

Abstract
Background
Immune checkpoint inhibitors (ICIs) targeting the PD‐1/PD‐L1 axis have changed the first‐line treatment of people with advanced non‐small cell lung cancer (NSCLC). Single‐agent pembrolizumab (a PD‐1 inhibitor) is currently the standard of care as monotherapy in patients with PD‐L1 expression ≥ 50%, either alone or in combination with chemotherapy when PD‐L1 expression is less than 50%. Atezolizumab (PD‐L1 inhibitor) has also been approved in combination with chemotherapy and bevacizumab (an anti‐angiogenic antibody) in first‐line NSCLC regardless of PD‐L1 expression. The combination of first‐line PD‐1/PD‐L1 inhibitors with anti‐CTLA‐4 antibodies has also been shown to improve survival compared to platinum‐based chemotherapy in advanced NSCLC, particularly in people with high tumour mutational burden (TMB). The association of ipilimumab (an anti CTLA4) and nivolumab (PD‐1 inhibitor) has been approved by the US Food and Drug Administration (FDA) in all patients with PD‐L1 expression ≥1%. Although these antibodies are currently used in clinical practice, some questions remain unanswered, such as the best‐treatment strategy, the role of different biomarkers for treatment selection and the effectiveness of immunotherapy according to specific clinical characteristics.
Objectives
To determine the effectiveness and safety of first‐line immune checkpoint inhibitors (ICIs), as monotherapy or in combination, compared to platinum‐based chemotherapy, with or without bevacizumab for people with advanced NSCLC, according to the level of PD‐L1 expression.
Search methods
We performed an electronic search of the main databases (Cochrane Central Register of Controlled Trials, MEDLINE, Embase) from inception until 31 December 2020 and conferences meetings from 2015 onwards.
Selection criteria
We included randomised controlled trials (RCTs) reporting on the efficacy or safety of first‐line ICI treatment for adults with advanced NSCLC who had not previously received any anticancer treatment. We included trials comparing single‐ or double‐ICI treatment to standard first‐line therapy (platinum‐based chemotherapy +/‐ bevacizumab). All data come from ‘international multicentre studies involving adults, age 18 or over, with histologically‐confirmed stage IV NSCLC.
Data collection and analysis
Three review authors independently assessed the search results and a fourth review author resolved any disagreements. Primary outcomes were overall survival (OS) and progression‐free survival (PFS); secondary outcomes were overall objective response rate (ORR) by RECIST v 1.1, grade 3 to 5 treatment‐related adverse events (AEs) (CTCAE v 5.0) and health‐related quality of life (HRQoL). We performed meta‐analyses where appropriate using the random‐effects model for hazard ratios (HRs) or risk ratios (RRs), with 95% confidence intervals (95% CIs), and used the I² statistic to investigate heterogeneity.
Main results
Main results
We identified 15 trials for inclusion, seven completed and eight ongoing trials. We obtained data for 5893 participants from seven trials comparing first‐line single‐ (six trials) or double‐ (two trials) agent ICI with platinum‐based chemotherapy, one trial comparing both first‐line single‐ and double‐agent ICsI with platinum‐based chemotherapy. All trials were at low risk of selection and detection bias, some were classified at high risk of performance, attrition or other source of bias. The overall certainty of evidence according to GRADE ranged from moderate‐to‐low because of risk of bias, inconsistency, or imprecision. The majority of the included trials reported their outcomes by PD‐L1 expressions, with PD‐L1 ≥ 50 being considered the most clinically useful cut‐off level for decision makers. Also, iIn order to avoid overlaps between various PDL‐1 expressions we prioritised the review outcomes according to PD‐L1 ≥ 50.
Single‐agent ICI In the PD‐L1 expression ≥ 50% group single‐agent ICI probably improved OS compared to platinum‐based chemotherapy (hazard ratio (HR) 0.68, 95% confidence interval (CI) 0.60 to 0.76, 6 RCTs, 2111 participants, moderate‐certainty evidence). In this group, single‐agent ICI also may improve PFS (HR: 0.68, 95% CI 0.52 to 0.88, 5 RCTs, 1886 participants, low‐certainty evidence) and ORR (risk ratio (RR):1.40, 95% CI 1.12 to 1.75, 4 RCTs, 1672 participants, low‐certainty evidence). HRQoL data were available for only one study including only people with PD‐L1 expression ≥ 50%, which suggested that single‐agent ICI may improve HRQoL at 15 weeks compared to platinum‐based chemotherapy (RR: 1.51, 95% CI 1.08 to 2.10, 1 RCT, 297 participants, low‐certainty evidence). In the included studies, treatment‐related AEs were not reported according to PD‐L1 expression levels. Grade 3‐4 AEs may be less frequent with single‐agent ICI compared to platinum‐based chemotherapy (RR: 0.41, 95% CI 0.33 to 0.50, I² = 62%, 5 RCTs, 3346 participants, low‐certainty evidence).
More information about efficacy of single‐agent ICI compared to platinum‐based chemotherapy according to the level of PD‐L1 expression and to TMB status or specific clinical characteristics is available in the full text.
Double‐agent ICI Double‐ICI treatment probably prolonged OS compared to platinum‐based chemotherapy in people with PD‐L1 expression ≥50% (HR: 0.72, 95% CI 0.59 to 0.89 2 RCTs, 612 participants, moderate‐certainty evidence). Trials did not report data on HRQoL, PFS and ORR according to PD‐L1 groups. Treatment related AEs were not reported according to PD‐L1 expression levels. The frequency of grade 3‐4 AEs may not differ between double‐ICI treatment and platinum‐based chemotherapy (RR: 0.78, 95% CI 0.55 to 1.09, I² = 81%, 2 RCTs, 1869 participants, low‐certainty evidence).
More information about efficacy of double‐agent ICI according to the level of PD‐L1 expression and to TMB status is available in the full text.
Authors' conclusions
Authors' conclusions
The evidence in this review suggests that single‐agent ICI in people with NSCLC and PD‐L1 ≥50% probably leads to a higher overall survival rate and may lead to a higher progression‐free survival and overall response rate when compared to platinum‐based chemotherapy and may also lead to a lower rate of adverse events and higher HRQoL. Combined ICI in people with NSCLC and PD‐L1 ≥50% also probably leads to a higher overall survival rate when compared to platinum‐based chemotherapy, but its effect on progression‐free survival, overall response rate and HRQoL is unknown due to a lack of data. The rate of adverse events may not differ between groups.
This review used to be a living review. It is transitioned out of living mode because current research is exploring ICI in association with chemotherapy or other immunotherapeutic drugs versus ICI as single agent rather than platinum based chemotherapy.
Plain language summary
Immunotherapy versus chemotherapy for people with advanced non‐small cell lung cancer who have not been not previously been treated
Review question
Is immunotherapy more effective and less toxic than chemotherapy for people diagnosed with non‐small cell lung cancer (a subtype of lung cancer) who have not previously been treated and who are not suitable for curative treatment?
Background
Lung cancer is the leading cause of cancer deaths and non‐small cell lung cancer represent more than 85% of all lung cancer cases. Curative surgery and radiotherapy are not treatment options when the disease is at an advanced stage and until recently these people were offered chemotherapy. Since 2016, immunotherapies (antibodies able to stimulate the immune system against cancer cells) have been shown to improve survival for these patients.
Side effects of immunotherapies are mainly inflammation of the tissues caused by the activation of the immune system against different organs, while chemotherapy usually causes a reduction in the white blood cells and red blood cells, hair loss, nausea and vomiting. In this Cochrane Review, we tried to find out how effective and safe immunotherapies (given alone or as combinations) are compared to standard chemotherapy for people with non‐small cell lung cancer who are not suitable for possibly curative treatment.
Study characteristics
We searched the main databases and records of conference meetings up to 31st December 2020. We included seven studies (5893 participants) comparing immunotherapies (antibodies that interact with specific proteins called immune checkpoints) with chemotherapy for people with non‐small cell lung cancer not previously treated.
Key results
We reported the results by PD‐L1 levels (a protein produced by the tumour or immune cells and bound by immune checkpoint inhibitors)
In people with more than 50% of tumour/immune cells expressing PD‐L1 protein, single immunotherapy might improve survival with fewer side effects. In addition, treatment with combined immunotherapies may improve survival in both people with high expression of PD‐L1 protein.The rate of toxic effects may be the same for people treated with combined immunotherapies or chemotherapy.
Certainty of evidence
Overall, the certainty of the evidence ranged from moderate to low.
Conclusions
For people with advanced non‐small cell lung cancer with a high expression of PD‐L1 protein, immunotherapies alone or combinations of immunotherapies prolonged life compared to chemotherapy. The frequency of side effects may be lower with the use of immunotherapies alone compared to chemotherapy. The frequency of side effects may not differ between combinations of immunotherapies and chemotherapy.
Summary of findings
Background
Description of the condition
Lung cancer is one of the leading causes of cancer death worldwide (Bray 2018). Non‐small cell lung cancer (NSCLC), which accounts for 85% to 90% of lung cancers (Novello 2016), includes two major histological types: non‐squamous carcinoma and squamous carcinoma.The expression of PD‐L1 (a protein produced by the tumour or immune cells and bound by immune checkpoint inhibitors (ICI)) on NSCLC cells may vary. Approximately 23% of NSCLCs have ≥ 50% tumour cells expressing PD‐L1 (PD‐L1 tumour proportional score (TPS) ≥ 50%), 38% have 1% to 49% of tumour cells expressing PD‐L1 (PD‐L1 TPS 1% to 49%), and 39% of NSCLCs do not express PD‐L1 (PD‐L1 TPS <1%) (Garon 2015).
Until recently, the standard first‐line treatments for NSCLC with no driver mutations (epidermal growth factor receptor (EGFR), anaplastic lymphoma kinase (ALK) or receptor tyrosine kinase (ROS1) genomic aberrations) were platinum‐based chemotherapy including platinum and a non‐pemetrexed third‐generation agent for squamous histology, and platinum‐pemetrexed or platinum‐paclitaxel with or without bevacizumab (an anti‐angiogenic agent) for non‐squamous histology, achieving median progression‐free survival (PFS) of five to six months, and median overall survival (OS) of 11 (squamous histology) to 17 (non‐squamous histology) months (Lopez‐Chavez 2012; Paz‐Ares 2013; Scagliotti 2008).
Description of the intervention
The advent of ICI, has dramatically changed the choice of first‐line treatment. Pembrolizumab, a programmed cell death protein‐1 (PD‐1) inhibitor, has been shown to prolong survival in people with NSCLC and PD‐L1 TPS ≥1% (Garon 2015). In 2016, pembrolizumab, was approved by the Food and Drug Administration (FDA) and the European Medicines Agency (EMA) as monotherapy in treatment‐naive metastatic NSCLC with a PD‐L1 TPS ≥ 50%. Moreover, in April 2019, the FDA expanded pembrolizumab indication to first‐line treatment of people with stage IV or stage‐III NSCLC, who are not candidates for surgical resection or definitive chemoradiation and who have no EGFR or ALK genomic aberrations and PD‐L1 TPS ≥ 1%. Similarly, first‐line cemiplimab (a PD‐1 inhibitor), and atezolizumab (a PD‐L1 inhibitor) given as single agents were associated with promising survival and responses compared to platinum‐based chemotherapy in NSCLC people with PD‐L1 expression on tumour cells or immune cells ≥50% (Sezer 2020) and ≥5% (Peters 2017), respectively.
Nivolumab, another PD‐1 inhibitor, has also been reported to lead to durable responses and interesting survival outcomes in first‐line setting, as monotherapy (Gettinger 2016); or in combination with ipilimumab, an anti‐cytotoxic T‐lymphocyte‐associated protein 4 (CTLA‐4) agent (Ready 2019), and this combination has been recently approved by the FDA in people with NSCLC and PD‐L1 expression ≥1%.
Among PD‐1/PD‐L1 inhibitors and anti‐CTLA‐4 agents combinations, tremelimumab, a CTLA4 inhibitor plus durvalumab, a PD‐1 inhibitor, showed anti‐tumour activity regardless of PD‐L1 expression in people with advanced NSCLC (Antonia 2016).
How the intervention might work
Checkpoint inhibitors are a class of humanised immunoglobulins that target and inhibit molecules responsible for the physiological 'off‐switch' of immune cells to prevent an excessive and uncontrolled immune response. Their inhibition activates T‐lymphocytes and enhances the adaptive anti‐cancer immune response.
Nivolumab,pembrolizumab and cemiplimab (immunoglobulin (Ig) G4 monoclonal antibodies) bind PD‐1 on immune cells, blocking their interaction with PD‐L1 and PD‐L2 expressed by tumour cells (Ishida 1992). Atezolizumab, durvalumab, and avelumab (IgG1 monoclonal antibodies) bind PD‐L1 on tumour cells, preventing interaction with PD‐1. Both classes of drugs counteract PD‐1 ‐mediated inhibition of the immune response.
CTLA‐4 is expressed by T cells and, after binding to CD80/CD86, activates an inhibitory downstream signal in human lymphocytes (Hathcock 1993). Ipilimumab (IgG1) and tremelimumab (IgG2) block human CTLA4, inducing T‐cell activation, proliferation, and intratumoural infiltration, with improved anti‐cancer immune response.
Combining PD‐L1 inhibitors and anti‐CTLA4 agents might improve antitumour immunity because PD‐1 and CTLA4 modulate effector T‐cell activation, proliferation, and function through distinct complementary mechanisms (Okazaki 2013). Double immune checkpoint blockade may have a relevant role in particular for tumours with elevated tumour mutation burden (TMB) (Lawrence 2013), which are known to be highly sensitive to immunotherapy. Furthermore, recent evidence has shown that the primary target of PD‐1 inhibition is the downstream pathway of CD28, a co‐stimulatory receptor that can bind to CD80/CD86 (Hui 2017). Considering that CD80/CD86 is also a ligand for CTLA‐4, the combination of CTLA‐4 and PD‐1 inhibitors may have a synergistic effect with high activation of CD80/CD86 ‐ CD28 axis and an increased antitumour immune response.
Why it is important to do this review
Recent advances in immunotherapy have led to the approval of immunotherapy alone or in combination with chemotherapy as first‐line treatment for NSCLC, according to PD‐L1 expression. Double immune checkpoint blockade is also emerging as a treatment option in NSCLC with high TMB. Some questions remain unanswered, such as the best treatment strategy (i.e. immunotherapy as single agent or in combination), the role of different biomarkers (i.e. PD‐L1 TPS, TMB) for treatment selection and the effectiveness of immunotherapy according to specific clinical characteristics. In fact, people with advanced NSCLC and uncontrolled brain metastases, auto‐immune disorders, steroid dependency, and poor performance status are usually excluded from clinical trials aiming to test treatment with single‐agent or combined immune checkpoint inhibitors. Furthermore, people with oncogene‐addicted (i.e. EGFR mutated or ALK rearranged) NSCLC have also been excluded from these clinical trials. Although this review did not attempt to address specific questions for these subgroups of patients, particular attention will be paid to potentially interesting clinical and pathological variables that may influence the outcomes of immune checkpoint inhibitors, such as gender, age, smoking status, histology and PD‐L1 expression. In fact, subgroup analyses from randomised clinical trials in people with pre‐treated NSCLC have raised doubts about the immunotherapy benefit for elderly people (Brahmer 2015) or those people who have never smoked (Borghaei 2015), or among those with NSCLC with low/negative PD‐L1 expression (Borghaei 2015). Furthermore, a recent meta‐analysis showed that in patients with cancer, the magnitude of benefit from ICI may be sex‐dependent, with worse outcomes reported for women (Conforti 2018).
Objectives
Primary
To determine the effectiveness and safety of first‐line immune checkpoint inhibitors, as monotherapy or in combination compared to platinum‐based chemotherapy with or without bevacizumab for people with advanced non‐small cell lung cancer (NSCLC), according to the level of PD‐L1 expression.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials (RCTs) reporting on the effectiveness or safety of immune checkpoint inhibitors (ICIs) as first‐line treatment for people with advanced NSCLC, with or without blinding. We applied no language or publication status restrictions, and when sufficient data was available, we included meeting abstracts and unpublished online data.
Types of participants
We included studies involving participants with metastatic NSCLC or locally advanced NSCLC not susceptible to curative treatment. People should have not received any first‐line systemic treatment. We did not apply any restrictions for age, gender, drug dosage, or treatment duration.
Types of interventions
We considered studies for inclusion if researchers reported one or more of the following comparisons.
Single‐agent immune checkpoint inhibitors (ICIs) versus standard first‐line therapy (doublet chemotherapy ± bevacizumab).
Doublet immune checkpoint inhibitors (ICIs) versus standard first‐line therapy (doublet chemotherapy ± bevacizumab).
A doublet chemotherapy regimen includes any platinum‐based doublet along with a third‐generation agent (i.e. gemcitabine, vinorelbine, taxanes, pemetrexed).
Although we acknowledge that a lot of evidence is available on the combination of first‐line immune checkpoint inhibitors and chemotherapy, this ongoing Cochrane systematic review examines the potential benefit of immunotherapy and chemotherapy combinations compared to first‐line chemotherapy or single‐agent ICI (Syn 2018).
Types of outcome measures
Primary outcomes
Overall survival (OS): defined as time from randomisation to death from any cause (https://www.cancer.gov/publications/dictionaries/cancer-terms/def/overall-survival).
Progression‐free survival (PFS): defined as time from randomisation to cancer progression or death from any cause. (https://www.cancer.gov/publications/dictionaries/cancer-terms/def/progression-free-survival).
Secondary outcomes
Overall objective response rate (ORR): measured by Response Evaluation Criteria in Solid Tumours (RECIST) v.1.1 (Eisenhauer 2009); guidelines for response criteria for use in trials testing immunotherapeutics (iRECIST) (Seymour 2017); or immune‐related RECIST (irRECIST) (Nishino 2013).
Health‐related quality of life (HRQoL): measured via validated generic or disease‐specific questionnaires, or validated items
Treatment‐related adverse events (AEs): any AEs as reported by the included trials individually. We investigated the incidence of grade 3 (severe or medically significant but not immediately life‐threatening; hospitalisation or prolongation of hospitalisation indicated; disabling; limiting self‐care activities of daily living) and grade 4 events (life‐threatening consequences; urgent intervention indicated) based on the Common Terminology Criteria for Adverse Events (CTCAE) and Patient‐Reported Outcomes CTCAE (PRO‐CTCAE) (Kluetz 2016). We also checked the included trials for incidence of grade 5 AEs (deaths related to adverse events).
Search methods for identification of studies
Electronic searches
We searched the following electronic databases from inception to 31st December 2020.
Cochrane Lung Cancer Group Trial Register.
Cochrane Central Register of Controlled Trials (CENTRAL), in the Cochrane Library.
MEDLINE, accessed via PubMed.
Embase.
We did not apply any restrictions on language of publication.
We have presented the search strategies for CENTRAL, MEDLINE, and Embase in Appendix 1, Appendix 2, and Appendix 3, respectively.
We searched all databases using both controlled vocabulary (namely, medical subject headings (MeSH) in MEDLINE and EMTREE in Embase) and a wide range of free‐text terms. We performed the MEDLINE search using the Cochrane highly sensitive search strategy and precision‐maximising version (2008 version), as described in the Cochrane Handbook for Systematic Reviews of Interventions (Chapter 6.4.11.1, and detailed in Box 6.4.b) (Higgins 2011b).
We also conducted searches in the following clinical trials registries to identify unpublished and ongoing trials until 22nd January 2020.
ClinicalTrials.gov.(Appendix 4)
WHO International Clinical Trials Registry Platform (ICTRP).
Searching other resources
We handsearched the references of eligible studies to identify additional studies for inclusion.
We searched the meeting abstracts of conferences from the following sources from 2015 onwards.
World Conference on Lung Cancer (WCLC).
European Society for Medical Oncology (ESMO).
European Society for Medical Oncology Immuno‐Oncology congress (ESMO IO).
European Lung Cancer Conference (ELCC).
American Society of Clinical Oncology (ASCO).
American Association of Cancer Research (AACR).
We retrieved clinical study reports about the checkpoint inhibitors from the European Medicines Agency (EMA) website.
Data collection and analysis
Selection of studies
Three review authors (CM, RM and MI) screened independently all titles and abstracts retrieved by electronic searches. These review authors obtained the full texts for all relevant studies and checked independently the eligibility of each study against review eligibility criteria. We pursued discordant evaluations by discussion to reach consensus. When necessary to reach consensus, we involved a fourth review author (RF).
Data extraction and management
The review authors developed a data extraction form. Two review authors (RM, RF) independently extracted relevant data. To reach consensus, we involved a third review author when necessary (MI). We were not blinded to the names of study authors nor to the institutions where studies were conducted and funded. When we encountered multiple publications for the same study, we choose the first publication dealing with the primary endpoint in this review as a study identifier (study ID).
We extracted the following details from each included study.
Source: citation, study name if applicable, and contact details.
Study details: study design, location, setting (type and stage of disease), sample size, and study start date and completion date, study follow‐up
Characteristics of participants: inclusion and exclusion criteria, number of participating centres, number of participants, participant and tumour characteristics (age, sex, ethnicity, smoking status, performance status, histology, molecular status, tumour‐node‐metastasis (TNM) stage, PD‐L1 expression, tumour mutational burden (TMB).
Characteristics of interventions (e.g. drugs, doses, cycle duration).
Outcomes: primary and secondary outcomes with definitions and time points.
Results: number of participants allocated to each group, and for each outcome of interest, sample size, summary data for each group, estimate of effect with confidence interval and P value and subgroup analyses, and whether analyses have been performed by intention‐to‐treat (ITT) or per‐protocol methods.
Miscellaneous: funding source.
Assessment of risk of bias in included studies
Two review authors (RM, RF) independently applied the Cochrane 'Risk of bias' tool per Chapter 8 of the Cochrane Handbook for Systematic Reviews of Interventions, to assess quality and potential biases across studies eligible for inclusion in this review (Higgins 2011). We rated each domain of the tool as having 'low', 'high', or 'unclear' risk of bias at study level and for each outcome if possible, and we supported the rating of each domain by providing a brief description. We summarised risk of bias for each outcome within a study by considering all domains relevant to the outcome (i.e. both study‐level entries, such as allocation sequence concealment, and outcome‐specific entries, such as blinding). We provided a figure to summarise the risk of bias, similar to Figure 8.6.C, as presented in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
When two review authors could not reach consensus, we consulted with a third review author (SPB).
Using the Cochrane 'Risk of bias' tool, we considered the following domains.
Selection bias: random sequence generation.
Selection bias: allocation concealment.
Performance bias: blinding of participants and personnel.
Detection bias: blinding of outcome assessment.
Attrition bias: incomplete outcome data for outcomes related to efficacy and safety.
Reporting bias: selective reporting of outcomes.
Other bias: such as inclusion of patients concordant to pre‐specified number of participants needed for calculation, unplanned interim analyses, and unbalanced baseline characteristics across arms.
Measures of treatment effect
For time‐to‐event outcomes ‐ OS and PFS ‐ we used hazard ratios (HRs) to measure treatment effects. We reported each HR along with the 95% confidence Interval (CI). An HR of one indicates that the hazard rate is equivalent between experimental and control groups, and an HR other than one indicates differences in hazard rates between the two groups. We extracted the HR from the included studies when it was available. When it was not reported in the included study, we tried to calculate the HR by using Kaplan‐Meier survival curves and the dedicated methods of Parmar and Tierney (Parmar 1998; Tierney 2007).
For dichotomous outcomes AE and ORR, we used risk ratios (RRs) and 95% CIs if possible.
For dichotomous outcomes related to OS and PFS at specific time points, we used survival rates and 95% CIs.
For continuous outcomes (HRQOL), we used mean differences (MDs) between treatment arms when a similar scale was implemented to measure outcomes, and planned to use standardised mean differences (SMDs) if different scales were used to measure the same outcome. We confirmed that higher scores for continuous outcomes have the same meaning for the particular outcome, explained the direction, and reported if directions were reversed.
Unit of analysis issues
The primary unit of analysis was the participant.
Studies with multiple treatment groups
For studies with multiple comparison groups that compared two or more intervention groups versus the same control group, we first tried to combine groups to create a single pair‐wise comparison. We calculated within‐study correlation as recommended in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
When studies employed a cross‐over design and provided sufficient reporting, we followed the recommendations detailed in Chapter 16.4.5 in the Cochrane Handbook for Systematic Reviews of Interventions (Elbourne 2002; Higgins 2011).
Dealing with missing data
When we identified missing or unclear data, we contacted the study author directly. We followed Cochrane recommendations when dealing with such data details, as provided in Chapter 16 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011), and we considered two approaches.
Analysing only available data.
Imputing the missing data using replacement values and treating these as if they were observed.
Assessment of heterogeneity
We followed Cochrane recommendations for assessment of heterogeneity (Deeks 2011). We visually investigated heterogeneity by using forest plots generated via RevMan 5.4 (RevMan 2020). We assessed statistical heterogeneity of treatment effects between pooled trials for each considered outcome by using the I² statistic to quantify the degree of heterogeneity (Higgins 2002). We considered an I² > 30% as showing moderate heterogeneity, with an I² > 75% signifying significant heterogeneity.
Assessment of reporting biases
We planned to generate funnel plots and to perform Egger's linear regression tests to investigate reporting biases for considered outcomes when the number of trials included in a single meta‐analysis was sufficient (at least 10 trials). We followed recommendations provided in Chapter 10 of the Cochrane Handbook for Systematic Reviews of Interventions (Sterne 2011).
Data synthesis
If sufficient clinically similar studies were available, we pooled their results in meta‐analyses and performed meta‐analyses based on ITT analyses when available, according to PD‐L1 tumour proportional score (TPS) or PD‐L1 expression on tumour cells (TC) or immune cells (IC). Considered PD‐L1 categories were as follows: "positive" (PD‐L1 TPS ≥1% or TC1‐2‐3/IC1‐2‐3), "negative" (PD‐L1 TPS <1% or TC0/IC0), "high" (PD‐L1 TPS ≥50% or PD‐L1 TC3/IC3).
We performed meta‐analyses according to recommendations given in Chapter 9 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). For meta‐analyses, we entered data into RevMan 5.4 (RevMan 2020). One review author (RM) entered the data, and a second review author (RF) double‐checked the data for accuracy.
We performed random‐effects model for all outcomes and fixed‐effect model for selected outcomes as sensitivity analyses.
We applied the inverse‐variance method for fixed‐effect model for time‐to‐event outcomes. We applied the Mantel‐Haenszel method for dichotomous outcomes and the inverse‐variance method for continuous outcomes. We planned to use Peto’s odds ratio (OR) method under the fixed‐effect model in cases of rare events (Brockhaus 2014). For random‐effects model, we applied the DerSimonian and Laird method (DerSimonian 1986).
Subgroup analysis and investigation of heterogeneity
We performed subgroup analyses, when data were adequate, to assess the effect on heterogeneity for each of the primary and secondary outcomes on the following subgroups.
TMB measured on tissue or blood (high versus low). For studies where different analyses of TMB were presented, we chose the TMB assessment and cut‐offs with more available data.
Clinical characteristics such as age, gender, performance status, smoking history, NSCLC histology.
Sensitivity analysis
We investigated the robustness of review by performing the following sensitivity analyses when appropriate.
Performing fixed‐effect models for selected outcomes.
Including only 'low risk of bias' outcomes, according to the summary assessment of risk of bias.
Including or not including results from studies with incomplete data, whether or not the data were imputed.
Summary of findings and assessment of the certainty of the evidence
We followed the GRADE approach when creating our ’Summary of findings’ tables, as suggested in Chapters 11 and 12 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
We used the five GRADE considerations to rate the certainty of evidence as ’high’, ’moderate’, ’low’, or ’very low’.
Risk of bias: serious or very serious.
Inconsistency: serious or very serious.
Indirectness: serious or very serious.
Imprecision: serious or very serious.
Publication bias: likely or very likely.
We created two ’Summary of findings’ tables.
Single immune checkpoint inhibitors (ICIs) compared to chemotherapy for people with advanced non‐small cell lung cancer (NSCLC) (Table 1)
Combined immune checkpoint inhibitors (ICIs) compared to chemotherapy for people with advanced non‐small cell lung cancer (NSCLC) (Table 2)
Summary of findings 1. Single‐immune checkpoint inhibitors (ICIs) compared to chemotherapy for people with advanced non‐small cell lung cancer (NSCLC).
| Single immune checkpoint inhibitors compared to chemotherapy for people with advanced non‐small cell lung cancer | |||||||
|
Patient or population: People with advanced non‐small cell lung cancer Setting: Hospital Intervention: Single immune checkpoint inhibitors Comparison: First‐line, platinum‐based chemotherapy | |||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | ||
| Risk with chemotherapy | Risk with Single immune checkpoint inhibitors | ||||||
| Overall survival (OS) | by PD‐L1 expression ≥ 50% | 470 per 1000 | 130 more per 1000 (90 more to 170 more) |
HR 0.68 (0.60 to 0.76) |
2111 (6 studies) |
Moderate1 ⊕⊕⊕⊝ |
Data from Carbone 2017 and Rizvi 2020 used to estimate the anticipated effect at 12 month. |
| Progression‐free survival (PFS) | PD‐L1 expression ≥ 50% | 50 per 1000 |
80 more per 1000 (20 more to 150 more) |
HR 0.68, (0.51 to 0.88) |
1886 (5 studies) |
Low1,2 ⊕⊕⊝⊝ |
Data from Mok 2019; Reck 2016; Herbst 2020 used to estimate the anticipated effect at 12 months |
| Overall response rate (ORR) | PDL1 expression ‐ PDL1≥50% | 287 per 1000 |
115 more per 1,000 (34 more to 215 more) |
RR 1.40, (1.12 to 1.75) |
1672 (4 studies) |
Low1,2 ⊕⊕⊝⊝ |
|
| Adverse Events grade 3‐4 | 414 per 1,000 |
244 fewer per 1,000 (217 fewer to 207 fewer) |
RR 0.41 (0.33 to 0.50) |
3346 (5 studies) |
Low1,2 ⊕⊕⊝⊝ |
Data presented as overall pooled result, as data were not available for this outcome by PD‐L1 expression or by TMB | |
| QOL‐C30 GHS/QOL (range 0‐100) ‐ change from baseline to week 15 | by PDL1 expression ‐ PDL1≥50% | 265 per 1,000 |
135 more per 1,000 (21 more to 146 more) |
RR 1.51 (1.08 to 2.10) |
297 (1 study) |
Low1,3 ⊕⊕⊝⊝ |
A high score indicates a good quality of life. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; HR: Hazard ratio; RR: Risk ratio | |||||||
|
GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | |||||||
1Downgraded one point due to risk of other bias (Carbone 2017 differences in baseline characteristics; Mok 2019 several protocol amendments), performance bias (Carbone 2017, Hellmann 2018, Reck 2016, Rizvi 2020, Sezer 2020), or of attrition bias (Hellmann 2018 and Rizvi 2020).
2Downgraded one point due to inconsistency.
3Downgraded one point due to imprecision. Results come from one single trial with relatively small sample size, or the confidence interval includes both clinically relevant values and clinically irrelevant values, thus limiting confidence to draw conclusions on an apparent lack of effect or a possible relevant effect.
Summary of findings 2. Combined immune checkpoint inhibitors (ICIs) compared to chemotherapy for people with advanced non‐small cell lung cancer (NSCLC).
| Combined immune checkpoint inhibitors compared to chemotherapy for people with advanced non‐small cell lung cancer | |||||||
| Patient or population: people with advanced non‐small cell lung cancer Setting: Hospital Intervention: Combined immune checkpoint inhibitors Comparison: First‐line, platinum‐based chemotherapy | |||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | ||
| Risk with Chemotherapy | Risk with combined immune checkpoint inhibitors | ||||||
| Overall Survival (OS) | by PD‐L1 expression ‐ PD‐L1≥50% | 510 per 1,000 | 110 more per 1,000 (40 more to 162 more) | HR 0.72 (0.59 to 0.89) | 612 (2 RCTs) |
Moderate1 ⊕⊕⊕⊝ |
Rizvi 2020 data used to calculate anticipated absolute effects |
| Progression‐free survival (PFS) | by PD‐L1 expression ‐ PD‐L1≥50% | None of the included trials reported this outcome. | |||||
| Overall response rate (OSR) | by PD‐L1 expression ‐ PD‐L1≥50% | None of the included trials reported this outcome | |||||
| Adverse Events grade 3 to 4 | 348 per 1,000 |
77 fewer per 1000 (157 fewer to 31 more) |
RR 0.78 (0.55 to 1.09) |
1869 (2 RCTs) |
Low1,2 ⊕⊕⊝⊝ |
Data presented as overall pooled result, as data was not available for this outcome by PD‐L1 expression or by TMB | |
| HRQoL | None of the included trials reported this outcome. | ||||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; HR: Hazard ratio; RR: Risk ratio; | |||||||
|
GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | |||||||
1Downgraded one point due to risk of bias in Hellmann 2018 high risk of attrition bias because of TMB analysis or of other bias because of the several protocol amendments affecting the outcomes measured, Rizvi 2020 high risk of attrition bias).
2Downgraded one point due to inconsistency
.
The following outcomes were included.
OS
PFS
ORR
AEs: grades 3, 4
HRQoL
Results
Description of studies
See Characteristics of included studies; Characteristics of excluded studies and Characteristics of ongoing studies.
Results of the search
A total of 12,557 records were retrieved from searching the databases electronically up to 31st December 2020. Three thousand and six from CENTRAL, 4935 records from MEDLINE and 4616 from Embase. After removing the duplicate references, 6886 records remained, and three review authors (CM, RM, MI) excluded 6866 records based on screening the titles and abstracts. We selected 20 records that appeared to be relevant on the basis of full‐text screening. Among these we identified seven completed studies (Carbone 2017; Hellmann 2018; Herbst 2020; Mok 2019; Reck 2016; Rizvi 2020; Sezer 2020) for inclusion in this review, and eight ongoing studies EMPOWER‐Lung 2; eNERGY; IPSOS; JAVELIN Lung 100; KEYNOTE‐598;MILES 5; NEPTUNE; PEARL). We excluded the other five studies (Huan 2019; Hui 2017a; Leighl 2019; Mok 2017; Ready 2019) (see Excluded studies and the PRISMA study flow diagram in Figure 1).
1.
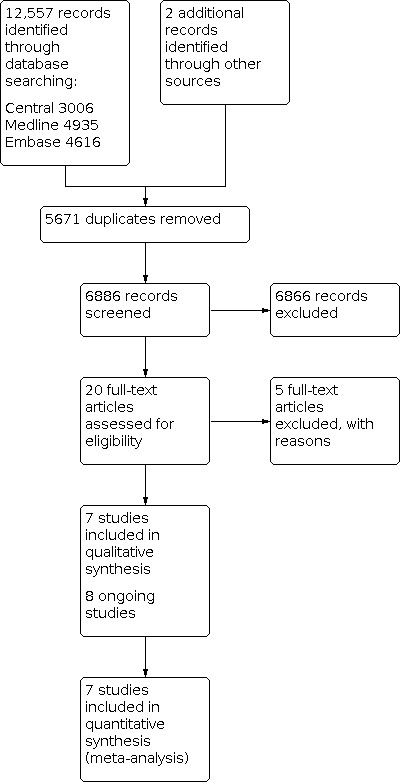
Study flow diagram.
Included studies
All seven included studies were randomised controlled trials (RCTs) published in English between 2016 and 2020. We divided the included studies by intervention strategies into two types :
single‐agent immune checkpoint inhibitor (ICI) compared with standard chemotherapy;
combination of ICI compared with standard chemotherapy.
Six of the seven included studies compared single checkpoint inhibitors versus standard chemotherapy and two studies compared a double checkpoint inhibitor strategy to standard chemotherapy.
Single‐agent immune checkpoint inhibitors (ICIs) versus standard platinum‐based chemotherapy
We identified six trials comparing a single checkpoint inhibitor agent with a standard chemotherapy for the treatment of NSCLC (Carbone 2017; Herbst 2020; Mok 2019; Reck 2016; Rizvi 2020; Sezer 2020). All six trials were randomised controlled phase 3 trials with an open‐label trial design. Overall, 3893 participants with stage IV or recurrent NSCLC were included in these trials.
Population
All six trials were international multicentre studies involving adults, age 18 or over, with histologically‐confirmed stage IV NSCLC who had not received any previous systemic anti‐cancer treatment for advanced disease. The inclusion criteria across the trials were similar and all included people with NSCLC without epidermal growth factor receptor (EGFR) mutations or anaplastic lymphoma kinase (ALK) translocations, with Eastern Cooperative Oncology Group (ECOG) performance score (PS) 0‐1 and with adequately treated brain metastases. Three trials (Carbone 2017; Herbst 2020; Mok 2019), included participants with PD‐L1 positive tumours (PDL1 tumour proportional score (TPS) ≥1% or PDL1 TC1/2/3 or IC 1/2/3). In two trials (Reck 2016; Sezer 2020), only participants with high expression of PD‐L1 (TPS ≥ 50%) were eligible for inclusion, whereas in Rizvi 2020, participants with negative PD‐L1 expression (PD‐L1 TPS <1%) were also included.
In Sezer 2020, PD‐L1 testing instruction changed in August 2018 and a population with confirmed PD‐L1 TPS ≥ 50% was defined as PD‐L1 ≥ 50% ITT population.
The number of randomised participants varied from 305 in Reck 2016 to 1274 in Mok 2019. Trial participants were stratified according to the tumour histology types across all the six trials (Carbone 2017;Herbst 2020; Mok 2019; Reck 2016; Rizvi 2020; Sezer 2020), PD‐L1 expression in four trials (Carbone 2017; Herbst 2020; Mok 2019; Rizvi 2020), ECOG PS in three trials (Herbst 2020; Mok 2019; Reck 2016), and the region of enrolment in three trials (Mok 2019; Reck 2016; Sezer 2020). In Carbone 2017, participants were also grouped according to their tumour mutational burden (TMB) level into low‐ (0 to <100 mutations), medium‐ (100 to 242 mutations) and high‐burden groups (> 243 mutations).
The proportion of women was lower in the nivolumab group and the proportion of people with a PD‐L1 expression level of 50% or more was also lower in Carbone 2017. In addition, participants in the nivolumab arm had a greater TMB. In Reck 2016, the proportion of people who never smoked was lower in the pembrolizumab arm and the proportion of people with brain metastases was higher in the pembrolizumab arm. In Herbst 2020, Mok 2019, and Sezer 2020, there were no differences in baseline characteristics.
Setting
All six included trials were multicentre and participants were enrolled from centres in North America, Europe, Asia and Australia.
Intervention
All included trials compared a single‐agent anti‐PD‐1 or anti‐PD‐L1 with platinum‐based chemotherapy. In Carbone 2017, nivolumab was given at 3 mg/kg of body weight every two weeks until disease progression, toxicity or withdrawal of consent. In Mok 2019 and Reck 2016, pembrolizumab was given at a flat dose of 200 mg every three weeks for up two years. In Rizvi 2020, durvalumab as monotherapy was given at 20 mg/kg of body weight every four weeks. In Herbst 2020, atezolizumab was given at 1200 mg every three weeks until disease progression or loss of clinical benefit. In Sezer 2020, cemiplimab was administered at a dose of 350 mg every three weeks until disease progression the maximum duration for 108 weeks .
Cross‐over was allowed in Carbone 2017Reck 2016 and Sezer 2020, but it was not permitted in Herbst 2020, Mok 2019 or Rizvi 2020. In Sezer 2020, participants progressing to cemiplimab had the opportunity to continue anti‐PD1 in association with chemotherapy.
Study duration
Median duration of follow‐up was 13.5 months in Carbone 2017, 25.2 months in Reck 2016, 11.2 months in Mok 2019, 30.2 months in Rizvi 2020, 10.8 months in the PD‐L1 ≥ 50% intention‐to‐treat (IIT) population in Sezer 2020 and 13.4 months for participants with positive (tumour cells (TC) or immune cells (IC) score ≥1%) PD‐L1 in Herbst 2020.
Outcomes
Primary outcomes
Overall survival (OS) was the primary outcome in three included trials Herbst 2020, Mok 2019 and Sezer 2020. In Mok 2019, the initial plan was to report OS for participants with PDL1 TPS ≥ 50%. However, after trial amendment OS was reported for people with both PD‐L1 TPS of ≥ 50% and ≥ 1%. In addition, in the same trial, OS for participants with PD‐L1 TPS ≥ 20% was also added as a primary outcome. In Rizvi 2020, the primary outcome was OS for durvalumab in monotherapy compared with chemotherapy in patients with PD‐L1 ≥ 25%. OS for PD‐L1 positive (TC or IC score ≥ 1%) participants was the primary outcome in Herbst 2020.
Progression‐free survival (PFS) was the primary outcome in two trials Carbone 2017 and Reck 2016, and was a primary outcome together with OS in Sezer 2020. In Carbone 2017, PFS was reported for participants with PDL1 TPS ≥ 5%. In Reck 2016, PFS was reported for all participants with PDL1 TPS ≥50%.
Secondary outcomes
PFS was the secondary outcome in Carbone 2017 for participants with PD‐L1 TPS ≥1%, in Mok 2019 for people with PD‐L1 TPS ≥1%, ≥20% and ≥50%, in Rizvi 2020 for people with PD‐L1 TPS ≥25%, and in Herbst 2020 for all PD‐L1 positive participants (TC or IC score ≥1%).
OS was a secondary outcome in Carbone 2017 and Reck 2016. OS was reported for people with PD‐L1 TPS ≥ 5% and PD‐L1 TPS ≥1% in Carbone 2017 and for participants with PD‐L1 TPS ≥50% in Reck 2016.
Objective response rate (ORR) was a secondary endpoint in Carbone 2017 for participants with PD‐L1 TPS ≥5%, in Reck 2016 for PD‐L1 TPS ≥50%, in Mok 2019 for people with PD‐L1 TPS ≥1%, ≥ 20% and ≥ 50%, in Rizvi 2020 for PD‐L1 TPS ≥ 25% subgroup, in Herbst 2020 for PD‐L1 positive participants (TC or IC score ≥1%) and in Sezer 2020, for PD‐L1 TPS ≥ 50%.
ORR, PFS and OS according to TMB status were exploratory analyses in Carbone 2017, Herbst 2020, Mok 2019 and Rizvi 2020.
Combined inhibitors (ICIs) versus standard platinum‐based chemotherapy
We only identified two trials Hellmann 2018 and Rizvi 2020 in which a different checkpoint inhibitor combination was compared with a standard chemotherapy treatment. Both were multicentre randomised controlled phase 3 open‐label design trials. Overall, 1910 participants were included in these two studies.
Population
Adults with histologically‐confirmed stage IV NSCLC who had not received previous systemic anticancer treatment for advanced disease were enrolled in both trials (Hellmann 2018; Rizvi 2020). Similarly, both trials involved participants with ECOG PS score 0‐1, adequately‐treated brain metastases and without EGFR mutations or ALK translocations. However, in part one of Hellmann 2018, only participants with PD‐L1 TPS ≥1% were included. On the other hand, the PD‐L1 positive status was not an inclusion criteria in Rizvi 2020.
Participants were stratified by tumour‐histology types in both trials. Participants were stratified by the PD‐L1 expression levels, < 25% versus ≥ 25% in Rizvi 2020. The TMB co‐primary survival analysis was conducted in the subgroup of participants who had a TMB of at least 10 mutations per megabase in Hellmann 2018.
People characteristics were well‐balanced among the groups in both Hellmann 2018 and Rizvi 2020.
Setting
Both were multicentre international trials and the recruitment centres were in North America, Europe, Asia and Australia.
Intervention
Anti‐CTLA‐4 agents were tested in both trials. The combination of nivolumab and ipilimumab was the intervention arm compared to chemotherapy in Hellmann 2018. The combination of durvalumab and tremelimumab was compared with the standard chemotherapy in Rizvi 2020. Nivolumab was given at 3 mg/kg of body weight every two weeks until disease progression in combination with ipilimumab at 1 mg/kg every six weeks for a maximum of four cycles in Hellmann 2018. Durvalumab was given at a dose of 20 mg/kg every four weeks until disease progression or in association to tremelimumab 1 mg/kg every four weeks for up to four doses in Rizvi 2020.
Cross‐over during the trial was not permitted in both trials.
Study duration
Median duration of follow up was 30.2 months for people with PD‐L1 expression ≥ 25% in Rizvi 2020. The minimum follow‐up for OS was 29.3 months in Hellmann 2018.
Outcomes
Primary outcomes
Primary endpoints were PFS (assessed by BIRC (Blinded, Independent Review Committee)) with nivolumab plus ipilimumab versus chemotherapy in participants with TMB ≥10 mutations per megabase and OS in PD‐L1 ≥1% participants in Hellmann 2018 . In Rizvi 2020, primary endpoints were PFS, (according to BIRC), and OS with durvalumab + tremelimumab compared to platinum‐based chemotherapy in patients with PD‐L1 ≥ 25%.
Secondary outcomes
Secondary endpoints included PFS among participants with TMB ≥ 10 mutations per megabase and a PD‐L1 ≥ 1% and OS among people with TMB ≥ 10 mutations per megabase in Hellmann 2018. In Rizvi 2020, key secondary endpoints were PFS and ORR with durvalumab + tremelimumab compared to platinum‐based chemotherapy in people with PD‐L1 ≥ 25%, in participants with PD‐L1 expression ≥ 1% and in the overall population. ORR in the overall population was a secondary endpoint for Hellmann 2018.
OS, PFS and ORR by TMB were exploratory analyses in Rizvi 2020.
Excluded studies
We excluded five studies for the following reasons (see Characteristics of excluded studies).
-
Wrong study design: three studies:
Huan 2019 because it is not an RCT;
Hui 2017a and Leighl 2019 refer to the same study: KEYNOTE 001 which is a multi‐cohort phase 1 trial.
-
Wrong intervention: two studies:
Mok 2017: evaluates ICI plus chemotherapy;
Ready 2019: does not compare ICI with chemotherapy.
Risk of bias in included studies
Figure 2 and Figure 3 are a visual representative of the 'Risk of bias' assessment across all included trials and also for each individual domain in the included trials. See Characteristics of included studies section 'Risk of bias' table for further explanations about the bias identified for each domain within each included studies.
2.

'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.

'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.
We assessed the risk of bias of five included studies using information published in full papers, one trial (Sezer 2020) was only available as abstract. The overall risk of bias was generally low or unclear, with much of the methodological information confirmed through a direct checking of the trial protocols to support the published information and to clarify the reasons for our rating.
Allocation
Six included trials (Carbone 2017; Hellmann 2018; Herbst 2020; Mok 2019; Reck 2016; Rizvi 2020) were at low risk of selection bias because of adequate methods of sequence generation and allocation concealment. Methods of randomisation and allocation concealment were stated across all the included trials. We rated Sezer 2020 as unclear as the method of randomisation and assignment to treatment were not reported.
Blinding
Blinding of participants and personnel (performance bias)
All seven included studies had an open‐label design being at potential risk of performance bias. However only Carbone 2017, Hellmann 2018; Reck 2016, Rizvi 2020 and Sezer 2020 where PFS was a primary endpoint were at high risk of performance bias due to absence of blinding of participants and personnel, whereas in Herbst 2020 and Mok 2019, where the primary endpoint was OS, performance bias influenced only results of secondary or exploratory outcomes.
Blinding of outcome assessors (detection bias)
Five included trials (Carbone 2017; Hellmann 2018; Mok 2019; Reck 2016; Rizvi 2020), were at low risk of detection bias because outcome assessors were blinded. In one study (Herbst 2020), the primary outcome measured was OS, therefore, lack of blinding of the outcome assessors had no influence. In Sezer 2020, relevant information regarding blinding of outcome assessors was missing.
Incomplete outcome data
Four trials (Carbone 2017;Herbst 2020; Mok 2019; Reck 2016) were at low risk of attrition bias, as no participants were lost to follow‐up and the total number of participants included in each outcome was reported. Two trials (Hellmann 2018; Rizvi 2020) were at high risk of attrition bias. In Hellmann 2018) only 229 (13%) out of 1739 initially randomised participants were included in the analysis of the trial primary outcome. In Rizvi 2020, the primary analysis population for the study was amended to include only people with PD‐L1 expression ≥ 25%, therefore only 488 (44%) of 1118 randomised people were included in the analysis of the trial primary outcome. In Sezer 2020, the information relevant to the number of dropouts was missing for most of the outcome, therefore we rated this domain as 'unclear'.
Selective reporting
Six trials (Carbone 2017; Hellmann 2018; Herbst 2020; Mok 2019; Reck 2016; Rizvi 2020) were at low risk of reporting bias as all prespecified outcomes were reported. Sezer 2020, was only available as a conference presentation with not enough details to make a judgment and risk of reporting bias was considered unclear.
Other potential sources of bias
All trials were at unclear risk of other source of bias because some authors had declared personal fees or other support from the pharmaceutical companies conducting the trials and it was impossible to know how these conflicts of interests have biased data collection and analysis. Furthermore, Carbone 2017, was at high risk of other source of bias because of some baseline differences between the groups. In the nivolumab group, the percentage of women was lower than that in the chemotherapy group (32% versus. 45%), as was the percentage of participants with a PD‐L1 expression level of 50% or more (32% versus 47%); the percentage of participants with liver metastases was slightly higher in the nivolumab group (20% versus. 13%). In addition, people in the nivolumab group had a lower tumour mutational burden than those in the chemotherapy group (30% versus 39%). Mok 2019 was at high risk of other source of bias due to the inclusion of a new PD‐L1 TPS cut‐off (20%) and the change of primary and secondary endpoint according to a new PD‐L1 TPS categorisation (≥ 1%, ≥ 20%, ≥ 50%). Hellmann 2018, was at high risk of other source of bias due to the amendment including TMB as a new biomarker and the modification of primary and secondary endpoint accordingly to TMB levels. We rated Sezer 2020, at high risk of other sources of bias because although PD‐L1≥ 50% was an inclusion criteria, 235 participants were retested and 88 of them had a confirmed PD‐L1 TPS ≥ 50% being included in the PD‐L1 ≥ 50% ITT population.
Publication bias
We did not have sufficient data to provide a funnel plot, nor to perform the Egger test. Publication bias was assessed only for the six trials testing single‐agent ICI versus platinum‐based chemotherapy, because for the comparison double ICI versus chemotherapy only two studies were available. Publication bias is unlikely to have occurred particularly for the PD‐L1 ≥1% category. In fact, trials comparing single‐agent ICI versus platinum‐based chemotherapy found mainly unfavourable results or no difference for the experimental arm (single‐agent ICI) compared to control arm (chemotherapy) for the PD‐L 1 ≥1% subgroup.
Effects of interventions
Comparison 1: First‐line single‐agent immune checkpoint inhibitor (ICI) versus platinum‐based chemotherapy
Primary outcome ‐ Overall survival (OS)
a. Main analysis: OS and PDL‐1 expressions
Information on OS was provided by all seven included trials comparing single‐agent immune checkpoint inhibitors (ICIs) (nivolumab, pembrolizumab durvalumab, atezolizumab, cemiplimab) to standard platinum‐based chemotherapy (Carbone 2017; Mok 2019; Reck 2016; Rizvi 2020; Sezer 2020; Herbst 2020). Rizvi 2020, reported OS data for participants with negative PD‐L1 expression < 1% as well as PD‐L1 level ≥ 1%. Carbone 2017; Mok 2019 and Herbst 2020 provided data for PD‐L1 at ≥ 1% and ≥ 50%,whereas, both Reck 2016 and Sezer 2020 only included participants with PD‐L1≥ 50%.
PD‐L1 expression <1% (negative PD‐L1): one trial only (Rizvi 2020) reported data for this subgroup as negative PD‐L1 expression was an exclusion criterion for the other five trials. There was no evidence of a difference in OS between ICI single‐agent (durvalumab) and standard platinum‐based chemotherapy (hazard ratio (HR): 1.18, 95% confidence interval (CI) 0.86 to 1.61, 1 RCT, 178 participants) Analysis 1.1 ; Figure 4
1.1. Analysis.

Comparison 1: Single immunecheckpoint inhibitors vs chemotherapy, Outcome 1: Overall survival by PD‐L1 expression
4.

Forest plot of comparison: Single Immuno versus Chemotherapy, outcome: 4.1 Overall survival by PD‐L1 expression.
PD‐L1 expression ≥ 1% (positive PD‐L1): four trials (Carbone 2017; Mok 2019; Rizvi 2020; Herbst 2020) provided data. There was no evidence of a difference between participants receiving single‐agent ICI or platinum‐based chemotherapy (HR 0.88, 95% CI 0.78 to 1.00, P = 0.05, I² = 40%, 4 RCTs, 2937 participants) Analysis 1.1 ; Figure 4. However, a considerable level of heterogeneity occurred, while excluding Carbone 2017 and pooling data from the remaining three trials (Mok 2019; Reck 2016; Herbst 2020) showed a difference in favour of single‐agent ICI group with no heterogeneity (HR 0.83, 95%CI 0.75 to 0.92, P = 0.0005, I² = 0%, 4 RCTs, 2937 participants) (analysis not shown). A sensitivity analysis applying a fixed‐effect model also showed evidence of a difference in favour of the group who received single‐agent ICI compared to platinum‐based chemotherapy (HR: 0.87, 95% CI 0.80 to 0.96, P = 0.004, I² = 40%, 4 RCTs, 2937 participants) (analysis not shown).
PD‐L1 expression ≥50% (high PD‐L1): All six trials (Carbone 2017; Mok 2019; Reck 2016; Rizvi 2020; Sezer 2020; Herbst 2020) reported results. There was probably evidence of a difference favouring the group who received single‐agent ICI compared to platinum‐based chemotherapy (HR: 0.68, 95% CI 0.60 to 0.76, P< 0.00001; I2 = 1%, 6 RCTs, 2111 participants; moderate certainty of evidence) Analysis 1.1; Figure 4. The certainty for this outcomes was downgraded one level due to study limitations.
There was evidence of a difference between subgroups (PD‐L1 negative, positive and high) (Test for subgroup differences: Chi² = 16.31, df = 2 (P = 0.0003), I² = 87.7%), Analysis 1.1; Figure 4.
b. Subgroup analyses: .
i. OS and Tumour Mutational Burden (TMB)
TMB was assessed on tissue in Carbone 2017 and Mok 2019 and thresholds to define high TMB were >243 mut/exome (Carbone 2017) and >175 mut/exome (Mok 2019), respectively. In both Rizvi 2020 and Herbst 2020, TMB was assessed on plasma cell‐free circulating tumour DNA and cut off to define high TMB was ≥ 20 Mut/Mb.
Four trials (Carbone 2017; Mok 2019; Rizvi 2020, Herbst 2020) reported OS results according to two TMB categories, low and high.
TMB ‐ Low, there was no evidence of a difference in OS between single‐agent ICI and platinum‐based chemotherapy was reported (HR: 1.01, 95% CI 0.88 to 1.15, P = 0.92, I2 = 0%, 4 RCTs, 1380 participants). Analysis 1.2 ; Figure 5
1.2. Analysis.

Comparison 1: Single immunecheckpoint inhibitors vs chemotherapy, Outcome 2: Overall Survival by Tumor Mutational Burden
5.

Forest plot of comparison: Single Immuno versus Chemotherapy, outcome: 4.4 Overall Survival by Tumor Mutational Burden.
TMB ‐ High, there was evidence of a difference in favour of single‐agent ICI recipients when compared with platinum‐based chemotherapy (HR: 0.72, 95% CI 0.57 to 0.90, P = 0.004, I2=,19%, 4 RCTs, 655 participants) Analysis 1.2 ; Figure 5.
Although the pooled survival data showed no evidence of a difference between participants receiving single‐agent ICI or platinum‐based chemotherapy, (HR 0.89, 95%CI 0.76,to 1.05, P = 0.17, I2 = 51%, 4 RCTs, 2035) the test for subgroup differences showed there was evidence of a difference according to TMB‐High and TMB‐Low categories (Test for subgroup differences: Chi² = 6.39, df = 1 (P = 0.01), I² = 84.4%) Analysis 1.2 ; Figure 5.
ii. OS by age and PD‐L1 expression ≥ 1%
Two trials (Carbone 2017 , Mok 2019) provided data for people < 65 and ≥ 65 years old and PD‐L1 expression ≥ 1%.
Age < 65 years and PD‐L1 expression ≥ 1%. There was no evidence of a difference in OS among < 65 years old participants who received single‐agent ICI compared to those who randomised to platinum‐based chemotherapy (HR: 0.93, 95% CI 0.68 to 1.29, P = 0.68, I2 = 69%, 2 RCTs, 988 participant) Analysis 1.3.
1.3. Analysis.

Comparison 1: Single immunecheckpoint inhibitors vs chemotherapy, Outcome 3: Overall survival by age (PDL1≥ 1%)
Age ≥ 65 years and PD‐L1 expression ≥1%. There was no evidence of a difference in OS among ≥ 65 years old people who received single‐agent ICI compared to platinum‐based chemotherapy (HR: 0.90, 95% CI 0.72 to 1.13, P = 0.36, I2 = 37% 2 RCTs, 827 participants) Analysis 1.3.
The pooled survival data showed no evidence of a difference in OS for people who received single‐agent ICI compared to platinum‐based chemotherapy (HR 0.90, 95% CI 0.78 to 1.03, P = 0.20, I2 = 38%, 2 RCTs,1815 participants) Analysis 1.5. When applying a fixed‐effect there was a difference favouring the single‐agent ICI group without affecting the heterogeneity level (HR 0.89, 95% CI 0.79 to 1.00; P = 0.04, I2 = 38%, 2 RCTS, participants = 1815) (analysis not shown). However, there was no evidence of a difference between people < 65 and ≥ 65 years old, who received the single‐agent ICI or the standard platinum‐based chemotherapy either applying a random‐effects model (Test for subgroup differences: Chi² = 0.04, df = 1 (P = 0.85), I² = 0%) or a fixed‐effect model (Test for subgroup differences: Chi²= 0.00, df = 1, (P = 0.98), I² = 0%) (analysis not shown), Analysis 1.3.
1.5. Analysis.

Comparison 1: Single immunecheckpoint inhibitors vs chemotherapy, Outcome 5: Overall survival by sex (PDL1≥ 1%)
iii. OS by age and PD‐L1 expression ≥ 50%
Three trials (Mok 2019; Reck 2016; Herbst 2020) provided data for this outcome.
Age < 65 years and PD‐L1 expression ≥ 50%. There was evidence of a difference in OS favouring among people < 65 years old who received single‐agent ICI compared to platinum‐based chemotherapy (HR: 0.72, 95%CI 0.57 to 0.90, P = 0.004, I2= 0%, 3 RCTs, 571 participants) Analysis 1.4
1.4. Analysis.

Comparison 1: Single immunecheckpoint inhibitors vs chemotherapy, Outcome 4: Overall survival by age (PDL1 ≥50%)
Age ≥ 65 years and PD‐L1 expression ≥ 50%. There was evidence of a difference in OS favouring ≥ 65 years old people who received single‐agent ICI compared to platinum‐based chemotherapy (HR: 0.60, 95% CI 0.47 to 0.78, P< 0.0001, I2= 0%, 2 RCTs, 435 participants) Analysis 1.4
The pooled survival data showed evidence of a difference in OS favouring participants receiving single‐agent ICI compared to platinum‐based chemotherapy (HR: 0.67, 95% CI 0.56 to 0.79, P < 0.00001, I2= 0%, 3 RCTs, 1006 participants) Analysis 1.4. No difference according to age subgroups (people < 65 and ≥65 years old with PD‐L1 expression ≥ 50%) was observed (Test for subgroup differences: Chi² = 1.06, df = 1 (P = 0.30), I² = 5.6%). Analysis 1.4.
iv. OS by gender and PD‐L1 expression ≥ 1%
Four trials (Carbone 2017; Mok 2019; Reck 2016; Herbst 2020) provided data for overall survival outcome according to PD‐L1 levels and gender.
Data were reported by Carbone 2017 and Mok 2019.
Males with PD‐L1 expression ≥ 1%.
There was no evidence of a difference in OS among males participants receiving single‐agent ICI compared to platinum‐based chemotherapy ( HR; 0.86, 95% CI 0.71 to 1.03, P = 0.09, I2 = 33%, 2 RCTS, 1234 participants) Analysis 1.5. When applying a fixed‐effect model there was a difference in favour of the single‐agent ICI group with no changes in the heterogeneity level (HR: 0.84, 95% CI 0.74 to 0.97, P = 0.02, I2 = 33%, 2 RCTS, 1234 participants).
Females with PD‐L1 expression ≥ 1%.
There was no evidence of a difference in OS among females who received single‐agent ICI compared to platinum‐based chemotherapy (HR: 0.98, 95% CI 0.77 to 1.25, P = 0.88, I2 =17%, 2 RCTs, 581 participants) Analysis 1.5
Moreover, there was no evidence of a difference between subgroups (Test for subgroup differences: Chi² = 0.78, df = 1 (P = 0.38), I² = 0%).
The overall pooled survival data showed no evidence of a difference in OS among males and females with PD‐L1 level ≥ 1% who received single‐agent ICI compared to platinum‐based chemotherapy (HR: 0.90 95% CI 0.78 to 1.03, P = 0.12, I2 = 23%, 2 RCTs, 1815 participants) Analysis 1.5. When applying a fixed‐effect model a difference in OS was observed favouring the single‐agent ICI group compared to platinum‐based chemotherapy (HR: 0.88 95% CI 0.78 to 0.99, P = 0.03, I2 = 23%, 2 RCTs, 1815 participants).
However, no difference according to gender subgroups (male or female with PD‐L1 expression ≥1%) was observed applying either a random‐effects model (Test for subgroup differences: Chi² =0.78, df = 1, P = 0.38, I2 = 0%) or a fixed‐effect model (Test for subgroup differences: Chi² =1.19, df = 1, P = 0.27, I2 = 16.3%) (analysis not shown) Analysis 1.5.
v. OS by gender and PD‐L1 expression ≥ 50%
Three trials (Mok 2019; Reck 2016; Herbst 2020) reported OS data by gender.
Males with PD‐L1 expression ≥ 50%. There was evidence of a difference in OS among male participants who received single‐agent ICI compared to platinum‐based chemotherapy (HR: 0.62, 95% CI 0.52 to 0.76, P < 0.00001. I2= 0%, 3 RCTs, 745 participants) Analysis 1.6
1.6. Analysis.

Comparison 1: Single immunecheckpoint inhibitors vs chemotherapy, Outcome 6: Overall survival by sex (PDL1≥50%)
Females with PD‐L1 expression ≥ 50%. There was no evidence of a difference for female participants who received single‐agent ICI compared to platinum‐based chemotherapy (HR: 0.81, 95% CI 0.61 to 1.08, P = 0.15, I2 =0%, 3 RCTs, 363 participants) Analysis 1.6.
The overall pooled results showed evidence of a difference in OS favouring the group who received single‐agent ICI compared to platinum‐based chemotherapy (HR 0.68, 95%CI 0.58 to 0.79, P < 0.00001, I2= 0%, 3RCTs, 1108 participants) Analysis 1.6.
Furthermore, there was no evidence of a difference between the two subgroups (Test for subgroup differences: Chi² = 2.17, df = 1 (P = 0.14), I² = 54.0%) Analysis 1.6..
vi. OS by smoking status and PD‐L1 expression ≥1%
Two trials (Carbone 2017 and Mok 2019) provided data for this outcome. We reported the OS and the smoking status results by the following categories.
Never smoked and PD‐L1 expression ≥1%. There was no evidence of a difference between never smokers who received single‐agent ICI compared to platinum‐ based chemotherapy (HR: 1.00, 95%CI 0.76 to 1.33, P = 0.98, fixed‐effect model or random‐effects model, I2= 0%, 2 RCTs, 341 participants) Analysis 1.7
1.7. Analysis.

Comparison 1: Single immunecheckpoint inhibitors vs chemotherapy, Outcome 7: Overall survival by smoking status (PDL1≥ 1%)
Former smokers and PD‐L1 expression ≥ 1%. There was no evidence of a difference in OS between former smokers who received single‐agent ICI compared to platinum‐based chemotherapy HR: 0.87, 95% CI 0.57 to 1.33, P = 0.52, I2= 85%, 2 RCTs, 1089 participants) Analysis 1.7 However, when applying a fixed‐effect model there was a difference favouring the single‐agent ICI group with no changes in the heterogeneity level (HR: 0.83, 95%CI 0.71 to 0.96, P = 0.01, I2= 85%, 2 RCTs, 1089 participants) (analysis not shown).
Current smokers and PD‐L1 expression ≥ 1%. There was no evidence of a difference in OS between current smokers who received single‐agent ICI compared to standard chemotherapy (HR: 0.98, 95% CI 0.75 to 1.27, P = 0.85, I2 = 0%, 2 RCTs, 378 participants) Analysis 1.7
The pooled data showed no evidence of a difference between people receiving single‐agent ICI or platinum‐based chemotherapy for people with PD‐L1 ≥1% regardless of their smoking status (HR: 0.92, 95%CI 0.78 to 1.10, P = 0.38, I2 = 44%, 2 RCTs, 1808 participants), Analysis 1.7.
Applying a fixed‐effect model did not alter the level of heterogeneity nor the overall effect (HR 0.89, 95% CI 0.79 to 1.00; I2 = 44%, 2 RCTs, participants = 1808) (analysis not shown). However, when applying a fixed‐effect model and excluding Carbone 2017 from the "former smokers" subgroup not only reduced the heterogeneity to I2 = 32%, but it also showed a difference in OS favouring the single‐agent ICI group regardless of the smoking status (HR: 0.84, 95% CI 0.73 to 0.96; I2 = 32%, P = 0.01, 2 RCTs, participants = 1440) (analysis not shown).
There was no evidence of a difference between smoking status subgroups either when applying a random‐effects model (Test for subgroup differences: Chi² = 0.31, df = 2 (P = 0.86), I² = 0%) Analysis 1.7, or a fixed‐effect model (Test for subgroup differences: Chi² = 2.09, df=2 (P = 0.35) I² = 4.1%) and also after exclusion of Carbone 2017 from former smokers subgroup (Test for subgroup differences: Chi² = 5.75, df = 2 (P = 0.06) I² = 65.2%) (analysis not shown). .
vii. OS by smoking status and PD‐L1 expression ≥50%
Three trials (Mok 2019; Reck 2016; Herbst 2020) contributed to this outcome.
Never smokers and PD‐L1 expression ≥50%. There was no evidence of a difference in OS between people who never smoked who received single‐agent ICI compared to platinum‐based chemotherapy (HR: 1.18, 95% CI 0.78 to 1.79, P = 0.44, I2 = 0%, 3 RCTs, 179 participants), Analysis 1.8.
1.8. Analysis.

Comparison 1: Single immunecheckpoint inhibitors vs chemotherapy, Outcome 8: Overall survival by smoking status (PDL1≥ 50%)
Former smokers and PD‐L1 expression ≥ 50%. There was evidence of a difference in OS favouring the former smoker group who received single‐agent ICI compared to platinum‐based chemotherapy (HR: 0.60, 95% CI 0.49 to 0.73, P < 0.00001, I2= 0%, 3 RCTs, 700 participants) Analysis 1.8.
Current smokers and PD‐L1 expression ≥ 50%. There was evidence of a difference in OS favouring the current smoker group who received single‐agent ICI compared to platinum‐based chemotherapy (HR: 0.65, 95% CI 0.44 to 0.97, P = 0.04 I2=11%, 3 RCTs, 230 participants), Analysis 1.8.
The pooled results showed a difference in OS between participants with PD‐L1 expression ≥ 50% who received single‐agent ICI compared to platinum‐based chemotherapy regardless of their smoking status (HR: 0.70, 95% CI 0.56 to 0.86, P = 0.001, I2 = 29%, 3 RCTs, 1109 participants), Analysis 1.8. In addition, there was evidence of a difference between smoking status subgroups (Test for subgroup differences: Chi² = 8.27, df = 2, P = 0.02, I² = 75.8%) Analysis 1.8.
viii. OS by ECOG PS and PD‐L1 expression ≥ 1%
Two trials (Carbone 2017; Mok 2019) provided OS data according to ECOG PS and PD‐L1 expression, ≥ 1%.
ECOG PS 0 and PD‐L1 expression ≥ 1%.. There was no evidence of a difference in OS between ECOG PS 0 people who received single‐agent ICI compared to platinum‐based chemotherapy (HR: 0.90, 95% CI 0.63 to 1.29, P = 0.57, I2=50%, 2 RCTs, 568 participants) Analysis 1.9.
1.9. Analysis.

Comparison 1: Single immunecheckpoint inhibitors vs chemotherapy, Outcome 9: Overall survival by ECOG PS (PDL1≥ 1%)
ECOG PS 1 and PD‐L1 expression >1%. There was no evidence of a difference in OS between ECOG PS 1 participant who received single‐agent ICI compared to platinum‐based chemotherapy (HR: 0.90, 95%CI 0.74, 1.09, P = 0.29, I2 = 43%, 2 RCTs, 1246 participants) Analysis 1.9. .
The pooled results showed no difference in OS between people receiving single‐agent ICI or platinum‐based chemotherapy (HR: 0.89, 95%CI 0.77 to 1.03, P = 0.11, I2 = 20%, 2 RCTs, 1814 participants) Analysis 1.9. When applying a fixed‐effect model there was a difference favouring single‐agent ICI compared to platinum‐based chemotherapy regardless ECOG PS in people with PD‐L1 expression >1% (HR: 0.88, 95% CI 0.78 to 1.00, P = 0.04, I2 = 20%, 2 RCTs, 1814 participants) (analysis not shown). There was also no evidence of a difference between the two ECOG PS subgroups either by random‐effects model (Test for subgroup differences: Chi² = 0.00, df = 1, P = 0.98, I² = 0%) Analysis 1.9 or fixed‐effect model (Test for subgroup differences: Chi² = 0.00, df = 1, P = 1.00, I² = 0%) (analysis not shown).
ix. OS by ECOG PS and PD‐L1 expression ≥ 50%
Three trials provided data for this outcome (Herbst 2020Mok 2019; Reck 2016).
ECOG PS 0 and PD‐L1 expression ≥ 50%. There was a evidence of a difference in OS favouring the single‐agent ICI recipients compared to the platinum‐based chemotherapy recipients (HR: 0.60, 95% CI 0.44 to 0.81, P< 0.0009, I2= 0%, 3 RCTs, 367 participants) Analysis 1.10.
1.10. Analysis.

Comparison 1: Single immunecheckpoint inhibitors vs chemotherapy, Outcome 10: Overall survival by ECOG PS (PDL1≥ 50%)
ECOG PS 1 and PD‐L1 expression ≥ 50%. There was evidence of a difference in OS favouring people who received single‐agent ICI compared to platinum‐based chemotherapy recipients (HR: 0.67, 95% CI 0.55 to 0.82, P = 0.0001, I2 =0%, 3 RCTs, 741 participants), Analysis 1.10.
The pooled results showed a difference in OS in favour of people receiving single‐agent ICI compared to platinum‐based chemotherapy (HR: 0.65, 95% CI 0.55 to 0.77, P< 0.00001, I2 = 0%, 3 RCTs, 1108 participants) Analysis 1.10.There was no evidence of a difference between the two subgroups, ECOG PS 0 and 1, (Test for subgroup differences: Chi² = 0.40, df = 1 (P = 0.52), I² = 0%) Analysis 1.10.
x. OS by histology and expression ≥ 1%
Two trials (Carbone 2017; Mok 2019) provided data for this outcome.
Squamous and PD‐L1 expression ≥ 1%. There was evidence of a difference in OS favouring the participants who received single‐agent ICI when compared to platinum‐based chemotherapy (HR: 0.76, 95% CI 0.63 to 0.93, P = 0.006, I2= 0%, 2 RCTs, 621 participants) Analysis 1.11.
1.11. Analysis.

Comparison 1: Single immunecheckpoint inhibitors vs chemotherapy, Outcome 11: Overall survival by histological type (PDL1≥ 1%)
Non‐squamous and PD‐L1 expression ≥ 1%. There was no evidence of a difference between the two groups, single‐agent ICI and standard chemotherapy, (HR: 0.99, 95% CI 0.73 to 1.33, P = 0.94, I2= 72%, 2 RCTs, 1194 participants). Analysis 1.11.
The pooled results showed no difference in OS between single‐agent ICI and standard chemotherapy, (HR: 0.89, 95% CI 0.73 to 1.07, P = 0.20, I2 = 56% 2 RCTs, 1815 participants) Analysis 1.11. However, when applying a fixed‐effect model we found evidence of a difference between the two groups and the level of heterogeneity remained unchanged (HR: 0.88, 95%CI 0.78 to 0.98, P = 0.03, I2 = 56% 2 RCTs, 1815 participants) (analysis not shown).
In addition, there was no evidence of any differences according to histology subgroups (squamous versus non‐squamous) either applying a random‐effects model (Test for subgroup differences: Chi² = 2.00, df = 1 (P = 0.16), I² = 50.0%) Analysis 1.11 or a fixed‐effect model (Test for subgroup differences: Chi² = 3.09, df = 1 (P = 0.08), I² = 67.7%) (analysis not shown).
xi. OS by histology and expression ≥ 50%
Three trials provided data for this outcome ( Herbst 2020; Mok 2019 ; Reck 2016).
Squamous and PD‐L1 expression ≥ 50%. There was evidence of a difference in OS among people receiving single‐agent ICI compared to participants treated with standard chemotherapy (HR: 0.57, 95% CI 0.43 to 0.76, P = 0.0001, I2=0%, 3 RCTs, 327 participants) Analysis 1.12
1.12. Analysis.

Comparison 1: Single immunecheckpoint inhibitors vs chemotherapy, Outcome 12: Overall survival by histological type (PDL1≥ 50%)
Non‐squamous and PD‐L1 expression ≥ 50%. Three trials ( Herbst 2020; (Mok 2019; Reck 2016) provided data for this subgroup. There was evidence of a difference in OS favouring the group who received single‐agent ICI compared to standard chemotherapy (HR:0.69, 95%CI 0.55 to 0.87, P = 0.002, I2=26%, 3 RCTs, 782 participants) Analysis 1.12.
The pooled results showed a difference in OS between single‐agent ICI and standard platinum‐based chemotherapy regardless of histology subtype in people with PD‐L1 expression ≥ 50% (HR: 0.66, 95% CI 0.56 to 0.77, P<0.00001, I2 = 0%, 3 RCTs, 1109 participants) Analysis 1.12. There was no evidence of a difference between the two histology subtypes (squamous and non‐squamous) (Test for subgroup differences: Chi² = 1.08, df = 1, P = 0.30, I² = 7.6%) Analysis 1.12.
2. Primary outcome ‐ Progression‐free survival (PFS)
a. Main analysis: PFS by PD‐L1 expression
Five trials provided data for this outcome (Carbone 2017; Herbst 2020; Mok 2019; Reck 2016; Sezer 2020).
PD‐L1 expression <1% (negative PD‐L1). None of the included trials reported PFS data by this subgroup.
PD‐L1 expression ≥ 1% (positive PD‐L1). Three trials (Carbone 2017; Herbst 2020; Mok 2019) provided data for this subgroup. There was no evidence of a difference in PFS between the group who received single‐agent ICI or platinum‐based chemotherapy (HR: 0.99, 95% CI 0.79 to 1.24, P = 0.95, I2 = 80% 3 RCTs, 2369 participants) Analysis 1.13.
1.13. Analysis.

Comparison 1: Single immunecheckpoint inhibitors vs chemotherapy, Outcome 13: Progression free survival by PD‐L1 expression
PD‐L1 expression ≥ 50% (high PD‐L1). Five trials (Carbone 2017; Herbst 2020; Mok 2019; Reck 2016; Sezer 2020) reported PFS results. There may be evidence of a difference in PFS favouring the group who received single‐agent ICI compared to platinum‐based chemotherapy (HR: 0.68, 95% CI 0.52 to 0.88, P = 0.003 I2 = 80%, 5 RCTs, 1886 participants; low certainty of evidence) Analysis 1.13. The certainty for this outcome was downgraded one level due to study limitations, and one level due to inconsistency.
The pooled results showed a difference in PFS favouring the single‐agent ICI compared to the platinum‐based chemotherapy, (HR: 0.79, 95% CI 0.63 to 0.99 P = 0.04 I2 = 79.2%, 5 RCTs, 4255 participants) Analysis 1.13. There was evidence of a difference between PD‐L1 ≥ 1% and ≥50% subgroups (Test for subgroup differences: Chi² = 4.82, df = 1, P = 0.03, I² = 79.2%) Analysis 1.13.
b. Subgroup analyses:
i. PFS by Tumour Mutaution Burden (TMB)
Four trials (Carbone 2017; Herbst 2020; Mok 2019; Rizvi 2020) provided data for this outcome.
TMB ‐ High. There was evidence of a difference in PFS favouring participants who received single‐agent ICI compared to platinum‐based chemotherapy (HR: 0.72, 95% CI 0.60 to 0.86, P = 0.0003, I2 = 0%, 4 RCTs, 655 participants) Analysis 1.14
1.14. Analysis.

Comparison 1: Single immunecheckpoint inhibitors vs chemotherapy, Outcome 14: Progression Free Survival by Tumor Mutational Burden
TMB ‐ Low. There was no evidence of a difference in PFS for people who received single‐agent ICI compared to platinum‐based chemotherapy (HR: 1.24, 95% CI 1.00 to 1.55, P = 0.05, I2= 69%, 4 RCTs, 1380 participants) Analysis 1.14. In contrast, when applying a fixed‐effect model there was a difference in favour of the chemotherapy group (HR: 1.22, 95% CI 1.08 to 1.37, P = 0.001, I2 = 69%, 4 RCTs, 1380 participants) (analysis not shown).
The pooled results showed no difference between people who received single‐agent ICI compared to those who received platinum‐based chemotherapy (HR: 0.97, 95% CI 0.76 to 1.22, P = 0.77, I2 = 80%, 4 RCTs, 2035 participants) Analysis 1.14. The pooled results and the level of heterogeneity did not change with fixed‐effect model (HR: 1.03, 95%CI 0.93 to 1.14, P = 0.54, I2 = 80%, 4 RCTs, 2035 participants) (analysis not shown). There was evidence of a difference between TMB‐high and TMB‐low subgroups either when applying a random‐effects model (Test for subgroup differences: Chi² = 14.47, df = 1 (P = 0.0001), I² = 93.1%) Analysis 1.14 or a fixed‐effect model (Test for subgroup differences: Chi² = 23.29, df = 1 (P < 0.00001), I² = 95.7%) (analysis not shown).
3. Secondary outcomes ‐ Overall response rate (ORR)
a. Main analysis: ORR by PD‐L1 expression
Three trials (Herbst 2020; Mok 2019; Reck 2016;Sezer 2020) provided data for this outcome.
PD‐L1 expression <1% (negative PD‐L1).: None of the included trials reported ORR for this subgroup.
PD‐L1 expression ≥ 1% (positive PD‐L1). Two trials (Herbst 2020; Mok 2019) reported ORR showing no evidence of a difference in the group who received single‐agent ICI compared to standard platinum‐based chemotherapy (RR: 0.99, 95% CI 0.86 to 1.15, P = 0.90, I2=0%, 2 RCTs, 1828 participants) Analysis 1.15.
1.15. Analysis.

Comparison 1: Single immunecheckpoint inhibitors vs chemotherapy, Outcome 15: Overall response rate by PDL1 expression
PD‐L1 expression ≥ 50% (high PD‐L1). Four trials (Herbst 2020; Mok 2019; Reck 2016; Sezer 2020 ) reported ORR. There may be evidence of a difference in favour of single‐agent ICI compared with platinum‐based chemotherapy (RR: 1.40, 95% CI 1.12 to 1.75, P = 0.003, I 2= 60%, 4 RCTs, 1672 participants, low‐certainty evidence) Analysis 1.15. The certainty for this outcomes was downgraded one level due to study limitations, and one level due to inconsistency.
There was evidence of a difference between the PD‐L1≥1% and PD‐L1 ≥50% subgroups (Test for subgroup differences: Chi² = 6.50, df = 1 (P = 0.01), I² = 84.6%) Analysis 1.15.
b. Subgroup analyses
i. ORR by Tumour Mutational Burden (TMB)
Three trials (Carbone 2017; Mok 2019, Rizvi 2020) provided data for this outcome.
TMB ‐ High. There was no evidence of a difference in ORR in the group of people who received single‐agent ICI compared to platinum‐based chemotherapy was reported (RR: 1.25, 95%CI 0.99 to 1.59, P = 0.06, I2=1%, 3 RCTs, 599 participants) Analysis 1.16.
1.16. Analysis.

Comparison 1: Single immunecheckpoint inhibitors vs chemotherapy, Outcome 16: Overall Response Rate by Tumor Mutational Burden
TMB ‐ Low. There was evidence of a difference in ORR favouring platinum‐based chemotherapy group (RR: 0.73, 95% CI 0.59 to 0.91, P = 0.004. I2= 0%, 3 RCTs, 1047 participants) Analysis 1.16.
The pooled results showed no difference in ORR between the two treatment groups (RR: 0.97, 95% CI: 0.73 to 1.27, P = 0.80, I2 = 64%, 1646 participants) Analysis 1.16. There was evidence of a difference between the two subgroups (TMB‐High and TMB‐Low) (Test for subgroup differences: Chi² = 10.80, df = 1 (P = 0.001), I² = 90.7%).
4. Secondary outcome ‐ Adverse events (AEs)
a. Main analysis ‐ AEs overall
None of the included trials provided data by PD‐L1 expression for AEs.
Five included trials (Carbone 2017; Herbst 2020; Mok 2019; Reck 2016; Rizvi 2020) reported grade three to five AEs. For overall population and not according to PD‐L1 expression levels.
Overall, there was evidence of reduced grade 3 to 5 adverse events in people who received single‐agent ICI compared with platinum‐based chemotherapy (RR: 0.43, 95% CI 0.36 to 0.52, P < 0.00001, I2= 41%, 5 RCTs, 6692 participants, low‐certainty evidence) Analysis 1.17 ; Figure 6.
1.17. Analysis.

Comparison 1: Single immunecheckpoint inhibitors vs chemotherapy, Outcome 17: Adverse Events grade 3‐5
6.

Forest plot of comparison: Single Immuno versus Chemotherapy, outcome: 4.7 Adverse Events grade 3‐5.
Adverse events grade 3 to 4. There was may be evidence of a difference with reduced adverse events grade 3 to 4 in favour of single‐agent ICIs compared to platinum‐based chemotherapy (RR: 0.41, 95% CI 0.33 to 0.50, P < 0.00001, I2= 62%, 5 RCTs, 3346 participants, low‐certainty evidence) Analysis 1.17 ; Figure 6. The certainty for this outcomes was downgraded one level due to study limitations, and one level due to inconsistency.
Removing data from Reck 2016, reduced the heterogeneity level to 13% without changing the results (RR: 0.38, 95%CI 0.33 to 0.44, P<0.00001, I2= 13%, 4 RCTs, 3042 participants) (analysis not shown).
Adverse events grade five (toxic deaths). There was no evidence of a difference in toxic deaths (grade 5 AEs) in participants who received single‐agent ICI compared to platinum‐based chemotherapy (RR: 0.78, 95% CI 0.43 to 1.41, P = 0.40, I2 = 0%, 5 RCTs, 3346 participants) Analysis 1.17 ; Figure 6.
In addition, there was evidence of subgroup differences between grade 3 to 4 and grade 5 adverse events (Test for subgroup differences: Chi² = 4.08, df = 1 (P = 0.04), I² = 75.5%).
5. Secondary outcome ‐ Health‐related quality of life (HRQoL)
HRQoL was measured in only one multicentre open‐label phase 3 trial (Reck 2016) comparing pembrolizumab to platinum‐based chemotherapy in advanced NSCLC.The quality of life was measured using the European Organisation for Research and Treatment of Cancer (EORCT) Quality of Life Questionnaire Core 30 items (QOL‐C30); and the EORTC quality of life questionnaire lung cancer 13 items (QLQ‐LC13) and the European Quality of Life 5 dimensions‐3 Level (EQ‐5D‐3L). The QOL‐C30 measures five functional demotions namely: physical, role, cognitive and social and three symptoms items fatigue, fatigue, nausea or vomiting and pain, six single items (dyspnoea, sleep disturbance, appetite loss, constipation, diarrhoea and financial impact. and a global health and quality of life scale (GHS/QOL) Osoba 1998.
The three instruments were administrated on day one of cycle one to three and every nine weeks thereafter, at the treatment discontinuation and at the 30 days safety checks. The compliance with the questionnaires was more than 90% at baseline and around 80% at week 15 for both groups.
Overall HRQoL using QOL‐C30 GHS/QOL. At week 15, there was evidence of improvement from baseline in favour of people who received pembrolizumab compared with platinum‐based chemotherapy group, the difference was 7.8 points ( MD: 7.80, 95% CI 2.45 to 13.15, 1 RCT, 297 participants) Analysis 1.18.
1.18. Analysis.

Comparison 1: Single immunecheckpoint inhibitors vs chemotherapy, Outcome 18: QOL‐C30 GHS/QOL ‐ change from baseline to week 15
Time to symptoms deterioration using quality of life questionnaire lung cancer 13 items (QLQ‐LC13)
Time to deterioration defined as the time to the first onset of 10 points or more decrease from baseline of the following symptoms: cough, chest pain or dyspnoea. There was evidence of a difference in time to deterioration of the composite symptoms favouring the pembrolizumab group (HR:0.66, 95% CI 0.440 to 99, 1 RCT, 194 participants) Analysis 1.19.
1.19. Analysis.

Comparison 1: Single immunecheckpoint inhibitors vs chemotherapy, Outcome 19: Time to deterioration ‐ QLQ‐LC13
Improvement on Quality of Life Questionnaire Core 30 items (QOL‐C30)
The proportion of individuals who improved on the global health score and quality of life (GHS/QOL), functional and symptoms scales on the QLQ‐C30 were reported at week 15. Improvement was defined as a 10‐point greater increase in functional scores or a 10‐point greater decrease in symptoms scores.
At week 15, a greater improvement in the GHS/QoL score from baseline was may be evident among pembrolizumab group (RR:1.51, 95% CI 1.08 to 2.10, 1 RCT, 297 participants, low‐certainty evidence) Analysis 1.22. The certainty for this outcomes was downgraded one level due to study limitations, and one level due to imprecision. Similarly, fewer individuals treated with pembrolizumab had deteriorated fatigue (RR:1.61, 95% CI 1.21 to 2.13, 1 RCT, 297 participants) and pain (RR 1.39, 95% CI 1.05 to 1.84, RCT, 297 participants), respectively Analysis 1.22. However, there was no evidence of a difference between the two groups in physical function (RR:1.09, 95% CI 0.74 to 1.61, 1 RCT, 297 participants), role function (RR:1.29, 95% CI 0.92, 1.81, 1 RCT, 297 participant), emotional (RR 0.81, 95% CI 0.60 to 1.10, 1 RCT, 297 participant), cognitive (RR 0.75, 95% CI 0.50 to 1.15,1 RCT, 297 participants) and social functions (RR 1.20, 95% CI 0.85 to 1.69,1 RCT, 297 participant) Analysis 1.22. There was no evidence of a difference between the group in nausea and vomiting (RR 0.90, 95% CI 0.55 to 1.50, 1 RCT, 297 participants), dyspnoea (RR 1.24, 95% CI 0.88 to 1.73, 1 RCT, 297 participants), sleep disturbance (RR 0.89, 95% CI 0.66 to 1.21,1 RCT, 297 participants), appetite loss (RR 1.12, 95% CI 0.82 to 1.54,1 RCT, 297 participants) and constipation (RR 1.33, 95% CI 0.84 to 2.12, 1 RCT, 297 participants) Analysis 1.22.
1.22. Analysis.

Comparison 1: Single immunecheckpoint inhibitors vs chemotherapy, Outcome 22: HRQoL‐QLQC30‐change from baseline at week 15 ‐improvement ‐ PD‐L1≥50%
There was evidence of a difference of the proportion of people who reported improved diarrhoea favouring chemotherapy (RR 0.49, 95% CI 0.25 to 0.94,1 RCT, 297 participants) Analysis 1.22.
The number of individuals who reported any improvement in their financial difficulties at week 15 did not differ between the groups (RR 0.88, 95% CI 0.54 to 1.42, RCT, 297 participants, Analysis 1.22.
Deterioration on Quality of Life Questionnaire Core 30 items (QOL‐C30)
The proportion of individuals who deteriorated on the global health score and quality of life (GHS/QOL), functional and symptoms scales on the QLQ‐C30 were reported at week 15. Deterioration was defined as a 10‐point greater decrease in functional scores or a 10‐point greater increase in symptoms scores.
At week 15, the deterioration in GHS/QoL scores were similar between the two groups (RR 0.71, 95% CI 0.48 to 1.06, 1 RCT, 297 participants) Analysis 1.21.
1.21. Analysis.

Comparison 1: Single immunecheckpoint inhibitors vs chemotherapy, Outcome 21: HRQoL‐QLQC30‐change from baseline at week 15 ‐deterioration ‐ PDL1≥50%
Fewer participants in the pembrolizumab group had deteriorated status on physical functioning (RR 0.55, 95% CI 0.38 to 0.82, 1 RCT, 297 participants), role functioning (RR 0.61, 95% CI 0.42 to 0.88,1 RCT, 297 participants) and social functioning scores (RR 0.61, 95% CI 0.41 to 0.89, 1 RCT, 297 participants) Analysis 1.21. However, there was no evidence of a difference in emotional (RR 1.20, 95% CI 0.67 to 2.14,1 RCT, 297 participants) and cognitive functioning scores (RR 1.03, 95% CI 0.70 to 1.51,1 RCT, 297 participants) Analysis 1.21. Among the pembrolizumab group, fewer individuals reported deterioration their in fatigue (RR 0.43, 95% CI 0.29 to 0.63,1 RCT, 297 participants), nausea and vomiting (RR 0.48, 95% CI 0.28 to 0.81,1 RCT, 297 participants), dyspnoea (RR 0.43, 95% CI 0.27 to 0.68, 1 RCT, 297 participants), appetite loss (RR 0.59, 95% CI 0.37 to 0.94, 1 RCT, 297 participants) and constipation symptoms (RR 0.63, 95% CI 0.41 to 0.97,1 RCT, 297 participants) Analysis 1.21. There was no evidence of a difference between the number of individuals in the pembrolizumab and chemotherapy groups who reported a worsening in their pain (RR 0.75, 95% CI 0.47 to 1.18, 1 RCT, 297 participants,) and diarrhoea symptoms (RR 0.73, 95% CI 0.44 to 1.23, 1 RCT, 297 participants) Analysis 1.21. The number of individuals who reported a deterioration in their financial difficulties did not differ between the groups (RR 0.75, 95% CI 0.49 to 1.15,1 RCT, 297 participants) Analysis 1.21.
Stable status on Quality of Life Questionnaire Core 30 items (QLQ‐C30)
The proportion of individuals who reported no changes or stable status on the global health score and quality of life (GHS/QOL), functional and symptoms scales on the QLQ‐C30 was reported at week 15 in Reck 2016. On the GHS/QOL scores the number of individuals who reported "stable status" did not differ between the two groups (RR 0.89, 95% CI 0.68 to 1.17, 1 RCT, 297 participants) Analysis 1.20. In contrast, more individuals in the pembrolizumab reported their physical function was stable at week 15 (RR 1.35, 95% CI 1.05 to 1.74, 1 RCT, 297 participants). However, there was no evidence of a difference between the two groups regarding number of individuals who reported " stable status" on role (RR 1.16, 95% CI 0.88 to 1.55, 1 RCT, 297 participants), emotional (RR 1.11, 95% CI 0.88 to 1.39,1 RCT, 297 participants), cognitive (RR 1.12, 95% CI 0.89 to 1.40,1 RCT, 297 participants) and social functions (RR 1.21, 95% CI 0.92 to 1.58, 1 RCT, 297 participants) Analysis 1.20.
1.20. Analysis.

Comparison 1: Single immunecheckpoint inhibitors vs chemotherapy, Outcome 20: HRQoL‐QLQC30‐change from baseline at week 15‐stable‐PD‐L1≥50%
There was evidence of a difference favouring pembrolizumab group in the proportion of individuals who reported "stable status" of nausea and vomiting (RR 1.24, 95% CI 1.05 to 1.47,1 RCT, 297 participants ), insomnia (RR 1.36, 95% CI 1.03 to 1.79, 1 RCT, 297 participants,) and diarrhoea symptoms (RR 1.21, 95% CI 1.04 to 1.40,1 RCT, 297 participants) Analysis 1.20. However, the two groups did not differ regarding number of individuals who reported "stable status" on fatigue (RR 1.19, 95% CI 0.82 to 1.71,1 RCT, 297 participants), pain (RR 0.82, 95% CI 0.62 to 1.10, 1 RCT, 297 participants), dyspnoea (RR 1.28, 95% CI 0.99 to 1.64,1 RCT, 297 participants), appetite loss (RR 1.16, 95% CI 0.90 to 1.49, 1 RCT, 297 participants), and constipation symptoms (RR 1.09, 95% CI 0.89 to 1.33, 1 RCT, 297 participants) Analysis 1.20.
There was no significant differences between pembrolizumab and platinum‐based chemotherapy in the number of people who were stable financially (RR 1.16, 95% CI 0.96 to 1.41, RCT, 297 participants) Analysis 1.20
Comparison 2: First‐line combined ICI versus platinum‐based chemotherapy
Primary outcome ‐ Overall survival (OS)
a. Main analysis: . OS and PDL‐1 expressions
Two trials (Hellmann 2018; Rizvi 2020) comparing double ICI (nivolumab plus ipilimumab or durvalumab plus tremelimumab) versus standard platinum‐based chemotherapy reported results. The OS data were reported according to the following PD‐L1 expression categories.
PD‐L1 expression < 1% (negative PD‐L1). There was evidence of a difference in OS favouring people who received the combination of ICI compared to platinum‐based chemotherapy (HR: 0.67, 95% CI 0.55 to 0.81, P<0.0001 I2 = 0%, 2 RCTs, 532 participants) Analysis 2.1 ; Figure 7.
2.1. Analysis.

Comparison 2: Combined immune checkpoint inhibitors vs chemotherapy, Outcome 1: Overall Survival by PD‐L1 expression
7.

Forest plot of comparison: Combined Immuno versus Chemotherapy, outcome: 5.1 Overall Survival by PD‐L1 expression.
PD‐L1 expression ≥ 1% (positive PD‐L1). There was no evidence of a difference in OS between double ICI and platinum‐based chemotherapy (HR: 0.89, 95% CI 0.70 to 1.13; P = 0.35, I2 = 75%, 2RCTs, participants = 1378) Analysis 2.1; Figure 7.
PD‐L1 expression ≥ 50% (high PD‐L1).There was probably evidence of a difference in favour of participants who received combination of ICI compared to people treated with platinum‐based chemotherapy (HR: 0.72, 95% CI 0.59 to 0.89, P = 0.002, I2= 0%, 2 RCTs, 612 participants, moderate‐certainty evidence) Analysis 2.1 ; Figure 7. The certainty for this outcomes was downgraded one level due to study limitations.
Although there was no difference according to PD‐L1 expression by random‐effects model (Test for subgroup differences: Chi² = 3.55, df = 2 (P = 0.17), I² = 43.7%) Analysis 2.1, Figure 7, evidence of a subgroup difference was observed by fixed‐effect model (Test for subgroup differences: Chi² = 7.51, df = 2 (P = 0.02), I² = 73.4%
b. Subgroup analyses
i. OS and Tumour Mutational Burden (TMB)
TMB was assessed on tissue in Hellmann 2018 and on plasma ctDNA in Rizvi 2020. Thresholds to define high TMB were ≥ 10 mut/Mb (Hellmann 2018) and ≥ 20 Mut/Mb (Rizvi 2020), respectively.
TMB ‐ Low. There was no evidence of a difference in OS between double ICI and platinum‐based chemotherapy groups (HR: 0.93, 95% CI 0.61 to 1.43, P = 0.75, I2=86%, 2 RCTs, 769 participants) Analysis 2.2 ; Figure 8.
2.2. Analysis.

Comparison 2: Combined immune checkpoint inhibitors vs chemotherapy, Outcome 2: Overall Survival by Tumor Mutational Burden
8.

Forest plot of comparison: Combined Immuno versus Chemotherapy, outcome: 5.2 Overall Survival by Tumor Mutational Burden.
TMB ‐ High. There was evidence of a difference in OS favouring people who were treated with combined ICI compared to platinum‐based chemotherapy (HR: 0.60, 95% CI 0.44 to 0.82, P = 0.001, I2 = 38%, 2 RCTs, 433 participants) Analysis 2.2, Figure 8.
The pooled results showed no difference between double ICI and platinum‐based chemotherapy (HR: 0.75, 95% CI 0.54 to 1.05, P = 0.09, I2 = 83%, 2 RCTs, 1202 participants) Analysis 2.2, Figure 8. When applying a fixed‐effect model there was evidence of a difference between treatments (HR: 0.82, 95% CI 0.72 to 0.94, P = 0.003, I2 = 83%, 2 RCTs, 1202 participants) (analysis not shown).
There was no evidence of a difference between the two TMB subgroups (Test for subgroup differences: Chi² = 2.72, df = 1 (P = 0.10), I² = 63.3%) Analysis 2.2, Figure 8 when using a random‐effects model. When applying a fixed‐effect model there was a difference between the TMB‐High and the TMB‐low subgroups (Test for subgroup differences: Chi² = 8.59, df = 1 (P = 0.03), I² = 88.4%) (analysis not shown).
ii. OS by age and PD‐L1 expression ≥ 1% and PD‐L1 expression >50%
None of the included trials reported this outcome.
iii. OS by gender and PD‐L1 expression ≥ 1% and PD‐L1 expression >50%
None of the included trials reported this outcome.
iv. OS by smoking status and PD‐L1 expression ≥ 1% and PD‐L1 expression >50%
None of the included trials reported this outcome.
v. OS by ECOG PS and PD‐L1 expression ≥ 1%.and PD‐L1 expression >50%
None of the included trials reported this outcome. .
vi. OS by histology and PD‐L1 expression ≥ 1%.and PD‐L1 expression >50%
None of the included trials reported this outcome.
2. Primary outcome ‐ Progression‐free survival (PFS)
a. Main analysis: PFS by PD‐L1 expression
None of the included trials reported this outcome.
b. Subgroup analyses:
i. Progression‐free survival (PFS) by Tumour Mutational Burden (TMB)
Two trials Hellmann 2018 and Rizvi 2020 reported PFS results by TMB.
TMB ‐ Low. There was no evidence of a difference on PFS outcome between double ICI and platinum‐based chemotherapy (HR: 1.29, 95% CI 0.90 to 1.85, P = 0.17, I2 = 80%, 2 RCTs, 769 participants) Analysis 2.3. When applying the fixed‐effect model, results changed with evidence of a difference in favour of platinum‐based chemotherapy and no changes in the heterogeneity level (HR: 1.30, 95% CI 1.10 to 1.52, P = 0.002, I2 = 80%, 2 RCTs, 769 participants) (analysis not shown).
2.3. Analysis.

Comparison 2: Combined immune checkpoint inhibitors vs chemotherapy, Outcome 3: Progression Free Survival by Tumor Mutational Burden
TMB ‐ High. There was evidence of a difference in PFS among people who were treated with double ICI compared to platinum‐based chemotherapy (HR: 0.56, 95% CI 0.43 to 0.73, P < 0.001, I2 = 0%, 2 RCTs, 433 participants) Analysis 2.3.
The pooled results showed no evidence of a difference on PFS between the two groups (HR: 0.86, 95% CI 0.53 to 1.40; P = 0.55, I2 = 91%, 2 RCTs, 1202 participants) Analysis 2.3. There was evidence of a difference between the two subgroups (Test for subgroup differences: Chi² = 13.29, df = 1 (P = 0.0003), I² = 92.5%) Analysis 2.3.
3. Secondary outcomes ‐ Overall response rate (ORR)
a. Main analysis: ORR by PD‐L1 expression
No studies found reported on this outcome.
b. Subgroup analyses:.
i. ORR by Tumour Mutational Burden (TMB)
Two included trials (Hellmann 2018; Rizvi 2020) provided data for this outcome.
TMB ‐ Low. Only Rizvi 2020, provided ORR data for people with blood TMB < 20 mut/Mb. There was evidence of a difference with a higher ORR with chemotherapy treatment compared to double ICI (RR: 0.53, 95% CI 0.37 to 0.77, P<0.001, 1 RCT, 389 participants).
TMB ‐ High. Both included trials provided ORR data for people with high TMB. There was evidence of a difference between the two groups with a lower ORR in people treated with platinum‐based chemotherapy compared to double ICI (RR 1.83, 95% CI 1.40 to 2.39, I2 = 0%, P < 0.0001, 2 RCTs, 433 participants) Analysis 2.4
2.4. Analysis.

Comparison 2: Combined immune checkpoint inhibitors vs chemotherapy, Outcome 4: Overall Response Rate by Tumor Mutational Burden
Overall, there was no evidence of a difference between the two groups (RR 1.25, 95% CI 0.53 to 2.96; I2 = 93%, P = 0.60, 2 RCTs, 822 participants) Analysis 2.4. There was evidence of a difference between TMB low and high subgroups (Test for subgroup differences: Chi² = 27.65, df = 1 (P < 0.00001) Analysis 2.4.
4.Secondary outcome ‐ Adverse events (AEs)
Two trials comparing double ICI to platinum‐based chemotherapy (Rizvi 2020; Hellmann 2018) reported grade 3 to 5 AEs. The pooled results showed no evidence of a difference between the two groups in grade 3 to 5 adverse events (RR 0.84, 95% CI 0.62 to 1.15; P = 0.28, I2 = 60%, 2 RCTs, 3738 participants) Analysis 2.5 ; Figure 9. Applying a fixed‐effect model there evidence of a difference between in favour of people treated with double ICI (RR 0.83, 95% CI 0.73 to 0.95; P = 0.007, I2 = 60%, 3738 participants) (analysis not shown).
2.5. Analysis.

Comparison 2: Combined immune checkpoint inhibitors vs chemotherapy, Outcome 5: Adverse Events grade 3‐5
9.

Forest plot of comparison: Combined Immuno versus Chemotherapy, outcome: 5.5 Adverse Events grade 3‐5.
Adverse Events grade 3 to 4. There was no evidence of a difference in grade 3 and 4 AEs among people receiving double ICI compared to platinum‐based chemotherapy (RR: 0.78, 95% CI 0.55 to 1.09, P = 0.15, I2=81%, 2 RCTs, 1869 participants, low certainty of evidence) Analysis 2.5 ; Figure 9. The certainty for this outcomes was downgraded one level due to study limitations, and one level due to inconsistency. However, there was evidence of a difference between the two groups favouring double ICI compared to platinum‐based chemotherapy when we applied a fixed‐effect model (RR: 0.81, 95% CI 0.71 to 0.93, P = 0.003, I2=81%, 2 RCTs, 1869 participants) (analysis not shown)
Adverse Events grade 5 (toxic deaths). There was no evidence of a difference in participants who received double‐agent ICI compared to platinum‐based chemotherapy in the risk of toxic death (RR: 1.51, 95% CI 0.65 to 3.48, P = 0.33, I2 = 0%, 2 RCTs, 1869 participants) Analysis 2.5 ; Figure 9.
There was no evidence of a difference between the two subgroups (Test for subgroup differences: Chi² = 2.07, df = 1 (P = 0.15), I² = 51.8%).
5. Secondary outcome ‐ Health‐related quality of life (HRQoL)
None of the included trials reported this outcome.
Discussion
Summary of main results
We obtained data for 5893 participants from seven studies comparing first‐line single‐agent (six trials) or double‐agent (two trials) immune checkpoint inhibitors (ICI) with platinum‐based chemotherapy; one study comparing both first‐line, single‐ and combined ICI with platinum‐based chemotherapy.
Single‐agent immune checkpoint inhibitors (ICIs) versus chemotherapy
Overall, the effect of first‐line, single‐agent ICI on survival (overall survival (OS) and progression‐free (PFS)) and responses (objective response rate (ORR)) were not consistent across different PD‐L1 subgroups (negative, positive, high). In fact, for people with advanced non‐small lung cell cancer (NSCLC) and PD‐L1 expression ≥ 50%, single‐agent ICI improved OS, PFS and ORR compared to platinum‐based chemotherapy, while for people with PD‐L1 expression < 1% or ≥ 1% no differences between single‐agent ICI and platinum‐based chemotherapy were observed. For some outcomes, such as OS, the absence of a survival difference in PD‐L1≥1% subgroup could be explained by the high heterogeneity coming from studies with imbalance in baseline characteristics between treatment groups or with a high cross‐over rate from platinum‐based chemotherapy to single‐agent ICI. Similarly, the high heterogeneity observed for PFS or ORR in PD‐L1≥1% and in PD‐L1≥50% subgroups, respectively, could be explained by the different methods used to determine PD‐L1 expression on tumour cells or immune cells, or by the repeated PD‐L1 testing performed in some trials.
Subgroups analysis according to PD‐L1 expression and baseline clinical characteristics of participants of the included studies showed absence of a difference according to age, gender, ECOG PS (Eastern Cooperative Oncology Group Performance Status) or histology categories. In fact, single‐agent ICI improved OS compared to platinum‐based chemotherapy in people with PD‐L1 ≥ 50% regardless of age, gender, ECOG PS and histology while no difference between the two treatments was found in people with PD‐L1 expression ≥ 1%. The only clinical characteristic with a potential impact on OS was smoking status in people with PD‐L1 expression ≥50%. In fact, in people who had never smoked with PD‐L1 expression ≥ 50% there was no difference in OS between single‐agent ICI and platinum‐based chemotherapy, while in current and former smokers single‐agent ICI improved OS compared to platinum‐based chemotherapy.
Overall, in people with evaluable tumour mutational burden (TMB), single‐agent ICI did not improve survival (OS and PFS) nor response (ORR) compared to platinum‐based chemotherapy. However, results were not consistent according to different TMB subgroups (high versus low) and in people with high TMB, single‐agent ICI improved OS and PFS compared to platinum‐based chemotherapy while in people with low TMB, platinum‐based chemotherapy was superior to single‐agent ICI in terms of ORR and PFS, and no difference between these two treatments was observed in OS.
Single‐agent ICI was associated with reduced grade 3 to 5 adverse events (AEs) compared to platinum‐based chemotherapy. However, a difference according to AEs grade subgroups (grade 3 to 4 versus grade 5) was observed and fewer grade 3 to 4 AEs were reported with single‐agent ICI while no difference in toxic deaths (grade 5 AEs) between single‐agent ICI and platinum‐based chemotherapy was found. AEs were not reported according to PD‐L1 expression and the high heterogeneity observed for grade 3 to 4 AEs may be due to the inclusion of people with different PD‐L1 expression levels who could also experience different degree of toxicities from single‐agent ICI.
Data regarding health‐related quality of life (HRQoL) came from one single study and showed no difference or a better performance in QOL‐C30 and QLQ‐LC13 domains for pembrolizumab compared to platinum‐based chemotherapy with the exception of diarrhoea showing a higher proportion of people with less diarrhoea at week 15 in the chemotherapy group compared to single‐agent ICI.
Combined ICIs versus chemotherapy
Overall, the effect of combined ICIs on OS compared to platinum‐based chemotherapy were not consistent across different PD‐L1 or TMB subgroups. double‐agent ICIs prolonged OS compared to platinum‐based chemotherapy in people with PD‐L1 expression ≥ 50% or <1% or with high TMB while no differences in OS between treatments were observed in people with PD‐L1 expression ≥1% or with low TMB. Overall, combined ICIs did not improve PFS and ORR compared to platinum‐based chemotherapy but a subgroup effect was observed with improved PFS and ORR in people with high TMB and no difference between treatments or improved ORR or PFS with chemotherapy compared to combined ICIs in people with low TMB.
No difference in grade 3 to 5 adverse events between combined ICIs and platinum‐based chemotherapy was observed.
Overall completeness and applicability of evidence
The following limitations may affect the strength of conclusions of this review.
There are no data available to explore the effect of single‐ or combined ICIs compared to platinum‐based chemotherapy in special subgroups such as people with brain or liver metastases or with common oncogenic drivers in NSCLC (i.e. KRAS, EGFR and BRAF mutations or ALK and ROS1 rearrangements).
We did not explore the role of chemotherapy‐immunotherapy combinations because another Cochrane Review is currently ongoing on this topic (Syn 2018).
Subgroup analyses according to clinical characteristics for the effectiveness of double‐ICI treatment compared to platinum‐based chemotherapy was not performed because data were only available for one trial (Hellmann 2018).
The subgroup analyses for the comparison of single‐agent ICI with chemotherapy according to other clinical characteristics (age, gender, histology, ECOG PS, smoking status) involved only a small number of people and could not lead to definitive conclusions.
Analysis of HRQoL was only reported in only one study (Reck 2016) and was missing for the studies comparing combined ICIs versus platinum‐based chemotherapy.
The applicability of the evidence about PD‐L1 as biomarker for ICI effectiveness is limited by the fact that PD‐L1 expression in one out of six studies (Herbst 2020) was measured on both tumour and immune cells and that in the same study the antibody for PD‐L1 staining (SP142) had lower sensitivity compared to the others (Tsao 2018). Moreover we could not draw any conclusions about the effectiveness of single‐agent ICI compared to platinum‐based chemotherapy because PD‐L1 expression <1% as negative PD‐L1 was an exclusion criterion in five out of six trials comparing single‐agent ICI versus platinum‐based chemotherapy. However, the survival data for this subgroup were available for the comparison of double‐ICI treatment with platinum‐based chemotherapy.
TMB analyses across different studies were mainly exploratory and included too few participants to produce generalisable results. In addition, the different samples (tissue in Carbone 2017, Mok 2019 and Hellmann 2018 and plasma in (Herbst 2020), methodologies (whole exome sequencing in Carbone 2017 and Mok 2019, Foundation one CDx tissue assay in Hellmann 2018, Guardant OMNI next generation sequencing (NGS) assay in Rizvi 2020. Foundation One plasma NGS assay in Herbst 2020) and thresholds used (≥ 243 mut/exome in Carbone 2017, ≥175 mut/exome in Mok 2019, ≥10 Mut/Mb in Hellmann 2018 and ≥20 Mut/Mb in Rizvi 2020 and in Herbst 2020) may limit the generalisability of the results. Another possible limitation of this meta‐analysis is that results for subgroups defined by TMB status were mainly exploratory and computed on sample sizes too small to derive definite and generalisable conclusions.
We could not provide enough information about potential harmful effects of single‐ or combined ICIs. In fact, recent evidence has shown a possible detrimental effect of ICI, known as hyper progressive disease (HPD) and characterised by tumour growth acceleration, rapid progression and deaths in a subgroup of people with advanced NSCLC treated with single‐agent ICI (Champiat 2018; Ferrara 2018). Although HPD has not been explored in clinical trials, the early mortality rate in the first three months could approximately estimate the potential detrimental effect of ICI in people with NSCLC (Ferrara 2020).
Quality of the evidence
All the included studies were randomised parallel controlled trials, and cross‐over was permitted in three trials (Carbone 2017, Reck 2016, Sezer 2020). Because of the lack of information about one trial (Sezer 2020), only available as an abstract, three domains of the risk of bias remain unclear. All trials were potentially affected by performance bias because of their open‐label design, but five trials (Carbone 2017; Hellmann 2018; Reck 2016; Rizvi 2020; Sezer 2020) in which PFS was a primary endpoint were at high risk of performance bias due to the lack of blinding of participants and personnel, whereas in two studies (Herbst 2020; Mok 2019 ) in which the primary endpoint was OS, performance bias influenced only the results of secondary or exploratory outcomes. Six trials were at low risk of detection bias because OS was the primary endpoint or when PFS was a primary endpoint, the outcome assessors were blinded to the treatment assignment. Two trials (Hellmann 2018; Rizvi 2020) were also at high risk of attrition bias because of the high dropout rate of people after randomisation. The risk of detection and of attrition bias was not clear for Sezer 2020, because it was available only as an abstract. Four studies were at high risk of other bias because of the imbalance of some baseline characteristics between treatment groups (Carbone 2017), because of amendments affecting primary or secondary endpoints (Carbone 2017; Hellmann 2018; Mok 2019), or because of changes in the primary target population (Sezer 2020). All trials were at unclear risk of other sources of bias because some authors had declared personal fees or other support from the pharmaceutical companies conducting the trials and it was impossible to know how these conflicts of interests have biased data collection and analysis. Publication bias is unlikely to have occurred for the four trials comparing single‐agent ICI compared with platinum‐based chemotherapy for both PD‐L1 ≥1% and ≥ 50% categories, while it was not possible to assess publication bias for the comparison double ICI versus chemotherapy because of the limited number of studies included.
Overall survival results were reliable, because of absence of detection bias and because performance bias affected only some trials despite the open‐label design of all the studies. Because of attrition bias and the high risk of other source of bias in some trials, we could not derive to definitive conclusions from the survival data for specific subgroups according to TMB status and PD‐L1 expression.
Using GRADE assessment, the certainty of the evidence ranged from moderate to low (see Table 1; Table 2). Moderate certainty of evidence was due to the high risk of attrition bias, performance bias or other sources of bias affecting some of the included studies. Certainty of evidence was downgraded for some outcomes because of inconsistency of results and a high heterogeneity score. Certainty of evidence for outcomes coming from one single study (i.e. HRQoL for single‐agent ICI compared to chemotherapy in Reck 2016 and ORR of combined ICI compared to chemotherapy by TMB in Rizvi 2020), or having results with wide confidence intervals was further downgraded because of the imprecision.
Potential biases in the review process
We implemented a wide search that included ongoing trial registry databases and contacted experts in the field. We did not apply any language or publication status restrictions. Therefore, we are certain that all the relevant trials were included in the review so far. It is unknown whether there are other reports of unpublished trials in other languages or presented at different meetings. We prespecified all the review outcomes and subgroups prior to the completion of the analysis. However, it was not feasible to investigate the risk of publication bias and complete a funnel plot due to the insufficient number of included studies.
Agreements and disagreements with other studies or reviews
Different systematic reviews and network meta‐analyses have recently investigated the use of first‐line chemotherapy and immunotherapy combinations in NSCLC (Chen 2019; Dafni 2019), or the effectiveness of ICI regardless to treatment type (alone or in combination) and line of therapy (Sun 2020; Yu 2019). This is the first meta‐analysis exploring single‐ and combined ICI treatment compared with platinum‐based chemotherapy in first‐line setting.
Other meta‐analyses including different cancer types or NSCLC people treated with ICI in different lines and combinations have explored the benefit of ICI according to clinical relevant characteristics. Unlike our meta‐analysis, single‐agent ICI did not significantly improve OS compared to first‐line chemotherapy in elderly (≥ 65 years) people (Raphael 2020) in one meta‐analysis. However data were not stratified according to PD‐L1 expression and only three studies using first‐line ICI were included. The same meta‐analysis showed no OS and PFS benefit for first‐line single‐agent ICI compared to chemotherapy among people who never smoked, being in line with our findings for this specific subgroup. In this regard, never/light smoker status has been recently associated with lower PFS and duration of response following single‐agent ICI compared to heavy smoking in people with advanced NSCLC and PD‐L1 expression ≥ 50% (Gainor 2020). Similarly, never smoking status is often associated with oncogenic drivers potentially targetable with active drugs and in a multicentric retrospective registry single‐agent ICI showed low clinical activity in patients with actionable molecular alterations (Mazieres 2019).
A recent meta‐analysis of three trials showed that pembrolizumab improved OS compared to platinum‐based chemotherapy in people with advanced NSCLC older than 75 years and similarly to the present study, the improvement was significant only in people with PD‐L1 expression ≥ 50%. This finding suggests that PD‐L1 status rather than age has an impact on survival upon single‐agent ICI (Nosaki 2019).
Although we did not find a subgroup effect according to sex, single‐agent ICI did not improve survival compared to platinum‐based chemotherapy in females with PD‐L1 expression >1%. In agreement with us, the lower magnitude of benefit of single‐agent ICI in women across different cancer types has been reported in a recent meta‐analysis (Conforti 2018). Furthermore, a meta‐analysis of eight trials comparing single‐agent ICI to chemotherapy in both treatment naive and previously treated people with advanced NSCLC have reported no significant benefit in PFS among females (El‐Osta 2019).This finding was not confirmed by another meta‐analysis (Wallis 2019), however, it also included studies testing combinations of ICI and anti‐CTLA‐4 agents or ICI and chemotherapy.
Finally, we reported that both high PD‐L1 or high TMB are associated with increased benefit from ICI as single agents or in combination compared to platinum‐based chemotherapy. Similarly, PD‐L1 and TMB were independently associated with better response and survival outcome in another meta‐analysis including ICI monotherapy or in combination with other treatment (Yu 2019).
Authors' conclusions
Implications for practice.
The evidence in this review suggests that single‐agent ICI in people with NSCLC and PD‐L1 ≥50% probably leads to a higher overall survival rate and may lead to a higher progression free survival and overall response rate when compared to platinum‐based chemotherapy and may also lead to a lower rate of adverse events and higher HRQoL. Combined ICI in people with NSCLC and PD‐L1 ≥50% also probably leads to a higher overall survival rate when compared to platinum‐based chemotherapy, but its effect on progression free survival, overall response rate and HRQoL is unknown due to a lack of data. The rate of adverse events may not differ between groups.
Implications for research.
The characterisation of both molecular profiles and tumour microenvironment from people who have never smoked could improve the understanding of mechanisms of resistance to ICI. In addition, our findings suggest to integrate multiple clinical and biological (i.e. TMB) parameters together with PD‐L1 expression in a common algorithm able to predict effectiveness of single‐ or double‐agent ICIs in people with advanced NSCLC.
What's new
| Date | Event | Description |
|---|---|---|
| 22 March 2021 | New search has been performed | This review used to be a living review but it has been transitioned out of living mode. Monthly literature search ran until 30st December 2020. |
| 22 March 2021 | New citation required but conclusions have not changed | No new study found. Conclusion unchanged |
History
Protocol first published: Issue 2, 2019 Review first published: Issue 12, 2020
Acknowledgements
We thank Joanne Brooker, Tari Turner, Nuala Livingston and Eve Tomlinson for methodological support.
We thank Giorgio Maria Agazzi for designing the Embase search strategy ; Cheryl Ho, Sarah Hodgkinson, Fergus Macbeth, Celine Mascaux, Nichole Taske, Marta Roque Figuls and Anne‐Claire Toffart for comments on our protocol, as well as Nicole Skoetz, Sign‐off Editor.
Appendices
Appendix 1. CENTRAL search strategy
#1, MeSH descriptor: [Carcinoma, Non‐Small‐Cell Lung] explode all trees #2, nsclc #3, lung cancer* #4, lung carcinom* #5, lung neoplasm* #6, lung tumor* #7, lung tumour* #8, non small cell* #9, nonsmall cell* #10, (#3 or #4 or #5 or #6 or #7) and (#8 or #9) #11, first line #12, naive #13, #11 or #12 #14, MeSH descriptor: [Programmed Cell Death 1 Receptor] explode all trees #15, Programmed Cell Death 1 #16, PD‐1 Receptor #17, CD279 Antigen* #18, PD1 Receptor #19, MeSH descriptor: [Programmed Cell Death 1 Ligand 2 Protein] explode all trees #20, CD 273 #21, PD L2 Ligand #22, B7 DC Ligand #23, B7 DC Antigen* #24, programmed death #25, pd l1 #26, pd l2 #27, pd 2 #28, MeSH descriptor: [Immunotherapy] explode all trees #29, Immunotherap* #30, durvalumab #31, MEDI4736 #32, MEDI‐4736 #33, Imfinzi #34, avelumab #35, MSB0010718C #36, atezolizumab #37, MPDL3280A #38, Tecentriq #39, RG7446 #40, RG 7446 #41, pembrolizumab #42, lambrolizumab #43, Keytruda #44, MK3475 #45, MK 3475 #46, nivolumab #47, MDX‐1106 #48, ONO‐4538 #49, BMS‐936558 #50, Opdivo #51, immune checkpoint inhibitor* #52, MeSH descriptor: [Ipilimumab] explode all trees #53, Ipilimumab #54, Yervoy #55, MDX010 #56, MDX 010 #57, tremelimumab #58, ticilimumab #59, CP 675* #60, CP675* #61, MeSH descriptor: [Drug Therapy] explode all trees #62, Drug Therap* #63, Chemotherap* #64, Pharmacotherap* #65, MeSH descriptor: [Antineoplastic Agents] explode all trees #66, Antineoplas* #67, Antitumo* #68, Anticancer #69, #14 or #15 or #16 or #17 or #18 or #19 or #20 or #21 or #22 or #23 or #24 or #25 or #26 or #27 or #28 or #29 or #30 or #31 or #32 or #33 or #34 or #35 or #36 or #37 or #38 or #39 or #40 or #41 or #42 or #43 or #44 or #45 or #46 or #47 or #48 or #49 or #50 or #51 or #52 or #53 or #54 or #55 or #56 or #57 or #58 or #59 or #60 or #61 or #62 or #63 or #64 or #65 or #66 or #67 or #68 #70, #10 and #13 and #69
Appendix 2. MEDLINE search strategy
#1,"Search Carcinoma, Non‐Small‐Cell Lung[MeSH Terms]" #2,"Search nsclc" #3,"Search lung cancer*" #4,"Search lung carcinom*" #5,"Search lung neoplasm*" #6,"Search lung tumor*" #7,"Search lung tumour*" #8,"Search non small cell*" #9,"Search nonsmall cell*" #10,"Search (#3 OR #4 OR #5 OR #6 OR #7) AND (#8 OR #9)" #11,"Search #1 OR #2 OR #10" #16,"Search (first line) OR naive" #19,"Search ((((programmed cell death 1 receptor[MeSH Terms]) OR Programmed Cell Death 1) OR PD‐1 Receptor) OR CD279 Antigen*) OR PD1 Receptor" #22,"Search ((((programmed cell death 1 ligand 2 protein[MeSH Terms]) OR CD273 Antigen) OR PD L2 Ligand) OR B7 DC Ligand) OR B7 DC Antigen*" #29,"Search (((programmed death) OR pd l1) OR pd l2) OR pd 2" #31,"Search (immunotherapy[MeSH Terms]) OR Immunotherap*" #34,"Search ((((((((((durvalumab) OR MEDI4736) OR MEDI‐4736) OR Imfinzi) OR avelumab) OR MSB0010718C) OR atezolizumab) OR MPDL3280A) OR Tecentriq) OR RG7446) #36,"Search ((((pembrolizumab) OR lambrolizumab) OR Keytruda) OR MK3475) OR MK 3475" #38,"Search ((((nivolumab) OR MDX‐1106) OR ONO‐4538) OR BMS‐936558) OR Opdivo" #41,"Search immune checkpoint inhibitor*" #43,"Search ((((Ipilimumab[MeSH Terms]) OR Yervoy) OR MDX 010) OR MDX010) OR MDX CTLA 4" #45,"Search tremelimumab OR ticilimumab OR CP 675* OR CP675*" #48,"Search (((Drug Therapy[MeSH Terms]) OR Drug Therap*) OR Chemotherap*) OR Pharmacotherap*" #51,"Search (((Antineoplastic Agents[MeSH Terms]) OR Antineoplastic*) OR Antitumo*) OR Anticancer" #52,"Search #19 OR #22 OR #29 OR #31 OR #34 OR #36 OR #38 OR #41 OR #43 OR #45 OR #48 OR #51" #53,"Search #11 AND #16 AND #52" #54,"Search ((((((randomized controlled trial[Publication Type]) OR randomized[Title/Abstract]) OR placebo[Title/Abstract]) OR drug therapy[MeSH Subheading]) OR randomly[Title/Abstract]) OR trial[Title/Abstract]) OR groups[Title/Abstract]" #55,"Search (animals[MeSH Terms]) NOT humans[MeSH Terms]" #56,"Search #54 NOT #55" #57,"Search #53 AND #56"
Appendix 3. Embase search strategy
| #5 | #1 AND #2 AND #3 AND #4 |
| #4 | 'crossover procedure'/exp OR 'double‐blind procedure'/exp OR 'randomized controlled trial'/exp OR 'single‐blind procedure'/exp OR random* OR factorial* OR crossover* OR (cross NEXT/1 over*) OR placebo* OR (doubl* NEAR/1 blind*) OR (singl* NEAR/1 blind*) OR assign* OR allocat* OR volunteer* |
| #3 | 'programmed death 1 receptor'/exp OR 'programmed death 1 ligand 2'/exp OR 'programmed death 1 ligand 1'/exp OR 'anti programmed death':ab,ti OR 'programmed death ligand':ab,ti OR 'programmed death 2':ab,ti OR 'programmed death 1':ab,ti OR 'target* pd‐l1':ab,ti OR 'target* pd‐l2':ab,ti OR 'target* pd‐1':ab,ti OR 'pd‐l1 block*':ab,ti OR 'pd‐l2 block*':ti,ab OR 'pd‐1 block*':ab,ti OR 'pd‐l1 inhibitor*':ab,ti OR 'pd‐l2 inhibitor*':ab,ti OR 'pd‐1 inhibitor*':ab,ti OR 'anti pd‐l1':ab,ti OR 'anti pd‐l2':ab,ti OR 'anti pd‐1':ab,ti OR 'immunotherap*':ab,ti OR 'immunotherapy'/exp OR 'durvalumab':ab,ti OR 'avelumab':ab,ti OR 'atezolizumab':ab,ti OR 'pembrolizumab':ab,ti OR 'nivolumab':ab,ti OR 'immune checkpoint inhibitor*':ab,ti OR 'ono‐4538':ti,ab OR 'bms‐936558':ti,ab OR 'ipilimumab':ti,ab OR 'tremelimumab':ti,ab OR 'ticilimumab':ti,ab OR 'pembrolizumab':ti,ab OR 'lambrolizumab':ti,ab OR 'cytotoxic t lymphocyte antigen 4'/exp OR ctla4:ti,ab OR keytruda:ti,ab OR mdx1106:ti,ab OR opdivo:ti,ab OR 'mk 3475':ti,ab OR mpdl3280a:ti,ab OR yervoy:ti,ab OR 'mdx 010':ti,ab OR 'mdx 101':ti,ab OR 'cp 675,206':ti,ab OR 'chemotherap*':ab,ti OR 'chemotherapy'/exp OR 'antineoplastic agent'/exp |
| #2 | 'first line':ti,ab OR 'first‐line':ti,ab OR naive:ti,ab |
| #1 | 'lung tumor'/exp OR 'non small cell lung cancer'/exp OR ((lung NEXT/1 carcinom*):ab,ti) OR ((lung NEXT/1 neoplasm*):ab,ti) OR 'lung cancer':ab,ti OR nsclc:ab,ti OR 'non small cell lung':ab,ti OR ((lung NEXT/1 tumour*):ab,ti) OR 'nonsmall cell lung':ab,ti OR 'non‐small cell lung':ab,ti OR 'lung carcinoma'/exp OR 'lung cancer'/exp |
Appendix 4. clinicaltrials.gov search strategy
#1 non small cell lung cancer #2 nsclc #3 immunotherapy #4 PD1 #5 PDL1 #6 CTLA
date of last search 22.01.2020
Data and analyses
Comparison 1. Single immunecheckpoint inhibitors vs chemotherapy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 Overall survival by PD‐L1 expression | 6 | Hazard Ratio (IV, Random, 95% CI) | Subtotals only | |
| 1.1.1 PD‐L1 TPS <1% | 1 | 178 | Hazard Ratio (IV, Random, 95% CI) | 1.18 [0.86, 1.61] |
| 1.1.2 PD‐L1 TPS≥1% | 4 | 2937 | Hazard Ratio (IV, Random, 95% CI) | 0.88 [0.78, 1.00] |
| 1.1.3 PD‐L1 TPS ≥50% | 6 | 2111 | Hazard Ratio (IV, Random, 95% CI) | 0.68 [0.60, 0.76] |
| 1.2 Overall Survival by Tumor Mutational Burden | 4 | 2035 | Hazard Ratio (IV, Random, 95% CI) | 0.89 [0.76, 1.05] |
| 1.2.1 Tumor Mutational Burden low | 4 | 1380 | Hazard Ratio (IV, Random, 95% CI) | 1.01 [0.88, 1.15] |
| 1.2.2 Tumor Mutational Burden high | 4 | 655 | Hazard Ratio (IV, Random, 95% CI) | 0.72 [0.57, 0.90] |
| 1.3 Overall survival by age (PDL1≥ 1%) | 2 | 1815 | Hazard Ratio (IV, Random, 95% CI) | 0.90 [0.78, 1.06] |
| 1.3.1 Age <65 years | 2 | 988 | Hazard Ratio (IV, Random, 95% CI) | 0.93 [0.68, 1.29] |
| 1.3.2 Age ≥65 years | 2 | 827 | Hazard Ratio (IV, Random, 95% CI) | 0.90 [0.72, 1.13] |
| 1.4 Overall survival by age (PDL1 ≥50%) | 3 | 1006 | Hazard Ratio (IV, Random, 95% CI) | 0.67 [0.56, 0.79] |
| 1.4.1 Age <65 years | 3 | 571 | Hazard Ratio (IV, Random, 95% CI) | 0.72 [0.57, 0.90] |
| 1.4.2 Age ≥65 years | 2 | 435 | Hazard Ratio (IV, Random, 95% CI) | 0.60 [0.47, 0.78] |
| 1.5 Overall survival by sex (PDL1≥ 1%) | 2 | 1815 | Hazard Ratio (IV, Random, 95% CI) | 0.90 [0.78, 1.03] |
| 1.5.1 Male | 2 | 1234 | Hazard Ratio (IV, Random, 95% CI) | 0.86 [0.71, 1.03] |
| 1.5.2 Female | 2 | 581 | Hazard Ratio (IV, Random, 95% CI) | 0.98 [0.77, 1.25] |
| 1.6 Overall survival by sex (PDL1≥50%) | 3 | 1108 | Hazard Ratio (IV, Random, 95% CI) | 0.68 [0.58, 0.79] |
| 1.6.1 Male | 3 | 745 | Hazard Ratio (IV, Random, 95% CI) | 0.62 [0.52, 0.76] |
| 1.6.2 Female | 3 | 363 | Hazard Ratio (IV, Random, 95% CI) | 0.81 [0.61, 1.08] |
| 1.7 Overall survival by smoking status (PDL1≥ 1%) | 2 | 1808 | Hazard Ratio (IV, Random, 95% CI) | 0.92 [0.78, 1.10] |
| 1.7.1 Never smoked | 2 | 341 | Hazard Ratio (IV, Random, 95% CI) | 1.00 [0.76, 1.33] |
| 1.7.2 Former smoker | 2 | 1089 | Hazard Ratio (IV, Random, 95% CI) | 0.87 [0.57, 1.33] |
| 1.7.3 Current smokers | 2 | 378 | Hazard Ratio (IV, Random, 95% CI) | 0.98 [0.75, 1.27] |
| 1.8 Overall survival by smoking status (PDL1≥ 50%) | 3 | 1109 | Hazard Ratio (IV, Random, 95% CI) | 0.70 [0.56, 0.86] |
| 1.8.1 Never smoked | 3 | 179 | Hazard Ratio (IV, Random, 95% CI) | 1.18 [0.78, 1.79] |
| 1.8.2 Former smoker | 3 | 700 | Hazard Ratio (IV, Random, 95% CI) | 0.60 [0.49, 0.73] |
| 1.8.3 Current smokers | 3 | 230 | Hazard Ratio (IV, Random, 95% CI) | 0.65 [0.44, 0.97] |
| 1.9 Overall survival by ECOG PS (PDL1≥ 1%) | 2 | 1814 | Hazard Ratio (IV, Random, 95% CI) | 0.89 [0.77, 1.03] |
| 1.9.1 ECOG PS 0 | 2 | 568 | Hazard Ratio (IV, Random, 95% CI) | 0.90 [0.63, 1.29] |
| 1.9.2 ECOG PS 1 | 2 | 1246 | Hazard Ratio (IV, Random, 95% CI) | 0.90 [0.74, 1.09] |
| 1.10 Overall survival by ECOG PS (PDL1≥ 50%) | 3 | 1108 | Hazard Ratio (IV, Random, 95% CI) | 0.65 [0.55, 0.77] |
| 1.10.1 ECOG PS 0 | 3 | 367 | Hazard Ratio (IV, Random, 95% CI) | 0.60 [0.44, 0.81] |
| 1.10.2 ECOG PS 1 | 3 | 741 | Hazard Ratio (IV, Random, 95% CI) | 0.67 [0.55, 0.82] |
| 1.11 Overall survival by histological type (PDL1≥ 1%) | 2 | 1815 | Hazard Ratio (IV, Random, 95% CI) | 0.89 [0.73, 1.07] |
| 1.11.1 Squamous | 2 | 621 | Hazard Ratio (IV, Random, 95% CI) | 0.76 [0.63, 0.93] |
| 1.11.2 Non‐squamous | 2 | 1194 | Hazard Ratio (IV, Random, 95% CI) | 0.99 [0.73, 1.33] |
| 1.12 Overall survival by histological type (PDL1≥ 50%) | 3 | 1109 | Hazard Ratio (IV, Random, 95% CI) | 0.66 [0.56, 0.77] |
| 1.12.1 Squamous | 3 | 327 | Hazard Ratio (IV, Random, 95% CI) | 0.57 [0.43, 0.76] |
| 1.12.2 Non‐squamous | 3 | 782 | Hazard Ratio (IV, Random, 95% CI) | 0.69 [0.55, 0.87] |
| 1.13 Progression free survival by PD‐L1 expression | 5 | Hazard Ratio (IV, Random, 95% CI) | Subtotals only | |
| 1.13.1 PDL1≥1% | 3 | 2369 | Hazard Ratio (IV, Random, 95% CI) | 0.99 [0.79, 1.24] |
| 1.13.2 PDL1≥50% | 5 | 1886 | Hazard Ratio (IV, Random, 95% CI) | 0.68 [0.52, 0.88] |
| 1.14 Progression Free Survival by Tumor Mutational Burden | 4 | 2035 | Hazard Ratio (IV, Random, 95% CI) | 0.97 [0.76, 1.22] |
| 1.14.1 Tumor Mutational Burden high | 4 | 655 | Hazard Ratio (IV, Random, 95% CI) | 0.72 [0.60, 0.86] |
| 1.14.2 Tumor Mutational Burden low | 4 | 1380 | Hazard Ratio (IV, Random, 95% CI) | 1.24 [1.00, 1.55] |
| 1.15 Overall response rate by PDL1 expression | 4 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 1.15.1 PDL1≥1% | 2 | 1828 | Risk Ratio (IV, Random, 95% CI) | 0.99 [0.86, 1.15] |
| 1.15.2 PDL1≥50% | 4 | 1672 | Risk Ratio (IV, Random, 95% CI) | 1.40 [1.12, 1.75] |
| 1.16 Overall Response Rate by Tumor Mutational Burden | 3 | 1646 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.73, 1.27] |
| 1.16.1 Tumor Mutational Burden high | 3 | 599 | Risk Ratio (M‐H, Random, 95% CI) | 1.25 [0.99, 1.59] |
| 1.16.2 Tumor Mutational Burden low | 3 | 1047 | Risk Ratio (M‐H, Random, 95% CI) | 0.73 [0.59, 0.91] |
| 1.17 Adverse Events grade 3‐5 | 5 | 6692 | Risk Ratio (M‐H, Random, 95% CI) | 0.43 [0.36, 0.52] |
| 1.17.1 Adverse Events grade 3‐4 | 5 | 3346 | Risk Ratio (M‐H, Random, 95% CI) | 0.41 [0.33, 0.50] |
| 1.17.2 Adverse Events grade 5 (toxic deaths) | 5 | 3346 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.43, 1.41] |
| 1.18 QOL‐C30 GHS/QOL ‐ change from baseline to week 15 | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.19 Time to deterioration ‐ QLQ‐LC13 | 1 | Hazard Ratio (IV, Random, 95% CI) | Totals not selected | |
| 1.20 HRQoL‐QLQC30‐change from baseline at week 15‐stable‐PD‐L1≥50% | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.20.1 GHS/QOL | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.68, 1.17] |
| 1.20.2 Physical functioning | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 1.35 [1.05, 1.74] |
| 1.20.3 Role functioning | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 1.16 [0.88, 1.55] |
| 1.20.4 Emotional functioning | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 1.11 [0.88, 1.39] |
| 1.20.5 Cognitive functioning | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 1.12 [0.89, 1.40] |
| 1.20.6 Social functioning | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 1.21 [0.92, 1.58] |
| 1.20.7 Fatigue | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 1.19 [0.82, 1.71] |
| 1.20.8 Nausea and vomiting | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 1.24 [1.05, 1.47] |
| 1.20.9 Pain | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.62, 1.10] |
| 1.20.10 Dyspnoea | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 1.28 [0.99, 1.64] |
| 1.20.11 Insomnia | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 1.36 [1.03, 1.79] |
| 1.20.12 Appetite loss | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 1.16 [0.90, 1.49] |
| 1.20.13 Constipation | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.89, 1.33] |
| 1.20.14 Diarrhoea | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 1.21 [1.04, 1.40] |
| 1.20.15 Financial difficulties | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 1.16 [0.96, 1.41] |
| 1.21 HRQoL‐QLQC30‐change from baseline at week 15 ‐deterioration ‐ PDL1≥50% | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.21.1 GHS/QOL | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.48, 1.06] |
| 1.21.2 Physical functioning | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 0.55 [0.38, 0.82] |
| 1.21.3 Role functioning | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 0.61 [0.42, 0.88] |
| 1.21.4 Emotional functioning | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 1.20 [0.67, 2.14] |
| 1.21.5 Cognitive functioning | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.70, 1.51] |
| 1.21.6 Social functioning | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 0.61 [0.41, 0.89] |
| 1.21.7 Fatigue | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 0.43 [0.29, 0.63] |
| 1.21.8 Nausea and vomiting | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 0.48 [0.28, 0.81] |
| 1.21.9 Pain | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.47, 1.18] |
| 1.21.10 Dyspnoea | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 0.43 [0.27, 0.68] |
| 1.21.11 Insomnia | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 0.68 [0.44, 1.05] |
| 1.21.12 Appetite loss | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 0.59 [0.37, 0.94] |
| 1.21.13 Constipation | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 0.63 [0.41, 0.97] |
| 1.21.14 Diarrhea | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 0.74 [0.44, 1.23] |
| 1.21.15 Financial difficulties | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.49, 1.15] |
| 1.22 HRQoL‐QLQC30‐change from baseline at week 15 ‐improvement ‐ PD‐L1≥50% | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.22.1 GHS/QOL | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 1.51 [1.08, 2.10] |
| 1.22.2 Physical functioning | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.74, 1.61] |
| 1.22.3 Role functioning | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 1.29 [0.92, 1.81] |
| 1.22.4 Emotional functioning | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.60, 1.10] |
| 1.22.5 Cognitive functioning | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.50, 1.15] |
| 1.22.6 Social functioning | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 1.20 [0.85, 1.69] |
| 1.22.7 Fatigue | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 1.61 [1.21, 2.13] |
| 1.22.8 Nausea and vomiting | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.55, 1.50] |
| 1.22.9 Pain | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 1.39 [1.05, 1.84] |
| 1.22.10 Dyspnoea | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 1.24 [0.88, 1.73] |
| 1.22.11 Insomnia | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.66, 1.21] |
| 1.22.12 Appetite loss | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 1.12 [0.82, 1.54] |
| 1.22.13 Constipation | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.84, 2.12] |
| 1.22.14 Diarrhea | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 0.49 [0.25, 0.94] |
| 1.22.15 Financial difficulties | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 0.88 [0.54, 1.42] |
Comparison 2. Combined immune checkpoint inhibitors vs chemotherapy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 Overall Survival by PD‐L1 expression | 2 | Hazard Ratio (IV, Random, 95% CI) | Subtotals only | |
| 2.1.1 PD‐L1 <1% | 2 | 532 | Hazard Ratio (IV, Random, 95% CI) | 0.67 [0.55, 0.81] |
| 2.1.2 PD‐L1 ≥1% | 2 | 1378 | Hazard Ratio (IV, Random, 95% CI) | 0.89 [0.70, 1.13] |
| 2.1.3 PD‐L1≥50% | 2 | 612 | Hazard Ratio (IV, Random, 95% CI) | 0.72 [0.59, 0.89] |
| 2.2 Overall Survival by Tumor Mutational Burden | 2 | 1202 | Hazard Ratio (IV, Random, 95% CI) | 0.75 [0.54, 1.05] |
| 2.2.1 Tumor Mutational Burden low | 2 | 769 | Hazard Ratio (IV, Random, 95% CI) | 0.93 [0.61, 1.43] |
| 2.2.2 Tumor Mutational Burden high | 2 | 433 | Hazard Ratio (IV, Random, 95% CI) | 0.60 [0.44, 0.82] |
| 2.3 Progression Free Survival by Tumor Mutational Burden | 2 | 1202 | Hazard Ratio (IV, Random, 95% CI) | 0.86 [0.53, 1.40] |
| 2.3.1 Tumor Mutational Burden low | 2 | 769 | Hazard Ratio (IV, Random, 95% CI) | 1.29 [0.90, 1.85] |
| 2.3.2 Tumor Mutational Burden high | 2 | 433 | Hazard Ratio (IV, Random, 95% CI) | 0.56 [0.43, 0.73] |
| 2.4 Overall Response Rate by Tumor Mutational Burden | 2 | 822 | Risk Ratio (M‐H, Random, 95% CI) | 1.25 [0.53, 2.96] |
| 2.4.1 Tumor Mutational Burden low | 1 | 389 | Risk Ratio (M‐H, Random, 95% CI) | 0.53 [0.37, 0.77] |
| 2.4.2 Tumor Mutational Burden high | 2 | 433 | Risk Ratio (M‐H, Random, 95% CI) | 1.83 [1.40, 2.39] |
| 2.5 Adverse Events grade 3‐5 | 2 | 3738 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.62, 1.15] |
| 2.5.1 Adverse events grade 3‐4 | 2 | 1869 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.55, 1.09] |
| 2.5.2 Adverse events grade 5 (toxic deaths) | 2 | 1869 | Risk Ratio (M‐H, Random, 95% CI) | 1.51 [0.65, 3.48] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Carbone 2017.
| Study characteristics | ||
| Methods |
Study design: randomised open‐label parallel controlled, phase 3 trial Study setting: multicentre trial, 141 study locations Country: 35 countries: USA, Argentina, Australia, Brazil, Belgium, Canada, Chile, China, Colombia, France, Germany, Greece, Hungary, Ireland, Israel, Italy, Japan, Mexico, the Netherlands, Peru, Poland, Romania, Russian Federetaion, Saudi Arabia, Spain, others Publication: full‐text paper Study power calculation: yes Study duration: study start date: March 2014, study completion date: October 2018 Ethical approval: yes, by ethics committee at each centre |
|
| Participants |
Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Intervention: nivolumab Solution for Injection 3 mg/kg Intravenous every 2 weeks until disease progression, discontinuation due to unacceptable toxicity, withdrawal of consent or study closure Control: investigator's choice chemotherapy administered in 3‐week cycles up to a maximum of 6 cycles of Intravenous injection until disease progression, unacceptable toxicity or completion of the 6 cycles, whichever comes first |
|
| Outcomes |
Primary outcomes
Secondary outcome measures
Participants characteristics Enrolled N = 1325 Randomised N = 541 Nivolumab group: N= 271. Age median, range: 63 (32 to 89) years. Age category n (%) ≥ 75: 30 (11). Female n (%): 87 (32). Ethnicity/Region n (%): not reported; ECOG performance n (%): 0 = 85 (31), 1= 183 (68), ≥2: 2(1); smoking status n (%): former smoker: 186 (69), current smoker: 52 (19), never smoked: 30 (11), unknown = 3 (1). Tumour histological type n (%): squamous: 66 (24); non‐squamous 205 (76); PD‐L1 expression level n (%): ≥5%: 208 (77), PD‐L1 expression level n (%) ≥ 50%: 88 (32) Chemotherapy group: N= 270. Age median, range: 65 (29 to 87) years. Age category n(%) ≥75: 32 (12). Female n(%): 122 (45); Ethnicity/Region n (%): not reported; ECOG performance n (%): 0 = 93 (34), 1 = 174 (67), ≥2: 3(1); smoking status n (%): former smoker: 182 (67), current smoker: 55 (20), never smoked: 29 (11), unknown: 4 (1). Tumour histological type n (%): squamous: 64 (24); non‐squamous 206 (76); PD‐L1 expression level n (%): ≥ 5%: 210 (78), PD‐L1 expression level n (%) ≥50%: 126 (47). |
|
| Notes |
Sponser source: Bristol‐Myers Squibb Author's email: david.carbone@osumc.edu ClinicalTrials.gov Identifier: NCT02041533 Protocol amendments: the study was amended to include as secondary objective the comparison of overall survival upon nivolumab or investigators' choice chemotherapy among all randomised patients with any PD‐L1 positive tumour expression. Other notes Treatment beyond progression allowed in the Nivolumab arm Cross‐over was optional |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants are enrolled using the Interective Voice Response System (IVRS) and randomised when a randomisation call is made into the IVRS |
| Allocation concealment (selection bias) | Low risk | Central allocation (through a telephone‐based randomisation). |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | It is an open‐label trial, so not blinded for participants and personnel. The absence of blinding could influence results of primary outcome (progression‐free survival). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The primary outcome (progression‐free survival) is assessed by blinded independent central review |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There was a dropout rate of 22% and progression‐free survival was measured on 423 patients instead of 541 randomised patients. However, progression‐free survival in all randomised patients (secondary outcome) confirmed the results observed in 423 patients limiting the risk of attrition bias. |
| Selective reporting (reporting bias) | Low risk | The study protocol is available and the published reports include all expected outcomes |
| Other bias | High risk | Some baseline differences in patients characteristics (gender, PD‐L1 expression, liver metastases). Some authors declared conflicts of interest related to the present study or received personal fees from pharmaceutical companies conducting the trial. |
Hellmann 2018.
| Study characteristics | ||
| Methods |
Study design: randomised parallel controlled trial Study setting: multicentre trial, 301 study locations Country: 35 countries: USA, Argentina, Australia, Brazil, Belgium, Canada, Chile, China, Colombia, France, Germany, Greece, Hungary, Ireland, Israel, Italy, Japan, Mexico, the Netherlands, Peru, Poland, Romania, Russian Federetaion, Saudi Arabia, Spain, others Publication: full‐text paper Study power calculation: yes Study duration: start date: August 5, 2015, estimated primary completion date November 8, 2020 (Final data collection date for primary outcome measure). Ethical approval: yes, by ethics committee at each centre |
|
| Participants |
Inclusion criteria
Exclusion criteria
Participants characteristics (subgroup with PD‐L1 expression ≥1%) Number randomised: 1189 Nivolumab + Ipilimumab N = 396. Age median, range: 64 (26 to 84) years. Age category n(%) : < 65 years = 199 (50.3); ≥65 to 75 years: 157 (39.6) years; ≥ 75 years 40 (10.1); Male n (%): 255 (64.4); ECOG performance n (%): 0 = 135 (34.1), 1= 260 (65.7); current or former smoker n(%) = 334 (84.3); never smoked n (n%) 56 (14.1), unknown = 6 (1.5); Squamous n(%) = 117 (29.5); Non‐squamous 279 (70.5); PD‐L1 expression 1‐49% = 191 (48.2), ≥50% = 205 (51.8), Tumour Mutation Burden (TMB) n (%): Evaluable =240 (60.6) / ≥10 Mut/Mb = 101 (42.1) / <10 Mut/Mb = 139 (57.9) Nivolumab N = 396. Age median, range = 64 years (27 to 85), Male n (%) = 272 (69); ECOG performance 0 n (%) = 142 (36); squamous n (%) = 117 (29.5), non‐squamous n (%) = 279 (70.5); PD‐L1 expression(%) < 1% / ≥1% = 0 (0) / 396 (100); current or former smoker n (%) = 342 (86); never smoked n (%) = 50 (13), unknown n (%) = 4 (1); Tumour Mutation Burden (TMB) n (%): Evaluable = 228 (57.6) / ≥10 Mut/mb = 102 (45) / <10 Mut/Mb = 126 (55). Chemotherapy N = 397. Age median, range: 64 (27 to 85) years, Age category n(%) : < 65 years = 207 (52.1); 65 to 75 years: 149 (37.5) years; ≥ 75 years 41 (10.3). Male n (%): 260 (65.5); ECOG performance n (%): 0 = 134 (33.8%), 1= 259 (65.2), other or missing = 4 (1); current or former smoker n(%) = 340 (85.6); never smoked 51 (12.8), unknown = 6 (1.5); Squamous n(%) = 116 (29.2); Non‐squamous 281 (70.8); PD‐L1 expression level n (%) 1% to 49% = 205 (51.6), ≥50%= 192 (48.4%). Tumour Mutation Burden (TMB) n (%): Evaluable = 242 (61) / ≥10 Mut/mb = 112 (46.3) / <10 Mut/Mb = 130 (53.7) |
|
| Interventions |
Randomisation ratio: 1:1:1 Intervention 1: Nivolumab: 3 mg per kg of body weight plus ipilimumab 1 mg per kg every 6 weeks Intervention 2: nivolumab (240 mg every 2 weeks) Control: Platinum doublet chemotherapy based on tumour histology type (pemetrexed maintenance permitted in eligible people) every 3 weeks for up to four cycles |
|
| Outcomes |
Secondary outcomes
|
|
| Notes |
Sponsorship source: Bristol‐Myers Squibb and Ono Pharmaceutical Author's email: hellmanm@mskcc.org ClinicalTrials.gov Identifier: NCT02477826 Protocol amendments: several amendments took place with regard to the planned trial population, biomarkers and primary objectives. Changes occurred after enrolment had been completed but before the locking the database and breaking the treatment codes. Other notes: treatment beyond progression allowed. Cross‐over during the trial was not permitted. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants are enrolled using the Interective Voice Response System (IVRS) and randomised when a randomisation call is made into the IVRS |
| Allocation concealment (selection bias) | Low risk | Central allocation (through a telephone based randomisation). |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial design.The absence of blinding could influence results of some primary outcomes (progression‐free survival). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Primary outcome (progression‐free survival) is assessed by blinded independent central review, co‐primary outcome (overall survival) is not influenced by blinding of assessment. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Although number of participants enrolled, randomised and analysed is clearly reported, there was a high dropout rate (randomised patients 1739, primary outcome reported in 299 patients) |
| Selective reporting (reporting bias) | Low risk | Protocol was available and all outcomes were reported |
| Other bias | High risk | Although tumour mutational burden analysis was performed before database lock and breaking of coded treatments, this amendment substantially affect the primary and secondary endpoints of the study. Some authors declared conflicts of interest related to the present study or received personal fees from pharmaceutical companies conducting the trial. |
Herbst 2020.
| Study characteristics | ||
| Methods |
Study design: randomised open‐label parallel controlled, phase 3 trial Study setting: multicentre trial, 191 planned study locations Country: 19 countries USA, Brazil, China, France, UK, Germany, Greece, Hungry, Italy, Poland, Romania, Russia, Serbia, Spain, Ukraine ,Japan, Korea, Thailand,Turkey) Publication: abstract |
|
| Participants | 572 participants Inclusion criteria
Exclusion criteria
Participants characteristics Number randomised: 554 Atezolizumab group N = 277. Age < 65 years, n (%): 143, (51.6). Male sex n (%): 196 (70.8); ECOG performance n (%): 0: 97 (35); never smoked 37 (13.4. Histology: non‐squamous n (%) = 192 (69.3) Chemotherapy group: N = 277. Age < 65 years n (%): 134, (48.4). Male sex n (%): 193 (69.7); ECOG performance n (%): 0: 102 (36.8); never smoked 35 (12.6). Histology: non‐squamous n (%) = 193 (69.7) |
|
| Interventions |
Intervention: atezolizumab, other names: MPDL3280A, RO5541267 Atezolizumab 1200 milligram (mg), intravenous infusion every 21 days until loss of clinical benefit (as assessed by the investigator), unacceptable toxicity, or death (maximum up to approximately 58 months). Comparators: (Carboplatin/Cisplatin) + (Pemetrexed/ Gemcitabine) Participants with non‐squamous NSCLC will receive chemotherapy with pemetrexed in combination with either cisplatin or carboplatin (per investigator discretion) on Day 1 of each 21‐day cycle for 4 or 6 cycles as per local standard of care, followed by maintenance therapy with pemetrexed alone as per local standard of care until disease progression (per RECIST v1.1), unacceptable toxicity, or death (maximum up to approximately 58 months). Participants with squamous NSCLC will receive chemotherapy with gemcitabine on Days 1 and 8 of each 21‐day cycle in combination with either cisplatin or carboplatin on Day 1 of each 21‐day cycle for 4 or 6 cycles as per local standard of care, followed by best supportive care as per local standard of care until disease progression, unacceptable toxicity, or death (maximum up to approximately 58 months). |
|
| Outcomes |
Primary outcome measures Overall survival (OS): time frame: from randomisation to death from any cause (maximum up to approximately 58 months) in PD‐L1 positive (tumour cell (TC) or immune cell score (IC) ≥ 1%) The primary endpoint was tested hierarchically in the following subgroups: PD‐L1 ≥50% on TC or IC (TC3 or IC3); PD‐L1 ≥5% on TC or IC (TC 2/3 or IC 2/3); and PD‐L1≥1% on TC or IC (TC1/2/3 or IC1/2/3). Secondary outcome measures
|
|
| Notes |
Sponsorship source: Roche ClinicalTrials.gov Identifier: NCT02409342 Study protocol: not published Other notes: Cross‐over during the trial was not permitted |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation number was obtained by a web‐based response system |
| Allocation concealment (selection bias) | Low risk | Central allocation was performed. Study sites obtained participants randomisation number and treatment assignment from interactive voice or web‐based response system |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | No blinding it is an open‐label study design. However, the absence of blinding unlikely influenced results of the primary outcome (OS). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Primary endpoint (overall survival) was not influenced by the absence of blinding assessment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Outcome reported as intention to treat (ITT) population. No drop outs |
| Selective reporting (reporting bias) | Low risk | Protocol was available and all outcomes were reported. |
| Other bias | Unclear risk | Some authors declared conflicts of interest related to the present study or received personal fees from pharmaceutical companies conducting the trial. |
Mok 2019.
| Study characteristics | ||
| Methods |
Study design: randomised open‐label parallel controlled, phase 3 trial Study setting: multicentre trial, 213 study locations Country: 32 countries: USA, Argentina, Australia, Brazil, Belgium, Canada, Chile, China, Colombia, France, Germany, Greece, Hungary, Ireland, Israel, Italy, Japan, Mexico, the Netherlands, Peru, Poland, Romania, Russian Federetaion, Saudi Arabia, Spain, others Publication: full‐text paper Study power calculation: yes Study duration: December 2014 to March 2017 Ethical approval: yes, by ethics committee at each centre |
|
| Participants |
Inclusion criteria
Exclusion criteria
Participants characteristics Number screened = 3428 Number with PD‐L1 expression = 3019, 1978 had PD‐L1 expression ≥1% Number included in Intertion to treat (ITT): 1274 (637 in each arm) Pembrolizumab group (Tumour proportional score PD‐L1 => 1%) N = 637. Age median, (range): 63, (56 to 69 years). Age category n (%): < 65: 359 (56); Male sex n (%): 450 (71). Ethnicity/Region n (%): East Asia: 185 (29), Europe :149 (23); Latin America: 78 (19); Other 111 (27); ECOG performance n(%): 0: 198 (31), 1: 439 (69); current or former smoker never smoked 125 (20), 370 (58), 142 (22). Histology: squamous n(%) = 243 (38); non‐squamous 394 (62); brain metastases n (%): 35 (5); Disease status n (%): locally advanced: 76 (12), metastatic 561 (88); PD‐L1 tumour proportional score n (%): 1% to 19%: 224 (35), 20% to 49%: 114 (18); ≥ 50%: 299 (47) Chemotherapy group N = 637.. Age median, (range): 63, (57 to 69 years). Age category n (%): < 65: 348 (55); Male sex n (%): 452 (71). Ethnicity/Region n (%): East Asia: 185 (29), Europe :137 (22); Latin America: 133 (21); Other 182 (29); ECOG performance n (%): 0: 192 (30), 1: 445 (70); current or former smoker never smoked 146 (23), 351 (55), 140 (22), Histology: squamous n (%) = 249 (39); non‐squamous 388 (61); brain metastases n (%): 35 (5); Disease status n (%): locally advanced: 84 (13), metastatic 553 (87); PD‐L1 tumour proportional score n (%): % to 19%: 232 (36), 20% to 49%: 105 (16); ≥ 50%: 300 (47) |
|
| Interventions |
Intervention: pembrolizumab 200 mg intravenous (IV) on Day 1 of every 21‐day cycle (every 3 weeks, or Q3W) for up to 35 treatments. Control: standard of care (SOC) platinum‐based chemotherapy (carboplatin + paclitaxel or carboplatin + pemetrexed for 4 to 6 21‐day cycles) |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
Sponsorship source: Merck Sharp & Dohme Corp ClinicalTrials.gov Identifier: NCT02220894 Study protocol: published Protocol amendments: primary endpoint was changed during the trial ( and overall survival in patients with PD‐L1 ≥ 1% on tumour cells was included as primary endpoint). In a second amendment a new cut‐off for PD‐L1 expression on tumour cells (TPS 20%) was tested and primary and secondary endpoints were amended to include OS, PFS and objective response, respectively, in people with PD‐L1 TPS ≥ 50%, ≥20% and ≥1%. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was computer generated, accessed via an interactive voice‐response and integrated web‐response system, and stratified by region of enrolment (east Asia vs rest of world) |
| Allocation concealment (selection bias) | Low risk | Enrolled participants were randomly assigned 1:1 in blocks of four per stratum (central allocation) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The study was open‐label due to quote: "differences in infusion durations, administration schedules and requirement for pre medications". However, the absence of blinding unlikely influenced results of primary outcome (OS). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Central radiological reviewers were unaware of treatment assignments |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Number of participants eligible of inclusion in the trial, number included in each arm and the number of participants who discontinued all clearly documented. Reasons for dropout were also documented. Dropout regarded only 1 patient. Outcomes were measured on randomised patients. |
| Selective reporting (reporting bias) | Low risk | Protocol was available and all outcomes were reported |
| Other bias | High risk | Protocol amended changing the primary and secondary endpoints and a new cut off point (PD‐L1 tumour proportional score 20%) was included. Some authors declared conflicts of interest related to the present study or received personal fees from pharmaceutical companies conducting the trial. |
Reck 2016.
| Study characteristics | ||
| Methods |
Study design: randomised open‐label parallel controlled, phase 3 trial Study setting: multicentre trial, 102 study locations Country: 16 countries, Publication: full‐text paper Study power calculation: yes Study duration: September 2014 to October 2015 Median follow‐up period: 25.2 months (data cut‐off in July 2017) Ethical approval: yes, by ethics committee at each centre |
|
| Participants |
Inclusion criteria
Exclusion criteria
Participants characteristics Number eligible: 1934 Number randomised: 305 Pembrolizumab group N= 154. Age median, (range): 64.5, (33 to 90 years). Male sex n (%): 92 (59.7), Ethnicity/Region n (%): East Asia: 21 (13.6), Non‐East Asia: 133 (86.4); ECOG performance n (%): 0: 54 (35.1), 1: 99 (64.3); current or former smoker never smoked 34 (22.1), 115 (74.7), 5 (3.2). Histology: squamous n (%) = 29 (18.8); non‐squamous 125 (81.2); brain metastases n (%): 18 (11.7) Chemotherapy group N = 151. Age median, (range): 66, (38 to 85 years). Male sex n (%): 95 (62.9). Ethnicity/Region n (%): East Asia: 19 (12.6), Non‐East Asia: 132 (87.4); ECOG performance n (%): 0: 53 (35.1), 1: 98 (64.9); current or former smoker never smoked 31 (20.5), 101 (66.9), 19 (12.6). Histology: squamous n(%) = 27 (17.9); non‐squamous 124 (82.1); brain metastases n (%): 10 (6.6) |
|
| Interventions | Randomisation ratio:1:1 Intervention group: pembrolizumab 200 mg every three weeks (35 cycles) Control group: platinum‐doublet chemotherapy 4 to 6 cycles |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes |
NCT02142738 Cross‐over phase: this is only applicable for participants randomised to receive SOC. Eligible participants were treated with pembrolizumab for the remainder of the study or until disease progression, unacceptable AEs, intercurrent illness that prevents further administration of treatment, investigator's decision to withdraw the participant, noncompliance with study treatment or procedures requirements, the participant receives 35 treatments of study treatment (pembrolizumab arm only), or administrative reasons. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random sequence generated using Interactive Voice Response System (IVRS) and integrated web response system (IWRS) |
| Allocation concealment (selection bias) | Low risk | Central allocation |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label design trial. The absence of blinding could influence results of primary outcome (progression‐free survival) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessment was done by blinded independent central radiologic review |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Primary and secondary endpoints were reported on the intention to treat population |
| Selective reporting (reporting bias) | Low risk | Outcomes reported for all groups |
| Other bias | Unclear risk | Some authors declared conflicts of interest related to the present study or received personal fees from pharmaceutical companies conducting the trial |
Rizvi 2020.
| Study characteristics | ||
| Methods |
Study design: randomised open‐label parallel controlled, phase 3 trial Study setting: multicentre trial, 167 study locations Country: 17 countries, USA, Canada, Europe, Russia, Australia and parts of Asia, including Japan, Korea, Thailand, Taiwan and Vietnam. Publication: Abstract |
|
| Participants |
Inclusion criteria
Exclusion criteria
Participants Characterisctics Number randomised: 1118 Durvalumab group (PD‐L1 TPS ≥25%) N = 163. Age median, (range): 64, (32 to 84 years). Male sex n (%): 113 (69.3); ECOG performance n (%): 0: 57 (35); current/former/never smoked 47 (28.8), 92 (56.4), 24 (14.7). Histology: squamous n (%) = 52 (31.9) Durvalumab plus tremelimumab group (PD‐L1 TPS ≥25%) N = 163. Age median, (range): 65, (34 to 87 years), Male sex n (%): 118 (72.4); ECOG performance n (%): 0: 65 (39.9); current/former/never smoked 42 (25.8), 96 (58.9), 25 (15.3). Histology: squamous n (%) = 53 (32.5) Chemotherapy group (PD‐L1 TPS ≥25%) N= 162. Age median, (range): 64.5, (35 to 85 years). Male sex n (%): 106 (65.4); ECOG performance n (%): 0: 70 (43.2); current/former/never smoked 39 (24.1), 102 (63), 22 (13). Histology: squamous n(%) = 52 (32.1) |
|
| Interventions | Randomisation ratio: 1:1:1 Intervention1: PD‐L1 monoclonal antibody monotherapy (durvalumab); (20 mg/kg i.v. q4w) Intervention 2 : durvalumab + tremelimumab combination therapy, (D: 20 mg/kg i.v. q4w; T: 1 mg/kg i.v. q4w (up to 4 doses) Control: chemotherapy (intended up to 6 cycles; pemetrexed maintenance permitted in eligible people) until disease progression |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
Sponsorship source: AstraZeneca AB ClinicalTrials.gov Identifier: NCT02453282 Study protocol: not published Other notes: Cross‐over during the trial was not permitted |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Using the Interactive Voice Response System (IVRS) and integrated web response system (IWRS) |
| Allocation concealment (selection bias) | Low risk | A blocked randomisation is used and all the centres use the same randomisation list in order to minimize any imbalance in the number of patients assigned to each treatment group. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | It is an open‐label study. The absence of blinding could influence results of some primary outcomes (progression‐free survival). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The primary endpoint is progression‐free survival by blinded independent central review assessment |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The primary analysis population for the study was amended to include only people with PD‐L1 expression ≥25%, therefore only 488 (44%) of 1118 randomised people were included in the analysis of the trial primary outcome |
| Selective reporting (reporting bias) | Low risk | Outcomes reported for all groups |
| Other bias | Unclear risk | Some authors declared conflicts of interest related to the present study or received personal fees from pharmaceutical companies conducting the trial. |
Sezer 2020.
| Study characteristics | ||
| Methods |
Study design: randomised open‐label parallel controlled, phase 3 trial Study setting: multicentre trial, 188 study locations Country: 26 countries, Publication: second interim analysis at a conference presentation Study power calculation: not reported Study duration: May 2017 ‐ ongoing Median follow‐up period: 10.8 months (data cut‐off in March 2020) Ethical approval: yes, by ethics committee at each centre |
|
| Participants | 710 participants, 563 with PD‐L1=> 50% Inclusion criteria
Exclusion Criteria
Note: Other protocol defined Inclusion/Exclusion criteria apply. |
|
| Interventions |
Intervention Cemiplimab (Anti‐PD‐1 Antibody) Comparator Standard of care platinum‐based chemotherapy |
|
| Outcomes |
Primary outcome measures
Secondary outcome measures
|
|
| Notes |
Sponsorship source: Regeneron Pharmaceuticals ClinicalTrials.gov Identifier: NCT03088540 Study protocol: no protocol published Other notes: 74% of people cross‐over from chemotherapy to receive cemiplimab |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation was not reported |
| Allocation concealment (selection bias) | Unclear risk | Method of assignment to intervention was not reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | It is an open‐label study. The absence of blinding could influence results of some primary outcomes (progression‐free survival). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No full publication was available, trial primary outcomes were presented in an abstract |
| Selective reporting (reporting bias) | Unclear risk | No information |
| Other bias | High risk | PD‐L1 level ≥50% was an inclusion criteria, however some participants were retested and had PD‐L1 expression < 50%. Only OS and PFS data were reported for people with ≥ 50% PD‐L1 level intention to treat population, the rest of participants characteristics were presented for all participants regardless of the PD‐L1 level. |
AEs; adverse events; ALK: anaplastic lymphoma kinase ; CNS: central nervous system; CT: computed tompography; ; IVRS: interactive voice response system; MRI: magnetic resonance imaging; NSCLC: non‐small cell lung cancer; ORR; objective response rate; OS; overall survival; PFS: progression‐free survival; TMB: tumour mutational burden.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Huan 2019 | wrong study design |
| Hui 2017a | wrong study design |
| Leighl 2019 | wrong study design |
| Mok 2017 | Wrong intervention |
| Ready 2019 | wrong intervention |
Characteristics of ongoing studies [ordered by study ID]
EMPOWER‐Lung 2.
| Study name | A randomized, phase 3, open‐label study of combinations of REGN2810 (Anti‐PD‐1 antibody), platinum‐based doublet chemotherapy, and ipilimumab (Anti‐CTLA‐4 Antibody) versus pembrolizumab monotherapy in first‐line treatment of patients with advanced or metastatic Non‐Small Cell Lung Cancer with tumors expressing PD‐L1 ≥50% |
| Methods | Open‐label, parallel assignment, Phase 3 RCT |
| Participants | 5 participants Inclusion criteria
Exclusion criteria
|
| Interventions |
Intervention REGN2810 plus ipilimumab Other name: cemiplimab Intervention REGN2810 plus chemotherapy plus Ipilimumab Other name: cemiplimab Comparator Pembrolizumab Reference drug administered IV infusion |
| Outcomes |
Primary outcome measures
Secondary outcome measures
|
| Starting date | Actual study start date: June 4, 2018 Estimated study completion date: November 18, 2020 |
| Contact information | |
| Notes | ClinicalTrials.gov Identifier: NCT03515629 |
eNERGY.
| Study name | Randomized phase III study testing nivolumab and ipilimumab versus a carboplatin based doublet in first line treatment of PS 2 or elderly (more than 70 years old). patients with advanced Non‐small Cell Lung Cancer |
| Methods | Open‐label, parallel assignment, Phase 3 RCT |
| Participants | 242 participants Inclusion criteria
Age and reproductive status
WOCBP should use an adequate method to avoid pregnancy :
Exclusion criteria
|
| Interventions |
Intervention Nivolumab + Ipilimumab. Nivolumab dosed intravenously over 30 minutes at 240 mg every 2 weeks combined with Ipilimumab dosed intravenously over 30 minutes at 1 mg/kg every 6 weeks until disease progression, unacceptable toxicity, or other reasons specified in the protocol. Comparator Chemotherapy. Doublet of chemotherapy according to standard of care carboplatin (AUC 5) with a dose that will be capped to 700 mg and pemetrexed (500 mg/m²) over 4 to 6 hours every three weeks (restricted to non‐squamous histology) or carboplatin (AUC 6) with a dose that will be capped to 700 mg and paclitaxel (90 mg/m²) D1 D8 D15 over 4 to 6 hours every 4 weeks, with a maximum of 4 cycles of carboplatin based doublet, and the possibility to use maintenance with pemetrexed. |
| Outcomes |
Primary outcome measures
Secondary outcome measures
|
| Starting date | Estimated study start date: December 2017 Estimated study completion date: June 2022 |
| Contact information | |
| Notes | ClinicalTrials.gov Identifier: NCT03351361 |
IPSOS.
| Study name | A phase III, open‐label, multicenter, randomized study to investigate the efficacy and safety of atezolizumab compared with chemotherapy in patients with treatment naïve advanced or recurrent (stage IIIb not amenable for multimodality treatment) or metastatic (stage IV) Non‐Small Cell Lung Cancer who are deemed unsuitable for platinum‐containing therapy |
| Methods | Open‐label, parallel assignment, Phase 3 RCT |
| Participants | 441 participants Inclusion criteria
Exclusion criteria Cancer‐specific exclusion criteria
|
| Interventions |
Interventions Atezolizumab Participants will receive atezolizumab 1200 milligrams (mg) intravenous (IV) infusion on Day 1 of each 21‐day cycle until loss of clinical benefit, unacceptable toxicity, participant or physician decision to discontinue, or death. Comparators Single‐agent chemotherapy (vinorelbine or gemcitabine) Participants will receive single‐agent chemotherapy; either vinorelbine oral or IV, or gemcitabine IV, according to the label based on investigator's choice. |
| Outcomes |
Primary outcomes
Secondary outcomes
|
| Starting date | Actual study start date: September 11, 2017 Estimated study completion date: January 20, 2021 |
| Contact information | |
| Notes | ClinicalTrials.gov Identifier: NCT03191786 |
JAVELIN Lung 100.
| Study name | A phase III, open‐label, multicenter trial of avelumab (MSB0010718C) versus platinum‐based doublet as a first‐line treatment of recurrent or stage IV PD‐L1+ Non‐small Cell Lung Cancer |
| Methods | Open‐label, parallel assignment, Phase 3 RCT |
| Participants | 1224 participants Inclusion criteria
ExclusioncCriteria
|
| Interventions |
Intervention Avelumab Participants will be administered with avelumab at a dose of 10 milligram per kilogram (mg/kg) 1‐hour intravenous (IV) infusion once every 2 weeks until disease progression or unacceptable toxicities. Other Names:Anti‐PD‐L1; MSB0010718C Comparator Platinum‐containing chemotherapy regimen Platinum‐containing chemotherapy regimen: Investigator's choice platinum containing chemotherapy regimen to be administered consisting of one of the following: non‐squamous tumour histology Pemetrexed (500 milligram per metre square [mg/m^2]) +cisplatin (75 mg/m^2) or Pemetrexed (500 mg/m^2) + carboplatin (AUC 6 mg/mL*min) Squamous tumour histology Paclitaxel (200 mg/m^2) +carboplatin (AUC 6 mg/mL*min) Gemcitabine (1250 mg/m^2)+ cisplatin (75 mg/m^2) Gemcitabine (1000 mg/m^2 )+carboplatin (AUC 5 mg/mL*min) |
| Outcomes |
Primary outcome measures
Secondary outcome measures
|
| Starting date | Actual study start date: October 29, 2015 Estimated study completion date: June 3, 2025 |
| Contact information | |
| Notes | https://clinicaltrials.gov/ct2/show/NCT02576574 |
KEYNOTE‐598.
| Study name | A phase 3, randomized, double‐blind study of pembrolizumab plus ipilimumab vs pembrolizumab plus placebo in previously untreated, stage IV, metastatic Non‐small Cell Lung Cancer subjects whose tumors are PD‐L1 positive (TPS ≥ 50%) (KEYNOTE‐598) |
| Methods | Open‐label, parallel assignment, Phase 3 RCT |
| Participants | 548 participants Inclusion criteria
Exclusion criteria
|
| Interventions |
Intervention pembrolizumab + ipilimumab Participants receive 200 mg of pembrolizumab by intravenous (IV) infusion on Day 1 of each 3‐week cycle for up to 35 cycles of treatment plus 1 mg/kg of ipilimumab by IV infusion on Day 1 of each 6‐week cycle for up to 18 cycles of treatment. Comparator: pembrolizumab + placebo Participants receive 200 mg of pembrolizumab by IV infusion on Day 1 of each 3‐week cycle for up to 35 cycles of treatment plus placebo by IV infusion on Day 1 of each 6‐week cycle for up to 18 cycles of treatment. |
| Outcomes |
Primary outcome measures
Secondary outcome measures
|
| Starting date | Actual study start date: December 14, 2017 Estimated study completion date: February 2024 |
| Contact information | |
| Notes | https://clinicaltrials.gov/ct2/show/NCT03302234 |
MILES 5.
| Study name | A study comparing immunotherapy with chemotherapy in the treatment of elderly patients with advanced NSCLC (MILES‐5) |
| Methods | Open‐label, parallel assignment, Phase 2 RCT |
| Participants | 460 participants Inclusion Criteria
Exclusion criteria Cancer related
|
| Interventions |
Interventions Durvalumab monotherapy (anti‐PDL1 antibody) followed at progression by standard of care chemotherapy Durvalumab (anti‐PDl1 antibody ) + tremelimumab (anti CTLA‐4 antibody) followed at progression by standard of care chemotherapy Comparator: Standard of care chemotherapy followed at progression by durvalumab 8anti‐PDL1 antibody) |
| Outcomes |
Primary outcome measures 12‐month overall survival |
| Starting date | December 20, 2018 Estimated study completion date: June 2023 |
| Contact information | |
| Notes | ClinicalTrials.gov Identifier: NCT03975114 |
NEPTUNE.
| Study name | Study of 1st line therapy study of durvalumab with tremelimumab versus SoC in Non Small‐Cell Lung Cancer (NSCLC) (NEPTUNE). |
| Methods | Open‐label, multicentre, Phase 3 RCT |
| Participants | 953 participants
NCT03191786 Inclusion criteria
Exclusion criteria Patients should not enter the study if any of the following exclusion criteria are fulfilled.
|
| Interventions |
Intervention Durvalumab (PD‐L1 monoclonal antibody) + Tremelimumab (monoclonal antibody directed against CTLA‐4) Comparator: Standard of Care chemotherapy treatment |
| Outcomes |
Primary outcomes Overall survival (OS), time frame up to 4 years after first patient randomised Secondary outcomes:
Other outcomes Treatment‐related adverse events as assessed by CTCAE v4.03 |
| Starting date | November 3, 2015 Estimated study completion date: August 22, 2019 |
| Contact information | |
| Notes | ClinicalTrials.gov Identifier: NCT02542293 |
PEARL.
| Study name | A phase III randomized, open‐label, multi‐center study of durvalumab (MEDI4736) versus standard of care (SoC) platinum‐based chemotherapy as first line treatment in patients with PD‐L1‐High Expression advanced Non Small‐Cell Lung Cancer |
| Methods | Open‐label, parallel assignment, Phase 3 RCT |
| Participants | 662 participants Inclusion criteria
Exclusion criteria
|
| Interventions |
Intervention Durvalumab (MEDI4736) Anti‐PD‐L1 monoclonal Antibody monotherapy Comparator: Standard of care: platinum‐based chemotherapy |
| Outcomes |
Primary outcome measures
Secondary outcome measures
Other outcome measures
|
| Starting date | Actual study start date: January 2, 2017 Estimated study completion date: December 31, 2020 |
| Contact information | |
| Notes | ClinicalTrials.gov Identifier: NCT03003962 |
Differences between protocol and review
Background
In the background section we modified the description of the interventions according to the characteristics of the included studies.
Methods
We removed doublet immune checkpoint inhibitors versus single‐agent immune checkpoint inhibitor in the first‐line setting as it was inconsistent with the tile of our review.
Although we initially developed a data extraction form on Covidence as indicated in the protocol, in the final review, we used an Excel sheet to extract data from included studies because it was easier and more intuitive than Covidence.
Although we specified in the methods to report PFS and OS survival rates at specific time points, we did not perform this analysis because survival rates were not available from the majority of included studies. If these data are available in the future we will report them as RRs.
In the "type of studies" of the methods section, we deleted the sentence considering studies with cross‐over designs. Indeed it was not relevant in the designs used in clinical trials in oncology setting.
'Summary of findings' tables
We initially planned to include the following OS, PFS, ORR, AEs grades 3 to 4 and 5, and HRQoL outcomes. The included trials in this review reported outcomes by different PDL‐1 levels. therefore we found it necessary to select PD‐L1 ≥ 50% as it was the most common used level. The AEs were not reported by PD‐L1 expression.
We provided two 'Summary of findings' tables instead of one:
1. Single immune checkpoint inhibitors compared to chemotherapy for people with advanced non‐small cell lung cancer (Table 1)
2. Combined immune checkpoint inhibitors compared to chemotherapy for people with advanced non‐small cell lung cancer (Table 2)
Subgroups
Due to lack of data we were unable to carry out the analysis for the subsequent subgroups that were inially described in the protocol.
Immune checkpoint inhibitor or immuno‐oncology therapy class (anti‐PD‐1, anti‐PD‐L1, anti‐CTLA4)
Inclusion or exclusion of participants with brain metastasis
Follow‐up duration
liver metastases
Presence of molecular alterations (EGFR mutation, ALK and ROS1 rearrangements)
The analysis of other subgroups reported in the protocol such as age, gender, performance status, smoking status, NSCLC histology and TMB expression (high versus low) were carried out.
In the protocol ≥ 50%, ≥ 20%, or ≥ 1% PD‐L1 expression thresholds were initially used, but in the review we chose to regroup PD‐L1 categories in "positive" (PD‐L1 TPS ≥1% or TC1‐2‐3/IC1‐2‐3), "negative" (PD‐L1 TPS <1% or TC0/IC0) or "high" (PD‐L1 TPS ≥50% or PD‐L1 TC3/IC3).
Update (March 2021): considering that current research is exploring ICI in association with chemotherapy or other immunotherapeutics drugs versus ICI as single agent rather than platinum based chemotherapy, we believe that platinum based chemotherapy alone will be no longer the proper standard arm of future randomised clinical trials in this setting. For this reason, this review has been transitioned out of living mode and will, from now on, be updated on a 2‐years basis like any other Cochrane systematic review.
Contributions of authors
Writing the protocol: RF, RM, MI, SPB, CM, VW.
Designing search strategies and monthly searches: FC
Selection of studies: RF, CM, RM, MI
Assessement of risk of bias: RF, RM
Extraction of data: MI, RF, SPB, RM
Conduct of the analysis: RF, SPB, MI, RM
Writing of the final review: RF, RM, MI, SPB, CM, VW
Critical appraisal of the final review paper: VW
Sources of support
Internal sources
No sources of support supplied
External sources
-
INCa, France
French National Cancer Institute (INCa n° 2017‐186)
Declarations of interest
Roberto Ferrara: none known.
Martina Imbimbo: received payment for development of educational presentations from BMS, case report pseudoprogression from immunotherapy and is Principal Investigator for phase I studies sponsored by Medimmune and BMS.
Sophie Paget‐Bailly: declares a payment from BMS for the development of an educational presentation and the travel and accomodations to attend the same presentation.
Reem Malouf: none known.
François Calais: none known.
Corynne Marchal: none known.
Virginie Westeel received payment for board membership from MSD, BMS, Takeda, Roche, Astra Zeneca, for lectures including service on speakers bureaus from BMS, Astra Zeneca, Roche, MSD, for Payment for development of educational presentations from BMS, Astra Zeneca, Roche, for travel accommodations and meeting expenses from BMS, Astra Zeneca, Roche, Pfizer, Boehringer Ingelheim.Virginie Westeel declares payment to institution for the conduct of clinical trials from Abbvie, Boehringer Ingelheim, Bristol Myers Squibb, Fresenius, Lilly, MSD, Merck Serono, Novartis, Roche.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Carbone 2017 {published data only}
- Carbone DP, Reck M, Paz-Ares L, Creelan B, Horn L, Steins M. First-line nivolumab in stage IV or recurrent non-small-cell lung cancer. New England Journal of Medicine 2017;376:2415-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT02041533. An open-label, randomized, phase 3 trial of nivolumab versus investigator's choice chemotherapy as first-line therapy for stage IV or recurrent PD-L1+ non-small cell lung cancer (CheckMate 026). https://clinicaltrials.gov/ct2/show/NCT02041533 accessed 27 May 2019.
Hellmann 2018 {published data only}
- Brahmer J, Schenker M, Lee KH, Provencio M, Nishio M, Lesniewski-Kmak K, et al. CheckMate 227: patient-reported outcomes of first-line nivolumab + ipilimumab in high tumor mutational burden advanced NSCLC. Journal of Thoracic Oncology 2018;13(suppl 10):S332. [Google Scholar]
- Hellmann MD, Ciuleanu TE, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. New England Journal of Medicine 2018 May 31;378(22):2093-104. [DOI: 10.1056/NEJMoa1801946] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmann MD, Paz‑Ares L, Bernabe Caro R, Zurawski B, Kim SW, Carcereny Costa E. Nivolumab plus ipilimumab in advanced non–small-cell lung cancer. New England Journal of Medicine 2019;381:2020-31. [DOI: 10.1056/NEJMoa1910231] [DOI] [PubMed] [Google Scholar]
- O'Byrne KJ, Lee KH, Kim SW, Park K, Nishio M, Sakai H, et al. First-line (1L) nivolumab (NIVO) plus ipilimumab (IPI) in Asian patients (pts) with advanced non-small cell lung cancer (aNSCLC) in CheckMate 227. Annals of Oncology 2020;31(suppl 4):S824. [Google Scholar]
- Paz-Ares L, Brahmer J, Hellmann MD, Reck M, O'Byrne K, Borghaei H, et al. CheckMate 227: a randomized, open-label phase 3 trial of nivolumab nivolumab plus ipilimumab, or nivolumab plus chemotherapy versus chemotherapy in chemotherapy-naive patients with advanced non-small cell lung cancer (NSCLC). Annals of Oncology 2017;28:iii50-1. [Google Scholar]
- Peters S, Ramalingam SS, Paz-Ares L, Bernabe Caro R, Zurawski B, Kim SW, et al. Nivolumab (NIVO) + low-dose ipilimumab (IPI) vs platinum-doublet chemotherapy (chemo) as first-line (1L) treatment (tx) for advanced non-small cell lung cancer (NSCLC): CheckMate 227 part 1 final analysis. Annals of Oncology 2019;30(suppl 5):v913-4. [Google Scholar]
- Ramalingam SS, Ciuleanu TE, Pluzanski A, Lee JS, Schenker M, Caro RB, et al. Nivolumab + ipilimumab versus platinum-doublet chemotherapy as first-line treatment for advanced non-small cell lung cancer: Three-year update from CheckMate 227 Part 1. Journal of Clinical Oncology May 20, 2020;38, no. 15_suppl:9500. [Google Scholar]
- Reck M, Hellmann MD, Paz-Ares LG, Ramalingam SS, Brahmer JR, O'Byrne KJ, et al. Nivolumab (nivo) + ipilimumab (Ipi) vs platinum-doublet chemotherapy (chemo) as first-line (1L) treatment (Tx) for advanced non-small cell lung cancer (NSCLC): safety analysis and patient-reported outcomes (PROs) from CheckMate 227. Journal of Clinical Oncology 2018;36(15):9020. [Google Scholar]
Herbst 2020 {published data only}
- A study of atezolizumab (MPDL3280A) compared with a platinum agent (cisplatin or carboplatin) + (pemetrexed or gemcitabine) in participants with stage IV non-squamous or squamous non-small cell lung cancer (NSCLC) [IMpower110]. https://clinicaltrials.gov/ct2/show/NCT02409342 accessed May 2019.
- De Marinis F, Jassem J, Spigel DR, Lam S, Mocci S, Sandler A, et al. 480TiP - IMpower110: Phase III study on 1L atezolizumab (atezo) in PD-L1–selected chemotherapy (chemo)-naive NSCLC patients (pts). In: Annals of Oncology. Vol. 27 (suppl 9). 2016:ix139-ix56.. [DOI: 10.1093/annonc/mdw594] [DOI]
- De Marinis F, Jassem J, Spigel DR, Lam S, Mocci S, Sandler A, et al. PIMpower110: Phase III study on 1L atezolizumab (atezo) in PD-L1–selected chemotherapy (chemo)-naive NSCLC patients (pts). Annals of Oncology 2016;27(Suppl 9):ix139-56. [Google Scholar]
- Herbst RS, Marinis F, Giaccone G, Reinmuth N, Vergnenegre A, Barrios CH. Clinical efficacy of atezolizumab (atezo) in biomarker subgroups by SP142, SP263 and 22C3 PD-L1 immunohistochemistry (IHC) assays and by blood tumour mutational burden (bTMB): results from the IMpower110 study. In: Annals of Oncology. Vol. Abstract presented at Proffered Paper session, ESMO Immunoncology Congress. 2019.
- Herbst RS, Giaccone G, Marinis F, Reinmuth N, Vergnenegre A, Barrios CH, et al. Atezolizumab for first-line treatment of PD-L1-selected patients with NSCLC. New England Journal of Medicine Oct 2020;383(14):1328-39. [DOI] [PubMed] [Google Scholar]
- Spigel D, Marinis F, Giaccone G, Reinmuth N, Vergnenegre A, Barrios CH. IMpower110: Interim overall survival (OS) analysis of a phase III study of atezolizumab (atezo) vs platinum-based chemotherapy (chemo) as first-line (1L) treatment (tx) in PD-L1–selected NSCLC. Annals of Oncology (2019);30 (suppl_5):v851-v934. 10.1093/annonc/mdz394. [Google Scholar]
Mok 2019 {published data only}
- Lopes G, Wu YL, Kudaba I, Kowalski D, Cho BC, Castro G, et al. Pembrolizumab (pembro) versus platinum based chemotherapy (chemo) as first-line therapy for advanced/metastatic NSCLC with a PD-L1 tumor proportion score (TPS) ≥ 1%: open-label, phase 3. Journal of Clinical Oncology 2018;36(18):LBA4. [Google Scholar]
- Mok TS, Wu YL, Kudaba I, Kowalski DM, Cho BC, Turna HZ, et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): a randomised, open-label, controlled, phase 3 trial. Lancet 2019;393(10183):1819-30. [DOI] [PubMed] [Google Scholar]
Reck 2016 {published data only}
- Brahmer J, Rodriguez-Abreu D, Robinson A, Hui R, Csoszi T, Fulop A, et al. Updated analysis of keynote-024: pembrolizumab vs platinum-based chemotherapy for advanced NSCLC with PD-L1 TPS >=50%. Journal of Thoracic Oncology 2017;12(11 suppl 2):S1793-4. [Google Scholar]
- Brahmer JR, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Health-related quality-of-life results for pembrolizumab versus chemotherapy in advanced, PD-L1-positive NSCLC (KEYNOTE-024): a multicentre, international, randomised, open-label phase 3 trial. The Lancet Oncology 2017;18(12):1600-9. [DOI] [PubMed] [Google Scholar]
- Brahmer JR, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. KEYNOTE-024 5-year OS update: first-line (1L) pembrolizumab (pembro) vs platinum-based chemotherapy (chemo) in patients (pts) with metastatic NSCLC and PD-L1 tumour proportion score (TPS) >=50%. Annals of Oncology 2020;31(suppl 4):S1181-2. [Google Scholar]
- Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, et al. KEYNOTE-024 Investigators. pembrolizumab versus chemotherapy for PD-L1-Positive non-small-cell lung cancer. New England Journal of Medicine 2016;375(19):1823-33. [DOI] [PubMed] [Google Scholar]
- Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, et al. Updated analysis of KEYNOTE-024: pembrolizumab versus platinum-based chemotherapy for advanced non-small-cell lung cancer with PD-L1 tumor proportion score of 50% or greater. Journal of Clinical Oncology 2019 March;37(7):537-46. [DOI] [PubMed] [Google Scholar]
- Satouchi M, Nosaki K, Takahashi T, Nakagawa K, Aoe K, Kurata T, et al. First-line pembrolizumab versus chemotherapy in metastatic non-small-cell lung cancer: KEYNOTE-024 Japan subset. Cancer Science 2020;00:1-10. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Rizvi 2020 {published data only}
- Cho BC, Lee KH, Ahn M-J, Lucien Geater S, Ngoc TV, Wang C-C, et al. Efficacy and safety of first-line durvalumab (D) ± tremelimumab (T) vs chemotherapy (CT) in Asian patients with metastatic NSCLC: results from MYSTIC. Annals of Oncology 2019;30:ix157-8. [Google Scholar]
- NCT02453282. Phase III open label first line therapy study of MEDI 4736 (durvalumab) with or without tremelimumab versus SOC in non small-cell lung cancer (NSCLC). (MYSTIC). available at https://clinicaltrials.gov/ct2/show/NCT02453282.
- Peters S, Chul Cho B, Reinmuth N, Lee KH, Luft A, Ahn MJ, et al. Tumor mutational burden (TMB) as a biomarker of survival in metastatic non-small cell lung cancer (mNSCLC): Blood and tissue TMB analysis from MYSTIC, a phase III study of first-line durvalumab ± tremelimumab vs chemotherapy. Cancer Research 2019;79 (13 Suppl):Abstract nr CT074. [Google Scholar]
- Rizvi NA, Cho BC, Reinmuth N, Lee KH, Luft A, Ahn MJ, et al. Durvalumab with or without tremelimumab vs standard chemotherapy in first-line treatment of metastatic non-small cell lung cancer: the MYSTIC Phase 3 randomized clinical trial. JAMA Oncology 2020;Apr 9:E1-14. [DOI: 10.1001/jamaoncol.2020.0237] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi NA, Chul Cho B, Reinmuth N, Lee KH, Ahn M, Luft A, et al. Durvalumab with or without tremelimumab vs platinum-based chemotherapy as first-line treatment for metastatic non-small cell lung cancer: MYSTIC. Annals of Oncology (2018);29 (suppl_10):x39-x43. 10.1093/annonc/mdy511. [Google Scholar]
Sezer 2020 {unpublished data only}
- NCT03088540. A global, randomised, phase 3, open-label study of REGN2810 (ANTI-PD 1 Antibody) versus platinum based chemotherapy in first line treatment of patients with advanced or metastatic PD L1+non-small cell lung cancer. https://clinicaltrials.gov/ct2/show/NCT03088540 accessed January 2020.
- Sezer A, Kilickap S, Gümüş M, Bondarenko I, Özgüroğlu M, Gogishvili M, et al. EMPOWERLung 1: Phase 3 first line (1L) Cemiplimab monotherapy vs platinum doublet chemotherapy (chemo) in advanced non small cell lung cancer (NSCLC) with programmed cell death ligand 1 (PD L1) ≥50%. In: ESMO Congress. 2020.
References to studies excluded from this review
Huan 2019 {published data only}
- Huang M, Pietanza MC, Samkari A, Pellissier J, Burke T, Chandwani S, et al. Q-TWiST analysis to assess benefit-risk of pembrolizumab in patients with PD-L1-positive advanced or metastatic non-small cell lung cancer. Pharmacoeconomics 2019;37:105-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hui 2017a {published data only}
- Hui R, Garon EB, Goldman JW, Leighl NB, Hellmann MD, Patnaik A, et al. Pembrolizumab as first-line therapy for patients with PD-L1-positive advanced non-small cell lung cancer: a phase 1 trial. Annals of Oncology 2017;28(4):874-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Leighl 2019 {published data only}
- Leighl NB, Hellmann MD, Hui R, Carcereny E, Felip E, Ahn MJ, et al. Pembrolizumab in patients with advanced non-small-cell lung cancer (KEYNOTE-001): 3-year results from an open-label, phase 1 study. Lancet 2019;7(4):347-57. [DOI] [PubMed] [Google Scholar]
Mok 2017 {published data only}
- Mok T, Johnson M, Garon E, Peters S, Soria J, Wang L, et al. Poseidon: a phase 3 study of first-line durvalumab +/- tremelimumab + chemotherapy vs chemotherapy alone in metastatic NSCLC. Journal of Thoracic Oncology 2017;12(11 suppl 2):S1975. [Google Scholar]
Ready 2019 {published data only}
- Ready N, Hellmann MD, Awad MM, Otterson GA, Gutierrez M, Gainor JF, et al. First-line nivolumab plus ipilimumab in advanced non-small-cell lung cancer (CheckMate 568): outcomes by programmed death ligand 1 and tumor mutational burden as biomarkers PST - aheadofprint DBP - 2019/02/21 06:00. Journal of clinical Oncology 2019;37(12):JCO1801042. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to ongoing studies
EMPOWER‐Lung 2 {published data only}
- NCT03515629. REGN2810 (Anti-PD-1 antibody), platinum-based doublet chemotherapy, and ipilimumab (anti-CTLA-4 antibody) versus pembrolizumab monotherapy in patients with lung cancer. https://clinicaltrials.gov/ct2/show/NCT03515629 accessed May 2019.
- Rizvi N, Lee S, Curtis P, Caldwell W, Gao B, Rietschel P. EMPOWER-Lung 3: a phase 3 study of cemiplimab, ipilimumab and chemotherapy in advanced NSCLC with PD-L1 <50%. Journal of Thoracic Oncology October 2018 Volume 13;Supplement(10):S931. [Google Scholar]
eNERGY {published data only}
- NCT03351361. Randomized phase III study testing nivolumab and ipilimumab versus a carboplatin based doublet in first line treatment of PS 2 or elderly patients with advanced non-small cell lung cancer (eNERGY). https://clinicaltrials.gov/ct2/show/NCT03351361 accessed May 2019.
IPSOS {published data only}
- Lee SM, Schulz C, Cardona A, Bartakova P, Peters S. Phase III study of atezolizumab (atezo) vs chemotherapy (chemo) in patients (pts) with treatment-naive advanced, recurrent or metastatic NSCLC unsuitable for platinum (plat)-based chemo. Annals of Oncology 2017;28(suppl 5):v460-96. [Google Scholar]
- NCT03191786. A study of atezolizumab compared with chemotherapy in treatment naïve participants with locally advanced or recurrent or metastatic non-small cell lung cancer who are deemed unsuitable for platinum-containing therapy (IPSOS). https://clinicaltrials.gov/ct2/show/NCT03191786 accessed May 2019.
JAVELIN Lung 100 {published data only}
- NCT02576574. Avelumab in first-line non-small cell lung cancer (JAVELIN Lung 100). https://clinicaltrials.gov/ct2/show/NCT02576574 accessed May 2019.
- Reck M, Yang C, Postmus PE, Barlesi F, Font EF, Thomas M, et al. JAVELIN Lung 100: updated design of a phase 3 trial of avelumab vs platinum doublet chemotherapy as first-line (1L) treatment for metastatic or recurrent PD-L11 non-small-cell lung cancer (NSCLC). Annals of Oncology 9 September 2017;28(suppl 5):v460-96. [Google Scholar]
KEYNOTE‐598 {published data only}
- NCT03302234. Study of pembrolizumab given with ipilimumab or placebo in participants with untreated metastatic non-small cell Lung cancer (MK-3475-598/KEYNOTE-598). https://clinicaltrials.gov/ct2/show/NCT03302234 accessed May 2019.
MILES 5 {published and unpublished data}
- A study comparing immunotherapy with chemotherapy in the treatment of elderly patients with advanced NSCLC (MILES-5). Ongoing study. December 20, 2018Estimated study completion date: June 2023. Contact author for more information.
NEPTUNE {published data only}
- Mok T, Schmid P, Aren O, Arrieta O, Gottfried M, Jazieh, et al. 192TiP: NEPTUNE: a global, phase 3 study of durvalumab (MEDI4736) plus tremelimumab combination therapy versus standard of care (SoC) platinum-based chemotherapy in the first-line treatment of patients (pts) with advanced or metastatic NSCLC. Journal of Thoracic Oncology 2016;11(4):S140-41. [Google Scholar]
- Mok T, Schmid P, Aren O, Arrieta O, Gottfried M, Jazieh AR, et al. Phase 3, randomised, open-label study of durvalumab (MEDI4736) in combination with tremelimumab versus platinum-based chemotherapy in first-line treatment of patients with advanced or metastatic NSCLC: NEPTUNE. Annals of Oncology 2015;26(Suppl_9):125-47. [Google Scholar]
- Mok T, Schmid P, Castro G, Syrigos K, Martin C, N amamoto Y, et al. Global, phase 3 study of first-line durvalumab (MEDI4736) + tremelimumab vs standard of care platinum-based chemotherapy in advanced/metastatic NSCLC: NEPTUNE. Annals of Oncology 2016;27(9):mdw594.046. [Google Scholar]
- Mok T, Schmid P, De Castro G, Syrigos K, Martin C, Yamamoto N, et al. P2.06-022 first-line durvalumab plus tremelimumab vs platinum-based chemotherapy for advanced/metastatic NSCLC: phase 3 NEPTUNE study. Journal of Thoracic Oncology 2017;12(1):S1084. [DOI] [PubMed] [Google Scholar]
- Study of 1st line therapy study of durvalumab with tremelimumab versus SoC in non small-cell lung cancer (NSCLC) (NEPTUNE). https://clinicaltrials.gov/ct2/show/NCT02542293 accessed May 2019.
PEARL {published data only}
- NCT03003962. Study of durvalumab alone or chemotherapy for patients with advanced non small-cell lung cancer. https://clinicaltrials.gov/ct2/show/NCT03003962 accessed May 2019.
Additional references
Antonia 2016
- Antonia S, Goldberg SB, Balmanoukian A, Chaft JE, Sanborn RE, Gupta A, et al. Safety and antitumour activity of durvalumab plus tremelimumab in non-small cell lung cancer: a multicentre, phase 1b study. Lancet Oncology 2016;Mar;17(3):299-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
Borghaei 2015
- Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. New England Journal of Medicine 2015;22, 373(17):1627-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
Brahmer 2015
- Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WE, Poddubskaya E, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. New England Journal of Medicine 2015;373(2):123-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bray 2018
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians 2018;68(6):394-424. [DOI] [PubMed] [Google Scholar]
Brockhaus 2014
- Brockhaus AC, Bender R, Skipka G. The Peto odds ratio viewed as a new effect measure. Statistics in Medicine 2014;33(28):4861-74. [DOI] [PubMed] [Google Scholar]
Champiat 2018
- Champiat S, Ferrara R, Massard C, Besse B, Marabelle A, Soria JC, et al. Hyperprogressive disease: recognizing a novel pattern to improve patient management. Nature Review Clinical Oncology 2018;15(12):748-62. [DOI] [PubMed] [Google Scholar]
Chen 2019
- Chen Y, Zhou Y, Tang L, Peng X, Jiang H, Wang G, et al. Immune-checkpoint inhibitors as the first line treatment of advanced non-small cell lung cancer: a meta-analysis of randomized controlled trials. Journal of Cancer 2019;10(25):6261-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Conforti 2018
- Conforti F, Pala L, Bagnardi V, De Pas T, Martinetti M, Viale G, et al. Cancer immunotherapy efficacy and patients' sex: a systematic review and meta-analysis. Lancet Oncology 2018;19(6):737-46. [DOI] [PubMed] [Google Scholar]
Dafni 2019
- Dafni U, Tsourti Z, Vervita K, Peters S. Immune checkpoint inhibitors, alone or in combination with chemotherapy, as first-line treatment for advanced non-small cell lung cancer. A systematic review and network meta-analysis. Lung Cancer 2019 Aug;134:127-40. [DOI] [PubMed] [Google Scholar]
Deeks 2011
- Deeks JJ, Higgins JP, Altman DG. Chapter 9. Analysing data and undertaking meta-analyses. In Higgins JP, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). Available from www.handbook.cochrane.org.
DerSimonian 1986
- DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clinical Trials 1986;7(3):177-88. [DOI] [PubMed] [Google Scholar]
Eisenhauer 2009
- Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). European Journal of Cancer 2009;45(2):228-47. [DOI] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne DR, Altman DG, Higgins JP, Curtin F, Worthington HV, Vail A. Meta-analyses involving cross-over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140-9. [DOI] [PubMed] [Google Scholar]
El‐Osta 2019
- El-Osta H, Jafri S. Predictors for clinical benefit of immune checkpoint inhibitors in advanced non-small-cell lung cancer: a meta-analysis. Immunotherapy 2019;11(3):189–99. [DOI] [PubMed] [Google Scholar]
Ferrara 2018
- Ferrara R, Mezquita L, Texier M, Lahmar J, Audigier-Valette C, Tessonnier L, et al. Hyperprogressive disease in patients with advanced non-small cell lung cancer treated with PD-1/PD-L1 inhibitors or with single-agent chemotherapy. JAMA Oncology 2018;4(11):1543-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ferrara 2020
- Ferrara R, Mezquita L, Texier M, Lahmar J, Audigier-Valette C, Tessonnier L, et al. Comparison of fast-progression, hyperprogressive disease, and early deaths in advanced non–small-cell lung cancer treated with PD-1/PD-L1 inhibitors or chemotherapy. Journal of Clinical Oncology Precision Oncology July 2020;4:829-40. [DOI] [PubMed] [Google Scholar]
Gainor 2020
- Gainor JF, Rizvi H, Jimenez Aguilar E, Skoulidis F, Yeap BY, Naidoo J, et al. Clinical activity of programmed cell death 1 (PD-1) blockade in never, light, and heavy smokers with non-small-cell lung cancer and PD-L1 expression ≥50%. Annals of Oncology 2020;31(3):404-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Garon 2015
- Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. New England Journal of Medicine 2015;372(21):2018-28. [DOI] [PubMed] [Google Scholar]
Gettinger 2016
- Gettinger S, Rizvi NA, Chow LQ, Borghaei H, Brahmer J, Ready N, et al. Nivolumab monotherapy for first-line treatment of advanced non-small-cell lung cancer. Journal of Clinical Oncology 2016;34(25):2980-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hathcock 1993
- Hathcock KS, Laszlo G, Dickler HB, Bradshaw J, Linsley P, Hodes RJ. Identification of an alternative CTLA-4 ligand costimulatory for T cell activation. Science 1993;262:905-7. [DOI] [PubMed] [Google Scholar]
Higgins 2002
- Higgins JP, Thompson SG. Quantifying heterogeneity in a meta‐analysis. Statistics in Medicine 2002;21(11):1539-58. [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JP, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration. Available from handbook.cochrane.org.
Hui 2017
- Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 2017;355(6332):1428-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ishida 1992
- Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO Journal 1992;11:3887-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kluetz 2016
- Kluetz PG, Chingos DT, Basch EM, Mitchell SA. Patient-Reported Outcomes in Cancer Clinical Trials: measuring symptomatic adverse events with the National Cancer Institute's Patient-Reported Outcomes version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE). American Society of Clinical Oncology Educational Book 2016;35:67-73. [DOI] [PubMed] [Google Scholar]
Lawrence 2013
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013;499(7457):214-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lopez‐Chavez 2012
- Lopez-Chavez A, Young T, Fages S, Leon L, Schiller JH, Dowlati A, et al. Bevacizumab maintenance in patients with advanced non-small-cell lung cancer, clinical patterns, and outcomes in the Eastern Cooperative Oncology Group 4599 Study: results of an exploratory analysis. Journal Thoracic Oncology 2012;7(11):1707-12. [DOI] [PubMed] [Google Scholar]
Mazieres 2019
- Mazieres J, Drilon A, Lusque A, Mhanna L, Cortot AB, Mezquita L, et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: results from the IMMUNOTARGET Registry. Annals of Oncology 2019;30(8):1321-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Nishino 2013
- Nishino M, Giobbie-Hurder A, Gargano M, Suda M, Ramaiya NH, Hodi FS. Developing a common language for tumor response to Immunotherapy: immune-related response criteria using unidimensional measurements. Clinical Cancer Research 2013;19(14):3936-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Nosaki 2019
- Nosaki K, Hosomi Y, Saka H, Baas P, Castro G, Reck M, et al. Safety and efficacy of pembrolizumab (pembro) monotherapy in elderly patients (pts) with PD-L1–positive advanced NSCLC: pooled analysis from KEYNOTE-010, -024, and -042. Annals of Oncology 2019;30 (suppl_2):ii38-ii68. 10.1093/annonc/mdz063. [DOI] [PubMed] [Google Scholar]
Novello 2016
- Novello S, Barlesi F, Califano R, Cufer T, Ekman S, Levra MG, et al. Metastatic non-small-cell lung cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Annals of Oncology 2016;27(Suppl 5):v1-v27. [DOI] [PubMed] [Google Scholar]
Okazaki 2013
- Okazaki T, Chikuma S, Iwai Y, Fagarasan S, Honjo T. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nature Immunology 2013;14(12):1212-8. [DOI] [PubMed] [Google Scholar]
Osoba 1998
- Osoba D, Rodrigues G, Myles J, Zee B, Pater J. Interpreting the significance of changes in health-related quality-of-life scores. Journal of Clinical Oncology 1998 Jan;16(1):139-44. [DOI] [PubMed] [Google Scholar]
Parmar 1998
- Parmar MK, Torri V, Stewart L. Extracting summary statistics to perform meta‐analyses of the published literature for survival endpoints. Statistics in Medicine 1998;17(24):2815‐34. [DOI] [PubMed] [Google Scholar]
Paz‐Ares 2013
- Paz-Ares LG, Marinis F, Dediu M, Thomas M, Pujol JL, Bidoli P, et al. PARAMOUNT: Final overall survival results of the phase III study of maintenance pemetrexed versus placebo immediately after induction treatment with pemetrexed plus cisplatin for advanced nonsquamous non-small-cell lung cancer. Journal of Clinical Oncology 2013;31(23):2895-902. [DOI] [PubMed] [Google Scholar]
Peters 2017
- Peters S, Gettinger S, Johnson ML, Jänne PA, Garassino MC, Christoph D, et al. Phase II trial of atezolizumab as first-line or subsequent therapy for patients with programmed death-ligand 1-selected advanced non-small-cell lung cancer (BIRCH). Journal of Clinical Oncology 2017, Aug 20;35(24):2781-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
Raphael 2020
- Raphael J, Batra A, Boldt G, Shah PS, Blachette P, Rodrigues G, et al. Predictors of survival benefit from immune checkpoint inhibitors in patients with advanced non-small-cell lung cancer: a systematic review and meta-analysis. Clinical Lung Cancer 2020;21(2):106–113.e5. [DOI] [PubMed] [Google Scholar]
RevMan 2020 [Computer program]
- The Cochrane Collaboration Review Manager (RevMan). Version 5.4. Copenhagen: Nordic Cochrane Centre: The Cochrane Collaboration, 2014.
Scagliotti 2008
- Scagliotti GV, Parikh P, Pawel J, Biesma B, Vansteenkiste J, Manegold C, et al. Phase III study comparing cisplatin plus gemcitabine with cisplatin plus pemetrexed in chemotherapy-naive patients with advanced-stage non-small-cell lung cancer. Journal of Clinical Oncology 2008;26(21):3543-51. [DOI] [PubMed] [Google Scholar]
Seymour 2017
- Seymour L, Bogaerts J, Perrone A, Ford R, Schwartz LH, Mandrekar S. iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncology 2017;18(3):e143-e152. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sterne 2011
- Sterne JA, Egger M, Moher D. Chapter 10. addressing reporting biases. In: Higgins JP, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). Available from handbook.cochrane.org.
Sun 2020
- Sun L, Zhang L, Yu J, Zhang Y, Pang X, Ma C, et al. Clinical efficacy and safety of anti-PD-1/PD-L1 inhibitors for the treatment of advanced or metastatic cancer: a systematic review and meta-analysis. Scientific Reports 2020;10(1):2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Syn 2018
- Syn NL, Roudi R, Wang LZ, Wang L, Loh M, Huang Y, et al. Immune checkpoint inhibitors plus chemotherapy versus chemotherapy or immunotherapy for first-line treatment of advanced non-small cell lung cancer: a generic protocol. Cochrane Database of Systematic Reviews 2018, Issue 4. Art. No: CD013009. [DOI: 10.1002/14651858.CD013009] [DOI] [Google Scholar]
Tierney 2007
- Tierney JF, Stewart LA, Ghersi D, Burdett S, Sydes MR. Practical methods for incorporating summary time‐to‐event data into meta‐analysis. Trials 2007;8:16. [DOI: 10.1186/1745-6215-8-16] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tsao 2018
- Tsao MS, Kerr KM, Kockx M, Beasley MB, Borczuk AC, Botling J, et al. PD-L1 Immunohistochemistry Comparability Study in Real-Life Clinical Samples: Results of Blueprint Phase 2 Project. Journal of Thoracic Oncology 2018;13(9):1302-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wallis 2019
- Wallis CJD, Butaney M, Satkunasivam R, Freedland SJ, Patel SP, Hamid O, et al. Association of patient sex with efficacy of immune checkpoint inhibitors and overall survival in advanced cancers: a systematic review and meta-analysis. JAMA Oncology 2019 Apr;5(4):529–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yu 2019
- Yu Y, Zeng D, Ou Q, Liu S, Li A, Chen Y et al. Association of survival and immune-related biomarkers with immunotherapy in patients with non-small cell lung cancer: a meta-analysis and individual patient-level analysis. JAMA Network Open 2019 Jul 3;2(7):e196879. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to other published versions of this review
Ferrara 2019
- Ferrara R, Imbimbo M, Paget‐Bailly S, Malouf R, Calais F, Agazzi GM, et al. Single or combined immune checkpoint inhibitors compared to first‐line chemotherapy with or without bevacizumab for people with advanced non‐small cell lung cancer. Cochrane Database of Systematic Reviews 2019, Issue 2. Art. No: CD013257. [DOI: 10.1002/14651858.CD013257] [DOI] [Google Scholar]