

Stabilization of HIV-1 Env state 2A has been shown to sensitize infected cells to ADCC. State 2A can be stabilized by a “cocktail” composed of CD4mc, anti-CoRBS, and anti-cluster A Abs.
KEYWORDS: HIV-1, envelope glycoproteins, Env conformation, State 2A, nonneutralizing antibodies, CD4-induced antibodies, cluster A, coreceptor binding site, gp120, small CD4 mimetics, soluble CD4
ABSTRACT
The HIV-1 envelope glycoprotein (Env) trimer [(gp120/gp41)3] is a metastable complex expressed at the surface of viral particles and infected cells that samples different conformations. Before engaging CD4, Env adopts an antibody-resistant “closed” conformation (State 1). CD4 binding triggers an intermediate conformation (State 2) and then a more “open” conformation (State 3) that can be recognized by nonneutralizing antibodies (nnAbs) such as those that recognize the coreceptor binding site (CoRBS). Binding of antibodies to the CoRBS permits another family of nnAbs, the anti-cluster A family of Abs which target the gp120 inner domain, to bind and stabilize an asymmetric conformation (State 2A). Cells expressing Env in this conformation are susceptible to antibody-dependent cellular cytotoxicity (ADCC). This conformation can be stabilized by small-molecule CD4 mimetics (CD4mc) or soluble CD4 (sCD4) in combination with anti-CoRBS Ab and anti-cluster A antibodies. The precise stoichiometry of each component that permits this sequential opening of Env remains unknown. Here, we used a cell-based enzyme-linked immunosorbent assay (CBE) to evaluate each component individually. In this assay, we used a “trimer mixing” approach by combining wild-type (wt) subunits with subunits impaired for CD4 or CoRBS Ab binding. This enabled us to show that State 2A requires all three gp120 subunits to be bound by sCD4/CD4mc and anti-CoRBS Abs. Two of these subunits can then bind anti-cluster A Abs. Altogether, our data suggest how this antibody-vulnerable Env conformation is stabilized.
IMPORTANCE Stabilization of HIV-1 Env State 2A has been shown to sensitize infected cells to ADCC. State 2A can be stabilized by a “cocktail” composed of CD4mc, anti-CoRBS, and anti-cluster A Abs. We present evidence that optimal State 2A stabilization requires all three gp120 subunits to be bound by both CD4mc and anti-CoRBS Abs. Our study provides valuable information on how to stabilize this ADCC-vulnerable conformation. Strategies aimed at stabilizing State 2A might have therapeutic utility.
INTRODUCTION
The HIV-1 envelope glycoprotein (Env) is a trimer formed by three heterodimers composed of a surface gp120 subunit associated noncovalently with a transmembrane gp41 subunit (1–3). Env mediates viral entry by binding first to its main receptor CD4 and then subsequently to its coreceptor (mainly CCR5 or CXCR4). The conformational changes in Env induced by these sequential interactions are required for the fusion of the viral and target cell membranes. Imaging by single-molecule Förster resonance energy transfer (smFRET) allowed the characterization of four conformational states (4–8): an antibody (Ab)-resistant “closed” conformation (State 1), an intermediate conformation (State 2), an “open” conformation (State 3), and an off-pathway conformation, which is susceptible to nonneutralizing antibody (nnAb) attack (State 2A) (reviewed in reference 9). Prior to CD4 engagement, Env preferentially adopts the closed antibody-resistant State 1. Binding to CD4 triggers Env causing it to sample downstream conformations (State 2/3) that expose epitopes that are recognized by easily elicited CD4-induced (CD4i) Abs, which are common in HIV-1-infected individuals (10, 11). Primary HIV-1 strains have Envs with higher State 1 occupancy (4, 6), consistent with their natural resistance to CD4i Abs. Additionally, HIV-1 prevents the exposure of CD4i epitopes by limiting the accumulation of Env at the cell surface via Vpu-mediated BST-2 downregulation (12–14) and by downregulating CD4 via the activities of both Nef and Vpu, thus effectively limiting Env-CD4 interaction (10, 14, 15).
It was previously reported that exposure of CD4i epitopes could also be accomplished with small-molecule CD4-mimetic compounds (CD4mc), resulting in the sensitization of infected cells to antibody-dependent cellular cytotoxicity (ADCC) (16, 17). However, among the different families of CD4i Abs, anti-cluster A Abs are responsible for the majority of ADCC activity against HIV-1 (10, 14, 16, 18, 19). CD4mc alone are unable to expose the cluster A epitopes, requiring instead a sequential “opening” of the trimer (20, 21). CD4mc initially triggers Env into more open conformations, which exposes the coreceptor binding site (CoRBS) and thus allows the binding of anti-CoRBS Abs. This in turn triggers further conformational changes, which leads to the exposure of cluster A epitopes (20). In addition, anti-CoRBS and anti-cluster A Ab binding to Env has been shown to engage Fc-gamma receptors in a synergistic fashion to mediate ADCC (21). Consistent with these findings, smFRET experiments recently showed that these two families of Abs in combination with soluble CD4 (sCD4) or CD4mc stabilize a new conformational state (State 2A) susceptible to ADCC responses (7). However, the precise stoichiometry of each component allowing this “sequential opening” of Env resulting in the stabilization of State 2A remains unknown. Here, we developed an assay to evaluate each component individually. In this assay, we used a “trimer mixing” approach in which we combined wild-type (wt) subunits with subunits impaired for either CD4 or CoRBS Ab binding at different ratios of the wt to the mutant. This enabled us to explore the relative contribution of CD4 and CoRBS Abs to the exposure of the gp120 inner domain cluster A region.
RESULTS
Impact of CD4 interaction on exposure of gp120 inner domain cluster A epitopes.
To study the stoichiometry of sCD4/CD4mc and CoRBS Abs required to stabilize State 2A and expose the anti-cluster A epitopes, we adapted a cell-based enzyme-linked immunosorbent assay (ELISA) (CBE) (22) to allow the generation of “mixed trimers” at the surface of transfected human osteosarcoma (HOS) cells. Briefly, different amounts of the tier 2 HIV-1JRFL wild type (wt) versus CD4 binding site (CD4BS)- or CoRBS-expressing Env mutants were cotransfected into HOS cells at wt/mutant ratios of 3:0, 2:1, 1:2, and 0:3, and antibody binding was measured by a CBE (as described in Materials and Methods) (22, 24).
Since previous observations indicate that the initial step in State 2A stabilization is dependent on CD4mc or sCD4 interaction (7, 20), we first selected a CD4BS mutant unable to bind to these ligands. While the D368R mutation abrogates CD4 interaction (Fig. 1A) (10, 19, 23), it still responds to CD4mc such as BNM-III-170 and exposes the CoRBS as measured by 17b binding by a CBE (Fig. 1B) (24). We therefore evaluated the capacity of another CD4BS mutant, E370R, which not only impairs CD4 interaction (Fig. 1A) but also was reported to abrogate CD4mc binding (24). Indeed, as shown in Fig. 1B, the E370R mutant does not expose the CoRBS upon BNM-III-170 addition. As expected, the lack of CD4 interaction abrogated infectivity for both mutants (Fig. 1C) despite normal Env processing and trimer stability (Fig. 1D).
FIG 1.

CD4 binding site mutant characterization. (A and B) Recognition of Env-expressing cells by a cell-based ELISA using CD4-Ig (A) or 17b (B) in the presence or absence of sCD4 or the CD4mc BNM-III-170. Data shown represent mean relative light unit (RLU) values ± standard errors of the means (SEM) from at least three independent experiments performed in quadruplicate, with the signal obtained from cells transfected with an empty pcDNA3.1 plasmid (no Env) subtracted, normalized to Env levels as determined by bNAb 2G12. (C) Infectivity of Env variants compared to the wild type (wt). Reverse-transcriptase-normalized amounts of recombinant luciferase-expressing HIV-1 pseudotyped with the wt or the D368R or E370R mutant were used to infect Cf2Th-CD4/CCR5 cells at 37°C for 48 h. (D) The trimer stability of Env variants was measured by immunoprecipitation of radiolabeled Env and calculated as described in Materials and Methods. The association index measures the association of mutant gp120 with the Env complex on expressing cells relative to the wt, and the processing index measures proteolytic processing of the mutant gp160 Env precursor to mature gp120 relative to the wt. Results represent the mean values from three independent experiments.
To better understand the impact of CD4 interaction on the exposure of cluster A epitopes, we evaluated by a CBE the binding of sCD4/CD4mc, anti-CoRBS Abs, and anti-cluster A Abs to the mixed wt:E370R trimers (Fig. 2A). The loss of CD4 binding observed in Fig. 2B is consistent with an increase in the number of gp120 subunits unable to interact with CD4 (E370R) as the wt/E370R ratio decreases. Indeed, each additional E370R subunit decreases CD4 interaction by approximately one-third, consistent with a model where the stoichiometry of CD4 is equal to the number of wt subunits in a trimer. Exposure of the CoRBS, as measured by 17b binding, in the presence of sCD4 or the CD4mc BNM-III-170 is proportional to the amount of wt Env present at each wt/E370R ratio and in good agreement with a model where CoRBS binding is proportional to the amount of CD4 competent subunits (Fig. 2C). To evaluate the impact of E370R titration on the exposure of cluster A epitopes, we used horseradish peroxidase (HRP)-conjugated anti-cluster A antibody N5-i5 (N5-i5-HRP) (25–28). This HRP-conjugated antibody is required since it is known that exposure of the cluster A region requires the combination of sCD4/CD4mc together with CoRBS Abs (7, 20). By using the N5-i5-HRP conjugate, we ensured that the signal measured is cluster A specific and not confounded by the potential detection of CoRBS Abs. As expected, the N5-i5 interaction was detected for the “pure” wt trimer (3:0) but not for E370R trimers (0:3), which are unable to bind CD4 or CD4mc (Fig. 2D). Indeed, the N5-i5-HRP signal falls below or close to the background threshold, which corresponds to the binding of N5-i5-HRP to the mutant trimer in the presence of sCD4. Interestingly, as we increased the amount of the E370R mutant, we observed that the decrease of the N5-i5 interaction differed from the one observed for both the CD4-immunoglobulin fusion protein (CD4-Ig) and 17b. This suggests that N5-i5 engages the trimer with a different stoichiometry than sCD4/CD4mc and CoRBS. Indeed, half of the N5-i5 signal was lost upon the addition of a single E370R subunit (2 wt:1 E370R) and reached background levels of detection with the addition of a second E370R subunit (1 wt:2 E370R). Altogether, these results are consistent with a model where optimal exposure of the cluster A region requires the three gp120 subunits to be engaged by sCD4/CD4mc in order to allow two subunits to be bound by anti-cluster A Abs.
FIG 2.
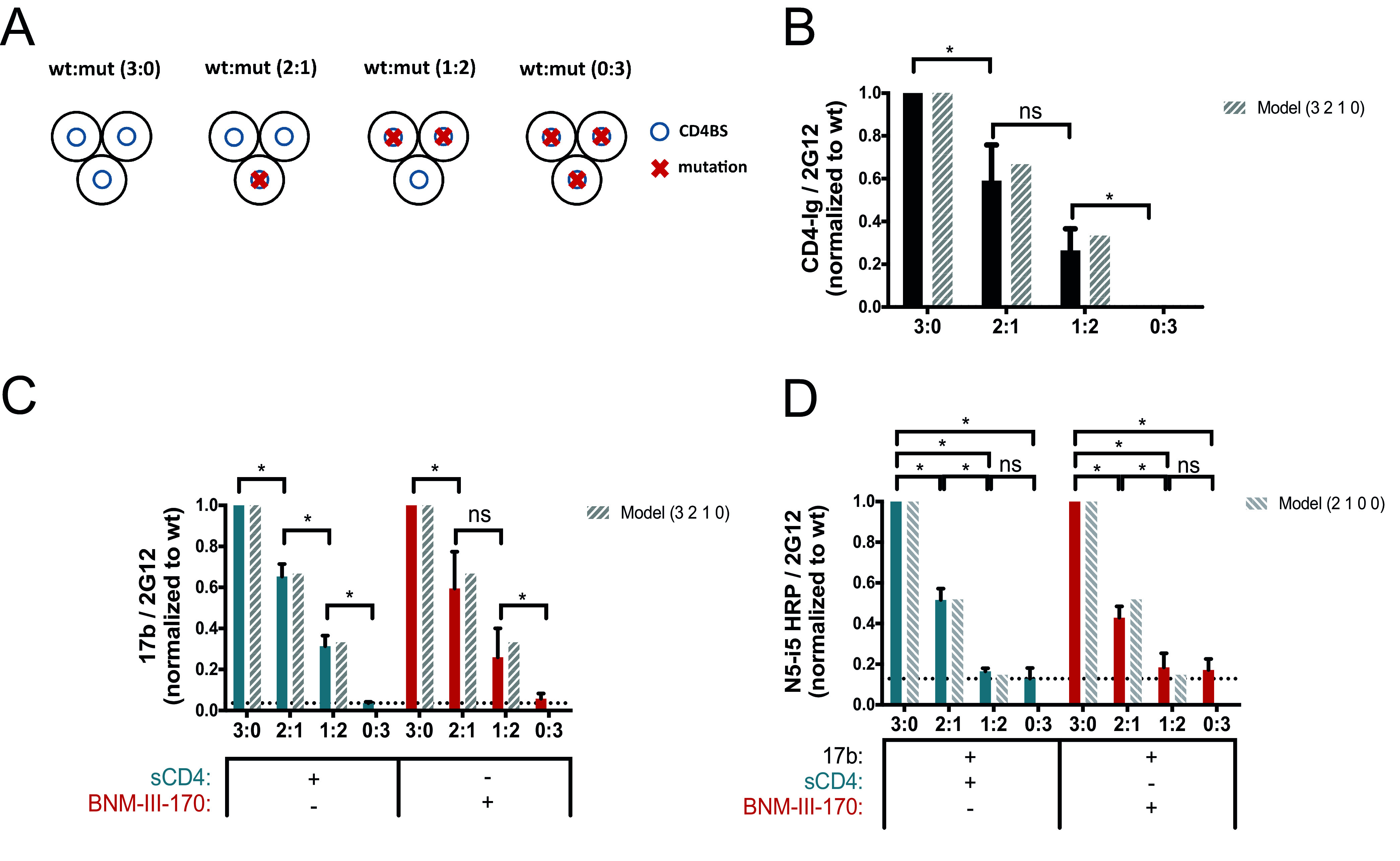
Impact of CD4 binding on anti-cluster A epitope exposure. (A) Exposure of the gp120 cluster A region was evaluated with a cell-based ELISA by varying the concentrations of the wild type (wt) and the E370R CD4 binding site (CD4BS) variant (mut) (wt/E370R ratios of 3:0, 2:1, 1:2, and 0:3), as described in Materials and Methods. The ratios correspond to the ratio of the Env subunit transfected into the cells and represent the composition of the predominant trimer expressed at the cell surface as shown on the scheme. Env trimers are shown as three black circles representing the gp120 subunits, blue circles represent the CD4BS, and red crosses indicate the presence of the E370R mutation impairing CD4 interaction. (B to D) Recognition of cell-expressed trimeric Env by CD4-Ig (B), anti-CoRBS Ab 17b (C), or the HRP-conjugated anti-cluster A antibody N5-i5 (N5-i5-HRP) (D) was evaluated in the presence of sCD4 (3 μg/ml) or the CD4mc BNM-III-170 (5 μM), alone or in combination with 17b (1 μg/ml). N5-i5-HRP was used to avoid codetection of 17b and N5-i5 by a secondary antibody. The dashed bars show the predicted values calculated from the theoretical composition of trimers following a model that represents the predicted proportional binding values of each trimer. Data shown represent mean RLU values ± SEM from at least three independent experiments performed in quadruplicate, with the signal obtained from cells transfected with an empty pcDNA3.1 plasmid (no Env) subtracted, normalized to Env levels as determined by bNAb 2G12, relative to the wt. The dotted line is the threshold representing the background signal level. It is defined by the signal obtained with the 0:3 ratio (wt/mutant) in the presence of sCD4. Statistical significance was tested using an unpaired t test or a Mann-Whitney U test based on statistical normality (*, P < 0.05; ns, not significant).
Impact of the CoRBS interaction on exposure of cluster A epitopes.
In addition to the sCD4/CD4mc interaction, previous observations indicate that State 2A stabilization requires the interaction of CoRBS Abs (7, 20). Therefore, we designed a CoRBS mutant unable to bind to 17b without altering its capacity to bind CD4. The 17b epitope (PDB accession number 1GC1) contains a number of strong hydrogen bonds and salt bridges in addition to hydrophobic van der Waals interactions. We chose to mutate two of the strongest interactions to destabilize the 17b interface on gp120. We mutated K121 in gp120 to D to remove the main-chain hydrogen bond that K121 makes to L54 on the 17b heavy chain and remove any aliphatic interactions that it has with its side chain to L54 and V56. We also mutated R419 to D on gp120 to disrupt salt bridges to 17b residues E100B and E100D on the heavy chain (Fig. 3). R419 is part of the coreceptor binding site and forms a distant salt bridge interaction with TYS10 of CCR5 (PDB accession number 6MET) (Fig. 3). The introduction of the K121D/R419D mutations did not affect CD4 binding (Fig. 4A). However, they completely abrogated the CoRBS 17b Ab interaction as shown by a lack of reactivity in the presence of sCD4 or CD4mc (Fig. 4B). As expected, they also abrogated viral infectivity (Fig. 4C), suggesting that in addition to preventing 17b binding, they can also impair coreceptor binding. Of note, these mutations affected Env processing without affecting trimer stability (Fig. 4D).
FIG 3.

Contribution of K121 and R419 to the 17b and CCR5 complex interfaces. (A) The 17b gp120 interface. K121 forms a hydrogen bond to the main-chain carbonyl of L54 of 17b’s heavy chain CDR H2 (PDB accession number 1GC1). The aliphatic portion of the K121 side chain packs against the L54 and V56 side chains. The change to D121 removes the hydrogen bond and prevents any favorable side chain interactions. R419 forms salt bridges to E100B and E100D in the third complementarity-determining region of the heavy chain (CDR H3) of 17b. D419 prevents both from occurring. (B) The CCR5 interface. R419 forms a long distant salt bridge (5.8 Å) with TYS10 on CCR5 (PDB accession number 6MET). The mutation to D419 removes this interaction.
FIG 4.

Coreceptor binding site mutant characterization. (A and B) Effects of the mutations K121D and R419D on the recognition of cell-expressed trimeric Env by CD4-Ig (A) or anti-CoRBS Ab 17b (B) in the presence or absence of sCD4 (3 μg/ml) or the CD4mc BNM-III-170 (5 μM) were examined by cell-based ELISAs. (C) Infectivity levels relative to the wild type (wt) were determined by infecting Cf2Th-CD4/CCR5 cells with HIV-1 pseudotyped with the wt or the K121D/R419D mutant normalized by RT. (D) Association and processing indices were calculated as described in Materials and Methods. Data shown are displayed as described in the legend of Fig. 1. Statistical significance was tested using an unpaired t test or a Mann-Whitney U test based on statistical normality (ns, not significant).
To better understand the role of the anti-CoRBS Ab interaction in the exposure of cluster A epitopes, we repeated our CBEs using mixed wt:K121D/R419D trimers. As observed in Fig. 5A, all titrations of the wt:K121D/R419D trimers had similar CD4 binding, consistent with the maintained CD4 binding capacity of this double mutant. We observed a stepwise decrease in 17b binding in the presence of sCD4 or CD4mc when we increased the concentration of the double mutant (Fig. 5B). Indeed, as we decreased the ratio of the wt to the K121D/R419D mutant, 17b binding was reduced by about one-third for each additional K121D/R419D subunit, suggesting that the stoichiometry of anti-CoRBS is also equal to the number of wt subunits in a trimer.
FIG 5.

Impact of CoRBS binding on anti-cluster A epitope exposure. Exposure of the gp120 cluster A region was evaluated by a cell-based ELISA by varying the concentrations of the wild type (wt) and the K121D/R419D CoRBS variant (mut) (ratios of the wt to the K121D/R419D mutant of 3:0, 2:1, 1:2, and 0:3), as described in Materials and Methods. The ratios correspond to the ratio of the Env subunit transfected into the cells and represent the composition of the predominant trimer expressed at the cell surface (the predicted values calculated from the theoretical composition of trimers corresponding to each transfection ratio are shown as dashed bars). Recognition of cell-expressed trimeric Env by CD4-Ig (A), anti-CoRBS Ab 17b (B), or the HRP-conjugated anti-cluster A antibody N5-i5 (N5-i5-HRP) (C) was evaluated in the presence of sCD4 (3 μg/ml) or the CD4mc BNM-III-170 (5 μM), alone or in combination with 17b (1 μg/ml). N5-i5-HRP was used to avoid codetection of 17b and N5-i5 by a secondary antibody. The dotted line is the threshold representing the background signal level. It is defined by the signal obtained with the 0:3 ratio (wt/mutant) in the presence of sCD4. Data shown represent mean RLU values ± SEM from at least three independent experiments performed in quadruplicate, with the signal obtained from cells transfected with an empty pcDNA3.1 plasmid (no Env) subtracted, normalized to Env levels as determined by bNAb 2G12, relative to the wt. Statistical significance was tested using an unpaired t test or a Mann-Whitney U test based on statistical normality (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant).
We then evaluated the contribution of the CoRBS to the exposure of the gp120 inner domain cluster A region. As described above for Fig. 2D, we used the anti-cluster A N5-i5-HRP conjugate to evaluate cluster A exposure. In agreement with the loss of the N5-i5 signal observed with additional E370R subunits, the addition of a single K121D/R419D subunit (2 wt:1 K121D/R419D) decreased the N5-i5 signal by half, while the addition of two K121D/R419D subunits (1 wt:2 K121D/R419D) decreased the signal to the background level (Fig. 5C). Taken together, these results support a “sequential opening” model where the optimal exposure of cluster A epitopes requires all three subunits to be bound by sCD4 or CD4mc and then by anti-CoRBS Ab, allowing two subunits to be bound by anti-cluster A Abs.
DISCUSSION
The HIV-1 envelope glycoprotein (Env) is the only virus-specific antigen present at the surface of virions and HIV-1-infected cells and represents the main target for antibodies. However, most antibodies elicited during natural infection recognize CD4-induced (CD4i) Env epitopes that are not readily exposed in the closed untriggered trimer (State 1). Soluble CD4 (sCD4) or small CD4 mimetics (CD4mc) can trigger Env into more open states and stabilize downstream State 2/3 conformations (4–6) that can be recognized by CD4i Abs targeting the CoRBS and the V3 loop. CoRBS binding upon sCD4/CD4mc was shown to allow interaction by anti-cluster A Abs and stabilize a fourth conformation, State 2A (7). By using mixed trimers containing subunits unable to interact with CD4 or anti-CoRBS Abs, we found that the optimal exposure of cluster A epitopes required sCD4/CD4mc engagement in all three gp120 subunits (Fig. 2B). Our results are consistent with the requirement of all gp120 subunits to be bound by CoRBS Abs (Fig. 5B), thus allowing two gp120 subunits to interact with anti-cluster A Abs (Fig. 2D and Fig. 5C). Additionally, our results suggest that a minimum of two gp120 subunits need to be bound by CD4 and anti-CoRBS Abs to allow one subunit to be bound by anti-cluster A Abs. Our results are in agreement with in silico modeling showing that an anti-cluster A and an anti-CoRBS Ab can bind to the same gp120 protomer (21).
It was previously reported that sCD4 engagement with a first gp120 subunit results in the stabilization of the two adjacent gp120 protomers in the State 2 conformation, creating an asymmetric trimer presenting protomers in distinct conformations (6). Similarly, State 2A-stabilized trimers were shown to adopt an asymmetric configuration (7). Interestingly, cluster A epitope exposure, a hallmark of State 2A, can be induced, in the presence of CD4mc, by the bivalent antigen binding fragment [F(ab′)2] of anti-CoRBS Abs or the full Ab but not the monovalent Fab (7, 20, 21), suggesting that subunit cross-linking might be necessary to stabilize State 2A. This is in good agreement with structural information available based on cryo-electron microscopy (cryo-EM) structures of HIV-1 virion-associated Env trimers triggered with soluble (d1d2 domain) CD4 and 17b Fab (29) and a recent computational model of the Env trimer-d1d2CD4-17b Fab ternary complex that was built based on available cryo-EM data, atomic-resolution structures of the g120 core, and information on CD4 and 17b binding interactions (30). As shown in Fig. 6A and B (left), the binding of the Fab of CoRBS-specific antibody 17b to the CD4-triggered Env trimer does not expose the cluster A region (highlighted in red). This region is still buried at the gp120-gp41 interface and is not accessible for antibody recognition. However, when 17b IgG is used, it could interact in a multivalent fashion (using both Fab arms) with two neighboring gp120 protomers to trigger asymmetric trimer opening and exposure of the cluster A region for anti-cluster A antibody binding (Fig. 6B, right). We cannot rule out the possibility that in addition to engaging the subunits of the same trimer (intraspike cross-linking), the CoRBS-specific antibody could cross-link gp120 protomers from two distinct trimers (to form interspike cross-links). While we observed that a minimum of two gp120 subunits need to be bound by anti-CoRBS Abs to expose cluster A epitopes (Fig. 5B and C), our data cannot differentiate between these two possibilities (intra- versus interspike cross-linking). A significant improvement in the resolution of current maps (7) will be required to unequivocally answer this question. Considering that multivalent antibody-antigen immune complexes are necessary to potently engage Fc-gamma receptors, as reviewed by Anand and Finzi (31), cross-linking of Env at the surface of infected cells (either intra- or interspike) might contribute to the robust ADCC responses observed when treating infected cells with CD4mc (20, 21). Additionally, recent data suggest that the angle at which the Fc portion of CD4-induced antibodies is exposed toward the effector cell could impact ADCC potency (32). A better understanding of how State 2A is stabilized could provide relevant information about the mechanism driving Fc-gamma receptor engagement. This in turn could lead to the optimization of strategies exploiting Fc-mediated functions to eliminate HIV-1-infected cells and potentially decrease the size of the viral reservoir.
FIG 6.
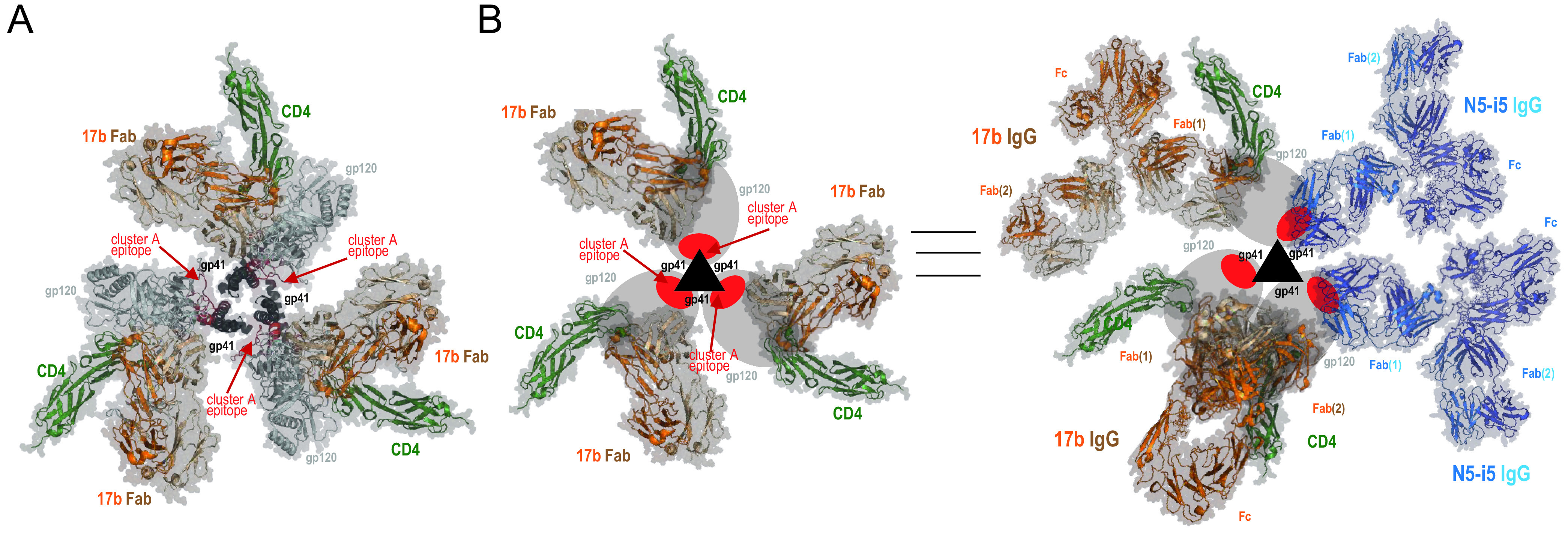
Model of asymmetric HIV-1 trimer opening induced by binding of soluble CD4 and CoRBS-specific antibody 17b. (A) Env trimer–CD4-17b Fab complex as in the structure under PDB accession number 3J70 (30). The epitope footprint of anti-cluster A antibody N5-i5 is highlighted in red. (B) Model of the asymmetric opening of the Env trimer upon binding to soluble CD4 and IgG of CoRBS-specific antibody 17b. Env trimer promoters are shown schematically, gp120 is shown as ovals, and gp41 is shown as triangles. The cluster A epitope region is highlighted in red. (Left) Env trimer–CD4-17b Fab complex as described above for panel A. The cluster A region is buried at the trimer interface and not accessible for antibody recognition. (Right) Structural rearrangements of the Env trimer upon CD4 and 17b IgG binding. The model involves two 17b IgG molecules bound to the CD4-triggered HIV-1 trimer, one with both Fab arms involved in an intraprotomer interaction and a second IgG interacting through one Fab arm only. The cluster A region becomes exposed by rearrangements of the gp120 subunits relative to gp41 in the trimer to expose two binding sites for N5-i5 IgG. Based on the binding data, two N5-i5 IgGs are bound per trimer (attached by a single Fab arm).
Altogether, by combining cell-based ELISA and trimer mixing approaches, here, we provide new insights into the mechanism underlying the stabilization of State 2A, an antibody-vulnerable conformation. Why is it important to study the State 2A conformation? The main reason resides in the vulnerability of infected cells exposing this Env conformation to ADCC. Ongoing studies will determine whether this translates into decreasing the size of the viral reservoir in vivo.
MATERIALS AND METHODS
Cell lines.
HEK293T (human embryonic kidney) cells, Cf2Th canine thymocytes, and HOS (human osteosarcoma) cells (American Type Culture Collection) were grown at 37°C with 5% CO2 in Dulbecco’s modified Eagle’s medium containing 5% fetal bovine serum and 100 μg/ml penicillin-streptomycin (Wysent), as previously described (16, 24). Cf2Th cells stably expressing human CD4 and CCR5 (37) were grown in medium supplemented with 0.4 mg/ml of G418 (Invitrogen) and 0.15 mg/ml of hygromycin B (Roche Diagnostics).
Plasmids and site-directed mutagenesis.
The sequence of full-length clade B HIV-1JRFL Env was codon optimized (GenScript) and cloned into the expression plasmid pcDNA3.1. Mutations were introduced into the plasmid expressing HIV-1JRFL Env using the QuikChange II site-directed mutagenesis protocol or the QuikChange multisite-directed mutagenesis kit (Stratagene). Truncation of the cytoplasmic tail (EnvΔCT variant) was made by adding a stop codon at position 711. The presence of the desired mutations was confirmed by Sanger DNA sequencing. All residues are numbered based on alignment with the HXBc2 prototype sequence, according to convention.
Antibodies.
The following monoclonal Abs (mAbs) were used: anti-HIV-1 gp120 mAbs recognizing the gp120 outer domain (2G12) (NIH AIDS Reagent Program), CD4-induced gp120 epitopes (17b and N5-i5), and the soluble CD4-immunoglobin fusion protein (CD4-Ig). Horseradish peroxidase (HRP)-conjugated goat anti-human IgG antibody (Invitrogen) and HRP-conjugated N5-i5 were used as secondary Abs.
Horseradish peroxidase antibody conjugation.
N5-i5-HRP was generated from N5-i5 IgG using the EZ-link plus activated peroxidase kit (Thermo-Fisher). Briefly, 1 mg of purified N5-i5 IgG dialyzed into phosphate-buffered saline (PBS) was added to 1 vial of EZ-link activated peroxidase from the kit, and the final volume was adjusted to 1 ml with PBS. Ten microliters of a 5 M sodium cyanoborohydride solution was added in a chemical fume hood, and the sample was left to incubate at room temperature for 1 h. Twenty microliters of 3 M ethanolamine (pH 9) was used to quench the reaction at the end of the incubation. N5-i5-HRP was purified from the unreacted HRP by gel filtration on a Superdex 200 16/60 column (GE Healthcare) with a solution containing 10 mM Tris-HCl (pH 7.2) and 100 mM ammonium acetate used as the running buffer. N5-i5-HRP was easily separated from unreacted HRP but not from unreacted IgG. Fractions corresponding to the correct molecular weight were combined and concentrated for later use.
Small molecules.
The CD4-mimetic small molecule BNM-III-170 was synthesized as described previously (33). The compounds were dissolved in dimethyl sulfoxide (DMSO) at a stock concentration of 10 mM, aliquoted, and stored at −20°C. The compound was then diluted to 5 μM in blocking buffer for cell-based ELISAs.
Recombinant luciferase viruses.
Recombinant viruses containing the firefly luciferase gene were produced by calcium phosphate transfection of 293T cells with the HIV-1 proviral vector pNL4.3 Env-Luc and the pcDNA3.1 plasmid expressing the wild-type (wt) or mutant HIV-1JRFL envelope glycoproteins at a ratio of 2:1. Two days after transfection, the cell supernatants were harvested, and the reverse transcriptase (RT) activities of all viruses were measured as described previously (34). The virus-containing supernatants were stored in aliquots at −80°C.
Infection by single-round luciferase viruses.
Cf2Th-CD4/CCR5 target cells were seeded at a density of 5 × 103 cells/well in 96-well luminometer-compatible tissue culture plates (Corning or PerkinElmer) 24 h before infection. Normalized amounts of viruses (as evaluated by reverse transcriptase activities of the viral stocks) were then added to the target cells, followed by spin infection at 800 × g for 1 h in 96-well plates at 25°C. After spin infection, cells were incubated for 48 h at 37°C, the medium was then removed from each well, and the cells were lysed by the addition of 30 μl of passive lysis buffer (Promega) and three freeze-thaw cycles. An LB 941 TriStar luminometer (Berthold Technologies) was used to measure the luciferase activity of each well after the addition of 100 μl of luciferin buffer (15 mM MgSO4, 15 mM KPO4 [pH 7.8], 1 mM ATP, and 1 mM dithiothreitol) and 50 μl of 1 mM d-luciferin potassium salt (Prolume).
Immunoprecipitation of envelope glycoproteins.
For pulse-labeling experiments, 3 × 105 HEK293T cells were transfected by the calcium phosphate method with HIV-1JRFL envelope expressors (EnvΔCT). One day after transfection, cells were metabolically labeled with 100 μCi/ml [35S]methionine-cysteine (35S protein labeling mix; Perkin-Elmer) in Dulbecco’s modified Eagle’s medium lacking methionine and cysteine and supplemented with 5% dialyzed fetal bovine serum. After 24 h, supernatants were recovered, and cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (140 mM NaCl, 8 mM Na2HPO4, 2 mM NaH2PO4, 1% IGEPAL CA-630, 0.05% sodium dodecyl sulfate [SDS], 0.5% sodium deoxycholate [DOC]). Precipitation of radiolabeled HIV-1JRFL envelope glycoproteins from cell lysates or supernatants was performed with a mixture of sera from HIV-1-infected individuals at 4°C for 1 h at 37°C in the presence of 45 μl of 10% protein A-Sepharose beads (American BioSciences). The beads were then washed twice with RIPA buffer. Laemmli buffer with β-mercaptoethanol was added to the beads before heating them at 100°C for 5 min and loaded onto SDS-PAGE polyacrylamide gels. After migration, gels were dried with a model 583 gel dryer (Bio-Rad, Hercules, CA, USA) and exposed in a storage phosphor cassette. Densitometry data were acquired with a Typhoon Trio variable-mode imager (Amersham Biosciences) in storage phosphor acquisition mode. Results were analyzed using ImageQuant 5.2 (Molecular Dynamics) (36).
Processing and association indices were determined by precipitation of radiolabeled cell lysates and supernatants with mixtures of sera from HIV-1-infected individuals. The association index is a measure of the ability of the mutant gp120 molecule to remain associated with the Env trimer complex on the expressing cell relative to that of the wt Env trimers. The association index was calculated with the following formula: association index = ([mutant gp120]cell/[mutant gp120]supernatant)/([wt gp120]cell/[wt gp120]supernatant). The processing index is a measure of the proteolytic processing of the mutant gp160 Env precursor to mature gp120 relative to that of the wild-type Env trimers. The processing index was calculated with the following formula: processing index = ([total gp120]mutant/[gp160]mutant)/([total gp120]wt/[gp160]wt).
Cell-based ELISA and trimer mixing.
The detection of trimeric HIV-1JRFL EnvΔCT at the surface of HOS cells was performed by a cell-based ELISA, as previously described (22, 24). Briefly, HOS cells were seeded in T-25 flasks (2 × 106 cells per flask) and transfected the next day with a total of 12 μg of pcDNA3.1 expressing codon-optimized HIV-1JRFL EnvΔCT per flask using the standard polyethylenimine (PEI; Polysciences Inc., PA, USA) transfection method. For the trimer mixing approach, transfection was done with 12 μg of wt Env (3:0 [wt/mutant]), 8 μg wt Env and 4 μg mutant Env (2:1), 4 μg wt Env and 8 μg mutant Env (1:2), or 12 μg mutant Env (0:3). Twenty-four hours after transfection, cells were plated in 384-well plates (2 × 104 cells per well). One day later, cells were incubated in blocking buffer (washing buffer [25 mM Tris {pH 7.5}, 1.8 mM CaCl2, 1.0 mM MgCl2, and 140 mM NaCl] supplemented with 10 mg/ml nonfat dry milk and 5 mM Tris [pH 8.0]) for 30 min and then coincubated for 1 h with either the broadly neutralizing Ab (bNAb) 2G12, the anti-CoRBS 17b Ab, the soluble CD4 (sCD4)-immunoglobin fusion protein (CD4-Ig) (1 μg/ml), or the HRP-conjugated anti-cluster A N5-i5 Ab (N5-i5-HRP) in the presence or absence of (+)-BNM-III-170 (50 μM), sCD4 (3 μg/ml), or the vehicle (DMSO and PBS) diluted in blocking buffer. Cells were then washed five times with blocking buffer and five times with washing buffer. A secondary HRP-conjugated antibody specific for the Fc region of human IgG (Pierce) was then incubated to reveal 17b and CD4-Ig binding. N5-i5 binding did not require the addition of a secondary antibody as it was already conjugated to HRP (N5-i5-HRP). Of note, the signals obtained with the N5-i5-HRP-conjugated antibody are lower than those obtained when using a secondary HRP-conjugated antibody to reveal binding (not shown).
Secondary antibody was added for 45 min and then washed again as described above. All incubations were done at room temperature. To measure the HRP enzyme activity, 20 μl of a 1:1 mix of Western Lightning oxidizing and enhanced luminol reagents (Perkin Elmer Life Sciences) was added to each well. The chemiluminescence signal was acquired for 1 s/well with the LB 941 TriStar luminometer (Berthold Technologies).
Purification of recombinant sCD4.
For four-domain sCD4 production, FreeStyle 293F cells (Invitrogen) were grown in FreeStyle 293F medium (Invitrogen) to a density of 1 × 106 cells/ml at 37°C with 8% CO2, with regular agitation (125 rpm). Cells were transfected with a codon-optimized plasmid expressing sCD4 using the 293Fectin reagent, as directed by the manufacturer (Invitrogen). One week later, the cells were pelleted and discarded. The supernatants were filtered (0.22-μm-pore-size filter) (Corning), and sCD4 was purified using nickel affinity columns, as directed by the manufacturer (Invitrogen) (35).
Statistical analyses.
Statistics were analyzed using GraphPad Prism version 6.01 (GraphPad, San Diego, CA, USA). Every data set was tested for statistical normality, and this information was used to apply the appropriate (parametric or nonparametric) statistical test. P values of <0.05 were considered significant (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
ACKNOWLEDGMENTS
This work was supported by NIH grant R01 AI150322-01 to A.F. This work was also supported by CIHR Foundation grant number 352417 to A.F., NIH grant R01 AI129769 to M.P. and A.F., grant R01 AI116274 to M.P., and grant P01-GM56550/AI150741 to A.B.S. and A.F. A.F. is the recipient of a Canada Research Chair on Retroviral Entry. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We have no conflicts of interest to report.
The views expressed in this presentation are those of the authors and do not reflect the official policy or position of the Uniformed Services University, the U.S. Army, the Department of Defense, or the U.S. Government.
REFERENCES
- 1.Helseth E, Olshevsky U, Furman C, Sodroski J. 1991. Human immunodeficiency virus type 1 gp120 envelope glycoprotein regions important for association with the gp41 transmembrane glycoprotein. J Virol 65:2119–2123. doi: 10.1128/JVI.65.4.2119-2123.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang X, Mahony E, Holm GH, Kassa A, Sodroski J. 2003. Role of the gp120 inner domain beta-sandwich in the interaction between the human immunodeficiency virus envelope glycoprotein subunits. Virology 313:117–125. doi: 10.1016/s0042-6822(03)00273-3. [DOI] [PubMed] [Google Scholar]
- 3.Finzi A, Xiang SH, Pacheco B, Wang L, Haight J, Kassa A, Danek B, Pancera M, Kwong PD, Sodroski J. 2010. Topological layers in the HIV-1 gp120 inner domain regulate gp41 interaction and CD4-triggered conformational transitions. Mol Cell 37:656–667. doi: 10.1016/j.molcel.2010.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Munro JB, Gorman J, Ma X, Zhou Z, Arthos J, Burton DR, Koff WC, Courter JR, Smith AB, III, Kwong PD, Blanchard SC, Mothes W. 2014. Conformational dynamics of single HIV-1 envelope trimers on the surface of native virions. Science 346:759–763. doi: 10.1126/science.1254426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herschhorn A, Ma X, Gu C, Ventura JD, Castillo-Menendez L, Melillo B, Terry DS, Smith AB, III, Blanchard SC, Munro JB, Mothes W, Finzi A, Sodroski J. 2016. Release of gp120 restraints leads to an entry-competent intermediate state of the HIV-1 envelope glycoproteins. mBio 7:e01598-16. doi: 10.1128/mBio.01598-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma X, Lu M, Gorman J, Terry DS, Hong X, Zhou Z, Zhao H, Altman RB, Arthos J, Blanchard SC, Kwong PD, Munro JB, Mothes W. 2018. HIV-1 Env trimer opens through an asymmetric intermediate in which individual protomers adopt distinct conformations. Elife 7:e34271. doi: 10.7554/eLife.34271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alsahafi N, Bakouche N, Kazemi M, Richard J, Ding S, Bhattacharyya S, Das D, Anand SP, Prevost J, Tolbert WD, Lu H, Medjahed H, Gendron-Lepage G, Ortega Delgado GG, Kirk S, Melillo B, Mothes W, Sodroski J, Smith AB, III, Kaufmann DE, Wu X, Pazgier M, Rouiller I, Finzi A, Munro JB. 2019. An asymmetric opening of HIV-1 envelope mediates antibody-dependent cellular cytotoxicity. Cell Host Microbe 25:578–587.e5. doi: 10.1016/j.chom.2019.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu M, Ma X, Castillo-Menendez LR, Gorman J, Alsahafi N, Ermel U, Terry DS, Chambers M, Peng D, Zhang B, Zhou T, Reichard N, Wang K, Grover JR, Carman BP, Gardner MR, Nikic-Spiegel I, Sugawara A, Arthos J, Lemke EA, Smith AB, III, Farzan M, Abrams C, Munro JB, McDermott AB, Finzi A, Kwong PD, Blanchard SC, Sodroski JG, Mothes W. 2019. Associating HIV-1 envelope glycoprotein structures with states on the virus observed by smFRET. Nature 568:415–419. doi: 10.1038/s41586-019-1101-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Q, Finzi A, Sodroski J. 2020. The conformational states of the HIV-1 envelope glycoproteins. Trends Microbiol 28:655–667. doi: 10.1016/j.tim.2020.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Veillette M, Coutu M, Richard J, Batraville LA, Dagher O, Bernard N, Tremblay C, Kaufmann DE, Roger M, Finzi A. 2015. The HIV-1 gp120 CD4-bound conformation is preferentially targeted by antibody-dependent cellular cytotoxicity-mediating antibodies in sera from HIV-1-infected individuals. J Virol 89:545–551. doi: 10.1128/JVI.02868-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Decker JM, Bibollet-Ruche F, Wei X, Wang S, Levy DN, Wang W, Delaporte E, Peeters M, Derdeyn CA, Allen S, Hunter E, Saag MS, Hoxie JA, Hahn BH, Kwong PD, Robinson JE, Shaw GM. 2005. Antigenic conservation and immunogenicity of the HIV coreceptor binding site. J Exp Med 201:1407–1419. doi: 10.1084/jem.20042510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arias JF, Heyer LN, von Bredow B, Weisgrau KL, Moldt B, Burton DR, Rakasz EG, Evans DT. 2014. Tetherin antagonism by Vpu protects HIV-infected cells from antibody-dependent cell-mediated cytotoxicity. Proc Natl Acad Sci U S A 111:6425–6430. doi: 10.1073/pnas.1321507111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alvarez RA, Hamlin RE, Monroe A, Moldt B, Hotta MT, Rodriguez Caprio G, Fierer DS, Simon V, Chen BK. 2014. HIV-1 Vpu antagonism of tetherin inhibits antibody-dependent cellular cytotoxic responses by natural killer cells. J Virol 88:6031–6046. doi: 10.1128/JVI.00449-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Veillette M, Desormeaux A, Medjahed H, Gharsallah NE, Coutu M, Baalwa J, Guan Y, Lewis G, Ferrari G, Hahn BH, Haynes BF, Robinson JE, Kaufmann DE, Bonsignori M, Sodroski J, Finzi A. 2014. Interaction with cellular CD4 exposes HIV-1 envelope epitopes targeted by antibody-dependent cell-mediated cytotoxicity. J Virol 88:2633–2644. doi: 10.1128/JVI.03230-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alsahafi N, Ding S, Richard J, Markle T, Brassard N, Walker B, Lewis GK, Kaufmann DE, Brockman MA, Finzi A. 2016. Nef proteins from HIV-1 elite controllers are inefficient at preventing antibody-dependent cellular cytotoxicity. J Virol 90:2993–3002. doi: 10.1128/JVI.02973-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Richard J, Veillette M, Brassard N, Iyer SS, Roger M, Martin L, Pazgier M, Schon A, Freire E, Routy JP, Smith AB, III, Park J, Jones DM, Courter JR, Melillo BN, Kaufmann DE, Hahn BH, Permar SR, Haynes BF, Madani N, Sodroski JG, Finzi A. 2015. CD4 mimetics sensitize HIV-1-infected cells to ADCC. Proc Natl Acad Sci U S A 112:E2687–E2694. doi: 10.1073/pnas.1506755112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richard J, Prevost J, von Bredow B, Ding S, Brassard N, Medjahed H, Coutu M, Melillo B, Bibollet-Ruche F, Hahn BH, Kaufmann DE, Smith AB, III, Sodroski J, Sauter D, Kirchhoff F, Gee K, Neil SJ, Evans DT, Finzi A. 2017. BST-2 expression modulates small CD4-mimetic sensitization of HIV-1-infected cells to antibody-dependent cellular cytotoxicity. J Virol 91:e00219-17. doi: 10.1128/JVI.00219-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ding S, Veillette M, Coutu M, Prevost J, Scharf L, Bjorkman PJ, Ferrari G, Robinson JE, Sturzel C, Hahn BH, Sauter D, Kirchhoff F, Lewis GK, Pazgier M, Finzi A. 2016. A highly conserved residue of the HIV-1 gp120 inner domain is important for antibody-dependent cellular cytotoxicity responses mediated by anti-cluster A antibodies. J Virol 90:2127–2134. doi: 10.1128/JVI.02779-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richard J, Veillette M, Batraville LA, Coutu M, Chapleau JP, Bonsignori M, Bernard N, Tremblay C, Roger M, Kaufmann DE, Finzi A. 2014. Flow cytometry-based assay to study HIV-1 gp120 specific antibody-dependent cellular cytotoxicity responses. J Virol Methods 208:107–114. doi: 10.1016/j.jviromet.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 20.Richard J, Pacheco B, Gohain N, Veillette M, Ding S, Alsahafi N, Tolbert WD, Prevost J, Chapleau JP, Coutu M, Jia M, Brassard N, Park J, Courter JR, Melillo B, Martin L, Tremblay C, Hahn BH, Kaufmann DE, Wu X, Smith AB, III, Sodroski J, Pazgier M, Finzi A. 2016. Co-receptor binding site antibodies enable CD4-mimetics to expose conserved anti-cluster A ADCC epitopes on HIV-1 envelope glycoproteins. EBioMedicine 12:208–218. doi: 10.1016/j.ebiom.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anand SP, Prevost J, Baril S, Richard J, Medjahed H, Chapleau JP, Tolbert WD, Kirk S, Smith AB, III, Wines BD, Kent SJ, Hogarth PM, Parsons MS, Pazgier M, Finzi A. 2019. Two families of Env antibodies efficiently engage Fc-gamma receptors and eliminate HIV-1-infected cells. J Virol 93:e01823-18. doi: 10.1128/JVI.01823-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Veillette M, Coutu M, Richard J, Batraville LA, Desormeaux A, Roger M, Finzi A. 2014. Conformational evaluation of HIV-1 trimeric envelope glycoproteins using a cell-based ELISA assay. J Vis Exp 2014:51995. doi: 10.3791/51995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richard J, Veillette M, Ding S, Zoubchenok D, Alsahafi N, Coutu M, Brassard N, Park J, Courter JR, Melillo B, Smith AB, III, Shaw GM, Hahn BH, Sodroski J, Kaufmann DE, Finzi A. 2016. Small CD4 mimetics prevent HIV-1 uninfected bystander CD4+ T cell killing mediated by antibody-dependent cell-mediated cytotoxicity. EBioMedicine 3:122–134. doi: 10.1016/j.ebiom.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ding S, Grenier MC, Tolbert WD, Vezina D, Sherburn R, Richard J, Prevost J, Chapleau JP, Gendron-Lepage G, Medjahed H, Abrams C, Sodroski J, Pazgier M, Smith AB, III, Finzi A. 2019. A new family of small-molecule CD4-mimetic compounds contacts highly conserved aspartic acid 368 of HIV-1 gp120 and mediates antibody-dependent cellular cytotoxicity. J Virol 93:e01325-19. doi: 10.1128/JVI.01325-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tolbert WD, Sherburn RT, Van V, Pazgier M. 2019. Structural basis for epitopes in the gp120 cluster A region that invokes potent effector cell activity. Viruses 11:69. doi: 10.3390/v11010069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tolbert WD, Gohain N, Alsahafi N, Van V, Orlandi C, Ding S, Martin L, Finzi A, Lewis GK, Ray K, Pazgier M. 2017. Targeting the late stage of HIV-1 entry for antibody-dependent cellular cytotoxicity: structural basis for Env epitopes in the C11 region. Structure 25:1719–1731.e4. doi: 10.1016/j.str.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gohain N, Tolbert WD, Acharya P, Yu L, Liu T, Zhao P, Orlandi C, Visciano ML, Kamin-Lewis R, Sajadi MM, Martin L, Robinson JE, Kwong PD, DeVico AL, Ray K, Lewis GK, Pazgier M. 2015. Cocrystal structures of antibody N60-i3 and antibody JR4 in complex with gp120 define more cluster A epitopes involved in effective antibody-dependent effector function against HIV-1. J Virol 89:8840–8854. doi: 10.1128/JVI.01232-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Acharya P, Tolbert WD, Gohain N, Wu X, Yu L, Liu T, Huang W, Huang CC, Kwon YD, Louder RK, Luongo TS, McLellan JS, Pancera M, Yang Y, Zhang B, Flinko R, Foulke JS, Jr, Sajadi MM, Kamin-Lewis R, Robinson JE, Martin L, Kwong PD, Guan Y, DeVico AL, Lewis GK, Pazgier M. 2014. Structural definition of an antibody-dependent cellular cytotoxicity response implicated in reduced risk for HIV-1 infection. J Virol 88:12895–12906. doi: 10.1128/JVI.02194-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu J, Bartesaghi A, Borgnia MJ, Sapiro G, Subramaniam S. 2008. Molecular architecture of native HIV-1 gp120 trimers. Nature 455:109–113. doi: 10.1038/nature07159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rasheed M, Bettadapura R, Bajaj C. 2015. Computational refinement and validation protocol for proteins with large variable regions applied to model HIV Env spike in CD4 and 17b bound state. Structure 23:1138–1149. doi: 10.1016/j.str.2015.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anand SP, Finzi A. 2019. Understudied factors influencing Fc-mediated immune responses against viral infections. Vaccines (Basel) 7:103. doi: 10.3390/vaccines7030103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tolbert WD, Sherburn R, Gohain N, Ding S, Flinko R, Orlandi C, Ray K, Finzi A, Lewis GK, Pazgier M. 2020. Defining rules governing recognition and Fc-mediated effector functions to the HIV-1 co-receptor binding site. BMC Biol 18:91. doi: 10.1186/s12915-020-00819-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Melillo B, Liang S, Park J, Schon A, Courter JR, LaLonde JM, Wendler DJ, Princiotto AM, Seaman MS, Freire E, Sodroski J, Madani N, Hendrickson WA, Smith AB, III. 2016. Small-molecule CD4-mimics: structure-based optimization of HIV-1 entry inhibition. ACS Med Chem Lett 7:330–334. doi: 10.1021/acsmedchemlett.5b00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rho HM, Poiesz B, Ruscetti FW, Gallo RC. 1981. Characterization of the reverse transcriptase from a new retrovirus (HTLV) produced by a human cutaneous T-cell lymphoma cell line. Virology 112:355–360. doi: 10.1016/0042-6822(81)90642-5. [DOI] [PubMed] [Google Scholar]
- 35.Finzi A, Pacheco B, Zeng X, Kwon YD, Kwong PD, Sodroski J. 2010. Conformational characterization of aberrant disulfide-linked HIV-1 gp120 dimers secreted from overexpressing cells. J Virol Methods 168:155–161. doi: 10.1016/j.jviromet.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coutu M, Finzi A. 2015. HIV-1 gp120 dimers decrease the overall affinity of gp120 preparations for CD4-induced ligands. J Virol Methods 215–216:37–44. doi: 10.1016/j.jviromet.2015.02.017. [DOI] [PubMed] [Google Scholar]
- 37.LaBonte JA, Patel T, Hofmann W, Sodroski J. 2000. Importance of membrane fusion mediated by human immunodeficiency virus envelope glycoproteins for lysis of primary CD4-positive T cells. J Virol 74:10690–10698. doi: 10.1128/jvi.74.22.10690-10698.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]