

Pharmacokinetics of drugs can be affected by physiologic changes during pregnancy. Our aim was to assess the influence of covariates on tenofovir (TFV) pharmacokinetics in pregnant and postpartum women receiving tenofovir disoproxil fumarate (TDF).
KEYWORDS: HIV, AIDS, TDF, population pharmacokinetics, postpartum, pregnancy, tenofovir, tenofovir disoproxil fumarate
ABSTRACT
Pharmacokinetics of drugs can be affected by physiologic changes during pregnancy. Our aim was to assess the influence of covariates on tenofovir (TFV) pharmacokinetics in pregnant and postpartum women receiving tenofovir disoproxil fumarate (TDF). Population pharmacokinetic parameter estimates and the influence of covariates were assessed using nonlinear mixed-effects modeling (NONMEM 7.4). Forty-six women had intensive pharmacokinetic evaluations during the second and third trimesters of pregnancy, with another evaluation postpartum. A two-compartment pharmacokinetic model with allometric scaling for body weight and first-order absorption best described the tenofovir plasma concentration data. Apparent oral clearance (CL/F) and volume of distribution at steady state (Vss/F) were increased during pregnancy. Weight, serum creatinine (SCr), pregnancy, albumin, and age were associated with TFV CL/F during univariate assessment, but in the multivariate analysis, changes in CL/F and Vss/F were only associated with increased body weight and enhanced renal function. Due to greater weight and lower SCr during pregnancy, CL/F was 28% higher during pregnancy than postpartum. In the final model, CL/F (liters per hour) was described as 2.07 × (SCr/0.6)0.65 × weight0.75, with a low between-subject variability (BSV) of 24%. The probability of target attainment (proportion exceeding area under the concentration-time curve of >1.99 μg·h/ml, the 10th percentile of average TFV exposure for nonpregnant historical controls) was 68%, 80%, 87%, and 93% above the target with 300 mg, 350 mg, 400 mg, and 450 mg of TDF, respectively, during pregnancy and 88%, 92%, 96%, and 98% above the target with same doses in postpartum women. Dose adjustment of TDF during pregnancy is not generally warranted, but any modification should be based on weight and renal function. (This study has been registered at ClinicalTrials.gov under identifier NCT00042289.)
TEXT
Tenofovir disoproxil fumarate (TDF), a diester prodrug of tenofovir (TFV), is a backbone component of combination antiretroviral (ARV) therapy currently recommended by the U.S. perinatal guidelines for the management of pregnant women living with HIV for preventing perinatal infection (1). Following oral administration and absorption, TFV is transported inside cells and phosphorylated by intracellular kinases to tenofovir-monophosphate (TFV-MP) and then its active metabolite, tenofovir-diphosphate TFV-DP (2). Tenofovir is predominantly eliminated unchanged in the urine by a combination of active tubular secretion and glomerular filtration (3, 4). Due to several physiological changes that occur during pregnancy (primarily increased renal clearance), plasma concentrations of TFV are decreased during the 2nd and 3rd trimesters of pregnancy, returning to baseline in the postpartum period (5). In addition, relative decreases in plasma TFV exposure significantly vary by age, weight, and other physiologic parameters (6, 7). An understanding of TFV between-subject (BSV) and within-subject variability patterns in pregnant and postpartum women is vital to determining the key factors influencing changes in TFV exposure.
A population pharmacokinetic (POPPK) approach allows for the development of a model that describes the time course of drug concentrations and integrates between-subject and within-subject variability. A POPPK analysis of TFV by Hirt and colleagues assessed TFV plasma concentrations in 38 women living with HIV who received 600 mg of TDF at delivery and 300 mg daily for 7 days postpartum, and they demonstrated an acceptable materno-fetal transport of TFV of approximately 60% (8). In another POPPK study of 186 women with HIV (including 46 pregnant women on 300 mg once daily TDF), Benaboud et al. reported that pregnant women had a 39% higher apparent TFV clearance during delivery than nonpregnant women. The Benaboud et al. POPPK model was unable to evaluate gestation-dependent changes on TFV clearance or measures of kidney function (serum creatinine concentrations or estimated glomerular filtration rate), limiting the ability to inform dosing recommendations (9). While these pregnancy POPPK studies of TFV provided critical insights into sources of intra- and interindividual variability in TFV disposition during pregnancy, both studies were conducted using sparse PK data. Intensive PK data may provide an improved understanding of intra- and interindividual variability in TFV disposition during pregnancy and postpartum.
Our objective was to evaluate the impact of clinical factors in pregnant and postpartum women living with HIV that affect TFV PK (when administered as 300 mg of TDF daily) within a large, well-characterized, and diverse population of women who had intensive TFV PK data during pregnancy and postpartum.
RESULTS
Study population.
The demographic characteristics are summarized in Table 1. A total of 46 subjects were included in the POPPK analysis. Briefly, median maternal age, prepregnancy weight, and weight gain were 31 years (interquartile range [IQR], 27 to 34), 76.4 kg (IQR, 66.3 to 96.9), and 6.85 kg (IQR, 4.15 to 11.40), respectively. Thirty-nine percent of participants in the analyzed data set were Hispanic, 35% were black, 20% white non-Hispanic, 2% Asian/Pacific Islander, 2% more than one race, and 2% unknown.
TABLE 1.
Baseline characteristics (n = 46)
| Characteristic | Median (% or IQR) value |
|---|---|
| Participant’s age at 1st visit (yr) | 30.94 (27, 34) |
| No. of women from each visit | |
| Second trimester | 7 (8.1) |
| Third trimester | 41 (47.7) |
| 2–3 wk postpartum | 3 (3.5) |
| 6–12 wk postpartum | 35 (40.7) |
| Race | |
| Hispanic | 18 (39.1) |
| Black | 16 (34.8) |
| White, non-Hispanic | 9 (19.6) |
| Asian/Pacific Islander | 1 (2.2) |
| More than 1 race | 1 (2.2) |
| Unknown | 1 (2.2) |
| Prepregnancy wt (kg) | 76.40 (66.30, 96.88) |
| Wt in the 3rd trimester (kg) | 80.6 (50.8, 121.9) |
| Wt gain in 3rd trimester (kg) | 6.85 (4.15, 11.40) |
| Concomitant medications | |
| Ritonavir boosted protease inhibitor | 39 (84.8) |
| Atazanavir-ritonavir | 24 (52.2) |
| Lopinavir-ritonavir | 12 (26.1) |
| Fosamprenavir-ritonavir | 1 (2.2) |
| Saquinavir-ritonavir | 2 (4.3) |
| Emtricitabine | 33 (71.7) |
| Lamivudine | 10 (21.7) |
| Zidovudine | 7 (15.2) |
| Efavirenz | 3 (8.7) |
| Nevirapine | 4 (6.5) |
POPPK structural base model.
A two-compartment model (a central and a peripheral maternal compartment) with first-order absorption, with elimination assumed to be from the central compartment (Fig. 1), best described our data. This two-compartment model structure (a central and a peripheral compartment) was used as the base model. Due to lack of adequate sampling time points during the absorption phase (days 0 and 1), complex absorption models could not be characterized, and a simpler first-order absorption model (Ka fixed to 6.93 h−1) was chosen. A Ka value of 6.93 h−1 was estimated based on the base model but subsequently fixed due to low precision of the estimate. The robustness of the fixed value was further verified using a sensitivity analysis by varying Ka from 0.1 to 10 h−1. Changes in the objective function value (OFV) and the variance model parameter values indicated the chosen value of 6.93 h−1 was appropriate. Between-individual variability was described by an exponential model and, with a residual power error model, provided the best description of the data (based on goodness-of-fit plots and lower objective function [OFV of >−51]). The addition of interoccasion variability (IOV) on apparent clearance (CL/F) and apparent volume of distribution (Vss/F) further led to a reduction in objective function values (P < 0.001) and improved model fit.
FIG 1.
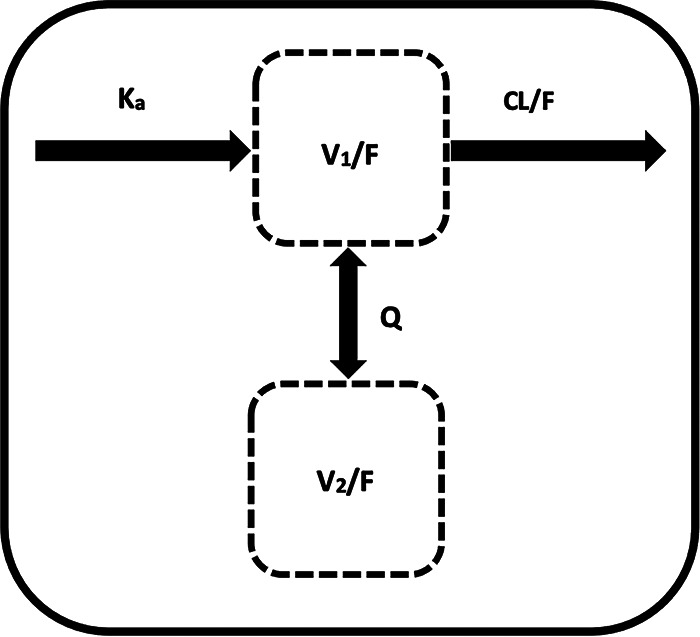
Model structure of tenofovir population pharmacokinetics. CL/F, V1/F, V2/F, Ka, and Q represent apparent clearance, volume of central compartment, volume of peripheral compartment, absorption rate constant, and intercompartmental clearance, respectively.
Population pharmacokinetic analysis.
A total of 688 plasma samples (8 plasma PK sample collections from 7 pregnant women in the second trimester, 8 plasma PK samples from 41 pregnant women in the third trimester, and 8 plasma samples from 38 women postpartum) were available for POPPK modeling. Most TFV peak plasma concentrations during pregnancy and postpartum were observed at 0.5 to 2 h postdose (Fig. 2 and 3), consistent with what is known about the time to maximum plasma concentration (Tmax) of TFV in participants taking TDF. No participant had TFV concentrations below the lower limit of detection for TFV of 10 ng/ml. During pregnancy, one patient received her postdose TDF late (Fig. 2).
FIG 2.
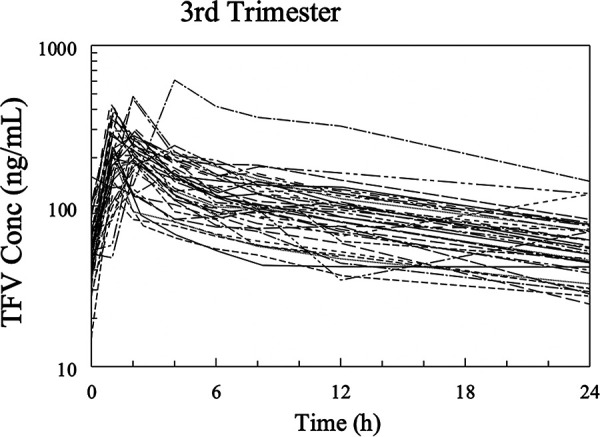
Spaghetti plots of tenofovir concentrations versus time (3rd trimester).
FIG 3.
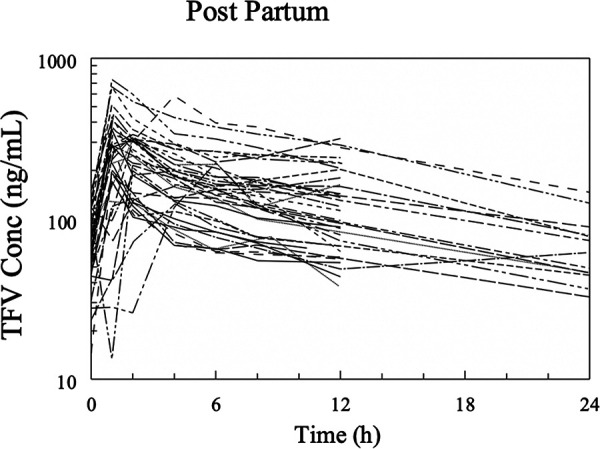
Spaghetti plots of tenofovir concentrations versus time (postpartum).
Table 2 shows the change in objective function values following the inclusion of covariates into the base population PK model. CL/F and Vss/F were increased during pregnancy. For univariate inclusion of covariates for CL/F, serum creatinine (ΔOFV = −16.174), pregnancy status (ΔOFV = −5.376), serum albumin concentration (ΔOFV = −5.051), gestational age (ΔOFV = −3.986), and maternal age (ΔOFV = −6.119) had a significant relationship with TFV CL/F. Insignificant covariates included the use of concomitant ritonavir (ΔOFV = −2.007). The univariate inclusion of covariates on Vss/F led to a decrease in the OFV for pregnancy status (ΔOFV = −6.474), gestational age (ΔOFV = −5.726), maternal age (ΔOFV = −5.849), serum creatinine (ΔOFV = −14.627), and serum albumin (ΔOFV = −7.401) (Table 2). The only significant covariates for CL/F obtained in the multivariate analyses was creatinine clearance (ΔOFV = −16.699; P < 0.001). There were no significant covariates for Vss/F in the multivariate analysis.
TABLE 2.
Change in objective function value by univariate and multivariate inclusion of covariates into base population pharmacokinetic model
| Covariatea | Univariate analysis |
Multivariate analysisb |
||
|---|---|---|---|---|
| Δ Objective function | Univariate estimate | Δ Objective function | Retained in model | |
| CL/F | ||||
| CovPREG | −5.376 | 1.18 | 0.707 | No |
| (GA/20)Cov | −3.986 | 0.25 | −2.114 | No |
| (Age/31)Cov | −6.119 | −0.33 | −1.977 | No |
| (SCR/0.6)Cov | −16.174 | −0.7 | −16.699 | Yes |
| (ALB/3.6)Cov | −5.051 | −0.43 | −2.343 | No |
| COVRTV | −2.007 | 0.932 | ND | NA |
| Vss/F | ||||
| CovPREG | −6.474 | 1.3 | −0.019 | No |
| (GA/20)Cov | −5.726 | 0.48 | −2.593 | No |
| (Age/31)Cov | −5.849 | −0.42 | −2.245 | No |
| (SCR/0.6)Cov | −14.627 | 1.04 | −1.913 | No |
| (ALB/3.6)Cov | −7.401 | −0.89 | −2.195 | No |
| COVRTV | −1.647 | 1.02 | ND | NA |
CovPREG, pregnancy status; (GA/20)Cov, gestational age; (age/31)Cov, age; (SCr/0.6)Cov, serum creatinine; (ALB/3.6)Cov, serum albumin; COVRTV, ritonavir.
ND, not determined; NA, not applicable.
The scatterplot of TFV clearance versus serum creatinine (a marker for renal function) (Fig. 4) demonstrated an inverse relationship (more marked in pregnancy than postpartum). Figure 5 shows box plots for TFV CL/F by pregnancy status, i.e., pregnant versus postpartum. The clearance of TFV was 28% higher in pregnant women than postpartum women.
FIG 4.
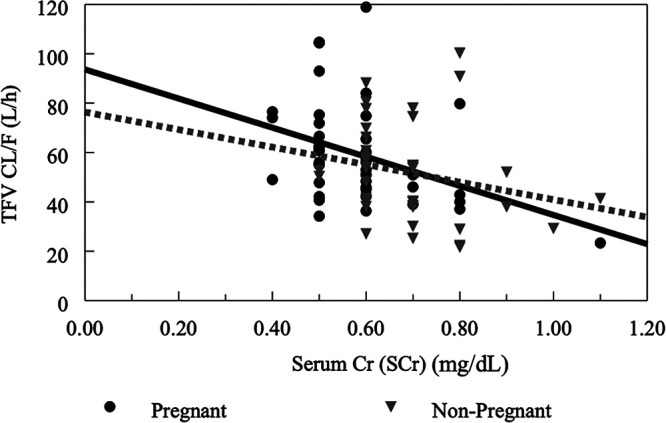
Apparent TFV clearance versus serum creatinine clearance (marker of renal function). Solid lines represent lines of identity for pregnant women, while interrupted lines represent lines of identity for nonpregnant (postpartum) women. Serum creatinine was measured in mg/dl.
FIG 5.
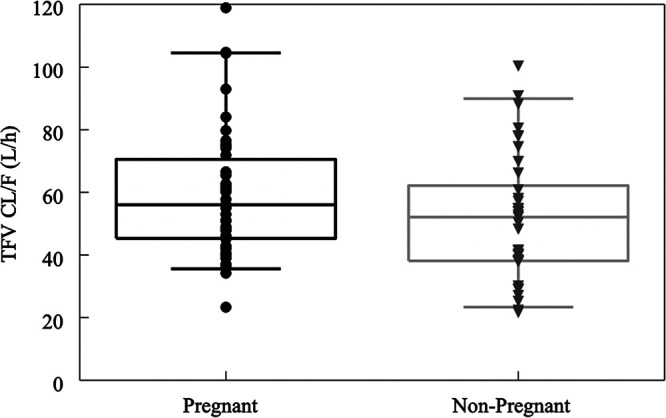
Boxplot of empirical Bayesian estimates of apparent TFV clearance in pregnant and nonpregnant (postpartum) women. Horizontal line, median; box, quartiles; whisker, range of the data (boxplots).
Model evaluation and validation.
Model performance was evaluated using various plots, including scatter goodness-of-fit plots of the individual predicted versus observed plasma TFV concentrations, conditional weighted residuals versus concentration/time, and plot of distribution of CL/F and Vss/F. The model-predicted and individual-predicted concentrations versus observed TFV concentrations using the final model are represented as scatterplots in Fig. 6 and 7, respectively. Model predictions were symmetrically distributed around the line of identity, indicating that the model adequately described TFV disposition. The model-predicted and individual-predicted values agreed with the observed values across the range of predicted concentration-time and the distributions of CL/F and Vss/F, and values followed a log-normal distribution. The plots of the conditional weighed residuals (CWRES) fit the data well (Fig. 8).
FIG 6.
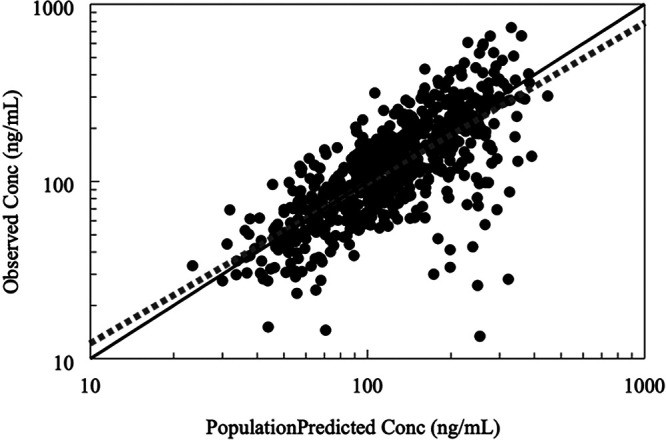
Goodness-of-fit diagnostic plots of observed TFV concentrations versus predicted values in the population. Solid lines represent lines of identity for pregnant women, while interrupted lines represent lines of identity for nonpregnant women. A symmetrical distribution around the line of identity is observed indicating the goodness of fit of the model.
FIG 7.
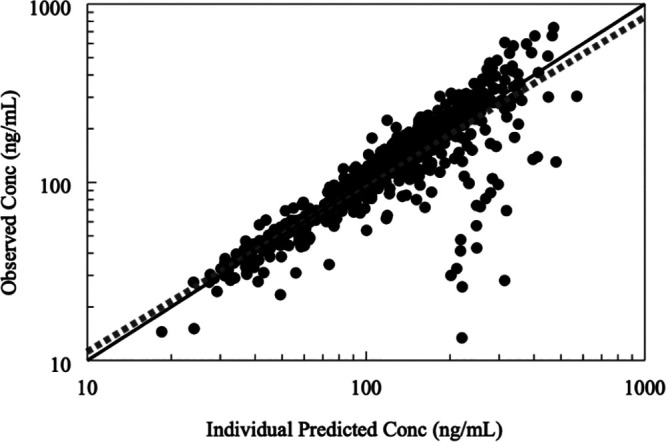
Goodness-of-fit diagnostic plots of observed TFV concentrations versus individual predicted TFV concentrations. Solid lines represent lines of identity for pregnant women, while interrupted lines represent lines of identity for nonpregnant women. A symmetrical distribution around the line of identity is observed indicating the goodness of fit of the model.
FIG 8.
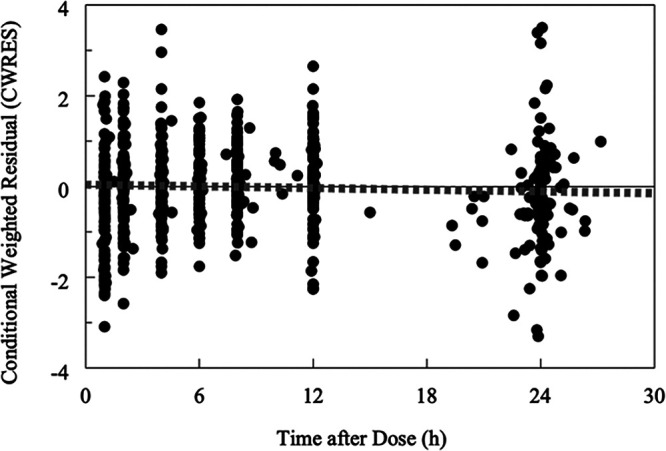
Goodness-of-fit diagnostic plots of conditional weighted residual versus time after dose for the final model. Solid lines represent lines of identity for pregnant women, while interrupted lines represent lines of identity for nonpregnant women. A symmetrical distribution around the line of identity is observed indicating the goodness of fit of the model.
Model stability, reliability, and validity were assessed by bootstrapping the final model, including 1,000 bootstrap replicates, minimized successfully with 998 successful runs. The estimated PK parameter values based on the original data set were in good agreement with the medians of the parameter values estimated from the bootstrap replicates (Table 3). The visual predictive check (VPC) plots during pregnancy (Fig. 9) and postpartum (Fig. 10) as a function of time indicate a good predictive performance of the model.
TABLE 3.
Final model parameter estimates and variability for tenofovir pharmacokineticsa
| Parameter | Final model |
Bootstrap analysis, final model |
|||
|---|---|---|---|---|---|
| Population median | %RSE | Median | 2.5% | 97.5% | |
| Structural (base) model | |||||
| CL/F (liter/h/kg0.75) | 2.07 | 0.09 | 2.00 | 1.85 | 2.16 |
| V1/F (liter/kg) | 6.37 | 1.07 | 6.34 | 4.87 | 8.70 |
| V2/F (liter/kg) | 9.55 | 1.20 | 9.60 | 8.12 | 12.30 |
| Ka (h−1) | 6.93 | NA | NA | NA | NA |
| Q (liter/h/kg0.75) | 3.14 | 0.95 | 3.15 | 0.05 | 4.69 |
| SCr factor | −0.65 | 0.13 | −0.66 | −0.90 | −0.36 |
| Statistical model | |||||
| BSV (%) | |||||
| Vss/F | 25 | 21 | 24 | 5 | 40 |
| CL/F | 24 | 12 | 23 | 17 | 29 |
| IOV-CL/F | 26 | 15 | |||
| Residual variability | |||||
| Proportional (% CV) | 3.0 | 2.6 | 3.0 | 1.1 | 6.0 |
| Power exponent | 1.43 | 0.08 | 1.38 | 1.25 | 1.54 |
Vss/F, volume of distribution at steady state; IOV, interoccasion (within-subject) variability; CL/F, apparent clearance; Ka, absorption rate constant; V1/F, apparent volume of distribution of the central compartment; V2/F, apparent volume of distribution of the peripheral compartment; Q, intercompartmental clearance; SCr, serum creatinine; NA, not applicable; %RSE, relative standard error of parameter estimate. Between-subject variability and residual (proportion) variability estimates are shown as percent coefficient of variation (% CV).
FIG 9.
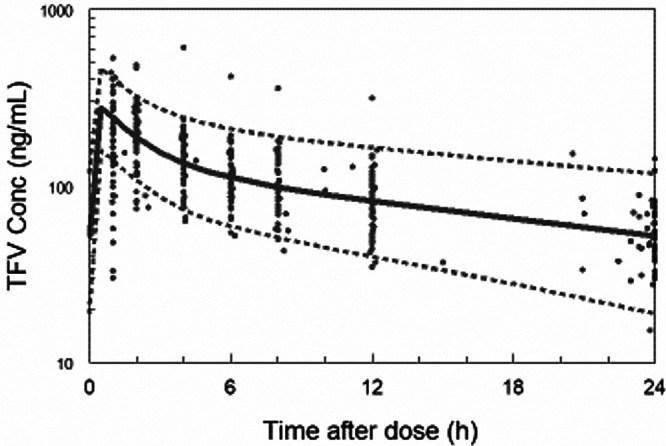
Visual predictive plot (VPC) for pregnancy, 5th, 50th, and 95th percentile.
FIG 10.
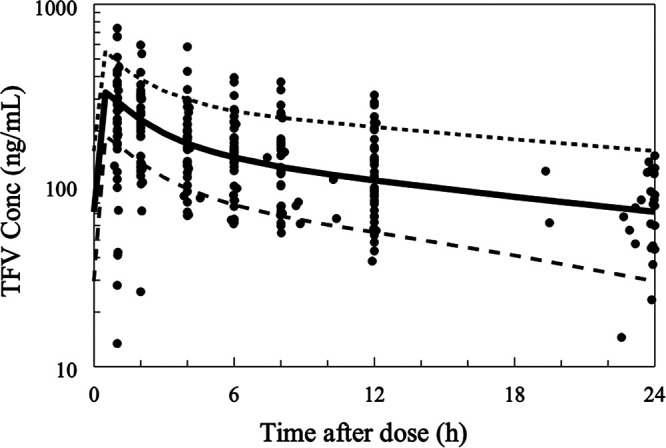
Visual predictive plot (VPC) for postpartum, 5th, 50th, and 95th percentile.
POPPK final model.
Typical values for clearance, intercompartmental clearance, and central and peripheral volumes of distribution are shown in Table 3. In the final model, the equation to estimate the apparent clearance of TFV was CL/F (liters/h) = 2.07 × (serum creatinine/0.6)0.65 × weight0.75, where serum creatinine was measured in milligrams per deciliter and weight in kilograms. The variability in the estimate of CL/F, expressed as percent coefficient of variation (% CV), was 24%, while the variability in the estimate for Vss/F was 25%. Interoccasion variability was modeled to CL/F (IOV-CL/F) and had a variability estimate of 26%.
Simulation of TDF dosage regimens and PTA.
The median concentration versus time profiles for simulated pregnant and postpartum women are shown in Fig. 11, and the proportions of TFV simulations in pregnant and postpartum women exceeding an area under the concentration-time curve (AUC) of >1.99 μg·h/ml (the 10th percentile of average TFV exposure in nonpregnant historical controls) (4) are shown in Fig. 12. While pregnancy was not an independent covariate in the final POPPK model, the greater weight and lower serum creatinine that occurred during normal pregnancy resulted in a TFV profile during pregnancy that was lower throughout the dose interval than that in postpartum women. This was associated with a lower AUC of 2.36 μg·h/ml versus 3.02 μg·h/ml and higher CL/F of 57.6 liters/h versus 44.9 liters/h in simulated pregnant versus postpartum women (Fig. 11). The probability of target attainment (PTA; exceeding AUC of >1.99 μg·h/ml) was 68%, 80%, 87%, and 93% above the target with 300 mg, 350 mg, 400 mg, and 450 mg of TDF, respectively, in pregnant women and 88%, 92%, 96%, and 98% above the target with the same doses in postpartum women (Fig. 12).
FIG 11.

Median concentration versus time profiles for simulated pregnant and nonpregnant women derived using the final population pharmacokinetic model. Solid lines represent lines of identity for pregnant women (PREG), while interrupted lines represent lines of identity for nonpregnant women.
FIG 12.
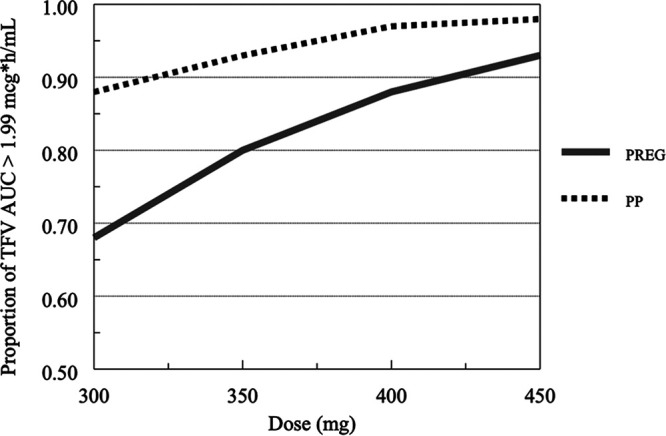
Simulations of tenovovir proportion exceeding AUC of >1.99 μg·h/ml in pregnant (PREG) and postpartum (PP) women.
DISCUSSION
Using a population PK approach, we leveraged rich, intensive TDF-derived TFV data to estimate individual PK parameters in pregnant women living with HIV. Among the investigated covariates, TFV CL/F and Vss/F both increased with increasing gestation, which is consistent with previous reports of TFV PK during pregnancy (5, 9, 10). The population PK parameters for TFV compared well with published values in the literature and demonstrated that TFV CL/F is related to renal function, reflected as serum creatinine in the final model (11–13).
A two-compartment model with first-order absorption and elimination, similar to those of Hirt et al. (8) and Benaboud et al. (9), best described the plasma concentrations of TFV in our study. Our POPPK model represents the first TDF-derived TFV pregnancy POPPK model from an intensive sampling (with paired evaluations, pregnancy and postpartum) of pregnant women living with HIV. This is important, because the use of intensive PK and paired evaluations are advantageous and can improve understanding of intra- and interindividual variability in PK parameter estimates needed for robust predictions.
Overall, the estimates for CL/F and V1/F and the between-subject variability compared well with those of previous studies. The population mean baseline V1/F of 6.37 liters/kg (446 liters for a 70-kg pregnant woman) is similar to those reported (343 to 552 liters) from other TDF-derived TFV pregnancy studies (5, 10). Furthermore, the overall variability (between-subject and interoccasion variability) of CL/F after accounting for variability in all covariates in our model was 35%. This variability in CL/F in our study was higher than the variability in the Hirt et al. TFV intrapartum and postpartum model (CL/F of 30%) and lower than the overall variability in the Benaboud et al. TFV model (CL/F of 43%). One advantage of our population PK study is the separation of CL/F variability into interoccasion (within-subject) variability in CL/F and between-subject variability in CL/F, a characteristic not reported and accounted for in other TDF-derived TFV pregnancy models. Accounting for within-subject variability in CL/F for TFV has advantages, as it explains some of the variability within subjects that cannot be explained by between-subject variability. Other factors, such as pharmacogenomic differences in single-nucleotide polymorphisms (SNPs) in the genes coding for biotransformation and enzyme transport, drug-drug interactions, and other patient-specific covariates, also may vary from one participant to another and account for additional variability.
In our study, serum creatinine had a significant influence on TFV CL/F in the final population PK model. Therefore, we investigated the influence of renal function on TDF-derived TFV PK. Tenofovir has the potential to cause nephrotoxicity (14). In fact, several studies demonstrate a mild to moderate decline in estimated glomerular filtration rate relative to the baseline in patients taking TDF-based combination antiretroviral therapy (15–17). Following oral administration of TDF, it is rapidly metabolized to TFV, which is then eliminated unchanged by a combination of glomerular filtration and active tubular secretion (18, 19). Approximately 70 to 80% of TDF-derived TFV is actively secreted from basolateral membranes of the kidneys into proximal renal tubular cells via the human organic anion transporter 1 (hOAT-1) and hOAT-3 and then extruded into the renal tubular lumen by the multidrug resistance transporter 2 (MRP-2) and MRP-4 luminal transmembrane transporters (20, 21). Therefore, increased accumulation of TFV in the renal tubular cells potentially can cause mitochondrial dysfunction and proximal tubular injury (22, 23). Thus, TDF fixed-dose antiretroviral regimens should be used with caution in patients with renal insufficiency (2). Our results showed that concomitant ritonavir use did not influence TFV PK, although decreased TFV clearance has been previously reported in nonpregnant women (24). While the inhibition of the MRP-2-mediated transport of TFV is a possible explanation for this PK interaction (19), the physiologic changes in the renal system during pregnancy resulting in a decrease in plasma TFV concentrations might explain our findings.
When TDF is used during pregnancy, it is recommended that renal function testing be done at baseline and then every 6 months (1), the same schedule should be used for nonpregnant adults, and renal dosing should be done in patients with renal insufficiency. As TFV has limited plasma protein binding (<1% bound), changes in protein binding during pregnancy is not expected to alter TFV clearance. Instead, as TFV is principally renally cleared, physiologic changes of pregnancy likely to alter TFV serum concentrations would be related to renal function. During pregnancy, renal blood flow rises by about 70 to 80% from its baseline value at 20 to 22 weeks of gestation, peaks around 32 to 34 weeks of gestation, and then falls to about 60 to 70% above prepregnancy levels toward the end of pregnancy. The glomerular filtration rate (GFR), on the other hand, rises in parallel by about 40 to 50% of its baseline values at 20 to 22 weeks, continues to increase through most of the 3rd trimester (exceeding its baseline by about 50% at 36 to 38 weeks of gestation), and then declines steadily until the time of delivery (25–27). These pregnancy-related changes result in mean serum creatinine of approximately 16%, 23%, and 20% lower in the first, second, and third trimesters of pregnancy compared to nonpregnant adults with normal kidney function (28). The significant relationship between serum creatinine (a biomarker for renal function) and TFV CL/F in the final POPPK model is likely related to these renal changes during pregnancy.
Our findings using this POPPK model are biologically and pharmacologically plausible and have important clinical implications. The results of TFV studies show that the efficacy of TDF-derived TFV was highly variable among individuals, which could be related to plasma TFV exposure (2). Morphologic and biological characteristics enabled us to explain some between-subject variability of TFV PK parameters (24% for CL/F and 25% for Vss/F), but these explain approximately half of the clinical variability of TDF-derived TFV seen during pregnancy in our model. Although interoccasion variability improves our understanding of TFV variability from this POPPK model by 26%, it cannot fully explain the variability in TDF-derived TFV exposure in pregnant women living with HIV. Other potential mechanisms, including variability in trans-membrane drug transporter mechanisms of TFV absorption, pharmacogenomic variation in drug transporters, and drug-metabolizing enzymes, need to be explored. Our results show that TFV exposure decreased by 28% during pregnancy compared to postpartum. Examining known pharmacokinetic-pharmacodynamic (PKPD) relationships of TFV (AUC and viral response) in the context of lower exposures and what a clinically relevant decrease means in relation to these targets is critical during pregnancy. A prior TFV noncompartmental PK study reported that a 25% decrease in TFV exposure was not associated with perinatal transmission or virologic failure (10). In addition, our simulated TFV AUC in pregnant women of 2.36 μg·h/ml is similar to TFV AUCs simulated from POPPK models in nonpregnant adults (2.62 μg·h/ml [29], 2.65 μg·h/ml [30], and 2.88 μg·h/ml [31]). Since TDF is available in 150-mg (lowest dose) and 300-mg (standard dose) tablets, we simulated pregnant and postpartum AUCs following the administration of TDF as 300 mg, 350 mg, 400 mg, and 450 mg of TDF. The probability of target attainment (proportion exceeding AUC of >1.99 μg·h/ml, the 10th percentile of average TFV exposure in nonpregnant historical controls) (5) was approximately 70% above this target with the standard dose (300 mg) of TDF, as shown in Fig. 12. It appears that the exposure of pregnant women to TDF doses of ≥400 mg during pregnancy and postpartum may be too high. The probability of target attainment simulations and the between-subject variability in TFV clearance of 24% suggest that the standard 300 mg of TDF is enough to reach target therapeutic thresholds for TFV during pregnancy and postpartum and should not warrant TDF dose modifications during pregnancy. A pregnancy POPPK study in pregnant and postpartum women living with hepatitis B reported similar findings and recommendations (32).
Our TFV pregnancy POPPK model has several strengths. First, our model described the PK of TFV with reasonable accuracy and precision in pregnant and postpartum patients. Second, our POPPK model examined the association between TFV concentrations and serum creatinine, which would be useful to optimize TDF dosing in pregnant women living with HIV with various degrees of renal impairment. Third, we utilized rich, intensive PK sampling data for the POPPK model, which is precise and thorough enough to define accurately the metabolic phase of drug elimination of TDF-derived TFV. Fourth, we accounted for individual within-subject variability PK parameters, including interoccasion variability. Studies have shown that interoccasion variability may change randomly between study occasions, and ignoring such interoccasion variability may result in biased population parameter estimates, a high incidence of statistically significant spurious period effects, and a falsely optimistic impression of the potential value of therapeutic drug monitoring (33).
Our study had limitations. We do not have first trimester PK data, so it was not possible to incorporate first trimester changes in TFV PK into our model. However, we do not think the lack of first trimester data had a substantial effect on our results, because pregnancy physiology usually changes little in the first trimester. We did not collect data on TFV-DP, the active moiety of TFV, so we could not investigate the relationship between plasma TDF-derived TFV and intracellular TFV-DP concentrations in our POPPK model. It would have been ideal to be able to predict intracellular TFV-DP concentrations, as the antiretroviral activity of TDF is a function of intracellular TFV-DP levels (13). The timing of TDF dose to meals was not controlled for in our POPPK model. Studies have demonstrated that TDF absorption is sensitive to high-fat diet, and differences in rate of absorption can account for PK differences in patients taking TDF, with Ka, maximum concentration of drug in serum (Cmax), and AUC of TFV about 2 to 3 times higher in fed compared to fasted participants (25% bioavailability in the fasting state and 39% bioavailability with a high-fat diet) (24). Using postpregnancy weight for missing prepregnancy weights potentially can bias POPPK analysis, because prepregnancy weight relies on maternal self-report, often ascertained by recalled weight. On average, women underreport their weight by approximately 1 to 3 kg. However, researchers and clinicians rely on self-reported prepregnancy weight, because most women do not have a preconception visit where weight is measured and also because it saves time in clinical and research settings.
In conclusion, the POPPK parameters of TDF-derived TFV in pregnant and postpartum women living with HIV was best described using a two-compartment model with first-order absorption and elimination. Our model suggests that changes in TDF-derived TFV PK can be predicted by clinical factors and, thus, any dose modifications for TFV during pregnancy and postpartum should be based on renal function and weight during the second and third trimesters of pregnancy as well as postpartum. While weight and serum creatinine accounted for variability in CL/F unexplained by between-subject variability for CL/F (26%), these typical changes in BSV do not need dose adjustments in TDF during pregnancy and postpartum, but more extreme weights or renal function differences may warrant dose adjustments in pregnant and postpartum women. In situations where dose adjustments are warranted for TDF, switching to other antiretrovirals would be another alternative. TDF is a cornerstone drug used in many antiretroviral fixed-dose combinations in pregnant women, and identification of other sources of variability would be vital to improving its safety and efficacy. Larger clinical studies with a range of doses exploring the impact of creatinine clearance and gene polymorphisms of TFV transporters would be critical.
MATERIALS AND METHODS
Data were collected as part of International Maternal Pediatric Adolescent AIDS Clinical Trials (IMPAACT) protocol P1026s, a multicenter, nonblinded, prospective phase IV study of the pharmacokinetics and safety of selected antiretrovirals (ARVs) in HIV-infected pregnant women. This analysis included pregnant women receiving 300 mg of tenofovir disoproxil fumarate (TDF) in several protocol arms. The study protocol, the informed consent documents, and all subsequent modifications were reviewed and approved by the local institutional review board (IRB)/Ethics Committee responsible for oversight of the study. The study followed all relevant human subject research guidelines. All participants provided signed informed consent before participation, and the study was registered at ClinicalTrials.gov (ClinicalTrials registration no. NCT00042289).
Pregnant women living with HIV were eligible for enrollment in the second and third trimesters if they were receiving TDF 300 mg once daily as part of clinical care. All ARVs were prescribed by the participants’ clinical care providers and dispensed by local pharmacies per local standard of care. Maternal exclusion criteria were current use of medications known to interfere with tenofovir metabolism (including acyclovir, cidofovir, ganciclovir, valacyclovir, valganciclovir, aminoglycosides, and nonsteroidal anti-inflammatory drugs), presence of renal disease or liver disease, and other clinical or laboratory toxicities that, per site investigators, would require a change in the antiretroviral regimen.
Clinical and laboratory data.
Maternal demographic and clinical information was abstracted from the medical record, including maternal age, gestational age, serum creatinine, serum albumin, maternal race and ethnicity, weight, and concomitant medications. Plasma HIV-1 RNA assays were performed locally. Study mothers and infants were monitored through 6 months after delivery. Gestational age at the time of delivery, birth weight, and HIV infection status data were collected from the infant’s medical record. Physical examinations were performed on neonates after delivery, and infant laboratory evaluations were performed as clinically indicated. Maternal clinical and laboratory toxicities were assessed through clinical and laboratory evaluations on each pharmacokinetic sampling day, at delivery, and at 24 weeks postpartum. Any additional toxicities noted as part of clinical care were also recorded. The study team reviewed toxicity reports on monthly conference calls, although each participant’s physician was responsible for toxicity management. The Division of AIDS (DAIDS) Table for Grading the Severity of Adult and Pediatric Adverse Events (August 1992) was used to grade adverse events for study participants. All toxicities were monitored through resolution or 24 weeks postpartum.
Sample collection and pharmacokinetic sampling schedule.
Intensive PK samples were collected at 20 to 26 weeks gestation for 2nd trimester PK evaluation, at 30 to 36 weeks gestation for 3rd trimester PK evaluation, and between 2 and 12 weeks after delivery for postpartum evaluation. Plasma samples were drawn immediately prior to an observed dose and at 1, 2, 4, 6, 8, 12, and 24 h postdose. Paired maternal and cord blood samples were collected at delivery. All plasma samples were collected at steady state. The steady-state PK profiles collected included 688 plasma tenofovir concentrations from 46 women during the second trimester (n = 7), third trimester (n = 41), and postpartum (n = 38) (Table 1).
Bioanalytical methods.
Plasma samples collected for PK assays were stored at ≤−70°C until analysis. Tenofovir concentrations were measured by a previously validated, liquid chromatography-tandem mass spectrometry (LC‐MS/MS) method (4). The linear range was 10 to 1,500 ng/ml, with a lower limit of detection for TFV of 10 ng/ml. Accuracy and precision were within ±20% at 10 ng/ml and ±15% at other quality control concentrations.
Covariate data.
The potential impact of clinical covariates, such as maternal age, trimester of pregnancy, maternal weight gain, gestational age, serum creatinine, serum albumin, and maternal race and ethnicity were evaluated for inclusion in the model. Baseline covariate parameters were obtained from the medical records and defined using the last recorded values before the first dose of TDF in pregnant women. There were 12 missing covariate laboratory values, and values across all subjects were used for them. For missing prepregnancy weight, the postpartum weight was used.
Population PK base structural model development.
Population pharmacokinetic analyses were constructed in the nonlinear mixed effects modeling (NONMEM) software (version 7.4; ICON Development Solutions, Ellicott City, MD). R software (version 3.0.1) was used for data set construction, graphical inspection, statistical analysis, model diagnostics, and final model development, and Wings for NONMEM 7.4 was used for bootstrap analysis. The first-order conditional estimation method with interaction (FOCE-I) was used for model fitting.
Multiple absorption mechanisms, including first-order, zero-order, and sequential zero- and first-order absorption with and without an absorption lag time, were modeled using a one-compartment and then a two-compartment approach that assumed plasma as the central compartment, from where blood samples for TFV concentration measurements are obtained and TFV is eliminated, and a peripheral compartment representative of less well-perfused tissues, such as muscle and fat. An exponential model was used to describe interindividual (between-subject) variability. All PK parameters were assumed to be log-normally distributed around the population mean value, θ (equation 1):
| (1) |
where Pa is the estimate of the PK parameter (e.g., TFV clearance) in individual a, θ is the population typical mean of the PK parameter, and ηa is the between-subject (interindividual) variability, describing the deviation from the typical population parameter for the a-th participant on TDF. Thus, as described in equation 1, the value is a function of the typical population value for the PK parameter and the random effect of the individual variance from the typical population mean. Interoccasion variability (IOV) was also assessed for inclusion in the model.
Power, proportional, additive, and combined (additive and proportional) residual error models were also evaluated. Competing PK models were selected based on minimization of objective function value, precision of parameter estimates, visual inspection of goodness-of-fit plots, and physiologically reasonable and/or statistically significant covariates. Model discrimination was based on relative objective function values (OFVs) computed in NONMEM as −2× log likelihood, and the number of estimable parameters (p) in the model was determined as Akaike information criterion (AIC) = OFV + 2p. The AIC is a test used to evaluate how well a model fits the data it describes. The AIC penalizes models that use more independent parameters to prevent overfitting.
Covariate model development.
The covariate model was developed after the basic model was constructed. Body weight (WT) was incorporated into the model with allometric scaling before assessment of other covariates. Standard allometric exponents were utilized, i.e., WT0.75 for apparent clearance (CL/F) and apparent intercompartmental clearance (Q/F) and isometric exponent (WT1.0) for central compartment (V1/F) and the peripheral compartment (V2/F). The sum of V1/F and V2/F in this two-compartment model equals the apparent volume of distribution at steady state (Vss/F). The impact of weight was evaluated as prepregnancy weight, current weight, and a hybrid approach with different influences of prepregnancy weight and weight gain. The hybrid approach included additional scalars added to the weight gain for CL/F and Vss/F such that the weight gain of pregnancy would not have the same impact as the prepregnancy weight. While the hybrid approach worked best in the initial analysis, the bootstrap of the final hybrid model showed unacceptable 95% confidence intervals (CIs) and was abandoned in favor of total weight, which performed better than prepregnancy weight. Covariate analysis was performed on CL/F and Vss/F, in a stepwise manner, using the changes in the objective function value, a reduction of ≥3.84 (P < ∼0.05) significance threshold for forward step, followed by a reduction of ≥10 (P <∼0.001) significance threshold for backward elimination. Biologically plausible covariates were evaluated, including maternal age, trimester of pregnancy, serum creatinine, albumin, gestational age, and concomitant ritonavir use. All potential covariates identified in the forward screen were combined into a single model to start the backward analysis. Using the resulting new model, individual covariates were removed one by one. If the objective function increased with removal of covariate by less than 10, it was eliminated from the model. The process was repeated until no additional factor could be removed without significant worsening of the model.
Continuous covariates (e.g., gestational age and serum creatinine) were modeled using a median-normalized power model (equation 2), where Pa represents the population prediction of the parameter, COVi represents the value of the i-th continuous covariate, COVmedian represents the median value of the covariate in the population, θ1 represents the population typical value of the parameter, and θ2 represents an estimated parameter describing the fixed effect of the covariate on the PK parameter.
| (2) |
Categorical covariates (e.g., ritonavir use; yes or no) were described using a power model (equation 3), where variables are indicated as binary (0 or 1). θ1 represents the parameter estimate for an individual with COV coded as 0, and θ2 represents the change in PK parameter relative to when COV is equal to 1.
| (3) |
After identifying significant covariates, additional assessment of the random error structure was investigated.
The ability of body size models to reduce the unexplained between-subject variability and improve the goodness of fit of the model was also explored. To investigate the effect of weight, based on visual inspection, goodness-of-fit plots, likelihood ratio testing (by a decrease in OFV), and model stability assessment, the covariate relationships between CL/F and Vss/F that includes an adjustment for the weight of each subject individually using normalization to weight and allometric scaling with an exponent (θ2) were defined as the following (equations 4 and 5):
| (4) |
| (5) |
where CL/Fi represents the body weight-normalized apparent clearance of the i-th individual, Vss/Fi represents the body weight-normalized apparent volume of distribution of the i-th individual, θ1 represents the typical values for apparent clearance, θ2 represents the allometric power exponent for clearance, θ3 represents the typical values for apparent volume of distribution, θ4 represents the allometric power exponent for apparent volume of distribution, ηi represents the between-subject variability random effect with a mean of 0 and variance of w2, WT represents the body weight of the i-th individual, and WTmedian represents the median weight. Weight was measured in kilograms. The body size model best described and fit the PK data.
Model checking and evaluation, diagnosing errors, validation, and reliability testing.
Goodness-of-fit plots generated for model evaluation and verification included observed versus predicted data plots and weighted residuals versus population predicted values for final model evaluation. Bootstrapping of the final model was performed by creating 1,000 simulated data sets by resampling with replacement from the original data set. Model parameters based on the original data set were compared to the bootstrap results. Visual predictive check (VPC) plots were generated on the basis of the 1,000 simulations. The VPC plot showed the 10th, 50th, and 90th percentiles of observed data over time calculated from 1,000 Monte Carlo samples (simulated using the model, the parameter estimates, and the design of the data set). A 30% variability in weight and serum creatinine among simulated subjects was used.
Simulation of dosage regimens.
A Monte Carlo simulation was performed using the final PK model with covariates to predict the distribution of plasma TFV concentrations. Four dosage regimens were studied, 300 mg, 350 mg, 400 mg, and 450 mg of TDF, with administration every 24 h. We simulated the profiles of 1,000 pregnant and 1,000 postpartum women with a set of covariates resampled among the observed covariates of included patients and a vector of random effects drawn from the estimated distribution. The concentration-time profiles at steady state for the four TDF dosage regimens were simulated throughout pregnancy and continued until 12 weeks postpartum based on the distribution of clinical characteristics from our study. The proportion of TFV simulations in pregnant and postpartum women exceeding an AUC of >1.99 μg·h/ml and the summary statistics (median, 2.5th, and 97.5th percentiles) were obtained.
ACKNOWLEDGMENTS
We thank the women who participated in the protocol, the staff of the participating IMPAACT centers, P1026s Protocol Team Members, and Lane R. Bushman and Brian L. Robbins from the Antiviral Pharmacology Laboratory for performing the tenofovir plasma concentration assays. In addition to the coauthors, P1026s Protocol Team members include Francesca Aweeka, Emily Barr, Nantasak Chotivanich, Tim Roy Cressey, Lisa M. Frenkel, Amita Gupta, Amy Jennings, Gonzague Jourdain, Regis Kreitchmann, Rita Patel, Kittipong Rungruengthanakit, David Shapiro, and Pra‐ornsuda Sukrakanchana. Participating sites and site personnel include University of Southern California Los Angeles NICHD Clinical Research Site (USC LA NICHD CRS) (Andrea Kovacs, James Homans, LaShonda Spencer, and Francoise Kramer); Texas Children's Hospital CRS (Shelley Buschur, Hunter Hammill, II, Mary E. Paul, and Chivon McMullen‐Jackson); Seattle Children's Hospital CRS (Ann Melvin, Corry Venema‐Weiss, Jenna Lane, and Jane Hitti; supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under award number UL1TR000423); St. Jude/University of Tennessee Health Science Center CRS (Katherine Knapp, Edwin Thorpe, Jr, L. Jill Utech, and Nina Sublette); University of California, San Francisco NICHD CRS (Diane Wara, Deborah Cohan, and Nicole Tilton); Bronx‐Lebanon Hospital IMPAACT CRS (Mary Elizabeth Vachon, Mirza Mahboobullah Baig, Murli Udharam Purswani, and Jenny Gutierrez); Western New England Maternal Pediatric Adolescent AIDS CRS (Katherine Luzuriaga, Sharon Cormier, and Margaret McManus); University of California San Diego Maternal, Child, and Adolescent HIV CRS (Andrew Hull, Mary Caffery, Kimberly Norris, and Stephen A. Spector); Harbor UCLA Medical Center NICHD CRS (Margaret A. Keller, Susan Ballagh, Judy Hayes, and Yolanda Gonzalez; supported by the National Center for Advancing Translational Sciences through UCLA CTSI grant UL1TR000124); University of Colorado Denver NICHD CRS (Emily Barr, Tara Kennedy, Alisa Katai, and Jenna Wallace; NIH/NCATS Colorado CTSI grant number UL1 TR000154); Rush University Cook County Hospital, Chicago NICHD CRS (James B. McAuley, Helen Cejtin, Maureen McNichols, and Julie Schmidt); University of Maryland Baltimore NICHD CRS (Douglas Watson, Judy Ference, and Corinda Hilyard; supported by the University of Maryland Clinical Translational Science Institute and the University of Maryland General Clinical Research Center); University of Alabama Birmingham NICHD CRS (Marilyn Crain, Tina Y. Simpson, and Alan T. N. Tita); University of Puerto Rico Pediatric HIV/AIDS Research Program CRS (Irma L. Febo, Carmen D. Zorrilla, Vivian Tamayo‐Agrait, and Ruth Santos).
Overall support for the International Maternal Pediatric Adolescent AIDS Clinical Trials Network (IMPAACT) was provided by the National Institute of Allergy and Infectious Diseases (NIAID) with cofunding from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) and the National Institute of Mental Health (NIMH), all components of the National Institutes of Health (NIH), under award numbers UM1AI068632 (IMPAACT LOC), UM1AI068616 (IMPAACT SDMC), and UM1AI106716 (IMPAACT LC), and by NICHD contract number HHSN275201800001I. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
REFERENCES
- 1.Panel on Treatment of Pregnant Women with HIV Infection and Prevention of Perinatal Transmission. 2020. Recommendations for the use of antiretroviral drugs in pregnant women with HIV infection, and interventions to reduce perinatal HIV transmission in the United States–August 26. https://clinicalinfo.hiv.gov/sites/default/files/guidelines/documents/PerinatalGL.pdf. Accessed 18 September 2020.
- 2.Gilead. 2004. TRUVADA (emtricitabine and tenofovir disoproxil fumarate) tablets for oral use—drug label. https://www.gilead.com/%7E/media/files/pdfs/medicines/hiv/truvada/truvada_pi.pdf. Accessed 16 September 2020.
- 3.Durand-Gasselin L, Van Rompay KK, Vela JE, Henne IN, Lee WA, Rhodes GR, Ray AS. 2009. Nucleotide analogue prodrug tenofovir disoproxil enhances lymphoid cell loading following oral administration in monkeys. Mol Pharm 6:1145–1151. doi: 10.1021/mp900036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Delahunty T, Bushman L, Robbins B, Fletcher CB. 2009. The simultaneous assay of tenofovir and emtricitabine in plasma using LC/MS/MS and isotopically labeled internal standards. J Chromatogr B Analyt Technol Biomed Life Sci 877:1907–1914. doi: 10.1016/j.jchromb.2009.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Best BM, Burchett S, Li H, Stek A, Hu C, Wang J, Hawkins E, Byroads M, Watts DH, Smith E, Fletcher CV, Capparelli EV, Mirochnick M, International Maternal Pediatric and Adolescent AIDS Clinical Trials (IMPAACT) P1026s Team. 2015. Pharmacokinetics of tenofovir during pregnancy and postpartum. HIV Med 16:502–511. doi: 10.1111/hiv.12252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nishijima T, Komatsu H, Gatanaga H, Aoki T, Watanabe K, Kinai E, Honda H, Tanuma J, Yazaki H, Tsukada K, Honda M, Teruya K, Kikuchi Y, Oka S. 2011. Impact of small body weight on tenofovir-associated renal dysfunction in HIV-infected patients: a retrospective cohort study of Japanese patients. PLoS One 6:e22661. doi: 10.1371/journal.pone.0022661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suzuki S, Nishijima T, Kawasaki Y, Kurosawa T, Mutoh Y, Kikuchi Y, Gatanaga H, Oka S. 2017. Effect of tenofovir disoproxil fumarate on incidence of chronic kidney disease and rate of estimated glomerular filtration rate decrement in HIV-1-infected treatment-naïve Asian patients: results from 12-year observational cohort. AIDS Patient Care STDS 31:105–112. doi: 10.1089/apc.2016.0286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hirt D, Urien S, Ekouevi DK, Rey E, Arrive E, Blanche S, Amani-Bosse C, Nerrienet E, Gray G, Kone M, Leang SK, McIntyre J, Dabis F, Treluyer JM, ANRS 12109. 2009. Population pharmacokinetics of tenofovir in HIV-1-infected pregnant women and their neonates (ANRS 12109). Clin Pharmacol Ther 85:182–189. doi: 10.1038/clpt.2008.201. [DOI] [PubMed] [Google Scholar]
- 9.Benaboud S, Hirt D, Launay O, Pannier E, Firtion G, Rey E, Bouazza N, Foissac F, Chappuy H, Urien S, Treluyer JM. 2012. Pregnancy-related effects on tenofovir pharmacokinetics: a population study with 186 women. Antimicrob Agents Chemother 56:857–862. doi: 10.1128/AAC.05244-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Colbers AP, Hawkins DA, Gingelmaier A, Kabeya K, Rockstroh JK, Wyen C, Weizsäcker K, Sadiq ST, Ivanovic J, Giaquinto C, Taylor GP, Moltó J, Burger DM, PANNA Network. 2013. The pharmacokinetics, safety and efficacy of tenofovir and emtricitabine in HIV-1-infected pregnant women. AIDS 27:739–748. doi: 10.1097/QAD.0b013e32835c208b. [DOI] [PubMed] [Google Scholar]
- 11.Jullien V, Tréluyer JM, Rey E, Jaffray P, Krivine A, Moachon L, Lillo-Le Louet A, Lescoat A, Dupin N, Salmon D, Pons G, Urien S. 2005. Population pharmacokinetics of tenofovir in human immunodeficiency virus-infected patients taking highly active antiretroviral therapy. Antimicrob Agents Chemother 49:3361–3366. doi: 10.1128/AAC.49.8.3361-3366.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greene SA, Chen J, Prince HMA, Sykes C, Schauer AP, Blake K, Nelson JAE, Gay CL, Cohen MS, Dumond JB. 2019. Population modeling highlights drug disposition differences between tenofovir alafenamide and tenofovir disoproxil fumarate in the blood and semen. Clin Pharmacol Ther 106:821–830. doi: 10.1002/cpt.1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baheti G, Kiser JJ, Havens PL, Fletcher CV. 2011. Plasma and intracellular population pharmacokinetic analysis of tenofovir in HIV-1-infected patients. Antimicrob Agents Chemother 55:5294–5299. doi: 10.1128/AAC.05317-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fernandez-Fernandez B, Montoya-Ferrer A, Sanz AB, Sanchez-Niño MD, Izquierdo MC, Poveda J, Sainz-Prestel V, Ortiz-Martin N, Parra-Rodriguez A, Selgas R, Ruiz-Ortega M, Egido J, Ortiz A. 2011. Tenofovir nephrotoxicity: 2011 update. AIDS Res Treat 2011:354908. doi: 10.1155/2011/354908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jose S, Hamzah L, Campbell LJ, Hill T, Fisher M, Leen C, Gilson R, Walsh J, Nelson M, Hay P, Johnson M, Chadwick D, Nitsch D, Jones R, Sabin CA, Post FA, UK Collaborative HIV Cohort Study Steering Committee. 2014. Incomplete reversibility of estimated glomerular filtration rate decline following tenofovir disoproxil fumarate exposure. J Infect Dis 210:363–373. doi: 10.1093/infdis/jiu107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fafin C, Pugliese P, Durant J, Mondain V, Rahelinirina V, De Salvador F, Ceppi C, Perbost I, Rosenthal E, Roger PM, Cua E, Dellamonica P, Esnault V, Pradier C, Moranne O. 2012. Increased time exposure to tenofovir is associated with a greater decrease in estimated glomerular filtration rate in HIV patients with kidney function of less than 60 ml/min/1.73 m2. Nephron Clin Pract 120:c205–c214. doi: 10.1159/000342377. [DOI] [PubMed] [Google Scholar]
- 17.Bonjoch A, Echeverría P, Perez-Alvarez N, Puig J, Estany C, Clotet B, Negredo E. 2012. High rate of reversibility of renal damage in a cohort of HIV-infected patients receiving tenofovir-containing antiretroviral therapy. Antiviral Res 96:65–69. doi: 10.1016/j.antiviral.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 18.Ray AS, Fordyce MW, Hitchcock MJ. 2016. Tenofovir alafenamide: a novel prodrug of tenofovir for the treatment of human immunodeficiency virus. Antiviral Res 125:63–70. doi: 10.1016/j.antiviral.2015.11.009. [DOI] [PubMed] [Google Scholar]
- 19.Ray AS, Cihlar T, Robinson KL, Tong L, Vela JE, Fuller MD, Wieman LM, Eisenberg EJ, Rhodes GR. 2006. Mechanism of active renal tubular efflux of tenofovir. Antimicrob Agents Chemother 50:3297–3304. doi: 10.1128/AAC.00251-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ho ES, Lin DC, Mendel DB, Cihlar T. 2000. Cytotoxicity of antiviral nucleotides adefovir and cidofovir is induced by the expression of human renal organic anion transporter 1. J Am Soc Nephrol 11:383–393. [DOI] [PubMed] [Google Scholar]
- 21.van Aubel RA, Smeets PH, Peters JG, Bindels RJ, Russel FG. 2002. The MRP4/ABCC4 gene encodes a novel apical organic anion transporter in human kidney proximal tubules: putative efflux pump for urinary cAMP and cGMP. J Am Soc Nephrol 13:595–603. [DOI] [PubMed] [Google Scholar]
- 22.Herlitz LC, Mohan S, Stokes MB, Radhakrishnan J, D'Agati VD, Markowitz GS. 2010. Tenofovir nephrotoxicity: acute tubular necrosis with distinctive clinical, pathological, and mitochondrial abnormalities. Kidney Int 78:1171–1177. doi: 10.1038/ki.2010.318. [DOI] [PubMed] [Google Scholar]
- 23.Del Palacio M, Romero S, Casado JL. 2012. Proximal tubular renal dysfunction or damage in HIV-infected patients. AIDS Rev 14:179–187. [PubMed] [Google Scholar]
- 24.Kearney BP, Flaherty JF, Shah J. 2004. Tenofovir disoproxil fumarate: clinical pharmacology and pharmacokinetics. Clin Pharmacokinet 43:595–612. doi: 10.2165/00003088-200443090-00003. [DOI] [PubMed] [Google Scholar]
- 25.Costantine MM. 2014. Physiologic and pharmacokinetic changes in pregnancy. Front Pharmacol 5:65. doi: 10.3389/fphar.2014.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheung KL, Lafayette RA. 2013. Renal physiology of pregnancy. Adv Chronic Kidney Dis 20:209–214. doi: 10.1053/j.ackd.2013.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lindheimer MD, Davison JM, Katz AI. 2001. The kidney and hypertension in pregnancy: twenty exciting years. Semin Nephrol 21:173–189. doi: 10.1053/snep.2001.20937. [DOI] [PubMed] [Google Scholar]
- 28.Wiles K, Bramham K, Seed PT, Nelson-Piercy C, Lightstone L, Chappell LC. 2019. Serum creatinine in pregnancy: a systematic review. Kidney Int Rep 4:408–419. doi: 10.1016/j.ekir.2018.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boffito M, Pozniak A, Kearney BP, Higgs C, Mathias A, Zhong L, Shah J. 2005. Lack of pharmacokinetic drug interaction between tenofovir disoproxil fumarate and nelfinavir mesylate. Antimicrob Agents Chemother 49:4386–4389. doi: 10.1128/AAC.49.10.4386-4389.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blum MR, Chittick GE, Begley JA, Zong J. 2007. Steady-state pharmacokinetics of emtricitabine and tenofovir disoproxil fumarate administered alone and in combination in healthy volunteers. J Clin Pharmacol 47:751–759. doi: 10.1177/0091270007300951. [DOI] [PubMed] [Google Scholar]
- 31.Ramanathan S, Shen G, Cheng A, Kearney BP. 2007. Pharmacokinetics of emtricitabine, tenofovir, and GS-9137 following coadministration of emtricitabine/tenofovir disoproxil fumarate and ritonavir-boosted GS-9137. J Acquir Immune Defic Syndr 45:274–279. doi: 10.1097/QAI.0b013e318050d88c. [DOI] [PubMed] [Google Scholar]
- 32.Cressey TR, Harrison L, Achalapong J, Kanjanavikai P, Patamasingh Na Ayudhaya O, Liampongsabuddhi P, Siriwachirachai T, Putiyanun C, Suriyachai P, Tierney C, Salvadori N, Chinwong D, Decker L, Tawon Y, Murphy TV, Ngo-Giang-Huong N, Siberry GK, Jourdain G, Sangsawang S, Jittayanun K, Matanasarawut W, Somsamai R, Suriyachai P, Nasomchai P, Achalapong J, Chanta C, Putiyanun C, Ek-Isariyaphorn R, Buranabanjasatean S, Yuthavisuthi P, Ngampiyaskul C, Kanjanavikai P, Phanomcheong S, Chotivanich N, Hongsiriwon S, Limtrakul A, Suwannarat A, Luvira A, Na Ayudhaya OP, Prommas S, Layangool P, Siriwachirachai T, Srirompotong U, Varadisai S, Krikajornkitti S, Sabsanong P, Wongngam P, Liampongsabuddhi P, Pongdetudom K, Puernngooluerm P, et al. 2018. Tenofovir exposure during pregnancy and postpartum in women receiving tenofovir disoproxil fumarate for the prevention of mother-to-child transmission of hepatitis B virus. Antimicrob Agents Chemother 62:e01686-18.doi: 10.1128/AAC.01686-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karlsson MO, Sheiner LB. 1993. The importance of modeling interoccasion variability in population pharmacokinetic analyses. J Pharmacokinet Biopharm 21:735–750. doi: 10.1007/BF01113502. [DOI] [PubMed] [Google Scholar]