

Abstract
Glioblastoma Multiforme (GBM) tumors contain a small population of glioma stem-like cells (GSCs) among the various differentiated GBM cells (d-GCs). GSCs drive tumor recurrence, and resistance to Temozolomide (TMZ), the standard of care (SoC) for GBM chemotherapy. In order to investigate a potential link between GSC specific mitochondria function and SoC resistance, two patient-derived GSC lines were evaluated for differences in their mitochondrial metabolism. In both the lines, GSCs had significantly lower mitochondrial -content, and -function compared to d-GCs. In vitro, the standard mitochondrial-specific inhibitors oligomycin A, antimycin A, and rotenone selectively inhibited GSC proliferation to a greater extent than d-GCs and human primary astrocytes. These findings indicate that mitochondrial inhibition can be a potential GSC-targeted therapeutic strategy in GBM with minimal off-target toxicity. Mechanistically the standard mitochondrial inhibitors elicit their GSC-selective cytotoxic effects through the induction of apoptosis or autophagy pathways. We tested for GSC proliferation in the presence of 3 safe FDA-approved drugs--trifluoperazine, mitoxantrone, and pyrvinium pamoate, all of which are also known mitochondrial-targeting agents. The SoC GBM therapeutic TMZ did not trigger cytotoxicity in glioma stem cells, even at 100 μM concentration. By contrast, trifluoperazine, mitoxantrone, and pyrvinium pamoate exerted antiproliferative effects in GSCs about 30–50 fold more effectively than temozolomide. Thus, we hereby demonstrate that FDA-approved mitochondrial inhibitors induce GSC-selective cytotoxicity, and targeting mitochondrial function could present a potential therapeutic option for GBM treatment.
Keywords: Glioblastoma multiforme, mitochondria, chemotherapy, cancer stem cells, drug repurposing, therapeutics
Graphical abstract
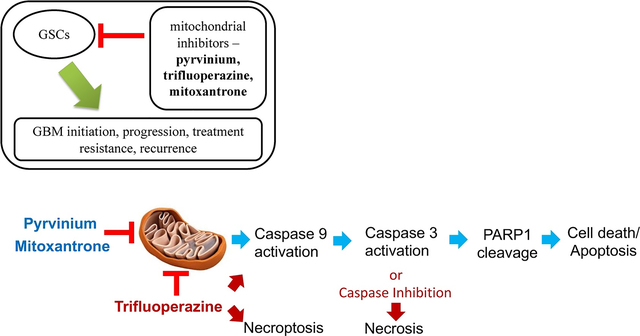
1.0. Introduction
Glioblastoma multiforme (GBM) is a lethal form of a brain tumor in adults, accounting for ~40% of total malignant brain tumors [1]. Every year approximately 14,000 new cases of GBM are diagnosed in the United States [2]. Standard of Care for GBM consist of maximal surgical resection of the tumor, followed by radiation therapy and adjuvant chemotherapy with Temozolomide (Temodar®) [3, 4]. Even after employing all possible treatments, the prognosis of GBM is poor with 15–18 months median survival [5]. A well-accepted hypothesis for the cause of poor prognosis is the development of radio- and chemotherapy resistance leading to tumor progression [6]. According to the current paradigm, the development of chemo- and radioresistance is derived from genetic and functional heterogeneity of GBM cells, which is thought to be driven by a small population of glioma stem-like cells (GSCs) [7]. The small population of GSCs expresses stem cell markers and exhibits unlimited self-renewal and tumorigenic properties. By contrast, the highly prevalent d-GCs exhibit limited self-renewal and tumor-initiating capacity [8].
GSCs can evade the immune system [9], infiltrate nearby healthy brain tissues [10] and show higher expression of ATP-binding cassette transporters, which confer them resistance to anticancer chemotherapy [11]. These GSCs can also repopulate a tumor by first converting into progenitor-like cells and then differentiating into d-GCs [12]. The infiltration of GSCs into adjacent tissues creates extreme difficultly for surgical resection and/or radiotherapeutic regression. GBM’s resistance to treatment seems driven by GSC-cell biology--as even a few GSCs evading treatment can repopulate the tumor and cause a malignant relapse [6]. Although GSCs have been identified as a major obstacle in improving GBM patient prognosis and survival, there is currently no GSC-targeted FDA-approved therapy in the clinic. The standard of care (SoC) chemotherapeutic Temozolomide exerts a short-term antiproliferative effect on the GSCs and extends patient survival for 6 months to 1-year. Despite the treatment, however, GBMs typically recur after one year, and the recurrent GBM is TMZ-resistant [13]. In addition, studies show that TMZ can increase GSC enrichment in vitro as well as in human patients and, therefore, potentially increase the chance of tumor relapse [14, 15]. Hence there is an urgent need to develop new drugs that selectively target GSCs.
In order to develop drugs that selectively kill GSCs and/or limit the off-target adverse effects, efforts should focus on targeting a biochemical property unique to the GSC or a metabolic pathway that should have a much higher impact on GSCs compared to the surrounding non-malignant cells [16]. Cancer cells often acquire unique metabolic rearrangement to suit their need for unlimited proliferation and tissue invasion [17–19]. A well-known metabolic change of some cancer cells, known as the Warburg effect, restricts aerobic (mitochondrial) metabolism and increases anaerobic metabolism, including glycolysis. However, this phenomenon is not universal among all cancers [20]. Metabolic targeting in vitro and in vivo has shown to be effective in reversing the stem-like properties of GSCs [21]. While it is ambiguous whether GSCs have a universal glycolytic or oxidative metabolic phenotype, the mitochondrial function has shown to be essential for GSC-survival and maintenance of stem-like properties [22]. In addition, mitochondrial heterogeneity among glioma cells has been reported to create resistance to TMZ through the loss of mitochondrial DNA (mtDNA) copy number in patients with relapsing GBMs [23]. MtDNA heteroplasmy and decreased mitochondrial complexes I and V activity with upregulation of a cytochrome c oxidase isoform has been observed in GBM. Apparent knock-down of this isoform restores TMZ sensitivity in resistant glioma cells. This indicates a metabolic vulnerability of GSCs, which could be targeted with small-molecule mitochondrial electron transport chain (ETC) inhibitors. We previously screened a library of 1600 safe FDA-approved drugs to identify novel mitochondrial inhibitors [24], and in this study, we tested the potency of three previously known mitochondrial inhibitors to kill GSCs.
In summary, in the current study, we demonstrate that patient-derived recurrent GSC lines show intertumoral mitochondrial heterogeneity, and have a lower mitochondrial number, lower mitochondrial gene expression, and lower mitochondrial function as compared to differentiated tumor cells. We also demonstrated that classical mitochondrial ETC inhibitors such as rotenone, antimycin A and oligomycin A decrease GSC viability about 100-fold more potently than temozolomide. Furthermore, three FDA-approved drugs, trifluoperazine, mitoxantrone, and pyrvinium pamoate, also reduce GSC viability about 50-fold more potently than temozolomide.
2.0. Materials and Methods
All the chemicals used for this study were obtained from Sigma-Aldrich (MO, USA) unless specified otherwise.
2.1. Cell culture:
Patient-derived primary GSCs (0827 and 0923) were cultured as neurospheres in neurobasal A medium supplemented with 20 ng/ml Epidermal Growth Factor, 20 ng/ml basic-fibroblast growth factor, 0.5x B27 (Gibco), 0.5x N2 (Gibco), 0.5x glutamax (Gibco), 100 μg/mL streptomycin and 100 unit penicillin G, under 5% CO2 at 37 °C. The mature GBM cells (d-GCs) were obtained by culturing the GSCs in an adherent monolayer in DMEM media supplemented with 10% fetal bovine Albumin (FBS, Corning), 50 μg/mL unit streptomycin and 50 unit/mL penicillin G, under 5% CO2 at 37 °C for 10 days. These cell lines were originally established from patient-derived tissue after the approval of the National Cancer Institute Institutional Review Board from patient samples [25, 26]. All procedures to generate the patient-derived primary GSCs were done in accordance with relevant guidelines and regulations. Informed consents were obtained from the patients, and all the experimental protocols were approved by the National Cancer Institute Institutional Review Board and University of California, Davis Biological Use Authorization (BUA#R2077).
2.2. Cellular O2 consumption assay:
Cellular O2 consumption assays were performed using an Oxytherm Clark-type electrode system (Hansatech) as described previously [27]. The O2 consumption for each experiment were recorded after putting 2.5 million cells in the Oxytherm chamber containing 1 mL of serum, antibiotic, and bicarbonate-free DMEM medium (Corning). The baseline O2 consumption was recorded for 10 min or until the O2 tension in the chamber reached 50 nM/mL of O2 tension.
2.3. DNA and RNA isolation and quantification:
Following manufacturer’s instructions, the DNeasy blood & tissue kit (Qiagen, Valencia, CA, USA) was employed to isolate total DNA from 0827 and 0923 GSCs and d-GCs. Quantification of total isolated DNA was performed by a NanoDrop 2000c or NanoDrop One Spectrophotometer (Thermo Scientific, Waltham, MA, USA).
RNeasy plus mini kit (Qiagen, Valencia, CA) was used to extract total RNA from 0827 and 0923 GSCs and d-GCs. RNA quantity and quality were measured by a NanoDrop 2000c Spectrophotometer (Thermo Scientific, Waltham, MA, USA) [28].
2.4. Quantitative PCR (qPCR):
iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA, USA) was used to synthesize cDNA from total mRNA using a C1000 Touch Thermal Cycler (Bio-Rad Laboratories, Hercules, CA, USA). SensiFAST SYBR No-ROX Kit (Bioline, Taunton, MA, USA) was used to run the qPCR using Roche Lightcycler 480 (Roche Diagnostics, Indianapolis, IN, USA). The cycle threshold (Ct) value was determined, and the data were analyzed by delta CT calculation [28]. Table 1 lists primer sets used in qPCR.
Table 1.
qPCR primer list.
| Species | Gene | Sequence (5′ → 3′) |
|---|---|---|
| Human | MT-TL1 (DNA) Forward | CACCCAAGAACAGGGTTTGT |
| MT-TL1 (DNA) Reverse | TGGCCATGGGTATGTTGTTA | |
| Human | B2M (DNA) Forward | TGCTGTCTCCATGTTTGATGTATCT |
| B2M (DNA) Reverse | TCTCTGCTCCCCACCTCTAAGT | |
| Human | MT-ND2 Forward | CATATACCAAATCTCTCCCTC |
| MT-ND2 Reverse | GTGCGAGATAGTAGTAGGGTC | |
| Human | MT-CYB Forward | ACCCCCTAGGAATCACCTCC |
| MT-CYB Reverse | GCCTAGGAGGTCTGGTGAGA | |
| Human | β-ACTB Forward | GCCAACACAGTGCTGTCTGG |
| β-ACTB Reverse | CTGCTTGCTGATCCACATCTGC |
2.5. Cell viability assays:
The GSCs (0827 and 0923) and their respective d-GCs were plated at 50,000 cells/well in a tissue culture-treated, 96-well, white opaque walled, clear flat bottom plate (Corning #3903) in 100 μL of their respective culture media. The cells were allowed to grow overnight, and the next day the medium in the d-GC plates were aspirated, and 100 μL of GSC culture medium was added. This was done to remove any protective effect of 10% FBS in the d-GC growth medium. The drug stock solutions (1000x) were made in DMSO, and the working drug solutions were prepared at 2x concentrations in the GSC growth medium, and 100 uL of the 2x drug working solution was added in each well. The vehicle wells got treated with DMSO at the same v/v concentration as the drugs. The DMSO levels were 0.1% or less in each well. The cells were incubated for 48 h with the respective drugs, and the cell viability was measured using CellTiterGlo® according to the manufacturer’s instructions.
Human primary astrocytes were purchased from Sciencell (#1800) and were cultured according to the manufacturer’s instructions. The astrocytes between passage 1 – 3 were plated at 50,000 cells/well in a tissue culture-treated, 96-well clear flat bottomed plate in 100 μL of culture medium. The cells were allowed to grow overnight, and the next day the medium in the plates were aspirated, and 100 μL of GSC culture medium was added. The drug stock solutions (1000x) were made in DMSO, and the working drug solutions were prepared at 2x concentrations in the GSC growth medium, and 100 uL of the 2x drug working solution was added in each well. The vehicle wells got treated with DMSO at the same v/v concentration as the drugs. The DMSO levels were 0.1% or less in each well. The cells were incubated for 48 h with the respective drugs, and the cell viability was measured using Sulforhodamine B assay [29].
2.6. Protein extraction and western blot analysis:
The methods described in this section are previously published elsewhere [28]. GBM stem-like cells (GSCs) or d-GCs were homogenized with 1x cell lysis buffer (Cell Signaling Technologies, Danvers, MA, USA) containing 1x Halt phosphatase and protease inhibitor cocktail (Thermo-Fisher, Waltham, MA, USA) and 1% PMSF (Sigma-Aldrich, St. Louis, MO, USA). 25 μg of lysates were loaded per lane into 4–12% Bis-Tris gels (Invitrogen, Waltham, MA, USA) [28]. Electrophoresis was achieved in accordance with the manufacturer’s recommendations. After the proteins were resolved by electrophoresis, the proteins were then transferred to nitrocellulose membranes by the Trans-Blot Turbo System (Bio-Rad, Hercules, CA, USA) and blocked with an Odyssey blocking buffer (LI-COR Biosciences, Lincoln, NE, USA) for 1 h. Membranes were labeled overnight with the following primary antibodies in blocking buffer: 1:10,000 dilution Monoclonal Anti-α-Tubulin (T5168 Millipore Sigma, MO, USA), 1:1000 dilution GAPDH (14C10 Rabbit mAb #2118, Cell Signaling Technologies, Danvers, MA, USA), 1:15,000 dilution GFAP (Z0334 Agilent-Dako, Santa Clara, CA, USA), 1:2500 dilution OLIG2 (AB9610 polycloncal rabbit, Chemicon, EMD Millipore, Hayward, CA, USA), 1:500 dilution mt-ND2 (LSC -C137113 LifeSpan Biosciences Inc. Seattle, WA, USA), 1:1,000 dilution COX IV (COX IV (3E11) Rabbit mAb #4850, Cell Signaling Technologies, Danvers, MA, USA), 1:1,000 dilution Cleaved caspase-9 (Rabbit mAb #20750 Cell Signaling Technologies, Danvers, MA, USA), 1:1,000 dilution Cleaved caspase-3 (Rabbit #9661 Cell Signaling Technologies, Danvers, MA, USA), 1:1,000 dilution Cleaved PARP (Rabbit #9542 Cell Signaling Technologies, Danvers, MA, USA), 1, 1000 dilution Bcl 2 (Rabbit mAb #2870 Cell Signaling Technologies, Danvers, MA, USA) and 1:200 dilution MCL1 (sc-12756 Mouse mAb Santa Cruz Biotechnology Inc., Dallas, TX, USA). Both corresponding IRDye 680CW and IRDye 800CW-coupled secondary antibodies (LI-COR Biosciences, Lincoln, NE, USA) were diluted 1:20,000, and, subsequently, added to the membranes for a 1 h incubation. Proteins were visualized with the Odyssey infrared imager and software (LI-COR Biosciences, Lincoln, NE, USA) according to the manufacturer’s instruction [28].
2.7. Apoptosis Assay:
The GSCs (0827 and 0923) were plated at 200,000 cells/well, and their d-GCs were plated at 150,000 cells in a tissue culture-treated, 24-well, clear flat bottom plate in 500 μL of culture media. The cells were allowed to grow overnight. The drug stock solutions (1000x) were made in DMSO, and the working drug solutions were prepared at 2x concentrations in the GSC or d-GC growth medium, and 500 uL of the 2x drug working solution was added in each well. The vehicle wells got treated with DMSO at the same v/v concentration as the drugs. The DMSO levels were 0.1% or less in each well. After 48 h incubation with drug or vehicle, the cells were trypsinized to form a single-cell suspension. Cells were then mixed with the Muse- Annexin V and Dead Cell Assay solution at a 1:1 ratio by volume and incubated at room temp in the dark for 30 min per the manufacturer’s recommendations. Finally, a Muse- Cell Analyzer (Muse-Cell Analyzer; Millipore, Billerica, MA, USA) was used for data acquisition (percent apoptotic cells).
3. Results
3.1. Patient-derived GSCs show tumorigenicity-dependent mitochondrial heterogeneity and lower mitochondrial function than d-GCs
We compared the mitochondrial function of the two GSC lines (0827 and 0923) cultured as 3D neurospheres and their respective d-GCs in vitro. Based on the tumorigenicity, invasion, and survival of mice injected with the GSCs, 0827 is considered a less lethal and less invasive phenotype (tumorigenic in ~66% mice, Supplementary Fig. S1A–B) while 0923 was considered as a representative of a more lethal and highly invasive GBM (100% tumor induction and killing in mice, Supplementary Fig. S1A–B). The d-GCs from 0827 and 0923 were generated by culturing the GSCs in growth factor-free 10% FBS supplemented medium. Significant reduction of the most specific glioblastoma stem-like cell marker oligodendrocyte lineage transcription factor 2 (OLIG2) [30] and upregulation of the Glial fibrillary acidic protein (GFAP) was indicative of differentiation of the GSCs (Supplementary Fig. S2). Mitochondrial function was significantly higher (~2 fold) in the less invasive/tumorigenic cell line (0827), than the more invasive/tumorigenic cell line (0923), indicating mitochondrial heterogeneity in patient-derived GSC lines (Fig. 1A). The GSCs from both the lines showed significantly lower mitochondrial oxygen consumption rate (OCR) when compared to d-GCs for each cell line. The basal mitochondrial function was specific for each GSC line, and differentiation significantly increased mitochondrial activity for both GSC lines but to a different extent. Differentiation increased mitochondrial function ~2 fold higher in less aggressive line, while the more aggressive line revealed only ~1.4. These data indicate impairment in the mitochondrial biogenetic pathway and are consistent with the previous reports that the GBMs follow a ‘Warburg effect’, i.e., ‘more tumorigenic, less mitochondrial’ [21, 31]. The effect on mitochondrial function for each cell line was confirmed by measuring mitochondrial content in the GSCs and d-GCs using mitochondrial copy number. Similar to the mitochondrial function, we saw a significant increase in mitochondrial copy number in the less invasive/tumorigenic line upon differentiation, while there was no significant increase in the mitochondrial copy number in the more aggressive cell line after differentiation (Fig. 1B). Furthermore, mitochondrial content in the less invasive/tumorigenic cell line (0827), especially in the d-GCs, was significantly higher than the more invasive/tumorigenic cell line (0923) suggesting induction of Warburg effect in GBM.
Figure 1: Glioma stem-like cells show much lower mitochondrial function and content than differentiated glioma cells.
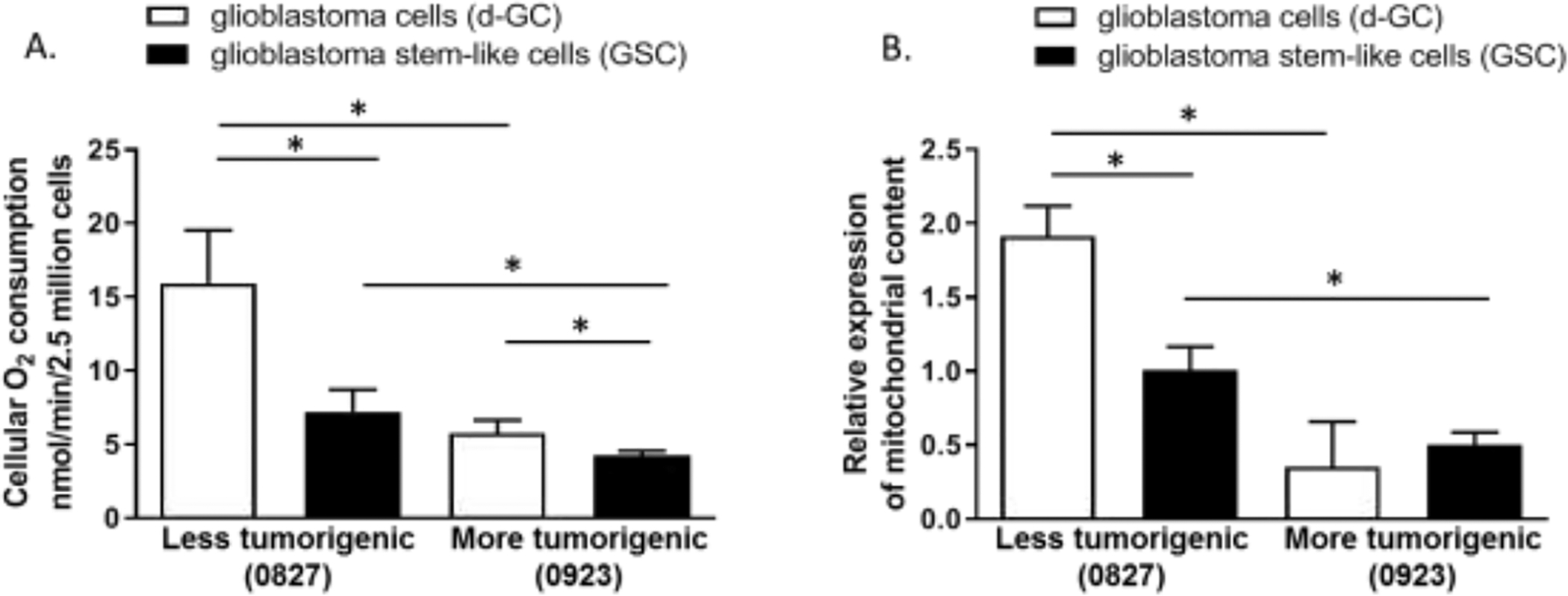
Glioma stem-like cells have significantly lower (A) mitochondrial function and (B) content (mitochondrial copy number). Differential glioma cells were generated by culturing glioma stem-like cells in growth factor-free, and 10% FBS supplemented medium. Mitochondrial O2 consumption was measured by the Clarke Electrode method. Mitochondrial content was measured by measuring mitochondrial DNA to nuclear DNA ratio by QPCR. Data are presented as average + std. dev from 3 independent observations (N=3). Data are analyzed with unpaired two-tailed student’s t-test. “*” denotes statistical significance (p < 0.05).
3.2. GSCs show lower mitochondrial gene expression
Our findings of reduced mitochondrial functions, and reduced mitochondrial DNA/cell, suggested that mitochondrial gene expression might also be reduced in Glioma stem cells. We evaluated the expression of mitochondrially encoded NADH:Ubiquinone Oxidoreductase Core Subunit 2 (ND2) and cytochrome b (CYB) at the RNA level. The difference of mitochondrially encoded gene expression between d-GCs and GSCs at the RNA level was statistically significant in the less tumorigenic cell line (0827) (Fig. 2) while in, the more tumorigenic cell line (0923) the d-GCs show an increasing trend in ND2 and CYB expression which was statistically not significant. The increase in the mitochondrial gene expression was also confirmed at the protein level by Western blot analysis using MT-ND2 and cytochrome oxidase IV (COX IV) subunit as markers. (Supplementary Fig. S3–S4).
Figure 2. Glioma stem-like cells have significantly lower mitochondrial gene expression than differentiated glioma cells.
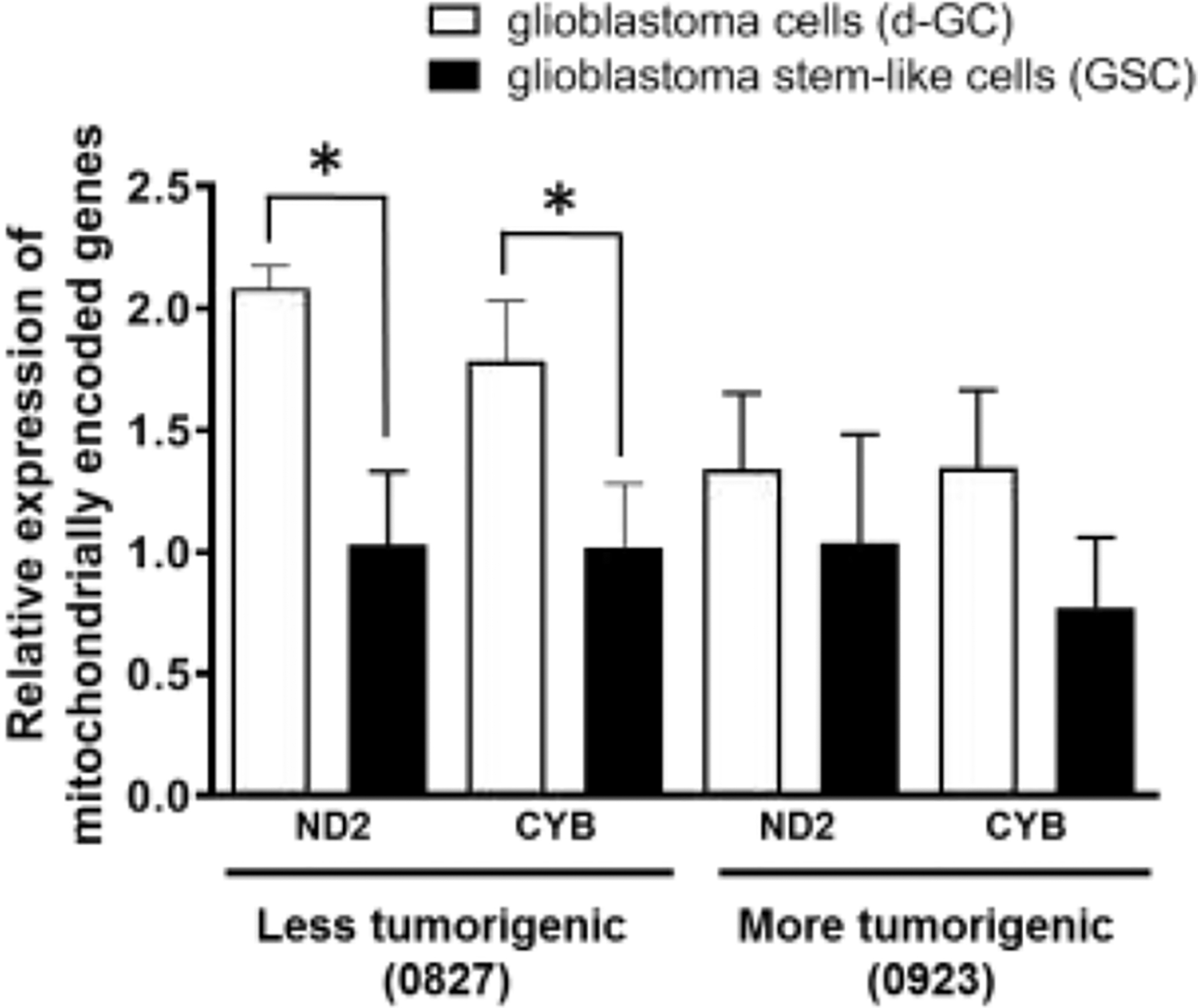
Differentiated glioma cells have significantly higher expression of mitochondrially encoded genes such as complex I subunit ND2 and Cytochrome B at the RNA level. Differential glioma cells were generated by culturing glioma stem-like cells in growth factor-free, and 10% FBS supplemented media. Mitochondrial gene expression at the RNA level was measured by QRTPCR. Data are presented as average + std. dev from 3 independent observations (N=3). Data are analyzed with unpaired two-tailed student’s t-test. “*” denotes statistical significance (p < 0.05).
3.3. Classical mitochondrial inhibitors inhibit patient-derived GSC-growth 100-fold more potently than the standard of care temozolomide.
Since we observe a mitochondrial heterogeneity between the GSCs and d-GCs, we hypothesized that there would be differential susceptibility of GSCs and d-GCs to the antiproliferative/cytotoxic effects of mitochondrial inhibitors. This hypothesis was tested by concentration-response experiments with the classical mitochondrial inhibitors oligomycin A (mitochondrial complex V inhibitor), antimycin A (mitochondrial complex III inhibitor), and rotenone (mitochondrial complex I inhibitor). We also used the SoC drug Temozolomide as a control. We observed two main findings. First, irrespective of the tumorigenic potential, GSCs were always significantly more sensitive to these classical mitochondrial inhibitors than the d-GC (Fig 3A–F). Secondly, mitochondrial inhibitors were about 100-fold more effective at reducing the GSC and d-GC numbers than temozolomide, which is the SoC in GBM. Thus, current GBM chemotherapy doesn’t appear to selectively target glioma stem cells at all (Supplementary Fig. S5), which is the rational basis of its long-term ineffectiveness.
Figure 3. Preferential antiproliferative effects of mitochondrial inhibitors on glioma stem-like cells.
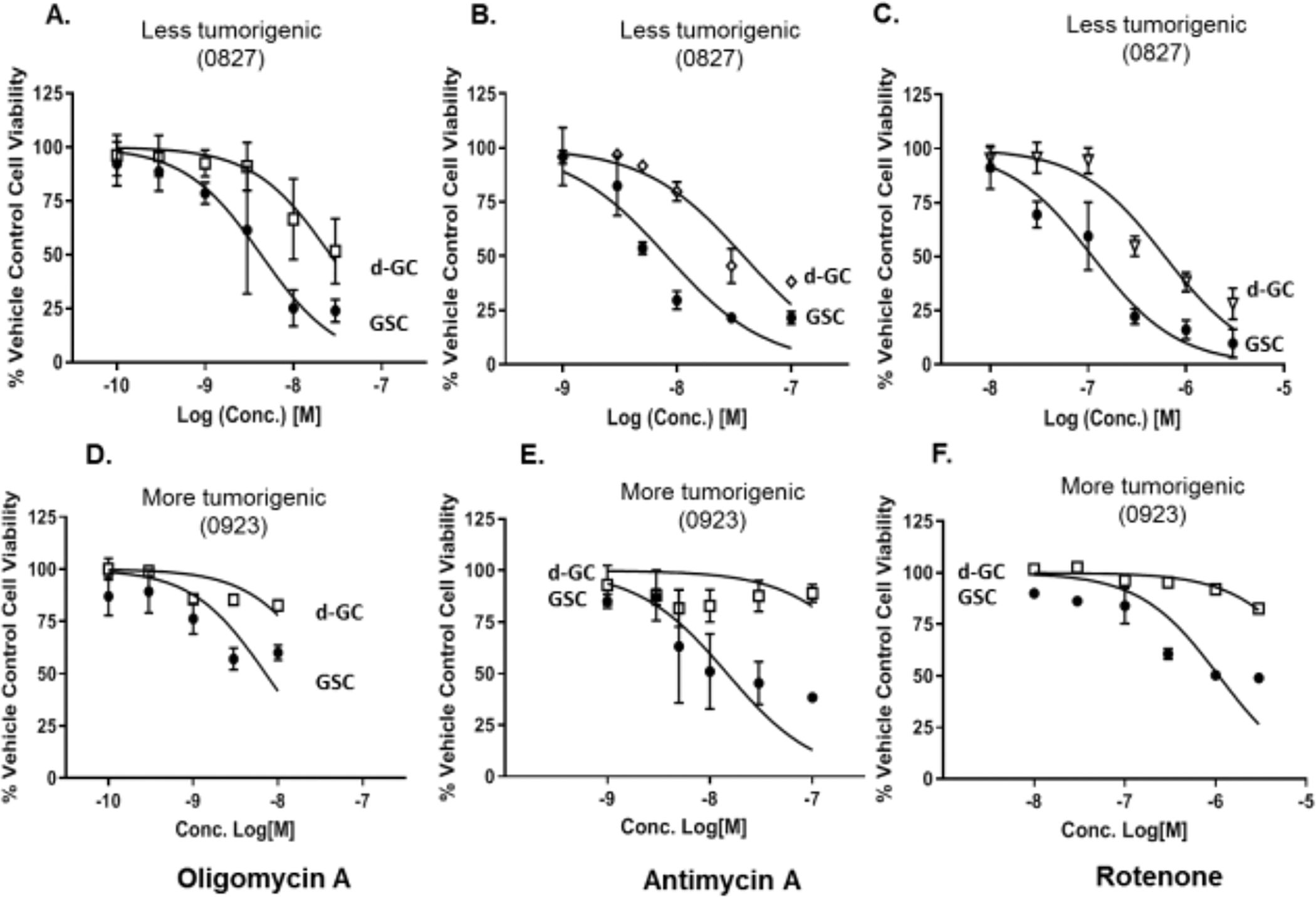
Glioma stem-like cells are significantly more sensitive to the antiproliferative effects of the mitochondrial inhibitors (A,D) oligomycin A; (B,E) antimycin A; and (C,F) rotenone. (A-C) 0827 glioma cell line. (D-F) 0923 glioma cell line. Furthermore, Classical mitochondrial inhibitors are about 100-fold more potent at killing GSCs than current GBM standard of care treatment, TMZ. Glioma stem-like cells were differentiated by the withdrawal of growth factors and the addition of 10% FBS. Cells were treated with specified concentrations of mitochondrial inhibitors for 48 h, and the cell viability was measured using the Cell titer-Glo™ method. Data are presented as average ± std. dev from 3 independent experiments (N=3). d-GC, differentiated glioma stem cells; GSC, Glioma stem cells.
3.4. Classical mitochondrial inhibitors induce apoptosis via the intrinsic apoptosis pathway in the less tumorigenic GSCs, whereas more tumorigenic GSCs are resistant to apoptosis induction.
Next, we investigated whether the selective reduction in GSC number by the mitochondrial inhibitors was due to cytotoxicity. We measured the percentage of apoptotic cells present 48 h after incubating with the mitochondrial inhibitors. All three mitochondrial inhibitors dose-dependently increased the percentage of annexin V positive cells in the less tumorigenic lines (0827) while the more tumorigenic cell line was resistant to apoptosis induction by the mitochondrial inhibitors (Fig. 4A–C, Supplementary Figure S6–S11), indicating that the standard mitochondrial inhibitors trigger cell death by induction of apoptosis in the less tumorigenic GSCs (0827). Differentiation of the less tumorigenic GSCs (0827) caused a marked decrease in apoptosis induction by the mitochondrial inhibitors, explaining their differential sensitivity to these agents (Supplementary Fig. S12). Cleavage of cysteine-dependent aspartate-directed protease-9 (caspase 9) (Fig. 4D and Supplementary Fig. S13) was detected in the less tumorigenic line (0827) after 24 h incubation with the mitochondrial inhibitors indicating the initiation of the intrinsic apoptosis pathway. Activation of the intrinsic apoptotic pathway was further confirmed by the detection of cleaved caspase 3 (Fig. 4D and Supplementary Fig. S14) and cleaved Poly [ADP-ribose] polymerase 1 (PARP1) (Fig. 4D and Supplementary Fig. S15) by immunoblotting. On the other hand, the more tumorigenic line (0923) was completely resistant to the induction of apoptosis by the mitochondrial inhibitors (Fig. 4A–C), suggesting that reduction of cell number observed in the 0923 GSC line is mediated by a non-apoptotic mechanism. We observed an upregulation of Microtubule-associated proteins 1A/1B light chain 3B (LC3) at the protein level in the more tumorigenic cell line (0923) in response to the mitochondrial inhibitors, suggesting that the process of autophagy might be involved in the GSC-specific antiproliferative effects of mitochondrial inhibitors (Supplementary Fig. S16) [32]. The more aggressive GSC lines showed a lower expression of B-Cell lymphoma 2 (Bcl 2) and Induced myeloid leukemia cell differentiation protein (Mcl-1) (Supplementary Fig. S17) indicating a general downregulation of Bcl 2 family proteins.
Figure 4. Concentration-dependent induction of apoptosis by mitochondrial inhibitors.
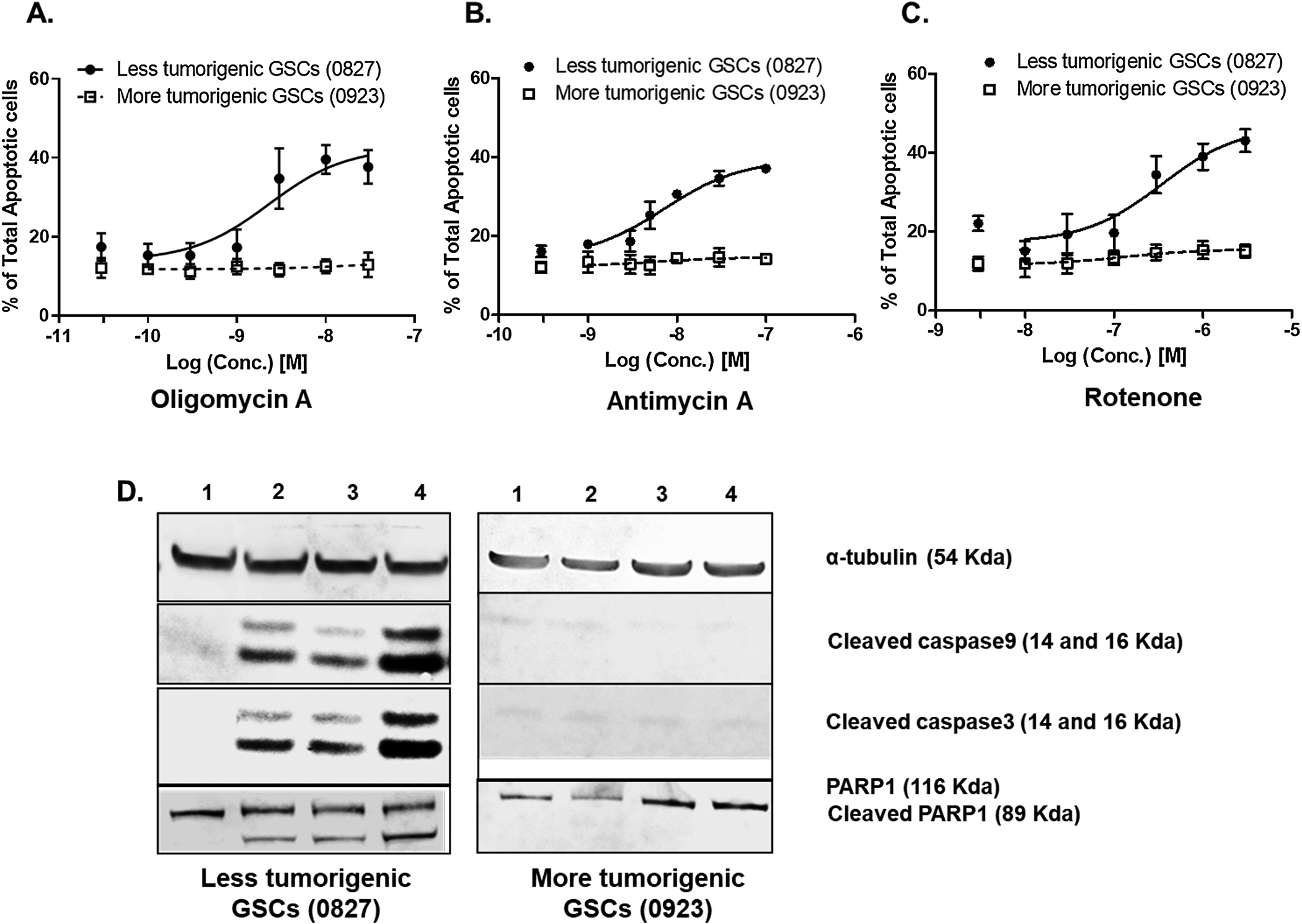
Mitochondrial inhibitors such as (A) Oligomycin A; (B) Antimycin A and (C) Rotenone concentration-dependently increase the percentage of total apoptotic cells in less tumorigenic GSCs (0827) whereas more tumorigenic GSCs are resistant. The GSCs were incubated with the mitochondrial inhibitors at the specified concentration for 48 h, and the percentage of annexin V positive cells was measured using a Muse Cell analyzer. Data are presented as average ± std. dev from 3 independent experiments (N=3). (D) A representative image of a cropped immunoblot showing cleavage of caspase 9 and caspase 3 and PARP1 indicating activation of intrinsic apoptosis pathway (Lane 1 = Vehicle; Lane 2 = Rotenone; Lane 3 = Antimycin; Lane 4 = Oligomycin A). The full immunoblots are shown in supplementary figures S13 (Caspase 9), S14 (Caspase 3), and S15 (PARP1).
3.5. Classical mitochondrial inhibitors do not affect human primary astrocytes at the GSC-inhibitory concentrations in vitro.
One of the major concerns for any cancer therapy is its specificity to target the tumor cells while sparing the normal cells in the body. Glioblastomas are thought to originate from glial cells such as astrocytes. Hence we tested the effects of the mitochondrial inhibitors on the human primary astrocytes. Oligomycin A, antimycin A, and rotenone did not affect human primary astrocyte numbers relative to the control at the concentrations they showed anti-GSC effects (≤ 15% reduction of human primary astrocytes at the highest dose) (Fig. 5A–F).
Figure 5: GSC-specificity of mitochondrial inhibitors compared to human primary astrocytes.
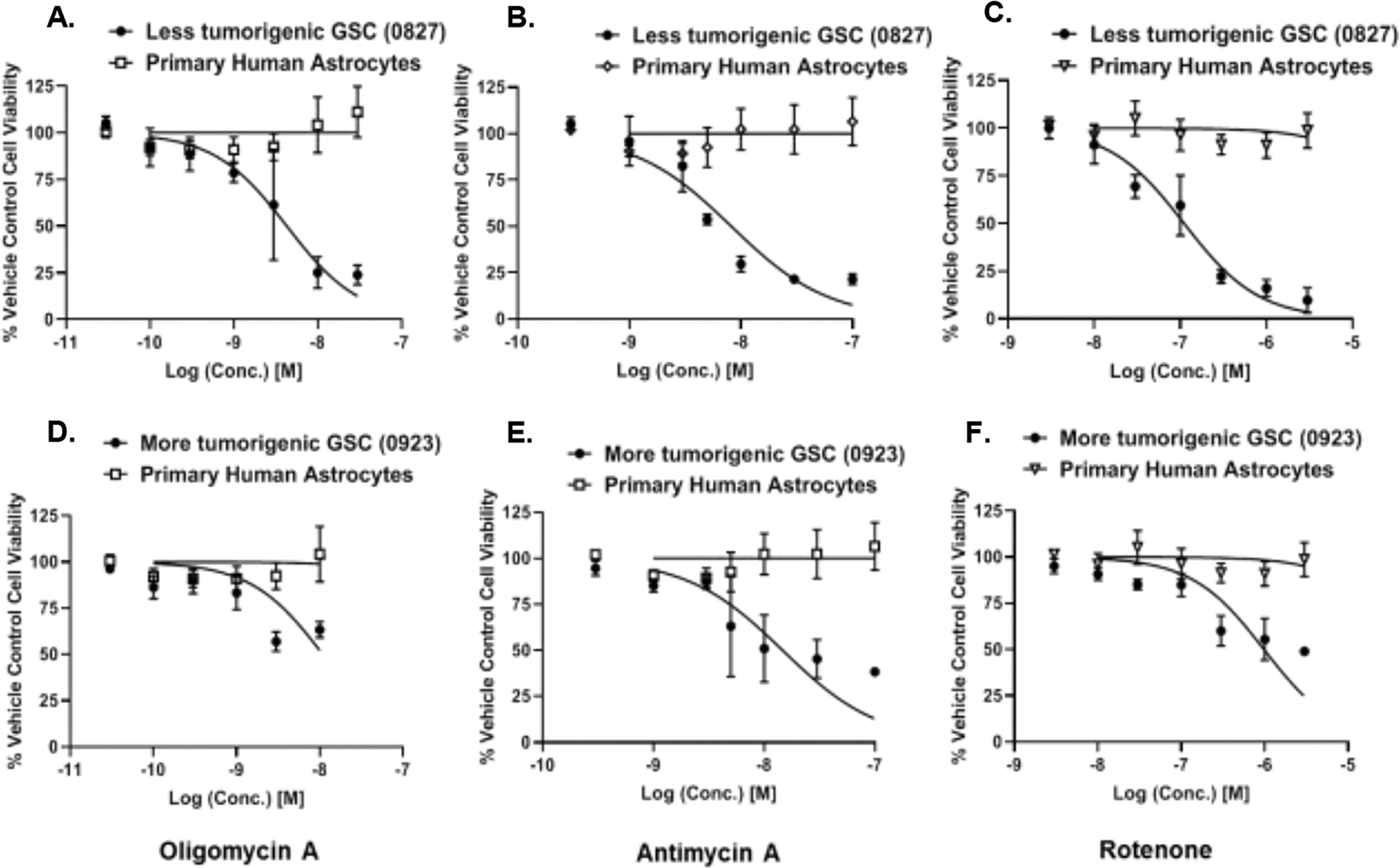
Human primary astrocytes do not show any prominent toxicity when incubated with mitochondrial inhibitors such as (A, D) Oligomycin A; (B, E) Antimycin A; and (C,F) Rotenone at the anti-GSC antiproliferative concentrations. (A-C) 0827 glioma cell line and astrocytes. (D-F) 0923 glioma cell line and astrocytes. Data are presented as average ± std. dev from 3 independent experiments (N=3).
3.6. Screening identifies milder mitochondrial inhibitors in a library of FDA-approved drugs that shows better GSC-selective antiproliferative activity than TMZ in vitro.
Although the classical mitochondrial inhibitors show GSC-specific cytotoxic activity and are 100-fold more potent towards GSCs than temozolomide, they, in general, are used for laboratory experiments in cells and animals and have not been evaluated in humans. We searched through our screen of 1600 clinically tried and FDA-approved drugs for their mitochondrial inhibitory properties, to identify three drugs that have been used in humans for other indications: pyrvinium pamoate, trifluoperazine, and mitoxantrone. Similar to the standard mitochondrial inhibitors, three FDA drugs showed GSC-specific reduction in cell number in the lesser tumorigenic GSC line (0827), with them being ≥ 5 fold more potent towards the GSCs vs. d-GCs (Fig.6A–C, Table 2). Pyrvinium pamoate and trifluoperazine maintained its GSC-specific activity in the more tumorigenic GSC line (Fig. 6D–E, Table 2). By contrast, mitoxantrone did not show any differential reduction in cell number between the GSC and d-GC state (Fig. 6F, Table 2). More importantly, these novel mitochondrial inhibitors, however, retained their potency compared to TMZ: they were about 50-fold more cytotoxic to GSCs than TMZ. When tested for cancer cell specificity, these three FDA-approved drugs showed a significant reduction of human primary astrocyte number (> 25% reduction in 48 h) (Supplementary Fig S18), indicating that these drugs, if used clinically, might have some ‘off-target’ toxicity. Among the three FDA-approved molecules, pyrvinium pamoate showed the best therapeutic selectivity followed by trifluoperazine. Mitoxantrone affected the GSCs and astrocytes equally, indicating that it may not be a good GSC-selective agent. Pyrvinium and mitoxantrone induced cleavage of Caspase9/3 in the less tumorigenic GSC line indicating that they are inducing mitochondria-mediated intrinsic apoptosis pathway similar to classical mitochondrial inhibitors. Trifluoperazine revealed an absence of Caspase 9/3 cleavage, which suggests a different mechanism (Supplementary Fig. 19–20). In Pancreatic ductal adenocarcinoma cells, trifluoperazine triggered mitochondrial stress, leading to decreased OXPHOS and activation of the unfolded protein response [33]. Both apoptosis and necroptosis were the cell death pathways initiated by trifluoperazine [33]. Similar findings showed that SH-SY5Y cells underwent apoptosis followed caspase inhibition and then necrosis induced by trifluoperazine [34]. Similarly, our data may suggest that trifluoperazine induces cell death favoring necroptosis over apoptosis, or apoptosis is initiated, but necrosis is the final mechanism in 0827 GSCs.
Figure 6.
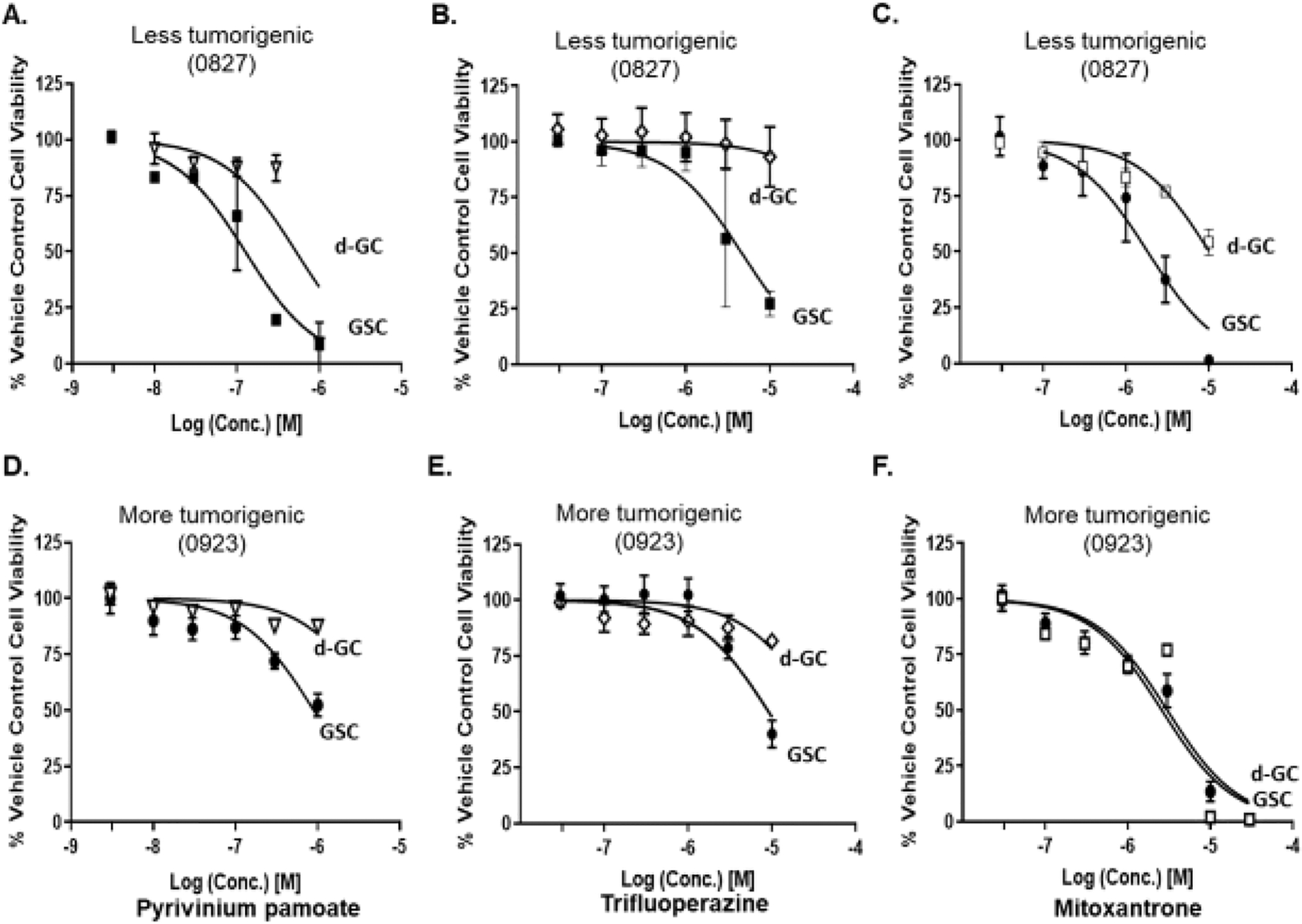
GSC-specific antiproliferative effects of mitochondria-targeted FDA approved drugs. An increase in GSC number is concentration-dependently inhibited by mitochondrially-targeted FDA-approved drugs (A, D) pyrvinium pamoate; (B, E) trifluoperazine; and (C, F) mitoxantrone. Glioma stem-like cells were treated with specified concentrations of FDA-approved drugs for 48 h, and the cell viability was measured using the Cell titre-Glo ™ method. Data are presented as average ± std. dev from 3 independent experiments (N=3).
Table 2.
The IC50s of the FDA-approved drugs and TMZ on GSC viabilities.
| Drugs | 0827 GSC IC50 (μM, 95% CI) | 0827 d-GC IC50 (μM, 95% CI) | 0923 GSC IC50 μM (95% CI) | 0923 d-GC IC50 μM (95% CI) |
|---|---|---|---|---|
| Pyrvinium pamoate | 0.1 (0.1 – 0.2) | 0.5 (0.3 – 0.9) | 0.9 (0.7 – 1.2) | 5.3 (3.5 – 7.9) |
| Trifluoperazine | 4.7 (3.4 – 6.5) | >100 | 9.1 (6.8 – 12.5) | 35.7 (22.8 – 66.5) |
| Mitoxantrone | 1.8 (1.4 – 2.4) | 10.2 (8.5–12.2) | 2.6 (2.1 – 3.2) | 3.0 (1.9 – 4.8) |
| Temozolomide | >100 | >100 | >100 | >100 |
These data suggest that mitochondrial inhibition elicit preferential cytotoxicity to GSCs within GBM and could be a potential adjuvant therapy to treat TMZ-resistant GBM. Thus, the in vitro data from both classical and novel mitochondrial inhibitors show about 100X and 50X more potency than TMZ, respectively.
4.0. Discussion
Cellular heterogeneity of GBM tumors has hindered advances in therapeutic intervention to promote tumor regression and increased patient survival [35]. Currently, DNA alkylating agent, TMZ, continues to remain the chemotherapeutic SoC for GBMs. However, since the FDA approval of TMZ in 2005, half of the GBM patients are shown to be nonresponsive to TMZ-treatment due to the high expression of O6-methylguanine methyltransferase (MGMT) [36]. Furthermore, under in vitro conditions, GBM cell lines revealed increased tumorigenicity (gauged by increased colony formation in soft agar) coupled with a corresponding significant increase of GSCs within their cell population after treatment with TMZ [37]. Our experiments indicate that TMZ does not target human patient-derived GSCs even up to concentrations of 100 μM.
Targeting metabolic pathways relevant to cancer cells has shown promising results both for in vitro and in vivo models for multiple cancers [38]. Additionally, interference of mitochondrial function by small-molecules in cancer cells has been shown to overcome radio/chemotherapeutic resistance in several types of cancers [39–42]. Inhibitors of mitochondrial ETC revealed selective depletion of cancer stem cell or tumor-initiating cell population in leukemia [43, 44], breast [45–47], pancreatic [48, 49] and colon cancers [50, 51]. In GBMs, mitochondrial inhibition by doxycycline has been shown to mitigate TMZ-induced enhancement of tumorigenicity in GBM cells determined by soft agar colony formation assay [37]. Metformin, a biguanide and mild complex I inhibitor, selectively targeted GSCs and was synergistic with TMZ to inhibit GSC-initiated tumor growth [52]. In general, small-molecule targeting of the mitochondrial oxidative phosphorylation pathway is potentially advantageous: 1) Glioblastoma stem-like cells show increased activation of hypoxia-inducible factor 1(HIF1) [53], which enhances cellular adaption of GSCs in a hypoxic (<5% O2) microenvironment, promotes drug resistance in GSCs [54], maintains tumorigenic properties of GSCs [55], and is also responsible for dedifferentiation of non-GSCs to GSCs [56]. It has been reported that inhibition of mitochondrial respiratory complexes inhibits hypoxic activation of HIF1 [57]. Hence, inhibition of HIF1 activation by the mitochondrial inhibitors can be an added advantage. 2) GSCs show an increased expression of ATP-binding cassettes of efflux transporters contributing to chemotherapy resistance [58]. Mitochondrial inhibition could lead to a reduction in ATP synthesis with decreasing drug efflux. 3) Mitochondrial heterogeneity between GSCs and d-GCs promotes precision targeting to eliminate GSCs. GBM relapse may be less likely to occur upon GSC eradication. Our results from this study show that, as a class, mitochondrial inhibitors could be useful as an adjuvant therapy to prevent tumor recurrence in certain GBM patients. Our data are in accord with multiple reports of mitochondrially active agents, causing selective GSC-depletion [59, 60]. It is, however, crucial that the potential novel anti-GBM therapeutic agents must be tested in multiple patient-derived GSC lines as we see significant variation in cellular signaling in the two patient-derived GSC lines tested in our study.
Irrespective of the advances in medical technologies, the drug discovery efforts against GBM have been mostly unsuccessful at the clinical level [61]. Loss of efficacy and poor blood-brain-barrier (BBB) penetration in preclinical in vivo models and clinical trials are the biggest challenges in the novel anti-GBM drug discovery efforts. Furthermore, the requirement of extensive evidence of the safety and efficacy of a novel therapeutic agent can take an extended period of time and remains a major obstacle to the rapid translation of novel therapies from bench to bedside. Drug repurposing or repositioning is an established method of rapidly increasing the availability of effective anti-GBM treatment options. Repurposed FDA-approved drug candidates with mitochondrial inhibitory effect could serve as alternate therapeutics to treat GBMs resistant to SoC chemo- and radiotherapies. This opens doors for hundreds of FDA-approved drugs with anti-mitochondrial effects to be tested on GBMs, and their repurposing could result in quicker entry into patient clinical trials due to their lack of documented adverse side effects. We previously screened a library of 1600 FDA-approved drugs and clinically-trialed molecules for their anti-mitochondrial effects [24]. We identified three FDA-approved drugs (trifluoperazine, mitoxantrone, and pyrvinium pamoate) with strong mitochondrial inhibitory effects. Earlier work showed that trifluoperazine promotes mitochondria stress with a decrease in OXPHOS and an increase in ROS [33]. Mitoxantrone decreases complex V activity and ATP production in cardiac cells [62] and reduces mitochondrial calcium uniporter activity [63]. In myeloma/erythroleukemia cells, pyrvinium pamoate inhibits mitochondrial complex I [64]. Pyrvinium pamoate impairs the reversal mitochondrial electron-transport chain complex, NADH-fumarate reductase system under hypoglycemic and hypoxic conditions found in tumor microenvironments [65]. In confirmation of our hypothesis, all three of these drugs exhibit cytotoxicity against both the GSC line tested in the current study. All three of these drugs have shown mitochondrial inhibition as well as efficacy in various GBM models in the past [66–69]. Our data reveal that mitochondrial inhibition is the mechanism of their cytotoxicity towards GSCs and, therefore, GBMs. Although out of the three drugs tested, only trifluoperazine is readily blood-brain-barrier permeable, local administration of pyrvinium pamoate through intracranial injection in an in vivo models show tumor xenograft regression [66]. In a clinical trial, locoregional chemotherapy with mitoxantrone increased the patient survival by 50% [67]. These results suggest that FDA-approved drugs with antimitochondrial effects can be considered a potential adjuvant therapy for TMZ-resistant GBM. Moreover, trifluoperazine has been shown to hinder the plasticity of the dGCs by preventing radiation therapy induced GSCs [70].
Hereby we report differential sensitivity of GSCs and d-GCs to the mitochondrial inhibitor and identification of clinically used drugs to overcome a major obstacle in GBM chemotherapy. In addition, our data suggest that in vitro drug discovery efforts using in vitro GSC cultures with serum may miss potential therapeutic agents due to a change in cellular signaling and subsequently a loss of sensitivity.
5. Conclusions
Efforts aimed at eradicating glioblastoma through mitochondrial inhibition led to the findings that GSCs were selectively targeted due to their lower mitochondrial content. GSCs exhibited the “Warburg effect” by showing a decrease in cellular respiration with increased tumorigenicity. Classical mitochondria inhibitors, Rotenone, Antimycin A, and Oligomycin A preferentially kill GSCs compared to dGCs. GSCs with low tumorigenicity appear to undergo apoptotic death, whereas autophagy appears to be the preferred death mechanism in more tumorigenic GSCs when GSCs were exposed to classical mitochondria inhibitors. FDA-approved drugs trifluoperazine, mitoxantrone, and pyrvinium pamoate have shown efficacy toward mitochondrial inhibition with GSCs target selectivity. The three FDA approved drugs are more effective than TMZ at eradicating GSCs and could be used to treat a patient’s GBMs and prevent tumor relapse.
Supplementary Material
Highlights.
Patient-derived glioma stem-like cells (GSCs) show mitochondrial heterogeneity
Classical mitochondrial inhibitors preferentially kill GSCs
Mitochondrial inhibitors are better than temozolomide in killing GSCs
Mitochondrial inhibitors induce intrinsic apoptosis pathway or autophagy in GSCs
Mitochondria-targeting FDA-approved drugs can selectively kill GSCs
Acknowledgements:
Funding Sources: This study was partly funded by National Institutes of Health National Institute of Neurological Diseases and Stroke (1R01NS083795) and the gift funds from University of California Davis Comprehensive Cancer Center Support Grant, number P30CA093373. We thank Mehdia Zaidi for critical reading of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Chemical compounds studied in this article: oligomycin A (PubChem CID: 5281899), antimycin A (PubChem CID: 6604296), rotenone (PubChem CID:6758), trifluoperazine (PubChem CID:5566), mitoxantrone (PubChem CID:4212), pyrvinium pamoate (PubChem CID:54680693).
Conflict of Interest
The authors do not declare a conflict of interest.
References
- [1].Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz-Sloan JS, CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011–2015, Neuro-oncology 20(suppl_4) (2018) iv1–iv86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].N.C. Institute, Glioblastoma—Unraveling the Threads: A Q&A with Drs. Mark Gilbert and Terri Armstrong of the NIH Neuro-Oncology Branch, 2017. https://www.cancer.gov/news-events/cancer-currents-blog/2017/glioblastoma-research-making-progress. (Accessed July 1st 2019).
- [3].Nam JY, de Groot JF, Treatment of Glioblastoma, Journal of oncology practice 13(10) (2017) 629–638. [DOI] [PubMed] [Google Scholar]
- [4].Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, Hau P, Brandes AA, Gijtenbeek J, Marosi C, Vecht CJ, Mokhtari K, Wesseling P, Villa S, Eisenhauer E, Gorlia T, Weller M, Lacombe D, Cairncross JG, Mirimanoff RO, Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial, The Lancet. Oncology 10(5) (2009) 459–66. [DOI] [PubMed] [Google Scholar]
- [5].Di Carlo DT, Cagnazzo F, Benedetto N, Morganti R, Perrini P, Multiple high-grade gliomas: epidemiology, management, and outcome. A systematic review and meta-analysis, Neurosurgical review 42(2) (2019) 263–275. [DOI] [PubMed] [Google Scholar]
- [6].Osuka S, Van Meir EG, Overcoming therapeutic resistance in glioblastoma: the way forward, The Journal of clinical investigation 127(2) (2017) 415–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Jackson M, Hassiotou F, Nowak A, Glioblastoma stem-like cells: at the root of tumor recurrence and a therapeutic target, Carcinogenesis 36(2) (2015) 177–85. [DOI] [PubMed] [Google Scholar]
- [8].Safa AR, Saadatzadeh MR, Cohen-Gadol AA, Pollok KE, Bijangi-Vishehsaraei K, Glioblastoma stem cells (GSCs) epigenetic plasticity and interconversion between differentiated non-GSCs and GSCs, Genes & diseases 2(2) (2015) 152–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Alvarado AG, Thiagarajan PS, Mulkearns-Hubert EE, Silver DJ, Hale JS, Alban TJ, Turaga SM, Jarrar A, Reizes O, Longworth MS, Vogelbaum MA, Lathia JD, Glioblastoma Cancer Stem Cells Evade Innate Immune Suppression of Self-Renewal through Reduced TLR4 Expression, Cell stem cell 20(4) (2017) 450–461. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Velpula KK, Rehman AA, Chelluboina B, Dasari VR, Gondi CS, Rao JS, Veeravalli KK, Glioma stem cell invasion through regulation of the interconnected ERK, integrin alpha6 and N-cadherin signaling pathway, Cellular signalling 24(11) (2012) 2076–84. [DOI] [PubMed] [Google Scholar]
- [11].Drean A, Rosenberg S, Lejeune FX, Goli L, Nadaradjane AA, Guehennec J, Schmitt C, Verreault M, Bielle F, Mokhtari K, Sanson M, Carpentier A, Delattre JY, Idbaih A, ATP binding cassette (ABC) transporters: expression and clinical value in glioblastoma, Journal of neuro-oncology 138(3) (2018) 479–486. [DOI] [PubMed] [Google Scholar]
- [12].Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN, Cancer stem cells in glioblastoma, Genes & development 29(12) (2015) 1203–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chamberlain MC, Temozolomide: therapeutic limitations in the treatment of adult high-grade gliomas, Expert review of neurotherapeutics 10(10) (2010) 1537–44. [DOI] [PubMed] [Google Scholar]
- [14].Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, Lu L, Irvin D, Black KL, Yu JS, Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma, Molecular cancer 5 (2006) 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kang MK, Kang SK, Tumorigenesis of chemotherapeutic drug-resistant cancer stem-like cells in brain glioma, Stem cells and development 16(5) (2007) 837–47. [DOI] [PubMed] [Google Scholar]
- [16].Ahmad F, Sun Q, Patel D, Stommel JM, Cholesterol Metabolism: A Potential Therapeutic Target in Glioblastoma, Cancers 11(2) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ward PS, Thompson CB, Metabolic reprogramming: a cancer hallmark even warburg did not anticipate, Cancer cell 21(3) (2012) 297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ahmad F, Dixit D, Sharma V, Kumar A, Joshi SD, Sarkar C, Sen E, Nrf2-driven TERT regulates pentose phosphate pathway in glioblastoma, Cell death & disease 7 (2016) e2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ahmad F, Patrick S, Sheikh T, Sharma V, Pathak P, Malgulwar PB, Kumar A, Joshi SD, Sarkar C, Sen E, Telomerase reverse transcriptase (TERT) - enhancer of zeste homolog 2 (EZH2) network regulates lipid metabolism and DNA damage responses in glioblastoma, 143(6) (2017) 671–683. [DOI] [PubMed] [Google Scholar]
- [20].Obre E, Rossignol R, Emerging concepts in bioenergetics and cancer research: metabolic flexibility, coupling, symbiosis, switch, oxidative tumors, metabolic remodeling, signaling and bioenergetic therapy, The international journal of biochemistry & cell biology 59 (2015) 167–81. [DOI] [PubMed] [Google Scholar]
- [21].Xing F, Luan Y, Cai J, Wu S, Mai J, Gu J, Zhang H, Li K, Lin Y, Xiao X, Liang J, Li Y, Chen W, Tan Y, Sheng L, Lu B, Lu W, Gao M, Qiu P, Su X, Yin W, Hu J, Chen Z, Sai K, Wang J, Chen F, Chen Y, Zhu S, Liu D, Cheng S, Xie Z, Zhu W, Yan G, The Anti-Warburg Effect Elicited by the cAMP-PGC1alpha Pathway Drives Differentiation of Glioblastoma Cells into Astrocytes, Cell reports 18(2) (2017) 468–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Garnier D, Renoult O, Alves-Guerra MC, Paris F, Pecqueur C, Glioblastoma Stem-Like Cells, Metabolic Strategy to Kill a Challenging Target, Front Oncol 9 (2019) 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Oliva CR, Nozell SE, Diers A, McClugage SG 3rd, Sarkaria JN, Markert JM, Darley-Usmar VM, Bailey SM, Gillespie GY, Landar A, Griguer CE, Acquisition of temozolomide chemoresistance in gliomas leads to remodeling of mitochondrial electron transport chain, The Journal of biological chemistry 285(51) (2010) 39759–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sahdeo S, Tomilov A, Komachi K, Iwahashi C, Datta S, Hughes O, Hagerman P, Cortopassi G, High-throughput screening of FDA-approved drugs using oxygen biosensor plates reveals secondary mitofunctional effects, Mitochondrion 17 (2014) 116–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nevo I, Woolard K, Cam M, Li A, Webster JD, Kotliarov Y, Kim HS, Ahn S, Walling J, Kotliarova S, Belova G, Song H, Bailey R, Zhang W, Fine HA, Identification of molecular pathways facilitating glioma cell invasion in situ, PLoS One 9(11) (2014) e111783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Son MJ, Woolard K, Nam DH, Lee J, Fine HA, SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma, Cell Stem Cell 4(5) (2009) 440–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Liu Y, Morgan JB, Coothankandaswamy V, Liu R, Jekabsons MB, Mahdi F, Nagle DG, Zhou YD, The Caulerpa pigment caulerpin inhibits HIF-1 activation and mitochondrial respiration, J Nat Prod 72(12) (2009) 2104–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hayashi G, Jasoliya M, Sahdeo S, Sacca F, Pane C, Filla A, Marsili A, Puorro G, Lanzillo R, Brescia Morra V, Cortopassi G, Dimethyl fumarate mediates Nrf2-dependent mitochondrial biogenesis in mice and humans, Human molecular genetics 26(15) (2017) 2864–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Vichai V, Kirtikara K, Sulforhodamine B colorimetric assay for cytotoxicity screening, Nature protocols 1(3) (2006) 1112–6. [DOI] [PubMed] [Google Scholar]
- [30].Trepant AL, Bouchart C, Rorive S, Sauvage S, Decaestecker C, Demetter P, Salmon I, Identification of OLIG2 as the most specific glioblastoma stem cell marker starting from comparative analysis of data from similar DNA chip microarray platforms, Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine 36(3) (2015) 1943–53. [DOI] [PubMed] [Google Scholar]
- [31].Poteet E, Choudhury GR, Winters A, Li W, Ryou MG, Liu R, Tang L, Ghorpade A, Wen Y, Yuan F, Keir ST, Yan H, Bigner DD, Simpkins JW, Yang SH, Reversing the Warburg effect as a treatment for glioblastoma, The Journal of biological chemistry 288(13) (2013) 9153–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ahmad F, Dixit D, Joshi SD, Sen E, G9a inhibition induced PKM2 regulates autophagic responses, The international journal of biochemistry & cell biology 78 (2016) 87–95. [DOI] [PubMed] [Google Scholar]
- [33].Huang C, Lan W, Fraunhoffer N, Meilerman A, Iovanna J, Santofimia-Castano P, Dissecting the Anticancer Mechanism of Trifluoperazine on Pancreatic Ductal Adenocarcinoma, Cancers (Basel) 11(12) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Seokheon Hong M.-y.L., Soon Shin Ki and Jung Kang Shin, Perphenazine and trifluoperazine induce mitochondria-mediated cell death in SH-SY5Y cells, Animal Cells and Systems 16(1) (2012) 20–26. [Google Scholar]
- [35].Dirkse A, Golebiewska A, Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment, 10(1) (2019) 1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kitange GJ, Carlson BL, Schroeder MA, Grogan PT, Lamont JD, Decker PA, Wu W, James CD, Sarkaria JN, Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts, Neuro-oncology 11(3) (2009) 281–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].William D, Walther M, Schneider B, Linnebacher M, Classen CF, Temozolomide-induced increase of tumorigenicity can be diminished by targeting of mitochondria in in vitro models of patient individual glioblastoma, PloS one 13(1) (2018) e0191511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Porporato PE, Filigheddu N, Pedro JMB, Kroemer G, Galluzzi L, Mitochondrial metabolism and cancer, Cell Res 28(3) (2018) 265–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Song KH, Kim JH, Lee YH, Bae HC, Lee HJ, Woo SR, Oh SJ, Lee KM, Yee C, Kim BW, Cho H, Chung EJ, Chung JY, Hewitt SM, Chung TW, Ha KT, Bae YK, Mao CP, Yang A, Wu TC, Kim TW, Mitochondrial reprogramming via ATP5H loss promotes multimodal cancer therapy resistance, J Clin Invest 128(9) (2018) 4098–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D, Korbel C, Laschke MW, Gimotty PA, Philipp SE, Krause E, Patzold S, Villanueva J, Krepler C, Fukunaga-Kalabis M, Hoth M, Bastian BC, Vogt T, Herlyn M, Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells, Cancer cell 23(6) (2013) 811–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Bosc C, Selak MA, Sarry JE, Resistance Is Futile: Targeting Mitochondrial Energetics and Metabolism to Overcome Drug Resistance in Cancer Treatment, Cell metabolism 26(5) (2017) 705–707. [DOI] [PubMed] [Google Scholar]
- [42].Wang H, Gao Z, Targeted production of reactive oxygen species in mitochondria to overcome cancer drug resistance, 9(1) (2018) 562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lee EA, Angka L, Rota SG, Hanlon T, Mitchell A, Hurren R, Wang XM, Gronda M, Boyaci E, Bojko B, Minden M, Sriskanthadevan S, Datti A, Wrana JL, Edginton A, Pawliszyn J, Joseph JW, Quadrilatero J, Schimmer AD, Spagnuolo PA, Targeting Mitochondria with Avocatin B Induces Selective Leukemia Cell Death, Cancer Res 75(12) (2015) 2478–88. [DOI] [PubMed] [Google Scholar]
- [44].Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, McAfoos T, Morlacchi P, Ackroyd J, An inhibitor of oxidative phosphorylation exploits cancer vulnerability, 24(7) (2018) 1036–1046. [DOI] [PubMed] [Google Scholar]
- [45].Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K, Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission, Cancer Res 69(19) (2009) 7507–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Fiorillo M, Lamb R, Tanowitz HB, Mutti L, Krstic-Demonacos M, Cappello AR, Martinez-Outschoorn UE, Sotgia F, Lisanti MP, Repurposing atovaquone: targeting mitochondrial complex III and OXPHOS to eradicate cancer stem cells, Oncotarget 7(23) (2016) 34084–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Banerjee A, Birts CN, Darley M, Parker R, Mirnezami AH, West J, Cutress RI, Beers SA, Rose-Zerilli MJJ, Blaydes JP, Stem cell-like breast cancer cells with acquired resistance to metformin are sensitive to inhibitors of NADH-dependent CtBP dimerization, Carcinogenesis 40(7) (2019) 871–882. [DOI] [PubMed] [Google Scholar]
- [48].Lonardo E, Cioffi M, Sancho P, Sanchez-Ripoll Y, Trabulo SM, Dorado J, Balic A, Hidalgo M, Heeschen C, Metformin targets the metabolic achilles heel of human pancreatic cancer stem cells, PloS one 8(10) (2013) e76518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Singh BN, Kumar D, Shankar S, Srivastava RK, Rottlerin induces autophagy which leads to apoptotic cell death through inhibition of PI3K/Akt/mTOR pathway in human pancreatic cancer stem cells, Biochemical pharmacology 84(9) (2012) 1154–63. [DOI] [PubMed] [Google Scholar]
- [50].Song IS, Jeong YJ, Han J, Mitochondrial metabolism in cancer stem cells: a therapeutic target for colon cancer, BMB reports 48(10) (2015) 539–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Zhang X, Fryknas M, Hernlund E, Fayad W, De Milito A, Olofsson MH, Gogvadze V, Dang L, Pahlman S, Schughart LA, Rickardson L, D’Arcy P, Gullbo J, Nygren P, Larsson R, Linder S, Induction of mitochondrial dysfunction as a strategy for targeting tumour cells in metabolically compromised microenvironments, Nature communications 5 (2014) 3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Valtorta S, Lo Dico A, Raccagni I, Gaglio D, Belloli S, Politi LS, Martelli C, Diceglie C, Bonanomi M, Ercoli G, Vaira V, Ottobrini L, Moresco RM, Metformin and temozolomide, a synergic option to overcome resistance in glioblastoma multiforme models, Oncotarget 8(68) (2017) 113090–113104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Heddleston JM, Li Z, McLendon RE, Hjelmeland AB, Rich JN, The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype, Cell cycle (Georgetown, Tex.) 8(20) (2009) 3274–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Lo Dico A, Martelli C, Diceglie C, Lucignani G, Ottobrini L, Hypoxia-Inducible Factor-1alpha Activity as a Switch for Glioblastoma Responsiveness to Temozolomide, Frontiers in oncology 8 (2018) 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, Shi Q, Cao Y, Lathia J, McLendon RE, Hjelmeland AB, Rich JN, Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells, Cancer cell 15(6) (2009) 501–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Lee G, Auffinger B, Guo D, Hasan T, Deheeger M, Tobias AL, Kim JY, Atashi F, Zhang L, Lesniak MS, James CD, Ahmed AU, Dedifferentiation of Glioma Cells to Glioma Stem-like Cells By Therapeutic Stress-induced HIF Signaling in the Recurrent GBM Model, Molecular cancer therapeutics 15(12) (2016) 3064–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Li J, Mahdi F, Du L, Datta S, Nagle DG, Zhou YD, Mitochondrial respiration inhibitors suppress protein translation and hypoxic signaling via the hyperphosphorylation and inactivation of translation initiation factor eIF2alpha and elongation factor eEF2, Journal of natural products 74(9) (2011) 1894–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Emery IF, Gopalan A, Wood S, Chow KH, Battelli C, George J, Blaszyk H, Florman J, Yun K, Expression and function of ABCG2 and XIAP in glioblastomas, Journal of neuro-oncology 133(1) (2017) 47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Leidgens V, Proske J, Rauer L, Moeckel S, Renner K, Bogdahn U, Riemenschneider MJ, Proescholdt M, Vollmann-Zwerenz A, Hau P, Seliger C, Stattic and metformin inhibit brain tumor initiating cells by reducing STAT3-phosphorylation, Oncotarget 8(5) (2017) 8250–8263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Park J, Shim JK, Kang JH, Choi J, Chang JH, Kim SY, Kang SG, Regulation of bioenergetics through dual inhibition of aldehyde dehydrogenase and mitochondrial complex I suppresses glioblastoma tumorspheres, Neuro-oncology 20(7) (2018) 954–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Tan SK, Jermakowicz A, Mookhtiar AK, Nemeroff CB, Schurer SC, Ayad NG, Drug Repositioning in Glioblastoma: A Pathway Perspective, Frontiers in pharmacology 9 (2018) 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Rossato LG, Costa VM, Dallegrave E, Arbo M, Silva R, Ferreira R, Amado F, Dinis-Oliveira RJ, Duarte JA, de Lourdes Bastos M, Palmeira C, Remiao F, Mitochondrial cumulative damage induced by mitoxantrone: late onset cardiac energetic impairment, Cardiovasc Toxicol 14(1) (2014) 30–40. [DOI] [PubMed] [Google Scholar]
- [63].Arduino DM, Wettmarshausen J, Vais H, Navas-Navarro P, Cheng Y, Leimpek A, Ma Z, Delrio-Lorenzo A, Giordano A, Garcia-Perez C, Medard G, Kuster B, Garcia-Sancho J, Mokranjac D, Foskett JK, Alonso MT, Perocchi F, Systematic Identification of MCU Modulators by Orthogonal Interspecies Chemical Screening, Mol Cell 67(4) (2017) 711–723 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Harada Y, Ishii I, Hatake K, Kasahara T, Pyrvinium pamoate inhibits proliferation of myeloma/erythroleukemia cells by suppressing mitochondrial respiratory complex I and STAT3, Cancer Lett 319(1) (2012) 83–8. [DOI] [PubMed] [Google Scholar]
- [65].Tomitsuka E, Kita K, Esumi H, An anticancer agent, pyrvinium pamoate inhibits the NADH-fumarate reductase system--a unique mitochondrial energy metabolism in tumour microenvironments, J Biochem 152(2) (2012) 171–83. [DOI] [PubMed] [Google Scholar]
- [66].Venugopal C, Hallett R, Vora P, Manoranjan B, Mahendram S, Qazi MA, McFarlane N, Subapanditha M, Nolte SM, Singh M, Bakhshinyan D, Garg N, Vijayakumar T, Lach B, Provias JP, Reddy K, Murty NK, Doble BW, Bhatia M, Hassell JA, Singh SK, Pyrvinium Targets CD133 in Human Glioblastoma Brain Tumor-Initiating Cells, Clinical cancer research : an official journal of the American Association for Cancer Research 21(23) (2015) 5324–37. [DOI] [PubMed] [Google Scholar]
- [67].Boiardi A, Eoli M, Salmaggi A, Lamperti E, Botturi A, Broggi G, Bissola L, Finocchiaro G, Silvani A, Systemic temozolomide combined with loco-regional mitoxantrone in treating recurrent glioblastoma, Journal of neuro-oncology 75(2) (2005) 215–20. [DOI] [PubMed] [Google Scholar]
- [68].Pinheiro T, Otrocka M, Seashore-Ludlow B, Rraklli V, Holmberg J, Forsberg-Nilsson K, Simon A, Kirkham M, A chemical screen identifies trifluoperazine as an inhibitor of glioblastoma growth, Biochemical and biophysical research communications 494(3–4) (2017) 477–483. [DOI] [PubMed] [Google Scholar]
- [69].Dunn PP, Slabas AR, Cottingham IR, Moore AL, Trifluoperazine inhibition of electron transport and adenosine triphosphatase in plant mitochondria, Archives of biochemistry and biophysics 229(1) (1984) 287–94. [DOI] [PubMed] [Google Scholar]
- [70].Bhat K, Saki M, Vlashi E, Cheng F, Duhachek-Muggy S, Alli C, Yu G, Medina P, He L, Damoiseaux R, Pellegrini M, Zemke NR, Nghiemphu PL, Cloughesy TF, Liau LM, Kornblum HI, Pajonk F, The dopamine receptor antagonist trifluoperazine prevents phenotype conversion and improves survival in mouse models of glioblastoma, Proc Natl Acad Sci U S A 117(20) (2020) 11085–11096. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.