

Abstract
Glioblastoma multiforme (GBM) is the most aggressive type of malignant brain tumor. Current FDA-approved treatments include surgical resection, radiation, and chemotherapy, while hyperthermia, immunotherapy, and most relevantly, nanoparticle (NP)-mediated delivery systems or combinations thereof have shown promise in preclinical studies. Drug-carrying NPs are a promising approach to brain delivery as a result of their potential to facilitate the crossing of the blood-brain barrier (BBB) via two main types of transcytosis mechanisms; adsorptive-mediated transcytosis (AMT) and receptor-mediated transcytosis (RMT) and their ability to accumulate in the brain thus providing local sustained release of tumoricidal drugs. NP-based drug delivery has the potential to significantly reduce drug-related toxicity, increase specificity and consequently improve the lifespan and quality of life of patients with GBM. Due to significant advances in the understanding of the molecular etiology and pathology of GBM, the efficacy of drugs loaded into vectors targeting this disease has increased in both preclinical and clinical settings. Multi-targeting NPs, such as those incorporating multiple specific targeting ligands, are an innovative technology that can lead to decreased off-target effects while simultaneously having increased accumulation and action specifically at the tumor site. Targeting ligands can include antibodies, or fragments thereof, and peptides or small molecules, which can result in a more controlled drug delivery system compared to conventional drug treatments. This review focuses on GBM treatment strategies, summarizing current options and providing a detailed account of preclinical findings with prospective NP-based approaches aimed at improving tumor targeting and enhancing therapeutic outcomes for GBM patients.
Keywords: Glioblastoma multiforme (GBM), brain tumor, glioblastoma treatments, nanoparticle-based therapy, nanotechnology
Introduction
There are two major types of glioma recognized in humans: diffuse glioma and circumscribed glioma. Diffuse gliomas are the most common primary central nervous system (CNS) neoplasm that include astrocytomas, oligodendrogliomas and oligoastrocytomas; and can become malignant and aggressive (1). In 2016, the World Health Organization updated their classification of brain tumors from the more conceptual definition used previously by including histology as well as features at the molecular level which led to the development of a grading scale (2, 3). As a result, diffuse gliomas are classified in the range of grade II – grade IV and include glioblastoma multiforme (GBM), a grade IV astrocytoma (4). The total incidence of both diffuse and circumscribed gliomas is approximately 18 per 100,000 persons in the United States and 7 per 100,000 persons worldwide; however, nearly all of these cases are diffuse gliomas (5, 6).
GBM is considered the most malignant and aggressive human brain tumor type as well as being the most common form of brain tumor in adults. Most glioblastomas (~90%) originate de novo from normal glial cells with no evidence of low-grade glioma and are clinically classified as primary glioblastomas; while secondary glioblastomas develop from low-grade gliomas (7, 8). Primary and secondary glioblastomas differ in both their oncogenic ontogeny and rate of tumor growth, with primary glioblastomas growing more aggressively than secondary glioblastomas (3, 4, 9); however, they both share similar morphological features and lead to similar clinical symptoms (10). Worldwide, GBM causes more than 100,000 deaths per year (11), and the incidence rate in men is higher than in women (12).
GBM most commonly occurs in the cerebral hemispheres of the brain, such as the frontal and temporal lobes (13–15); however in some patients, GBM has been found in the cerebellum, brainstem and spinal cord (15, 16). Unlike other rapidly growing tumor types, GBM possesses low metastatic potential (17–21).
GBM tumors are often highly vascularized in order to receive an adequate blood supply and nutrients which facilitate tumor progression. The most common clinical signs of GBM, namely headache, vomiting, focal or progressive neurologic deficits, seizure, vision disturbance and frequent syncope, are dependent on the size and location of the tumor as well as the rate of increasing intracranial pressure (22). For diagnosis, computed tomography, and magnetic resonance imaging (MRI) scans are performed.
Since the 1930’s, an increasing number of therapeutic strategies have been introduced to treat GBM, the timeline of which is outlined in Figure 1 (23). These treatment strategies range from physical and chemical to monoclonal antibody-based therapies (23). Although such treatments may improve patient quality of life and prolong survival, many have limitations such as high expense, the need for specialized and precise surgical tools (e.g. surgery) or have adverse side-effects (e.g. radiation and chemotherapy). In addition, the blood-brain barrier (BBB) can prevent access of drugs to the tumor, and thus methods for drug delivery across the BBB need to be developed.
Figure 1.
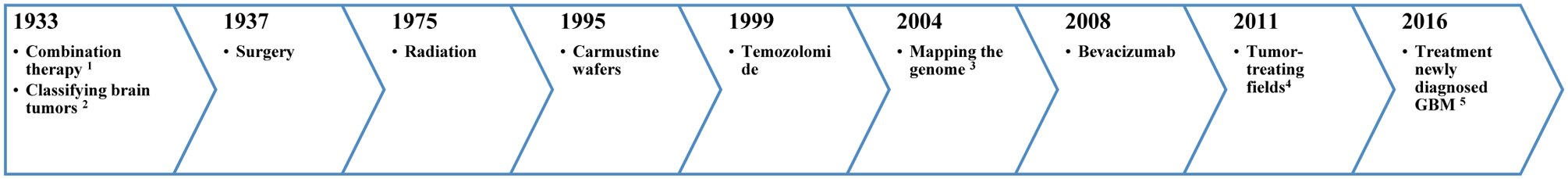
Timeline showing the various treatment strategies for GBM employed over the past several decades.
1 combination of chemotherapy and radiation, 2 WHO develops universal system for classifying brain tumors, 3 mapping the genome of glioblastoma (28), 4 FDA approval: the treatment of recurrent GBM (29, 30) and 5 TTFs have been approved (29, 30).
Nanoparticles (NPs) can potentially facilitate the delivery of drugs across the BBB. In addition, due to their physicochemical properties, ability to self-assemble, biocompatibility, and tunability with respect to surface decoration for tumor targeting, NPs have great potential in combatting GBM (24–26). NPs are also able to provide controlled drug release thereby reducing the number of required doses (27).
Here, we review treatment strategies currently used, or being investigated as approaches, to combat GBM. Particular focus is placed on novel NP-based drug delivery systems and the preclinical data demonstrating their capacity to overcome the BBB and specifically target glioma cells as well as reduce off-target side effects, improve survival rates, and enable efficient drug delivery to GBM tumors.
GBM treatment strategies
The range of standard and prospective strategies for GBM treatments are summarized in Figure 2. These all have advantages and disadvantages and are discussed further in this section.
Figure 2.
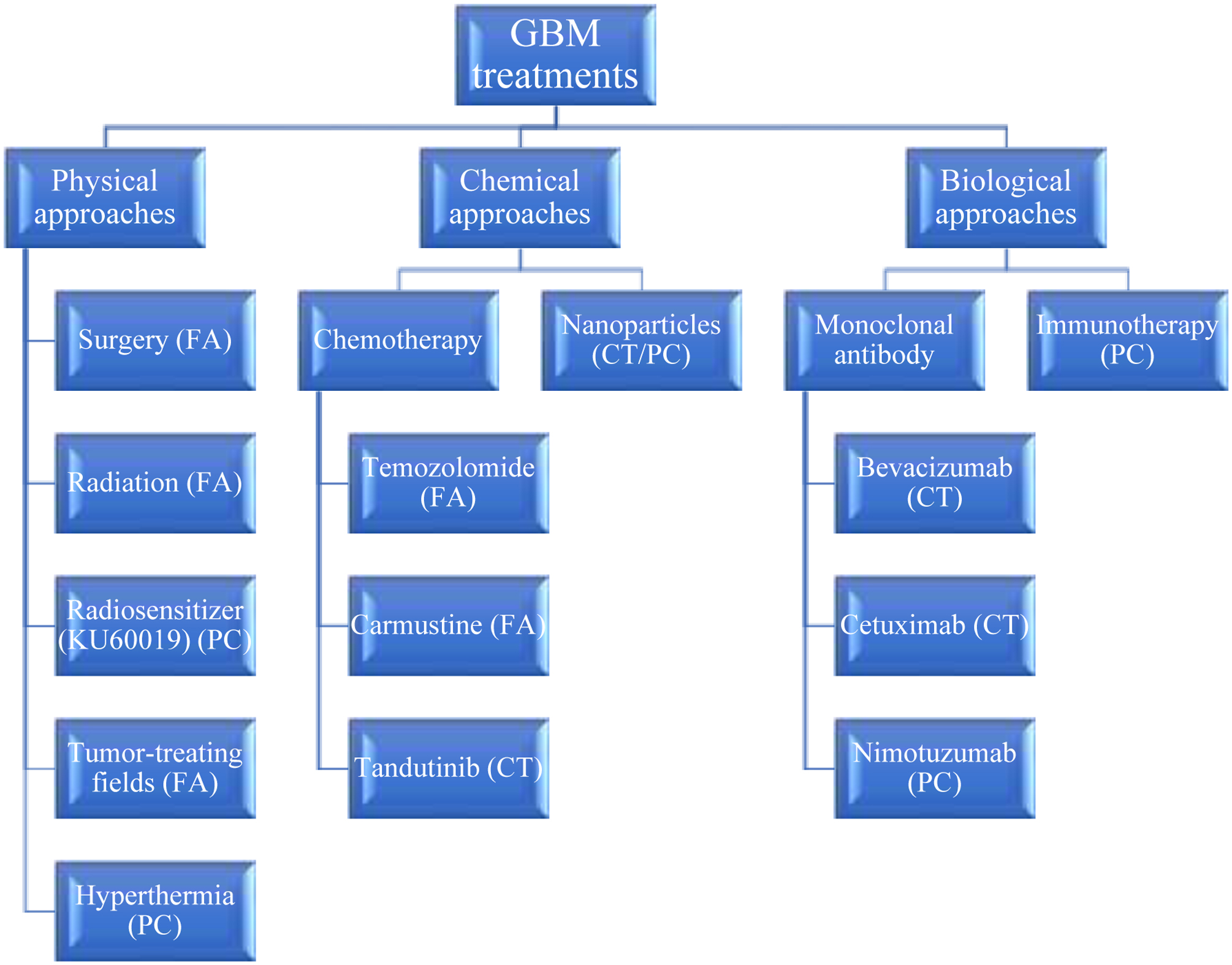
The different types of physical, chemical, and biological GBM treatment approaches (PC = preclinical testing stage, CT = reached clinical trials and FA = FDA-approved).
Surgery
Surgery is usually the initial treatment and maximal safe surgical resection is performed in more than 50% of all GBM patients (31); however, the procedure may result in damage to surrounding healthy brain tissue, negatively affecting movement and language. Surgical resection can be used in combination with imaging techniques and/or radiation therapy (32) to improve surgical efficiency. The benefit of imaging techniques such as MRI (33) and navigated transcranial magnetic stimulation are that they can be used to map and prevent damage to the brain areas responsible for motor control (34). Surgery performed in combination with MRI to map the functional cortex reduces the risk of postoperative motor deficit (35, 36). Although the overall goal of surgery for GBM patients is maximal safe resection, there are limitations to its effectiveness due to a variety of issues such as tumor size, inadequate tumor removal, location of tumor, as well as patients’ condition (age, gender or comorbidities) (37). Therefore, surgery is sometimes used in combination with radiation or chemotherapy to increase median survival times (32).
Radiotherapy
Patients with GBM who receive radiotherapy are exposed to fractionated localized radiation using a standard dose of radiation of 60 Gy with a range from 30.6 to 74 Gy (38). Radiotherapy can be conducted using an external source, an internal source or radioactive monoclonal antibodies (39–42).
External beam radiation therapy, the most frequently used method, produces high energy beams applied externally to the brain to cover the whole tumor volume (39) whereas internal radiation therapy, or brachytherapy, delivers radiation via a radioactive substance positioned near or in the GBM tumor thereby reducing damage to more distal healthy brain tissue (42). Certain strategies, such as three dimensional conformational radiation therapy or image guided radiation therapy (43, 44), stereotactic radiosurgery (45) or proton radiation therapy (46) apply a focused delivery of energy directly to the tumor, resulting in less damage to healthy brain tissue as well as increased patient survival times. Tumor treating fields are a U.S. Food and Drug Administration (FDA)-approved noninvasive antimitotic therapy that delivers alternating electrical fields of intermediate frequency (200 kHz) and low intensity (47). These devices can improve median survival time of patients with recurrent GBM and increase the apoptosis rate of human GBM cells (U87MG cells) in vitro (47, 48).
Cotara® is an example of a radioactive monoclonal antibody therapy, more specifically a 131I-labeled chimeric monoclonal antibody (specific for DNA and histone H1) that delivers cytotoxic radiation to sites of necrosis (i.e. GBM tumors). In a phase II clinical trial (NCT00677716), Cotara® showed an excellent safety profile and improved survival times of GBM patients (40, 41, 49).
Supplemental to radiotherapy, KU60019, a kinase inhibitor, has been shown to have radiosensitizing activity which increases the anticancer efficacy of radiation (50). In vivo mouse studies using KU60019 in combination with radiation showed an enhancement of the radiation effects, delayed GBM tumor progression, and prolonged median survival times compared to radiation alone (51).
Although radiotherapy is typically an effective GBM treatment, there are some situations where tumor response is poor, such as when the tumor is too large upon diagnosis or is resistant to radiotherapy. In addition, there can also be undesirable side effects of radiotherapy such as damage to the epithelial surfaces of the mouth and throat, hair loss, lymphedema and heart disease (52).
Monoclonal antibodies
Bevacizumab is a human monoclonal antibody that inhibits vascular endothelial growth factor (VEGF) to prevent tumor angiogenesis and is used as a standard of care for several tumor types (53). It has been approved for GBM treatment in the US since 2009 (54). Due to the immunosuppressive nature of VEGF (e.g. VEGF inhibits dendritic cell maturation), bevacizumab may have the potential to alter the tumor microenvironment to favor tumor-specific immune responses and potentially enhance tumor regression when used in combination with immune checkpoint inhibitors. An ongoing phase II trial (NCT02336165) using bevacizumab in combination with the immune checkpoint inhibitor, durvalumab (an anti-PD-L1 antibody), was designed to assess the potential synergy between these antibodies in GBM patients (55). Even though the impact of bevacizumab on glioma tumor immunity remains unclear at this stage, studies in other tumor types suggest that bevacizumab may have immune stimulating effects and could be a candidate regimen in synergistic therapy with immune-checkpoint inhibitors in non-small-cell lung cancer (NSCLC) patients (56–58). Studies have also been performed in order to test the efficacy of bevacizumab when combined with chemotherapeutic drugs. For example, a phase II clinical trial (NCT00921167) combining bevacizumab with irinotecan showed that the median survival time of patients who received the combination treatment was higher than those that received bevacizumab alone. Specifically, using bevacizumab and irinotecan showed a 6-month increase in progression free survival rates compared to bevacizumab alone (59). Aside from the potential therapeutic benefit that may be obtained by combining bevacizumab with immune checkpoint inhibitors, bevacizumab has also been investigated in combination with other antibodies. Cetuximab is a monoclonal antibody that acts as an epidermal growth factor receptor (EGFR) antagonist. Cetuximab was used in a phase II clinical trial in combination with bevacizumab and irinotecan to treat recurrent GBM patients after first line standard temozolomide (TMZ) treatment failed. Despite there being well-tolerated toxicity, the response rate was not found to be improved compared to treatment with bevacizumab alone or bevacizumab + irinotecan (60). More promisingly, nimotuzumab, another EGFR antagonist, has been used to treat newly diagnosed glioblastoma patients and was found to increase overall survival significantly compared to standard radiochemotherapy (TMZ plus radiation) (61). In an in vivo human GBM mouse model (i.e. xenogeneic model using nude mice), treatment with nimotuzumab and TMZ resulted in downregulation of VEGF expression, reduced angiogenesis as well as enhanced antitumor activity and survival when compared to either treatment alone (62). Though monoclonal antibodies have high specificity and high affinity for their targets, access to the brain is limited by the BBB and thus the effectiveness of these treatments is hindered (40, 63). This hindrance is further exacerbated by the fact that monoclonal antibodies have a large molecular size and low BBB permeability (40). Similar to chemotherapies, these constraints mean monoclonal antibodies require higher doses in order to be effective which causes damage to healthy off-target tissues (63).
Chemotherapy
TMZ is the primary gold standard chemotherapeutic drug used for the treatment of GBM (64). It is an orally delivered alkylating agent that exerts its cytotoxic effects through DNA methylation (65). TMZ can be used with radiotherapy to improve the median survival of GBM patients (66). TMZ can also be combined with O6-benzylguanine (O6GB) (NCT00613093), an O6-methylguanine-DNA methyltransferase inhibitor, in order to restore drug sensitivity in TMZ resistant tumor cells (67). A completed phase II trial showed that adding O6GB one day before dosing with TMZ restored sensitivity in TMZ-resistant anaplastic glioma patients, but it was not significantly effective in GBM patients with TMZ resistance (68). The authors suggested that the reason for the difference in effectiveness may have been due to GBM tumors possessing greater interstitial pressures than anaplastic gliomas. Due to the physiological restriction of the BBB, TMZ alone barely increases survival times in GBM patients; therefore, combining TMZ with other therapies such as radiotherapy or chemotherapy is needed to cause significant anticancer activity of TMZ and prolong patient survival (52, 69).
Carmustine (bis-chloroethylnitrosourea, BCNU), an alkylating agent of DNA and RNA, can be used to treat newly diagnosed glioblastoma and can improve median survival of patients (70). BCNU wafers consist of biodegradable polymers containing 3.85% BCNU that, when placed in the resection cavity of patients with primary or recurrent GBM, improve median survival times (71). Glioblastoma cells overexpress T-type calcium channels involved in promoting angiogenesis and invasion of tumor cells. Mibefradil, a novel chemotherapeutic agent, can be used to block T-type calcium channels in patients with recurrent GBM and has been shown to prolong progression free survival of GBM patients and increase overall survival (72, 73).
Tandutinib is an inhibitor of type III receptor tyrosine kinases (e.g. platelet-derived growth factor (PDGF) receptor-b, Fms-like tyrosine kinase 3, and c-Kit) that can be used to treat patients with recurrent GBM. Unfortunately, the combination of tandutinib with bevacizumab has been shown to exacerbate the commonly seen side effects such as hypertension, fatigue and diarrhea without improving efficacy compared with bevacizumab alone (74, 75). Irinotecan is an inhibitor of topoisomerase I and has demonstrated toxicity against glioblastoma cells in preclinical studies due to its ability to cross the BBB (76). The combination of irinotecan with other medications such as TMZ and bevacizumab have demonstrated promising results for treating GBM. These combination treatments showed increased anticancer activity, prolonged median survival times and improved tumor response rates (59, 76, 77).
The chemotherapeutic agents have low BBB permeability, high liver accumulation, faster renal clearance and low specific to GBM cells resulting in low therapeutic effects and more off-target effects. In addition, multidrug resistance (MDR) mechanisms on the BBB (i.e. P-glycoprotein or MDR proteins) could affect the efficiency of drugs for GBM treatment (27, 67, 78–81). NPs are a promising drug delivery system for GBM treatment to not only to load anticancer agents and deliver them to the target GBM tumor cells, but also to reduce the systemic toxicity of those therapeutic agents. Anticancer agents such as doxorubicin (DOX), paclitaxel (PTX) and docetaxel can be entrapped into delivery systems such as NPs for treating GBM even though these drugs are not used as first-line glioma treatments. This gives encouragement for researchers to develop novel therapies and investigate GBM treatments of this type both in vitro and in vivo. Although chemotherapy is typically an effective GBM treatment, there are several obstacles that still need to be addressed: (1) the majority of GBM tumors are unresponsive to TMZ due to O6-methylguanine-DNA methyltransferase which limits its cytotoxic effects (67); (2) TMZ has low solubility and is rapidly hydrolyzed under physiological conditions (78); (3) the function of the BBB renders the majority of chemotherapeutic agents and targeted agents ineffective (27, 79); and (4) due to anticancer compounds limited ability to cross the BBB, patients require multiple doses which increases the toxicity and undesirable side effects (67, 80, 81).
Hyperthermia
Hyperthermia can be used to treat GBM patients (82, 83) and eradicate tumor cells by generating heat using methods such as direct heating (84), focused ultrasound (FUS) (85), electromagnetic methods at the target site (86, 87), or laser-induced interstitial heating (88). The combination of NPs and hyperthermia is a novel approach that allows for controlled heating of tumor tissue (87). Hyperthermia induces physiological changes in tumor cells that results in their apoptosis. Temperatures ranging from 41–46° C activate many intra- and extracellular degradation mechanisms such as aggregation of denatured proteins at the nuclear matrix and protein misfolding both of which ultimately leads to apoptosis, membrane dissolution, and cell necrosis (89). However, hyperthermia may also have a detrimental effect on healthy tissues such as induction of cell death in neural cells and increasing oxyhemoglobin saturation in red blood cells (87, 90).
Immunotherapy
The current standard treatments for GBM patients such as surgery, chemotherapy or radiotherapy have limitations due to damage to surrounding healthy brain tissue and/or other adverse off-target effects. In addition, these conventional treatments have demonstrated only minor enhancements in GBM patients’ overall survival. Thus, researchers have developed immunotherapeutic strategies for GBM patients which have demonstrated evidence of immunostimulatory effects in clinical settings (91). There are several immunotherapeutic approaches that can be used including: autologous stimulated lymphocytes (92), immune check-point inhibition (54), cytokine therapy (93), peptide vaccines and dendritic cell therapy (94). Using immunotherapy, mean overall survival can be almost tripled (e.g. 38.4 months, compared to current standard of care; 14.6 months), as has been the case in clinical trials on GBM patients receiving autologous dendritic cell vaccinations (94). For, as yet unknown reasons, patient responses to these approaches can vary greatly; and it is likely, as is also apparent for other tumor types, that immunotherapy of GBM patients will need to be given in conjunction with other types of therapy if longer survival outcomes or “cures” are to be achieved. For this strategy, the limitations are almost the same as monoclonal antibodies. Antibody-based therapy and immunotherapy have utility as treatments for GBM and are FDA approved; however, they are limited by the large molecular size of antibodies which hinders its ability to cross the BBB and thus efficacy (40, 63).
Therefore, it is essential that researchers find alternative approaches to overcome the limitations of current GBM treatment strategies and improve survival outcomes. NPs-based therapy has emerged as an effective and promising alternative strategy.
Nanoparticles (NPs)
Desirable physicochemical characteristics for NP-based GBM treatments
Nanomedicines, and specifically NPs, are being investigated as potential approaches to enhance the delivery of tumoricidal drugs in patients with GBM (26). NPs ranging from 1 to 1000 nm in diameter (d.) can be used for different purposes aside from drug delivery, including gene delivery and diagnostics (95). Many types of NPs exist that differ in terms of shape, size, charge, composition, and functionality; and they can be fabricated using a range of techniques including nanoprecipitation, double emulsion solvent evaporation or lithography (96). Using a nanoprecipitation method, NPs can be synthesized using biodegradable and biocompatible polyester homopolymers such as polylactic acid (PLA), polylactic-co-glycolic acid (PLGA) and polycaprolactone (PCL) that entrap/adsorb drug compounds and, upon appropriate functionalization, can improve the delivery of hydrophobic or hydrophilic small drug molecules to specific target sites (97). The use of biodegradable polymers can result in controlled drug release lasting several days or weeks. NPs can decrease the side effects of some drug compounds, improve solubility and permeability and protect drugs from enzymatic and chemical degradation (98).
When synthesizing NP formulations, certain parameters such as particle size, surface charge and composition may significantly affect brain uptake, cytotoxicity and therapeutic response (99, 100). Jallouli et al. studied the ability of cationic versus neutral 60 nm porous NPs (with maltodextrin backbones) to traverse the BBB in an in vitro BBB model previously shown to correlate well with in vivo findings. Neutral NPs were found to traverse endothelial cells via caveolae-dependent transcytosis. The authors proposed that the uptake may be mediated by the glucose transporter (GLUT-1) and/or lectins. Both the cationic and neutral NPs successfully traversed the model BBB via transcytosis using a lectin-dependent mechanism; however, transcytosis of the cationic NPs was less efficient (101). These results indicate that surface charge could influence both binding to, and traversing through the endothelial cells, and that both cationic and neutral porous NPs might be potential candidates for drug delivery to the brain. One technique commonly used to modify the surface of NPs is the conjugation of polyethylene glycol (PEG) to the surface, a process known as PEGylation. PEGylation has been shown to decrease opsonization and, consequently, uptake by the reticuloendothelial system (RES), resulting in prolonged circulating half-lives of the PEGylated NPs (102). In a study by Zhao et al., they used a GBM mouse model to demonstrate the safety and efficacy of PEGylated polyamidoamine (PAMAM; 5th generation) dendrimer NPs when conjugated to the CREKA peptide. They showed that these PEGylated NPs has a prolonged in vivo circulation time compared to uncoated NPs, decreased the inherent toxicity of PAMAM and deeply penetrated GBM tissue (103–105). There have been several in vivo studies using both healthy rats and mice as well as a mouse breast cancer model that support the claim that PEGylated-NPs can effectively prolong nanocarrier circulation in the blood and increase the stability of NP formulations compared to uncoated NPs (106–108).
As stated previously, the treatment strategies for GBM patients such as surgery, radiotherapy and hyperthermia are typically effective and safe GBM treatments (31, 38, 82, 83). The issue with current GBM treatments stems from the fact that they are often not particularly efficacious in the clinical setting (32, 52, 80, 81, 89). The reasons for their ineffectiveness include: (1) invasive GBM cells can prevent the complete surgical removal of GBM tissue making recurrence highly probable (32); (2) larger tumors have a poor response and often develop a resistance to radiotherapy (52); and (3) these treatments cause damage to surrounding healthy brain tissue and to the epithelial surfaces of the soft organs (i.e. mouth and throat) (32, 52, 89). The use of NPs offers the ability to load anticancer agents into a vehicle which allows them to have prolonged blood circulation, avoid the RES, and provide protection from degradation all of which means the compounds reach the target GBM tumors and maintain their anticancer activity (96). The composition, size and surface characteristics of NPs can be easily modified to achieve drug delivery to brain tumors (95–97).
An ideal NP system must have several physicochemical properties to be effective for drug delivery to the brain such as being nontoxic, biodegradable, biocompatible and having a particles size of less than 200 nm. Effective NPs are ones that do not produce an immune response and have a controllable release profile. NPs which have been modified to selectively target the brain are ones that will have the greatest potency (97, 98, 102, 109, 110). NPs are a useful approach to delivering drug compounds across the BBB for the treatment of brain diseases such as GBM where oftentimes therapies have limited access to the target disease tissue (111). However, there are several challenges of NP based delivery for GBM treatment that should be addressed. NPs might have a broad size distribution (112–114), be problematic to scale up (115), and can result in unwanted drug distribution and accumulation in off-target tissues (i.e. liver) (116). In addition, using surfactants during the NP fabrication process can cause toxicity (117, 118). In order to address these issues, researchers have determined what properties of NPs are desirable for a successful GBM treatment formulation. The advantages and disadvantages of NPs as therapeutic vectors for GBM treatment are summarized in Table I.
Table I.
Advantages and disadvantages of NPs used for GBM treatment.
| Advantages | Disadvantages |
|---|---|
| - Easily prepared (26) | - Scaling up can be problematic (115) |
| - Readily modified for targeting (103, 112, 119, 120) | - Surfactant can cause toxicity (117, 118) |
| - Versatile vector for drugs, biological agents, and nucleic acids (26, 103, 112, 119, 120) | - Can result in unwanted drug distribution and accumulation in off-target tissues such as the liver and kidneys (116) |
| - High therapeutic efficacy (121–123) | - Broad size distribution (112–114) |
| - Increases drug circulation half-life (102,124–126) | - Can be cytotoxic per se(99) |
| - Allows for controlled release (127) | |
| - Reduces drug-mediated toxicity (113, 122) | |
| - Smaller size NPs can take advantage of the EPR effect which lends to its ability to accumulate in the tumor tissues (128) |
Mechanisms of uptake for GBM NPs
The major properties of NPs that control their brain targeting ability are their size, surface charge, whether their composition is made of hydrophilic and/or hydrophobic polymers, and the addition of targeting ligands on the NP surface. The nanostructure of the polymers used for NP synthesis, such as PCL, PLA, or PLGA copolymers, enable anticancer agents to cross the BBB, reach the target GBM tumors, and protect the drug from degradation and RES which allows for more drug to reach the target and enhances the anticancer activity (123, 129, 130). Additionally, smaller NPs with a ~90 nm d. can penetrate through the leaky blood-brain tumor barrier and accumulate in the GBM tumor via the enhanced permeability and retention (EPR) effect. This process does not require energy owing to the extravasation at the site which propels the NPs by means of the intravascular and interstitial pressure differential (128).
When discussing the mechanisms by which NPs cross the BBB, researchers have found that there are two main types of transcytosis mechanism that NPs use namely adsorptive-mediated transcytosis (AMT) and receptor-mediated transcytosis (RMT). To enhance the transport of NPs via AMT across the BBB, NP conjugated with either cationic proteins or PAMAM dendrimers were reported to enhance brain concentration in comparison to unconjugated NPs (26, 125). Cationic NPs are attractive carriers because they may cross the BBB by AMT (26, 125), however some researchers have suggested that both cationic and neutral NPs cross the BBB via transcytosis using a lectin-dependent mechanism (101). Thus, the effect of surface charge on AMT uptake needs further investigation. The issue with AMT is that it is a non-specific process meaning cationic NPs will be taken up by off-target organs leading to toxicity (103, 104). RMT, in contrast, is a more specific mechanism than AMT making it an attractive alternative target for NP delivery to the brain. RMT is activated by the use of specific ligands and other surface modifiers on the NP surface, which are substrates for specific BBB receptors for example transferrin receptor (TfR)-specific monoclonal antibody (OX26) (119), glutathione (131), low-density lipoprotein receptor-related proteins (LRP) (122, 132), or interleukin-13 on the GBM cells (123). Surface modification of NPs using CREKA peptides (103, 133) or PEGylation (103, 131) have also been shown to improve their ability to cross the BBB via RMT and enhance their anticancer activity against GBM. After NPs interact with the specific cell membrane receptors, they are endocytosed into the cells via clathrin-dependent transcytosis or caveolae-dependent transcytosis. Depending on the transcytosis mechanism used, either clathrin-1 proteins will form clathrin coated vesicles or caveolin proteins will form caveolar vesicles. These vesicles are responsible for encapsulating the NPs and moves them into the cell (134). Larger NPs with a mean greater than 1 μm d. can be delivered via a non-selective uptake mechanism (i.e. macropinocytosis) which forms large endocytic vacuoles using an actin-dependent process to enter the cells (134, 135).
NPs in GBM treatment
There has been a significant amount of research performed regarding the development of NP formulations to treat GBM and Table II summarizes their various physicochemical characteristics as well as salient findings. In pre-clinical studies, NPs carrying anticancer agents often prolonged median survival times in pre-clinical GBM animal models compared to soluble anticancer agents. NPs can be made from naturally sourced materials (e.g. gelatin, chitosan, albumin or polysaccharides) or synthetic, and preferably biodegradable and biocompatible, polymers such as PLA, PLGA and PCL (97). These polymeric NPs can encapsulate chemotherapeutic drugs useful for GBM treatment and induce selective toxicity at the target site.
Table II.
Preclinical findings and physicochemical characteristics of NPs and nanocarrier formulations used to treat GBM.
| Nanocarrier | Average diameter (nm) ± SD | ζ-potential (mV) ± SD | Drug | Salient Outcome(s) | Model | References |
|---|---|---|---|---|---|---|
| Polysorbate 80 coated PBCA | 240 ± 40 | −13.0 ± 2.1 | DOX |
|
Healthy rat model | (113) |
| Lecithin-PLGA- HSA-P188 | 468 ± 19 | −11.2 | DOX |
|
GBM rat model | (141) |
| Glutathione PEGylated | 96–119 | N/A | Methylprednisolone |
|
Multiple sclerosis rat model | (126) |
| Glutathione PEGylated | 95 | N/A | DOX |
|
GBM mouse model | (131) |
| MPEG-PCL | 72.5 ± 2.2 | −3.08 ± 0.94 | PTX |
|
GBM mouse model | (148) |
| IL-13 coated PEG-PCL | 113.4 | −3.56 | Docetaxel |
|
Glioma mouse model | (123) |
| Angiopep-2-conjugated PEG-PCL | 90 | N/A | PTX |
|
Glioma mouse model | (122) |
| RGD/IL-13 PEG-PCL | 121.3± 59.8 | −10.29 | Coumarin-6 |
|
GBM mouse model | (114) |
| PLGA | 71 ± 13 | N/A | PTX |
|
Glioma rat model | (149) |
| Angiopep-2-PLGA | 30 ± 1.24 | −48 ± 1.27 | DOX |
|
GBM mouse model and U87MG (GBM) cell lines | (170) |
| Transferrin coated PLGA | 145.7 ± 25.0 | N/A | PTX |
|
C6 glioma cell lines | (112) |
| Transferrin-PEG-PLA | 153.3 ± 28.2 | −9.6 ± 0.4 | Resveratrol |
|
C6 glioma rat model | (120) |
| OX26-PLGA | 194 ± 1 | −20 ± 2 | TMZ |
|
U87MG (GBM) cell lines | (119) |
| Anti-EGFR-PLGA | 254 ± 10 | −6.6 ± 4.1 | Curcumin |
|
DKMG/EGFRvIII cell lines (EGFRvIII overexpressed human glioblastoma cell line) | (171) |
| Anti-miR-21-PLA | 153 ± 3 | −22 | TMZ |
|
Glioma rat model | (155) |
| PEGylated PAMAM | 7.52 ± 0.35 | 3.39 ± 0.37 | CREKA peptide |
|
GBM mouse model | (103) |
| Transferrin and tamoxifen conjugated PEGylated PAMAM | 117 | N/A | DOX |
|
C6 glioma cell lines and BMVECs cells | (127) |
| PEG nanocarrier | 270 ± 20 | N/A | DOX |
|
GBM rat model | (137) |
| PEGylated dimethacrylate | 35 ± 1.5 | −5.2 ± 12.4 | TMZ |
|
U87MG mouse model and U87MG cell lines | (172) |
| CREKA PEG-PLGA | 93.2 ± 2.4 | −20.3 ± 3.1 | PTX |
|
U87MG moue model | (129) |
| tLyp-1-PEG-PLA | 111.3 ± 15.6 | −24.3 ± 3.36 | PTX |
|
C6 glioma mouse model | (173, 174) |
| Cyclic RGD-PEG-PLA | 73 | N/A | PTX |
|
GBM (U87MG) mouse model | (175) |
| CK peptide PEG-PLA | 117.36 | −26.72 | PTX |
|
U87MG mouse model | (130) |
N/A: Not applicable, PBCA: Poly(butyl cyanoacrylate), PLGA: polylactic-co-glycolic acid, - HAS: human serum albumin, P188: poloxamer 188, PEG: polyethylene glycol, , MPEG-PCL: Methyl poly(ethylene glycol)-poly(e-caprolactone), IL-13: interleukin-13, TMZ: temozolomide, DOX: doxorubicin, PTX: paclitaxel, PCL: polycaprolactone, RDG: a tripeptide of arginine-glycine-aspartic acid, OX26: a monoclonal antibody specific for the transferrin receptor (TfR), EGFR: epidermal growth factor receptor, miR-21: an oncogenic microRNA, PLA: polylactic acid, PAMAM: polyamidoamine, CREKA: a peptide of cysteine-arginine-glutamic-lysine-alanine, tLYP-1: a peptide specific for tenascin C (extracellular matrix component) and neutropilin-1 on neovasculature and glioma cells, CK peptide: composition of a human sonic hedgehog (SHH) targeting CVNHPAFAC peptide and a KDR targeting peptide (K237), C6: rat glioma cell lines, U87MG cells: human glioblastoma cells, and BMVECs: murine brain microvascular endothelial cells.
One drawback of NPs as delivery systems for GBM is that, due to the phagocytic macrophages located in the spleen and liver, many systemically delivered NP formulations do not accumulate sufficiently in the brain to have an effect on tumor growth. Thus, formulation modifications such as coating the NPs with hydrophilic surfactants have proven to be beneficial in altering the biodistribution of the administered NPs, enhancing accumulation in the brain, and also increasing circulation half-life of the NPs. Poly(n-butyl cyanoacrylate) (PBCA) NPs loaded with DOX and coated with the surfactant, polysorbate 80 (Tween® 80), have been investigated as a potential therapy for GBM. Using these NPs in a rat glioma model, Ambruosi et al. showed that the NPs delivered the drug across the BBB after IV injection, and that these NPs increased median survival times with 35% of the animals living for the entire length of the study (180 days) (136). Soluble DOX was less effective with only 10% of the animals surviving for greater than 65 days, while untreated animals survived for 18 – 24 days (136).
Steiniger et al. developed DOX-loaded PBCA NPs (270 ± 20 nm d.) with or without a polysorbate 80 coating, and investigated their therapeutic potential in a GBM tumor rat model (137). The median survival time of animals treated with DOX-loaded PBCA-polysorbate 80 coated NPs (injected IV) increased by 85% and 24% compared to the untreated control and DOX solution groups, respectively. NPs without the polysorbate 80 coating had a median survival time that increased by 38% compared with the untreated control group (137). Pereverzeva et al. conducted a tolerance profile and toxicity study of DOX-loaded PBCA NPs (240 ± 40 nm d.; injected IV) and DOX-loaded human serum albumin (HSA) NPs (404 ± 24 nm d.; injected IV) in healthy rats, demonstrating that both NP formulations were safer and reduced testicular and cardiological toxicity (measured on days 15 and 30) compared to soluble DOX (injected IV). The dosing regimen of DOX was 3× 1.5 mg/kg spaced 3 days apart (113). In a different study using healthy rats, Gulyaev et al. demonstrated that DOX-loaded PBCA NPs coated with polysorbate 80 (270 nm d.; injected IV) increased DOX concentrations in the brain by more than 60-fold compared to the uncoated equivalent indicating that the surfactant-coated NPs reached the brain more efficiently (138). Borchard et al. performed an in vitro study using bovine microvessel endothelial cells and found that coating poly(methyl methacrylate) NPs with polysorbate 80 led to a 5-fold increase in uptake into brain tissue compared to the uncoated NP control group (139). Although the mechanism by which polysorbate 80-coated NPs traverse the BBB has not been definitively established, their ability to effectively deliver drugs to brain tissue and significantly increase pharmacological effects compared to uncoated NPs and free drug has been demonstrated in rodent models as well as in vitro (138–140).
Another study using the rat 101/8 GBM model showed that DOX-loaded lecithin-PLGA-HSA NPs coated with the surfactant, poloxamer 188 (P188) (468 ± 19 nm d.; injected IV), delivered DOX across the BBB at a therapeutically relevant concentration and significantly reduced tumor growth compared to soluble DOX (141). HSA in this formulation functioned as a NP stabilizer and was used instead of polyvinyl alcohol (PVA). Lecithin was used to enhance efficacy of the NPs by increasing the amount of surface attached Pluronic copolymer, a phenomenon also reported for other NP formulations (141–143). Previous studies have demonstrated that using Pluronic copolymers such as P85 and P188 as surfactant coatings on NPs enhances delivery to cancer cells although the mechanism by which this occurs is uncertain (142, 144).
Aside from surfactants, specific ligands and other surface modifiers can be conjugated onto polymeric NPs to enhance anticancer activity toward GBM, prolong blood circulation (124) and reduce undesired off-target cytotoxicity of the drugs they carry (125). One example is glutathione, an endogenous tripeptide that possesses antioxidant-like properties and is actively transported across the BBB (145, 146). Moreover, glutathione has been used as a targeting ligand by coupling it to PEGylated liposomes to enhance their uptake into brain tissue via the glutathione transporter (126, 147). An in vivo study using female athymic Friend leukemia virus B mice challenged with human glioblastoma cells (U87MG) showed that DOX-loaded glutathione PEGylated liposomes (95 nm d.; injected IV) increased the median survival time by 38.5% compared with saline treated mice. This formulation increased the solubility of DOX, enhanced DOX activity as well as reduced side effects. For these reasons, this formulation was used in clinical phase I/IIa trials (NCT01386580) (131).
Methoxy poly(ethylene glycol)-poly(e-caprolactone) (MPEG-PCL) NPs have been investigated as nanocarriers to deliver anticancer agents to treat GBM. Xin et al. showed that PTX-loaded MPEG-PCL NPs (72.5 ± 2.2 nm d.; injected IV) had antitumor activity in a nude mouse human GBM model. The results showed that the mean survival time when treated with PTX-loaded MPEG-PCL NPs was significantly longer (28 days) than mice treated with Taxol (20 days) or PTX-loaded PCL NPs (23 days) (148). In another study, Zhou et al. developed PTX-loaded PLGA NPs (71 ± 13 nm d.; delivered intracranially) to treat GBM and showed that they significantly increased median survival time in a tumor-bearing rat model compared to free drug. These NPs also demonstrated the ability to deeply penetrate the brain tissue as opposed to larger NPs (147 ± 27 nm d.) and provided controlled drug release (149). This enhanced delivery/penetration (under conditions of convection-enhanced delivery) was reported to be due to the size of the NPs (71 ± 13 nm d.) since the U87MG cells characteristically have a pore cutoff size range of 7 – 100 nm (150), occluding NPs with greater than 100 nm d.. Because the NPs were delivered intracranially, the issue of the BBB was circumvented. Hobbs et al. reported that long-circulating NPs in the 100 – 200 nm size range exhibited a more diffuse extravasation along the vessel while, in comparison, NPs which were 380 – 780 nm in size extravasated in a more focal manner (150). In one particular study, transferrin was used as a specific ligand on the surface of PTX-loaded NPs (injected IV) and resulted in increased cytotoxicity toward, and enhanced intracellular uptake by, a C6 rat glioma cell line compared to free drug and uncoated PLGA NPs (112). The results confirmed that the therapeutic effects of PTX depended on transport of PTX across the BBB and its ability to reach the glioma site.
TMZ-loaded PLGA NPs (194 ± 1 nm d.) conjugated with a transferrin receptor (TfR)-specific monoclonal antibody (OX26) demonstrated enhanced ability to cross the BBB, resulting in increased anticancer activity of TMZ in in vitro studies. The TfR is highly overexpressed on glioblastoma cells (151) compared to most normal tissues, thus making this receptor an attractive active target for tumoricidal agents (119). By functionalizing the NPs with anti-TfR monoclonal antibodies anticancer activity was enhanced by not only improving their capacity to cross the BBB but also triggering enhanced uptake by GBM cells.
Another target that has been exploited to localize NPs to glioma cells has been interleukin-13 (IL-13) receptor alpha 2 (IL-13Rα2) which is highly overexpressed by glioma cells compared to normal cells (152). Gao et al. developed a glioma treatment utilizing docetaxel-loaded PEGylated-PCL NPs (113.4 nm d.) decorated with an IL-13 peptide ligand (123). They found that the IL-13 peptide resulted in significantly increased anticancer activity of docetaxel versus free drug and saline groups in a glioma-bearing murine model. It was demonstrated that IL-13 functionalization of NPs resulted in increased cellular uptake by glioma cells, altering the major uptake pathway from macropinocytosis to clathrin-dependent endocytosis and increasing glioma localization (123).
Another example of a surface modification for targeting purposes is angiopep-2, a ligand for LRP. NPs targeting LRP are transported across the BBB via LRP-mediated transcytosis and are also taken up by glioma cells via LRP-mediated endocytosis (122, 132). These transport mechanisms are illustrated in Figure 3. Xin et al. conjugated angiopep-2 to PEG-PCL NPs to target the BBB and glioma cells in glioma-challenged mice (122). The mean survival time of mice treated with PTX-loaded angiopep-2-PEG-PCL NPs (90 nm d.; injected IV) was significantly higher (35.8 days) than mice treated with saline (21.7 days), Taxol (24.1 days) or PTX-loaded PEG-PCL NPs (29.7 days) (122). In addition to enhancing antitumor efficacy, preliminary studies with the PTX-loaded angiopep-2-PEG-PCL NPs demonstrated that this GBM treatment was safe compared to free drug solutions and showed no acute toxicity to the brain, liver, kidney, or hematological system in mice (122).
Figure 3.
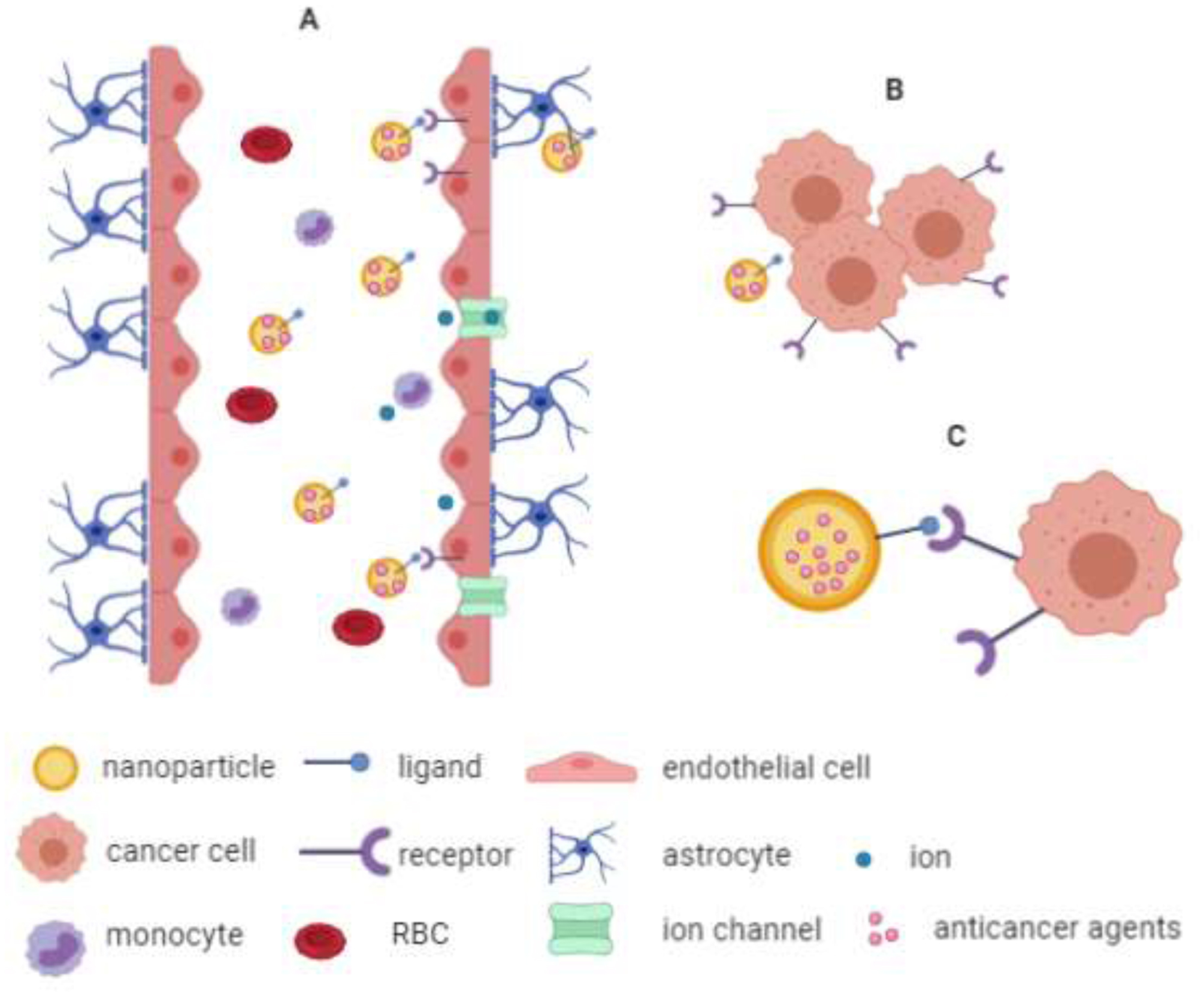
Schematic of the general mode of action of GBM targeting NP treatments. (A) NPs in blood circulation cross the BBB via RMT. (B and C) NPs with specific-targeting ligands (e.g. transferrin or angiopep-2) conjugated to their surface reach the GBM cells where they can be taken up by receptor-mediated endocytosis which will ultimately lead to GBM cell death (not shown).
Another target that has been researched for GBM treatment is miR-21, an oncogenic microRNA (miRNA) that is overexpressed in glioma cells compared to normal brain tissue (153). miR-21 plays an important role in cancer development because it is an apoptosis suppressor (154). Seo et al. used anti-miR-21-loaded PLA-hyperbranched polyglycerol (HPG) NPs (153 ± 3 nm d.; intracranial convection-enhanced delivery) and demonstrated promising findings upon treating malignant brain tumors in a rat model (155). The results showed that the median survival of rats treated with anti-miR-21-loaded PLA-HPG NPs was 29 days compared to 24 days for the untreated group. This formulation was also used in combination with TMZ solution and it was found that the median survival time was prolonged to 50 days compared to 41 days for TMZ solution alone. This corresponded to a 108% increase in median survival compared to the untreated group and a 22% increase compared to TMZ solution alone group (155).
Some researchers have used dendrimers to generate drug delivery NPs to treat GBM. Dendrimers are composed of repeating monomeric units, have extensive branching chains, multivalent functional groups and possess an internal cavity surrounded by reactive terminal groups (156). Their biocompatibility and biochemical properties could be beneficial as a controlled and specifically targeted drug delivery system and to improve the pharmacokinetic properties of drugs (156, 157). PAMAM dendrimer-based nanoplexes (or dendriplexes) can be loaded with drugs and macromolecules such as DNA or small interfering RNA (siRNA). Due to the high density of cationic charges on PAMAM, dendriplexes form when they are mixed with negatively charged DNA or siRNA. Dendriplexes can protect the nucleic acids from degradation and deliver them to their target sites. In addition, modification of PAMAM by adding specific ligands can enhance targeting to tumor cells overexpressing cognate receptors (158). In one study, PEGylated PAMAM (5th generation) dendrimer NPs were conjugated to the peptide, CREKA, which can bind to fibrin, an abundant component in GBM tumors. It was found that these small sized NPs (7.52 ± 0.35 nm d.; injected IV) demonstrated high accumulation and deep penetration into GBM tissue compared to normal tissues (103, 133). It should be noted however that no therapeutic study was performed, and one potential issue could be delivering sufficient quantities of a tumoricidal agent using such small NPs.
In another study by Li et al., DOX was coupled to the exterior of PAMAM (4th generation) via acid labile hydrazone bonds [154]. Hydrazones are organic molecules similar to ketones and aldehydes except that the oxygen is substituted with the N=NH2 functional group. The PAMAM-DOX conjugate was then PEGylated; as it has been found that PAMAM conjugated with PEG can decrease the inherent cytotoxicity of PAMAM (159). Finally, transferrin was conjugated to the PEGylated PAMAM dendrimers, and tamoxifen solution was encapsulated into the transferrin-PEGylated PAMAM-DOX dendrimers resulting in a dendriplex with dual targeting properties due to the presence of transferrin and tamoxifen, the latter of which has been shown to inhibit MDR as well as promote transport across the BBB (127, 160, 161). This formulation showed a capacity to deliver drugs across an in vitro BBB model as well as reduce the growth of avascular murine C6 glioma spheroids (in vitro) (127). In the dendriplex formulation used by Li et al, the hydrazone linkages resulted in 32% DOX release under weak acidic conditions (pH 4.5) and 6% at physiological pH 7.4 over a period of 24 hours. This study indicated that acidic environments triggered a more rapid release of DOX as well as demonstrating the stability of the dendriplex at physiological pH (127). These results were corroborated by Lu et al. who recently reported the conjugation of DOX and lactose-PEG via an acid labile hydrazone linkage, and found that the release of DOX was significantly higher at pH 5.0 than pH 7.4 (162).
Combination of NPs with other strategies for GBM treatment
In a review by Choi et al., they summarized that using radiotherapy with high atomic number (high-Z) metal NPs (i.e. gold or iron oxide) could significantly inhibit tumor growth in comparison to radiation alone in an in vivo mouse model (163). The therapeutic effects of radiation were enhanced by the oxidative stress and DNA damage caused by these high-Z NPs (164). In another study by Kievit et al., they showed that the majority of radiation-induced DNA damage is a result of abasic lesions or single strand breaks. During the cell repair process, apurinic endonuclease 1 (Ape1) will cleave these abasic lesions via the base excision repair (BER) pathway. Ape1 is a critical species for the proper function of the BER making it a promising target to enhance the effectiveness of radiotherapy. Ape1 can be delivered by NPs to sensitize GBM cells to radiation. In their work, Kievit et al. created Ape1:NPs complexes (48.5 ± 4.0 nm d.; injected IV) and found that the combination of these complexes with radiation significantly extended the progression free survival time to 48 days compared to 32 days for radiation alone in a genetic GBM mouse model (165). In this case, using Ape-1 NPs with radiation is a promising strategy for increasing the effectiveness of radiotherapy. Additionally, NPs loaded with therapeutic agents can be used in combination with non-invasive and reversible BBB disruption strategies (i.e. FUS, ultrasound-targeted microbubble destruction (UTMD) or MRI-guided delivery) to enhance their ability to cross the BBB and improve survival time in animal GBM models (166–168). In a study by Timbie et al., chemotherapy agent cisplatin loaded into NPs (45.3 ± 2.5 nm d.; injected IV) delivered with MRI-guided FUS improved survival time of F98 glioma rats by 61% and 64% compared to untreated control and NPs alone, respectively (166). Another study utilized a C6 GBM rat model treated with cilengitide, a cyclic arginine–glycine-aspartic acid (RGD) pentapeptide antagonist for the integrin receptors that are over-expressed in GBM and tumor-invaded endothelial cells. Cilengitide-loaded NPs (105 ± 1.78 nm d.; injected IV) combined with UTMD significantly prolonged survival time (81.2 days) compared to the normal saline group (16.5 days) and NPs alone group (30.4 days) (167). This UTMD has been shown to increase permeability of NPs delivery across the biological barrier into the brain without causing cellular damage (169) and to enhance glioma-targeted therapy of cilengitide (167). Although FUS and UTMD have no therapeutic effects as GBM treatments on their own, they can be used as a synergistic strategy when combined with drug-loaded NPs.
Challenges to NP-based drug delivery to GBM
NP drug delivery systems are a promising means of treating various types of cancers, particularly GBM. As described above, modifying NP surfaces such that they specifically target membrane receptors can enhance their ability to cross the BBB and reach target tissues. Using drug-loaded NPs versus soluble drugs can enhance cellular uptake, favor deep penetration into tissues and decrease the off-target effects of the drug. Problematically, NPs designed to cross biological membranes can cause undesired toxicities towards healthy tissues. This can be due to excessive disruption of the integrity of intracellular/cellular membranes by the NPs, potentially resulting in cell death (176). Although some NP formulations possess the ability to deliver genes into the nucleus by passing through the nuclear membrane, certain NPs can damage DNA often resulting from NP-induced oxidative stress (117, 118, 177). Nanotoxicity can result from various physicochemical properties of NPs such as size, shape and surface traits, which may significantly affect biological interactions and chemical absorption in a non-specific manner (178).
Biodistribution is an important consideration when it comes to administering NPs for drug delivery purposes. Even though targeted-nanocarriers can improve the delivery of drug compounds directly to target sites compared to soluble drug or non-targeting NPs, unwanted drug distribution and accumulation in off-target tissues such as the liver and kidneys is still a significant drawback (116). Another consideration is the biodegradability of the NP formulation, since it is desired that the NPs are eliminated in a timely manner and without generating toxic biproducts.
Currently, many researchers are developing NP formulations for GBM (and other diseases) which do not have defined toxicity profiles or for which there is a paucity of toxicity data. This is particularly the case when considering long term toxicity resulting from NPs that may be eliminated more slowly from the body. Thus, standardized in vitro and in vivo toxicity assays need to be performed in order to determine the nanotoxicity of NP-based drug delivery systems.
Conclusion
Malignant brain tumors such as GBM possess an added degree of difficulty when it comes to treatment compared to tumors in other parts of the body due to the BBB. To overcome this and other challenges associated with brain tumors, various novel drug delivery systems such as NPs have been designed, synthesized and evaluated for GBM treatment and have resulted in prolonged median survival times in pre-clinical studies compared with the soluble therapeutic agents. NPs have also been shown to reduce off-target effects compared to soluble drugs. NPs can be designed to promote drug accumulation at the site of the brain tumor and result in synergistic effects when combined with other treatments, not only for brain tumors or GBM, but also for CNS related diseases. The significance of NPs as drug delivery systems is increasing and from the results described here, a promising new era in treatment of brain diseases, especially GBM, has the potential to unfold.
Acknowledgements
K.W acknowledges support from the Government Pharmaceutical Organization (GPO) scholarship. J.C.Q. acknowledges support from the Alfred P. Sloan Foundation, the University of Iowa Graduate College, and the American Association for University Women. A.K.S acknowledges support from the Cancer Center support grant (P30 CA086862) and the Lyle and Sharon Bighley Chair in Pharmaceutical Sciences.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
References
- 1.Aldape K, Zadeh G, Mansouri S, Reifenberger G, von Deimling A. Glioblastoma: pathology, molecular mechanisms and markers. Acta neuropathologica. 2015;129(6):829–48. [DOI] [PubMed] [Google Scholar]
- 2.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta neuropathologica. 2007;114(2):97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta neuropathologica. 2016;131(6):803–20. [DOI] [PubMed] [Google Scholar]
- 4.Ghosh D, Nandi S, Bhattacharjee S. Combination therapy to checkmate Glioblastoma: clinical challenges and advances. Clinical and Translational Medicine. 2018;7(1):33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vigneswaran K, Neill S, Hadjipanayis CG. Beyond the World Health Organization grading of infiltrating gliomas: advances in the molecular genetics of glioma classification. Annals of Translational Medicine. 2015;3(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Claes A, Idema AJ, Wesseling P. Diffuse glioma growth: a guerilla war. Acta neuropathologica. 2007;114(5):443–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tso C-L, Freije WA, Day A, Chen Z, Merriman B, Perlina A, et al. Distinct Transcription Profiles of Primary and Secondary Glioblastoma Subgroups. Cancer research. 2006;66(1):159. [DOI] [PubMed] [Google Scholar]
- 8.Kabat GC, Etgen AM, Rohan TE. Do steroid hormones play a role in the etiology of glioma? Cancer Epidemiol Biomarkers Prev. 2010;19(10):2421–7. [DOI] [PubMed] [Google Scholar]
- 9.Zhang YY, Ruan LX, Zhang S. Rapid progression of glioblastoma multiforme: A case report. Oncol Lett. 2016;12(6):4803–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kleihues P, Ohgaki H. Primary and secondary glioblastomas: from concept to clinical diagnosis. Neuro-oncology. 1999;1(1):44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bush NAO, Chang SM, Berger MS. Current and future strategies for treatment of glioma. Neurosurgical Review. 2017;40(1):1–14. [DOI] [PubMed] [Google Scholar]
- 12.Mahvash M, Hugo H-H, Maslehaty H, Mehdorn HM, Stark AM. Glioblastoma Multiforme in Children: Report of 13 Cases and Review of the Literature. Pediatric Neurology. 2011;45(3):178–80. [DOI] [PubMed] [Google Scholar]
- 13.Larjavaara S, Mäntylä R, Salminen T, Haapasalo H, Raitanen J, Jääskeläinen J, et al. Incidence of gliomas by anatomic location. Neuro-oncology. 2007;9(3):319–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krex D, Klink B, Hartmann C, von Deimling A, Pietsch T, Simon M, et al. Long-term survival with glioblastoma multiforme. Brain. 2007;130(10):2596–606. [DOI] [PubMed] [Google Scholar]
- 15.Nakada M, Kita D, Watanabe T, Hayashi Y, Teng L, Pyko IV, et al. Aberrant signaling pathways in glioma. Cancers (Basel). 2011;3(3):3242–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mishra SS, Behera SK, Dhir MK, Senapati SB. Cerebellar giant cell glioblastoma multiforme in an adult. J Neurosci Rural Pract. 2014;5(3):295–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robert M, Wastie M. Glioblastoma multiforme: a rare manifestation of extensive liver and bone metastases. Biomed Imaging Interv J. 2008;4(1):e3–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ray A, Manjila S, Hdeib AM, Radhakrishnan A, Nock CJ, Cohen ML, et al. Extracranial metastasis of gliobastoma: Three illustrative cases and current review of the molecular pathology and management strategies. Mol Clin Oncol. 2015;3(3):479–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Romero-Rojas AE, Diaz-Perez JA, Amaro D, Lozano-Castillo A, Chinchilla-Olaya SI. Glioblastoma metastasis to parotid gland and neck lymph nodes: fine-needle aspiration cytology with histopathologic correlation. Head Neck Pathol. 2013;7(4):409–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Widjaja A, Mix H, Golkel C, Flemming P, Egensperger R, Holstein A, et al. Uncommon metastasis of a glioblastoma multiforme in liver and spleen. Digestion. 2000;61(3):219–22. [DOI] [PubMed] [Google Scholar]
- 21.Tysnes BB, Mahesparan R. Biological mechanisms of glioma invasion and potential therapeutic targets. J Neurooncol. 2001;53(2):129–47. [DOI] [PubMed] [Google Scholar]
- 22.Davis ME. Glioblastoma: Overview of Disease and Treatment. Clin J Oncol Nurs. 2016;20(5 Suppl):S2–S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alphandéry E Glioblastoma Treatments: An Account of Recent Industrial Developments. Frontiers in Pharmacology. 2018;9(879). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Michael JS, Lee B-S, Zhang M, Yu JS. Nanotechnology for Treatment of Glioblastoma Multiforme. J Transl Int Med. 2018;6(3):128–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saucier-Sawyer JK, Deng Y, Seo Y-E, Cheng CJ, Zhang J, Quijano E, et al. Systemic delivery of blood-brain barrier-targeted polymeric nanoparticles enhances delivery to brain tissue. Journal of drug targeting. 2015;23(7–8):736–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saraiva C, Praça C, Ferreira R, Santos T, Ferreira L, Bernardino L. Nanoparticle-mediated brain drug delivery: Overcoming blood–brain barrier to treat neurodegenerative diseases. Journal of Controlled Release. 2016;235:34–47. [DOI] [PubMed] [Google Scholar]
- 27.Chen Y, Liu L. Modern methods for delivery of drugs across the blood–brain barrier. Advanced drug delivery reviews. 2012;64(7):640–65. [DOI] [PubMed] [Google Scholar]
- 28.Zinn PO, Colen RR. Imaging Genomic Mapping in Glioblastoma. Neurosurgery. 2013;60(CN_suppl_1):126–30. [DOI] [PubMed] [Google Scholar]
- 29.Rick J, Chandra A, Aghi MK. Tumor treating fields: a new approach to glioblastoma therapy. Journal of Neuro-Oncology. 2018;137(3):447–53. [DOI] [PubMed] [Google Scholar]
- 30.Bomzon Ze, Wenger C, Proescholdt M, Mohan S. Tumor-Treating Fields at EMBC 2019: A Roadmap to Developing a Framework for TTFields Dosimetry and Treatment Planning. 2021. p. 3–17. [PubMed] [Google Scholar]
- 31.Stark AM, van de Bergh J, Hedderich J, Mehdorn HM, Nabavi A. Glioblastoma: clinical characteristics, prognostic factors and survival in 492 patients. Clinical neurology and neurosurgery. 2012;114(7):840–5. [DOI] [PubMed] [Google Scholar]
- 32.Serventi J, Behr J. Surgery and Evidence-based Treatments in Patients with Newly Diagnosed High-grade Glioma. Seminars in Oncology Nursing. 2018;34(5):443–53. [DOI] [PubMed] [Google Scholar]
- 33.Kazda T, Bulik M, Pospisil P, Lakomy R, Smrcka M, Slampa P, et al. Advanced MRI increases the diagnostic accuracy of recurrent glioblastoma: Single institution thresholds and validation of MR spectroscopy and diffusion weighted MR imaging. NeuroImage: Clinical. 2016;11:316–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frey D, Schilt S, Strack V, Zdunczyk A, Rösler J, Niraula B, et al. Navigated transcranial magnetic stimulation improves the treatment outcome in patients with brain tumors in motor eloquent locations. Neuro-oncology. 2014;16(10):1365–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mueller WM, Yetkin FZ, Hammeke TA, Morris GL 3rd, Swanson SJ, Reichert K, et al. Functional magnetic resonance imaging mapping of the motor cortex in patients with cerebral tumors. Neurosurgery. 1996;39(3):515–20; discussion 20–1. [DOI] [PubMed] [Google Scholar]
- 36.Pallud J, Rigaux-Viode O, Corns R, Muto J, Lopez Lopez C, Mellerio C, et al. Direct electrical bipolar electrostimulation for functional cortical and subcortical cerebral mapping in awake craniotomy. Practical considerations. Neurochirurgie. 2017;63(3):164–74. [DOI] [PubMed] [Google Scholar]
- 37.Sankey EW, Tsvankin V, Grabowski MM, Nayar G, Batich KA, Risman A, et al. Operative and peri-operative considerations in the management of brain metastasis. Cancer Med. 2019;8(16):6809–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fuller CD, Choi M, Forthuber B, Wang SJ, Rajagiriyil N, Salter BJ, et al. Standard fractionation intensity modulated radiation therapy (IMRT) of primary and recurrent glioblastoma multiforme. Radiat Oncol. 2007;2:26-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mann J, Ramakrishna R, Magge R, Wernicke AG. Advances in Radiotherapy for Glioblastoma. Front Neurol. 2017;8:748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sousa F, Moura RP, Moreira E, Martins C, Sarmento B. Chapter Two - Therapeutic Monoclonal Antibodies Delivery for the Glioblastoma Treatment. In: Donev R, editor. Advances in Protein Chemistry and Structural Biology. 112: Academic Press; 2018. p. 61–80. [DOI] [PubMed] [Google Scholar]
- 41.Patel SJ, Shapiro WR, Laske DW, Jensen RL, Asher AL, Wessels BW, et al. Safety and Feasibility of Convection-enhanced Delivery of Cotara for the Treatment of Malignant Glioma: Initial Experience in 51 Patients. Neurosurgery. 2005;56(6):1243–53. [DOI] [PubMed] [Google Scholar]
- 42.Barbarite E, Sick JT, Berchmans E, Bregy A, Shah AH, Elsayyad N, et al. The role of brachytherapy in the treatment of glioblastoma multiforme. Neurosurgical Review. 2017;40(2):195–211. [DOI] [PubMed] [Google Scholar]
- 43.Tanaka M, Ino Y, Nakagawa K, Tago M, Todo T. High-dose conformal radiotherapy for supratentorial malignant glioma: a historical comparison. The Lancet Oncology. 2005;6(12):953–60. [DOI] [PubMed] [Google Scholar]
- 44.Thibouw D, Truc G, Bertaut A, Chevalier C, Aubignac L, Mirjolet C. Clinical and dosimetric study of radiotherapy for glioblastoma: three-dimensional conformal radiotherapy versus intensity-modulated radiotherapy. J Neurooncol. 2018;137(2):429–38. [DOI] [PubMed] [Google Scholar]
- 45.Yanagihara TK, Saadatmand HJ, Wang TJC. Reevaluating stereotactic radiosurgery for glioblastoma: new potential for targeted dose-escalation. Journal of Neuro-Oncology. 2016;130(3):397–411. [DOI] [PubMed] [Google Scholar]
- 46.Adeberg S, Harrabi SB, Verma V, Bernhardt D, Grau N, Debus J, et al. Treatment of meningioma and glioma with protons and carbon ions. Radiat Oncol. 2017;12(1):193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kinzel A, Ambrogi M, Varshaver M, Kirson ED. Tumor Treating Fields for Glioblastoma Treatment: Patient Satisfaction and Compliance With the Second-Generation Optune(®) System. Clin Med Insights Oncol. 2019;13:1179554918825449-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kessler AF, Frömbling GE, Gross F, Hahn M, Dzokou W, Ernestus R-I, et al. Effects of tumor treating fields (TTFields) on glioblastoma cells are augmented by mitotic checkpoint inhibition. Cell Death Discov. 2018;4:12-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Buonerba C, Di Lorenzo G, Marinelli A, Federico P, Palmieri G, Imbimbo M, et al. A comprehensive outlook on intracerebral therapy of malignant gliomas. Critical Reviews in Oncology/Hematology. 2011;80(1):54–68. [DOI] [PubMed] [Google Scholar]
- 50.Levrero F, Daga A, Ravetti JL, Corv’o R, Fella M, Marcello D, et al. 224. Irradiation of glioma initiating cells-driven orthotopic glioblastoma after delivering of ATM inhibitor KU60019 as a radiosensitizer. Physica Medica. 2018;56:200. [Google Scholar]
- 51.Vecchio D, Daga A, Carra E, Marubbi D, Raso A, Mascelli S, et al. Pharmacokinetics, pharmacodynamics and efficacy on pediatric tumors of the glioma radiosensitizer KU60019. International Journal of Cancer. 2015;136(6):1445–57. [DOI] [PubMed] [Google Scholar]
- 52.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. New England Journal of Medicine. 2005;352(10):987–96. [DOI] [PubMed] [Google Scholar]
- 53.Tamura R, Tanaka T, Miyake K, Yoshida K, Sasaki H. Bevacizumab for malignant gliomas: current indications, mechanisms of action and resistance, and markers of response. Brain Tumor Pathology. 2017;34(2):62–77. [DOI] [PubMed] [Google Scholar]
- 54.Riccione KA, Gedeon P, Sanchez-Perez L, Sampson JH. Chapter 11 - Checkpoint Blockade Immunotherapy for Glioblastoma: Progress and Challenges. In: Sampson JH, editor. Translational Immunotherapy of Brain Tumors. San Diego: Academic Press; 2017. p. 261–300. [Google Scholar]
- 55.Reardon DA, Kaley TJ, Dietrich J, Lim M, Dunn GP, Gan HK, et al. Phase 2 study to evaluate the clinical efficacy and safety of MEDI4736 (durvalumab) in patients with glioblastoma (GBM). Journal of Clinical Oncology. 2016;34(15_suppl):TPS2080–TPS. [Google Scholar]
- 56.Martino EC, Misso G, Pastina P, Costantini S, Vanni F, Gandolfo C, et al. Immune-modulating effects of bevacizumab in metastatic non-small-cell lung cancer patients. Cell Death Discov. 2016;2:16025-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kandalaft LE, Motz GT, Busch J, Coukos G. Angiogenesis and the tumor vasculature as antitumor immune modulators: the role of vascular endothelial growth factor and endothelin. Curr Top Microbiol Immunol. 2011;344:129–48. [DOI] [PubMed] [Google Scholar]
- 58.Elamin YY, Rafee S, Toomey S, Hennessy BT. Immune effects of bevacizumab: killing two birds with one stone. Cancer Microenviron. 2015;8(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27(28):4733–40. [DOI] [PubMed] [Google Scholar]
- 60.Hasselbalch B, Lassen U, Hansen S, Holmberg M, Sørensen M, Kosteljanetz M, et al. Cetuximab, bevacizumab, and irinotecan for patients with primary glioblastoma and progression after radiation therapy and temozolomide: a phase II trial. Neuro-oncology. 2010;12(5):508–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Westphal M, Heese O, Steinbach JP, Schnell O, Schackert G, Mehdorn M, et al. A randomised, open label phase III trial with nimotuzumab, an anti-epidermal growth factor receptor monoclonal antibody in the treatment of newly diagnosed adult glioblastoma. European Journal of Cancer. 2015;51(4):522–32. [DOI] [PubMed] [Google Scholar]
- 62.Nitta Y, Shimizu S, Shishido-Hara Y, Suzuki K, Shiokawa Y, Nagane M. Nimotuzumab enhances temozolomide-induced growth suppression of glioma cells expressing mutant EGFR in vivo. Cancer Med. 2016;5(3):486–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Glaser T, Han I, Wu L, Zeng X. Targeted Nanotechnology in Glioblastoma Multiforme. Frontiers in Pharmacology. 2017;8(166). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Verma J, Lal S, Van Noorden CJF. Nanoparticles for hyperthermic therapy: synthesis strategies and applications in glioblastoma. Int J Nanomedicine. 2014;9:2863–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Baumann F, Bjeljac M, Kollias SS, Baumert BG, Brandner S, Rousson V, et al. Combined Thalidomide and Temozolomide Treatment in Patients with Glioblastoma Multiforme. Journal of Neuro-Oncology. 2004;67(1):191–200. [DOI] [PubMed] [Google Scholar]
- 66.Wong HL, Bendayan R, Rauth AM, Li Y, Wu XY. Chemotherapy with anticancer drugs encapsulated in solid lipid nanoparticles. Advanced drug delivery reviews. 2007;59(6):491–504. [DOI] [PubMed] [Google Scholar]
- 67.Sarkaria JN, Kitange GJ, James CD, Plummer R, Calvert H, Weller M, et al. Mechanisms of Chemoresistance to Alkylating Agents in Malignant Glioma. Clinical Cancer Research. 2008;14(10):2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Quinn JA, Jiang SX, Reardon DA, Desjardins A, Vredenburgh JJ, Rich JN, et al. Phase II trial of temozolomide plus o6-benzylguanine in adults with recurrent, temozolomide-resistant malignant glioma. J Clin Oncol. 2009;27(8):1262–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stupp R, Taillibert S, Kanner AA, Kesari S, Steinberg DM, Toms SA, et al. Maintenance Therapy With Tumor-Treating Fields Plus Temozolomide vs Temozolomide Alone for Glioblastoma: A Randomized Clinical Trial. Jama. 2015;314(23):2535–43. [DOI] [PubMed] [Google Scholar]
- 70.Reithmeier T, Graf E, Piroth T, Trippel M, Pinsker MO, Nikkhah G. BCNU for recurrent glioblastoma multiforme: efficacy, toxicity and prognostic factors. BMC Cancer. 2010;10:30-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chaichana KL, Zaidi H, Pendleton C, McGirt MJ, Grossman R, Weingart JD, et al. The efficacy of carmustine wafers for older patients with glioblastoma multiforme: prolonging survival. Neurol Res. 2011;33(7):759–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Holdhoff M, Ye X, Supko JG, Nabors LB, Desai AS, Walbert T, et al. Timed sequential therapy of the selective T-type calcium channel blocker mibefradil and temozolomide in patients with recurrent high-grade gliomas. Neuro-oncology. 2017;19(6):845–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Keir ST, Friedman HS, Reardon DA, Bigner DD, Gray LA. Mibefradil, a novel therapy for glioblastoma multiforme: cell cycle synchronization and interlaced therapy in a murine model. J Neurooncol. 2013;111(2):97–102. [DOI] [PubMed] [Google Scholar]
- 74.Batchelor TT, Duda DG, di Tomaso E, Ancukiewicz M, Plotkin SR, Gerstner E, et al. Phase II study of cediranib, an oral pan-vascular endothelial growth factor receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28(17):2817–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Odia Y, Iwamoto FM, Moustakas A, Fraum TJ, Salgado CA, Li A, et al. A phase II trial of enzastaurin (LY317615) in combination with bevacizumab in adults with recurrent malignant gliomas. J Neurooncol. 2016;127(1):127–35. [DOI] [PubMed] [Google Scholar]
- 76.Vredenburgh JJ, Desjardins A, Reardon DA, Friedman HS. Experience with irinotecan for the treatment of malignant glioma. Neuro-oncology. 2009;11(1):80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gruber ML, Buster WP. Temozolomide in combination with irinotecan for treatment of recurrent malignant glioma. Am J Clin Oncol. 2004;27(1):33–8. [DOI] [PubMed] [Google Scholar]
- 78.Gürten B, Yenigül E, Sezer A, Malta S. Complexation and enhancement of temozolomide solubility with cyclodextrins. Brazilian Journal of Pharmaceutical Sciences. 2018;54. [Google Scholar]
- 79.Cardoso FL, Brites D, Brito MA. Looking at the blood–brain barrier: Molecular anatomy and possible investigation approaches. Brain Research Reviews. 2010;64(2):328–63. [DOI] [PubMed] [Google Scholar]
- 80.van der Vring JA, Bernink PJ, van der Wall EE, van Velhuisen DJ, Braun S, Kobrin I. Evaluating the safety of mibefradil, a selective T-type calcium antagonist, in patients with chronic congestive heart failure. Clin Ther. 1996;18(6):1191–206. [DOI] [PubMed] [Google Scholar]
- 81.Nance E, Zhang C, Shih TY, Xu Q, Schuster BS, Hanes J. Brain-penetrating nanoparticles improve paclitaxel efficacy in malignant glioma following local administration. ACS Nano. 2014;8(10):10655–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pourgholi F, hajivalili M, Farhad J-N, Kafil HS, Yousefi M. Nanoparticles: Novel vehicles in treatment of Glioblastoma. Biomedicine & Pharmacotherapy. 2016;77:98–107. [DOI] [PubMed] [Google Scholar]
- 83.Sun J, Guo M, Pang H, Qi J, Zhang J, Ge Y. Treatment of malignant glioma using hyperthermia. Neural Regen Res. 2013;8(29):2775–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dewhirst M, Stauffer PR, Das S, Craciunescu OI, Vujaskovic Z. Chapter 21 - Hyperthermia. In: Gunderson LL, Tepper JE, editors. Clinical Radiation Oncology (Fourth Edition). Philadelphia: Elsevier; 2016. p. 381–98.e6. [Google Scholar]
- 85.Coluccia D, Fandino J, Schwyzer L, O’Gorman R, Remonda L, Anon J, et al. First noninvasive thermal ablation of a brain tumor with MR-guided focused ultrasound. J Ther Ultrasound. 2014;2:17-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gong W, Wang Z, Liu N, Lin W, Wang X, Xu D, et al. Improving Efficiency of Adriamycin Crossing Blood Brain Barrier by Combination of Thermosensitive Liposomes and Hyperthermia. Biological and Pharmaceutical Bulletin. 2011;34(7):1058–64. [DOI] [PubMed] [Google Scholar]
- 87.Baronzio Gian Franco M.D. Hager E. Dieter M.D. PD, D.Sc. Hyperthermia in Cancer Treatment: A Primer 2006. [Google Scholar]
- 88.Lagman C, Chung LK, Pelargos PE, Ung N, Bui TT, Lee SJ, et al. Laser neurosurgery: A systematic analysis of magnetic resonance-guided laser interstitial thermal therapies. Journal of Clinical Neuroscience. 2017;36:20–6. [DOI] [PubMed] [Google Scholar]
- 89.Hildebrandt B, Wust P, Ahlers O, Dieing A, Sreenivasa G, Kerner T, et al. The cellular and molecular basis of hyperthermia. Critical Reviews in Oncology/Hematology. 2002;43(1):33–56. [DOI] [PubMed] [Google Scholar]
- 90.Khan VR, Brown IR. The effect of hyperthermia on the induction of cell death in brain, testis, and thymus of the adult and developing rat. Cell Stress Chaperones. 2002;7(1):73–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nature Reviews Clinical Oncology. 2018;15(7):422–42. [DOI] [PubMed] [Google Scholar]
- 92.Thomas AA, Ernstoff MS, Fadul CE. Immunotherapy for the treatment of glioblastoma. Cancer J. 2012;18(1):59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yang L, Guo G, Niu X-y, Liu J. Dendritic Cell-Based Immunotherapy Treatment for Glioblastoma Multiforme. Biomed Res Int. 2015;2015:717530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Polyzoidis S, Ashkan K. Dendritic cell immunotherapy for glioblastoma. Expert Review of Anticancer Therapy. 2014;14(7):761–3. [DOI] [PubMed] [Google Scholar]
- 95.Wang EC, Wang AZ. Nanoparticles and their applications in cell and molecular biology. Integr Biol (Camb). 2014;6(1):9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Alphandéry E Nano-Therapies for Glioblastoma Treatment. Cancers (Basel). 2020;12(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Guterres SS, Alves MP, Pohlmann AR. Polymeric nanoparticles, nanospheres and nanocapsules, for cutaneous applications. Drug Target Insights. 2007;2:147–57. [PMC free article] [PubMed] [Google Scholar]
- 98.Huile Gao XJ. Brain Delivery Using Nanotechnology. In: Li Di EHK, editor. Blood‐Brain Barrier in Drug Discovery 2015. [Google Scholar]
- 99.Yildirimer L, Thanh NTK, Loizidou M, Seifalian AM. Toxicology and clinical potential of nanoparticles. Nano Today. 2011;6(6):585–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jingyan L, Cristina S. PLA/PLGA nanoparticles for delivery of drugs across the blood-brain barrier. Nanotechnology Reviews. 2013;2(3):241–57. [Google Scholar]
- 101.Jallouli Y, Paillard A, Chang J, Sevin E, Betbeder D. Influence of surface charge and inner composition of porous nanoparticles to cross blood-brain barrier in vitro. Int J Pharm. 2007;344(1–2):103–9. [DOI] [PubMed] [Google Scholar]
- 102.Li S-D, Huang L. Nanoparticles evading the reticuloendothelial system: role of the supported bilayer. Biochim Biophys Acta. 2009;1788(10):2259–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhao J, Zhang B, Shen S, Chen J, Zhang Q, Jiang X, et al. CREKA peptide-conjugated dendrimer nanoparticles for glioblastoma multiforme delivery. Journal of Colloid and Interface Science. 2015;450:396–403. [DOI] [PubMed] [Google Scholar]
- 104.Jevprasesphant R, Penny J, Jalal R, Attwood D, McKeown NB, D’Emanuele A. The influence of surface modification on the cytotoxicity of PAMAM dendrimers. International Journal of Pharmaceutics. 2003;252(1–2):263–6. [DOI] [PubMed] [Google Scholar]
- 105.Gref R, Domb A, Quellec P, Blunk T, Müller RH, Verbavatz JM, et al. The controlled intravenous delivery of drugs using PEG-coated sterically stabilized nanospheres. Advanced drug delivery reviews. 1995;16(2):215–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tan JS, Butterfield DE, Voycheck CL, Caldwell KD, Li JT. Surface modification of nanoparticles by PEO/PPO block copolymers to minimize interactions with blood components and prolong blood circulation in rats. Biomaterials. 1993;14(11):823–33. [DOI] [PubMed] [Google Scholar]
- 107.Klibanov AL, Maruyama K, Torchilin VP, Huang L. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett. 1990;268(1):235–7. [DOI] [PubMed] [Google Scholar]
- 108.Laginha KM, Verwoert S, Charrois GJ, Allen TM. Determination of doxorubicin levels in whole tumor and tumor nuclei in murine breast cancer tumors. Clin Cancer Res. 2005;11(19 Pt 1):6944–9. [DOI] [PubMed] [Google Scholar]
- 109.Karim R, Palazzo C, Evrard B, Piel G. Nanocarriers for the treatment of glioblastoma multiforme: Current state-of-the-art. Journal of Controlled Release. 2016;227:23–37. [DOI] [PubMed] [Google Scholar]
- 110.Koo YEL, Reddy GR, Bhojani M, Schneider R, Philbert MA, Rehemtulla A, et al. Brain cancer diagnosis and therapy with nanoplatforms. Advanced drug delivery reviews. 2006;58(14):1556–77. [DOI] [PubMed] [Google Scholar]
- 111.Tzeng SY, Green JJ. Therapeutic nanomedicine for brain cancer. Therapeutic Delivery. 2013;4(6):687–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shah N, Chaudhari K, Dantuluri P, Murthy RSR, Das S. Paclitaxel-loaded PLGA nanoparticles surface modified with transferrin and Pluronic®P85, an in vitro cell line and in vivo biodistribution studies on rat model. Journal of Drug Targeting. 2009;17(7):533–42. [DOI] [PubMed] [Google Scholar]
- 113.Pereverzeva E, Treschalin I, Bodyagin D, Maksimenko O, Langer K, Dreis S, et al. Influence of the formulation on the tolerance profile of nanoparticle-bound doxorubicin in healthy rats: focus on cardio-and testicular toxicity. Int J Pharm. 2007;337(1–2):346–56. [DOI] [PubMed] [Google Scholar]
- 114.Gao H, Xiong Y, Zhang S, Yang Z, Cao S, Jiang X. RGD and Interleukin-13 Peptide Functionalized Nanoparticles for Enhanced Glioblastoma Cells and Neovasculature Dual Targeting Delivery and Elevated Tumor Penetration. Molecular Pharmaceutics. 2014;11(3):1042–52. [DOI] [PubMed] [Google Scholar]
- 115.Paliwal R, Babu RJ, Palakurthi S. Nanomedicine scale-up technologies: feasibilities and challenges. AAPS PharmSciTech. 2014;15(6):1527–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.He C, Hu Y, Yin L, Tang C, Yin C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials. 2010;31(13):3657–66. [DOI] [PubMed] [Google Scholar]
- 117.Gou N, Onnis-Hayden A, Gu AZ. Mechanistic Toxicity Assessment of Nanomaterials by Whole-Cell-Array Stress Genes Expression Analysis. Environmental Science & Technology. 2010;44(15):5964–70. [DOI] [PubMed] [Google Scholar]
- 118.Bhabra G, Sood A, Fisher B, Cartwright L, Saunders M, Evans WH, et al. Nanoparticles can cause DNA damage across a cellular barrier. Nature Nanotechnology. 2009;4(12):876–83. [DOI] [PubMed] [Google Scholar]
- 119.Ramalho MJ, Sevin E, Gosselet F, Lima J, Coelho MAN, Loureiro JA, et al. Receptor-mediated PLGA nanoparticles for glioblastoma multiforme treatment. International Journal of Pharmaceutics. 2018;545(1):84–92. [DOI] [PubMed] [Google Scholar]
- 120.Guo W, Li A, Jia Z, Yuan Y, Dai H, Li H. Transferrin modified PEG-PLA-resveratrol conjugates: In vitro and in vivo studies for glioma. European Journal of Pharmacology. 2013;718(1):41–7. [DOI] [PubMed] [Google Scholar]
- 121.Zhao Z, Hu Y, Hoerle R, Devine M, Raleigh M, Pentel P, et al. A nanoparticle-based nicotine vaccine and the influence of particle size on its immunogenicity and efficacy. Nanomedicine. 2017;13(2):443–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Xin H, Sha X, Jiang X, Zhang W, Chen L, Fang X. Anti-glioblastoma efficacy and safety of paclitaxel-loading Angiopep-conjugated dual targeting PEG-PCL nanoparticles. Biomaterials. 2012;33(32):8167–76. [DOI] [PubMed] [Google Scholar]
- 123.Gao H, Yang Z, Zhang S, Cao S, Shen S, Pang Z, et al. Ligand modified nanoparticles increases cell uptake, alters endocytosis and elevates glioma distribution and internalization. Scientific Reports. 2013;3(1):2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sunoqrot S, Bugno J, Lantvit D, Burdette JE, Hong S. Prolonged blood circulation and enhanced tumor accumulation of folate-targeted dendrimer-polymer hybrid nanoparticles. Journal of Controlled Release. 2014;191:115–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Santos SD, Xavier M, Leite DM, Moreira DA, Custodio B, Torrado M, et al. PAMAM dendrimers: blood-brain barrier transport and neuronal uptake after focal brain ischemia. Journal of controlled release : official journal of the Controlled Release Society. 2018;291:65–79. [DOI] [PubMed] [Google Scholar]
- 126.Gaillard PJ, Appeldoorn CC, Rip J, Dorland R, van der Pol SM, Kooij G, et al. Enhanced brain delivery of liposomal methylprednisolone improved therapeutic efficacy in a model of neuroinflammation. Journal of controlled release : official journal of the Controlled Release Society. 2012;164(3):364–9. [DOI] [PubMed] [Google Scholar]
- 127.Li Y, He H, Jia X, Lu W-L, Lou J, Wei Y. A dual-targeting nanocarrier based on poly(amidoamine) dendrimers conjugated with transferrin and tamoxifen for treating brain gliomas. Biomaterials. 2012;33(15):3899–908. [DOI] [PubMed] [Google Scholar]
- 128.Yuan F, Leunig M, Huang SK, Berk DA, Papahadjopoulos D, Jain RK. Mirovascular Permeability and Interstitial Penetration of Sterically Stabilized (Stealth) Liposomes in a Human Tumor Xenograft. Cancer research. 1994;54(13):3352. [PubMed] [Google Scholar]
- 129.Wang X, Zhang Q, Lv L, Fu J, Jiang Y, Xin H, et al. Glioma and microenvironment dual targeted nanocarrier for improved antiglioblastoma efficacy. Drug Delivery. 2017;24(1):1401–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Feng X, Yao J, Gao X, Jing Y, Kang T, Jiang D, et al. Multi-targeting Peptide-Functionalized Nanoparticles Recognized Vasculogenic Mimicry, Tumor Neovasculature, and Glioma Cells for Enhanced Anti-glioma Therapy. ACS Applied Materials & Interfaces. 2015;7(50):27885–99. [DOI] [PubMed] [Google Scholar]
- 131.Gaillard PJ, Appeldoorn CCM, Dorland R, van Kregten J, Manca F, Vugts DJ, et al. Pharmacokinetics, Brain Delivery, and Efficacy in Brain Tumor-Bearing Mice of Glutathione Pegylated Liposomal Doxorubicin (2B3–101). PLOS ONE. 2014;9(1):e82331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Demeule M, Régina A, Ché C, Poirier J, Nguyen T, Gabathuler R, et al. Identification and Design of Peptides as a New Drug Delivery System for the Brain. Journal of Pharmacology and Experimental Therapeutics. 2008;324(3):1064. [DOI] [PubMed] [Google Scholar]
- 133.Song Y, Huang Z, Xu J, Ren D, Wang Y, Zheng X, et al. Multimodal SPION-CREKA peptide based agents for molecular imaging of microthrombus in a rat myocardial ischemia-reperfusion model. Biomaterials. 2014;35(9):2961–70. [DOI] [PubMed] [Google Scholar]
- 134.Kou L, Sun J, Zhai Y, He Z. The endocytosis and intracellular fate of nanomedicines: Implication for rational design. Asian Journal of Pharmaceutical Sciences. 2013;8(1):1–10. [Google Scholar]
- 135.Borgognoni C, Kim J, Zucolotto V, Harald F, Riehemann K. Human macrophage responses to metal-oxide nanoparticles: a review. Artificial Cells, Nanomedicine, and Biotechnology. 2018;46:1–10. [DOI] [PubMed] [Google Scholar]
- 136.Ambruosi A, Gelperina S, Khalansky A, Tanski S, Theisen A, Kreuter J. Influence of surfactants, polymer and doxorubicin loading on the anti-tumour effect of poly(butyl cyanoacrylate) nanoparticles in a rat glioma model. Journal of Microencapsulation. 2006;23(5):582–92. [DOI] [PubMed] [Google Scholar]
- 137.Steiniger SC, Kreuter J, Khalansky AS, Skidan IN, Bobruskin AI, Smirnova ZS, et al. Chemotherapy of glioblastoma in rats using doxorubicin-loaded nanoparticles. Int J Cancer. 2004;109(5):759–67. [DOI] [PubMed] [Google Scholar]
- 138.Gulyaev AE, Gelperina SE, Skidan IN, Antropov AS, Kivman GY, Kreuter J. Significant transport of doxorubicin into the brain with polysorbate 80-coated nanoparticles. Pharm Res. 1999;16(10):1564–9. [DOI] [PubMed] [Google Scholar]
- 139.Borchard G, Audus KL, Shi F, Kreuter J. Uptake of surfactant-coated poly(methyl methacrylate)-nanoparticles by bovine brain microvessel endothelial cell monolayers. International Journal of Pharmaceutics. 1994;110(1):29–35. [Google Scholar]
- 140.Kreuter J, Alyautdin RN, Kharkevich DA, Ivanov AA. Passage of peptides through the blood-brain barrier with colloidal polymer particles (nanoparticles). Brain Research. 1995;674(1):171–4. [DOI] [PubMed] [Google Scholar]
- 141.Wohlfart S, Khalansky AS, Gelperina S, Maksimenko O, Bernreuther C, Glatzel M, et al. Efficient chemotherapy of rat glioblastoma using doxorubicin-loaded PLGA nanoparticles with different stabilizers. PloS one. 2011;6(5):e19121–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Alakhov VY, Moskaleva EY, Batrakova EV, Kabanov AV. Hypersensitization of Multidrug Resistant Human Ovarian Carcinoma Cells by Pluronic P85 Block Copolymer. Bioconjugate Chemistry. 1996;7(2):209–16. [DOI] [PubMed] [Google Scholar]
- 143.Zhirnov AE, Demina TV, Krylova OO, Grozdova ID, Melik-Nubarov NS. Lipid composition determines interaction of liposome membranes with Pluronic L61. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2005;1720(1):73–83. [DOI] [PubMed] [Google Scholar]
- 144.Gelperina S, Maksimenko O, Khalansky A, Vanchugova L, Shipulo E, Abbasova K, et al. Drug delivery to the brain using surfactant-coated poly(lactide-co-glycolide) nanoparticles: influence of the formulation parameters. Eur J Pharm Biopharm. 2010;74(2):157–63. [DOI] [PubMed] [Google Scholar]
- 145.Kannan R, Chakrabarti R, Tang D, Kim KJ, Kaplowitz N. GSH transport in human cerebrovascular endothelial cells and human astrocytes: evidence for luminal localization of Na+-dependent GSH transport in HCEC. Brain Res. 2000;852(2):374–82. [DOI] [PubMed] [Google Scholar]
- 146.Kannan R, Kuhlenkamp JF, Jeandidier E, Trinh H, Ookhtens M, Kaplowitz N. Evidence for carrier-mediated transport of glutathione across the blood-brain barrier in the rat. J Clin Invest. 1990;85(6):2009–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lindqvist A, Rip J, Gaillard PJ, Bjorkman S, Hammarlund-Udenaes M. Enhanced brain delivery of the opioid peptide DAMGO in glutathione pegylated liposomes: a microdialysis study. Mol Pharm. 2013;10(5):1533–41. [DOI] [PubMed] [Google Scholar]
- 148.Xin H, Chen L, Gu J, Ren X, wei Z, Luo J, et al. Enhanced anti-glioblastoma efficacy by PTX-loaded PEGylated poly(ɛ-caprolactone) nanoparticles: In vitro and in vivo evaluation. International Journal of Pharmaceutics. 2010;402(1):238–47. [DOI] [PubMed] [Google Scholar]
- 149.Zhou J, Patel TR, Sirianni RW, Strohbehn G, Zheng M-Q, Duong N, et al. Highly penetrative, drug-loaded nanocarriers improve treatment of glioblastoma. Proc Natl Acad Sci U S A. 2013;110(29):11751–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hobbs SK, Monsky WL, Yuan F, Roberts WG, Griffith L, Torchilin VP, et al. Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Natl Acad Sci U S A. 1998;95(8):4607–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Calzolari A, Larocca LM, Deaglio S, Finisguerra V, Boe A, Raggi C, et al. Transferrin Receptor 2 Is Frequently and Highly Expressed in Glioblastomas. Translational Oncology. 2010;3(2):123–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Mintz A, Gibo DM, Slagle-Webb B, Christensen ND, Debinski W. IL-13Ralpha2 is a glioma-restricted receptor for interleukin-13. Neoplasia. 2002;4(5):388–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kumarswamy R, Volkmann I, Thum T. Regulation and function of miRNA-21 in health and disease. RNA Biol. 2011;8(5):706–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Liu C, Li B, Cheng Y, Lin J, Hao J, Zhang S, et al. MiR-21 plays an important role in radiation induced carcinogenesis in BALB/c mice by directly targeting the tumor suppressor gene Big-h3. Int J Biol Sci. 2011;7(3):347–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Seo Y-E, Suh H-W, Bahal R, Josowitz A, Zhang J, Song E, et al. Nanoparticle-mediated intratumoral inhibition of miR-21 for improved survival in glioblastoma. Biomaterials. 2019;201:87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Abbasi E, Aval SF, Akbarzadeh A, Milani M, Nasrabadi HT, Joo SW, et al. Dendrimers: synthesis, applications, and properties. Nanoscale Res Lett. 2014;9(1):247-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Duncan R, Izzo L. Dendrimer biocompatibility and toxicity. Advanced drug delivery reviews. 2005;57(15):2215–37. [DOI] [PubMed] [Google Scholar]
- 158.Abedi-Gaballu F, Dehghan G, Ghaffari M, Yekta R, Abbaspour-Ravasjani S, Baradaran B, et al. PAMAM dendrimers as efficient drug and gene delivery nanosystems for cancer therapy. Appl Mater Today. 2018;12:177–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Luong D, Kesharwani P, Deshmukh R, Mohd Amin MCI, Gupta U, Greish K, et al. PEGylated PAMAM dendrimers: Enhancing efficacy and mitigating toxicity for effective anticancer drug and gene delivery. Acta Biomaterialia. 2016;43:14–29. [DOI] [PubMed] [Google Scholar]
- 160.Ferretti C, Blengio M, Ghi P, Racca S, Genazzani E, Portaleone P. Tamoxifen counteracts estradiol induced effects on striatal and hypophyseal dopamine receptors. Life Sciences. 1988;42(24):2457–65. [DOI] [PubMed] [Google Scholar]
- 161.Kayyali R, Marriott C, Wiseman H. Tamoxifen decreases drug efflux from liposomes: Relevance to its ability to reverse multidrug resistance in cancer cells? FEBS Letters. 1994;344(2):221–4. [DOI] [PubMed] [Google Scholar]
- 162.Lu C, Xing MMQ, Zhong W. Shell cross-linked and hepatocyte-targeting nanoparticles containing doxorubicin via acid-cleavable linkage. Nanomedicine: Nanotechnology, Biology and Medicine. 2011;7(1):80–7. [DOI] [PubMed] [Google Scholar]
- 163.Choi J, Kim G, Cho SB, Im H-J. Radiosensitizing high-Z metal nanoparticles for enhanced radiotherapy of glioblastoma multiforme. Journal of Nanobiotechnology. 2020;18(1):122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Rosa S, Connolly C, Schettino G, Butterworth KT, Prise KM. Biological mechanisms of gold nanoparticle radiosensitization. Cancer Nanotechnol. 2017;8(1):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kievit FM, Wang K, Ozawa T, Tarudji AW, Silber JR, Holland EC, et al. Nanoparticle-mediated knockdown of DNA repair sensitizes cells to radiotherapy and extends survival in a genetic mouse model of glioblastoma. Nanomedicine. 2017;13(7):2131–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Timbie KF, Afzal U, Date A, Zhang C, Song J, Wilson Miller G, et al. MR image-guided delivery of cisplatin-loaded brain-penetrating nanoparticles to invasive glioma with focused ultrasound. Journal of Controlled Release. 2017;263:120–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Zhao Y-Z, Lin Q, Wong HL, Shen X-T, Yang W, Xu H-L, et al. Glioma-targeted therapy using Cilengitide nanoparticles combined with UTMD enhanced delivery. Journal of Controlled Release. 2016;224:112–25. [DOI] [PubMed] [Google Scholar]
- 168.Kovacs Z, Werner B, Rassi A, Sass JO, Martin-Fiori E, Bernasconi M. Prolonged survival upon ultrasound-enhanced doxorubicin delivery in two syngenic glioblastoma mouse models. Journal of Controlled Release. 2014;187:74–82. [DOI] [PubMed] [Google Scholar]
- 169.Bekeredjian R, Kroll RD, Fein E, Tinkov S, Coester C, Winter G, et al. Ultrasound Targeted Microbubble Destruction Increases Capillary Permeability in Hepatomas. Ultrasound in Medicine & Biology. 2007;33(10):1592–8. [DOI] [PubMed] [Google Scholar]
- 170.Luo Z, Jin K, Pang Q, Shen S, Yan Z, Jiang T, et al. On-Demand Drug Release from Dual-Targeting Small Nanoparticles Triggered by High-Intensity Focused Ultrasound Enhanced Glioblastoma-Targeting Therapy. ACS Applied Materials & Interfaces. 2017;9(37):31612–25. [DOI] [PubMed] [Google Scholar]
- 171.Jamali Z, Khoobi M, Hejazi SM, Eivazi N, Abdolahpour S, Imanparast F, et al. Evaluation of targeted curcumin (CUR) loaded PLGA nanoparticles for in vitro photodynamic therapy on human glioblastoma cell line. Photodiagnosis and Photodynamic Therapy. 2018;23:190–201. [DOI] [PubMed] [Google Scholar]
- 172.Fourniols T, Randolph LD, Staub A, Vanvarenberg K, Leprince JG, Préat V, et al. Temozolomide-loaded photopolymerizable PEG-DMA-based hydrogel for the treatment of glioblastoma. Journal of Controlled Release. 2015;210:95–104. [DOI] [PubMed] [Google Scholar]
- 173.Hu Q, Gao X, Gu G, Kang T, Tu Y, Liu Z, et al. Glioma therapy using tumor homing and penetrating peptide-functionalized PEG–PLA nanoparticles loaded with paclitaxel. Biomaterials. 2013;34(22):5640–50. [DOI] [PubMed] [Google Scholar]
- 174.Kang T, Zhu Q, Jiang D, Feng X, Feng J, Jiang T, et al. Synergistic targeting tenascin C and neuropilin-1 for specific penetration of nanoparticles for anti-glioblastoma treatment. Biomaterials. 2016;101:60–75. [DOI] [PubMed] [Google Scholar]
- 175.Zhan C, Wei X, Qian J, Feng L, Zhu J, Lu W. Co-delivery of TRAIL gene enhances the anti-glioblastoma effect of paclitaxel in vitro and in vivo. Journal of Controlled Release. 2012;160(3):630–6. [DOI] [PubMed] [Google Scholar]
- 176.Elsaesser A, Howard CV. Toxicology of nanoparticles. Advanced drug delivery reviews. 2012;64(2):129–37. [DOI] [PubMed] [Google Scholar]
- 177.Godbey WT, Wu KK, Mikos AG. Tracking the intracellular path of poly(ethylenimine)/DNA complexes for gene delivery. Proc Natl Acad Sci U S A. 1999;96(9):5177–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Stapleton PA. Toxicological considerations of nano-sized plastics. AIMS Environ Sci. 2019;6(5):367–78. [DOI] [PMC free article] [PubMed] [Google Scholar]