

Abstract
BACKGROUND:
Preeclampsia is characterized by a new onset of hypertension during pregnancy and is associated with autoantibodies against the angiotensin II type 1 receptor and oxidative stress. There is growing evidence for mitochondrial dysfunction in preeclampsia, however, the culprits for mitochondrial dysfunction are still being defined. We previously demonstrated that angiotensin II type 1 autoantibodies cause renal, placental, and endothelial mitochondrial dysfunction in pregnant rats. However, the role of the angiotensin II type 1 autoantibodies in endothelial mitochondrial function in response to sera from preeclamptics is unknown. Thus, we hypothesized that circulating factors, such as the angiotensin II type 1 autoantibodies, during preeclampsia would negatively impact the vascular endothelial mitochondrial function in human umbilical vein endothelial cells.
OBJECTIVE:
The objective of the study was to determine a role for circulating angiotensin II type 1 autoantibodies to cause endothelial mitochondrial reactive oxygen species and dysfunction in preeclampsia compared to normal pregnant controls.
STUDY DESIGN:
Immediately after delivery, sera was collected from preeclamptic patients and normal pregnant controls. The mitochondrial reactive oxygen species were determined from the cells treated overnight with 10% sera from either the control or preeclamptic patients with and without the antiotension II type 1 autoantibodies inhibitor peptide (‘n7AAc’).
RESULTS:
Preeclampsia patients at <34 weeks’ gestation exhibited an elevated mean arterial blood pressure. Cells treated with serum from the preeclampsia patients at <34 weeks gestational age showed significantly (P<0.05) greater mitochondrial oxidative stress and reduced respiration than cells treated with the control sera, and these abnormalities were restored with ‘n7AAc’.
CONCLUSION:
This study demonstrates that endothelial mitochondrial dysfunction occurs in response to circulating factors, especially in response to serum from preterm preeclampsia patients, and can be restored by blocking circulating angiotensin II type 1 autoantibodies, thereby indicating a potential new therapeutic target for preeclampsia.
Keywords: electron transport chain, endothelial cells, mitochondria, oxidative stress, placenta, preeclampsia, reactive oxygen species
Introduction
Preeclampsia (PE) is a pregnancy-specific syndrome that is primarily defined by the development of new-onset hypertension and organ dysfunction, with or without proteinuria, after 20 weeks of gestation.1-4 Approximately 10% of pregnancies in the United States and worldwide are affected by PE annually.1-4 There are no effective treatments available for PE except for delivering the fetus and placenta. Despite a considerable amount of research done in the field, the molecular mechanisms underlying the pathogenesis of PE are still unclear. Oxidative stress, a state of imbalance between reactive oxygen species (ROS) and the antioxidant defenses, has been shown to be positively associated with the pathology.5-9 ROS is an umbrella term used to describe a wide range of oxidant molecules such as the free radicals superoxide (O2.−), the hydroxyl radical (·OH), and peroxynitrite (ONOO−).6 When ROS and hydrogen peroxide (H2O2) (a nonradical) are released in excess, protein, DNA, and RNA could be damage, which ultimately leads to cellular dysfunction and death.9 Mitochondria are dynamic organelles known as the powerhouses of cells because they account for 90% of the energy production within the cell. In addition, mitochondria play an important role in calcium signaling, apoptosis, and redox signaling.10 ROS produced within the mitochondria play a crucial role in physiological signaling. However, an excessive generation of ROS could cause oxidative stress and cell death.11 The major site of ROS production within the mitochondria is the electron transport chain (ETC) located on the inner mitochondrial membrane. With the ETC being the site where electron transfer occurs through the complexes, any abnormality within the chain would cause leakage of the electrons, thereby affecting respiration which could lead to excessive ROS production.12
Agonistic autoantibodies against the angiotensin II type 1 (AT1) receptor were found to be elevated in pre-eclamptic patients and in reduced uterine perfusion pressure (RUPP) rats, a model of PE.13,14 Both rat and human AT1 autoantibodies (AT1-AAs) have a high binding affinity to an amino acid sequence composed of 7 residues (AFHYESQ) that are located on the second extracellular loop of the AT1 receptor. Therefore, we recently generated an inhibitory peptide (‘n7AAc’) to prevent AT1-AAs from binding to and activating the AT1 receptor.15 This inhibitory peptide contains the 7 amino acid sequence along with protein capping of the N and C terminus of the peptide, a process that is commonly used to increase a peptide half-life and to protect exogenous peptides from protein lysis and degradation. In previous studies we showed that ‘n7AAc’ binds to circulating AT1-AAs to inhibit the AT1-AAs from binding to the AT1 receptor.15 We have shown recently that ‘n7AAc’ decreases blood pressure, vasoactive factors, cytolytic natural killer cells, and mitochondrial oxidative stress, thereby providing an improved outcome in response to placental ischemia in the pregnant RUPP rat model of PE.16 Although ‘n7AAc’ improved the outcomes in animal models of PE, it is unknown if it could improve factors associated with PE in patients.
The evidence for mitochondrial oxidative stress in PE has been reported in a number of studies that demonstrated placental mitochondrial morphologic changes, reduced ETC activity, and alterations in the mitochondrial DNA and mitochondrial biogenesis in trophoblasts17-23 Moreover, changes in the H2O2 levels and respiration have been noted among either early onset or late onset PE.24,25 Collectively, these studies suggest an important role for impaired placental mitochondrial function in contributing to the oxidative stress in PE. However, the mechanisms linking placental mitochondrial dysfunction to endothelial mitochondrial dysfunction caused by factors found in the circulation of women with PE, such as the AT1-AAs, are not clearly defined. Our previous work indicates that mitochondrial dysfunction and oxidative stress cause hypertension in response to placental ischemia in the RUPP rat model of PE.26 Moreover, we have recently reported a role for AT1-AAs in mitochondrial dysfunction and hypertension in response to placental ischemia.16 Thus, the objective of the current study was to determine if circulating factors, such as the AT1-AAs, from the PE patients impair vascular endothelial cell mitochondrial function, indicated by mitochondrial oxidative stress and impaired respiration, and if this could be attenuated with ‘n7AAc’.
Materials and Methods
Reagents
OIigomycin (O4876), FCCP (C2920), rotenone (R8875), antimycin A (A8674), and M199 (M4530) were purchased from Sigma Aldrich (St. Louis, MO). DMEM (10566-016), fetal bovine serum (FBS) (16000044), antimycotic and antibiotic (15240062), and MitoSOX Red (M36008) were purchased from Thermo Fisher Scientific (Waltham, MA). Human umbilical vein endothelial cells (HUVECs) were purchased from ATCC (Manassas, VA).
Patient recruitment and sample collection
The study participants were recruited from the Department of Obstetrics and Gynecology, University of Mississippi Medical Center, Jackson, Mississippi. The research protocol was approved by the University of Mississippi Medical Center Institutional Review Board, and all participants provided informed consent.
All the women that were included were pregnant with singleton gestations and underwent a cesarean or vaginal delivery performed for the usual obstetrical indications. PE was defined as the development of de novo hypertension (ie, a blood pressure of ≥140/90 mm Hg) and proteinuria (ie, protein at a concentration of >300 mg/24 h or a +1 classification on a repeat dipstick). We excluded women with multiple gestations, fetal anomalies, those diagnosed with gestational diabetes or other preexisting medical conditions such as diabetes, chronic hypertension, sickle cell disease, lupus, other inflammatory disorders, or sexually transmitted infections, and those that used tobacco or other abused substances during pregnancy.
The obstetrical patients were admitted to the Winfred Wiser Hospital for Women and Infants for delivery because of usual obstetrical indications. All patients consented to have their blood drawn before delivery, and for use of their discarded placental tissue. The patients underwent blood collection by means of venipuncture into BD Vacutainer whole blood collection tubes (Fisher) for serum. Following the manufacturer instructions, the samples were allowed to clot and then centrifuged for 10 minutes at 3200 rpm at 4°C and the resultant serum was stored at −80°C.26,27 Subsequently, each patient’s serum was individually stored in 1 mL aliquots in microcentrifuge tubes for future use in the assays.
Human umbilical vein endothelial cell culture
To determine if circulating factors from the PE patients impair vascular endothelial cell mitochondrial function, HUVECs were grown to 70% confluency on gelatin coated T25 culture flasks in a humidified atmosphere of 5% CO2 at 37°C in HUVEC media (Medium 199: DMEM [50:50], 10% FBS, and 1% antimycotic and antibiotic). At passage 4, the cells were cultured in the appropriated T25 flasks or 6-well plates for additional experiments.
Mitochondrial respiration in human umbilical vein endothelial cells
Respiration was measured in the HUVECs following overnight incubation with 10% serum from either the control or PE groups. After the HUVECs were grown to 70% confluency in T25 culture flasks, the cells were serum starved for 4 hours before overnight incubation with 10% serum from either the control or PE groups. Serum is a complex mix of growth factors and hormones involved in growth promotion and specialized cell function, and therefore the role of serum in cell culture is very complex, but typically normal growth media contains 2% to 10% of serum.28 Castells-Sata et al29 tested a human plasma supplement as a substitute for FBS in primary human vascular cells culture, and the results indicated that they were able to support proliferation, preserve cellular morphology, and functionality similarly to FBS at a concentration of 5% and 10% human plasma or FBS in viability assays. Moreover, we have previously shown that this concentration of serum from RUPP rats causes activation of HUVECs, resulting in increased endothelin 1 and mitochondrial ROS.26,30 Therefore, 10% human serum was used in the experimental media for each patients respective wells used in this assay. The following day, the experimental serum was removed, and serum-free medium was added for an additional 4 hours. The cells were harvested and counted and used to measure respiration using the Oxygraph-2K (Innsbruck, Austria). Basal (cells only), leak (induced by oligomycin at a dose of 2 μg/mL), and maximal respiration (induced by 2 μL of a 1 mM solution of carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone [FCCP]; titrations were performed until the maximal response was achieved) were measured, and after FCCP titration, rotenone (0.5 μM) and antimycin A (2.5 μM) were injected to measure nonmitochondrial respiration. All the data were normalized to 1×106 cells.15,16,26
Mitochondrial mediated reactive oxygen species production in human umbilical vein endothelial cells
Mitochondrial-specific ROS production was measured using MitoSOX red, a fluorogenic dye specifically targeted to the mitochondria in live cells. Briefly, HUVECs were grown to 70% confluency in 6-well culture plates. The cells were serum starved for 4 hours before overnight incubation with 10% serum from either the control or PE groups with or without the AT1-AA inhibitory peptide (ie, ’n7AAc’ at a dose of 2 μg/mL in saline).15,26 The experimental serum was removed and the cells were incubated with MitoSOX red (5 μM) for 30 minutes at 37°C. Antimycin A (100 μM) was included in the experiment as a positive control. Serum-free medium was added and the cells were incubated for an additional 4 hours. The cells were collected and analyzed in the FL2 and FL3 channels of a Gallios flow cytometer (Beckman Coulter, Brea, CA). At least 5000 events for each sample were collected for analysis.
Statistical analysis
All data are expressed as mean±standard error (SEM), whereas the table and figures depict the mean±standard deviation. One-way analysis of variance or Student’s t tests were performed for statistical comparisons between the groups. A value of P<.05 was considered statistically significant.
Results
Patient demographics
We have included 8 normal pregnant women (control) and 10 women with PE (ie, 6 patients with PE at <34 weeks and 4 patients with PE at >34 weeks gestational age). The demographic and delivery information can be found in the Table. The maternal age was all within a similar range of 26 to 27 years old across all the groups. The maternal body mass index was not significantly different among the groups. Importantly, the gestational age was significantly lower in the PE of <34 weeks’ gestation group than the control women and the PE of >34 weeks’ gestation group. The fetal weight decreased in the PE of <34 weeks’ gestation group when compared with the control women or the PE of >34 weeks’ gestation group. The mean arterial pressure in the patients with PE was significantly higher both at admittance and delivery in the PE of <34 weeks’ gestation group (110±6 mm Hg; P<.05; n=6) than the control group (95±2 mm Hg; n=8) (Figure 1).
TABLE.
Patient demographics
| Demographic | Control (n=8) | PE<34 (n=6) | PE>34 (n=4) |
|---|---|---|---|
| Age, (y) | 26.0±2.03 | 26.33±2.06 | 26.0±2.04 |
| Ethnicity, n (%) | |||
| White | — | 2 (33.3) | 1 (25.0) |
| African American | 8 (100) | 4 (66.7) | 3 (75.0) |
| BMI, (kg/m2) | 31.16±0.94 | 36.31±3.91 | 33.67±3.24 |
| Delivery | |||
| Gestational age, (wk) | 39.06±0.03 | 32.13±2.21a | 38.48±0.42 |
| Platelets (1 × 103/mL) | 251.0±21.0 | 153.0±4.0a | 203.7±27.39 |
| Fetal weight, (g) | 3183±184.9 | 1715±353.6a | 2941 ±154.0 |
| Route of deliver, n (%) | |||
| Vaginal delivery | 1 (12.5) | 2 (33.3) | 4 (100) |
| Cesarean delivery | 7 (87.5) | 4 (66.7) | — |
| Fetal gender, n (%) | |||
| Male | 3 (37.5) | 2 (33.3) | 2 (50.0) |
| Female | 5 (62.5) | 4 (66.7) | 2 (50.0) |
| Blood pressure (mm Hg) | |||
| MAP at presentation | 94.5±2.67 | 110±6.04a | 100.2±4.52 |
| MAP at delivery | 93.5±3.77 | 104.3±3.62 | 97.0±5.12 |
The data for each group are expressed as mean±standard deviation. One-way analysis of variance or Student’s t test were performed for statistical comparisons between the groups.
MAP, mean arterial blood pressure; PE, preeclampsia.
A value of P<.05 was considered statistically significant.
FIGURE 1. Differences in the MAP at presentation.
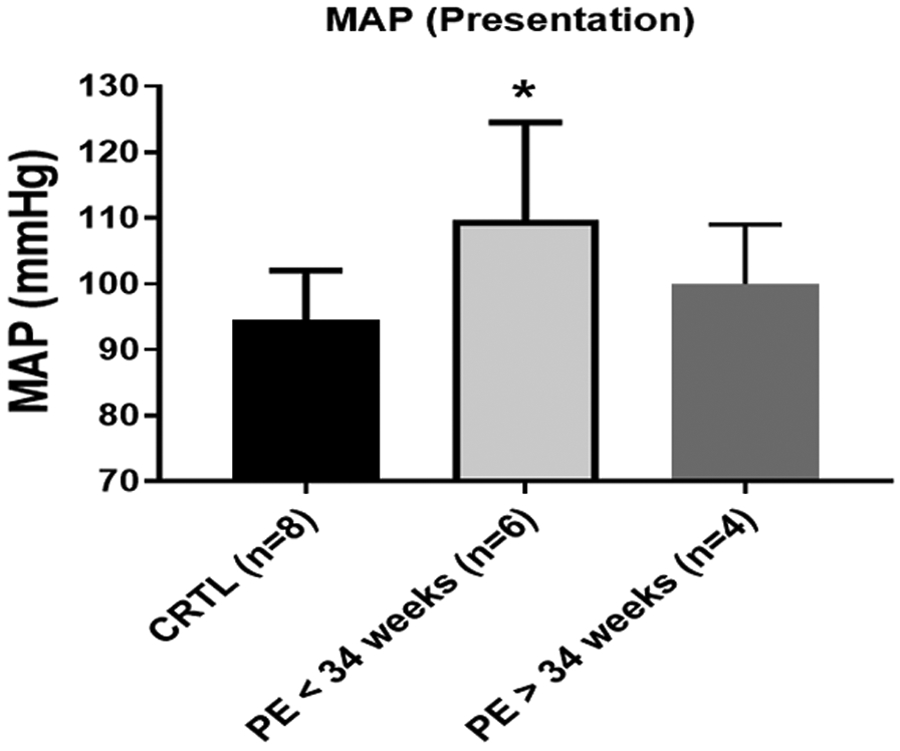
The control patients (n=8) and PE patients (n=10) (PE patients at <34 weeks [n=6] and PE patients at >34 weeks [n=4]), as separated by the serum samples used for the cell studies, displayed significant differences in the mean arterial blood pressure at presentation. The data are presented as mean ± standard deviation. The asterisk indicates a significant (P<.05) difference from the control group.
CRTL, control; MAP, mean arterial blood pressure; PE, preeclampsia.
Sera from the preeclampsia patients reduced endothelial cell respiration and increased mitochondrial reactive oxygen species production in vitro
Overnight incubation of the HUVECs with 10% serum from PE of <34 weeks’ gestation group significantly reduced the maximal respiration rate to 20.40±3.83 pmol O2 per million cells (n=6; P<.05), compared with the rate of 49.11±8.61 pmol O2 per million cells (n=8) that was seen in the HUVECs treated with the serum from the control patients (Figure 2, A). HUVECs incubated with 10% serum from PE patients of >34 weeks’ gestation reduced the maximal respiration rate to 46.5±17.13 pmol O2 per million cells (n=4), which was not significantly different when compared with the HUVECS treated with serum from the control patients (Figure 2, A). Likewise, HUVECs treated with 10% serum from the PE patients of <34 weeks’ gestation showed increased mitochondrial ROS production (93.25±1.54 percent gated; n=6; P<0.05) when compared with the treatment of the HUVECs with the serum from the control patients (83.13±3.38 percent gated; n=8). HUVECs treated with 10% serum from the PE patients of >34 weeks’ gestation showed increased mitochondrial ROS production (88.25±1.71 percent gated; n=4), which was not significantly greater than the serum from the control patients (Figure 2, B). Inhibition of the AT1-AA activity was tested with a specifically designed AT1-AA inhibitor peptide, ‘n7AAc’ to determine if the AT1-Aas were the cause of the increased mitochondrial ROS.24,31 The mitochondrial ROS was reduced with ‘n7Aac’ treatment in the HUVECS treated with serum from the PE patients at < 34 weeks’ gestation to 82.7±0.17 percent gated (n=3; P<.05), when compared with the HUVECS treated with serum from the PE patients at < 34 weeks’ gestation. The mitochondrial ROS was reduced with ‘n7Aac’ treatment in the HUVECS treated with serum from the PE patients at >34 weeks’ gestation to 77.4±1.21 percent gated (n=3; P<.05) in comparison with HUVECS with serum from the PE patients at >34 weeks’ gestation (Figure 3).
FIGURE 2. HUVECs exhibit impaired respiration with elevated mitochondrial ROS.

A, Cell respiration is reduced in HUVECs treated with PE patient serum (PE at <34 weeks’ gestation [n=6]; PE at >34 weeks’ gestation [n=4]). HUVECs treated with 10% preeclamptic serum from PE patients at <34 weeks’ gestation show a significant reduction in the uncoupled respiration rate vs the cells that were treated with nonpreeclamptic serum (n=8). B, The mitochondrial ROS in HUVECs treated with preeclamptic serum (PE at <34 weeks’ gestation [n=6]; PE at >34 weeks’ gestation [n=4]) or with nonpreeclamptic serum (n=8). HUVECs treated with 10% serum from preeclamptic patients at <34 weeks’ gestation showed a significant increase in the mitochondrial ROS production when compared with nonpreeclamptic serum. Data are presented as the mean±standard deviation. The asterisk indicates a significant (P<.05) difference from the control group.
CRTL, control; ET, electron transport; PE, preeclampsia; ROS, reactive oxygen species.
FIGURE 3. AT1-AA blockade attenuates mt ROS in response to PE sera.
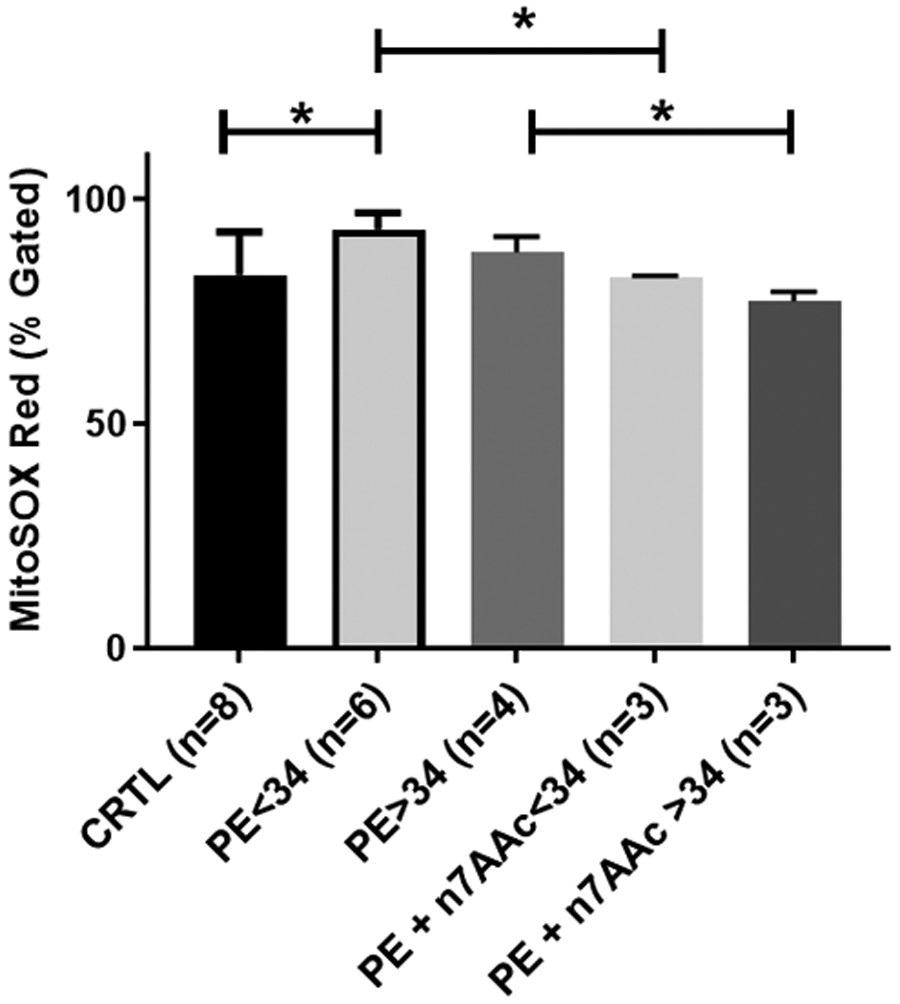
Preeclamptic sera increases mitochondrial ROS when incubated with HUVECs and coincubation with ‘n7AAc’ reverses the mitochondrial ROS production. HUVECs coincubated with pre-eclamptic (at <34 weeks’ gestation or >34 weeks’ gestation) serum and ‘n7AAc’ significantly reverses the mitochondrial ROS production when compared with treatment with preeclamptic (at <34 weeks’ gestaion or >34 weeks’ gestation) serum alone. Data are presented as mean±standard deviation. The asterisk indicates a significant (P<.05) difference from the control group and from the treatments with preeclamptic sera alone.
AT1-AA, angiotensin II type 1 autoantibodies; CRTL, control; mtROS, mitochondrial reactive oxygen species; PE, preeclampsia.
Discussion
Principle findings
Although the relevance of oxidative stress in the pathology of PE has been studied extensively, the results from this study link the reduced vascular endothelial mitochondrial respiration and mitochondrial ROS to the presence of circulating agonistic autoantibodies against the AT1 receptor. In conjunction with the data recently published by McCarthy et al,32 we demonstrated that soluble factors released in association with the placental mitochondrial dysfunction causes vascular mitochondrial ROS accumulation and decreased respiration in HUVECs exposed to the serum from PE patients. Importantly, we demonstrated that blockade of the circulating AT1-Aas in the serum of PE patients completely attenuated the mitochondrial ROS in HUVECs treated with the sera of PE patients. These data support the hypothesis that the endothelial dysfunction associated with PE is caused by AT1-Aas within the circulation, thereby suggesting an additional avenue and treatment option for PE.
Oxidative stress is defined as a state of imbalance between the ROS and antioxidant defenses in the cell.33 ROS are highly reactive free radicals that could damage cellular contents, thereby causing cellular dysfunction and ultimately cell death. Oxidative stress has been shown to be associated with the physiology of normal pregnancies.5,7 However, the magnitude of oxidative stress is further elevated during pregnancy complications such as PE.5,7
Results
In this study, we demonstrate both a decrease in the endothelial cell respiration and an increase in the mitochondrial ROS when HUVECs were exposed to sera from PE women who delivered at <34 weeks gestational age when compared with the cells exposed to sera from nonpreeclamptic women. This is in contrast to the respiration and mitochondrial ROS when endothelial cells were exposed to sera from PE women who delivered at >34 weeks’ gestation, which was not different from what was seen in cells exposed to sera from the nonpreeclamptic women.
Previously published studies reported that ROS can be generated by various sources such as nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, mitochondria, and xanthine oxidase in PE placenta.23,24 The role of NADPH oxidase in causing oxidative stress in PE patients or animal models has been shown in other studies.10,34-39 In fact, we previously published that in the RUPP rat model of PE, the placenta exhibits an elevated NADPH oxidase-mediated ROS production.39 As such, we have started exploring mitochondrial oxidative stress in the preeclamptic pathophysiology. We have previously published similar findings for placental and renal mitochondria from the RUPP rat model of PE and showed that administration of drugs that improved mitochondrial function lowered the blood pressure, thus indicating the importance of proper mitochondrial function during pregnancy.27
The existing literature indicate that mitochondrial dysfunction is evident in the preeclamptic placenta.16-21,31,40-46, In addition, endothelial dysfunction has been well established in PE. Because the endothelium is the innermost layer in the vasculature, it is the first organ system to be exposed to the circulating pathologic factors that are released from other organs including the placenta. Thus, we sought to examine if the circulating AT1-Aas in PE patient sera would alter the mitochondrial function in endothelial cells when treated in vitro. We have previously demonstrated that infusion of AT1-Aas into normal pregnant rats causes excessive mitochondrial ROS production.15 Moreover, blocking the activity of the AT1-Aas with the ‘n7Aac’-specific epitope binding peptide significantly lowered the blood pressure and mitochondrial ROS in response to the AT1-Aas during pregnancy. This novel role of AT1-Aas was further confirmed in a subsequent study, which demonstrated that preeclamptic rats (RUPP) treated with ‘n7Aac’ showed improved placental and renal mitochondrial function and reduced mitochondrial ROS production.26,40 Moreover, when we incubated the HUVECs with serum obtained from the ’n7Aac’ treated RUPP rats, we saw a significant reduction in the mitochondrial ROS when compared with the cells incubated with the RUPP rat serum.16
Clinical implications
These findings identify AT1-Aas as 1 of the circulating factors in the pre-eclamptic sera that drive mitochondrial oxidative stress in the vascular cells, which could be indicative of the peripheral vascular function.
Strengths and limitations
In this study, we showed that the maximal respiration rate was significantly reduced in the HUVECs incubated overnight with 10% serum from the PE patients at <34 weeks’ gestation (Figure 2, A). However, we did not find any significant differences in the basal respiration rate, suggesting that only the maximal capacity of the ETC is compromised in the endothelial cells that are exposed to the circulating factors in the patient sera. In agreement with our findings, McCarthy et al32 reported that 3% PE plasma treatment reduced endothelial cell respiration.32 We utilized MitoSOX red, a fluorogenic dye that measures real-time mitochondrial ROS in live HUVECs when analyzed by flow cytometry. We demonstrate that HUVECs treated with sera from pre-eclamptic patients at <34 weeks’ gestation showed a significant increase in MitoSOX fluorescence in comparison with the cells treated with the nonpreeclamptic sera (Figure 2, B). The mitochondrial ROS in the HUVECs treated with sera from preeclamptic patients at >34 weeks’ gestation was not significantly greater than those treated with the sera from the nonpreeclamptic patients, indicating that preterm PE has an impact on the vascular endothelium, which may be more pathologic when compared with women at an older gestational age. Importantly, similar findings of elevated mitochondrial ROS in HUVECs incubated with 3% patient plasma were published by McCarthy et al,32 in which a different technique was used. Thus, both studies demonstrate that increased mitochondrial ROS is stimulated by circulating factors in response to placental ischemia in PE. Furthermore, based on our previous studies on the role of AT1-AAs in mediating mitochondrial oxidative stress, 15-30,10,31-58 we asked whether blocking AT1-AAs in the serum with ’n7AAc’ would reverse the mitochondrial ROS production in the cells incubated with PE patient serum. We demonstrated that cells coincubated with preeclamptic sera from either <34 weeks or >34 weeks’ gestation and ‘n7AAc’ resulted in a significant attenuation of the MitoSOX fluorescence (Figure 3), suggesting that AT1-AAs mediate mitochondrial ROS production in the endothelial cells, and thereby offering a future treatment avenue for further investigations.
In summary, our study findings, in agreement with others, suggest that soluble factors from preeclamptic patients cause impaired endothelial mitochondrial function when compared with normal pregnancies.
Conclusion
Importantly, we extended the previous findings of McCarthy et al32 by showing that circulating factors from PE patients, such as AT1-AAs, cause endothelial cell mitochondrial oxidative stress and that inhibition of AT1-AA activity is a potential therapeutic strategy for improving the peripheral endothelial function during this disease. This supports the idea that PE is a multifaceted disease resulting from the dysfunction of many organs. Moreover, these studies indicate the importance of finding novel therapies that might affect mitochondrial health and function by preventing upstream factors from causing cellular stress and dysfunction.
Perspectives
We showed that AT1-AAs is an important factor present in the circulation of PE patients that impair mitochondrial function in the vascular endothelium. The existing literature on the role of mitochondrial ROS in the pathology of PE is compelling, however, the knowledge on the pathologic factors or mechanisms that drive mitochondrial ROS is limited. In the previous few years, we and other investigators uncovered the potential of circulating factors like AT1-AAs and natural killer cells in causing mitochondrial dysfunction and oxidative stress using animal models of PE. In addition, this study addresses an important question as to whether blocking circulating AT-AA signaling with ’n7AAc’ peptide would attenuate the mitochondrial ROS in endothelial cells. We were successful in showing that coincubation of ’n7AAc’ with PE patient sera significantly reverses the mitochondrial oxidative stress in the endothelial cells of human origin (HUVECs). Although exploring strategies to target mitochondrial oxidative stress offers benefits by reducing mitochondrial ROS and improving mitochondrial function, blockade of AT1-AAs would target and potentially prevent a wide range of downstream effects of AT1-AAs including mitochondrial ROS, which might offer a better therapeutic profile in both the mother and fetus. Finally, our study unveils exciting findings on 1 of the important mechanisms of mitochondrial ROS in PE which will be of great value to the PE scientific community.
AJOG MFM at a Glance.
Why was this study conducted?
Sera from preterm (at <34 weeks’ gestation) preeclampsia (PE) patients cause endothelial mitochondrial dysfunction, which is in contrast with what was seen for sera from PE patients at >34 weeks’ gestation or normal pregnant controls.
Key findings
Blocking peptides specifically designed to inhibit the angiotensin II type 1 autoantibodies (AT1-AAs) from binding to the angiotensin II type 1 (AT 1) receptor restored the endothelial cell mitochondrial function.
What does this add to what is known?
These data suggest that blockade of the AT1-AAs during PE could improve the vascular endothelial cell mitochondrial function.
Acknowledgments
We would like to thank Drs Robert Kramer, Jon Hosler, and Michael Puskarich at the University of Mississippi Medical Center for their assistance with the mitochondrial experiments.
The authors report receiving financial support for the study material from the Department of Pharmacology and Toxicology, University of Mississippi Medical Center, Jackson, Mississippi. E.D. reports receiving a T32 Trainee Grant from the American Heart Association (AHA) under grant number T32-HL105324. V.R.V. reports receiving a Predoctoral fellowship from the AHA under award number 17PRE33660592. D.C.C. reports receiving funding from the National Institutes of Health (NIH) under grant numbers R00HL130456 and P20GM104357. L.M.A. reports receiving financial support from the NIH under grant number P20GM121334 and an early career award from the AHA under award numer 19CDA34670055. M.W.C. reports receiving an early career award from the AHA under award number 18CDA34110264. B.L. reports receiving financial support from the NIH under grant numbers RO1HD067541-06 and P20GM121334.
Footnotes
The authors report no conflict of interest.
Contributor Information
Evangeline Deer, Department of Pharmacology and Toxicology, University of Mississippi Medical Center, Jackson, MS.
V. Ramana Vaka, Department of Pharmacology and Toxicology, University of Mississippi Medical Center, Jackson, MS.
Kristen M. McMaster, Department of Obstetrics and Gynecology, University of Mississippi Medical Center, Jackson, MS.
Kedra Wallace, Department of Obstetrics and Gynecology, University of Mississippi Medical Center, Jackson, MS.
Denise C. Cornelius, Department of Emergency Medicine, University of Mississippi Medical Center, Jackson, MS.
Lorena M. Amaral, Department of Pharmacology and Toxicology, University of Mississippi Medical Center, Jackson, MS.
Mark W. Cunningham, Department of Pharmacology and Toxicology, University of Mississippi Medical Center, Jackson, MS.
Babbette LaMarca, Department of Pharmacology and Toxicology, University of Mississippi Medical Center, Jackson, MS.
References
- 1.American College of Obstetricians and Gynecologists. Task Force on Hypertension in Pregnancy. Washington (DC): The American College of Obstetricians and Gynecologists; 2013:1–99. [DOI] [PubMed] [Google Scholar]
- 2.Roberts JM, Pearson G, Cutler J, Lindheimer M. NHLBI Working Group on Research on Hypertension During pregnancy. Summary of the NHLBI Working Group on research on hypertension during pregnancy. Hypertension 2003;41:437–45. [DOI] [PubMed] [Google Scholar]
- 3.Sibai B, Dekker G, Kupferminc M. Pre-eclampsia. Lancet 2005;365:785–99. [DOI] [PubMed] [Google Scholar]
- 4.Sibai BM, Caritis S, Hauth J. National Institute of Child Health and Human Development Maternal-Fetal Medicine Units Network. What we have learned about preeclampsia. Semin Perinatol 2003;27:239–46. [DOI] [PubMed] [Google Scholar]
- 5.Aouache R, Biquard L, Vaiman D, Miralles F. Oxidative stress in preeclampsia and placental diseases. Int J Mol Sci 2018;19:1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsubara K, Higaki T, Matsubara Y, Nawa A. Nitric oxide and reactive oxygen species in the pathogenesis of preeclampsia. Int J Mol Sci 2015;16:4600–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roberts JM, Hubel CA. Oxidative stress in preeclampsia. Am J Obstet Gynecol 2004;190:1177–8. [DOI] [PubMed] [Google Scholar]
- 8.Sánchez-Aranguren LC, Prada CE, Riaño-Medina CE, Lopez M. Endothelial dysfunction and preeclampsia: role of oxidative stress. Front Physiol 2014;5:372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol 2014;24:R453–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sherratt HS. Mitochondria: structure and function. Rev Neurol (Paris) 1991;147:417–30. [PubMed] [Google Scholar]
- 11.Redza-Dutordoir M, Averill-Bates DA. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta 2016;1863:2977–92. [DOI] [PubMed] [Google Scholar]
- 12.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J 2009;417:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wallukat G, Homuth V, Fischer T, et al. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J Clin Invest 1999;103:945–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.LaMarca B, Wallukat G, Llinas M, Herse F, Dechend R, Granger JP. Autoantibodies to the angiotensin type I receptor in response to placental ischemia and tumor necrosis factor alpha in pregnant rats. Hypertension 2008;52:1168–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cunningham MW Jr, Vaka VR, McMaster K, et al. Renal natural killer cell activation and mitochondrial oxidative stress; new mechanisms in AT1-AA mediated hypertensive pregnancy. Pregnancy Hypertens 2019;15:72–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vaka VR, Cunningham MW, Deer E, et al. Blockade of endogenous angiotensin II type I receptor agonistic autoantibody activity improves mitochondrial reactive oxygen species and hypertension in a rat model of preeclampsia. Am J Physiol Regul Integr Comp Physiol 2020;318:R256–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beyramzadeh M, Dikmen ZG, Erturk NK, Tuncer ZS, Akbiyik F. Placental respiratory chain complex activities in high risk pregnancies. J Matern Fetal Neonat Med 2017;30:2911–7. [DOI] [PubMed] [Google Scholar]
- 18.Illsinger S, Janzen N, Sander S, et al. Pre-eclampsia and HELLP syndrome: impaired mitochondrial function in umbilical endothelial cells. Reprod Sci 2010;17:219–26. [DOI] [PubMed] [Google Scholar]
- 19.Mando’ C, Miriam F, Palma CD, Borelli M, et al. OS048 Mitochondrial content and function in placental cells and tissues of preeclampsia and IUGR. Pregnancy Hypertens 2012;2:203. [DOI] [PubMed] [Google Scholar]
- 20.Matsubara S, Minakami H, Sato I, Saito T. Decrease in cytochrome c oxidase activity detected cytochemically in the placental trophoblast of patients with pre-eclampsia. Placenta 1997;18:255–9. [DOI] [PubMed] [Google Scholar]
- 21.Medrano Rodríguez JC, Yahuaca Mendoza P, Presno Bernal M, Alvarado Acosta JL. [Oxidative stress level and placental histological changes during preeclampsia]. Ginecol Obstet Mex 2008;76:319–26. [PubMed] [Google Scholar]
- 22.Rani N, Dhingra R, Arya DS, Kalaivani M, Bhatla N, Kumar R. Role of oxidative stress markers and antioxidants in the placenta of preeclamptic patients. J Obstet Gynaecol Res 2010;36:1189–94. [DOI] [PubMed] [Google Scholar]
- 23.Vishnyakova PA, Volodina MA, Tarasova NV, et al. Alterations in antioxidant system, mitochondrial biogenesis and autophagy in preeclamptic myometrium. BBA Clin 2017;8:35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zsengellér ZK, Rajakumar A, Hunter JT, et al. Trophoblast mitochondrial function is impaired in preeclampsia and correlates negatively with the expression of soluble fms-like tyrosine kinase 1. Pregnancy Hypertens 2016;6:313–9. [DOI] [PubMed] [Google Scholar]
- 25.Holland OJ, Cuffe JSM, Dekker Nitert M, et al. Placental mitochondrial adaptations in preeclampsia associated with progression to term delivery. Cell Death Dis 2018;9:1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vaka VR, McMaster KM, Cunningham MW, et al. Role of mitochondrial dysfunction and reactive oxygen species in mediating hypertension in the reduced uterine perfusion pressure rat model of preeclampsia. Hypertension 2018;72:703–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vishnyakova PA, Volodina MA, Tarasova NV, et al. Mitochondrial role in adaptive response to stress conditions in preeclampsia. Sci Rep 2016;6:32410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arora M. Cell culture media: a review. Mater Methods 2013;3:24. [Google Scholar]
- 29.Castells-Sala C, Martorell J, Balcells M. A human plasma derived supplement preserves function of human vascular cells in absence of fetal bovine serum. Cell Biosci 2017;7:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roberts L, LaMarca BB, Fournier L, Bain J, Cockrell K, Granger JP. Enhanced endothelin synthesis by endothelial cells exposed to sera from pregnant rats with decreased uterine perfusion. Hypertension 2006;47:615–8. [DOI] [PubMed] [Google Scholar]
- 31.Shi Z, Long W, Zhao C, Guo X, Shen R, Ding H. Comparative proteomics analysis suggests that placental mitochondria are involved in the development of pre-eclampsia. PLoS One 2013;8:e64351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCarthy C, Kenny LC. Therapeutically targeting mitochondrial redox signalling alleviates endothelial dysfunction in preeclampsia. Sci Rep 2016;6:32683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O. Oxidative stress and antioxidant defense. World Allergy Organ J 2012;5:9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Myatt L, Cui X. Oxidative stress in the placenta. Histochem Cell Biol 2004;122:369–82. [DOI] [PubMed] [Google Scholar]
- 35.Cui XL, Brockman D, Campos B, Myatt L. Expression of NADPH oxidase isoform 1 (Nox1) in human placenta: involvement in preeclampsia. Placenta 2006;27:422–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dechend R, Viedt C, Müller DN, et al. AT1 receptor agonistic antibodies from preeclamptic patients stimulate NADPH oxidase. Circulation 2003;107:1632–9. [DOI] [PubMed] [Google Scholar]
- 37.Matsubara S, Sato I. Enzyme histochemically detectable NAD(P)H oxidase in human placental trophoblasts: normal, pre-eclamptic, and fetal growth restriction-complicated pregnancy. Histochem Cell Biol 2001;116:1–7. [DOI] [PubMed] [Google Scholar]
- 38.Raijmakers MT, Peters WH, Steegers EA, Poston L. NAD(P)H oxidase associated superoxide production in human placenta from normotensive and pre-eclamptic women. Placenta 2004;25(SupplA):S85–9. [DOI] [PubMed] [Google Scholar]
- 39.Parrish MR, Wallace K, Tam Tam KB, et al. Hypertension in response to AT1-AA: role of reactive oxygen species in pregnancy-induced hypertension. Am J Hypertens 2011;24:835–40. [DOI] [PubMed] [Google Scholar]
- 40.Shanklin DR, Sibai BM. Ultrastructural aspects of preeclampsia. II. Mitochondrial changes. Am J Obstet Gynecol 1990;163:943–53. [DOI] [PubMed] [Google Scholar]
- 41.Shibata E, Nanri H, Ejima K, et al. Enhancement of mitochondrial oxidative stress and up-regulation of antioxidant protein peroxiredoxin III/SP-22 in the mitochondria of human pre-eclamptic placentae. Placenta 2003;24:698–705. [DOI] [PubMed] [Google Scholar]
- 42.Wang Y, Walsh SW. Placental mitochondria as a source of oxidative stress in pre-eclampsia. Placenta 1998;19:581–6. [DOI] [PubMed] [Google Scholar]
- 43.Torbergsen T, Oian P, Mathiesen E, Borud O. Pre-eclampsia—a mitochondrial disease? Acta Obstet Gynecol Scand 1989;68:145–8. [DOI] [PubMed] [Google Scholar]
- 44.He L, Wang Z, Sun Y. Reduced amount of cytochrome c oxidase subunit I messenger RNA in placentas from pregnancies complicated by preeclampsia. Acta Obstet Gynecol Scand 2004;83:144–8. [DOI] [PubMed] [Google Scholar]
- 45.Gao L, Laude K, Cai H. Mitochondrial pathophysiology, reactive oxygen species, and cardiovascular diseases. Vet Clin North Am Small Anim Pract 2008;38:137–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qiu C, Hevner K, Enquobahrie DA, Williams MA. A case-control study of maternal blood mitochondrial DNA copy number and preeclampsia risk. Int J Mol Epidemiol Genet 2012;3:237–44. [PMC free article] [PubMed] [Google Scholar]
- 47.Muralimanoharan S, Maloyan A, Mele J, Guo C, Myatt LG, Myatt L. MIR-210 modulates mitochondrial respiration in placenta with pre-eclampsia. Placenta 2012;33:816–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Salgado SS, Salgado MKR. Structural changes in pre-eclamptic and eclamptic placentas—an ultrastructural study. J Coll Phys Surg Pak 2011;21:482–6. [PubMed] [Google Scholar]
- 49.Williams MA, Sanchez SE, Ananth CV, Hevner K, Qiu C, Enquobahrie DA. Maternal blood mitochondrial DNA copy number and placental abruption risk: results from a preliminary study. Int J Mol Epidemiol Genet 2013;4:120–7. [PMC free article] [PubMed] [Google Scholar]
- 50.Busnelli A, Lattuada D, Ferrari S, et al. Mitochondrial DNA copy number in peripheral blood in the first trimester of pregnancy and different preeclampsia clinical phenotypes development: a pilot study. Reprod Sci 2019;26:1054–61. [DOI] [PubMed] [Google Scholar]
- 51.Lattuada D, Colleoni F, Martinelli A, et al. Higher mitochondrial DNA content in human IUGR placenta. Placenta 2008;29:1029–33. [DOI] [PubMed] [Google Scholar]
- 52.Mandò C, De Palma C, Stampalija T, et al. Placental mitochondrial content and function in intrauterine growth restriction and preeclampsia. Am J Physiol Endocrinol Metab 2014;306:E404–13. [DOI] [PubMed] [Google Scholar]
- 53.McCarthy CM, Kenny LC. Immunostimulatory role of mitochondrial DAMPs: alarming for pre-eclampsia? Am J Reprod Immunol 2016;76:341–7. [DOI] [PubMed] [Google Scholar]
- 54.Kiondo P, Wamuyu-Maina G, Wandabwa J, Bimenya GS, Tumwesigye NM, Okong P. The effects of vitamin C supplementation on pre-eclampsia in Mulago Hospital, Kampala, Uganda: a randomized placebo controlled clinical trial. BMC Preg Childbirth 2014;14:283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Poston L, Briley AL, Seed PT, Kelly FJ, Shennan AH. Vitamins in Pre-eclampsia (VIP) Trial Consortium. Vitamin C and vitamin E in pregnant women at risk for pre-eclampsia (VIP trial): randomised placebo-controlled trial. Lancet 2006;367:1145–54. [DOI] [PubMed] [Google Scholar]
- 56.Popova TA, Perfilova VN, Zhakupova GA, Verovsky VE, Ostrovskij OV, Tyurenkov IN. The effect of sulodexide on placental mitochondria function in rats with experimental preeclampsia. Biomed Khim 2016;62:572–6. [DOI] [PubMed] [Google Scholar]
- 57.Watson M, van Leer L, Vanderlelie JJ, Perkins AV. Selenium supplementation protects trophoblast cells from oxidative stress. Placenta 2012;33:1012–9. [DOI] [PubMed] [Google Scholar]
- 58.Khera A, Vanderlelie JJ, Perkins AV. Selenium supplementation protects trophoblast cells from mitochondrial oxidative stress. Placenta 2013;34:594–8. [DOI] [PubMed] [Google Scholar]