

Bacteria have evolved distinct molecular mechanisms as a defense against oxidative stress. The foremost regulator of the oxidative stress response has been found to be OxyR.
KEYWORDS: osaR, PA0056, dsbM, oxyR, oxidative stress regulation, antibiotic tolerance regulation, antibiotic resistance regulation
ABSTRACT
Bacteria have evolved distinct molecular mechanisms as a defense against oxidative stress. The foremost regulator of the oxidative stress response has been found to be OxyR. However, the molecular details of regulation upstream of OxyR remain largely unknown and need further investigation. Here, we characterize an oxidative stress and antibiotic tolerance regulator, OsaR (PA0056), produced by Pseudomonas aeruginosa. Knocking out of osaR increased bacterial tolerance to aminoglycoside and β-lactam antibiotics, as well as to hydrogen peroxide. Expression of the oxyR regulon genes oxyR, katAB, and ahpBCF was increased in the osaR mutant. However, the OsaR protein does not regulate the oxyR regulon genes through direct binding to their promoters. PA0055, osaR, PA0057, and dsbM are in the same gene cluster, and we provide evidence that expression of those genes involved in oxidant tolerance is controlled by the binding of OsaR to the intergenic region between osaR and PA0057, which contain two divergent promoters. The gene cluster is also regulated by PA0055 via an indirect effect. We further discovered that OsaR formed intramolecular disulfide bonds when exposed to oxidative stress, resulting in a change of its DNA binding affinity. Taken together, our results indicate that OsaR is inactivated by oxidative stress and plays a role in the tolerance of P. aeruginosa to aminoglycoside and β-lactam antibiotics.
INTRODUCTION
Pseudomonas aeruginosa is an important opportunistic Gram-negative pathogen that can cause serious infections, especially in immunocompromised individuals. Furthermore, infections caused by P. aeruginosa result in increased mortality rates for patients with cystic fibrosis or cancer (1). However, current methods to cure these infections in clinical practice are limited because of the resistance of P. aeruginosa to antibiotics (2).
A common bactericidal mechanism involving production of reactive oxygen species (ROS) is shared by three kinds of antibiotics (3, 4), namely, β-lactams, aminoglycosides (AGs), and quinolones. Blocked cell wall synthesis caused by β-lactams causes increased intracellular glucosamine-6-phosphate levels (5), resulting in increased glycolytic flux, providing substrates for ROS generation (6). Binding of AGs to ribosomes results in protein mistranslation (7–9), which triggers envelope stress via envelope (Cpx) and redox-responsive (Arc) two-component systems. Accordingly, membrane-coupled energy generation is accelerated, leading to ROS accumulation (10, 11). Some research showed no hypersensitivity for strains with ROS-related mutations in sodAB, oxyR, or recA, compared with the wild-type (WT) strain, in Escherichia coli, and there is no survival difference with ampicillin or kanamycin treatment under aerobic or anaerobic conditions (12–14). Therefore, the conclusion that ROS plays a role in antimicrobial lethality is still under debate at present.
Previous studies indicated that a mutation in dsbM, which encodes a disulfide oxidoreductase, contributes to a high level of resistance to several AGs in P. aeruginosa (15). The crystal structures revealed that DsbM consists of a thioredoxin domain containing the active site CXXC motif and a lid domain (16). Moreover, previous studies confirmed that DsbM and OxyR interact (17). The oxidation state of OxyR can be reduced by DsbM, and reduced OxyR cannot positively modulate the expression of the antioxidant genes katAB and ahpBCF to counteract ROS originating from antibiotic exposure. The oxidized DsbM is glutathionylated by glutathione (GSH) (16). Thus, DsbM is involved in AG resistance in P. aeruginosa.
The osaR gene is a conserved gene in P. aeruginosa strains, including PAO1, PAK, and PA14. Similar to OxyR, the transcriptional regulator OsaR belongs to the LysR family. Recent work established that there is a direct interaction between the OsaR and DsbM proteins (18). Therefore, OsaR might be involved in oxidative stress and resistance to AGs in P. aeruginosa like DsbM.
Here, we provide evidence that OsaR is involved in the tolerance of P. aeruginosa to AGs and β-lactams. Our results suggest that OsaR functions as a redox regulator that inhibits transcription of oxyR and the response to oxidative stress. In addition, OsaR regulates the operon consisting of PA0055, osaR, PA0057, and dsbM.
RESULTS
Deletion of osaR increases the tolerance of P. aeruginosa to AGs and β-lactams.
Deletion of osaR had no effect on the PAK growth rate (see Fig. S1 in the supplemental material). The P. aeruginosa WT (PAK), osaR mutant, and osaR overexpression (56OE) strains were subjected to drug sensitivity tests to compare their susceptibility to AGs and β-lactams. Table 1 provides the experimental data on the MICs of AGs, which indicate that the osaR mutant strain displays greater tolerance to several AGs. The gentamicin, kanamycin, streptomycin, and neomycin MIC values of the osaR mutant were largely 2-fold higher than those displayed by the WT strain. In addition, when osaR was overexpressed, the AG MICs of the 56OE strain were reduced, with the neomycin MIC reaching one-half that of the WT strain.
TABLE 1.
AG MICs for P. aeruginosa PAK, ΔosaR, and 56OE strains
| Bacterial strain | Relevant genotype | MIC (g liter−1) of: |
|||
|---|---|---|---|---|---|
| Gentamycin | Streptomycin | Kanamycin | Neomycin | ||
| PAK | WT | 5 | 40 | 80 | 64 |
| ΔosaR | ΔosaR | 10 | 80 | 160 | 128 |
| PAK/pUCP19 | WT | 5 | 40 | 80 | 64 |
| 56OE | WT/osaR | 3.2 | 25.6 | 68 | 32 |
Drug sensitivity tests also indicated that the osaR mutant displayed increased MIC values for the β-lactams ampicillin, carbenicillin, and ceftazidime (Table 2). To corroborate these results, we performed a carbenicillin killing assay (Fig. 1). The WT strain and osaR mutant were grown to an optical density (OD600) of 0.4 and treated with a concentration of carbenicillin of 320 μg ml−1. Figure 1 shows that the osaR mutant displayed less susceptibility than the WT strain. The susceptibility of the osaR mutant and WT strain was also investigated for several non-AGs and non-β-lactam antibiotics, but no difference between the two strains was observed (Table S3). In conclusion, the osaR mutant strain displayed greater AG and β-lactam tolerance than the WT strain, suggesting that osaR is involved in tolerance to these two types of antibiotics.
TABLE 2.
β-Lactam antibiotic MICs for P. aeruginosa PAK, ΔosaR, and 56OE strains
| Bacterial strain | Relevant genotype | MIC (g liter−1) of: |
||||
|---|---|---|---|---|---|---|
| Imipenem | Meropenem | Ampicillin | Carbenicillin | Ceftazidime | ||
| PAK | WT | 2 | 1 | 4 × 103 | 32 | <0.8 |
| ΔosaR | ΔosaR | 2 | 1 | 8 × 103 | 40 | 1.9 |
| PAK/pUCP19 | WT | 2 | 1 | |||
| 56OE | WT/pUCP19-osaR | 1 | 0.3 | |||
FIG 1.
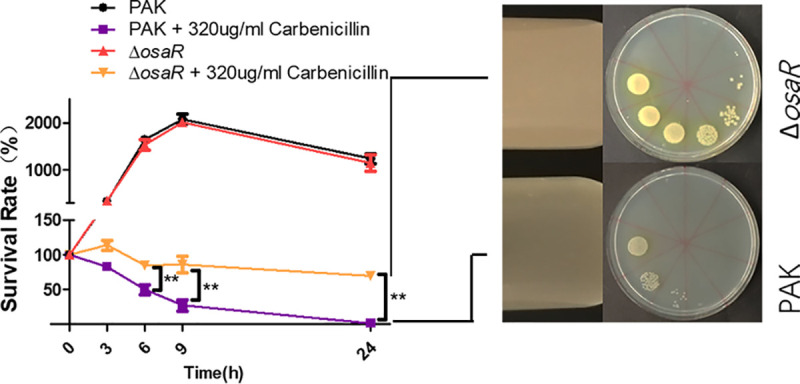
Role of OsaR in the bacterial tolerance to carbenicillin. Bacteria were treated with carbenicillin at an OD600 of 0.4 in LB medium at 37°C. Bacteria were collected at the indicated time points, and the CFU were determined by serial dilution (1:10) and plating. Images on the right show the growth state of PAK and ΔosaR strains in tubes after exposure to carbenicillin (320 μg ml−1) for 24 h; the treated cultures were then dropped on LB plates by serial dilution (1:10). All data were obtained from three independent experiments with at least three replicates. Error bars represent standard deviations (SDs). **, P < 0.01 by Student's t test.
Mutation of osaR reduces ROS accumulation and increases tolerance of P. aeruginosa to oxidative stress.
A large number of studies have shown that AGs and β-lactams can kill bacteria by affecting the bacterial respiratory chain, resulting in ROS production. Therefore, we investigated the tolerance of the WT strain, osaR mutant, and complemented strain to H2O2. As shown in Fig. 2A, the osaR promoter lacZ translation fusion reporter displayed the highest galactosidase activity around the stationary phase. Therefore, OsaR could play a role mainly during the stationary phase. The osaR mutant showed increased survival to H2O2 treatment at an OD600 of 2.0, whereas the complemented strain showed decreased survival (Fig. 2B). The catalase activity was measured with or without the presence of H2O2. Consistent with the results shown in Fig. 2B, the results shown in Fig. 2C indicate that the mutant has a higher catalase activity and stronger ability to degrade H2O2 than the WT strain.
FIG 2.

Role of osaR in bacterial tolerance to oxidative stresses. (A) The osaR promoter activity throughout growth was determined with the PosaR-lacZ fusion reporter. (B) Indicated strains were treated with H2O2 for 10 min, and the numbers of live bacteria were determined by serial dilution and plating. (C) The indicated strains were grown in LB medium to an OD600 of 2.0 and then incubated with or without 2 mM H2O2 for 30 min. The total intracellular catalase activity was measured using a catalase assay kit. (D) Carboxy-H2DCFDA was used as an indicator of ROS in cells together with gentamicin (15 μg ml−1). The ROS levels are indicated by the fluorescence density. (E and F) Thiourea at 150 mM was added or not 10 min before gentamicin (15 μg ml−1) (E) or kanamycin (60 μg ml−1) (F) treatment for the PAK and ΔosaR strains. Live bacteria were determined at different time points. All data were obtained from at least two independent experiments with at least three replicates. Error bars indicate SDs. **, P < 0.01; *, P < 0.05, compared to PAK, by Student's t test. NS, not significant.
Quantification of ROS levels using carboxy-2′,7′-dichlorodihydrofluorescein diacetate (carboxy-H2DCFDA) was used to further uncover the relationship between increased antioxidant capacity and AGs tolerance in the osaR mutant. The results shown in Fig. 2D indicate that the osaR mutant displayed the same level of ROS accumulation as the WT strain during treatment with 15 μg ml−1 gentamicin for 30 min. However, a significant difference in the ROS levels appeared 1 h later, indicating faster ROS scavenging by the osaR mutant.
It was reported previously that quenching of antibiotic-induced ROS production by thiourea protects E. coli cells from killing (19). Indeed, thiourea, a scavenger of hydroxyl radicals, decreased toxicity (12). We compared the impact of thiourea on the osaR mutant and the WT strain during treatment with 15 μg ml−1 gentamicin (Fig. 2E) or 60 μg ml−1 kanamycin (Fig. 2F). The osaR mutant showed greater tolerance than the WT strain, as expected. However, when exposed to both thiourea and antibiotic, the WT strain and osaR mutant both displayed increased survival rates (Fig. 2E and F). The results indicated that thiourea exerted a stronger effect on the WT strain due to its poorer ROS clearance ability, compared to the osaR mutant.
Together, these results suggest that deletion of the osaR gene increases the tolerance of P. aeruginosa to oxidative stress. As a result, the osaR mutant displays increased ROS clearance and enhanced tolerance to AGs and β-lactams.
OsaR indirectly represses the OxyR regulon.
P. aeruginosa contains catalases (KatA and KatB) and alkyl hydroperoxide reductases (AhpB and AhpCF), which contribute to bacterial tolerance to oxidative stress (20, 21). As a global transcription regulator, OxyR mainly regulates the antioxidant system that protects bacteria from ROS. Transcription of katA, katB, ahpB, and ahpCF can be activated by oxidized OxyR (22). Therefore, we determined the expression of the oxyR regulon (oxyR, katA, katB, ahpB, and ahpC) in the osaR mutant and the WT strain by quantitative PCR (qPCR). Statistically significantly increased expression levels of oxyR, katA, katB, ahpB, and ahpC were observed in the osaR mutant, in comparison to the WT strain (Fig. 3A). These results support the increased antioxidative capability of the osaR mutant. Similar results were observed in the appearance of H2O2 (Fig. 3B) and gentamicin (Fig. 3C). These observations highlight that the oxidative stress response arising from antibiotic treatments may be similar to but not equivalent to that induced by H2O2.
FIG 3.

Expression of oxidative stress response genes. (A to C) Total RNA was isolated from the indicated strains that had been treated with H2O2 (1 mM) for 10 min (B) or gentamicin (15 μg ml−1) (C) or had not received any treatment (A). mRNA levels of the oxyR regulon were determined by qPCR. (D) The promoter-gfp fusion reporter for expression of the oxyR, katA, and katB genes in various P. aeruginosa strains is shown. 56OE indicates the PA0056 (osaR) overexpression strain, and 58OE indicates the PA0058 (dsbM) overexpression strain. All data were obtained from at least three independent experiments with three replicates. Error bars indicate SDs. **, P < 0.01; *, P < 0.05 by Student's t test. NS, not significant.
The OxyR mRNA levels differed between the osaR mutant and the WT strain, which is not consistent with the assumption that OxyR regulates the downstream genes via changes in its oxidative reduction state without changes in its gene expression (23, 24). Therefore, we hypothesized that osaR might repress the transcription of oxyR. Promoter-gfp fusion reporter assays were carried out to confirm this. The promoter activity of PoxyR in the osaR mutant was found to be slightly higher than that of the WT strain during the stationary phase (Fig. 3D). In addition, PkatA-gfp and PkatB-gfp showed greater fluorescence in the osaR mutant at the late stationary phase (Fig. 3D). However, no difference in fluorescence of the PkatA-gfp and PkatB-gfp strains was observed after knockout of oxyR in the osaR mutant (Fig. 3D). These results suggest that OsaR affects transcription of katA and katB via OxyR.
In order to further study how OsaR affects expression of oxyR, katA, katB and ahpBCF, we performed electrophoretic mobility shift assays (EMSAs). The results strongly suggest that the OsaR protein does not bind to the region upstream of oxyR, katA, katB, and ahpBCF (Fig. S2A and B), indicating that the transcriptional regulation of oxyR mediated by OsaR is potentially indirect. Taken together, these results suggest that OsaR indirectly represses transcription of oxyR and therefore the expression of the OxyR regulon is increased in the osaR mutant.
OsaR and DsbM affect the oxidative stress response and antibiotic resistance via distinct mechanisms.
Previous work suggested that the dsbM mutant has a higher expression level of the OxyR regulon than the WT strain (15). Furthermore, evidence has been presented that reduction of oxidized OxyR is impaired in the absence of DsbM. This is consistent with the results shown in Fig. 3A, B, and D. Figure 3A shows that the change of OxyR regulon expression in the osaR mutant or overexpression strain is more modest than that in the dsbM mutant or overexpression strain (overexpression of OsaR is soluble in PAK [data not shown]). Similar results can be seen in Fig. 3B for the osaR mutant and dsbM mutant with H2O2. This suggests that DsbM represses the OxyR regulon more strongly than OsaR. We found that knockout of oxyR restored the AG sensitivity of the osaR and dsbM mutants (Table 3). The hypothesis that OsaR repression of oxyR is mediated by DsbM was put forward.
TABLE 3.
AG MICs for P. aeruginosa strains
| Bacterial straina | Relevant genotype | MIC (g liter−1) of: |
||
|---|---|---|---|---|
| Gentamycin | Streptomycin | Kanamycin | ||
| PAK | WT | 5 | 40 | 102.4 |
| ΔosaR | ΔosaR | 10 | 80 | 160 |
| ΔdsbM | ΔdsbM | 10 | 80 | 160 |
| ΔoxyR | ΔoxyR | 6.4 | 80 | 160 |
| ΔoxyR ΔosaR | ΔoxyR ΔosaR | 5 | 40 | 102.4 |
| ΔoxyR ΔdsaM | ΔoxyR ΔdsbM | 5 | 40 | 102.4 |
Catalase (25 U ml−1) was added to sustain bacterial growth after knockout of oxyR.
We identified a new gene cluster consisting of the four genes PA0055, osaR, PA0057, and dsbM in P. aeruginosa and designated it the LMD (LysR-type regulator, Metallo-β-lactamase, Disulfide oxidoreductase) gene cluster. PA0055 and osaR are located on the minus strand, whereas PA0057 and dsbM are located on the plus strand of the P. aeruginosa genome. Promoter prediction using BPROM suggests that there are only two putative promoters located between gene osaR and PA0057. There are no transcription terminators or promoters between osaR and PA0055 or between PA0057 and dsbM according to bioinformatics analysis (Fig. 4A). The overlap of the two putative promoters indicates the possible existence of a complex regulatory system involving osaR and dsbM in the LMD gene cluster (Fig. 4A). We hypothesized that PA0055 is transcribed together with osaR and that dsbM is transcribed together with PA0057. To test this hypothesis, the PA0055-R primer was used to generate PA0055-specific cDNA that served as a template to amplify the upstream gene osaR (Fig. 4B). Similarly, the PA0058-R primer was used to generate PA0058-specific cDNA that served as a template to amplify the upstream gene PA0057 (Fig. 4C). The osaR gene was present in the PA0055-specific cDNA and PA0057 was present in the PA0058-specific cDNA, suggesting that PA0055 and osaR are cotranscribed from a promoter upstream of osaR as part of a polycistronic mRNA and that PA0057 and dsbM are cotranscribed from a promoter upstream of PA0057.
FIG 4.

Regulation of the LMD gene cluster. (A) Organization of the LMD gene cluster. Arrows indicate the direction of transcription of genes. There are only two putative promoters located between gene osaR and PA0057, which have a cross in the −35 box. (B) Cotranscription of osaR and PA0055 determined by RT-PCR. Lanes 1 and 2, PA0055 (181 bp) and osaR (122 bp) cDNA as the template, respectively; lanes 3 and 4, no-reverse transcriptase controls with PA0055 and osaR primers, respectively; lanes 5 and 6, genome template as a positive control. (C) Cotranscription of PA0057 and dsbM by RT-PCR. Lanes 1 and 2, PA0057 (176 bp) and dsbM (110 bp) cDNA as the template, respectively; lanes 3 and 4, no-reverse transcriptase controls with PA0057 and dsbM primers, respectively; lanes 5 and 6, genome template as a positive control. Lane M, 2-kb DNA ladder (from the bottom up, 100 bp, 250 bp, 500 bp, 750 bp, 1,000 bp, and 2,000 bp). The transcriptional activity of PA0055 and osaR was assessed. (D and E) Effects of ΔosaR (D) and osaR overexpression and PA0055 overexpression (E) on LMD gene cluster transcription. (F and G) β-Galactosidase activity assay of the PosaR-lacZ (F) and PPA0057-lacZ (G) transcriptional fusion in the overexpression of PA0055 or osaR strains. 55OE indicates the PA0055 overexpression strain. All data were obtained from at least three independent experiments with three replicates. Error bars indicate SDs. **, P < 0.01; *, P < 0.05 by Student's t test.
In order to study regulation of the LMD gene cluster by OsaR, qPCR was performed to measure the transcription level of PA0055-dsbM. The transcription level of PA0055 decreased in the osaR mutant (Fig. 4D), whereas knockout of osaR resulted in higher expression levels of dsbM and PA0057. Expression levels of PA0057 and dsbM were reduced in the osaR overexpression strain (Fig. 4E). Interestingly, the results indicate that the transcription levels of osaR, PA0057, and dsbM were significantly decreased (Fig. 4E) in the PA0055 overexpression strain. It seems that PA0055 can regulate the LMD gene cluster together with OsaR. However, the PA0055 protein was estimated to contain no DNA binding domain.
There are two promoters that are assumed to be in opposite directions and have partial overlap in the interval between osaR and PA0057. These two putative promoters are named PosaR and PPA0057 (Fig. 4A). We investigated whether the PA0055 and OsaR proteins affect PosaR and PPA0057 activity, which might further explain PA0055-osaR-mediated regulation of LMD gene cluster expression. The DNA region between osaR and PA0057 was fused with lacZ in the forward and backward directions, respectively, in plasmid pDN19lacΩ. Forward is PPA0057 and backward is PosaR. Compared to PAK/PosaR-lacZ, 55OE/PosaR-lacZ and 56OE/PosaR-lacZ displayed significantly different β-galactosidase activity, suggesting low promoter activity in the PA0055 overexpression strain but higher activity in the OsaR overexpression strain for PosaR (Fig. 4F). This clearly demonstrates that PA0055 represses but OsaR actives PosaR promoter expression. For 55OE/PPA0057-lacZ and 56OE/PPA0057-lacZ, compared with the control PAK/PPA0057-lacZ, the results show that the overexpression of PA0055 and OsaR leads to a decrease in β-galactosidase activity (Fig. 4G). These results suggest that the PA0055 and OsaR proteins may inhibit the transcription of PA0057 and dsbM. In addition, β-galactosidase activity in PAK/PosaR-lacZ was >4-fold higher than in PAK/PPA0057-lacZ. On the whole, our data indicate that PA0055 represses PosaR and PPA0057 promoter activity and that OsaR positively regulates PosaR promoter activity but represses PPA0057 promoter activity.
DsbM expression levels increased about 1.5-fold in the osaR mutant but it could inactive OxyR. Further experiments were carried out to examine the effect of overexpression of DsbM. Increased osaR expression levels (Fig. 5A) and H2O2/AG susceptibility (Fig. 5B and Table 4) were displayed in the dsbM overexpression strain. Furthermore, the bacteria displayed a cumulative tolerance to H2O2/AGs after simultaneous knockout of osaR and dsbM (Fig. 5C and Table 4). Therefore, OsaR repression of the OxyR regulon is not through DsbM, considering that the increased expression of DsbM and OxyR regulon is inconsistent in the osaR mutant. It is the modest upregulation of dsbM in the osaR mutant that explains lower OxyR regulon expression (Fig. 3A and B) and lower H2O2 tolerance (Fig. 5C), compared with the dsbM mutant.
FIG 5.

DsbM overexpression can enhance oxidant susceptibility. (A) mRNA levels of PA0055, osaR, and PA0057 were determined by qPCR in the WT strain, dsbM mutant, and overexpression strain. (B and C) The indicated strains were then treated with H2O2 for 10 min, and the numbers of live bacteria were determined by serial dilution and plating. All data were obtained from at least three independent experiments with three replicates. Error bars indicate SDs. **, P < 0.01; *, P < 0.05 by Student's t test.
TABLE 4.
AG MICs for P. aeruginosa strains
| Bacterial strain | Relevant genotype | MIC (g liter−1) of: |
||
|---|---|---|---|---|
| Gentamycin | Streptomycin | Kanamycin | ||
| PAK | WT | 5 | 40 | 80 |
| ΔdsbM | ΔdsbM | 6.4 | 80 | 160 |
| 58OE | WT/pUCP19-dsbM | 2.5 | 25.6 | 51.2 |
| ΔdsbM/58OE | ΔdsbM/dsbM | 3.2 | 25.6 | 80 |
| ΔosaR ΔdsbM | ΔosaR ΔdsbM | >10 | 160 | 204.8 |
Disulfide bond and multimer formation in the oxidized OsaR.
As a disulfide bond redox enzyme, DsbM can catalyze oxidic OxyR into a reduced state, terminating OxyR activation of antioxidant genes (17). Recent research indicates that there is a physical interaction between DsbM and OsaR (18). As a hypothesis, DsbM could catalyze oxidized OsaR (OsaR-OX) into reduced OsaR (OsaR-RE). Changes in OsaR redox status are likely to determine its activity of regulation.
The tertiary structure prediction of the OsaR protein indicates that it is composed of a DNA binding domain and a regulatory domain (Fig. S3B), which is in accord with the description of the tertiary structure of the LTTR family proteins (25). Based on the sequence of the osaR gene, the OsaR protein contains cysteines in positions C69, C161, C166, and C189. Like OxyR, OsaR is likely S-S-bonded between C-166 and C-189 in the regulatory domain (Fig. S3A). Soluble expression of OsaR was finally achieved in the form of a maltose-binding protein (MBP) fusion in E. coli (Fig. S3C).
We performed SDS-PAGE of inclusion body OsaR protein expressed in the BL21(DE3) strain with pET28a vector for analysis of the protein under nonreducing conditions. The two heavy bands between 34 kDa and 43 kDa likely represent the OsaR protein in the reduced state and the oxidized state, respectively (Fig. S3D). Analytical ultracentrifugation was carried out to measure the aggregated form of OsaR with MBP-OsaR. Figure 6A shows that fresh purified MBP-OsaR exists mainly in the aggregated form under nonreducing conditions, which is similar to MBP-OsaR-RE exposed to mercaptoethanol (ME). It is worth mentioning that analytical ultracentrifugation has certain errors in the evaluation of molecular weight, but it can be determined that MBP-OsaR exists as a polymer in the reduced state (MBP-OsaR trimer of 236.442 kDa and MBP-OsaR tetramer of 315.256 kDa). MBP-OsaR-OX treated with H2O2 is mainly in the form of 44.9-kDa MBP (possibly due to the break of the MBP-OsaR linker) and 100-kDa MBP-OsaR monomer. MBP is a monomer and has no cysteine residues. OsaR-OX is a monomer and OsaR-RE most likely exists as a tetramer, in reference to the characterization of LysR family proteins. Free thiol measurements revealed that OsaR-OX has an intramolecular disulfide (Fig. 6B).
FIG 6.

Change of OsaR protein redox state with H2O2 treatment. Purified MBP-OsaR (40 μM) was incubated with 1% ME for 20 min to reduce MBP-OsaR (MBP-OsaR-RE) or with 10 mM H2O2 for 20 min to obtain oxidized MBP-OsaR (MBP-OsaR-OX). (A) Analytical ultracentrifugation was employed to detect the state of MBP-OsaR aggregation. The blue arrow indicates the polymer, and the pink arrow indicates the MBP-OsaR monomer or MBP broken away from MBP-OsaR. OsaR aggregates into pellets due to its instability and cannot be shown on the curve. (B) Measurement of free thiol contents in MBP-OsaR-RE and MBP-OsaR-OX (MBP has no cysteine residues). (C to E) Nonreducing SDS-PAGE (8%) of MBP-OsaR (2 μM) reduced with ME (1%) or oxidized with the indicated concentration of H2O2 in vitro. A total of 225 μg of protein was loaded per lane. Protein mobility was visualized using Coomassie brilliant blue staining. PAK WT bacteria containing osaR-His fusion were treated with 0, 0.1, 1, 10, or 100 mM H2O2 for 20 min (D) or the strain was treated with 1 mM H2O2 every 10 min (E) and then aliquots were taken at the given time points. After that, the samples were precipitated with TCA and then treated with AMS. Again, separation and detection of OsaR-His were achieved by nonreducing SDS-PAGE (10%) and immunoblotting. OsaR-RE or RE, the reduced form; OsaR-OX or OX, the oxidized form; C, the absence of ME or H2O2. All data were obtained from at least three independent experiments with at least three replicates.
When MBP-OsaR was incubated in vitro with different concentrations of H2O2 ranging from 0.1 mM to 100 mM, a new band, MBP-OsaR-OX, appeared, illustrating that MBP-OsaR is oxidized in the presence of H2O2 (Fig. 6C). However, the electrophoretic migration of OsaR was different when bacterial cultures were exposed to different concentrations of H2O2 followed by nonreducing SDS-PAGE (Fig. 6D). Using the 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS) alkylation method, we assessed the kinetics of OsaR oxidation after treatment with 1 mM H2O2 in vivo. OsaR was in a reduced state in the absence of H2O2 (Fig. 6D) and was oxidized after H2O2 was added to the bacteria (Fig. 6E). These experiments turned out to be technically challenging, giving rise to only weak Western blot bands, likely due to low native expression of the OsaR protein. The experiments described above suggest that OsaR exists as a polymer in the reduced state under physiological conditions, whereas upon oxidation a disulfide bond is formed, changing OsaR into a monomeric state.
osaR autoregulates its response to oxidative stress.
In order to further investigate whether the OsaR protein directly binds to the intergenic region (IR) between osaR and PA0057 to regulate transcription, we performed EMSA analysis. The DNA fragments used in the EMSA are shown in Fig. 7A. OsaR was shown to specifically bind to the F1 region (Fig. 7B). Subsequently, OsaR was incubated with the F2IR (Fig. 7C) or F3 (Fig. 7D) fragments, and the binding complex is shown in Fig. 7C.
FIG 7.

Transcription factor binding properties of OsaR identified by EMSA analysis. (A) DNA fragments used in the EMSA analysis. (B to D) Validation of binding of OsaR-RE (with 1% ME) to selected target regions by EMSA. (E and F) Ability of the reduced state (OsaR-RE) (with ME) and the oxidized state (OsaR-OX) (with H2O2) to bind the target fragment at a protein concentration of 200 nM. (G) Reduction of OsaR (1000 nM) with 1% ME for 60 min followed by dialysis to remove ME in an anaerobic chamber. Oxidation of OsaR-RE was performed with various concentrations of H2O2 before application to the shift assay. DNA-protein complexes were separated upon migration on a native PAGE gel. OsaR was purified under aerobic conditions and used at the following concentrations: 0 nM, 50 nM, 100 nM, 200 nM, or 400 nM. The protein is a mixture of OsaR and MBP, because OsaR is unstable and easy to precipitate when more purification steps are performed after MBP is cut off. The proportion of OsaR was calculated using ImageJ software according to the SDS-PAGE picture. Red arrows indicate the protein-DNA complex and black arrows indicate free DNA. MBP (400 nM plus the DNA fragment) and C (OsaR-RE at 400 nM plus flpJG) are negative controls.
The protein OsaR contains disulfide bonds, which affect its aggregation state. Addition of H2O2 oxidizes the sulfhydryl groups in OsaR to form disulfide bonds, while addition of ME opens the oxidized disulfide bonds in OsaR. No shift band appears when H2O2 increases in the EMSA system (OsaR purified under nonreducing conditions) (Fig. 7E). Figure 7F shows that a binding complex appears as the concentration of ME is increased to maintain OsaR in the reduced state. Addition of H2O2 into the EMSA system to oxidize into an oxidized state the OsaR-RE protein that was reduced with ME in advance resulted in disappearance of the binding complex (Fig. 7G). These results indicate that OsaR binds to DNA in a reduced state rather than in an oxidized state.
A DNase I footprinting assay was performed to identify the binding box of OsaR. A 6-fluorescein amidite (FAM)-labeled DNA fragment (F2IR) was incubated with purified OsaR-RE; subsequently, these complexes were treated with DNase I, followed by DNA fragment analysis by sequencing. A nearly continuous 40-bp region from position 120 to position 160 in F2IR was protected by OsaR-RE (Fig. 8A and B). Previous research suggested that the T-N11-A motif is a LTTR consensus binding sequence. Eleven T-N11-A boxes were retrieved in F2IR (Fig. 8C). T-N11-A boxes 5 to 7 are located in the region of the osaR gene that was protected in the DNase I footprinting assay (Fig. 8C).
FIG 8.

Identification of the OsaR-protected cis elements in the IR promoter region using a DNase I footprinting assay and EMSA. (A and B) The probes used were labeled with FAM dye, and the control products (red) together with the digestion products (blue) were analyzed with peak scanner software v1.0 (Applied Biosystems) (A). These two sequencing results are indicated by different colors separately and are then merged together (B). The whole region of the coding strand DNA is shown in panel A, and the region protected by OsaR from DNase I cleavage was enlarged and is shown separately in panel B. (C) The region protected by OsaR from DNase I digestion is the OsaR binding site, which is in the black dotted box; other salient features named 1 to 13 are all T-N11-A boxes in the DNA fragment identified by a program. (D) F2IR was cut from 238 bp to 90 bp (designated F2IRC); in the F2IRC site mutant, the four bases in bold are mutation sites (designated F2IRM). EMSAs of OsaR-RE binding with the site mutant DNA fragment F2IRM were performed; F2IR and F2IRC are positive controls, and OsaR-RE concentrations were 0 nM, 200 nM, or 400 nM. (E) For cold competition, OsaR-RE was first incubated with the DNA fragment F2IRC and then F2IRC marked with biotin (F2IRCb) was added. The OsaR-RE concentrations were 0 nM, 200 nM, and 400 nM, F2IRCb was a positive control, with OsaR-RE concentrations of 0 nM, 200 nM, 400 nM, and 800 nM. (F) Promoter activity was analyzed in the WT strain and OsaR overexpression strains, with four base mutations of the osaR promoter PosaRM in the putative OsaR binding site as F2IRM. The PosaR-lacZ reporter was a control. Red arrows indicate the protein-DNA complex, and black arrows indicate free DNA. NS, not significant. **, P < 0.01 by Student's t test.
To obtain a more detailed picture of OsaR interacting with its specific binding site, OsaR binding with F2IR was examined by EMSA with mutated DNA fragments. In the OsaR protein binding region, box 5, which is similar to the T-N11-A motif, is ATTCGCTCGATCCAT, AT-N11-AT. A fragment including only boxes 5 to 7, named F2IRC, was derived from F2IR that was cut from 238 bp to 90 bp (Fig. 7A). Considering the physical structures of the nucleotides, the base transition, which interchanges either two purine nucleotides (A and G) or two pyrimidine nucleotides (C and T), is less likely to introduce large changes in the DNA structure (26). We introduced point mutations in DNA fragment F2IRC to change the AT-TCGCTCGATCC (N11)-AT sequence into GC-TCGCTCGATCC (N11)-CG (Fig. 7A), and the mutated DNA fragment was designated F2IRM. F2IRM was not bound by OsaR-RE, compared to the positive-control F2IRC (Fig. 8D). OsaR-RE was incubated first with unlabeled F2IRC and biotin-labeled F2IRC was added subsequently; the EMSA did not show a band shift (Fig. 8E). A mutant osaR promoter in osaR binding sites (AT-N11-AT into GC-N11-GC)-lacZ translation fusion was constructed. Unlike the WT osaR promoter, the PosaRM mutant could not be induced in the osaR overexpression strain. PosaRM present lower activity then PosaR in PAK due to lacking of OsaR induction (Fig. 8F). This suggests that AT-TCGCTCGATCC (N11)-AT is the OsaR binding box in the IR between osaR and PA0057.
Despite PA0055 overexpression having no obvious effect on the AG and β-lactam MICs (data not shown), EMSAs were carried out to test the interaction of PA0055 protein (expressed with pET28a) (Fig. S3E) with the IR and F2IR. As shown in Fig. 9A and Fig. S4, no diffuse bands were formed. Combined with the β-galactosidase reporter results shown in Fig. 4F and G, this tentatively suggests that PA0055 could regulate promoters PosaR and PPA0057 indirectly. When OsaR-RE was incubated together with F2IR in the presence of PA0055, we observed a rapid decrease of the binding complex of OsaR to F2IR (Fig. 9B). Microscale thermophoresis (MST) was used to determine the dissociation constant (Kd) between protein PA0055 and OsaR (Fig. 9C). The results indicate that PA0055 showed binding affinity for OsaR (Kd = 5.85 nM). Therefore, PA0055 serves as an antiregulator that might negatively regulate the LMD gene cluster by interacting with OsaR to change its affinity to the regulatory region.
FIG 9.

Binding of PA0055 to promoter DNA and OsaR. (A) EMSA of PA0055 protein with the IR between osaR and PA0057. PA0055 used in the following concentrations: 0 nM, 50 nM, 100 nM, and 200 nM. (B) EMSA of OsaR-RE (200 nM) with the F2IR DNA fragment in the presence or absence of protein PA0055 (200 nM). The numbers 1 and 2 represent the order in which the corresponding proteins are added to the EMSA system. (C) Binding affinity of OsaR-RE for PA0055 protein assessed by MST analysis. The Kd was 5.85 nM. Red arrows indicate the protein-DNA complex, and black arrows indicate free DNA.
Finally, a PPA0057-gfp reporter was constructed to reveal whether the LMD gene cluster would be induced by antibiotics. Figure S5 shows that the relative fluorescence level of an osaR mutant is higher than that of the WT strain, which is in agreement with the result shown in Fig. 4G. Relative fluorescence levels increased similarly when carbenicillin was added in both the WT and osaR mutant strains, but a more remarkable increase was observed in the WT strain. Therefore, promoter PPA0057 in the LMD gene cluster can be induced by carbenicillin, and OsaR plays a part role in this pathway.
DISCUSSION
OsaR represses the OxyR system.
Work in E. coli has shown that OxyR acts as a transcriptional autorepressor that represses its own activity under noninducing conditions (27). oxyR transcription was also upregulated in response to polymyxin B, norfloxacin, and rifampin in Burkholderia cenocepacia (28). Evidence showing that the amounts of the OxyR protein change after H2O2 treatment in Xanthomonas oryzae (29) and in P. aeruginosa (30) has been provided. However, EMSA analysis did not reveal binding of purified OxyR protein to DNA upstream of oxyR in P. aeruginosa (22). We found that the oxyR expression levels increased in the osaR mutant and dsbM mutant without H2O2 treatment (Fig. 3A), suggesting that both OsaR and DsbM could regulate oxyR at the transcription level in P. aeruginosa.
Several classes of antibiotics can result in a rise in H2O2 production; accordingly, OxyR is also activated by antibiotics such as gentamicin (AG) (31, 32), cefotaxime (β-lactam), and levofloxacin (fluoroquinolone) in E. coli (33). Oxidized OxyR is deactivated by the glutaredoxin (GrxA) system and the thioredoxin system (22, 24). Moreover, OxyR could be reduced by DsbM with the participation of GSH (17). P. aeruginosa has three different types of catalase isoenzyme genes, katA, katB, and katE, which are not controlled only by OxyR. Overexpression of KatG inhibits killing by gentamicin, and addition of external catalase or overexpression of KatE/SodA can prevent cell death by spectinomycin in E. coli (31, 34). An increase in the catalase activity was also demonstrated in Burkholderia cenocepacia in response to antibiotics targeting the cell wall (28).
Various antibiotics boost production of ROS, although it is still controversial. Contradictory results against ROS in antibiotic-mediated killing were explained in several research papers and reviews (35). In brief, they could be attributable to the experimental setup, the genetic background of the strains, the concentrations of antibiotics, or other factors (36–40). Numerous studies confirmed the involvement of metabolic changes, including the tricarboxylic acid cycle, in ROS-mediated antibiotic killing of various bacterial species, except for stimulating defense systems against ROS. Conceivably, this study provides new evidence that the activated OxyR regulon could protect bacteria from ROS damage and improve the bacterial response to antibiotics.
Gene regulation in the LMD gene cluster.
In the LMD gene cluster, the genes PA0055, osaR, PA0057, and dsbM form an operon. OxyR can be reduced by DsbM; when DsbM is absent, oxidized OxyR cannot be reduced in a timely manner and consequently the expression of the OxyR regulon increases, causing AG resistance to increase (17). Thus, DsbM plays a role in oxidative stress and AG resistance in P. aeruginosa. It can end the OxyR response when the oxidative pressure is eliminated. PA0055 and OsaR can both repress PPA0057. However, the expression levels of PA0057 and dsbM are not particularly high in the osaR mutant. This indicates that there is likely a positive regulator for PPA0057, which deserves further study. OsaR has DNA binding domains and autoregulates its expression by binding to specific sites in the IR. β-Galactosidase activity assays indicated that overexpression of OsaR enhances the promoter activity of PosaR. However, the transcription level of PA0055 did not change significantly in the osaR overexpression strain (Fig. 4E). In the osaR knockout strain, the transcription level of PA0055 was significantly downregulated (Fig. 4D). This might be due to the autoinduction of OsaR, along with the negative feedback regulation of PA0055 in gene transcription. The β-galactosidase activity experiments suggest that PA0055 can inhibit the PosaR and PPA0057 promoters (Fig. 4F and G). We speculate that PA0055 could be a vital gene that cannot be knocked out (data not shown). Therefore, in the operon, PA0055 and osaR are cotranscribed under the regulation of promoter PosaR, while PA0057 and dsbM are cotranscribed under the regulation of promoter PPA0057. It is the overlap of the promoters PosaR and PPA0057 that makes the internal regulation of this operon orderly. PA0055 and OsaR form an oscillator in which OsaR-RE positively induces expression of PA0055 and osaR. As a result, PA0055 can exert feedback to stop OsaR-RE autoinduction when it accumulates to a certain level.
PA0057 is induced by cell wall stress and ROS.
Lactamases can be divided into four categories, i.e., class B, which requires Zn2+, and classes A, C, and D, which are not dependent on metal but have active serine sites (41). Although these four classes of β-lactamases are all found in P. aeruginosa, only two types, AmpC and PoxB, were reported to be encoded chromosomally (42). PA0057 is a class B β-lactamase encoded chromosomally. Most β-lactams, including carbapenems, are preferred substrates of type B lactamases (43). PA0057 is also able to hydrolyze imipenem and other lactamide antibiotics (except those with a single ring) (44). Accordingly, we found that increased expression of PA0057 in the osaR mutant plays a nonnegligible role in β-lactam resistance. Furthermore, when there are β-lactams or β-lactamase inhibitors in the environment that can bind to penicillin-binding proteins, cell wall synthesis is blocked and the level of intracellular wall peptides increases, disrupting the respiratory chain and leading to a rise in ROS. This may explain why PA0057 and dsbM are located in the same operon. PA0057 is responsible for β-lactam resistance and DsbM is involved in the defense against ROS.
Role of DsbM in oxidative stress.
Protein inactivation is one of the disastrous results of oxidative stress. Many types of amino acids may be covalently modified by oxidative damage. Among them, cysteine and methionine are the most sensitive to ROS. The thiol (–SH) functional groups in the cysteines of proteins can accept electrons from ROS and thereby be oxidized to sulfenic acids (–SOH). Subsequently, these sulfenic acids can either form a disulfide bond (S–S) or be irreversibly oxidized to sulfinic acid (–SO2H) and sulfonic acid (–SO3H) (45). Proteins in the periplasm that are sulfenylated by ROS may be repaired by DsbG and DsbC, and then the OX-DsbG and OX-DsbC are reduced by DsbD (45–47). In the cytoplasm, oxidized Cys residues are reduced to the free thiol state by various oxidoreductases, including thioredoxins and glutaredoxins (48, 49). The present study provides evidence that upregulation of DsbM located in the cytoplasm may involve one of the Dsb family oxidoreductases that repair oxidation-damaged cysteine in cytoplasmic proteins, including OsaR, and can deactivate OxyR after ROS stress is relieved.
The combination of all these data led us to formulate a hypothetical model, which is summarized in Fig. 10. OsaR is inactivated into an oxidized state and the OxyR regulon is activated when exposed to oxidative stress. The metallo-β-lactamase PA0057 enhances tolerance to β-lactams. The activated OxyR-OX regulon enhances tolerance to AGs by scavenging ROS. Finally, OxyR-OX and OsaR-OX are catalyzed into a reduced state by DsbM. The redox regulator OsaR plays a role in tolerance to AGs and β-lactams in P. aeruginosa.
FIG 10.
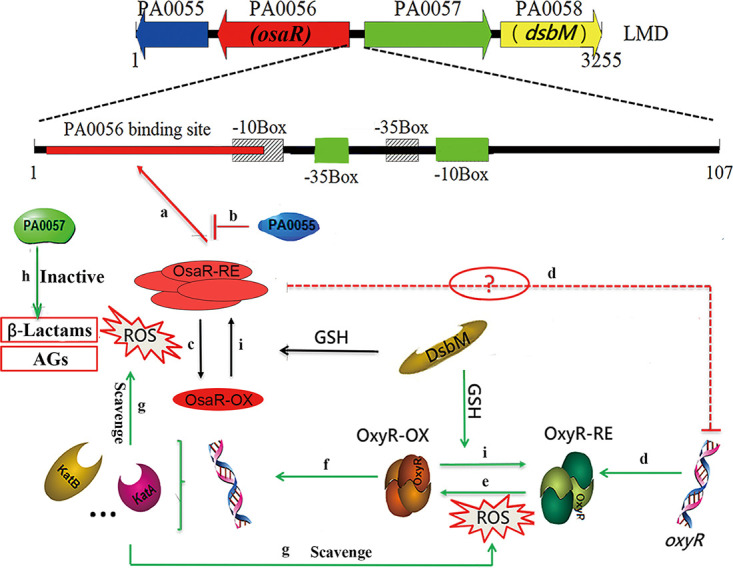
Proposed model for oxidative stress responses regulated by OsaR in P. aeruginosa. In this hypothetical model, the bacteria have a reduced internal milieu under normal physiological conditions. Therefore, reduced OsaR protein binds to its binding site in the form of a polymer, which facilitates transcriptional initiation of osaR (a). The negative feedback regulation of PA0055 protein keeps the positive autoregulation of osaR at a relative level (b). The transcription of PA0057 and dsbM is normally repressed. When exposed to antibiotics (β-lactams or AGs) or oxidative stress, OsaR is oxidized and forms monomers (c) and OxyR is activated (e), resulting in increased expression of antioxidant genes to restore the redox balance. OsaR protein is oxidized, forming a disulfide bond, and is converted into a nonbinding state (c). The expression levels of PA0055 and osaR are downregulated, but PA0057, dsbM, and oxyR (d) (the dotted line indicates indirect regulation) are upregulated. Among them, the metallo-β-lactamase expressed by PA0057 protein can hydrolyze the lactamide ring of β-lactams and enhance the tolerance of P. aeruginosa to lactams (h). Oxidized OxyR can continuously activate the transcription of downstream antioxidant genes to scavenge ROS (f and g). Thus, the influence of AGs on bacteria can be alleviated. Oxidized OxyR and oxidized OsaR could be catalyzed to a reduced state by DsbM when the oxidation stress is released (i). Finally, the LMD gene cluster is restored to the initial physiological state. The model marked with green lines integrates previous published data.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
Bacteria were grown in Luria-Bertani (LB) liquid medium or plated onto solid LB medium containing 1.5% (wt/vol) agar and cultured at 37°C with aeration. Antibiotics were used at the following concentrations for plasmid selection and maintenance: 25 mg liter−1 kanamycin, 10 mg liter−1 tetracycline, 10 mg liter−1 gentamicin, or 50 mg liter−1 ampicillin for E. coli and 100 mg liter−1 gentamicin, 10 mg liter−1 tetracycline, or 100 mg liter−1 carbenicillin for P. aeruginosa. The bacterial strains and plasmids used in this study are listed in Table S1 in the supplemental material, and the primers used in this study are listed in Table S2. All of the mutants were constructed by markerless homologous recombination.
MICs of antibiotics.
The MICs of antibiotics were determined using microdilution assays (50). Briefly, overnight cultures were diluted to the final desired inoculum of 5 × 105 CFU ml−1, and then 200 μl of the diluted culture was added to the first two columns of a 96-well plate; others received 100 μl. Two initial concentrations that were prepared for the same antibiotic to obtain more precise MIC values were added to the first and second wells in each row and then gradient diluted as indicated in Table 5. The 96-well plate was incubated at 37°C, and the MICs were measured after 18 h. All of the data were obtained from at least three independent experiments with at least three replicates.
TABLE 5.
Gradient dilution for MIC determinations
| Dilution step | Concentrationa |
|---|---|
| 1 | C1 |
| 2 | C2 |
| 3 | C1/2 |
| 4 | C2/2 |
| 5 | C1/4 |
| 6 | C2/4 |
| 7 | C1/8 |
| 8 | C2/8 |
| 9 | C1/16 |
| 10 | C2/16 |
| 11 | C1/32 |
| 12 | 0 |
C2 equals (C1 + C1/2)/2 equals 3C1/4 or C2 is a special concentration (C1 < C2 < C1/2) from pilot experiments.
H2O2 susceptibility assay.
H2O2 susceptibility was determined using the same method as that detailed by Weng et al. (51) but with slight modifications; bacteria at an OD600 of 2.0 were collected and washed with sterile phosphate-buffered saline (PBS). The strains were then treated with H2O2 (10 mM or 20 mM) for 10 min at 37°C.
Cellular ROS assay.
ROS were detected using the agent carboxy-H2DCFDA as described previously, with slight modifications (36). After treatment with high concentrations of antibiotic for 30 min at an OD of 2.0, the control and cells were stained with carboxy-H2DCFDA (dissolved in ethanol [final concentration, 20 μM]; Genecopoeia) at 37°C for 30 min. The stained cells were then washed with PBS, and the fluorescence intensity (FLU) (excitation wavelength, 488 nm; emission wavelength, 520 nm) was detected by a fluorescent microplate reader (PerkinElmer, USA). The relative fluorescence density of each sample was calculated as FLU divided by the cell density (OD600) to evaluate intracellular ROS levels.
Catalase activity assay.
Bacteria were cultured to an OD600 of 2.0 and then incubated with or without 2 mM H2O2 for 30 min. The bacteria were collected by centrifugation, followed by sonication for lysis. The total intracellular catalase activity was measured with a catalase assay kit (Beyotime, Shanghai, China). A bicinchoninic acid (BCA) kit was used for total protein concentration measurements (CWBio, China).
Killing experiments with antibiotic and thiourea.
For the killing assay with carbenicillin, overnight cultures were diluted 1:100 into 20 ml of LB broth and grown to an OD600 of 0.4 in flasks. The cultures were then divided into 5 ml per tube, and different concentrations of carbenicillin were added. The CFU were counted every 3 h.
Samples were analyzed as reported previously (52). Overnight cultures were diluted 1:100 into 20 ml of LB broth and grown to an OD600 of 1.0 in flasks as described above; 0.1 ml of either LB broth or LB broth with 150 mM thiourea (Sigma-Aldrich) was added, and shaking was continued for 10 min. After addition of antibiotic, the cultures were put back on the shaker and the OD600 was measured every 1 h. Before antibiotic addition, a sample was removed to detect the initial OD600. Cell survival was determined as the OD600 divided by the initial OD600.
Reverse transcription-PCR.
Reverse transcription (RT)-PCR was based on a previously described procedure (53). Total RNA was isolated as before. The RNA was treated with 20 U DNase (Thermo Fisher Scientific) for 30 min at 37°C. DNase was removed by adding 30 mM EDTA at 65°C for 10 min. Total RNA was then reverse transcribed with specific primers (PA0055SRTR or PA0058SRTR) using the StarScript II first-strand cDNA synthesis kit according to the manufacturer’s instructions. PA0055, osaR, PA0057, and dsbM were amplified with the first-strand cDNA reaction product as the template. Reactions without the reverse transcriptase enzyme were used as negative controls. The PCR positive control was prepared using the genomic DNA as the template.
RT-qPCR.
Bacteria were harvested at an OD of 2.0, and RNA was extracted using the RNA preparation pure cell/bacteria kit (Tiangen). The extracted RNAs were reverse transcribed into cDNAs with the Transcriptor first-strand cDNA synthesis kit (Roche). RT-qPCR analysis was performed with the SYBR green premix ex Taq kit (Tli RNase H Plus; TaKaRa), and rspL was used to normalize gene expression.
β-Galactosidase activity assay.
The strain was cotransformed with the reporter plasmid and the overexpression plasmid. Cultures inoculated with 1% diluted overnight cultures grew to an OD of 2.0 and were subjected to the β-galactosidase activity assay as reported (54); results were expressed in Miller units. PAK cells with the reporter plasmid and empty plasmid pUCP19 served as controls.
Biosensor assay.
Cultivation of P. aeruginosa was carried out at 37°C in ABTrace minimal medium supplemented with 0.5% (wt vol−1) glucose, 0.5% (wt vol−1) Casamino Acids, and 1 μM ferric chloride (ABTGCAAFe medium). Overnight cultures of P. aeruginosa strains containing promoter-gfp biosensors were diluted to an OD600 of 0.025, as described before (55). Briefly, a total of 100 μl of culture was transferred into each well of a 96-well microplate. OD600 and green fluorescent protein (GFP) fluorescence readings (in relative fluorescence units [RFU]) were measured every 30 min for 24 h using a microplate reader (Tecan Infinite 2000).
Amino acid sequence alignment and protein structure prediction.
The amino acid sequence alignment between OsaR and the P. aeruginosa OxyR protein (EcYcjZ [GenBank accession number EOQ56594.1]) was analyzed using the ClustalW2 server (http://www.ebi.ac.uk/Tools/msa/clustalw2). Structural modeling prediction of OsaR was performed using SWISS-MODEL (https://swissmodel.expasy.org). Promoter prediction of osaR and PA0057 used BPROM (http://linux1.softberry.com/berry.phtml?topic=bprom&group=programs&subgroup=gfindb).
Expression and purification of protein.
Expression and purification of protein PA0055 and inclusion OsaR were based on the pET system manual (Novagen user protocol TB055) and the HisTrap FF crude protocol (GE Healthcare). Transetta(DE3) was used for PA0055 expression, and BL21(DE3) was used for OsaR expression. The purified proteins were dialyzed with TGE buffer (50 mM Tris-HCl [pH 7.5], 10% glycerol, 1 mM EDTA) using an ultrafiltration tube (Millipore). The E. coli expression plasmid pETMALc-H, which is a MBP fusion expression vector, was used for expression of osaR. The plasmid was introduced into BL21(DE3). OsaR expression was performed as described previously with some modifications (56). The overnight culture was diluted 1% into 1 liter of fresh LB medium plus 0.2% glucose medium containing 25 μg ml−1 kanamycin. The culture was grown at 37°C until the OD600 reached 1.0, and then expression was induced with 0.3 mM isopropyl-β-d-thiogalactopyranoside (IPTG). The cells were grown at 28°C for a total of 2 h after induction. The cells were harvested and suspended in CB buffer according to the pMAL protein fusion and purification system manual (number E8000; New England Biolabs) to acquire and to purify OsaR protein. Finally, the pellets from ultrasonication were resuspended in 8 M urea to acquire the inclusion body in the supernatant.
The enzyme reactions were carried out. Affinity-purified fusion proteins were diluted to a working concentration of <2 mg ml−1 in PBS (pH 8.0), and 10% glycerol was added. Thrombin (Macklin, China) at 1:50 (quality ratio) was subsequently applied, and the reactions were performed for 12 h at 30°C (57). Cleavage was terminated with 1 mM benzamidine. After centrifugation of the samples for 5 min at 12,000 × g, the soluble fractions were extracted and analyzed by 10% SDS-PAGE, followed by Coomassie blue staining and Western blotting using peroxidase-linked monoclonal anti-His.
MST measurements.
MST experiments were performed as described by Jerabek-Willemsen et al. (58). Sixteen samples with 50 nM labeled OsaR protein probe and increasing concentrations of nonlabeled PA0055 protein (from 0.9 nM to 30 μM) were loaded on standard treated silicon capillaries (Monolith NT.115 series capillaries). The measurements were carried out using a Monolith NT.115 instrument (NanoTemper) at 30°C with 40% excitation power and medium LED power. Data analyses were performed using NanoTemper Analysis software. The dissociation constants (Kd) were calculated as described previously (59).
Analytical ultracentrifugation and DTNB assay.
ME (1%) was used to obtain the reduced protein, and 10 mM H2O2 was used for the oxidized form. Then, analytical ultracentrifugation was performed with a ProteomeLab XL-I ultracentrifuge (Beckman). The 5,5-dithiobis-2-nitrobenzoic acid (DTNB) assay was used to determine the free thiol contents (60). The ME or H2O2 used for reducing or oxidizing the protein was removed by dialysis with the DTNB assay buffer (200 mM KH2PO4/K2HPO4, 200 mM NaCl, 2 mM EDTA). After addition of DTNB, the A412 was recorded. The BCA kit (CWBio) was used for protein concentration measurements. Free thiol contents were determined as the free thiol concentration divided by the protein concentration.
Determination of OsaR redox status.
This experiment for determination of the OsaR redox status in vivo mainly followed the assay presented by Åslund et al. (61). Briefly, PAK-OsaR-His cultures at an OD of 2.0 were treated with 1 mM H2O2 for different times (or PAK-OsaR-His cultures were treated with different concentrations of H2O2 for 20 min), and then aliquots were taken and mixed with 10% trichloroacetic acid (TCA). Proteins were precipitated at 4°C for 1 h and collected by centrifugation (10 min at 10,000 × g). Pellets were washed once with cold acetone after thorough removal of the supernatant. The pellets were acquired and air dried for 30 min. Next, the pellets were dissolved in alkylation buffer (1% SDS, 0.67 M Tris-HCl [pH 8], 15 mM AMS [Molecular Probes, Sigma]). Alkylations were performed at 37°C for 1 h. All of the aforementioned samples were analysis via Western blotting.
For the in vitro assays, MBP-OsaR was reduced with ME (1%) and oxidized with given concentrations of H2O2. Subsequently, TCA was added to remove ME (1%) and H2O2 as described above. Theses pellets were dissolved in alkylation buffer, with or without ASM, for 1 h. Coomassie brilliant blue staining was performed after nonreducing SDS-PAGE (62).
Gel retardation assays.
Gel retardation assays were conducted like those in a previous study with some modifications (63). The DNA fragments were amplified by PCR. Purified DNA fragments and protein PA0055 or OsaR were mixed and incubated in buffer (20 mM Tris-HCl [pH 7.5], 50 mM KCl, 50 mM MgCl2, 10% glycerol) for 20 min at 30°C to allow complex formation. The complexes were detected by 5% (wt/vol) PAGE at 80 V and then stained with ethidium bromide.
EMSAs were performed as described previously with minor modifications (22, 64). DNA fragments generated by PCR amplification were 5′-end labeled with biotin. All binding reactions were performed at 30°C for 20 min in binding buffer (20 mM Tris-HCl [pH 7.5], 50 mM KCl, 50 mM MgCl2, 10% glycerol, 25 mg ml−1 sonicated herring sperm DNA). The protein-DNA migration was carried out at 8 V cm−1 in TBE buffer in 5% polyacrylamide gels. The samples were then transferred to a positively charged nylon membrane. Cross-linking was performed in an 80°C oven for 20 min, followed by streptavidin-horseradish peroxidase hybridization color development.
DNase I footprinting assay with FAM-labeled primers.
DNase I footprinting assays were performed according to the method described by Wang et al. (65). A 238-bp promoter region of osaR was PCR amplified with a FAM-labeled sense primer. Probes (250 ng) were incubated with 3.5-μg mixtures of recombinant protein or bovine serum albumin in a total volume of 40 μl in the same buffer as for the EMSAs described previously. After incubation for 20 min at 30°C, 0.075 U of DNase I (Invitrogen) was added, followed by further incubation for 1 min at 25°C. The reaction was stopped by the addition of 30 mM EDTA and incubation at 65°C for 10 min. Samples were purified with the PCR purification kit (TaKaRa), and the pellets were eluted in 10 μl of 2 mM Tris buffer (pH 8.0). The sequencing results were analyzed with Peak Scanner software v1.0 (Applied Biosystems, CA, USA) to determine the exact regions that were protected (63).
Supplementary Material
ACKNOWLEDGMENTS
We thank Xiaofei Song and Hesuiyuan Wang for their invaluable assistance with figure designing. We also thank Morten T. Rybtke for his critical help.
This work was supported by the Sino-Swiss Scientific and Technological Cooperation program supported by the Ministry of Science and Technology of China (grant 2015DFG32140) and the National Natural Science Foundation of China (grant 31770102). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Mustafa MH, Chalhoub H, Denis O, Deplano A, Vergison A, Rodriguez-Villalobos H, Tunney MM, Elborn JS, Kahl BC, Traore H, Vanderbist F, Tulkens PM, Van Bambeke F. 2016. Antimicrobial susceptibility of Pseudomonas aeruginosa isolated from cystic fibrosis patients in northern Europe. Antimicrob Agents Chemother 60:6735–6741. doi: 10.1128/AAC.01046-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shirani K, Ataei B, Roshandel F. 2016. Antibiotic resistance pattern and evaluation of metallo-beta lactamase genes (VIM and IMP) in Pseudomonas aeruginosa strains producing MBL enzyme, isolated from patients with secondary immunodeficiency. Adv Biomed Res 5:124. doi: 10.4103/2277-9175.186986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dwyer DJ, Kohanski MA, Hayete B, Collins JJ. 2007. Gyrase inhibitors induce an oxidative damage cellular death pathway in Escherichia coli. Mol Syst Biol 3:91. doi: 10.1038/msb4100135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yeom J, Imlay JA, Park W. 2010. Iron homeostasis affects antibiotic-mediated cell death in Pseudomonas species. J Biol Chem 285:22689–22695. doi: 10.1074/jbc.M110.127456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Winkler WC, Nahvi A, Roth A, Collins JA, Breaker RR. 2004. Control of gene expression by a natural metabolite-responsive ribozyme. Nature 428:281–286. doi: 10.1038/nature02362. [DOI] [PubMed] [Google Scholar]
- 6.Kawai Y, Mercier R, Wu LJ, Dominguez-Cuevas P, Oshima T, Errington J. 2015. Cell growth of wall-free L-form bacteria is limited by oxidative damage. Curr Biol 25:1613–1618. doi: 10.1016/j.cub.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karimi R, Ehrenberg M. 1994. Dissociation rate of cognate peptidyl-tRNA from the A-site of hyper-accurate and error-prone ribosomes. Eur J Biochem 226:355–360. doi: 10.1111/j.1432-1033.1994.tb20059.x. [DOI] [PubMed] [Google Scholar]
- 8.Fourmy D, Recht MI, Blanchard SC, Puglisi JD. 1996. Structure of the A site of Escherichia coli 16S ribosomal RNA complexed with an aminoglycoside antibiotic. Science 274:1367–1371. doi: 10.1126/science.274.5291.1367. [DOI] [PubMed] [Google Scholar]
- 9.Pape T, Wintermeyer W, Rodnina MV. 2000. Conformational switch in the decoding region of 16S rRNA during aminoacyl-tRNA selection on the ribosome. Nat Struct Biol 7:104–107. doi: 10.1038/72364. [DOI] [PubMed] [Google Scholar]
- 10.Liu X, De Wulf P. 2004. Probing the ArcA-P modulon of Escherichia coli by whole genome transcriptional analysis and sequence recognition profiling. J Biol Chem 279:12588–12597. doi: 10.1074/jbc.M313454200. [DOI] [PubMed] [Google Scholar]
- 11.Malpica R, Franco B, Rodriguez C, Kwon O, Georgellis D. 2004. Identification of a quinone-sensitive redox switch in the ArcB sensor kinase. Proc Natl Acad Sci U S A 101:13318–13323. doi: 10.1073/pnas.0403064101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Y, Imlay JA. 2013. Cell death from antibiotics without the involvement of reactive oxygen species. Science 339:1210–1213. doi: 10.1126/science.1232751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ezraty B, Vergnes A, Banzhaf M, Duverger Y, Huguenot A, Brochado AR, Su SY, Espinosa L, Loiseau L, Py B, Typas A, Barras F. 2013. Fe-S cluster biosynthesis controls uptake of aminoglycosides in a ROS-less death pathway. Science 340:1583–1587. doi: 10.1126/science.1238328. [DOI] [PubMed] [Google Scholar]
- 14.Keren I, Wu Y, Inocencio J, Mulcahy LR, Lewis K. 2013. Killing by bactericidal antibiotics does not depend on reactive oxygen species. Science 339:1213–1216. doi: 10.1126/science.1232688. [DOI] [PubMed] [Google Scholar]
- 15.Wang X, Li M, Liu L, Mou R, Zhang X, Bai Y, Xu H, Qiao M. 2012. DsbM, a novel disulfide oxidoreductase affects aminoglycoside resistance in Pseudomonas aeruginosa by OxyR-regulated response. J Microbiol 50:932–938. doi: 10.1007/s12275-012-2177-3. [DOI] [PubMed] [Google Scholar]
- 16.Jo I, Park N, Chung IY, Cho YH, Ha NC. 2016. Crystal structures of the disulfide reductase DsbM from Pseudomonas aeruginosa. Acta Crystallogr D Struct Biol 72:1100–1109. doi: 10.1107/S2059798316013024. [DOI] [PubMed] [Google Scholar]
- 17.Li M, Guan X, Wang X, Xu H, Bai Y, Zhang X, Qiao M. 2014. DsbM affects aminoglycoside resistance in Pseudomonas aeruginosa by the reduction of OxyR. FEMS Microbiol Lett 352:184–189. doi: 10.1111/1574-6968.12384. [DOI] [PubMed] [Google Scholar]
- 18.Han X, Liu Y, Ma Y, Xu H, Qiao M. 2019. Interaction between disulfide oxidoreductase (DsbM) and LysR type transcriptional regulator (PA0056) in Pseudomonas aeruginosa. Acta Sci Nat Univ Nankaiensis 52:1–6. [Google Scholar]
- 19.Foti JJ, Devadoss B, Winkler JA, Collins JJ, Walker GC. 2012. Oxidation of the guanine nucleotide pool underlies cell death by bactericidal antibiotics. Science 336:315–319. doi: 10.1126/science.1219192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hassett DJ, Charniga L, Bean K, Ohman DE, Cohen MS. 1992. Response of Pseudomonas aeruginosa to pyocyanin: mechanisms of resistance, antioxidant defenses, and demonstration of a manganese-cofactored superoxide dismutase. Infect Immun 60:328–336. doi: 10.1128/IAI.60.2.328-336.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma JF, Ochsner UA, Klotz MG, Nanayakkara VK, Howell ML, Johnson Z, Posey JE, Vasil ML, Monaco JJ, Hassett DJ. 1999. Bacterioferritin A modulates catalase A (KatA) activity and resistance to hydrogen peroxide in Pseudomonas aeruginosa. J Bacteriol 181:3730–3742. doi: 10.1128/JB.181.12.3730-3742.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wei Q, Minh PNL, Dötsch A, Hildebrand F, Panmanee W, Elfarash A, Schulz S, Plaisance S, Charlier D, Hassett D, Häussler S, Cornelis P. 2012. Global regulation of gene expression by OxyR in an important human opportunistic pathogen. Nucleic Acids Res 40:4320–4333. doi: 10.1093/nar/gks017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng M, Aslund F, Storz G. 1998. Activation of the OxyR transcription factor by reversible disulfide bond formation. Science 279:1718–1721. doi: 10.1126/science.279.5357.1718. [DOI] [PubMed] [Google Scholar]
- 24.Storz G, Tartaglia LA, Ames BN. 1990. Transcriptional regulator of oxidative stress-inducible genes: direct activation by oxidation. Science 248:189–194. doi: 10.1126/science.2183352. [DOI] [PubMed] [Google Scholar]
- 25.Maddocks SE, Oyston PC. 2008. Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology (Reading) 154:3609–3623. doi: 10.1099/mic.0.2008/022772-0. [DOI] [PubMed] [Google Scholar]
- 26.Makino K, Amemura M, Kawamoto T, Kimura S, Shinagawa H, Nakata A, Suzuki M. 1996. DNA binding of PhoB and its interaction with RNA polymerase. J Mol Biol 259:15–26. doi: 10.1006/jmbi.1996.0298. [DOI] [PubMed] [Google Scholar]
- 27.Schell MA. 1993. Molecular biology of the LysR family of transcriptional regulators. Annu Rev Microbiol 47:597–626. doi: 10.1146/annurev.mi.47.100193.003121. [DOI] [PubMed] [Google Scholar]
- 28.El-Halfawy OM, Valvano MA. 2014. Putrescine reduces antibiotic-induced oxidative stress as a mechanism of modulation of antibiotic resistance in Burkholderia cenocepacia. Antimicrob Agents Chemother 58:4162–4171. doi: 10.1128/AAC.02649-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu C, Wang N, Wu M, Tian F, Chen H, Yang F, Yuan X, Yang CH, He C. 2016. OxyR-regulated catalase CatB promotes the virulence in rice via detoxifying hydrogen peroxide in Xanthomonas oryzae pv. oryzae. BMC Microbiol 16:269. doi: 10.1186/s12866-016-0887-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ochsner UA, Vasil ML, Alsabbagh E, Parvatiyar K, Hassett DJ. 2000. Role of the Pseudomonas aeruginosa oxyR-recG operon in oxidative stress defense and DNA repair: OxyR-dependent regulation of katB-ankB, ahpB, and ahpC-ahpF. J Bacteriol 182:4533–4544. doi: 10.1128/jb.182.16.4533-4544.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dwyer DJ, Belenky PA, Yang JH, MacDonald IC, Martell JD, Takahashi N, Chan CTY, Lobritz MA, Braff D, Schwarz EG, Ye JD, Pati M, Vercruysse M, Ralifo PS, Allison KR, Khalil AS, Ting AY, Walker GC, Collins JJ. 2014. Antibiotics induce redox-related physiological alterations as part of their lethality. Proc Natl Acad Sci U S A 111:E2100–E2109. doi: 10.1073/pnas.1401876111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choi H, Lee DG. 2012. Synergistic effect of antimicrobial peptide arenicin-1 in combination with antibiotics against pathogenic bacteria. Res Microbiol 163:479–486. doi: 10.1016/j.resmic.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 33.Tkachenko AG, Akhova AV, Shumkov MS, Nesterova LY. 2012. Polyamines reduce oxidative stress in Escherichia coli cells exposed to bactericidal antibiotics. Res Microbiol 163:83–91. doi: 10.1016/j.resmic.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 34.Kolodkin-Gal I, Sat B, Keshet A, Engelberg-Kulka H. 2008. The communication factor EDF and the toxin-antitoxin module mazEF determine the mode of action of antibiotics. PLoS Biol 6:e319. doi: 10.1371/journal.pbio.0060319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Imlay JA. 2015. Diagnosing oxidative stress in bacteria: not as easy as you might think. Curr Opin Microbiol 24:124–131. doi: 10.1016/j.mib.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dwyer DJ, Collins JJ, Walker GC. 2015. Unraveling the physiological complexities of antibiotic lethality. Annu Rev Pharmacol Toxicol 55:313–332. doi: 10.1146/annurev-pharmtox-010814-124712. [DOI] [PubMed] [Google Scholar]
- 37.Dridi B, Lupien A, Bergeron MG, Leprohon P, Ouellette M. 2015. Differences in antibiotic-induced oxidative stress responses between laboratory and clinical isolates of Streptococcus pneumoniae. Antimicrob Agents Chemother 59:5420–5426. doi: 10.1128/AAC.00316-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lobritz MA, Belenky P, Porter CBM, Gutierrez A, Yang JH, Schwarz EG, Dwyer DJ, Khalil AS, Collins JJ. 2015. Antibiotic efficacy is linked to bacterial cellular respiration. Proc Natl Acad Sci U S A 112:8173–8180. doi: 10.1073/pnas.1509743112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao X, Drlica K. 2014. Reactive oxygen species and the bacterial response to lethal stress. Curr Opin Microbiol 21:1–6. doi: 10.1016/j.mib.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Malik M, Hussain S, Drlica K. 2007. Effect of anaerobic growth on quinolone lethality with Escherichia coli. Antimicrob Agents Chemother 51:28–34. doi: 10.1128/AAC.00739-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chagas TPG, Seki LM, Cury JC, Oliveira JAL, Dávila AMR, Silva DM, Asensi MD. 2011. Multiresistance, beta-lactamase-encoding genes and bacterial diversity in hospital wastewater in Rio de Janeiro, Brazil. J Appl Microbiol 111:572–581. doi: 10.1111/j.1365-2672.2011.05072.x. [DOI] [PubMed] [Google Scholar]
- 42.Kong KF, Jayawardena SR, del Puerto A, Wiehlmann L, Laabs U, Tummler B, Mathee K. 2005. Characterization of PoxB, a chromosomal-encoded Pseudomonas aeruginosa oxacillinase. Gene 358:82–92. doi: 10.1016/j.gene.2005.05.027. [DOI] [PubMed] [Google Scholar]
- 43.Drawz SM, Bonomo RA. 2010. Three decades of β-lactamase inhibitors. Clin Microbiol Rev 23:160–201. doi: 10.1128/CMR.00037-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li M, Guan X, Du X, Xu H, Bai Y, Zhang X, Qiao M. 2014. Cloning and expression of PA0057 gene in E. coli and purification and primary studies on characterization of recombinant protein. Acta Sci Nat Univ Nankaiensis 47:1–7. [Google Scholar]
- 45.Nagy P. 2013. Kinetics and mechanisms of thiol-disulfide exchange covering direct substitution and thiol oxidation-mediated pathways. Antioxid Redox Signal 18:1623–1641. doi: 10.1089/ars.2012.4973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Denoncin K, Vertommen D, Arts IS, Goemans CV, Rahuel-Clermont S, Messens J, Collet JF. 2014. A new role for Escherichia coli DsbC protein in protection against oxidative stress. J Biol Chem 289:12356–12364. doi: 10.1074/jbc.M114.554055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rietsch A, Bessette P, Georgiou G, Beckwith J. 1997. Reduction of the periplasmic disulfide bond isomerase, DsbC, occurs by passage of electrons from cytoplasmic thioredoxin. J Bacteriol 179:6602–6608. doi: 10.1128/jb.179.21.6602-6608.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Collet JF, Messens J. 2010. Structure, function, and mechanism of thioredoxin proteins. Antioxid Redox Signal 13:1205–1216. doi: 10.1089/ars.2010.3114. [DOI] [PubMed] [Google Scholar]
- 49.Fernandes AP, Holmgren A. 2004. Glutaredoxins: glutathione-dependent redox enzymes with functions far beyond a simple thioredoxin backup system. Antioxid Redox Signal 6:63–74. doi: 10.1089/152308604771978354. [DOI] [PubMed] [Google Scholar]
- 50.Wiegand I, Hilpert K, Hancock REW. 2008. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat Protoc 3:163–175. doi: 10.1038/nprot.2007.521. [DOI] [PubMed] [Google Scholar]
- 51.Weng Y, Chen F, Liu Y, Zhao Q, Chen R, Pan X, Liu C, Cheng Z, Jin S, Jin Y, Wu W. 2016. Pseudomonas aeruginosa enolase influences bacterial tolerance to oxidative stresses and virulence. Front Microbiol 7:1999. doi: 10.3389/fmicb.2016.01999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. 2007. A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130:797–810. doi: 10.1016/j.cell.2007.06.049. [DOI] [PubMed] [Google Scholar]
- 53.Sillanpaa J, Nallapareddy SR, Qin X, Singh KV, Muzny DM, Kovar CL, Nazareth LV, Gibbs RA, Ferraro MJ, Steckelberg JM, Weinstock GM, Murray BE. 2009. A collagen-binding adhesin, Acb, and ten other putative MSCRAMM and pilus family proteins of Streptococcus gallolyticus subsp. gallolyticus (Streptococcus bovis group, biotype I). J Bacteriol 191:6643–6653. doi: 10.1128/JB.00909-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu H, Lin W, Xia H, Xu S, Li Y, Yao H, Bai F, Zhang X, Bai Y, Saris P, Qiao M. 2005. Influence of ptsP gene on pyocyanin production in Pseudomonas aeruginosa. FEMS Microbiol Lett 253:103–109. doi: 10.1016/j.femsle.2005.09.027. [DOI] [PubMed] [Google Scholar]
- 55.Tan SY, Liu Y, Chua SL, Vejborg RM, Jakobsen TH, Chew SC, Li Y, Nielsen TE, Tolker-Nielsen T, Yang L, Givskov M. 2014. Comparative systems biology analysis to study the mode of action of the isothiocyanate compound iberin on Pseudomonas aeruginosa. Antimicrob Agents Chemother 58:6648–6659. doi: 10.1128/AAC.02620-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun W, Xie J, Lin H, Mi S, Li Z, Hua F, Hu Z. 2012. A combined strategy improves the solubility of aggregation-prone single-chain variable fragment antibodies. Protein Expr Purif 83:21–29. doi: 10.1016/j.pep.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 57.Li Y, Sousa R. 2012. Novel system for in vivo biotinylation and its application to crab antimicrobial protein scygonadin. Biotechnol Lett 34:1629–1635. doi: 10.1007/s10529-012-0942-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jerabek-Willemsen M, Wienken CJ, Braun D, Baaske P, Duhr S. 2011. Molecular interaction studies using microscale thermophoresis. Assay Drug Dev Technol 9:342–353. doi: 10.1089/adt.2011.0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lippok S, Seidel SAI, Duhr S, Uhland K, Holthoff H-P, Jenne D, Braun D. 2012. Direct detection of antibody concentration and affinity in human serum using microscale thermophoresis. Anal Chem 84:3523–3530. doi: 10.1021/ac202923j. [DOI] [PubMed] [Google Scholar]
- 60.Aitken A, Learmonth M. 1996. Estimation of disulfide bonds using Ellman’s reagent, p 487–488. In Walker JM (ed), The protein protocols handbook. Humana Press, Totowa, NJ. doi: 10.1007/978-1-60327-259-9_82. [DOI] [Google Scholar]
- 61.Åslund F, Zheng M, Beckwith J, Storz G. 1999. Regulation of the OxyR transcription factor by hydrogen peroxide and the cellular thiol-disulfide status. Proc Natl Acad Sci U S A 96:6161–6165. doi: 10.1073/pnas.96.11.6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Da Q, Wang P, Wang M, Sun T, Jin H, Liu B, Wang J, Grimm B, Wang H-B. 2017. Thioredoxin and NADPH-dependent thioredoxin reductase C regulation of tetrapyrrole biosynthesis. Plant Physiol 175:652–666. doi: 10.1104/pp.16.01500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zheng RP, Feng XM, Wei XY, Pan XL, Liu C, Song RP, Jin YX, Bai F, Jin SG, Wu WH, Cheng ZH. 2018. PutA is required for virulence and regulated by PruR in Pseudomonas aeruginosa. Front Microbiol 9:548. doi: 10.3389/fmicb.2018.00548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Caldara M, Minh PN, Bostoen S, Massant J, Charlier D. 2007. ArgR-dependent repression of arginine and histidine transport genes in Escherichia coli K-12. J Mol Biol 373:251–267. doi: 10.1016/j.jmb.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 65.Wang Y, Cen XF, Zhao GP, Wang J. 2012. Characterization of a new GlnR binding box in the promoter of amtB in Streptomyces coelicolor inferred a PhoP/GlnR competitive binding mechanism for transcriptional regulation of amtB. J Bacteriol 194:5237–5244. doi: 10.1128/JB.00989-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.