

Abstract
Aberrant changes in gene expression underlie the pathogenesis and progression of pressure-overload heart failure, leading to maladaptive cardiac hypertrophy, ventricular remodeling, and contractile dysfunction. Signaling through the G protein Gq triggers maladaptation and heart failure, in part through the activation of G protein-coupled receptor kinase-5 (GRK5). Hypertrophic stimuli induce the accumulation of GRK5 in the nuclei of cardiomyocytes, where it regulates pathological gene expression through multiple transcription factors including NFAT. The nuclear targeting of GRK5 is mediated by an amino-terminal (NT) domain that binds to calmodulin (CaM). Here, we sought to prevent GRK5-mediated pathology in pressure-overload maladaptation and heart failure by expressing in cardiomyocytes a peptide encoding the GRK5 NT (GRK5nt) that encompasses the CaM binding domain. In cultured cardiomyocytes, GRK5nt expression abrogated Gq-coupled receptor-mediated hypertrophy, including attenuation of pathological gene expression and the transcriptional activity of NFAT and NF-κB. We confirmed that GRK5nt bound to and blocked Ca2+-CaM from associating with endogenous GRK5, thereby preventing GRK5 nuclear accumulation after pressure overload. We generated mice that expressed GRKnt in a cardiac-specific fashion (TgGRK5nt mice), which exhibited reduced cardiac hypertrophy, ventricular dysfunction, pulmonary congestion, and cardiac fibrosis after chronic transverse aortic constriction. Together, our data support a role for GRK5nt as an inhibitor of pathological GRK5 signaling that prevents heart failure.
Introduction
Canonically, G protein-coupled receptor (GPCR) kinases (GRKs) regulate GPCR signaling through phosphorylation of the activated receptor, leading to the recruitment of β-arrestins and termination of the GPCR signal, either through receptor degradation or recycling(1–3). Seven members of the GRK family have been identified, with GRK2 and GRK5 being the most abundant in the heart. GRK2 and GRK5 appear to be crucial in the pathophysiology of heart failure (HF), because elevation of either GRK2 or GRK5 leads to exacerbated cardiac dysfunction in stress (4–8). Consistent with these findings, cardiac ablation of either GRK2 or GRK5 attenuates HF in response to stress (9–11).
Although both GRK2 and GRK5 have shared GPCR phosphorylation roles, the overall mechanisms by which they contribute to cardiac signaling and pathology are distinct. GRK2 is largely cytoplasmic in localization, and in addition to being an important regulator of β-adrenergic receptor signaling and contractility in the heart, is also involved in mitochondrial biology under stress conditions (5). In contrast, GRK5 is constitutively localized to the plasma membrane of cells through an amphipathic helix and PIP2 binding domain at the carboxyl-terminus (CT)(12). Mutation of residues 540–590 in the CT of GRK5 leads to improper membrane localization and a reduction in catalytic activity(13).
In addition to the CT domain-mediated regulation of localization, GRK5 contains an N-terminal calmodulin (CaM) binding domain that regulates its localization and kinase activity(14), as well as a nuclear localization (NLS) and export sequence (NES) within its catalytic domain (residues 186–467)(15). In the heart, GRK5 responds to selective hypertrophic stress by binding to CaM through its NT domain, disengaging from the membrane, and accumulating in the nuclei of cardiomyocytes(16) in a manner dependent upon its NLS. Elevated GRK5 in cardiomyocytes results in exacerbated cardiac hypertrophy and accelerated heart failure (HF) after ventricular pressure overload with transverse aortic constriction (TAC)(17). Mice with cardiac overexpression in mice of a mutant GRK5 that lacks the NLS do not show pathology post-TAC, demonstrating that this cardiac dysfunction and HF depends on the nuclear localization and accumulation of GRK5 after hypertrophic stress(17, 18). Similarly, the loss of GRK5 expression in the heart attenuates the hypertrophic response and delays HF progression after TAC in mice(9). This may be clinically important because both GRK2 and GRK5 are upregulated in failing human myocardium(5, 6). However, GRK2 does not have an NLS and has no role in the nucleus, unlike GRK5, so the mechanisms that mediate protection upon cardiac ablation of GRK2 or GRK5 are distinct.
Mechanistic studies of GRK5-mediated pathology post-TAC have revealed that nuclear GRK5 has effects on hypertrophic gene transcription. First, nuclear GRK5 acts as a histone deacetylase (HDAC) kinase, specifically phosphorylating HDAC5 after TAC, which releases repression on myocyte enhancer factor 2 (MEF2) and its hypertrophic gene transcriptional activity(17). Further, nuclear GRK5 facilitates nuclear factor of activated T-cells (NFAT) activity in the hypertrophying cardiomyocyte either by direct binding to NFAT or nearby DNA(19). These non-canonical actions of GRK5 in the nucleus depend on the calcium (Ca2+)-CaM (Ca2+-CaM) binding mentioned above because mutating the CaM-binding domain(14) within the NT of GRK5 prevents nuclear localization, HDAC5 kinase activity, and NFAT activation(16, 19).
CaM constitutes a critical signaling node in nearly every cell type, including cardiomyocytes, after elevation of intracellular Ca2+ which changes CaM’s conformation to enable binding to several interacting partners(20). Among these in the heart are the phosphatase calcineurin (CaN) and calmodulin kinase II (CAMKII). After TAC, the G protein Gq is the crucial trigger for the hypertrophic response(21) by activating Ca2+-CaM which stimulates CaN-NFAT(22), and which also promotes the nuclear translocation of GRK5 to direct hypertrophic gene transcription. Preventing the activation of Ca2+-CaM targets is protective against stress in the heart (23); however, no studies have investigated whether interfering with the inducible nuclear translocation of GRK5 alters response to stress in vitro and in vivo. In this study, we utilized an NT peptide from GRK5 (GRK5nt) to determine whether the nuclear translocation and subsequent pathology after hypertrophic stress of this kinase could be inhibited. Our results demonstrate that the pathological activity of GRK5 can be therapeutically targeted in the heart.
Results
Cardiac-Specific Transgenic GRK5nt Mice Exhibit Normal Baseline Cardiac Physiology
We included GRK5 residues 2–188 in the NT-peptide (Supp. Fig. 1a) (GRK5nt) to test whether potential competition of GRK5nt with endogenous GRK5 for Ca2+-CaM binding could affect cardiac physiology and pathophysiology. We generated transgenic mice expressing HA-tagged GRK5nt under the control of the ⍺MHC promoter(18) (TgGRK5nt). Immuno-blotting revealed positive HA expression only in the hearts of adult TgGRK5nt mice of the F1 generation and no expression in the liver as expected (Supp. Fig. 1b). Isolated adult cardiomyocytes from non-transgenic littermate control (NLC) and TgGRK5nt mice did not differ in morphology or cell size (Supp. Fig 2a–b). Furthermore, we found that the presence of GRK5nt did not affect the subcellular distribution or total levels of endogenous GRK5, assessed by western blotting of subcellular fractions (Supp. Fig. 2c–d). To assess whether chronic expression of the GRK5nt peptide in the heart had a physiological impact, we performed terminal hemodynamics and standard M-Mode echocardiography after an isoproterenol stress test on adult (10–14 weeks old) mice. We found that TgGRK5nt mice exhibited a similar hemodynamic response to isoproterenol compared to and non-transgenic littermate control (NLC) mice (Supp. Fig. 2e–g) and that cyclic adenosine monophosphate (cAMP) generation following isoproterenol stimulus in neonatal rat ventricular myocytes in vitro was not affected by adenoviral GRK5nt expression (Supp. Fig. 1c). We observed no significant differences in basic echocardiographic parameters, including cardiac function and dimensions (Supp. Fig. 2h–l), and gravimetrics revealed no differences in heart weight to tibial length (HW/TL) between TgGRK5nt and NLC mice (Supp. Fig. 2M), suggesting that this peptide does not alter heart growth and development up to adulthood when expressed in mice.
TgGRK5nt Mice Exhibit Attenuated Maladaptive Hypertrophy and HF Following Chronic TAC
Because GRK5 signaling is critically involved in cardiac hypertrophy and HF development after pressure-overload(16, 17), we tested whether GRK5nt expression in the hearts of mice altered maladaptive hypertrophy and HF development after TAC. Accordingly, we assessed cardiac function 8 weeks after TAC surgery or a sham procedure. No mice died following sham surgery, and a total of two mice per genotype died following TAC within the same day or 1 day following surgery, which we attribute to surgical complications. We found no statistically significant differences in heart rate among groups during image acquisition (Supp. Fig. 3a). Aortic pressure gradients as measured by echocardiography revealed that the amount of LV pressure overload and therefore the amount of mechanical stress imposed was similar between the two groups (Supp. Fig. 3b). Assessment of cardiac function by echocardiography revealed that NLC mice had significantly impaired systolic function, as measured by LV ejection fraction and fractional shortening, 8 weeks post-TAC compared to sham controls; however, this LV dysfunction was not observed in TgGRK5nt mice post-TAC (Fig. 1a–b). Further, we found that NLC post-TAC mice exhibited the expected increase in left ventricular posterior wall (LVPW) thickness compared to corresponding sham mice consistent with hypertrophy, an increase that was significantly attenuated in TgGRK5nt mice (Fig. 1c).
Fig. 1. Transgenic GRK5nt mice exhibit attenuated heart failure following chronic TAC.
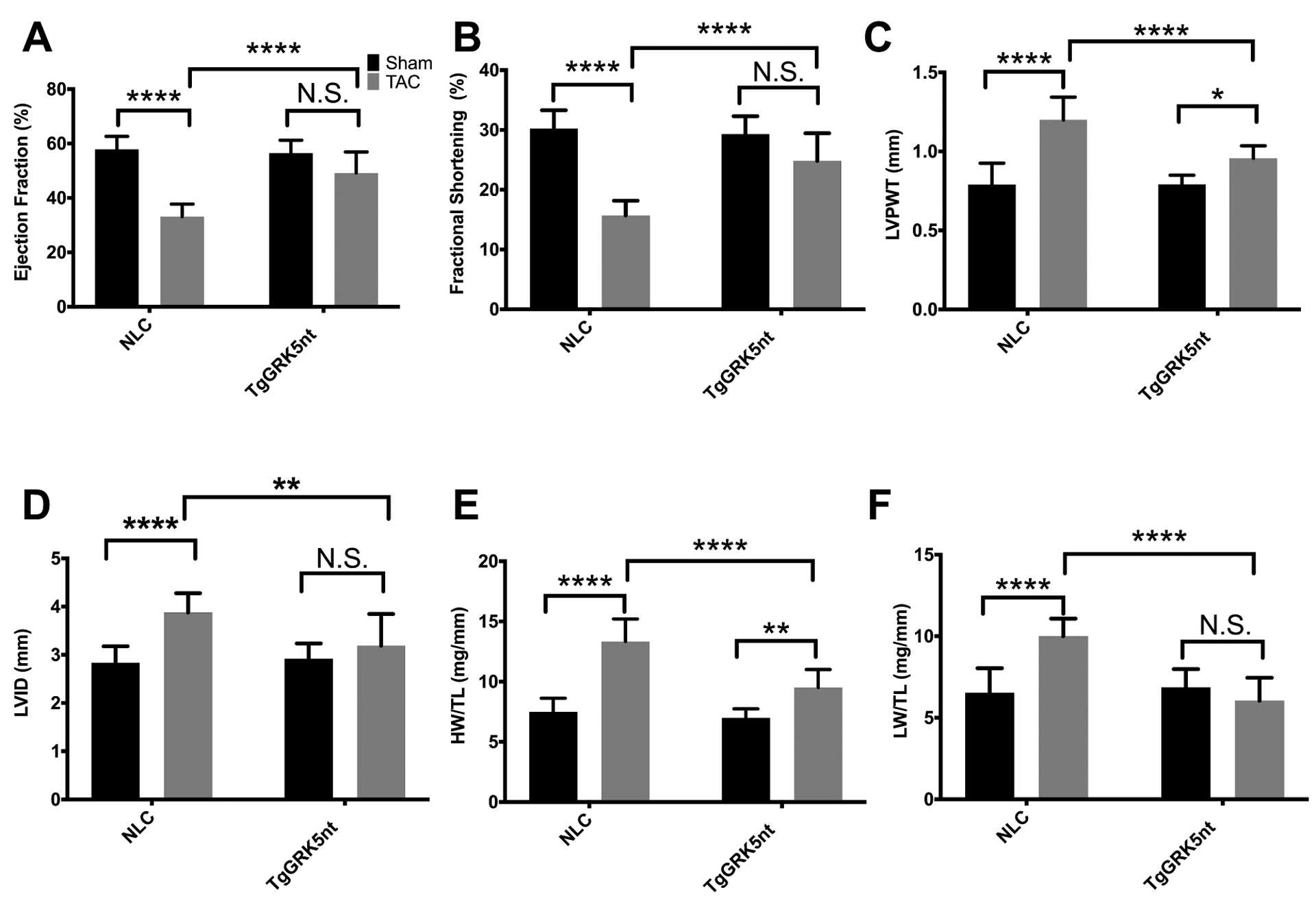
(A–D) Echocardiographic analysis of cardiac function in NLC and TgGRK5nt mice 8 weeks post- TAC or sham operation. Ejection fraction % (A), fractional shortening % (B), posterior wall thickness (C), and left-ventricular internal diameter (LVID) (D). (E and F) Heart weight (E) and lung weight (F) indexed to tibia length (HW/TL, LW/TL). N=10 NLC Sham mice, N=11 NLC TAC mice, N=7 TgGRK5nt Sham mice, N=7 TgGRK5nt TAC mice for (A) to (F). (* p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001, two-way ANOVA with Tukey’s post-hoc testing for multiple comparisons).
We found that, 8 weeks post-TAC, TgGRK5nt mice did not exhibit LV dilatation, an additional echocardiographic parameter that indicates a HF phenotype post-TAC, unlike NLC TAC mice (Fig. 1d). In agreement with echocardiographic measurements of posterior wall thickness, gravimetric determination of HW/TL also suggested attenuated TAC-induced hypertrophy in TgGRK5nt compared to NLC mice subjected to TAC (Fig. 1e). Another common symptom of HF is pulmonary congestion, so we assessed the wet lung weight to tibial length (LW/TL) ratios in our mice. Consistent with the above data, we found significant increases in LW/TL in post-TAC NLC mice compared to sham-operated mice that were not evident in TgGRK5nt mice (Fig. 1f). Together ,these data support a protective role of GRK5nt expressed in the cardiomyocyte against maladaptive hypertrophy and HF chronically after pressure overload stress.
Expression of GRK5nt Attenuates Cardiomyocyte Hypertrophy and Fibrosis Following Chronic TAC
To assess the attenuation of hypertrophy and HF post-TAC in TgGRK5nt mice histologically, we fixed and stained hearts with wheat germ agglutinin conjugate to highlight the borders of the cardiomyocytes. We found the expected increase in cardiomyocyte cross-sectional area (CSA) in NLC post-TAC mice compared to sham-operated control mice, and consistent with our in vivo findings, cardiomyocyte CSA in TgGRK5nt mice 8-week post-TAC was significantly lower than that in NLC TAC group (Fig. 2a–b). Moreover, we assessed cardiac fibrosis, another feature of maladaptation post-TAC, and found that 8 weeks after TAC, TgGRK5nt mice had significantly less myocardial fibrosis compared to post-TAC NLC mice (Fig. 2c–e). Together, these data suggest that cardiomyocyte expression of GRK5nt attenuates the development of fibrosis and cardiomyocyte hypertrophy following chronic TAC.
Fig. 2. Transgenic GRK5nt mice exhibit attenuated hypertrophy and fibrosis following chronic TAC.
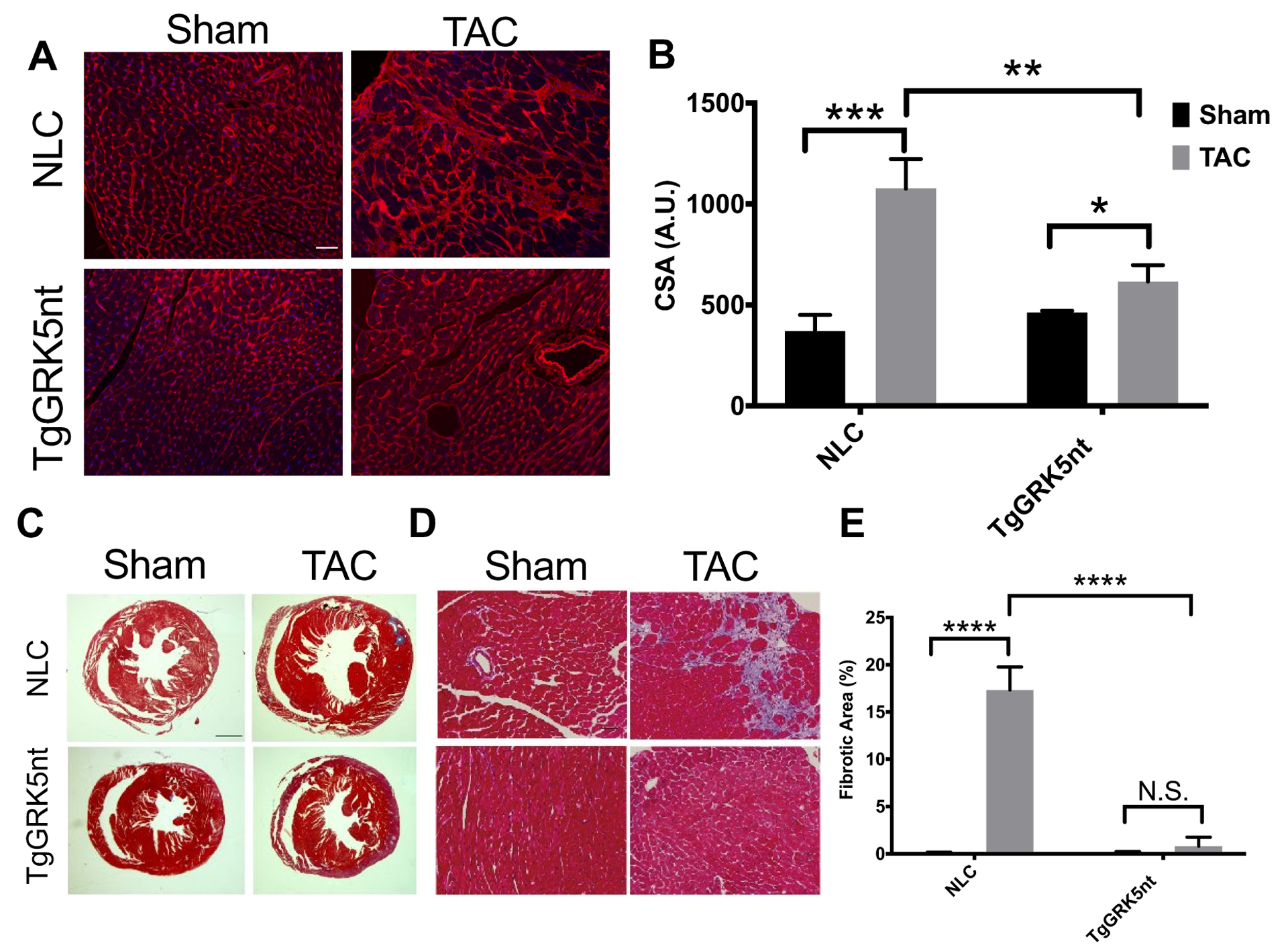
(A) Wheat germ agglutinin staining of heart sections from NLC and TgGRK5nt mice. (B) Planimetric assessment of cell surface area from wheat germ agglutinin-stained sections. (C–E) Masson Trichrome staining of heart sections from NLC and TgGRK5nt mice following sham operation or TAC surgery (C and D). Fibrotic area was normalized to total field tissue area to derive relative fibrotic area (E). Scale bar in (A,D) 100μm, (C) 1000μm. N=10 NLC Sham mice, N=11 NLC TAC mice, N=7 TgGRK5nt Sham mice, N=7 TgGRK5nt TAC mice for (A) to (D). (* p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001, two-way ANOVA with Tukey’s post-hoc testing for multiple comparisons in (B,E).
Expression of GRK5nt reduces early hypertrophic response and gene expression following TAC
Because GRK5 signaling contributes to both maladaptive hypertrophy and HF, we wanted to assess the hypertrophic response in TgGRK5nt in the absence of systolic dysfunction and HF. We therefore assessed cardiac function at an earlier time point after subjecting mice to TAC surgery or a sham procedure. 2 weeks after TAC, we observed no differences in systolic function as assessed by LV ejection fraction and fractional shortening, nor did we observe chamber dilatation as assessed by LV internal diameter (Fig. 3a–c). Although they did not show systolic dysfunction, NLC TAC mice exhibited a significant elevation in LV posterior wall thickness characteristic of hypertrophy compared to sham-operated controls (Fig. 3d). This hypertrophic response was not observed in TgGRK5nt TAC mice, which showed a significant reduction in posterior wall thickness compared to NLC TAC mice. Consistent with echocardiographic assessment, NLC TAC mice exhibited elevated HW/TL ratios compared to sham-operated controls that was not observed in TgGRK5nt mice (Fig. 3e), although the difference between NLC TAC and TgGRK5nt TAC HW/TL was not significant. Another characteristic of cardiac hypertrophy is the expression of fetal genes. We observed a significant increase in the expression of ANKRD1, MYH7, and a trend towards an increase in expression of BNP in NLC mice 2 weeks after TAC compared to sham-operated controls (Fig. 3f–h). The induction of MYH7 and ANKRD1 was significantly reduced in TgGRK5nt TAC mice compared to NLC TAC mice, whereas there was a similar elevation in BNP. We also found a small but statistically significant elevation in GRK5 expression in both genotypes (Fig. 3i), but no differences in GRK2 expression 2 weeks after TAC (Fig. 3j). Together, these data suggest that expression of GRK5nt reduces, but does not entirely prevent, early hypertrophic response following TAC.
Fig. 3. GRK5nt expression reduces the early hypertrophic response following TAC.
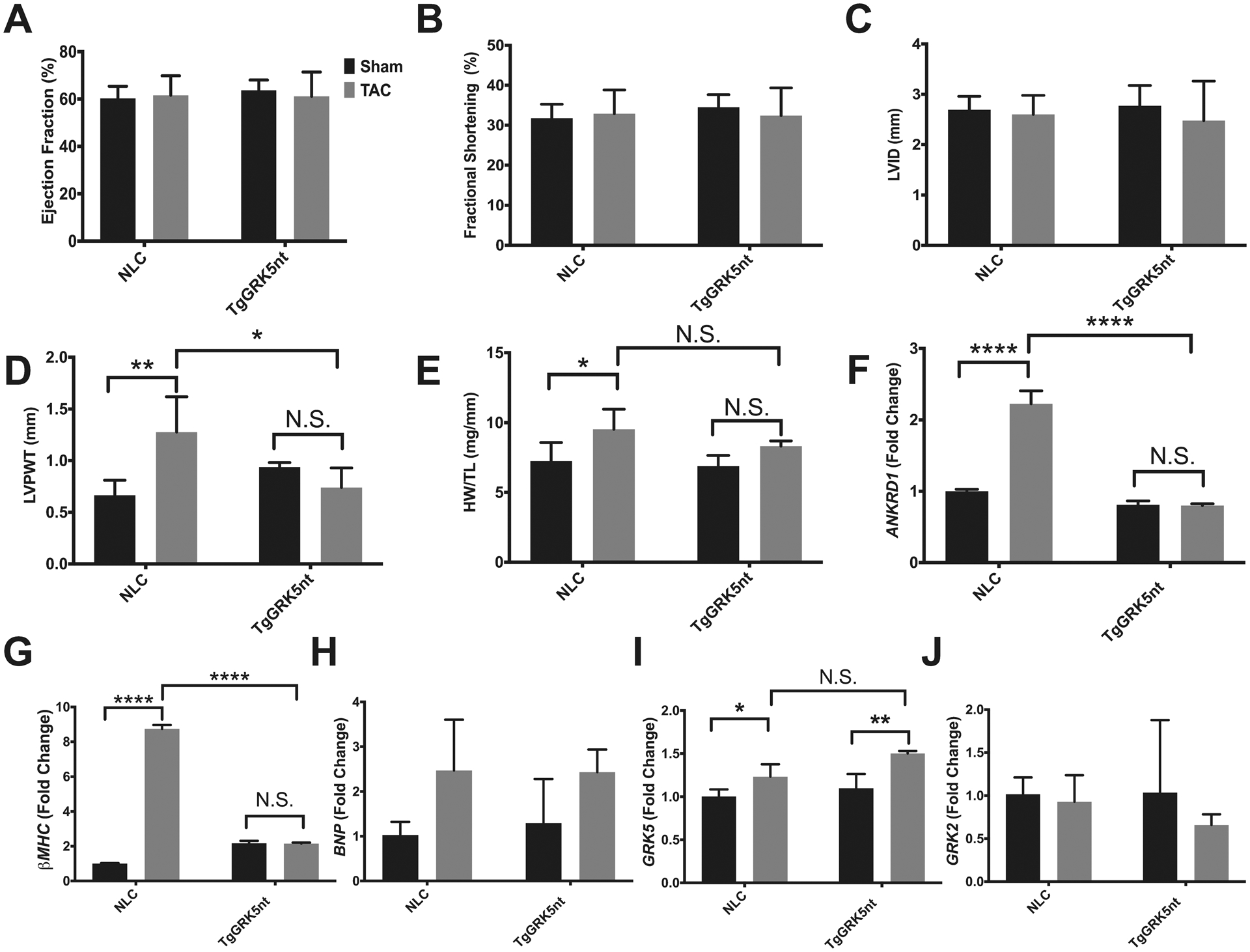
(A–D) Echocardiographic assessment of cardiac function 2 weeks post-TAC or sham operation. Ejection fraction % (A), fractional shortening % (B), left-ventricular internal diameter (LVID) (C), and posterior wall thickness (D). (E) heart weight to tibia length. (F to J) Ankrd1 (F), βMHC (G), BNP (H), GRK5 (I), and GRK2 (J) expression relative to 18s rRNA. N=4 NLC Sham mice, N=5 NLC TAC mice, N=4 TgGRK5nt Sham mice, N=4 TgGRK5nt TAC mice for (A) to (J). (* p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001, two-way ANOVA with Tukey’s post-hoc testing).
Expression of GRK5nt in vitro protects against cardiomyocyte hypertrophy and hypertrophic gene expression
We expressed FLAG-tagged GRK5nt in cultured cardiomyocytes to investigate the mechanisms by which this peptide may interrupt hypertrophy. GRK5nt expressed in neonatal rat ventricular myocytes (NRVMs) exhibited a broad subcellular distribution, as assessed by Western blotting of subcellular fractions and immunocytochemistry for FLAG (Fig. 4 a–b). We found that ⍺-adrenergic-mediated cardiomyocyte hypertrophy, induced by 48 hours of phenylephrine (PE), was significantly attenuated when the GRK5nt peptide was present compared to control cardiomyocytes (Fig. 4c–d). In addition, the presence of GRK5nt in NRVMs reduced the PE-induced increase in the expression of ANP, a fetal gene marker of hypertrophy (Fig. 4e). Further, we found that GRK5nt expression prevented a PE-induced increase in protein to DNA ratio, an index of cell size (Fig. 4f). Thus, the GRK5nt peptide is effective in vitro as well as in vivo to limit the consequences of hypertrophic stress.
Figure 4. GRK5nt expression in neonatal rat ventricular myocytes attenuates phenylephrine-induced cellular hypertrophy.
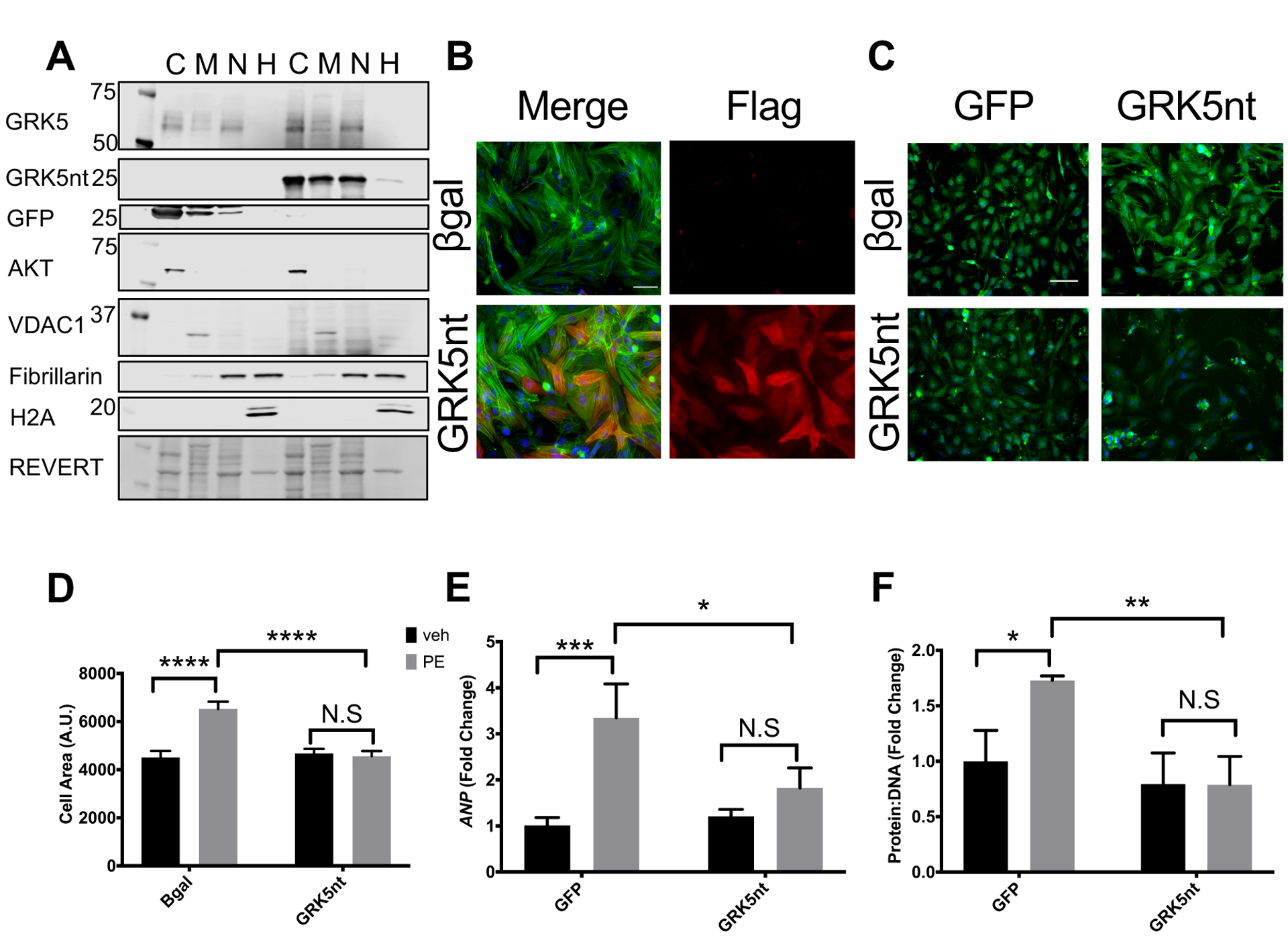
(A and B) Western blotting of GRK5 in subcellular fractions (C=cytosol, M=membrane, N=soluble nuclear, H=chromatin) (A) or Flag immunocytochemistry (B) of NRVMs infected with control (βGal or GFP) or GRK5nt adenovirus for 48 hrs. (C and D) Immunocytochemistry of α-sarcomeric actin (C) and quantified cell area (D) of NRVMs expressing control virus or GRK5nt stimulated with 100μM phenylephrine for 48 hrs. (E and F) Expression of ANP (E) and protein to DNA ratio (F) in control or GRK5nt-expressing NRVMs following 48 hrs of phenylephrine stimulation. Scale bar in (B) 100 μm, (C) 200 μm. N=3 independent cell isolations for each condition for (A) to (F). (* p<0.05, **** p<0.0001, two-way ANOVA with Tukey’s post-hoc testing (D–F)).
GRK5nt binds to Ca2+-calmodulin and alters NFAT signaling and GRK5 nuclear accumulation following in vivo hypertrophic stress
The NT of GRK5 contains a binding site for CaM that alters GRK5 association with the plasma membrane when Ca2+-CaM is bound(24). The GRK5nt sequence encompasses this CaM-binding region of GRK5, so we sought to determine whether the GRK5nt construct bound to CaM. CaM-conjugated beads were added to lysates of NRVMs expressing GFP as a control or GRK5nt. We found that in the presence of Ca2+, CaM-conjugated beads captured the GRK5nt peptide, but not when EDTA was present, suggesting that the interaction between GRK5nt and calmodulin depended on Ca2+-CaM and not apo-CaM (Fig. 5a–b). Because GRK5 is a known target of Ca2+-CaM, we also assessed the pulldown of full-length endogenous GRK5 in the presence of GFP or GRK5nt. We found that less Ca2+-CaM bound to the endogenous pool of full-length GRK5 from GRK5nt-expressing cell lysates than to that from GFP-expressing cell lysates (Fig. 5c), suggesting that GRK5nt competes with full-length GRK5 for binding to CaM. In agreement with our observations using NRVM lysates, GRK5nt from heart lysates from TgGRK5nt mice exhibited Ca2+-selective binding to Ca2+-CaM, and less endogenous GRK5 from heart lysates from TgGRK5nt mice compared to those from NLC mice bound to Ca2+-CaM (Fig. 5d–f). However, the binding of CaM kinase II (CaMKII) to CaM was not affected (Fig. 5g), suggesting that GRK5nt does not disrupt the binding of all Ca2+-calmodulin binding partners. GRK5-CaM binding triggers the disengagement of GRK5 from the membrane and its nuclear translocation and facilitation of NFAT activity(19). We next sought to determine whether the observed interference in binding of Ca2+-CaM to endogenous GRK5 in the presence of GRK5nt affected downstream signaling. We found that GRK5nt expression did not affect the acute response to PE-stimulation, with similar levels of phosphorylation of ERK observed in GRK5nt-expressing NRVMs compared to GFP-expressing NRVMs (Fig. 5h–i). Furthermore, to determine whether CaMKII signaling was affected by GRK5nt, we assessed CaMKII-mediated phosphorylation of CREB at Ser133 (25). PE-induced phosphorylation of CREB was similar in GRK5nt- and GFP-expressing NRVM(Fig. 5h–j), suggesting a similar CaMKII response in vitro. We found that GRK5nt expression prevented the PE-induced increase in NFAT activity as assessed with a luciferase reporter (Fig. 5k), in agreement with our data showing GRK5nt binds to CaM, a prerequisite for GRK5 to facilitate NFAT activity. GRK5 promotes NF-κB activity (26), and we found that GRK5nt reduced the PE-induced increase in NF-κB activity as assessed with a luciferase reporter (Fig. 5l). We next sought to determine whether Ca2+-CaM binding to GRK5nt prevented the nuclear accumulation of GRK5 after TAC. Two weeks after TAC, we saw that NLC mice but not TgGRK5nt TAC mice exhibited an increase in GRK5 in the nuclear fraction compared to sham controls (Fig. 5m–n). Together, our data show that GRK5nt acts to suppress CaM and GRK5 signaling through direct binding to CaM, thereby preventing the binding of endogenous GRK5 to CaM and the transduction of non-canonical GRK5 signaling in hypertrophic stress.
Fig. 5. GRK5nt binds to Ca2+-CaM selectively and alters NFAT signaling and GRK5 nuclear accumulation following hypertrophic stress.
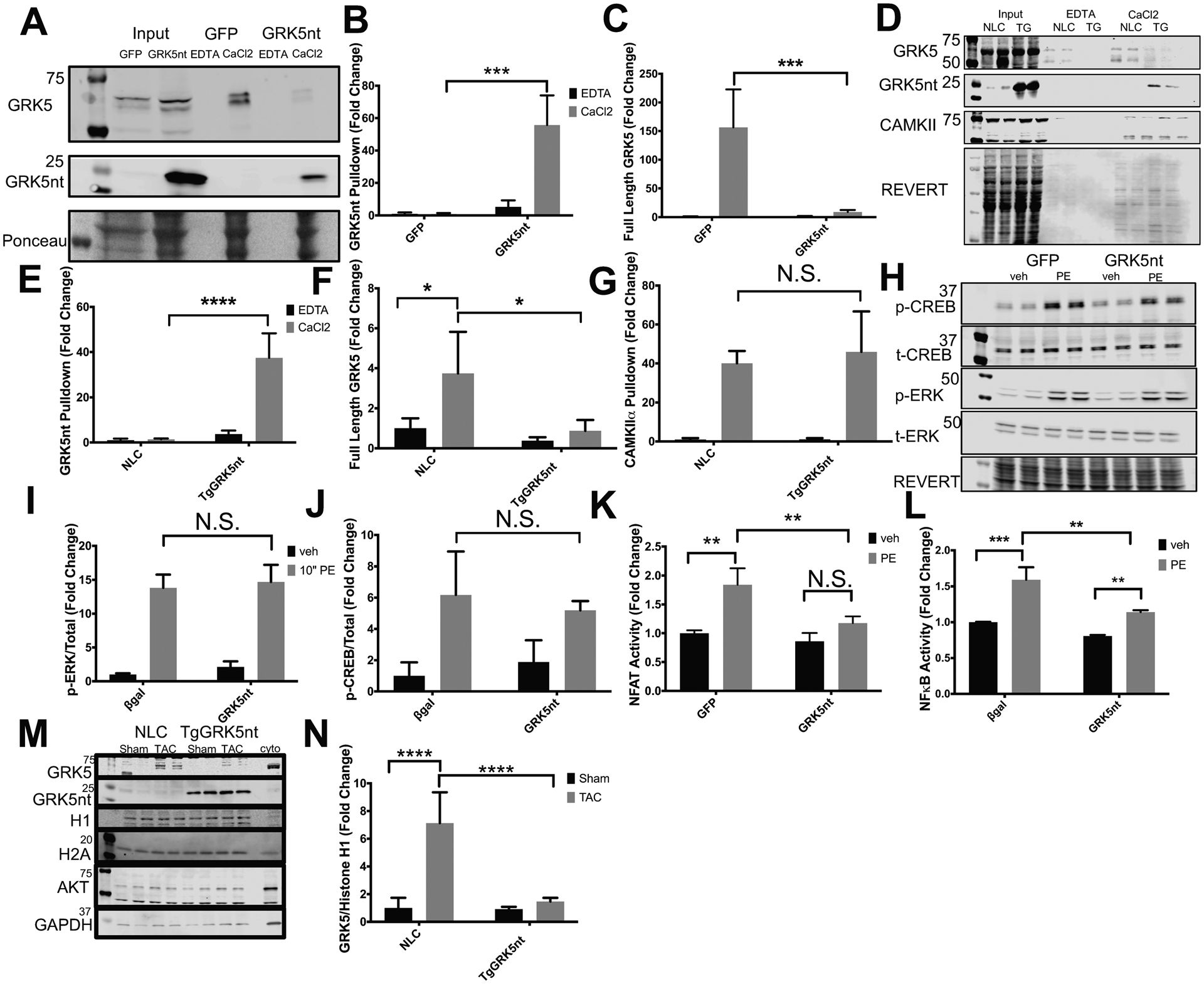
(A) Western blot of GFP and GRK5nt lysates with CaM pulldown in the presence of EDTA or CaCl2. (B and C) Quantification of CaM pulldown of GRK5nt (B) or full-length GRK5 (C). (D) Western blot of CaM pulldown in NLC and TgGRK5nt lysates. (E–G) Quantification of CaM pulldown of GRK5nt (E), full-length GRK5 (F), or CAMKII (G). (H) Western blot of response to 10 min of PE stimulation in NRVMs expressing GFP or GRK5nt. Quantification of phospho-ERK (I) and phospho-CREB (J). Assessment of NFAT (K) or NFkB (L) luciferase activity in GFP or GRK5nt-expressing NRVMs following 24 hrs of 100μM PE treatment. (M–N) Western blotting (M) and quantification (N) of nuclear GRK5 in fractions from NLC or TgGRK5nt hearts two weeks after TAC. Data were normalized to the nuclear marker Histone H1. N=3 lysates per infection condition from independent cell isolations for (A–C). N=3 NLC heart lysates and N=4 TgGRK5nt heart lysates for (D–G). N=3 independent cell isolations for (H–L). N=4 NLC Sham mice, N=5 NLC TAC mice, N=4 TgGRK5nt Sham mice, N=4 TgGRK5nt TAC mice for (M–N). (* p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001, two-way ANOVA with Tukey’s post-hoc testing).
Discussion
Following its discovery, GRK5 has been extensively characterized as a kinase responsible for the phosphorylation and subsequent desensitization of GPCRs including important receptors in the heart such as β-adrenergic receptors (βARs) (3, 27–29). This canonical activity of GRK5 leads to GPCR signal termination through β-arrestin recruitment to the phosphorylated receptor (30). GRK5 appears to be a key molecule in the heart because it is upregulated in human HF and in animal models (5). Moreover, increased GRK5 expression in the heart could contribute to loss of βAR-mediated cardiac contractility (31), and thus its up-regulation in models of ventricular dysfunction could contribute to loss of inotropic reserve, a hallmark of HF through excessive desensitization of βARs. Animal models of HF induction appear to support this mechanism of GRK5-mediated pathology because cardiac-specific GRK5 overexpressing transgenic mice have exaggerated hypertrophy and early HF after LV pressure overload induced by TAC (17) and this post-TAC chronic pathology is attenuated in GRK5 knockout (KO) mice (9). However, these studies with genetically engineered GRK5 mice have revealed that the canonical actions of GRK5 are not responsible for post-TAC pathology and HF and non-canonical, nuclear localization and activity of GRK5 appears to be the underlying mechanism (9, 16, 17, 19). As mentioned above, nuclear accumulation of GRK5 in a manner dependent on its NLS occurs in cardiomyocytes after hypertrophic stress (17). The ultimate importance of this non-canonical mechanism in post-TAC maladaptation and HF has been demonstrated in transgenic mice with cardiac-specific overexpression of a form of GRK5 with a defective NLS which do not show hypertrophy and HF after TAC (17).
In addition to TAC, GRK5 accumulates in the nucleus of cardiomyocytes of spontaneously hypertensive heart failure (SHHR) rats (32). Further, treatment of adult rat cardiomyocytes with the PKC activator PMA leads to an increase in the nuclear accumulation of GRK5 (32, 33), suggesting that PKC may be part of the mechanism for GRK5 nuclear accumulation. Indeed, we found that the binding of Ca2+-CaM to a region in the GRK5 NT domain, which can occur after Gq activation downstream of hypertrophic stress, mediates nuclear accumulation (16). Nuclear GRK5 has several non-canonical actions, which we have found to contribute to HF development after hypertrophic stress. This includes GRK5 being a Class II HDAC kinase, which following phosphorylation of HDAC5 causes the de-repression of the hypertrophic gene transcription factor MEF2(17). In addition, GRK5 facilitates NFAT activity after hypertrophic stress(19). Enhancement of NFAT activity in cardiomyocytes may not be kinase-dependent(19), which is supported by the ability of GRK5 to directly bind to DNA (15) and may indeed may not only be a co-factor for NFAT activity but its own transcriptional regulator. Moreover, GRK5 interacts with IκB(34), α-synuclein (35), p53(36, 37), Hip(38), as well as NFAT(20), interactions that suggest non-GPCR actions. Further, the subcellular localization of GRK5 outside of plasma membrane/sarcolemmal distribution not only includes the nucleus, but also within Lewy Bodies(35) and centrosomes(36).
Regardless of exact nuclear mechanisms responsible for maladaptive non-canonical actions of GRK5 promoting HF after TAC, non-canonical actions of GRK5 in the heart appear to depend on the nuclear localization of this kinase after stress. Thus, how GRK5 accumulates in the nucleus of cardiomyocytes is crucial for potentially targeting this pathology therapeutically. Because we have discovered that the nuclear localization of GRK5 requires Ca2+-CaM binding to a region within the NT domain of GRK5, we posited that expression of a peptide incorporating this region of GRK5 may compete with endogenous GRK5 for Ca2+-CaM binding after hypertrophic stress and prevent the pathological mechanisms of this kinase. We therefore expressed GRK5 residues 2–188 (Supplemental Fig. 1), which includes the known NT binding site for Ca2+-CaM, in cardiomyocytes in vitro and mouse hearts in vivo. We found that the GRK5nt peptide specifically bound only to Ca2+-activated CaM and that GRK5nt could compete off Ca2+-CaM bound to endogenous GRK5. GRK5nt expression in cardiomyocytes prevented adrenergic-mediated cell hypertrophy and we found that a contributing mechanism included blocking NFAT activity downstream of hypertrophic stimuli. These molecular actions of GRK5nt expression during cardiomyocyte hypertrophy appeared to be due in part to inhibition of GRK5 nuclear translocation because substantial nuclear accumulation of GRK5 occurred in NLC hearts after TAC as expected and as previously shown(17), but not in TgGRK5nt hearts after TAC. Moreover, GRK5nt expression in transgenic mouse hearts did not alter baseline cardiac contractile or morphological parameters, which may also mean that any disruption of basal Ca2+-CaM signaling due to this peptide does not lead to an overt phenotype.
In contrast to the innocuous action of GRK5nt expression throughout the developing mouse to adulthood, TgGRK5nt mice showed a substantially different phenotype compared to NLC mice after in vivo hypertrophic stress induced by TAC. We found that, as in cardiomyocytes in vitro, the GRK5nt peptide blocked hypertrophy in vivo and maladaptation after TAC and the critically important transition from hypertrophy to HF. Indeed, TgGRK5nt mice subjected to chronic TAC were substantially protected against several characteristics of HF, including reduced contractility, ventricular hypertrophy, chamber dilatation, fibrosis, and pulmonary congestion. The primary mechanism of this peptide in vivo appears to be blocking Ca2+-CaM binding and nuclear translocation of endogenous GRK5 preventing maladaptive gene transcription. This is important because potential gene therapy use of the GRK5nt does not appear to interfere with the classical GPCR activity of GRK5, as determined by βAR inotropy in adult mice and also cAMP accumulation after βAR stimulation, but prevents its non-canonical pathology activity in the nucleus. Together, these data suggest that GRK5nt may be selective in inhibiting GRK5’s actions in the heart. Future studies will need to investigate its gene therapy potential.
Our results are consistent with the hypothesis that GRK5nt suppresses cardiac pathology by preventing the transduction of Ca2+-CaM and GRK5-dependent actions in the nucleus, including activation of NFAT and NFkB. Other GRK5 targets that could also contribute are HDAC5 phosphorylation and subsequent de-repression of MEF2. Expression of an NT peptide of GRK5 using an adenovirus and myocardial injections into spontaneously hypertensive rats reduces cardiac hypertrophy and p65 activation and NFAT de-phosphorylation following phenylephrine stimulation in H9c2 myoblasts (39, 40). Although these results are consistent with our observed anti-hypertrophic phenotype in TgGRK5nt mice post-TAC, these rat studies were limited in scope due to the lack of overt cardiac dysfunction and HF in the spontaneously hypertensive rats.
One thing not established in our study is whether the Ca2+-CaM binding of the GRK5nt peptide in vivo in cardiomyocytes is GRK5 specific or whether other Ca2+-CaM events are disrupted. Some of the possible targets downstream of Ca2+-CaM would be CaMKII and calcineurin, both of which have been implicated in pathological hypertrophy (22, 23). We found that CaM binding to CaMKII was not disrupted by the presence of GRK5nt, and phosphorylation of CREB, which is directly downstream of CaMKII in Gq signaling, was also not affected by GRK5nt expression, consistent with a prior report (25). Thus, although GRK5nt does disrupt CaM binding to GRK5 and reduces its nuclear accumulation in hypertrophic stress, it may not disrupt CaMKII signaling, suggesting some selectivity of GRK5nt for disruption of GRK5 over other CaM substrates. In addition, the pool of Ca2+-CaM that appears to bind to GRK5 during hypertrophy may be a distinct pool from that which binds to the cardiac ryanodine receptor(41), although we did not identify the in vivo subcellular pool of CaM that is targeted by GRK5nt. Finally, regardless of other potential mechanisms, we showed that the GRK5nt peptide blocked nuclear accumulation of GRK5 in cardiomyocytes in vivo after hypertrophic stress, a mechanism that leads to delayed HF post-TAC.
Previous studies have used peptides to block hypertrophy and HF post-TAC by targeting Gq interactions, which is the nodal upstream mediator of cardiac hypertrophy. We have used a peptide that targeted the activation of G⍺q through activated hypertrophic GPCRs(21). We have used the RGS domain of GRK2 that binds to and inhibits Gαq activation(42). Cardiac-specific transgenic mice expressing each of these peptides presented with similar phenotype after TAC as our present TgGRK5nt mice(21, 42), which is unsurprising because Gq-activation leads to Ca2+-CaM activation directing GRK5 nuclear translocation.
In summary, we found that transgenic expression of GRK5nt results in substantial reduction of maladaptive cardiac hypertrophy and its transition to HF chronically in a mouse model of pressure overload. This included attenuation of functional, morphological and molecular phenotypes associated with chronic HF in TgGRK5nt mice. We established that GRK5nt blocked hypertrophy and HF chronically after TAC, in part by competing for Ca2+-CaM binding to endogenous GRK5 (Supp. Fig. 4). This study supports the potential use of GRK5nt as a gene therapy strategy to limit HF development in conditions and models of pressure-overload stress such as hypertensive heart disease or valvular insufficiency. Our findings may also enable technology and development strategies to modulate non-canonical actions of GRK5 to a greater extent than its canonical GPCR desensitization activity.
Materials and Methods
Experimental Animals and Procedures
All animal procedures were carried out according to National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Animal Care and Use Committee of Temple University. To generate cardiac-specific GRK5nt transgenic mice (TgGRK5nt), the DNA encoding human GRK5 residues 2–188 was subcloned from mammalian expression plasmid pcDNA-HsGRK5 into a vector driven by the MYH6 (ɑMHC) promoter(43) with an HA-epitope tag on the CT. For in vivo experiments, non-transgenic littermate control (NLC) mice were used as controls. Baseline echocardiographic studies were performed with a mixed male and female population, whereas adult 8–10-week-old males were used for surgeries. Echocardiography was performed as described previously(44) using a VevoSonic 2100 imaging system with M550D transducer for assessment of cardiac function. Mice were anesthetized by 3% v/v isoflurane inhalation and were placed on a pre-heated stage. Heart rate was maintained above 450 beats per min (bpm) and short axis images were acquired at the midventricular papillary muscle level for M-Mode imaging.
Hemodynamic measurements were performed as described(45) with a catheter-based pressure recording system. Briefly, mice were anesthetized with Avertin (2% Tribromoethanol) and the right carotid artery was cannulated with a catheter/pressure transducer (1.4 French micro-manometer Millar Instruments, Houston, TX), and the catheter was advanced into the left ventricle. Another catheter was inserted into the jugular vein for administration of isoproterenol. After recording the baseline hemodynamic parameters, myocardial responses to increasing doses of isoproterenol (0.1 ng, 0.5 ng, 1 ng, 5 ng, 10 ng) were performed. Data were recorded and analyzed in PowerLab software (AD Instruments).
Transverse aortic constriction (TAC) was performed as described previously(17). To measure transaortic pressure gradients to assess success of TAC surgery, echocardiography was performed at 8 weeks post-surgery for all sham and TAC groups, using a M250D probe to obtain an image of the complete descending aorta. Pulse-wave doppler echocardiography was performed using a pulse-wave angle of 15–20 degrees. Blood velocity across the descending aorta was measured and extrapolated to pressure using VisualSonics analysis software. TAC animals with peak transaortic pressure gradients below 45 mmHg were excluded from analysis. Animals were sacrificed at the end of each study and gravimetrics for both heart and wet lung weight were assessed and indexed to tibial length prior to organ preservation.
Histological Sectioning and Staining
Hearts from 8-week TAC studies were harvested for analyses of CSA and fibrosis. Masson’s trichrome staining was performed as previously described(45). Briefly, mice were euthanized, and hearts were fixed overnight in 4% paraformaldehyde at 4°C. Hearts were dehydrated and paraffinized followed by embedding in paraffin blocks. Blocks were sectioned at 5 μm and stained with Weigert’s iron hematoxylin and Masson trichrome (Sigma-Aldrich) according to the manufacturer’s instructions. Alternatively, tissue sections were deparaffinized and rehydrated according to the trichrome staining protocol, but after the wash with deionized water, heart sections were stained with Alexa Fluor 594–conjugated wheat germ agglutinin (10μg/mL in 1× PBS) (Invitrogen) for 1 hr at room temperature in a humidified chamber in the dark. Sections were washed 3 times for 5 min with PBS followed by mounting with coverslips using Fluoromount-G mounting media containing DAPI nuclear stain (Southern Biotech). CSA was measured at 20x magnification by tracing individual cells in ImageJ. Fibrosis was assessed by a thresholding method in ImageJ as described previously(46).
Cell Culture Experiments and Adenoviral Infection
Neonatal rat ventricular myocytes (NRVMs) were isolated as described previously(47). NRVMs were plated in Ham’s F10 medium supplemented with 10% horse serum and 5% FBS with antibiotics. The day after plating, NRVMs were maintained in serum free medium with antibiotics. For experiments involving adenoviral infection, viral particles were introduced at a multiplicity of infection (MOI) of 10 after replacing plating medium. Adult cardiomyocytes were isolated as described previously (48), and were plated in M199 medium containing 10% FBS and 10mM 2,3-butanedione monoxime (Sigma, B0753), or collected for subsequent subcellular fractionation.
For determination of cyclic adenosine monophosphate (cAMP) generation, NRVMs were infected with control AdGFP or AdGRK5nt-Flag at a MOI of 10 per virus. Following 48 hours of infection, NRVMs were treated with 50μM 3-isobutyl-1-methylxanthine (IBMX) for 30 minutes followed by stimulation with vehicle or 10μM isoproterenol for 10 minutes. Cells were lysed in cell lysis buffer (Cell Signaling Technology) followed by assessment of cyclic AMP generation using Cyclic AMP XP Assay Kit (Cell Signaling Technology, #4339S) according to manufacturer’s instructions.
NFAT activity was assessed using an adenovirus containing firefly luciferase under the control of repeats of the NFAT DNA-binding element (Seven Hills Bioreagents). Alternatively, an NFkB luciferase virus under the control of NFkB response elements was used (Vector Biolabs). NRVMs were co-infected with luciferase adenovirus and either AdGFP or AdGRK5nt-Flag at a MOI of 10 per virus. Following 48 hrs of infection, NRVMs were treated with either vehicle or 100 μM PE for 24 hrs. Cells were lysed in Promega passive lysis buffer and luciferase activity was measured using Dual-Glo luciferase reporter system on a luminometer (Tecan Group). All luminescence values were normalized to micrograms of protein in the luciferase reaction.
For myocyte hypertrophy experiments, NRVMs were infected with Adβ-galactosidase/AdGFP or AdGRK5nt at a MOI of 10. Following 24 hours of infection, NRVMs were treated with vehicle or 50 μM PE for 48 hrs. Cells were then either fixed in 3.7% formaldehyde in phosphate buffered saline (Sigma, PBS) for immunocytochemistry, frozen as pellets for quantification of protein and DNA, or frozen in Trizol prior to RNA analysis.
RNA Isolation and Quantitative PCR
RNA was isolated using Zymo Research Direct-zol RNA mini-prep according to manufacturer’s instructions. RNA was reverse-transcribed (one microgram) to cDNA using BioRad’s iScript cDNA synthesis kit. Quantitative PCR was performed using Applied Biosystems’ Sybr Green select master mix with 5 ng of cDNA per reaction and 400nM of each primer as described(42). Ribosomal RNA 18s was used as a reference gene in order to calculate ΔCt values. The ΔΔCt method was used to represent relative gene expression. All data were analyzed by comparing delta Ct values and normalized to the control treatment or surgery and infection or genotype.
Immunostaining and Immuno-Blotting
Fixed cells were permeabilized in 0.1% Triton X-100 in phosphate-buffered saline (PBS) for 5 mins followed by blocking for 1 hr at room temperature in PBS+1% BSA. Immunocytochemistry was performed using anti-sarcomeric actin (Sigma, 1:100 in blocking buffer) overnight at 4°C. Cells were washed 3x with PBS followed by incubation with anti-mouse AlexaFluor-488 or AlexaFluor-594 secondary antibody (1:500) for 1 hour at room temperature in the dark. Following 3 washes with PBS, actin-stained cells were counterstained with 10μg/mL DAPI and mounted in Fluoromount-G. Images were acquired using a Nikon-TiE epifluorescence microscope at 20x magnification.
Tissue and cells were lysed unless otherwise noted in RIPA buffer (25 mM Tris, 0.5% sodium deoxycholate, 150 mM sodium chloride, 1 mM sodium fluoride, 1% IGEPAL) plus protease inhibitors (set V, EDTA-free, Millipore). Protein determination for SDS-PAGE or hypertrophy experiments was performed using bicinchoninic acid assay (ThermoFisher). DNA isolation was performed using ChIP DNA isolation kit (Cell Signaling Technologies). DNA concentration for hypertrophy experiments was determined using Sybr Green I dye assay (ThermoFisher). Briefly, a standard curve was generated using dilutions of lambda DNA in the presence of Sybr I dye. Dilutions of DNA isolated from hypertrophy experiments were then assayed for Sybr I fluorescence and the standard curve was used to extrapolate the DNA concentration for indexing protein concentration. For SDS-PAGE, 20–30 μg samples of protein were resolved on 4–20% gradient gels (Invitrogen), followed by transfer to nitrocellulose membrane using TransBlot Turbo transfer stacks (BioRad).
For immunoblotting, membranes were stained with either REVERT 680 (Licor Technologies) or Ponceau S (Sigma) for 5 min, rinsed, and imaged to assess transfer efficiency and total protein. Membranes were then de-stained in wash buffer (PBS+0.5% Tween-20) for Ponceau-stained membranes or REVERT reversal solution (Licor) and blocked for 1 hr at room temperature with blocking buffer in PBS (Licor). Primary antibodies were diluted 1:1000 in blocking buffer and incubated overnight at 4°C. After primary antibody incubation, membranes were washed three times and incubated with secondary antibody (1:10,000 in Licor blocking buffer) for 1 hr at room temperature. Membranes were washed again and imaged using an Odyssey Licor scanner. Primary antibodies used were as follows: mouse monoclonal GRK5 (Santa Cruz Biotechnology, sc-518005), mouse monoclonal GAPDH (sc-32233), mouse monoclonal VDAC1 (sc-390996), mouse monoclonal total CREB (sc-377154), rabbit monoclonal HA (Cell Signaling Technology, C29F4), rabbit polyclonal Akt (CST, 9272), pan CaMKII (CST, 3362), phospho-CREB Ser133 (CST, 9198), rabbit phospho-ERK (CST, 4370), mouse total ERK (CST, 9107), rabbit polyclonal histone H1 (Invitrogen, PA5–30055), mouse monoclonal FLAG (Sigma, F1804), rabbit histone H2A (ActiveMotif, 39591), and mouse monoclonal sarcomeric actin (Sigma, SAB4200689). Secondary antibodies used were as follows: Goat anti-rabbit IRDye 800 (Licor Technologies, 926–32211), Goat anti-mouse IRDye 680 (Licor, 926–68078), AlexaFluor-488 phalloidin (ThermoFisher, A12379), Goat anti-mouse AlexaFluor-594 (ThermoFisher, A-11032), and Goat anti-mouse AlexaFlour-488 (Thermo Fisher, A-11001). The monoclonal GRK5 antibody used throughout the manuscript exhibited a singlet to triplet banding pattern, depending on sample type, all of which were absent in lysates from GRK5 knockout samples, suggesting possible splicing forms, post-translational modifications, or degradation products of GRK5.
Calmodulin Capture Assay
Calmodulin (CaM) capture assays were performed using agarose beads conjugated to recombinant bovine calmodulin (Sigma). A bead volume equivalent to 1μg calmodulin per pulldown was washed with either 2 mM EDTA or 2 mM CaCl2 in immunoprecipitation (IP) Buffer (50 mM Tris, 150 mM sodium chloride) before adding to 150 μg of either GFP or GRK5nt NRVM lysates, or NLC or TgGRK5nt heart lysates diluted in IP buffer. Every sample underwent CaM-agarose pulldown in the presence of EDTA or CaCl2 to determine the Ca2+-dependence of binding. Following overnight rotation, calmodulin beads were centrifuged and washed in EDTA or CaCl2 wash buffer three times. After the final wash, CaM beads were boiled in 2x Laemmli buffer (BioRad) for 5 min. Pulldown fractions were resolved by SDS-PAGE to analyze captured proteins.
Subcellular Fractionation
For assessment of nuclear GRK5, heart tissues were harvested in PBS and subsequently homogenized using a dounce homogenizer (Sigma). Cells were centrifuged at 500 × g for 5 min. Pellets were processed to generate cytoplasmic and nuclear fractions using a kit according to the manufacturer’s instructions (BioVision, K266). Briefly, cell pellets were re-suspended in hypotonic buffer (CEB-A) and incubated for 10 min. Detergent (CEB-B) was added to the suspensions, which were then vortexed to release non-nuclear proteins. Lysates were centrifuged and the supernatant was collected as the non-nuclear fraction. The pellets were lysed in nuclear lysis buffer (NEB) to extract nuclear proteins. Nuclear fractions were intermittently vortexed for 30 minutes on ice and clarified by centrifugation. Alternatively, NRVMs or isolated adult cardiomyocytes were fractionated using the ThermoFisher subcellular fractionation kit for cultured cells (ThermoFisher, 78840) Histone H1, fibrillarin, and histone H2A were used as molecular markers of the nuclear and chromatin fractions, and GAPDH and Akt were used as markers of non-nuclear or cytoplasmic fractions. VDAC1 was used as a marker of the membrane/organellar fractions.
Statistics
All bar graphs in figures are represented as mean +/− standard deviation from an N of at least 3 mice, independent cell cultures, or organ tissue extracts for every experiment. For all experiments with two independent variables with levels (for example, GRK5nt and control virus or NLC and TgGRK5nt), two-way ANOVA with Tukey’s post-hoc testing for multiple comparisons were used to determine statistical significance. In hemodynamic studies, two-way ANOVA with repeated measures was used to assess statistical significance, with Sidak post-hoc testing to assess time (isoproterenol boluses) and genotype interaction effects. For comparison of two groups, Student’s two-tailed t-test assuming equal variance was used. A p-value<0.05 was considered statistically significant.
Supplementary Material
Fig. S1. Design of the transgenic GRK5nt mouse line and confirmation of heart tissue-specific expression.
Fig. S2. Transgenic GRK5nt mice have normal baseline cardiac physiology and heart size.
Fig. S3. TgGRK5nt and NLC mice exhibited similar heart rate and peak pressure gradients after TAC or Sham surgery.
Fig. S4. GRK5nt mechanism of action in hypertrophic stress.
Acknowledgments:
We would like to thank Sudarsan Rajan of our viral vector core for propagation and titering of adenoviruses used in our studies. We also thank the contribution of Zuping Qu and Kevin Luu for animal husbandry, colony maintenance, and genotyping of mice.
Funding:
All studies were funded by National Institute of Health (NIH) grant P01 HL091799 (W.J.K.), P01 HL075443 (W.J.K.), and a pre-doctoral AHA fellowship 17PRE33661220 (R.C.C.).
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. Acquisition of mice will require an MTA from the Lewis Katz School of Medicine Institutional Animal Care and Use Committee (IACUC).
References and Notes
- 1.Rockman HA, Koch WJ, Lefkowitz RJ, Seven-transmembrane-spanning receptors and heart function. Nature 415 (2002), pp. 206–212. [DOI] [PubMed] [Google Scholar]
- 2.Shukla AK, Xiao K, Lefkowitz RJ, Emerging paradigms of β-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem. Sci 36 (2011), pp. 457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang D, Zhao Q, Wu B, Structural studies of G protein-coupled receptors. Mol. Cells 38, 836–842 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang ZM, Gold JI, Koch WJ, G protein-coupled receptor kinases in normal and failing myocardium. Front. Biosci. (Landmark Ed. 16, 3047–60 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sato PY, Chuprun JK, Schwartz M, Koch WJ, The evolving impact of g protein-coupled receptor kinases in cardiac health and disease. Physiol. Rev 95, 377–404 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pfleger J, Gresham K, Koch WJ, G protein-coupled receptor kinases as therapeutic targets in the heart. Nat. Rev. Cardiol 16 (2019), pp. 612–622. [DOI] [PubMed] [Google Scholar]
- 7.Penela P, Murga C, Ribas C, Tutor AS, Peregrín S, Mayor F, Mechanisms of regulation of G protein-coupled receptor kinases (GRKs) and cardiovascular disease. Cardiovasc. Res 69 (2006), pp. 46–56. [DOI] [PubMed] [Google Scholar]
- 8.Maning J, McCrink KA, Pollard CM, Desimine VL, Ghandour J, Perez A, Cora N, Ferraino KE, Parker BM, Brill AR, Aukszi B, Lymperopoulos A, Antagonistic roles of GRK2 and GRK5 in cardiac aldosterone signaling reveal GRK5-mediated cardioprotection via mineralocorticoid receptor inhibition. Int. J. Mol. Sci 21 (2020), doi: 10.3390/ijms21082868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gold JI, Gao E, Shang X, Premont RT, Koch WJ, Determining the absolute requirement of G protein-coupled receptor kinase 5 for pathological cardiac hypertrophy: Short communication. Circ. Res 111, 1048–1053 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y, Matkovich SJ, Duan X, Gold JI, Koch WJ, Dorn GW, Nuclear effects of G-protein receptor kinase 5 on histone deacetylase 5-regulated gene transcription in heart failure. Circ. Hear. Fail 4, 659–668 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raake PW, Vinge LE, Gao E, Boucher M, Rengo G, Chen X, Degeorge BR, Matkovich S, Houser SR, Most P, Eckhart AD, Dorn GW, Koch WJ, G protein-coupled receptor kinase 2 ablation in cardiac myocytes before or after myocardial infarction prevents heart failure. Circ. Res 103, 413–422 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thiyagarajan MM, Stracquatanio RAP, Pronin AN, Evanko DS, Benovic JL, Wedegaertner PB, A Predicted Amphipathic Helix Mediates Plasma Membrane Localization of GRK5. J. Biol. Chem 279, 17989–17995 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Xu H, Jiang X, Shen K, Fischer CC, Wedegaertner PB, The regulator of G protein signaling (RGS) domain of G protein-coupled receptor kinase 5 (GRK5) regulates plasma membrane localization and function. Mol. Biol. Cell 25, 2105–2115 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levay K, Satpaev DK, Pronin AN, Benovic JL, Slepak VZ, Localization of the sites for Ca2+-binding proteins on G protein- coupled receptor kinases. Biochemistry 37, 13650–13659 (1998). [DOI] [PubMed] [Google Scholar]
- 15.Johnson LR, Robinson JD, Lester KN, Pitcher JA, Distinct Structural Features of G Protein-Coupled Receptor Kinase 5 (GRK5) Regulate Its Nuclear Localization and DNA-Binding Ability. PLoS One 8 (2013), doi: 10.1371/journal.pone.0062508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gold JI, Martini JS, Hullmann J, Gao E, Chuprun JK, Lee L, Tilley DG, Rabinowitz JE, Bossuyt J, Bers DM, Koch WJ, Nuclear Translocation of Cardiac G Protein-Coupled Receptor Kinase 5 Downstream of Select Gq-Activating Hypertrophic Ligands Is a Calmodulin-Dependent Process. PLoS One 8 (2013), doi: 10.1371/journal.pone.0057324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martini JS, Raake P, Vinge LE, DeGeorge B, Chuprun JK, Harris DM, Gao E, Eckhart AD, Pitcher JA, Koch WJ, Uncovering G protein-coupled receptor kinase-5 as a histone deacetylase kinase in the nucleus of cardiomyocytes. Proc. Natl. Acad. Sci. U. S. A 105, 12457–12462 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rockman HA, Choi DJU, Rahman NU, Akhter SA, Lefkowitz RJ, Koch WJ, Receptor-specific in vivo desensitization by the G protein-coupled receptor kinase-5 in transgenic mice. Proc. Natl. Acad. Sci. U. S. A 93, 9954–9959 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hullmann JE, Grisanti LA, Makarewich CA, Gao E, Gold JI, Chuprun JK, Tilley DG, Houser SR, Koch WJ, GRK5-mediated exacerbation of pathological cardiac hypertrophy involves facilitation of nuclear NFAT activity. Circ. Res 115, 976–985 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Swulius MT, Waxham MN, Ca2+/calmodulin-dependent protein kinases. Cell. Mol. Life Sci 65 (2008), pp. 2637–2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akhter SA, Luttrell LM, Rockman HA, Iaccarino G, Lefkowitz RJ, Koch WJ, Targeting the receptor-G(q) interface to inhibit in vivo pressure overload myocardial hypertrophy. Science (80-.) 280, 574–577 (1998). [DOI] [PubMed] [Google Scholar]
- 22.Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN, A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 93, 215–228 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sussman MA, Lim HW, Gude N, Taigen T, Olson EN, Robbins J, Colbert MC, Gualberto A, Wieczorek DF, Molkentin JD, Prevention of cardiac hypertrophy in mice by calcineurin inhibition. Science (80-.) 281, 1690–1693 (1998). [DOI] [PubMed] [Google Scholar]
- 24.Pronin AN, Satpaev DK, Slepak VZ, Benovic JL, Regulation of G protein-coupled receptor kinases by calmodulin and localization of the calmodulin binding domain. J. Biol. Chem 272, 18273–18280 (1997). [DOI] [PubMed] [Google Scholar]
- 25.Sun P, Enslen H, Myung PS, Maurer RA, Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev 8, 2527–2539 (1994). [DOI] [PubMed] [Google Scholar]
- 26.Sorriento D, Ciccarelli M, Santulli G, Campanile A, Altobelli GG, Cimini V, Galasso G, Astone D, Piscione F, Pastore L, Trimarco B, Iaccarino G, The G-protein-coupled receptor kinase 5 inhibits NFκB transcriptional activity by inducing nuclear accumulation of IκBα. Proc. Natl. Acad. Sci. U. S. A 105, 17818–17823 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Venkatakrishnan AJ, Deupi X, Lebon G, Heydenreich FM, Flock T, Miljus T, Balaji S, Bouvier M, Veprintsev DB, Tate CG, Schertler GFX, Babu MM, Diverse activation pathways in class A GPCRs converge near the G-protein-coupling region. Nature 536, 484–487 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Inglese J, Freedman NJ, Koch WJ, Lefkowitz RJ, Structure and mechanism of the G protein-coupled receptor kinases. J. Biol. Chem 268 (1993), pp. 23735–23738. [PubMed] [Google Scholar]
- 29.Hata JA, Koch WJ, Phosphorylation of G protein-coupled receptors: GPCR kinases in heart disease. Mol. Interv 3 (2003), pp. 264–272. [DOI] [PubMed] [Google Scholar]
- 30.Premont RT, Gainetdinov RR, Physiological Roles of G Protein–Coupled Receptor Kinases and Arrestins. Annu. Rev. Physiol 69, 511–534 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Ben-Yehuda O, Rockman HA, Regulation of myocardial contractility: Insights from transgenic mice. Trends Cardiovasc. Med 6 (1996), pp. 95–99. [DOI] [PubMed] [Google Scholar]
- 32.Yi XP, Gerdes AM, Li F, Myocyte redistribution of GRK2 and GRK5 in hypertensive, heart-failure-prone rats. Hypertens. (Dallas, Tex. 1979) 39, 1058–63 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Xian PY, Zhou J, Baker J, Wang X, Gerdes AM, Li F, Myocardial expression and redistribution of GRKs in hypertensive hypertrophy and failure. Anat. Rec. - Part A Discov. Mol. Cell. Evol. Biol 282, 13–23 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Patial S, Luo J, Porter KJ, Benovic JL, Parameswaran N, G-protein-coupled-receptor kinases mediate TNFα-induced NF-κB signalling via direct interaction with and phosphorylation of IκBα. Biochem. J 425, 169–178 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu P, Wang X, Gao N, Zhu H, Dai X, Xu Y, Ma C, Huang L, Liu Y, Qin C, G protein-coupled receptor kinase 5, overexpressed in the α-synuclein up-regulation model of Parkinson’s disease, regulates bcl-2 expression. Brain Res 1307, 134–141 (2010). [DOI] [PubMed] [Google Scholar]
- 36.Michal AM, So CH, Beeharry N, Shankar H, Mashayekhi R, Yen TJ, Benovic JL, G protein-coupled receptor kinase 5 is localized to centrosomes and regulates cell cycle progression. J. Biol. Chem 287, 6928–6940 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen X, Zhu H, Yuan M, Fu J, Zhou Y, Ma L, G-protein-coupled receptor kinase 5 phosphorylates p53 and inhibits DNA damage-induced apoptosis. J. Biol. Chem 285, 12823–12830 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barker BL, Benovic JL, G protein-coupled receptor kinase 5 phosphorylation of hip regulates internalization of the chemokine receptor CXCR4. Biochemistry 50, 6933–6941 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sorriento D, Santulli G, Fusco A, Anastasio A, Trimarco B, Iaccarino G, Intracardiac injection of AdGRK5-NT reduces left ventricular hypertrophy by inhibiting NF-kappaB-dependent hypertrophic gene expression. Hypertens. (Dallas, Tex. 1979) 56, 696–704 (2010). [DOI] [PubMed] [Google Scholar]
- 40.Sorriento D, Santulli G, Ciccarelli M, Maione AS, Illario M, Trimarco B, Iaccarino G, The amino-terminal domain of GRK5 inhibits cardiac hypertrophy through the regulation of calcium-calmodulin dependent transcription factors. Int. J. Mol. Sci 19 (2018), doi: 10.3390/ijms19030861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM, Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ. Res 97, 1314–22 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Schumacher SM, Gao E, Cohen M, Lieu M, Chuprun JK, Koch WJ, A peptide of the RGS domain of GRK2 binds and inhibits Gα(q) to suppress pathological cardiac hypertrophy and dysfunction. Sci. Signal 9, ra30 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koch WJ, Rockman HA, Samama P, Hamilton R, Bond RA, Milano CA, Lefkowitz RJ, Cardiac function in mice overexpressing the β-adrenergic receptor kinase or a βARK inhibitor. Science (80-.) 268, 1350–1353 (1995). [DOI] [PubMed] [Google Scholar]
- 44.Brinks H, Boucher M, Gao E, Chuprun JK, Pesant S, Raake PW, Huang ZM, Wang X, Qiu G, Gumpert A, Harris DM, Eckhart AD, Most P, Koch WJ, Level of G protein-coupled receptor kinase-2 determines myocardial ischemia/reperfusion injury via pro-and anti-apoptotic mechanisms. Circ. Res 107, 1140–1149 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gao E, Lei YH, Shang X, Huang ZM, Zuo L, Boucher M, Fan Q, Chuprun JK, Ma XL, Koch WJ, A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ. Res 107, 1445–1453 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Emde B, Heinen A, Gödecke A, Bottermann K, Wheat germ agglutinin staining as a suitable method for detection and quantification of fibrosis in cardiac tissue after myocardial infarction. Eur. J. Histochem 58, 315–319 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Simpson P, McGrath A, Savion S, Myocyte hypertrophy in neonatal rat heart cultures and its regulation by serum and by catecholamines. Circ. Res 51, 787–801 (1982). [DOI] [PubMed] [Google Scholar]
- 48.Powell T, Twist VW, A rapid technique for the isolation and purification of adult cardiac muscle cells having respiratory control and a tolerance to calcium. Biochem. Biophys. Res. Commun 72, 319–326 (1976). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Design of the transgenic GRK5nt mouse line and confirmation of heart tissue-specific expression.
Fig. S2. Transgenic GRK5nt mice have normal baseline cardiac physiology and heart size.
Fig. S3. TgGRK5nt and NLC mice exhibited similar heart rate and peak pressure gradients after TAC or Sham surgery.
Fig. S4. GRK5nt mechanism of action in hypertrophic stress.