

HIV-1 infection is a health problem of enormous importance that still causes significant mortality. Antiretroviral treatment (ART) has demonstrated efficacy in the control of HIV-1 replication, decreasing the morbidity and mortality of the infection, but it cannot eradicate the virus.
KEYWORDS: CTL response, galectin-9, HIV-1 latency, TIM-3
ABSTRACT
Reactivation of latent HIV-1 is a necessary step for the purging of the viral reservoir, although it does not seem to be sufficient. The stimulation of HIV-1-specific cytotoxic T lymphocytes (CTL) may also be essential for this purpose. In this study, we aimed to show the effect of galectin-9 (Gal-9), known to revert HIV-1 latency, in combination with the blockade of T cell immunoglobulin and mucin domain-containing molecule 3 (TIM-3), a natural receptor for Gal-9 and an exhaustion marker. We confirmed the ability of Gal-9 to reactivate latent HIV-1 in Jurkat-LAT-GFP cells, as well as in an interleukin 7 (IL-7)-based cellular model. This reactivation was mediated not via the TIM-3 receptor but rather by the recognition of the Gal-9 of a specific oligosaccharide pattern of resting memory CD4+ T cells’ surfaces. The potency of Gal-9 in inducing transcription of latent HIV-1 was equal to or greater than that of other latency-reversing agents (LRA). Furthermore, the combination of Gal-9 with other LRA did not show synergistic effects in the reactivation of the latent virus. To evaluate the impact of TIM-3 inhibition on the CTL response, different coculture experiments with CD4+ T, CD8+ T, and NK cells were performed. Our data showed that blocking TIM-3 was associated with control of viral replication in both in vitro and ex vivo models in cells from people living with HIV-1 (PLWH) on antiretroviral therapy. A joint strategy combining the use of Gal-9 to reactivate latent HIV-1 and the inhibition of TIM-3 to enhance the HIV-1 CTL-specific response was associated with control of the replication of the virus that was being reactivated, thus potentially contributing to the elimination of the viral reservoir. Our results suggest this strategy as a promising approach to be tested in future studies. Reactivation of latent-HIV-1 by Gal-9 and reinvigoration of CD8+ T cells by TIM-3 blockade could be used separately or in combination.
IMPORTANCE HIV-1 infection is a health problem of enormous importance that still causes significant mortality. Antiretroviral treatment (ART) has demonstrated efficacy in the control of HIV-1 replication, decreasing the morbidity and mortality of the infection, but it cannot eradicate the virus. In our work, we tested a protein, galectin-9 (Gal-9), an HIV-1 latency-reversing agent, using an in vitro cellular model of latency and cells from people living with HIV-1 (PLWH) on antiretroviral therapy. Our results confirmed the potential role of Gal-9 as a molecule with a potent HIV-1 reactivation capacity. More importantly, using a monoclonal antibody against the receptor TIM-3 (T cell immunoglobulin and the mucin domain-containing molecule 3), we were able to enhance the HIV-1 cytotoxic T lymphocyte (CTL) specific response to eliminate the CD4+ T cells in which the virus had been reactivated. When Gal-9 and TIM-3 blockade were used together, control of the replication of HIV-1 was observed, suggesting a decrease in the cellular reservoir.
INTRODUCTION
Antiretroviral treatment (ART) has demonstrated great efficacy in HIV-1 suppression and clinical improvement of people living with HIV-1 (PLWH). However, ART does not achieve viral eradication, and the cure for HIV-1 infection remains an elusive goal. Although several strategies have been proposed to achieve a functional cure, none has produced encouraging results. The shock-and-kill strategy has possibly been the most frequently tested approach (1). It consists of the use of latency-reversing agents (LRA) that reactivate HIV-1 from latently infected resting memory CD4+ T cells (2–7). This strategy is based on the assumption that latently infected cells will be eliminated after the reactivation of latent proviruses, either by the cytopathic effect of HIV-1 gene expression or by the lytic effect of cytotoxic T lymphocytes (CTL) HIV-1 response. However, it has been documented that this is not the case and that LRA are not sufficient to eliminate the cellular reservoirs; this depends to a large extent on the presence of specific functional CTL. Therefore, this implies the need to achieve a means of restoring this activity of CTL, in addition to the use of reactivating agents (8).
Galectin-9 (Gal-9) has been identified as a molecule with the capacity to reverse HIV-1 latency (9). Gal-9 is a very versatile immunomodulatory molecule that functions in cell adhesion, cell surface recognition, and chemoattraction and as a modulator of important signaling between growth and apoptosis (10). The biological effects of Gal-9 are exerted through its binding to various receptors, such as T cell immunoglobulin and mucin domain-containing molecule 3 (TIM-3), cell surface protein disulfide isomerase (PDI), IgE, and CD44, among others. Due to its interaction with multiple receptors, Gal-9 modulates different immunological functions depending on different circumstances (11–16). It was recently shown that Gal-9 can increase the replication and reactivation of latent HIV-1 in primary CD4+ T cells. This effect would be carried out not by the binding of Gal-9 to its known receptors (TIM-3, PDI, and CD44) but by interaction with a set of oligosaccharides of the membrane of the T cells, which would lead to the transduction of intracellular signals for the transcription of HIV-1. It is noteworthy that activated CD4+ T cells and those that are latently infected with HIV-1 have an altered cell surface glycosylation pattern with respect to noninfected resting cells recognized by Gal-9 (9).
Of special interest is the interaction between Gal-9 and the receptor TIM-3, a type of transmembrane protein expressed in multiple cells of the immune system, including activated/depleted T cells and monocytes (17, 18). TIM-3 is the most recently described T cell exhaustion marker in HIV-1 infection. Among the known ligands of TIM-3, Gal-9 has been identified. Recently, the role of the Gal-9/TIM-3 pathway in the regulation of the acute CD8+ T cell response and memory in response to viral infections was investigated (19). Also, the Gal-9/TIM-3 interaction in CD8+ T cells seems to be associated with an increase in the exhaustion of T cells during HIV-1 chronic infection (20). Immune exhaustion markers, such as TIM-3 receptor, can also be detected in other immune cells, such as natural killer (NK) cells.
Thus, Gal-9 seems to play a role in the reactivation of HIV-1 from latently infected CD4+ T cells, though the reactivation of latent HIV-1 does not seem sufficient for the elimination of the viral reservoir, and stimulation of the response of the HIV-1-specific CTLs may be essential. The blockade of the receptor TIM-3 could improve the specific cytotoxic activity of CD8+ T lymphocytes. Thus, the purpose of this study was to evaluate the efficacy of the use of Gal-9 in reversing HIV-1 latency and the effect of TIM-3 inhibition on the control of viral replication, with special emphasis on the interaction of the two strategies should they be used together for the cure of HIV-1 infection.
RESULTS
Galectin-9 reverses HIV-1 latency in Jurkat-LAT-GFP cells through a TIM-3-independent pathway.
First, we used a Jurkat-LAT-GFP model (n = 4) to assess HIV-1 reactivation by Gal-9. This model cell contains a latent but transcriptionally competent HIV-1 provirus with a gene that encodes green fluorescent protein (GFP) as a reactivation marker. Viral reactivation after stimulation is associated with an increase in GFP, and its expression can be measured by flow cytometry. Significant differences were observed when we compared the Gal-9 reactivation alone or with an antibody against TIM-3 (anti-TIM-3) (5 μg/ml) with the negative control (uninfected primary Jurkat cell line) (P = 0.002 and P = 0.001, respectively). Uninfected Jurkat cells that expressed GFP constitutively (GFP+ control) showed a significant increase in GFP values compared with reactivation mediated by Gal-9 (P = 0.014). We observed that Gal-9 reactivates latent HIV-1 with a potency similar to that of phorbol-12-myristate-13-acetate (PMA), used as a positive control. The percentage of GFP expression with Gal-9 at a concentration of 2 nM was 28%, compared to 38.3% with PMA after 18 h of incubation (P = 0.065). When anti-TIM-3 (5 μg/ml) was added, the GFP expression was 31% (P = 0.089) (Fig. 1). The difference in the reactivation percentage with Gal-9 with and without anti-TIM-3 was not significant (P = 0.3). Our results demonstrate that Gal-9 induces HIV-1 transcription by a pathway independent of TIM-3, as TIM-3 inhibition did not affect the percentage of viral reactivation.
FIG 1.

Galectin-9 (Gal-9) reactivates latent HIV-1 in Jurkat-LAT-GFP cells. Bars show HIV-1 reactivation, calculated as percent GFP expression. After Gal-9 (2 nM) treatment (18 h) in a model of Jurkat-LAT-GFP cells (n = 4), we did not observe significant differences compared with the PMA (positive control). Similar results were obtained with the combination of Gal-9 (2 nM) with an antibody against TIM-3 (anti-TIM-3) (5 μg/ml). Significant differences were observed when we compared the Gal-9 reactivation alone or with anti-TIM-3 to the negative control (uninfected primary Jurkat cell line). Uninfected Jurkat cells that expressed GFP constitutively (Control GFP+) showed significantly higher GFP values than reactivation mediated by Gal-9. An increase (not significant) in GFP expression was observed after Gal-9 treatment compared to expression without Gal-9 addition (Jurkat cells). The same comparison achieved a significant difference when anti-TIM-3 was added (Gal-9 2 nM+ αTIM3 5 μg/ml). Statistical significance was calculated using the Mann-Whitney U test. P values of <0.05 were considered statistically significant. *, P < 0.05; NS, not significant.
A wide range of galectin-9 concentrations reverses HIV-1 latency in a cellular model by interaction with a specific pattern of oligosaccharides on the T cell surface membrane.
To further test HIV-1 reactivation by Gal-9, we used an in vitro latency model based on interleukin 7 (IL-7) (n = 8), the cytokine responsible for the homeostatic proliferation of memory T cells. This model of HIV-1 latency in CD4+ T cells was recently described (21); it allows viral integration with low levels of viral expression. Isolated resting memory CD4+ T cells were used to develop the in vitro latency model. After 5 days of incubation with IL-7 (1 nM), an infection with the X4-tropic NL4.3 HIV-1 strain (10 pg) was performed, and infected cells were cultured for 5 additional days. After this time Gal-9 was used for HIV-1 reactivation over a 24-h period (Fig. 2). To confirm the correct HIV-1 integration in the cellular model, a specific quantitative PCR was done (see Materials and Methods) (Fig. 3). Different concentrations of Gal-9 (2 nM, 5 nM, 200 nM, and 500 nM) were used to check differences in the reactivation potency. As determined by an enzyme-linked immunosorbent assay (ELISA) technique, the concentration of antigen p24 (p24Ag) was 29 ng/ml after reactivation of HIV-1 with Gal-9 at 2 nM, compared to 32 ng/ml with PMA (P = 0.64). No significant differences were observed between the reactivating capacity of each Gal-9 concentration and that of PMA: Gal-9 (5 nM), 24 ng/ml (P = 0.21); Gal-9 (200 nM), 27.3 ng/ml (P = 0.36); and Gal-9 (500 nM), 23 ng/ml (P = 0.41). No significant differences were detected when Gal-9 was added together with anti-TIM-3 (5 μg/ml) compared to PMA, as previously shown in the Jurkat-LAT-GFP model: Gal-9 (2 nM) plus anti-TIM-3, 3.24 ng/ml (P = 0.39); Gal-9 (5 nM) plus anti-TIM-3, 3.23 ng/ml (P = 0.38); and Gal-9 (200 nM) plus anti-TIM-3, 36.6 ng/ml (P = 0.41) (Fig. 4). Again, Gal-9 was shown to be a potent HIV-1 latency-reversing agent that used a TIM-3-independent pathway, with no differences among the concentrations used.
FIG 2.

Scheme of the HIV-1 primary CD4+ T cell latency model. Resting memory CD4+ T cells (CD4+ HL-DR− CD25−) were treated with IL-7 for 5 days and then infected with an X4-tropic NL4.3 strain and cultured for 5 additional days. Galectin-9 (Gal-9) was used for HIV-1 reactivation measured by quantification of the HIV-1 p24 antigen in culture supernatants. After 24 h of Gal-9 treatment, supernatants were collected for p24 antigen determination.
FIG 3.
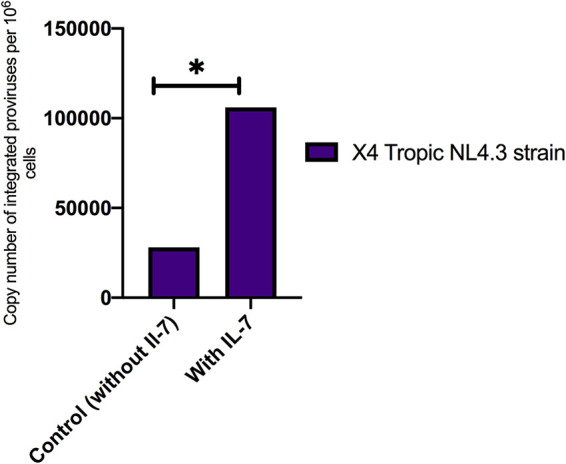
qPCR analysis of HIV-1 proviral integration. Data are copy numbers of integrated provirus per 106 cells. IL-7 allowed viral integration in the CD4+ T model using an X4-tropic NL4.3 strain compared with the control (without Il-7). *, P < 0.05.
FIG 4.

Galectin-9 (Gal-9) reverse HIV-1 latency in a cellular model based on IL-7. Bars show HIV-1 reactivation, expressed as p24 antigen production (n = 8). No significant differences were observed between p24 antigen concentrations with the positive control (PMA) and the different Gal-9 concentrations with or without an antibody against TIM-3 (anti-TIM-3). The comparison with the cellular model alone (Control) showed in all cases a significant difference in the p24 antigen values. Statistical significance was calculated using the Mann-Whitney U test. P values of <0.05 were considered statistically significant. *, P < 0.05; NS, not significant.
To further evaluate the mechanism involved in the reversal of HIV-1 latency by Gal-9, a deglycosylation assay was performed. Other authors have proposed that Gal-9 could act by signaling through specific glycans on the cell surface. We used an antibiotic (tunicamycin) which chemically degrades the pattern of oligosaccharides by inhibiting glycan synthesis. This pattern is recognized by Gal-9 in resting memory CD4+ T cells, leading to subsequent viral reactivation. It was therefore expected that HIV-1 reactivation would be lower after tunicamycin treatment. Again using the latency model based on IL-7, we observed a decreased reactivation of HIV-1 by Gal-9 in combination with tunicamycin. The reactivation of HIV-1 was performed in the IL-7 cellular model in incubation with Gal-9 alone or in combination with tunicamycin and in the presence or absence of anti-TIM-3. The p24Ag level with Gal-9 (2 nM) alone was 29.04 ng/ml, and that with Gal-9 (2 nM) plus tunicamycin was 15 ng/ml (P = 0.045). No differences were observed with the presence of anti-TIM-3; the p24Ag level with Gal-9 (2 nM) plus anti-TIM-3 plus tunicamycin was 15.2 ng/ml (P = 0.047). These results confirmed that for an adequate reactivation of HIV-1 in latently infected CD4+ T cells, the recognition of the specific pattern of oligosaccharides presented in these cells by Gal-9 appears to be necessary (Fig. 5).
FIG 5.

Deglycosylation assay with tunicamycin (TNC). Bars show HIV-1 reactivation, expressed as p24 antigen production (n = 4) in a cellular model based on IL-7. Comparison of p24 antigen values between Gal-9 alone and Gal-9 with TNC showed a reduction, although it was not significant. Similar results were observed with the presence of an antibody against TIM-3 (anti-TIM-3). Comparison with the cellular model alone (Control) showed a significant difference in the p24 antigen values in the cases of Gal-9 alone, Gal-9 with TNC, and Gal-9 with TNC and anti-TIM-3 (αTIM3). Statistical significance was calculated using the Mann-Whitney U test. P values of <0.05 were considered statistically significant. *, P < 0.05; NS, not significant.
Galectin-9 does not show in vitro synergism with other LRA.
We felt that it was of particular interest to assess the effect of the combination of Gal-9 with other LRA evaluated in previous studies and clinical trials. The possible synergistic effect of any of the combinations could help enhance the effect of Gal-9. In the IL-7 latency model, we first checked the reactivation of the latent virus with individual LRA. Isolated resting memory CD4+ T cells were used to develop the in vitro latency model. After 5 days of incubation with IL-7 (1 nM), an infection with the NL4.3 HIV-1 strain (10 pg) was performed. After 5 additional days, we carried out synergistic experiments with Gal-9, 2 nM; disulfiram (DS), 500 nM; romidepsin (RMD), 40 nM; bryostatin-1 (Bryo), 100 nM; vorinostat (Vor), 500 nM; JQ1, 1 µM; and maraviroc (MVC), 5 µM. Incubation for 24 h with the different LRA was performed. Mean p24Ag concentrations after reactivation with PMA and Gal-9 were 18.43 ng/ml and 17.87 ng/ml, respectively. Gal-9 reactivation capacity compared to those observed with the rest of the LRA was as follows: DS, 0.45 ng/ml (P = 0.001); RMD, 11.96 ng/ml (P = 0.07); Bryo, 13.04 ng/ml (P = 0.18); Vor, 14.57 ng/ml (P = 0.28); JQ1, 5.09 ng/ml (P = 0.001); and MVC, 7.20 ng/ml (P = 0.001). This demonstrates that, as a single agent, Gal-9 has a reactivating potency of latent HIV-1 similar to or greater than that of other LRA in this particular model (Fig. 6A).
FIG 6.
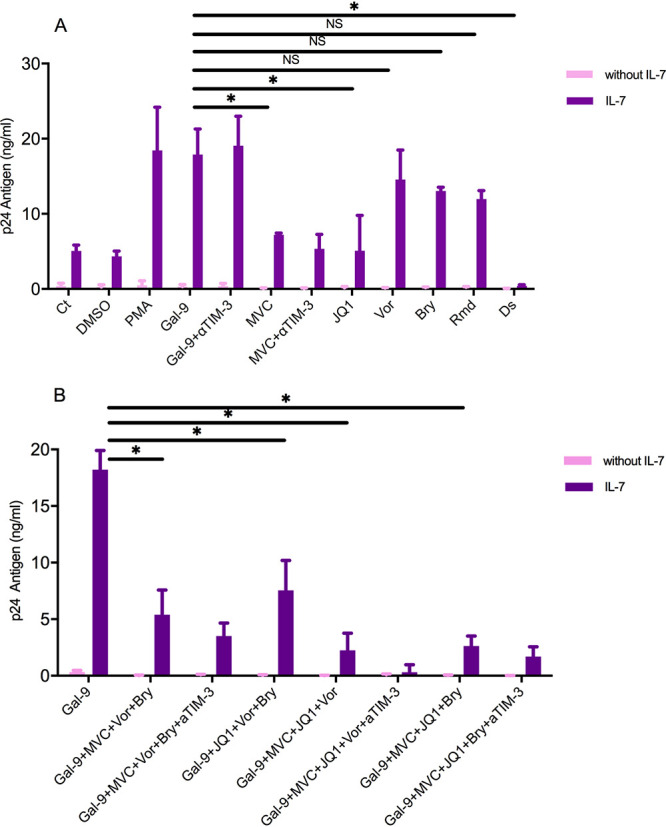
HIV-1 latency reactivation by Galectin-9 (Gal-9) and other latency reversal agents (LRA) in a cellular model based on IL-7. The bars show HIV-1 reactivation, expressed as p24 antigen production (n = 4) in a cellular model based on IL-7. No synergism between Gal-9 and other LRA was observed. (A) Reactivation mediated by Gal-9 alone or Gal-9 with an antibody against TIM-3 (αTIM-3) compared with the cellular model alone (control [Ct]) showed a significant increase in p24 antigen values. p24 antigen values for Gal-9 with or without anti-TIM-3 were significantly different from those for MVC, JQ1, and Ds and not significantly different from those for Vor, Bryo, and Rmd. (B) Several combinations of at least two LRA with Gal-9 did not reach the p24 antigen values obtained with Gal-9 alone. A significant difference was observed for the combinations Gal-9+MVC+Vor+Bryo; Gal-9+JQ1+Vor+Bryo; Gal-9+MVC+Vor+Bryo, and Gal-9+MVC+JQ1+Bryo. Statistical significance was calculated using Mann-Whitney U test. P values of <0.05 were considered statistically significant. *, P < 0.05; NS, not significant. MVC, maraviroc; Vor, vorinostat; DS, disulfiram; Rmd, romidepsin; Bryo, bryostatin-1.
No significant effect was shown after Gal-9 was combined with other LRA. In fact, with respect to the administration of Gal-9 alone, no significant differences were found with Gal-9-based dual combinations: Gal-9 plus Vor, 12.49 ng/ml (P = 0.4); Gal-9 plus Bryo, 14.27 ng/ml (P = 0.37); and Gal-9 plus RMD, 11.60 ng/ml (P = 0.17). Moreover, when combinations included more than two drugs, far from showing synergism or an additive effect, a certain antagonism was observed. In all the combinations analyzed, p24Ag concentrations fell considerably below the values obtained with each of them individually (Fig. 6B). Our experiments demonstrated that Gal-9 alone at 2 nM results in the highest latent-HIV-1 reactivation potency.
TIM-3 blockade improves CTL response in CD8+ T cells and has no significant effect on NK cells.
We further evaluated the improvement of the CTL response by the inhibition of the TIM-3 receptor. For this purpose, different types of in vitro cocultures were performed.
(i) Cocultures of total CD4± T cells with CD8± T cells and NK cells.
An improvement in the CTL activity of CD8+ T lymphocytes was observed with the lowest concentration of anti-TIM-3 (5 μg/ml) (Fig. 7A to C). CD4+ T, CD8+ T, and NK cells were purified from buffy coats of negative donors by magnetic separation (n = 10). The different types of cells isolated were grown in RPMI medium with IL-2 (10 µl/ml) and phytohemagglutinin (PHA) (1 µl/ml). The different cocultures were treated for 20 h with Gal-9 2 nM and anti-TIM-3 (5 μg/ml). An infection with the HIV-1 BaL virus (5 μl/million cells) was carried out. The supernatant cocultures collected on day 7 were used for p24Ag measurement.
FIG 7.

TIM-3 blockade improves CTL response in CD8+ T cells. Bars show HIV-1 reactivation, expressed as p24 antigen production (n = 10) in different coculture conditions. (A) In the CD4:CD8 cocultures, no significant differences were observed between the p24 antigen values in infected CD4 cells alone and CD4 cells with CD8 cells. However, a significant reduction in the p24 antigen values in the cocultures with an antibody against TIM-3 receptor (anti-TIM-3) at different concentrations (5 μg/ml and 7.5 μg/ml) was observed. No significant differences (NS) were detected with or without the addition of Gal-9 at 2 nM concentration in the coculture. (B) In the CD4:NK cocultures, no significant differences were observed between the p24 antigen values for infected CD4 cells alone and CD4 cells with NK cells. Although not significant, a reduction in the p24 antigen values in the cocultures with anti-TIM-3 at different concentrations (5 μg/ml and 7.5 μg/ml) was observed. No significant differences were detected with and without the addition of Gal-9 at 2 nM in the coculture. (C) In the CD4:CD8:NK cocultures, no significant differences were observed between the p24 antigen values in the infected CD4 cells alone and those in CD4 cells with CD8:NK cells. However, a significant reduction in the p24 antigen values in the cocultures with anti-TIM-3 was observed. No significant differences were detected with and without the addition of Gal-9 at 2 nM in the coculture. Statistical significance was calculated using the Mann-Whitney U test. P values of <0.05 were considered statistically significant. *, P < 0.05; NS, not significant.
A significant decrease in the production of p24Ag was detected when the Gal-9/TIM-3 interaction in total CD4+ T cells and CD8+ T coculture cells was blocked by anti-TIM-3 compared to that without TIM-3 blocking (P = 0.014). Moreover, there was no significant difference between the samples analyzed with and those without Gal-9 addition (P = 0.41) (Fig. 7A to C). Under the two conditions, we observed control of viral replication with the use of the anti-TIM-3 in the CD4+ T cell and CD8+ T cell cocultures. Altogether, these findings suggest that TIM-3 inhibition by a specific antibody leads to the control of viral replication irrespective of the increase in viral transcription induced by Gal-9. In contrast, no improvement in the control of viral replication was observed when total CD4+ T cells were cocultured with NK cells and the Gal-9/TIM-3 interaction was blocked by the addition of anti-TIM-3 (P = 0.5) (Fig. 7A to C). These results indicate that the reduction of p24 production was mediated by an improvement in the CTL response, rather than by NK cell activity. Additionally, there were no significant differences with the addition of Gal-9 to the coculture (P = 0.46) (Fig. 7A to C).
It has been suggested that the inhibition of TIM-3 in NK cells could have a negative effect, decreasing the NK cell capacity to control viral replication. To assess the possibility that the beneficial effect on CTL activity of CD8+ T cells might be at least partially offset by a negative effect on NK cells, CD4+ T cells were cocultured along with both CD8+ T and NK cells. TIM-3 blocking in NK cells did not appear to have a negative effect on the activity of the CTL response as a whole, enhanced by TIM-3 inhibition with a significant decrease in p24 antigen concentrations (P = 0.002) (Fig. 7A to C). The results were the same regardless of the antibody concentrations used (P = 0.028) or the addition of Gal-9 (P = 0.35) (Fig. 7A to C). These results showed the improvement of the CTL response and the control of viral replication despite the blockade of TIM-3 on NK cells.
(ii) Cocultures of resting CD4± T cells with CD8± T cells.
Next, to validate the previous finding using a complementary approach, we repeated the experiments using latently infected resting CD4+ T cells obtained through the IL-7 latency model. We performed a coculture with resting memory CD4+ T cells and CD8+ T cells. We used 2 nM Gal-9, which was added to the coculture to reactivate the latent HIV-1, and also anti-TIM-3 (5 μg/ml) to improve HIV-1 CTL activity. As expected, the p24Ag concentration increased from 13 ng/ml before to 27.5 ng/ml after the addition of Gal-9. After TIM-3 inhibition, the p24Ag concentration was 9 ng/ml (P = 0.036), suggesting that the reactivation of HIV-1 from latently infected resting CD4+ T cells was counteracted by the improvement in the HIV-1 CTL-specific response associated with the TIM-3 receptor blockade (Fig. 8).
FIG 8.

Galectin-9 (Gal-9) and TIM-3 inhibition as a joint strategy in HIV-1 latency. Bars show HIV-1 reactivation, expressed as p24 antigen production (n = 3) in a cellular model based on IL-7. Significant differences were observed after Gal-9 (2 nM) addition compared with CD4r:CD8 alone. The TIM-3 inhibition with and without Gal-9 resulted in a significant reduction in p24 antigen values. Statistical significance was calculated using the Mann-Whitney U test. P values of <0.05 were considered statistically significant. *, P < 0.05; NS, not significant. CD4r, resting CD4+ T cells of the cellular model.
(iii) Cocultures of total CD4± T cells with CD8± T cells and NK cells from PLWH.
Finally, the effect of the inhibition of TIM-3, with or without Gal-9 in the medium, was tested in an ex vivo model. CD4+ T cells, CD8+ T cells, and NK cells were isolated by magnetic separation from peripheral blood mononuclear cells (PBMCs) obtained from 10 PLWH (all men). At the time of blood extraction for this purpose, all the PLWH were on ART, with undetectable plasma HIV RNA; the mean CD4+ T cell count was 661 cells/mm3, the mean CD8+ T cell count was 920 cells/mm3, and the mean CD4+ T cell nadir was 327 cells/mm3. The mean time on ART was 88 months (Table 1).
TABLE 1.
Baseline characteristics of PLWH upon study entry
| Characteristic | Value for patient: |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
| Age (yrs) | 50 | 63 | 33 | 57 | 53 | 33 | 38 | 61 | 26 | 52 |
| CD4+ nadir (cells/mm3) | 392 | 365 | 479 | 272 | 250 | 186 | 50 | 633 | 234 | |
| CD4+ (cells/mm3) | 1,029 | 504 | 522 | 970 | 773 | 700 | 603 | 502 | 774 | 417 |
| CD4+/CD8+ | 1.07 | 1.04 | 0.8 | 0.7 | 0.99 | 1.2 | 0.4 | 0.6 | 0.8 | 0.4 |
| HIV-1 RNA (copies/ml) | <37 | <37 | <37 | <37 | <37 | <37 | <37 | <37 | <37 | <37 |
| ART duration (months) | 41 | 185 | 48 | 324 | 43 | 72 | 72 | 270 | 12 | 12 |
| ARTa | RPV, TAF, FTC | RPV, DTG | DTG, ABC, 3TC | DTG, ABC, 3TC | RPV, DTG | DRV, COBI, FTC, TAF | EVG, COBI, FTC, TAF | DRV, RTV, 3TC | DTG, ABC, 3TC | EVG, COBI, FTC, TAF |
RPV, rilpivirine; TAF, tenofovir alafenamide; FTC, emtricitabine; DTG, dolutegravir; ABC, abacavir; 3TC, lamivudine; DRV, darunavir; COBI, cobicistat; EVG, elvitegravir.
The different isolated cellular subpopulations were resuspended in RPMI medium and incubated for 2 days. Treatment of CD8+ T cells and NK cells with anti-TIM-3 (5 μg/ml) was carried out before coculturing with total CD4+ T cells for 24 h. This was a critical step. After this time, the different cocultures were performed in a 1:1 ratio. Total CD4+ T cells were combined with CD8+ T cells, with NK cells, and with CD8+ T cells and NK cells. A superinfection with an R5 HIV-1 virus (BaL virus) was carried out. Coculture supernatants were collected on days 7 and 10 to measure p24Ag. The HIV-1-specific CTL response of CD8+ T cells was based on levels of p24Ag production.
In CD4+ T cell cocultures in combination with CD8+ T cells, a low CD8+ T cell suppressive capacity (mean p24Ag level of 0.9 log) was observed, but this improved significantly after TIM-3 inhibition (mean p24Ag level of 2.4 log) (P = 0.007). In the case of total CD4+ T cells in combination with NK cell coculture, blocking TIM-3 did not cause a significant decrease in p24Ag level (mean, 0.8 log versus 1.2 log with antibody; P = 0.41). However, in total CD4+ T cell cocultures with CD8+ T cells and NK cells, TIM-3 blockade in NK cells did not affect CTL activity and a significant decrease in the p24Ag level was observed (mean, 1 log versus 2.3 log with the antibody) (P = 0.011) (Table 2). No significant differences were found in the measurements on day 7 or 10 or after the addition of Gal-9 (Fig. 9A and B). These findings reinforce our previous results in the in vitro experiments indicating that TIM-3 inhibition results in a decrease of HIV-1 replication that is mediated by an enhancement of CTL responses and independent of Gal-9 signaling or NK activity.
TABLE 2.
Absolute values of the HIV-1 p24 antigen in culture supernatants in cells cocultured from PLWH
| Cell culture | Mean p24Ag concn (ng/ml) |
|||
|---|---|---|---|---|
| Without Gal-9 |
With Gal-9 |
|||
| Day 7 | Day 10 | Day 7 | Day 10 | |
| Uninfected CD4 | −0.015 | −0.037 | −0.023 | −0.011 |
| Infected CD4 | 56.40 | 99.45 | 61.35 | 100.59 |
| CD4:CD8 | 19.92 | 31.52 | 36.76 | 38.303 |
| CD4:CD8 + anti-TIM-3 | 2.97 | 1.68 | 0.14 | 0.411 |
| CD4:NK | 21.15 | 33.36 | 20.49 | 29.89 |
| CD4:NK + anti-TIM-3 | 11.29 | 23.95 | 6.38 | 9.22 |
| CD4:CD8:NK | 15.85 | 17.14 | 23.67 | 28.63 |
| CD4:CD8:NK + anti-TIM-3 | 1.07 | 7.1 | 1.029 | 2.478 |
FIG 9.

TIM-3 blockade improves CTL response in CD8+ T cells from PLWH. Bars show mean p24 antigen reduction (log) (n = 10). (A) Without Gal-9 addition, significant increases (*, P < 0.05) in p24 of ≥2 log with TIM-3 inhibition (α-TIM-3) (dark green bars) in the CD4:CD8 and CD4:CD8:NK cocultures were observed; no significant difference was seen in the case of CD4:NK alone. No differences were detected between results on day 7 and day 10. (B) With Gal-9 addition, significant increases in p24 of ≥2 log with TIM-3 inhibition (dark green bars) in the CD4:CD8 and CD4:CD8:NK cocultures were observed; no significant difference was seen in the case of CD4:NK alone. No differences were detected between results on day 7 and day 10. Statistical significance was calculated using the Mann-Whitney U test. P values of <0.05 were considered statistically significant. *, P < 0.05; NS, not significant.
(iv) Cytokines and granzyme/perforin activity.
To assess the mechanism involved in the improvement of the control of viral replication following the inhibition of TIM-3, we measured cytokine levels as well as the production of granzyme and perforin on day 7 of each coculture. In the different experiments, a panel of Th1/Th2/Th17 cytokines was used. Using the supernatants from the different coculture conditions, we measured the different cytokines included in the panel. No differences were observed in the levels of cytokines analyzed in the different cocultures from buffy coat cells (in vitro) (total CD4+ T cells with CD8+ T cells, total CD4+ T cells with NK cells, and total CD4+ T cells with CD8+ T and NK cells) before and after the addition of anti-TIM-3. No differences were found in the production of either granzyme or perforin. The lack of an effect was also observed in the supernatants taken in the ex vivo experiments in which we used cells from PLWH (Fig. 10 and 11).
FIG 10.

Cytokine levels at day 7 from supernatants of CD4:CD8:NK coculture (in vitro model). Each graph presents expression levels for one cytokine. The control infection (purple) had only cells without Gal-9 or an antibody against TIM-3 (αTIM3). The addition of Gal-9 (2 nM) alone (green) or with an antibody against TIM-3 at 5 μg/ml and 7.5 μg/ml (orange and blue) was evaluated. Anti-TIM-3 alone at 5 μg/ml and 7.5 μg/ml (red and pink) was also tested. No differences were observed in the levels of all cytokines analyzed before and after the addition of anti-TIM-3.
FIG 11.

Cytokine levels at day 7 in supernatants of CD4:CD8:NK coculture (ex vivo model). Each graph represents the expression levels for one cytokine. CD4:CD8:NK cocultures (green) had only cells without Gal-9 or an antibody against TIM-3 (anti-TIM-3). The addition of Gal-9 (2 nM) alone (orange) to the CD4:CD8:NK coculture was evaluated. No differences were observed in the levels of all cytokines analyzed after the addition of anti-TIM-3 without Gal-9 (blue) or with Gal-9 (pink) to the CD4:CD8:NK coculture.
DISCUSSION
In this study, we have shown the ability of Gal-9 to reactivate latent HIV-1 both in Jurkat-LAT-GFP cells and in a latency cellular model based on IL-7. Unfortunately, the combination of Gal-9 with other LRA was not associated with positive effects in the reactivation of the latent virus and was rather antagonistic. Of note, our data showed that TIM-3 blockade in CD8+ T cells was associated with control of viral replication in both in vitro and ex vivo models, suggesting an increase in the CTL response needed for viral eradication.
The results obtained with Gal-9 for HIV-1 reactivation in Jurkat-LAT-GFP cells confirmed the previous findings of Abdel-Mohsen et al. (9), with similar reactivation percentages. The ability of Gal-9 to reactivate latent HIV-1 in resting memory CD4+ T cells was further confirmed with the experiments in the cellular model based on IL-7. An important finding was that similar results were obtained with concentrations of Gal-9 (2 nM) lower than those used by other groups (9). We performed all subsequent experiments with this lower concentration so that potential dose-dependent side effects of Gal-9 could be avoided should the drug be used in vivo.
The reactivation of latent HIV-1 in resting memory CD4+ T cells took place by a route alternative to the TIM-3 receptor used by Gal-9. The recognition of a specific pattern of oligosaccharides seems to be confirmed, which would include the loss of sialic acid molecules on the surface of these cells with which Gal-9 would interact (22). Leitner et al. (23) performed a series of experiments to study a potential interaction between Gal-9 and the TIM-3 receptor. They also found that the interaction of Gal-9 with the oligosaccharides with beta-galactoside was independent of the TIM-3 receptor, affirming that Gal-9 is not a ligand of TIM-3.
This TIM-3-independent pathway was demonstrated with the use of the antibiotic tunicamycin, which destroys this pattern. Compared to previous reports, the decrease in the percentage of reactivation of HIV-1 was not as striking (9), although the differences could be explained by the different cell models used in the experiments. Recognition of this pattern in latently infected CD4+ T cells would lead to intracellular signal transduction that would ultimately mediate latent HIV-1 transcription. The possible participation in the signaling of classic transcription factors such as NFAT, AP1, and NF-κB was known, but the intermediate pathways that, in turn, lead to their activation were unknown. Knowledge of these routes could explain the results that we obtained when we assessed the effect of combining Gal-9 with other LRA previously tested by us and other groups (4–7, 24–26).
None of the LRA analyzed exhibited, individually, a reactivation capacity higher than that of Gal-9. The different combinations of the LRA with Gal-9 did not lead to an increase in the reactivation of the latent virus either, since, far from finding synergism or at least an additive effect, we observed some antagonism. Gal-9 had a greater reactivating potential alone than any of the combinations tested. Abdel-Mohsen et al. found that the combination of Gal-9 with JQ1 and Gal-9 with vorinostat did manage to overcome mediated reactivation by Gal-9 alone. Perhaps the explanation could be found in the different concentrations of LRA and in the use of a cell model rather than cells taken directly from PLWH. However, we believe that the key to this possible antagonistic effect lies in the knowledge of these intermediate signaling pathways mediated by Gal-9 and that they deserve to be studied to explain these results.
Of special interest, we found that blockade of TIM-3, an inhibitory immune checkpoint that is a receptor for Gal-9, is associated with improved CTL activity. The use of inhibitors against immune checkpoints to improve the CTL response has become especially important in the field of oncology (27), where these receptors are among the most promising targets in the treatment of solid tumors. Specifically for HIV-1, blocking PD1 and PDL1, as well as CTLA-4, has been investigated (28–31). The recovery of the effector functions and the proliferative capacity of memory T cells is among the major purposes to be achieved with the strategies employed.
The interaction of TIM-3 with Gal-9 in the CD8+ T cells is associated with an increase in the exhaustion of T cells during HIV-1 chronic infection (20). In this work, we have demonstrated that TIM-3 inhibition leads to an improvement in the control of HIV-1 replication in both in vitro and ex vivo models by restoring the CTL activity of CD8+ T cells. Of particular importance, we have also shown that blockage of the receptor in NK cells was not associated with a partial or total loss in the control of viral replication. NK cells also express the TIM-3 receptor (32, 33), and a worsening of their function by blocking this receptor had been postulated (34). Therefore, we were concerned with the fact that the reinvigoration of CD8+ T cells would be offset by the negative impact on NK cells. The same result was obtained when an ex vivo assay directly using cells from PLWH was performed. Importantly, CD8+ T cells must first be treated with the anti-TIM-3 to achieve good virological control. These results were of great importance for the use of Gal-9 as the LRA and the blockade of TIM-3 to enhance the immune response as a joint strategy. We tested this strategy using a cellular model based on IL-7 and observed the replication control of the virus that was being reactivated. In our experiments, we used the clone of antibody against TIM-3 receptor that was previously used by Abdel-Mohsen (9). Different TIM-3 antibodies were used in other studies (18, 35). The study by Jones et al. (18) found similar results to ours; however, Grabmeier-Pfistershammer et al. (35) observed that TIM-3 antibodies were especially effective only in combination with PD-1 blockade.
CD8+ T cell exhaustion leads to a progressive loss of effector functions, such as cytotoxicity and cytokine production, in response to antigenic stimulation (16). In the case of our assays, we expected that the modification of the improvement in CTL activity after TIM-3 blockade would result in a change in the levels of certain cytokines, as well as an increase in granzyme and perforin. However, we did not find any difference in the values of any of them after the modulation of TIM-3. One possible explanation is that antigenic stimulation by infecting cells with a virus could be so strong that it would mask the effect of TIM-3 blocking itself. Alternatively, directly, our results could point to the fact that the control of viral replication by these modulated CD8+ T cells does not imply the secretion of cytokines. Possibly, the virological control mechanism is much more complex and a greater number of factors are involved. In a recent report, Angin et al. (36) analyzed genetic and metabolic profiles and their effect on the functionality of CD8+ T cells in PLWH progressors and PLWH controllers. They found differentiation in the expression of certain sets of genes that led to activation of either the mTORC1 pathway (in the case of progressors) or mTORC2 (in the case of controllers). The regulation of the effector function of CD8+ T cells would be associated with the mTORC2 pathway, whose expression would be marked by genes that are preferentially expressed in controller PLWH. In the case of our work, we do not know how much of this regulation would be conditioned by TIM-3 inhibition. It would be something worth studying.
In summary, our results confirm Gal-9 as a promising molecule for HIV-1 eradication studies. It requires more precise evaluations of the possible adverse effects that its modulation could have, as it is involved in a large number of biological processes. Most significantly, TIM-3 inhibition with a monoclonal antibody increases the CD8+ T cell capacity of controlling viral replication, thus potentially contributing to the elimination of the viral reservoir. Again, TIM-3 inhibition might not be exempt from toxicity, and the issue of toxicity in humans needs to be tested. The results regarding the safety, dosage, and effectiveness of the AMBER study (37) will inform future studies in the field of HIV-1 cure. We have explored two components of the shock-and-kill strategy: reactivation of latent HIV-1 by galectin-9 and reinvigoration of CD8+ T cells by TIM-3 blockade, which could be used separately or in combination.
MATERIALS AND METHODS
Ethics statement.
This study was performed in the Infectious Diseases Department of the Ramón y Cajal University Hospital. All subjects were properly informed and provided written informed consent. The study was conducted according to the Helsinki principles and was approved by the Ethics Committee (CEIC) of the Ramón y Cajal University Hospital, IRYCIS, Madrid, Spain.
Samples and subjects.
For the in vitro study, we used peripheral mononuclear cells (PBMCs) from the Transfusion Center of Madrid Community. They kindly provided buffy coats from healthy anonymous volunteers.
The ex vivo study included PLWH who were recruited in the HIV-1 outpatient clinic of the Infectious Diseases Department of the Ramón y Cajal University Hospital.
HIV-1 reactivation by galectin-9 in Jurkat-LAT-GFP cells.
The role of galectin-9 (Gal-9) (R&D Systems, Minneapolis, MN, USA) in reversing HIV-1 latency in Jurkat-LAT-GFP cells (clone A72 from the NIH AIDS Reagent Program, USA) was evaluated. This cell line bears the retroviral construct LTR-Tat-IRES-GFP, containing the gene that encodes the green fluorescent protein (GFP), which is used as an activation marker. As a positive control, we used phorbol-12-myristate-13-acetate (PMA) at 100 nM (Sigma-Aldrich, St. Louis, MO, USA). As a negative control, we used dimethyl sulfoxide (DMSO) (Sigma-Aldrich, St. Louis, MO, USA). Jurkat-LAT-GFP cells were cultured in RPMI medium (Gibco, Thermo Fisher Scientific Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco, Thermo Fisher Scientific Inc.), l-glutamine at 2 mM (PAN Biotech, Aidenbach, Germany), penicillin at 100 U/ml, and streptomycin at 100 µg/ml (Gibco, Thermo Fisher Scientific Inc.). Cells were plated at a density of 900,000 cells/well in a 24-well flat-bottom plate. The positive control (PMA) and negative control (DMSO) were added to the appropriate well with Jurkat-LAT-GFP cells. Gal-9 at a 2 nM concentration and antibody against TIM-3 (anti-TIM-3) (clone 344801; R&D Systems, Minneapolis, MN, USA) at a concentration of 5 μg/ml was added to each culture condition for 18 h at 37°C in a 5% CO2 atmosphere. After 18 h, GFP expression was measured by flow cytometry. Data acquisition was performed in a FACSCalibur Flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA) to assess the mean fluorescence intensity of GFP expression after stimulation with Gal-9. Data analysis was done using CellQuest (BD Biosciences, Franklin Lakes, NJ, USA) software.
HIV-1 reactivation by galectin-9 in the HIV-1 cellular model.
From the blood of healthy donors (buffy coats from the Madrid Transfusion Center), PBMCs were separated using a density gradient with Ficoll-Hypaque (Rafer SL, Zaragoza, Spain) by centrifugation at 2,000 rpm for 30 min. The intermediate phase was collected and washed with phosphate-buffered saline (PBS) (Lonza, Verviers, Belgium) by centrifugation at 1,500 rpm for 10 min. The cells obtained were resuspended and cultured at 37°C, 5% CO2, and 90% humidity in complete RPMI medium at a concentration of 5 × 106 cells/ml.
Resting memory CD4+ T lymphocytes were obtained from these previously isolated PBMCs by magnetic separation carried out in three steps. A negative selection was made to prevent the activation of CD4+ T cells. We used a cocktail of biotin-labeled monoclonal antibodies lacking anti-CD4 that recognized different surface antigens (anti-CD8, -CD14, -CD16, -CD19, -CD36, -CD56, -CD123, and -CD235a, anti-gamma/delta T cell receptor [TCR-γ/δ], and anti-glycophorin A) (Miltenyi Biotec, Bergisch Galdbach, Germany). A second separation was carried out using a cocktail of antibiotin and anti-CD61 monoclonal antibodies. Finally, a third magnetic separation was performed by negative selection. Anti-HLA-DR and anti-CD25 antibodies were used to finally obtain CD4+ CD25− HLA-DR− T lymphocytes.
Isolated resting memory CD4+ T cells were used to develop an in vitro cellular model based on interleukin 7 (IL-7) (R&D Systems, Minneapolis, MN, USA) (21) (Fig. 2). Briefly, resting memory CD4+ T cells were cultured with complete RPMI at 37°C, 5% CO2, and 90% humidity for 5 days in the presence of IL-7 (1 nM) to facilitate the entry of the virus and without the addition of IL-7 as an experimental control. After this incubation period, an infection with the NL4.3 HIV-1 strain was performed. A treatment with DNase (Qiagen, Hilden, Germany) and MgCl2 was carried out before infection. Cells were infected with 10 pg of p24 of NL4.3 HIV-1 strain per million cells, incubated by rotation for 2 h at room temperature, and centrifuged for 2 h at 2,000 rpm at 25°C. After a series of PBS (1×) washes, the infected cells were cultured with complete RPMI supplemented with IL-7 (1 nM) and without IL-7, plus IL-2 (Sigma-Aldrich, St. Louis, MO, USA) (10 IU/ml), and incubated at 37°C, 5% CO2, and 90% humidity for 5 days.
The correct HIV-1 integration was confirmed with the quantification of the integrated DNA. An integration PCR was performed using 20 ng of DNA obtained with the QIAamp DNA blood minikit (Qiagen, Hildenger, Germany) and mixed with several primers (Table 3) with TaqDNA in a final volume of 20 μl. The PCR conditions were as follows: 8 min at 95°C, 1 min at 95°C, 1 min at 60°C, 10 min at 72°C, and 15 min at 72°C for 12 cycles. Alu sequences are repetitive DNA sequences that are widely dispersed within the human genome. Alu sequences were used as markers to quantify the integrated HIV-1 DNA. Primers flanking the Alu and gag sequences of the HIV-1 structural gene were used to amplify this gene sequence. Only the integrated form of genomic copies of HIV-1 was exponentially amplified. In a second PCR, quantitative PCR (qPCR), the direct primer R and the reverse primer U5 were used, which weakens the viral LTR region (38). A standard DNA curve integrated from the 8E5 cell line with the Lambda T, AA55M, and MH603 primers and the CCR5 gene was used as maintenance with the forward and reverse primers and was quantified using a LightCycler 480 Instrument II (Roche Applied Science, Penzberg, Germany).
TABLE 3.
Primers used for PCR integrationa
| Primer | Sequence |
|---|---|
| Alu 1 | TCCCAGCTACTGGGGAGGCTGAG |
| Alu 2 | GCCTCCCAAAGTGCTGGGATTAC |
| M667 | ATGCCACGTAAGCGAAACTCTGGCTAACTAGGGAACCCACTG |
| MH603 | ACACTACTTTGAGCACTCAA |
| Lambda T | ATGCCACGTAAGCGAAACT |
| AA55M | GCTAGAGATTTTCCACACTGACTAA |
| CCR5_F | GCTGTTTGCGTCTCTCCCAGGA |
| CCR5_R | CTCACAGCCCTGTGCCTCTTCTTC |
Primers used in the amplification reaction to confirm the correct HIV-1 integration in the cellular model based on IL-7.
HIV-1 reactivation was carried out using the latently infected resting memory CD4 T cells obtained from the above-described latency model. In a 96-well plate (800,000 cells/well in RPMI), Gal-9 was added at different concentrations (2 nM, 5 nM, 200 nM, and 500 nM), alone or in combination with anti-TIM-3 at a concentration of 5 μg/ml. As a positive control, 100 nM PMA was used, while the negative control was 1 μl/ml DMSO. We incubated cells at 37°C, 5% CO2, and 90% humidity for 18 h. HIV-1 reactivation was measured by quantification of the HIV-1 p24 antigen (p24Ag) levels in culture supernatants after 24 h of Gal-9 addition.
A cell deglycosylation assay was performed to demonstrate that the reactivation carried out by Gal-9 was not mediated through TIM-3. The antibiotic tunicamycin (R&D Systems, Minneapolis, MN, USA), which deglycosylated the oligosaccharide surface pattern of the resting memory CD4+ T cells, was used. The reactivation of the virus was carried out with Gal-9 alone or in combination with tunicamycin and in the presence or absence of anti-TIM-3. After incubation for 18 h at 37°C, 5% CO2, and 90% humidity, the levels of p24Ag were quantified using an ELISA technique (Innotest HIV antigen monoclonal antibody [MAb] test; Innogenetics, Barcelona, Spain) according to the manufacturer´s instructions.
Gal-9 potency reactivation in combination with other latency reactivation agents (LRA) was analyzed. Disulfiram (DS) (500 nM; Sigma-Aldrich, St. Louis, MO, USA), romidepsin (RMD) (40 nM; Sigma-Aldrich), bryostatin-1 (Bryo) (100 nM; Sigma-Aldrich), vorinostat (Vor) (500 nM; Sigma-Aldrich), JQ1 (1 µM), and maraviroc (MVC) (5 µM; Sigma-Aldrich) were tested alone and in single and multiple combinations with Gal-9 using the same latency model described above based on IL-7 and the technique previously described. The concentrations of the different LRA were selected according to previously published data (7, 9, 26).
TIM-3 blockade in CD8± T and NK cells in vitro.
We obtained PBMCs from buffy coats using a density gradient with Ficoll-Hypaque (Rafer SL, Zaragoza, Spain) by centrifugation at 2,000 rpm for 30 min. We purified total CD4+ T cells, CD8+ T cells, and NK cells by magnetic separation (Miltenyi Biotec, Bergisch Galdbach, Germany). Anti-CD4 antibodies were used to isolate CD4+ T cells, anti-CD56 antibodies for NK cells, and anti-CD8 antibodies for CD8+ T lymphocytes. The different isolated cell types were grown in complete RPMI medium at 37°C, 5% CO2, and 90% humidity at a concentration of 5 × 106 cells/ml. IL-2 (10 μl/ml) and PHA (1 μl/ml) (Sigma-Aldrich, St. Louis, MO, USA) were added to CD4+ T cells for 2 days for activation. Afterward, cocultured combinations (1:1 ratio) of total CD4+ T cells with CD8+ T cells, total CD4+ T cells with NK cells, and total CD4+ T with CD8+ T cells and NK cells were cocultured in 96-well plates (800,000 cells/well) in complete RPMI medium supplemented with IL-2 (10 μl/ml) and treated for 20 h with Gal-9 alone or in combination with anti-TIM-3 at 5-μg/ml and 7.5-μg/ml concentrations. After this incubation, an infection with the HIV-1 BaL virus (5 μl/million cells) was carried out by centrifugation at 2,500 rpm at 22°C for 1 h. The plate was incubated for 1 h at 37°C, 5% CO2, and 90% humidity. After the incubation time, two washes with RPMI medium were performed. Finally, the cells were resuspended with complete RPMI medium and supplemented with IL-2 (10 μl/ml). Supernatants were collected at 3, 7, and 10 days. HIV-1-specific CTL activity was measured by p24 antigen production (Innotest HIV antigen MAb test; Innogenetics, Barcelona, Spain) with the supernatants of cocultures collected on day 7 according to the manufacturer´s instructions.
Cytokine measurement was also carried out, using a BD cytometric bead array (CBA) with human Th1/Th2/Th17 cytokines (BD Biosciences, San Jose, CA, USA), which allowed us to quantify interleukin-2 (IL-2), IL-4, IL-6, IL-10, tumor necrosis factor alpha (TNF-α), gamma interferon (IFN-γ), and IL-17A. Data acquisition was performed in a FACSCalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
A coculture with latently infected resting memory CD4+ T cells (using the latency model described above) and CD8+ T cells was performed. In 96-well plates (800,000 cells/well), latently infected resting CD4+ T cells and CD8+ T cells in a 1:1 ratio were added and treated with anti-TIM-3 for 18 h at 37°C, 5% CO2, and 90% humidity. After this incubation time, Gal-9 was added. Supernatants were collected at 3, 7, and 10 days. The CTL activity of CD8+ T lymphocytes was analyzed by p24 antigen production (Innotest HIV antigen MAb; Innogenetics, Barcelona, Spain) according to the manufacturer´s instructions.
TIM-3 blockade in CD8± T and NK cells ex vivo.
For the TIM-3 blockade in CD8+ T and NK cells, we selected PLWH among those treated in the HIV Clinics of the Infectious Diseases Department at the Ramón y Cajal University Hospital. All subjects were properly informed and gave written informed consent. PLWH had been receiving antiretroviral treatment (ART) and had exhibited suppressed viral load (VL) (<37 copies/ml) for at least 12 months. They underwent venipuncture to obtain 200 ml of whole blood in 10-ml tubes with EDTA as an anticoagulant (BD Vacutainer; Becton, Dickinson, Franklin Lakes, NJ, USA). As described above, we isolated the PBMCs from the blood obtained and then isolated the different subpopulations of CD4+ T cells, CD8+ T cells, and NK cells, which were suspended in total RPMI medium and incubated at 37°C, 5% CO2, and 90% humidity for 2 days. Only in the case of CD4+ T cells was the medium supplemented with IL-2 (10 μl/ml) and PHA (1 μl/ml) for activation. After this time, CD8+ T cells and NK cells were treated with anti-TIM-3 and incubated for 24 h. Following this incubation, according to a modification of the protocol of Sáez-Cirión et al. (39), cocultures of total CD4+ T cells with CD8+ T cells, total CD4+ T cells with NK cells, and total CD4+ T cells with CD8+ T and NK cells (in a 1:1 ratio) were carried out in 96-well plates (800,000 cells/well) with complete RPMI medium supplemented with IL-2 (10 μl/ml). An infection with the HIV-1 BaL virus (5 μl/million cells) was performed by centrifugation at 2,500 rpm at 22°C for 1 h. The plate was incubated for 1 h at 37°C, 5% CO2, and 90% humidity. After the incubation time, two washes were performed with RPMI medium. Finally, the cells were resuspended with complete RPMI and supplemented with IL-2 (10 μl/ml). Gal-9 was added to the appropriate wells. Supernatants were collected at 3, 7, and 10 days.
With the collected supernatants, p24 antigen levels were measured using the Zeptometrix kit p24 2.0 (Zeptometrix, Buffalo, NY, USA) according to the manufacturer´s indications. We analyzed cytokine production with the ProcartaPlex multiplex immunoassay kit (Thermo Fisher Scientific Inc., Waltham, MA, USA) according to manufacturer´s instructions. This technique allowed us to quantify IL-2, IL-17A, IL-4, IL-6, IL-10, granzyme A, granzyme B, perforin, TNF-α, and IFN-γ.
Statistical analysis.
Data analysis was carried out using GraphPad Prism v5.0 software and the SPSS statistical package IBM version 21.0 (GraphPad Software, Inc., CA, USA). The statistical analysis used to examine the differences between the different samples analyzed was the Mann-Whitney U test. P values of <0.05 were considered statistically significant.
ACKNOWLEDGMENTS
We greatly appreciate the technical assistance of Laura Luna and Manuela Beltrán. We thank Centro de Transfusiones Comunidad de Madrid (Spain) for providing the buffy coats.
This study was funded by Instituto de Salud Carlos III through the project PI17/01636 (cofunded by European Regional Development Fund/European Social Fund “A way to make Europe"/"Investing in your future”). This study was also supported by the Spanish AIDS Research Network RD16/0025/0001 (Spanish I + D + I Plan). The Spanish AIDS Research Network supported the work of Nadia Madrid-Elena, and Carolina Gutiérrez. Marta Sanz’s work was supported by Fondo Social Europeo (Programa Operativo de Empleo Juvenil y la Iniciativa de Empleo Juvenil).
Conceived and designed the experiments: S.M. and C.G. Performed the experiments: M.S., N.M.E., A.V. Selection of the PLWH: S.S.V. Analyzed the data: S.M., C.G., and M.S. Wrote the first draft of the manuscript: C.G., M.S., and S.M. Reviewed and approved the final manuscript: all authors.
REFERENCES
- 1.Siliciano JD, Siliciano RF. 2013. HIV-1 eradication strategies: design and assessment. Curr Opin HIV AIDS 8:318–325. doi: 10.1097/COH.0b013e328361eaca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Archin NM, Bateson R, Tripathy MK, Crooks AM, Yang KH, Dahl NP, Kearney MF, Anderson EM, Coffin JM, Strain MC, Richman DD, Robertson KR, Kashuba AD, Bosch RJ, Hazuda DJ, Kuruc JD, Eron JJ, Margolis DM. 2014. HIV-1 expression within resting CD4+ T cells after multiple doses of vorinostat. J Infect Dis 210:728–735. doi: 10.1093/infdis/jiu155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rasmussen TA, Tolstrup M, Brinkmann CR, Olesen R, Erikstrup C, Solomon A, Winckelmann A, Palmer S, Dinarello C, Buzon M, Lichterfeld M, Lewin SR, Østergaard L, Søgaard OS. 2014. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: a phase 1/2, single group, clinical trial. Lancet HIV 1:e13–e21. doi: 10.1016/S2352-3018(14)70014-1. [DOI] [PubMed] [Google Scholar]
- 4.Elliott JH, McMahon JH, Chang CC, Lee SA, Hartogensis W, Bumpus N, Savic R, Roney J, Hoh R, Solomon A, Piatak M, Gorelick RJ, Lifson J, Bacchetti P, Deeks SG, Lewin SR. 2015. Short-term administration of disulfiram for reversal of latent HIV infection: a phase 2 dose-escalation study. Lancet HIV 2:e520–e529. doi: 10.1016/S2352-3018(15)00226-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Søgaard OS, Graversen ME, Leth S, Olesen R, Brinkmann CR, Nissen SK, Kjaer AS, Schleimann MH, Denton PW, Hey-Cunningham WJ, Koelsch KK, Pantaleo G, Krogsgaard K, Sommerfelt M, Fromentin R, Chomont N, Rasmussen TA, Østergaard L, Tolstrup M. 2015. The depsipeptide romidepsin reverses HIV-1 latency in vivo. PLoS Pathog 11:e1005142. doi: 10.1371/journal.ppat.1005142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gutiérrez C, Serrano-Villar S, Madrid-Elena N, Pérez-Elías MJ, Martín ME, Barbas C, Ruipérez J, Muñoz E, Muñoz-Fernández MA, Castor T, Moreno S. 2016. Bryostatin-1 for latent virus reactivation in HIV-infected patients on antiretroviral therapy. AIDS 30:1385–1392. doi: 10.1097/QAD.0000000000001064. [DOI] [PubMed] [Google Scholar]
- 7.Madrid-Elena N, García-Bermejo ML, Serrano-Villar S, Díaz-de Santiago A, Sastre B, Gutiérrez C, Dronda F, Coronel Díaz M, Domínguez E, López-Huertas MR, Hernández-Novoa B, Moreno S. 2018. Maraviroc is associated with latent HIV-1 reactivation through NF-κB activation in resting CD4+ T cells from HIV-infected individuals on suppressive antiretroviral therapy. J Virol 92:e01931-17. doi: 10.1128/JVI.01931-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shan L, Deng K, Shroff NS, Durand CM, Rabi SA, Yang HC, Zhang H, Margolick JB, Blankson JN, Siliciano RF. 2012. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity 36:491–501. doi: 10.1016/j.immuni.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abdel-Mohsen M, Chavez L, Tandon R, Chew GM, Deng X, Danesh A, Keating S, Lanteri M, Samuels ML, Hoh R, Sacha JB, Norris PJ, Niki T, Shikuma CM, Hirashima M, Deeks SG, Ndhlovu LC, Pillai SK. 2016. Human galectin-9 is a potent mediator of HIV transcription and reactivation. PLoS Pathog 12:e1005677. doi: 10.1371/journal.ppat.1005677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heusschen R, Griffioen AW, Thijssen VL. 2013. Galectin-9 in tumor biology: a jack of multiple trades. Biochim Biophys Acta 1836:177–185. doi: 10.1016/j.bbcan.2013.04.006. [DOI] [PubMed] [Google Scholar]
- 11.John S, Mishra R. 2016. Galectin-9: from cell biology to complex disease dynamics. J Biosci 41:507–534. doi: 10.1007/s12038-016-9616-y. [DOI] [PubMed] [Google Scholar]
- 12.Elahi S, Dinges WL, Lejarcegui N, Laing KJ, Collier AC, Koelle DM, McElrath MJ, Horton H. 2011. Protective HIV-specific CD8+ T cells evade Treg cell suppression. Nat Med 17:989–995. doi: 10.1038/nm.2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jayaraman P, Sada-Ovalle I, Beladi S, Anderson AC, Dardalhon V, Hotta C, Kuchroo VK, Behar SM. 2010. Tim3 binding to galectin-9 stimulates antimicrobial immunity. J Exp Med 207:2343–2354. doi: 10.1084/jem.20100687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kashio Y, Nakamura K, Abedin MJ, Seki M, Nishi N, Yoshida N, Nakamura T, Hirashima M. 2003. Galectin-9 induces apoptosis through the calcium-calpain-caspase-1 pathway. J Immunol 170:3631–3636. doi: 10.4049/jimmunol.170.7.3631. [DOI] [PubMed] [Google Scholar]
- 15.Nagahara K, Arikawa T, Oomizu S, Kontani K, Nobumoto A, Tateno H, Watanabe K, Niki T, Katoh S, Miyake M, Nagahata S, Hirabayashi J, Kuchroo VK, Yamauchi A, Hirashima M. 2008. Galectin-9 increases Tim-3+ dendritic cells and CD8+ T cells and enhances antitumor immunity via galectin-9-Tim-3 interactions. J Immunol 181:7660–7669. doi: 10.4049/jimmunol.181.11.7660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, Zheng X, Strom TB, Kuchroo VK. 2005. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol 6:1245–1252. doi: 10.1038/ni1271. [DOI] [PubMed] [Google Scholar]
- 17.Anderson AC, Joller N, Kuchroo VJ. 2016. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity 44:989–1004. doi: 10.1016/j.immuni.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones RB, Ndhlovu LC, Barbour JD, Sheth PM, Jha AR, Long BR, Wong JC, Satkunarajah M, Schweneker M, Chapman JM, Gyenes G, Vali B, Hyrcza MD, Yue FY, Kovacs C, Sassi A, Loutfy M, Halpenny R, Persad D, Spotts G, Hecht FM, Chun TW, McCune JM, Kaul R, Rini JM, Nixon DF, Ostrowski MA. 2008. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J Exp Med 205:2763–2779. doi: 10.1084/jem.20081398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Finney CA, Ayi K, Wasmuth JD, Sheth PM, Kaul R, Loutfy M, Kain KC, Serghides L. 2013. HIV infection deregulates Tim-3 expression on innate cells: combination antiretroviral therapy results in partial restoration. J Acquir Immune Defic Syndr 63:161–167. doi: 10.1097/QAI.0b013e318285cf13. [DOI] [PubMed] [Google Scholar]
- 20.Merani S, Chen W, Elahi S. 2015. The bitter side of sweet: the role of galectin-9 in immunopathogenesis of viral infections. Rev Med Virol 25:175–186. doi: 10.1002/rmv.1832. [DOI] [PubMed] [Google Scholar]
- 21.Coiras M, Bermejo M, Descours B, Mateos E, García-Pérez J, López-Huertas MR, Lederman MM, Benkirane M, Alcamí J. 2016. IL-7 induces SAMHD1 phosphorylation in CD4+ T lymphocytes, improving early steps of HIV-1 life cycle. Cell Rep 14:2100–2107. doi: 10.1016/j.celrep.2016.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bi S, Earl LA, Jacobs L, Baum LG. 2008. Structural features of galectin-9 and galectin-1 that determine distinct T cell death pathways. J Biol Chem 283:12248–12258. doi: 10.1074/jbc.M800523200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leitner J, Rieger A, Pickl W, Zlabinger G, Grabmeier-Pfistershammer W, Steinberger P. 2013. TIM-3 does not act as a receptor for galectin-9. PLoS Pathog 9:e1003253. doi: 10.1371/journal.ppat.1003253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gutiérrez C, Díaz L, Vallejo A, Hernández-Novoa B, Abad M, Madrid N, Dahl V, Rubio R, Moreno AM, Dronda F, Casado JL, Navas E, Pérez-Elías MJ, Zamora J, Palmer S, Muñoz E, Muñoz-Fernández MA, Moreno S. 2011. Intensification of antiretroviral therapy with a CCR5 antagonist in patients with chronic HIV-1 infection: effect on T cells latently infected. PLoS One 6:e27864. doi: 10.1371/journal.pone.0027864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Archin NM, Liberty AL, Kashuba AD, Choudhary SK, Kuruc JD, Crooks AM, Parker DC, Anderson EM, Kearney MF, Strain MC, Richman DD, Hudgens MG, Bosch RJ, Coffin JM, Eron JJ, Hazuda DJ, Margolis DM. 2012. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 487:482–485. doi: 10.1038/nature11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.López-Huertas MR, Jiménez-Tormo L, Madrid-Elena N, Gutiérrez C, Rodríguez-Mora S, Coiras M, Alcamí J, Moreno S. 2017. The CCR5-antagonist maraviroc reverses HIV-1 latency in vitro alone or in combination with the. Sci Rep 7:2385. doi: 10.1038/s41598-017-02634-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anderson AC, Xiao S, Kuchroo VK. 2007. Tim protein structures reveal a unique face for ligand binding. Immunity 26:273–275. doi: 10.1016/j.immuni.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trautmann L, Janbazian L, Chomont N, Said EA, Gimmig S, Bessette B, Boulassel M-R, Delwart E, Sepulveda H, Balderas RS, Routy J-P, Haddad EK, Sekaly R-P. 2006. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med 12:1198–11202. doi: 10.1038/nm1482. [DOI] [PubMed] [Google Scholar]
- 29.Freeman GJ, Wherry EJ, Ahmed R, Sharpe AH. 2006. Reinvigorating exhausted HIV-specific T cells via PD-1-PD-1 ligand blockade. J Exp Med 203:2223–2227. doi: 10.1084/jem.20061800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cecchinato V, Tryniszewska E, Ma ZM, Vaccari M, Boasso A, Tsai WP, Petrovas C, Fuchs D, Heraud JM, Venzon D, Shearer GM, Koup RA, Lowy I, Miller CJ, Franchini G. 2008. Immune activation driven by CTLA-4 blockade augments viral replication at mucosal sites in simian immunodeficiency virus infection. J Immunol 180:5439–5447. doi: 10.4049/jimmunol.180.8.5439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Callahan MK, Postow MA, Wolchok JD. 2014. CTLA-4 and PD-1 pathway blockade: combinations in the clinic. Front Oncol 4:385. doi: 10.3389/fonc.2014.00385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garrido C, Allard B, Soriano-Sarabia N, Margolis DM. 2017. Characterization of immune exhaustion in natural killer cells and role in HIV infection, abstr 512. In 8th International Workshop on HIV Persistence during Therapy.
- 33.Anderson AC, Anderson DE, Bregoli L, Hastings WD, Kassam N, Lei C, Chandwaskar R, Karman J, Su EW, Hirashima M, Bruce JN, Kane LP, Kuchroo VK, Hafler DA. 2007. Promotion of tissue inflammation by the immune receptor Tim-3 expressed on innate immune cells. Science 318:1141–1143. doi: 10.1126/science.1148536. [DOI] [PubMed] [Google Scholar]
- 34.Ndhlovu LC, Lopez-Vergès S, Barbour JD, Jones RB, Jha AR, Long BR, Schoeffler EC, Fujita T, Nixon DF, Lanier LL. 2012. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood 119:3734–3743. doi: 10.1182/blood-2011-11-392951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grabmeier-Pfistershammer K, Stecher C, Zettl M, Rosskopf S, Rieger A, Zlabinger GJ, Steinberger P. 2017. Antibodies targeting BTLA or TIM-3 enhance HIV-1 specific T cell responses in combination with PD-1 blockade. Clin Immunol 183:167–173. doi: 10.1016/j.clim.2017.09.002. [DOI] [PubMed] [Google Scholar]
- 36.Angin M, Volant S, Passaes C, Lecuroux C, Monceaux V, Dillies MA, Valle-Casuso JC, Pancino G, Vaslin B, Le Grand R, Weiss L, Goujard C, Meyer L, Boufassa F, Müller-Trutwin M, Lambotte O, Sáez-Cirión A. 2019. Metabolic plasticity of HIV-specific CD8+ T cells is associated with enhanced antiviral potential and natural control of HIV-1 infection. Nat Metab 1:704–716. doi: 10.1038/s42255-019-0081-4. [DOI] [PubMed] [Google Scholar]
- 37.Clinical Trials.gov. 2016. A phase 1 study of TSR-022 an anti TIM-3 monoclonal antibody in patients with advanced solid tumors (AMBER). ClinicalTrials NCT02817633.
- 38.Brady T, Kelly BJ, Male F, Roth S, Bailey A, Malani N, Gijsbers R, O'Doherty U, Bushman FD. 2013. Quantitation of HIV DNA integration: effects of differential integration site distributions on Alu-PCR assays. J Virol Methods 189:53–57. doi: 10.1016/j.jviromet.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sáez-Cirión A, Shin SY, Versmisse P, Barré-Sinoussi F, Pancino G. 2010. Ex vivo T cell-based HIV suppression assay to evaluate HIV-specific CD8+ T-cell responses. Nat Protoc 5:1033–1041. doi: 10.1038/nprot.2010.73. [DOI] [PubMed] [Google Scholar]