMT phenotype, this strengthens the interpretation of our data that GSK346294A affects the EMT axis through PLK1 signaling. LOK expression was also elevated in TU-BcX-49S, and PDGFRB had elevated expression in TU-BcX-2K1, suggesting GSK346294A expression can also affect EMT through LOK expression. RET had low baseline endogenous expression in TU-BcX-2K1 and −49S but RET was inhibited by GSK346294A. This suggests GSK346294A was less likely to reverse EMT through this kinase-mediated signaling pathway.
Drug treatment experiments using GSK346294A versus vehicle control demonstrated the efficacy of this compound in the in vivo setting with implanted TU-BcX-4IC tumors. GSK346294A did not suppress tumorigenesis in this PDX model that exhibits rapid rates of tumor growth at baseline. Further demonstration of the aggressive behavior of TU-BcX-4IC is that implanted tumors can propagate in murine models independently of Matrigel™ and is highly metastatic to both lungs and liversGSK346294A did suppress the total number of lung and liver metastases, albeit not significantly. However, GSK346294A treatment alone was not sufficient to suppress the average areas of the metastatic lesions.
There is a known association between acquisition of a mesenchymal phenotype and the presence of CSC populations, and the combined presence of these two cellular features contributes to more aggressive behaviors in various cancer systems [33–35]. GSK346294A suppressed the circulating cancer stem-like cell (CSC) population in the peripheral blood of TU-BcX-4IC-implanted mice. This finding led us to evaluate the role of GSK346294A, as well as the other PKIS compounds, in suppression of the CSC phenotype. GSK346294A and GSK448459A significantly suppressed areas of spheres derived from TU-BcX-4IC cells, but none of the PKIS compounds reduced sphere area in MDA-MB-231 cells. We partly attribute these findings to the fact that MDA-MB-231 cells do not form true ‘spheres’ due to the very low endogenous protein expression of CDH1 and other cell surface adhesion proteins. Instead, MDA-MB-231 and other basal-B cell lines form aggregates in 3D culture. In contrast, TU-BcX-4IC cells form true spheres in 3D culture and is a better cell line to evaluate sphere area. Because cellular toxicity effects of drugs can affect sphere formation, we also performed a live/dead immunofluorescent stain and found that GSK346294A was not cytotoxic to TU-BcX-4IC patient-derived organoids (PDOs). The application of PDOs in therapeutic research is important because they more directly translate in vitro findings in cell systems to in vivo observations, and predict clinical response to therapeutics [36, 37]. TNBC cells pre-treated with GSK346294A were incapable of forming tumors when implanted in murine models, independent of the number of cells injected, while MDA-MB-231 DMSO-pre-treated cells formed tumors. Furthermore, GSK346294A decreased lung metastases, and only liver metastases in the MDA-MB-231 sphere-derived xenografts. These data show that GSK346294A suppressed the cancer stem cell-like and tumor initiating cell populations in TNBC.
Potential mechanisms regarding which downstream signaling pathways were responsible for the reversal of the EMT phenotype due to GSK346294A was determined using RNA-sequencing of the TNBC cell lines (MDA-MB-231 and TU-BcX-4IC) treated with GSK346294A or DMSO controls. As expected, because GSK346294A targets the PLK family, there was downregulation of cell cycle-associated pathways, specifically the G2-mitosis and metaphase pathways. Notably, GSK346294A also suppressed cytoskeletal organization pathways, including actin rearrangement, mitotic spindle assembly, and apical surface pathways. These results are consistent with our observations that GSK346294A altered cell morphology in various TNBC cell lines. TGFβ-dependent induction of EMT was downregulated in TNBC cell lines after treatment with GSK346294A, and breast cancer cell invasion pathways were downregulated in MDA-MB-231. Extracellular matrix genes were also consistently downregulated in both TU-BcX-4IC and MDA-MB-231 cells treated with GSK346294A compared to DMSO controls. These genes include collagen family members (COL1A1, COL8A1, COL12A1, COL1A2) and matrix-associated proteins (WNK4, JPH2, PDGFβ, SERPINB2). In the TU-BcX-4IC sequencing analysis, CSC pathways were also downregulated (Frizzled, WNT5A, FZD2), confirming our in vitro findings that GSK346294A suppressed the CSC population in TNBC.
Although GSK346294A is a narrow spectrum inhibitor, it demonstrated an interesting phenotype in our TNBC screens and warranted further exploration; we used it in this study as a screening modality. We show that this compound suppresses the mesenchymal and migratory phenotype of TNBC cells both in vitro and in vivo. As a future direction, candidate kinase(s) or signaling pathways responsible for driving the mesenchymal and migratory phenotype in breast cancer that we preliminarily identified in this project will be verified. Furthermore, because each of the PKIS compounds exerted unique effects on different aspects of the mesenchymal phenotype, through cell morphology, cell proliferation/viability, epithelial/mesenchymal gene expression changes, cell migration, or the cancer stem-cell like phenotype, these findings suggest there are unique kinases that are inhibited by the compounds responsible for the observed phenotypic EMT reversal. In conclusion, our primary aim in this manuscript was to use phenotypically active kinase inhibitors from a PLK1 scaffold to discover new targets and signaling pathways that can be targeted in TNBC that suppress the mesenchymal phenotype. Here we demonstrated the utility of a biology-based comparative analysis of structurally similar compounds that have unique kinase targets; we show these kinases are responsible for different EMT-driven mechanisms in TNBC. This approach can be applied to all cancer types and is not limited to breast cancer. Future studies are necessary to pursue identified individual kinase targets and pathways, in order to characterize the specific roles of these targets in breast cancer.
Materials and Methods
Reagents
Dulbecco’s modified Eagle’s medium (DMEM), Dulbecco’s phosphate-buffered saline (DPBS), phenol-red free DMEM, fetal bovine serum (FBS), minimal essential amino acids (MEMAA), non-essential amino acids (NEAA), antibiotic/anti-mitotic penicillin/streptomycin (pen/strep), sodium pyruvate, L-glutamine, trypsin/EDTA, trypan blue stain (0.4%) and ethylenediaminetetraacetic acid (EDTA 0.5 M, pH 8) were obtained from GIBCO (Invitrogen; Carlsbad CA). Insulin was purchased from Sigma-Aldrich (St. Louis MO) and charcoal stripped (CS) FBS from HyClone (Thermo Scientific; Logan UT). Dimethyl sulfoxide (DMSO) was obtained from Research Organics, Inc (Cleveland OH).
Cell Culture
Human MDA-MB-231, BT549 and MDA-MB-157 cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and are characterized as triple-negative/basal B mammary carcinoma. Cells were maintained in DMEM supplemented with 10% FBS, non-essential amino acids (NEAA), MEM amino acids, anti-anti (100 U/mL), sodium pyruvate and porcine insulin (1×10−10 mol/L) at 37 °C in humidified 5% CO2. For treatment studies, cells were grown in phenol red-free DMEM supplemented with 5% charcoal-stripped FBS and supplemented with NEAA, MEM amino acids, Gluta-Max and penicillin (100 U/mL).
The Published Kinase Inhibitor Sets (PKIS)
The PKIS library typically provides 10 μL of a 10mM solution in DMSO, dispensed in 96-well plates. Drugs were received as 10 mM DMSO stock solutions, diluted in culture media and used at 1 μM concentrations, as determined by dose-response studies. Larger aliquots of requested drugs were delivered as solids and dissolved in DMSO as a 1 mM stock solution and kept at −20 °C.
Kinase inhibition assay
The kinase assay for GSK346294A and GSK448459A was performed by Reaction Biology Corporation (Malvern, PA, USA) and contains 350 kinases of interest, as described previously [38].
Crystal violet staining
MDA-MB-231, MCF7, BT549 and MDA-MB-157 cells were plated at 5,000 cells per well in 96-well TC plate in 5% charcoal stripped (CS) DMEM. After 48 hours of exposing the cells to CS DMEM media, cells were treated with the vehicle or selective GSK library compounds for 72 hours and the plate was incubated in 37 °C, 5% CO2. The plate was then harvested by adding 10 μL glutaraldehyde to each well for 20 minutes. After rinsing and drying the plate, the cells were stained with 50 μM 0.1% crystal violet in 90% methanol for 20 minutes. After another rinse, the cells were left overnight to dry, and the following day morphological alterations of the cells were visualized with an inverted microscope and images were recorded. Cells were lysed with 33% acetic acid and absorbance was measured at 620 nm excitation to quantify proliferation after treatment.
Flow cytometric assay for CD44/CD24 cell populations:
Cells were plated in 10cm plates and the next day treatment added in 10mL fresh medium. After 48 hour incubation, cells were PBS wash, trypsinized to detached cells from the plates, trypsin was neutralized with fresh medium, spin down and wash with PBS. Counted and used 1×106 cells in 50μL PBS, stained with using 4μL of mouse antihuman CD24-FITC (BD Pharmingen Cat# 555427) and 1μL of mouse antihuman CD44-PE (BD Pharmingen Cat# 555479) protected from light on a slow shaker at room temperature for 30 min. Washed once with 4mL PBS and analyzed using Gallios flow cytometer (Beckman Coulter) and Kaluza Flow Cytometry Analysis Software.
Mammosphere formation and quantification
MDA-MB-231 and TU-BcX-4IC cells were plated in suspension conditions in low-attachment 96-well plates (1,000 cells per well), with serum-free F12/DMEM (Invitrogen, Carlsbad CA) media supplemented with B-27 (Invitrogen, Carlsbad CA). Cells were maintained in cell culture conditions for 24 hours to facilitate sphere formation. After 24 hours, suspended cells/spheres were treated with drug and DMSO controls for one week, with treatment frequencies of every three days. Area of spheres were used one endpoint to quantify drug effects on sphere formation. For these quantifications, images were obtained of spheres at both 40X and 100X magnifications, depending on the average size of the spheres. The lengths and widths of individual spheres after treatments were measured using the Aperio ImageScope program. Area was calculated and recorded and compared to DMSO treatment controls.
RNA isolation and quantitative real time PCR
MDA-MB-231 cells were plated in 5% CS DMEM and pre-treated for 72 hours with DMSO (control), or PKIS compounds (1 μM). Cells were harvested, and total RNA was isolated using RNeasy (Quiagen, Valencia, CA) following manufacturer’s protocol, and the quantity and quality determined by absorbance (260, 280 nm). Total RNA (2 μg) was reverse-transcribed (iScript kit, BioRad Laboratories, Hercules, CA) and analyzed by qRT-PCR. Primer sequences are as follows (Invitrogen, Carlsbad, CA): β-actin F-5’-TGAGCGCGGCTACAGCTT-3’; β-actin R-5’-CCTTAATGTCACACACGATT-3’; CDH1 F-5’-AGGTGACAGAGCCTCTGGATAGA-3’, CDH1 R-3’-TGGATGACACAGCGTGAGAGA-3’; VIM F-5’-CGTCCACCCGCACCTACAGC-3’; VIM R-5’-GCCAGCGAGAAGTCCACCGAG-3’; SNAI1 F-5’-ACCACTATGCCGCGCTCTT-3’, SNAI1 R-5’-GGTCGTAGGGCTGCTGGAA-3’; SLUG F-5’-TGTTGCAGTGAGGGCAAGAA-3’, SLUG R-5’- GACCCTGGTTGCTTCAAGGA-3’; TWIST F-5’- TGTCCGCGTCCCACTAGC-3’, TWIST R-5’-TGTCCATTTTCTCCTTCTCTGGA-3’. Quantitative reverse transcription-PCR was conducted as previously published [40]. Data represented as normalized fold expression compared with DMSO control of biological triplicate samples ± S.E.M.
Transwell migration assay
Migration assays were performed following the manufacturer’s instructions (BD Biosciences, San Jose, CA, USA). MDA-MB-231 cells were grown in charcoal stripped DMEM (5% FBS) for 48h, then pretreated for 72h (1 μM) with DMSO, GSK346294A and GSK448459A. Transwell inserts (8 μM; BD biosciences; San Jose, CA) were placed into each well containing 1 mL 10% FBS DMEM media. Pretreated cells in opti-MEM suspension was placed on each insert (500 μL, 25,000 cells per well). After 24 hours, membranes were scrubbed to remove non-migrated cells and membranes were removed and mounted on glass slides. Migrated cells were visualized by microscopy and counted. Data is represented as number of migrated cells per field of view for triplicate experiments.
Western blot of PDX tumors
Intact tumor pieces from the TU-BcX-4IC PDX model maintained in mice for three passages were treated with GSK346294A (1 μM) or DMSO for 24 hours in 10% FBS DMEM. Treated PDX tumor explant pieces were minced with scissors and lysed with mammalian protein extraction reagent (MPER) supplemented with 1% protease inhibitor and 1% phosphatase inhibitors (I/II) (Invitrogen, Grand Isles, NY). Lysed specimens were centrifuged at 12,000 RPM at 4°C and supernatant isolated, which contained the protein extracts. The NanoDrop ND-1000 was used to determine protein concentration of samples by absorbance at 260 and 280 nm. Protein specimens were normalized to the lowest concentration and mixed with NuPage LDS loading buffer (Thermo Fisher Scientific) and 2-mercaptoethanol (MP Biomedicals, LLC) and heat-denatured at 100°C on a heating block. Protein was loaded on Bis-Tris-NuPAGE gel (Invitrogen, Grand Isles NY). Protein was then transferred to nitrocellulose membranes using iBlot and iBlot transfer stacks per manufacturer’s instructions (Invitrogen, Grand Isles, NY). Membrane was incubated at room temperature with 5% bovine serum albumin (BSA) in 1% Tris-buffered saline, 0.1% Tween 20 (TBS-T) for CDH1 and Odyssey Blocking Buffer (Licor) for MMP9 and ZEB1 for 1 hour to block non-specific binding followed by 4°C incubation overnight with primary antibodies (CDH1: 1:1000 Cell Signaling Technology, Cat. No. 3195S; MMP9 (G657), 1:500, Cell Signaling Technology Cat. No. 2270: ZEB1: 1:1000, Santa Cruz Antibodies Cat. No. sc-25388). After three 10-minute washes in 1% TBS-T, membranes were incubated with appropriate secondary antibodies for at least one hour. IR-tagged secondary antibodies were purchased from LiCor Biosciences (Lincoln, NE) and used at a 1:10,000 dilution in 5% BSA. Following incubation with secondary antibodies, membranes were washed three times for 15 minutes per wash in 1% TBS-T, and blots were analyzed by the Image Lab Imaging System (BioRad). Band density was quantified by the Image Lab™ software. Data were normalized to β-actin.
Patient Derived Xenograft Models
Tissues were obtained through the Louisiana Cancer Research Consortium Biospecimen Core and were processed in compliance with NIH regulations and institutional guidelines and approved by the institutional review board at Tulane University. All animal procedures were reviewed and approved by the institutional animal care and use committee at Tulane University. TNBC PDX models utilized in this study include: TU-BcX-2K1, TU-BcX-2O0, TU-BcX-49S and TU-BcX-4IC. TU-BcX-2K1 and TU-BcX-2O0 were established from biopsy specimens of African-American women who did not receive chemotherapy before biopsy. TU-BcX-49S was established from the mastectomy specimen of an African-American woman who did receive chemotherapy. TU-BcX-4IC was established from the mastectomy specimen of an African-American woman and represented a metaplastic breast carcinoma; the tumor was chemo-refractive at the time of surgery. SCID/Beige (CB17.Cg-PrkdcscidLyst bg/Crl) were purchased from Charles River and are used to prevent rejection of the xenografted human tumors. The autosomal recessive SCID (Prkdcscid) mutation results in severe combined immunodeficiency affecting both the B and T lymphocytes. The autosomal recessive beige (Lystbg) mutation results in defective natural killer (NK) cells. Tumor tissues from each patient were cut into 3 × 3 mm2 pieces under aseptic sterile conditions, coated with full factor Matrigel (Cat No. 354234, Fisher Scientific, Waltham, MA, USA) and implanted bilaterally into the mammary fat pads (mfp) of mice under isoflurane and oxygen. Tumors were measured using a digital caliper after ostensible tumor take was established. Tumors were passaged when tumor volume achieved 750–1000 mm3. To passage, mice were euthanized by CO2 and tumors were removed, dissected, coated in full factor Matrigel, and then implanted bilaterally into new mice that were anesthetized using a mix of isoflurane and oxygen delivered by mask. Before surgery, mice were given Meloxicam (5 mg/kg/day, for 3 days post-surgery) for pain. For ex vivo treatments tumor pieces were plated in 24-well adherent plates and covered with DMEM media. Tumors were treated in-well with control (DMSO) or PKIS inhibitors (1μM). Explants were collected after 72 hours, and RNA was extracted using QIAzol Lysis Reagent (Cat No. 79306; Qiagen, Valencia, CA, USA) and dissection of the tumor with scissors. Total RNA was isolated and cDNA was made as described above. mRNA was analyzed by qRT-PCR. Primers (Invitrogen, Carlsbad, CA) were generated with sequences as follows: β-actin F-5’- GGCACCCAGCACAATGAAGA-3’; β-actin R-5’- ACTCCTGCTTGCTGATCCAC −3’; CDH1 F-5’-AGGTGACAGAGCCTCTGGATAGA-3’, CDH1 R-3’-TGGATGACACAGCGTGAGAGA-3’, cFOS F-5’- GAATGCGACCAACCTTGTGC-3’; cFOS R-5’- AGGGATCAGACAGAGGGTGT-3’; MMP2 F-5’-CGAGTGGATGCCGCCTTA-3’, MMP2 R-5’-CCAGGTTATCGGGGATGGC-3’; ; MMP9 F-5’-AGTCCACCCTTGTGCTCTTC-3’, MMP9 R-5’-GCCACCCGAGTGTAACCATA-3’ VIM F-5’-GAGAACTTTGCCGTTGAAGC-3’, VIM R-5’-GCTTCCTGTAGGTGGCAATC-3’; PLK1 F-5’- CGAGAGACAGGTGAGGTGGTCG-3’, PLK1 R-5’- CGAGGGCTTGGAGGCATTGAC-3’; LOK F-5’- GAGGTGTGGGAGATCGTGGG-3’, LOK R-5’- CACAATGTAGGGGTGGTCGC-3’; NEK2 F-5’- GGATGGCAAGCAAAACGTCA-3’, NEK2 R-5’- TACAGCAAGCAGCCCAATGA-3’; TNK1 F-5’-TTCCTGCGAGAGGTATCGGT-3’, TNK1 R-5’- ATTGTGGAGTCGGGGCTGTG-3’; PDGFRA F-5’- GACGGTCTTGGAAGTGAGCA-3’, PDGFRA R-5’- AACAGCACAGGTGACCACAA-3’; PDGFRB F-5’- ACAGACTCCAGGTGTCATCC-3’, PDGFRB R-5’- GGTGCGGTTGTCTTTGAACC-3’; RET F-5’- TGAAGCTCGTTCATCGGGAC-3’, RET R-5’- TCGTGAGTGGTACAGGACTCT-3’; HIPK4 F-5’- CAATGCGGTCTCCGACATGA-3’, HIPK4 R-5’- TCTGGCCTCTCAGCTTCCAT-3’; FLT4 F-5’- TGCTCGGAACATTCTGCTGT-3’, FLT4 R-5’- TAAAACACCTGGCCTCCTCG-3’; MST2 F-5’- GCCGGCGCCTAAGAGTAAA-3’; MST2 F-5’- TCGGCCACACAGTTATAGCC-3’. Notably, due to limited tissue availability of original tumor samples, in Figure 4 for the TU-BcX-49S tumor endogenous kinase expression panel, only duplicate samples could be analyzed for TNK1, PDGFRA, PDGFRB, RET, HIPK4, FLT4 and MST2.
Generation of PDX-derived cell lines
To generate cell lines derived from patient tumors, after resection small (2 × 2 mm2) explants of the primary (TU-BCx-4IC) or secondary tumor (passaged once in mice; TU-BCx-49S) were plated in adherent culture conditions in a 6-well plate with DMEM supplemented with 10% FBS. Cells grew out from the explants for 2–3 weeks; media was changed every 72 hours. When cell populations were observed, single populations were cloned into 96-well plates and expanded.
Flow cytometry for cancer stem cell populations
Circulating tumor cells were collected in whole blood with 0.5M EDTA (Gibco Invitrogen, Carlsbad CA), incubated in red blood cell lysis buffer (0.008% NH4Cl, pH 7.2–7.4; Sigma-Aldrich, St. Louis MO), and washed with PBS. Collected cells from the tumor and blood samples were placed in staining solution containing 1% Bovine Serum Albumin (BSA; Sigma-Aldrich) and 1% CD16/CD32 Mouse BD Fc BlockTM (BD Biosciences) in PBS. The following primary antibodies were used: Anti-human CD24 (APC), anti-human CD326 (EpCAM; PerCP-eFluor710) and anti-human/mouse CD44 (PE-Fluor 610) purchased from eBiosciences (San Diego, CA, USA). All cells from the blood were analyzed with a Galios Flow Cytometer (Beckman Coulter, Brea, CA, USA) running Kaluza software (Beckman Coulter). At least 5000 events were analyzed and reported as the mean ± SEM.
Generation of patient-derived organoids and live/dead fluorescence stain
When SCID/Beige mice implanted with TU-BcX-4IC tumors were passaged to maintain the tumor, small (2 × 2 mm2) explants were plated in 3D culture conditions. Cells were allowed to grow out from the explants for 7 days, which contained mixed cell and stromal populations. At that time, PDOs were transferred to a 96-well ULA spheroid plate (Corning, NY, Cat. No. 4515) and treated with GSK346294A or DMSO control. After 72 hours, media was removed and spheres were stained using the PromoKine live/dead staining kit (New York, USA). A mixture of Calcein-AM (2μM) and Ethidium homodimer (EthD)-III (5μM) was used to highlight live and dead cells. Stained cells were imaged with confocal microscopy and images were captured (8 images per well of adherent cells, 5 images per well of low-suspension cells). The 588nM excitation channel was used to identify red, ‘dead’ cells, and the 420nM excitation channel was used to visualize green, ‘live’ cells. Representative images were taken at 100x magnification.
In vivo treatment of TU-BcX-4IC implants
Female SCID/Beige mice (n=5/group) were inoculated with TU-BcX-4IC tumor implants (3 × 3 mm2) in the MFPs. For these studies, mice were treated with GSK346294A (125 mg/kg) or DMSO vehicle control twice a day. This dose was generated from pharmacokinetic and pharmacodynamic data. Treatments were initiated immediately after tumor implantation (after 24 hours) due to the rapid growth rate and tumor take of this PDX model. After tumors reached 850 mm3 in volume. Twenty days after survival surgery, mice were euthanized, and lungs, livers and peripheral blood was harvested. Peripheral blood removed from the intracardiac region and stored in EDTA at −20 °C until analyzed for CSC populations with flow cytometry. Lungs and livers were formalin fixed, paraffin-embedded, sectioned and stained with H & E to visualize metastatic lesions. Metastases were quantified using ImageScope and recorded. Resected tumors were flash frozen and saved for qRT-PCR analyses. RNA was extracted from the tumors as previously mentioned.
In vivo inoculation of spheres
TU-BcX-4IC and MDA-MB-231 generation M2 spheres were pre-treated with GSK346294A (1 μM) or DMSO vehicle for 72 hours in low-attachment conditions. Single cell suspensions were created using TrypLe (Invitrogen), and cell viability was quantified before inoculation using the Trypan Blue stain and hemocytometer set-up. Only viable cells were used for inoculations; this step was necessary to exclude non-viable cells that may have been affected by potential anti-proliferative effects of the PKIS inhibitors. MDA-MB-231 spheres were inoculated in the MFP of SCID/Beige mice in a cell concentration-dependent manner (N = 5). Cell concentrations used were 1,000 cells, 10,000 cells, 50,000 cells, 100,000 cells and 200,000 cells. The mouse injected with the DMSO-pretreated cells at the 200,000 cells concentration did not survive and died 3 days after inoculation. TU-BcX-4IC sphere-derived cells were inoculated in 5 mice per group at the 100,000 cell concentration.
Hematoxylin & Eosin Staining
Livers, lungs and tumor tissues were processed by the Department of Histology and Pathology at Tulane University. As per standard protocol, formalin-fixed tissues were paraffin-embedded and sectioned at 4 μM and mounted on glass slides. Mounted sections were then exposed to xylene, ethanol, and acetic acid with intermittent washings with water before being stained with hematoxylin and eosin. After staining, slides were then again exposed to ethanol and xylene to complete the protocol.
Immunofluorescence Staining
GSK346294A- or DMSO-treated MDA-MB-231 and TU-BcX-4IC cells were fixed in formalin (10% buffered formalin phosphate, Fischer Scientific, Hampton NH) and permeabilized with Triton-X100 (MP Biomedicals, St. Ana CA). Cytoskeletal components were stained with phalloidin conjugated to AlexaFluor 555 (Cell Signaling, clone 8953, 1:200, Danvers MA). For proliferation studies, cells were stained with a primary conjugated antibody against Ki-67 (BD Biosciences; 1:200, San Jose CA). For both experiments, cells were counterstained with DAPI (NucBlue Fixed Cell Stain ReadyProbe, Life Technologies, Carlsbad CA). ApoTome (commercial structure illumination microscopy by Zeiss, Thornwood, NY) fluorescent images were captured on an inverted microscope (Zeiss) and digitally filtered to obtain optical slices. 5 images per well were captured at 400x, n = 3. For Ki-67 staining, quantified results are represented as percent positive Ki-67 staining (red) of total number of cells visualized by DAPI nuclear stain (blue).
Image-based morphometric analysis
Polygonal outline and length measurement tools provided in the AxioVision software (Zeiss) formed the basis for morphometric analysis. Based on information obtained from these tools, four metrics for cellular morphology were identified and defined (see below). Aspect ratio was determined through the use of the length measurement tool, and two perpendicular length measurements were reported. Cellular area coverage, nucleus: cytoplasm area ratio, and circularity assessments were determined from area and perimeter measurements obtained by the polygonal outline tool [20]. A total of 45 individual cells were measured from each treatment group, including the DMSO control. Selection criteria for cells included: 1) well-defined border (eliminates most cells in aggregates or dimly stained cells) and 2) must contain only one nucleus (eliminates dividing cells and cells out of the plane of focus).
RNA Sequencing
TU-BcX-4IC and MDA-MB-231 cells were treated with DMSO vehicle or GSK346294A for 48 hours (TU-BcX-4IC) or 24 hours (MDA-MB-231) at 1μM. Total RNA was extracted as previously described, and changes in gene expression were determined using next generation sequencing [41]. FASTQ files were aligned to the Homo sapiens/hg19 reference genome using the RNA-seq Alignment tool v1.1.1 in the Illumina’s BaseSpace. Raw counts were extracted and used to find genes differentially expressed using DESEQ2 v1.16.1 in R-Studio 1.1.383. The data was normalized using the Variant Stabilizing Transformation and log2. A p value of <0.05 was considered significant and was further corrected by FDR < 0.05.
Microscopy Imaging
The Nikon eclipse TE2000-s inverted fluorescence microscope and camera with x-cite series 120 illuminator (Nikon; Melville, NY), in conjunction with IP Lab version 3.7 software (Rockville, MD) were used in the visualization of crystal violet-treated cells to observe morphological changes.
Statistical analysis
Studies run in triplicate were analyzed by unpaired Student’s t-test (Graph Pad Prism V.4). p-Values < 0.05 were considered statistically significant. A one-way ANOVA test was used to compare data in the mammosphere area quantification experiments.
Supplementary Material
Acknowledgements
Thank you to our collaboration with Indiana University for performing the RNA sequencing performed in this project. Thank you to the Sequencing Core at Tulane University under the direction of Dr. Erik Flemington for analysis of the RNA sequencing results. We would also like to thank the Histology Core facilities at Tulane University for the hematoxylin and eosin stains of the tissue from the mice experiments. We would also like to thank the animal vivarium staff at Tulane University for their advice and assistance with the mice experiments. We are always grateful for the support of the Krewe de Pink organization in New Orleans. Finally, but most importantly, we thank the patients who donated their breast cancer tissue specimens to benefit breast cancer research; their contributions are valued and appreciated.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2018. CA Cancer J Clin. 2018;68:7–30. [DOI] [PubMed] [Google Scholar]
- 2.Yates LR, Knappskog S, Wedge D, Farmery JHR, Gonzalez S, Martincorena I et al. Genomic evolution of breast cancer metastasis and relapse. Cancer Cell 2017; 32(2): 169–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. New England Journal of Medicine 2010; 33:1938–48. [DOI] [PubMed] [Google Scholar]
- 4.Andreopoulou E, Kelly CM, McDaid HM. Therapeutic advances and new directions for triple-negative breast cancer. Breast Care 2017; 12:21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abramson VG, Mayer IA. Molecular heterogeneity of triple negative breast cancer. Curr Breast Cancer Rep 2014; 6(3):154–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bareche Y, Venet D, Ignatiadis M, Afitmos P, Piccart M, Rothe F, Sotiriou C. Unravelling triple-negative breast cancer molecular heterogeneity using an integrative multiomic analysis. Annals Oncol 2018; 29(4):895–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gross S, Rahal R, Stransky N, Lengauer C, Hoeflich KP. Targeting cancer with kinase inhibitors. J Clin Invest 2015; 125(5):1780–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cohen P, Alessi DR. Kinase drug discovery – what’s next in the field? ACS Chem Biol 2013; 8:96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garcia-Aranda M, Redondo M. Protein kinase targets in breast cancer. Int J Mol Sci 2017; 18(12):2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamaguchi H, Condeelis J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim Biophys Acta 2007; 1773(5):642–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olson MF, Sahai E. The actin cytoskeleton in cancer cell motility. Clin & Exp Metastasis 2009; 26:273. [DOI] [PubMed] [Google Scholar]
- 12.Ridley AJ. Life at the leading edge. Cell 2011; 145(7):1012–22. [DOI] [PubMed] [Google Scholar]
- 13.Al Moustafa AE. Epithelial-mesenchymal transition and its regulators are major targets of triple-negative breast cancer. Cell Adh Migr 2013; 7(5):424–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crown J, O’Shaughnessy J, Gullo G. Emerging targeted therapies in triple-negative breast cancer. Annals Oncol 2012; 23(6):vi56–vi65. [DOI] [PubMed] [Google Scholar]
- 15.Duong-Ly KC, Peterson JR. The human kinome and kinase inhibition as a therapeutic strategy. Curr Protoc Pharmacol 2013. Chapter 2: Unit 2.9. DOI: 10.1002/0471141755.ph0209s60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhullar KS, Lagaron NO, McGowan E, Parmar I, Jha A, Hubbard BP, Vasantha Rupasinghe HP. Kinase-targeted cancer therapies: progress, challenges and future directions. Mol Cancer 2018; 17(48). DOI: 10.1186/s12943-018-0804-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang L-C, Ross KE, Baffi TR, Drabkin H, Kochut KJ, Ruan Z, D’Eustachio P, McSkimming D, Arighi C, Chen C et al. Integrative annotation and knowledge discovery of kinase post-translational modifications and cancer-associated mutations through federated protein ontologies and resources. Sci Rep 2018; 8:6518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nguyen D-T, Mathias S, Bologa C, Brunak S, Fernandez N, Gaulton A, Hersey A, Holmes J, Jensen LJ, Karlsson A et al. Pharos: Collating protein information to shed light on the druggable kinome. Nucleic Acids Res 2017; 45:D995–D1002. DOI: 10.1093/nar/gkw1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jeon J, Nim S, Teyra J, Datti A, Wrana JL, Sidhu SS, Moffat J, Kim PM. A systematic approach to identify novel cancer drug targets using machine learning, inhibitor design and high-throughput screening. Genome Med 2014; 6(7):57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matossian MD, Elliott S, Hoang VT, Burks HE, Phamduy TB, Chrisey DB, Zuercher WJ, Drewry DH, Wells C, Collins-Burow B, Burow ME. Novel application of the Published Kinase Inhibitor Set to identify therapeutic targets and pathways in triple negative breast cancer subtypes. PLOS ONE 2017; 12(8):e0177802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Emmitte KA, Adjebang GM, Andrews CW, Badiang Alberti JG, Bambal R, Chamberlain SD, Davis-Ward RG, Dickson HD, Hassler DF, Hornberger KR et al. Design of potent thiophene inhibitors of polo-like kinase 1 with improved solubility and reduced protein binding. Bioorganic & Med Chem Let 2009; 19(6):1694–1697. [DOI] [PubMed] [Google Scholar]
- 22.Rosfjord E, Lucas J, Li G, Gerber H-P. Advances in patient-derived tumor xenografts: From target identification to predicting clinical response rates in oncology. Biochem Pharmacol 2014; 91(2):135–43. [DOI] [PubMed] [Google Scholar]
- 23.Aparicio S, Hidalgo M, Kung AL. Examining the utility of patient-derived xenograft mouse models. Nat Rev Cancer 2015; 15:311–16. [DOI] [PubMed] [Google Scholar]
- 24.Matossian MD, Burks HE, Elliott S, Hoang VT, Bowles AC, Sabol RA, Bunnell BA, Martin EC, Burow ME, Collins-Burow BM. Panobinostat suppresses the mesenchymal phenotype in a novel claudin-low triple negative patient-derived breast cancer model. Oncoscience 2018; 5(3–4): 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol 2000; 182(3):311–22. [DOI] [PubMed] [Google Scholar]
- 26.Lenart P, Petronczki M, Steegmaier M, Di Fiore B, Lipp JJ, Hoffmann M, Rettig WJ, Kraut N, Peters J-M. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr Biol 2007; 17(4):304–15. [DOI] [PubMed] [Google Scholar]
- 27.Kumar S, Kim J. PLK-1 targeted inhibitors and their potential against tumorigenesis. BioMed Res Int 2015. DOI: 10.1155/2015/705745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumar S, Sharma AR, Sharma G, Chakraborty C, Kim J. PLK-1: Angel or devil for cell cycle progression. Biochem Biophys Acta 2016; 1865(2):190–203. [DOI] [PubMed] [Google Scholar]
- 29.Miller I, Min M, Yang C, Tian C, Gookin S, Carter D & Spencer SL. Ki67 is a graded rather than a binary marker of proliferation versus quiescence. Cell Reports 2018; 24:1105–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee WC, Shi H, Poon Z, Nyan LM, Kaushik T, Shivashankar GV, Chan JK, Lim CT, Han J, Van Vliet KJ. Multivariate biophysical markers predictive of mesenchymal stromal cell multipotency. Proc Natl Acad Sci USA 2014; 111(42):e4409–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ball SG, Shuttleworth A, Kielty CM. Inhibition of platelet-derived growth factor receptor signaling regulates Oct4 and Nanog expression, cell shape, and mesenchymal stem cell potency. Stem Cells 2012; 30(3):548–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matossian MD, Burks HE, Bowles A, Elliott S, Hoang VT, Sabol RA, Pashos N, O’Donnell B, Miller K, Wahba B et al. A novel patient-derived xenograft in claudin-low triple negative breast cancer systems. Breast Cancer Res & Treat 2018; 169(2):381–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Findlay VJ, Wang C, Watson DK, Camp ER. Epithelial to mesenchymal transition and the cancer stem cell phenotype: Insights from cancer biology with therapeutic implications for colorectal cancer. Cancer Gene Ther 2014; 21(5):181–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abell AN, Johnson GL. Implications of mesenchymal cells in cancer stem cell populations: Relevance to EMT. Curr Pathobiol Rep 2014; 2(1):21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang S-S, Jiang J, Liang X-H, Tang Y-L. Links between cancer stem cells and epithelial-mesenchymal transition. Onco Targets Ther 2015. DOI: 10.2147/OTT.S91863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abbasi J Patient-derived organoids predict cancer treatment response. JAMA 2018; 319(4):1427. [DOI] [PubMed] [Google Scholar]
- 37.Vlachogiannis G, Hedayat S, Vatsiou A, Jamin Y, Fernandez-Mateo J, Khan K, Lampis A, Eason K, Huntingford I, Burke R et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018; 359(6378):920–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anastassiadis T, Deacon SW, Devarajan K, Ma H & Peterson JR. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat Biotechnol 2011; 29:1039–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elkins JM, Fedele V, Szklarz M, Azeez KRA, Salah E, Mikolajczyk J et al. Comprehensive characterization of the Published Kinase Inhibitor Set. Nat Biotechnol 2016; 34(1): 95–103 [DOI] [PubMed] [Google Scholar]
- 40.Rhodes LV, Antoon JW, Muir SE, Elliott S, Beckman BS, Burow ME. Effects of human mesenchymal stem cells on ER-positive human breast carcinoma cells mediated through ER-SDF-1/CSCR4 crosstalk. Mol Cancer 2010; 9:295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miller DF, Yan PS, Buechlein A, Rodriguez BA, Yilmaz AS, Goel S, Lin H, Collins-Burow B, Rhodes LV, Braun C et al. A new method for stranded whole transcriptome RNA-seq. Methods 2013; 63(2):126–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.