

Polyketide synthase 13 (Pks13) is an important enzyme found in Mycobacterium tuberculosis that condenses two fatty acyl chains to produce α-alkyl β-ketoesters, which in turn serve as the precursors for the synthesis of mycolic acids that are essential building blocks for maintaining the cell wall integrity of M. tuberculosis. Coumestan derivatives have recently been identified in our group as a new chemotype that exerts its antitubercular effects via targeting of Pks13.
KEYWORDS: chemotherapy, Mycobacterium tuberculosis, Pks13 inhibitor, mouse model
ABSTRACT
Polyketide synthase 13 (Pks13) is an important enzyme found in Mycobacterium tuberculosis that condenses two fatty acyl chains to produce α-alkyl β-ketoesters, which in turn serve as the precursors for the synthesis of mycolic acids that are essential building blocks for maintaining the cell wall integrity of M. tuberculosis. Coumestan derivatives have recently been identified in our group as a new chemotype that exerts its antitubercular effects via targeting of Pks13. These compounds were active on both drug-susceptible and drug-resistant strains of M. tuberculosis and showed low cytotoxicity to healthy cells and a promising selectivity profile. No cross-resistance was found between the coumestan derivatives and first-line tuberculosis (TB) drugs. Here, we report that treatment of M. tuberculosis bacilli with 15 times the MIC of compound 1, an optimized lead coumestan compound, resulted in a CFU reduction from 6.0 log10 units to below the limit of detection (1.0 log10 units) per ml of culture, demonstrating a bactericidal mechanism of action. Single-dose (10 mg/kg of body weight) pharmacokinetic studies revealed favorable parameters with a relative bioavailability of 19.4%. In a mouse infection and chemotherapy model, treatment with compound 1 showed dose-dependent monotherapeutic activity, whereas treatment with 1 in combination with rifampin showed clear synergistic effects. Together, these data suggest that coumestan derivatives are promising agents for further TB drug development.
INTRODUCTION
Tuberculosis (TB), as a communicable disease, continues to be a global public health threat, with 10.0 million people falling ill in 2018. It is one of the top 10 causes of death worldwide and—at this writing—is the leading cause of death from a single infectious agent ranked above HIV/AIDS (1). Combinatorial chemotherapy remains the major public health tool to contain TB, but the emergence of multidrug-resistant (MDR) and extensively drug-resistant TB (XDR-TB) threaten the activity of the aging pool of first-line drugs. Four decades after the first-line TB drugs and regimen were established, only two new TB drugs, namely, bedaquiline and pretomanid, have been approved by FDA for treatment of drug-resistant TB (2). Without a doubt, new TB drugs with new targets remain an urgent need to achieve the End TB Strategy adopted by the WHO.
The unique mycobacterial cell wall consists of a peptidoglycan layer, an arabinogalactan polysaccharide layer, and the mycomembrane. The latter is mainly composed of very long-chain fatty acids, namely, mycolic acids that are ester bonded to arabinogalactan which is covalently anchored to peptidoglycan (3–5). The mycobacterial cell wall plays an important role in mycobacterial viability. Its lipids, mainly mycolic acids, form a permeability barrier that prevents entry of many environmental solutes and confers natural resistance to many antibiotics (6). The mycobacterial cell wall has been a rich source of novel targets for TB drug development (3, 6, 7). There are approximately 60 potential targets in the cell wall of Mycobacterium tuberculosis (3). Indeed, multiple antibiotics that target the M. tuberculosis cell envelope are in clinical use for TB treatment, such as isoniazid (INH), ethambutol (ETB), and the recent European Medicines Agency (EMA)-approved delamanid.
Polyketide synthase 13 (Pks13) is responsible for the last condensation step of the biosynthesis of mycolic acids, which are essential for mycobacterial cellular architecture and impermeability (8–10). The thioesterase domain of Pks13 also plays a role in lipid transport, which is also an essential function in M. tuberculosis (11). Recent studies have shown that Pks13 is a qualified drug target for M. tuberculosis (12–14); and different structural classes of compounds, such as benzofurans (12, 15), thiophenes (13), β-lactones (16), and coumestans (17, 18), have been found to target this druggable protein. Mutations in pks13 indeed conferred resistance to their cognate inhibitors, such as D1607N, D1644G, and D1644Y to benzofurans (12, 15); A1667V, D1644G, N1640K, and N1640S to bezofurans and coumestans (17); and F79S to thiophenes (13). Interestingly, the coumestan structural class of compounds is active on both drug-susceptible and drug-resistant M. tuberculosis, with attractive properties such as a bactericidal mode of action, low toxicity, and high bioavailability, as characterized by the serum inhibition titration assay (17, 18). In this study, we set out to further explore the pharmacokinetic parameters and in vivo efficacy of a hit-to-lead representative coumestan compound.
RESULTS
In vitro activity and target verification.
Coumestan derivatives as Pks13 inhibitors were shown to be active on both drug-susceptible and drug-resistant M. tuberculosis, with a bactericidal mode of action (17, 18) (Fig. 1). Here, we confirmed that the resistant mutants due to single nucleotide polymorphisms in pks13 were also resistant to the recently optimized lead coumestan compounds 1 and 2, with MIC shifts from 16-fold (2 on MTB5A [A1667V]) to 8,000-fold (1 on MTB4 [N1640K] or 1 on MTB3 [D1644G]) (Table 1). These data verified that Pks13 is the target for both compounds 1 and 2. In addition, these Pks13 mutants were also resistant to the benzofuran scaffold Pks13 inhibitors developed previously (compound 6) (15) (Table 1). The molecular interaction between the Pks13 inhibitors and the thioesterase domain of Pks13 was characterized by thermal stability analysis using the nano differential scanning fluorimetry (nanoDSF) method. Indeed, all six inhibitors (compounds 1 to 6) significantly right-shifted the melting temperature, with a trend observed in the DSF study in agreement with the observed MIC values, while the noninhibitor control compound had a minimal effect (see Table S1 in the supplemental material). However, no cross-resistance between the Pks13 mutants and the first-line TB drugs (INH, rifampin [RIF], and ETB) was found (Table 1).
FIG 1.

Chemical structures of Pks13 inhibitors compound 1 (designated 48 in reference 18), compound 2 (designated 50 in reference 18), compound 3 (designated 41 in reference 17), compound 4 (designated 42 in reference 17), compound 5 (designated 27 in reference 17), and compound 6 (designated TAM16 in in reference 15).
TABLE 1.
In vitro activity of selected Pks13 inhibitors and first-line TB drugs on Pks13 mutants of M. tuberculosis
| Strain | Mutation | MIC (μg/ml [MIC fold change]) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| INH | RIF | EMB | Compound |
|||||||
| 1 | 2 | 3 | 4 | 5 | 6 | |||||
| H37Rv | NAa | 0.04 | 0.125 | 1 | 0.0039 | 0.0039 | 0.125 | 0.25 | 0.5 | 0.0313 |
| MTB5B | N1640S | 0.04 | 0.125 | 1 | 0.0625 (16) | 0.0625 (16) | 16 (128) | 8 (32) | 4 (8) | 8−16 (256–512) |
| MTB4 | N1640K | 0.04 | 0.125 | 1 | 32 (8,000) | 4 (1,000) | 16 (128) | 16 (64) | 4 (8) | >64 (>2,048) |
| MTB3 | D1644G | 0.04 | 0.125 | 1 | 32 (8,000) | 4 (1,000) | 32 (256) | 16 (64) | 4 (8) | >64 (>2,048) |
| MTB5A | A1667V | 0.04 | 0.125 | 1 | 0.5 (125) | 0.0625 (16) | 16 (128) | 8 (32) | 4 (8) | 8 (256) |
NA, not applicable.
Kill kinetics.
In liquid media, compound 1 showed bactericidal activity in a dose-dependent manner (Fig. 2). Treatment with 15 times the MIC (0.06 μg/ml) of compound 1 demonstrated a biphasic kill curve, as follows: an initial rapid CFU reduction phase and a subsequent slower CFU reduction phase. From day 0 to day 3, CFU in the culture reduced rapidly, while from day 3 to day 10, the CFU reduction curve was less steep but the CFU in the culture reached the limit of detection (10 CFU per ml) (Fig. 2). Treatment with 5 times the MIC of compound 1 (0.02 μg/ml) achieved progressive CFU reduction during the experimental period but at a lower reduction rate than treatment with 15 times the MIC. From day 6 to day 10, the CFU reduction curve of 5 times the MIC treatment was slightly leveled off, and the CFU in the culture did not reach the limit of detection. Treatment with 2 times the MIC of isoniazid (0.08 μg/ml) achieved similar CFU reduction kinetics as treatment of 5 times the MIC of compound 1 before day 6. However, after day 6, significant rebound was observed for treatment with 2 times the MIC of isoniazid, which was probably associated with differential resistance frequency (Fig. 2). Treatment with 1 times the MIC of compound 1 achieved only a flat line from day 0 to day 3, but outgrowth was apparent afterward, possibly due to compound degradation within the culture (Fig. 2).
FIG 2.
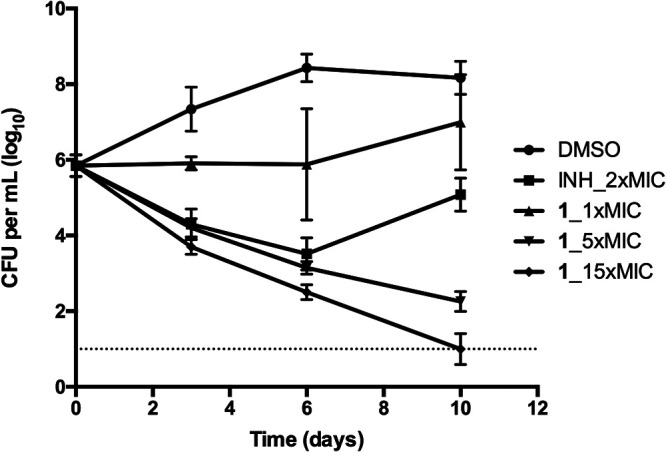
Kill kinetics of compound 1. INH_2x, isoniazid at 2 times the MIC as positive control; DMSO, negative control; compound 1 at 1, 5, and 15 times the MIC was tested on M. tuberculosis H37Rv. Data are shown as mean ± SD of four replications within one experiment. Similar results were obtained in a second independent experiment.
Pharmacokinetic study.
We previously showed that compound 1 was bioavailable by oral administration in a serum inhibition titration study (18). Here, we further pursued a pharmacokinetic study in BALB/c mice. Compound 1 was readily bioavailable by oral administration of 10 mg/kg of body weight and reached a maximum concentration of 98.4 ng/ml in the plasma at 0.833 h after administration (Table 2). Area under the concentration-time curve from 0 to t (AUC0–t) was 246 h·ng·ml−1 with a half-life of 4.54 h. The mean residence time was 6.55 h, and the overall relative bioavailability was 19.4% (Table 2).
TABLE 2.
Pharmacokinetic parameters of compound 1 in BALB/c mice by single oral administration of 10 mg/kg
| Parametera | Unit | Mean | SD |
|---|---|---|---|
| kel | h−1 | 0.166 | 0.06 |
| t1/2 | h | 4.54 | 1.5 |
| Tmax | h | 0.833 | 1.0 |
| Cmax | ng·ml−1 | 98.4 | 69 |
| AUC0–t | h·ng·ml−1 | 246 | 125 |
| AUC0–∞ | h·ng·ml−1 | 297 | 109 |
| AUMC0–t | h·ng·ml−1 | 1,099 | 982 |
| AUMC0–∞ | h·ng·ml−1 | 2,040 | 1,238 |
| MRT | h | 6.55 | 2.1 |
| F | % | 19.4 | 7.1 |
kel, elimination rate constant; t1/2, half-life; Tmax, time to maximum concentration of drug in serum; Cmax, maximum concentration of drug in serum; AUMC0–t, area under the first moment of the concentration-time curve from 0 to t; MRT, mean residence time.
In vivo efficacy study of the lead compound in mouse infection models.
To elucidate the in vivo efficacy of compound 1, we performed two mouse infection and chemotherapy modeling studies. In the acute monotherapy model, compound 1 demonstrated dose-dependent activity, for treatment with 50, 25, and 12.5 mg/kg achieved 2.21-, 1.07-, and 0.12-log10 CFU reduction in the mouse lungs, respectively (Fig. 3A). This dose-dependent CFU reduction was also in agreement with lung gross pathology and lung weight (see Fig. S1 in the supplemental material).
FIG 3.
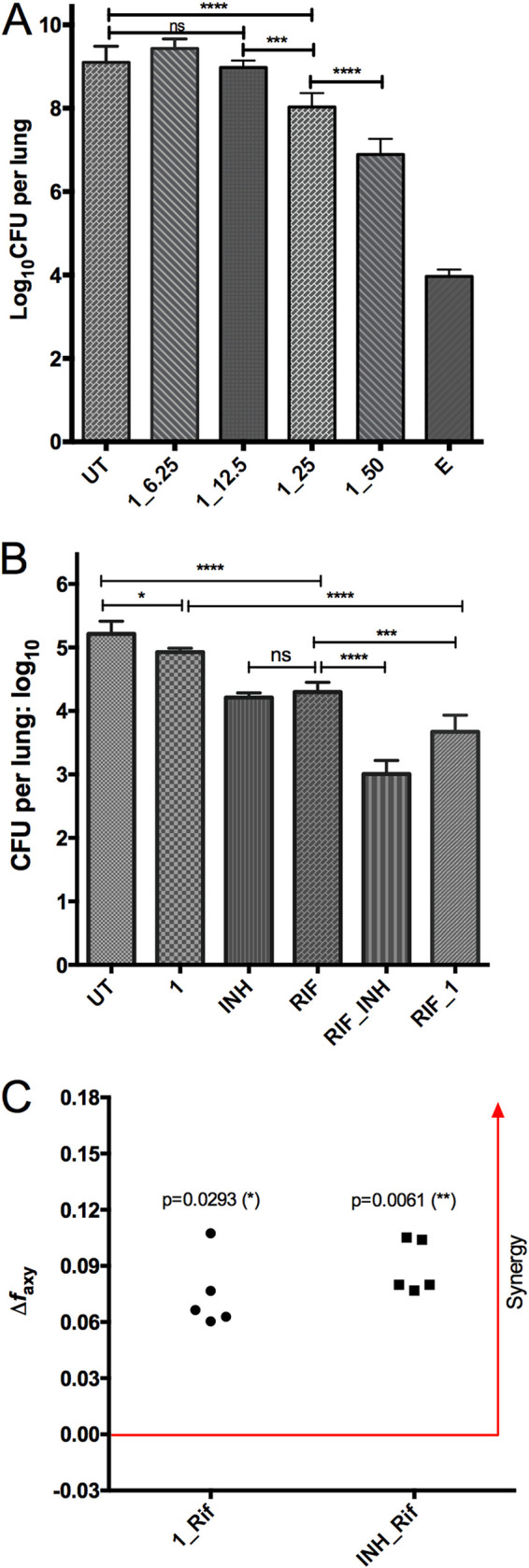
In vivo activity of compound 1, as shown in an acute monotherapy model (A) and a chronic combination therapy model (B, C). For the acute model, mice were infected with 3.2 log10 CFU by the aerosol route and treatment was initiated on day 1 after infection. For the chronic model, mice were infected with 2.0 log10 CFU by the aerosol route and treatment was initiated 28 days after infection. CFU in the lungs were determined after 4 weeks (A) or 8 weeks (B) of treatment. UT, untreated; E, ethambutol (100 mg/kg); RIF, rifampin (10 mg/kg); INH, isoniazid (10 mg/kg); 1_50, compound 1 at 50 mg/kg. All treatment was daily, 5 days a week. Data are shown as mean ± SD (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 by one-way ANOVA and Tukey’s multiple-comparison test). (C) Bliss independence model demonstrated synergistic effects of the combination of 1 and rifampin and INH and rifampin (unpaired t test applied).
In the chronic model, compound 1 at 25 mg/kg was effective, as shown by a reduction of 0.3-log10 CFU burden in the mouse lungs (P < 0.05). When compound 1 was combined with rifampin at 10 mg/kg, the combination achieved a significantly lower CFU burden in the lungs than either rifampin alone (3.67 versus 4.30, P < 0.001), isoniazid alone (3.67 versus 4.21, P < 0.001), or compound 1 alone (3.67 versus 4.93, P < 0.0001) (Fig. 3B). The combination of RIF and INH yielded a better CFU reduction than RIF and compound 1. When the combination therapy data were subjected to Bliss independence modeling, compound 1 and rifampin achieved a synergistic effect for CFU reduction in the mouse lungs, as shown by a larger observed fraction affected by combination (faxy,O) than predicted fraction affected by combination (faxy,P) (Fig. 3C). Similarly, synergy was also found for the combination of INH and rifampin (Fig. 3C). It is worth mentioning that in this chronic model, INH and RIF achieved a similar CFU reduction in the mouse lungs (1.00 versus 0.92, P > 0.05) (Fig. 3B).
DISCUSSION
Pks13 is comprised of the following five domains: an N-terminal acyl carrier protein (ACP) domain, a ketosynthase (KS) domain, an acyltransferase (AT) domain, an C-terminal ACP domain, and an C-terminal thioesterase (TE) domain (10). It was evident that a critical site for coumestan action against Pks13 resides in the TE domain (15). Indeed, all of our Pks13 mutants harbor single nucleotide polymorphisms in the TE domain (Table 1). It was recently shown that D1644, F1670, Y1674, and G1644 constitute a binding pocket (18) wherein Pks13 inhibitors may block the active site (15). However, there was a clear mutation- and compound-specific molecular interaction pattern. When the polar asparagine (N) at 1640 was mutated to polar serine (S), the MIC shift for both compounds 1 and 2 was only 16 times. However, when N at 1640 was mutated to the positively charged lysine (K), the MIC shift increased to 8,000 times for compound 1 and 1,000 times for compound 2 (Table 1). This result may be explained by molecular docking modeling using the solved crystal structures of Pks13-TE (PDB ID 5V3Y) for compound 1. Protonated N in the piperidine ring of compound 1 has an intermolecular hydrogen bond with the side chain oxygen of N1640. When N at 1640 was mutated into S or K, both of them lose the effect. At the same time, the longer side chain of hydrophilic K may prevent compound 1 from inserting into the hydrophobic pocket of Pks13-TE (see Fig. S3 in the supplemental material). Nevertheless, the drastic difference between S and K at 1640 was not observed with compounds 3 and 5, as no difference was seen in terms of MIC shift (Table 1). We speculate that the hydroxyl group at the benzene played an important role for the drastic MIC shift, as compounds 3 and 5 did not have the hydroxyl group at that position.
The coumestan class compound 1 worked by a bactericidal mode of action, with the minimum bactericidal concentration (MBC) at 0.0039 to 0.0078 μg/ml (18). In this study, we showed that at 5 or 15 times the MIC, compound 1 progressively killed M. tuberculosis in liquid culture and reached the limit of detection after 10 days for 15 times the MIC treatment (Fig. 2). Bactericidal killing was also reported for the benzofuran class of Pks13 inhibitors (15), most likely due to the essentiality of mycolic acid biosynthetic pathway (8–10, 19, 20).
Compounds 1 and 2 were orally bioavailable, as shown in our previous serum inhibition titration assay (18) and the pharmacokinetic study presented in this work (compound 1, Table 2). The relative bioavailability of compound 1 (19.4%) was not as high as we hoped. One possible reason could be that the propylene glycol (PG):Tween 80 (4:1) formulation might not be optimal for compound 1, even though it was suitable for an indole-2-carboxamide (21). However, the in vivo activity of compound 1 was verified by lung CFU reduction in both the acute monotherapy and the chronic combination therapy mouse modeling, and a favorable synergistic effect was observed for compound 1 combined with rifampin in the chronic model (Fig. 3; Fig. S1; see Fig. S2 in the supplemental material). We noticed that in the chronic model, 8 weeks of monotherapy with compound 1 resulted in higher lung weight but lower CFU load in the lungs (Fig. 3B; Fig. S2). More studies are needed to find out the reason behind this discordance, but we think the CFU data are a more important parameter for TB therapeutic efficacy. Worth mentioning is that when compound 1 was combined with rifampin, the lung weight increase disappeared completely (Fig. S2). While no Pks13 inhibitors have been clinically evaluated or licensed, the target is considered druggable because it is known to be essential for bacterial survival in vitro. The essentiality of Pks13 (mycolic acid biosynthesis pathway of M. tuberculosis) and the activities of the coumestan class Pks13 inhibitors on MDR- and XDR-TB and their in vivo efficacy justify further development of this interesting chemical entity.
MATERIALS AND METHODS
Mycobacterial strain and culture conditions.
M. tuberculosis H37Rv wild-type and congenic Pks13 mutant strains were cultured in 7H9 broth supplemented with 0.2% glycerol, 10% oleic acid-albumin-dextrose-catalase (OADC) (Becton, Dickinson, USA), and 0.05% Tween 80. CFUs were determined by plating diluted mouse lung homogenates on 7H11 agar containing 50 μg/ml cycloheximide, 25 μg/ml polymixin B, 50 μg/ml carbenicillin, and 20 μg/ml trimethoprim. Incubation was carried out at 37°C.
Target verification and cross-resistance characterization.
Four Pks13 mutants harboring N1640S (MTB5B, clone 2 from compound 27), N1640K (MTB4, clone 1 from compound 42), D1644G (MTB3, clone 2 from compound 41), and A1667V (MTB5A, clone 1 from compound 27) (17), respectively, were tested to characterize cross-resistance among previously reported Pks13 inhibitors, including both benzofurans (15, 17) and coumestans (17, 18). The recently optimized coumestan lead compounds 1 and 2 were tested against this panel of mutants to verify Pks13 as their target. Those mutants were also tested for susceptibility to the first-line TB drugs isoniazid, rifampin, and ethambutol. Microplate alamarBlue Assay (MABA) was used to determine the MIC as reported previously (18).
Kill kinetics.
Mid-log-phase (optical density at 600 nm [OD600], 0.9) M. tuberculosis H37Rv culture was diluted 100 times. Diluted culture was aliquoted to 10 ml into 50-ml conical tubes. Compound 1 was added to corresponding tubes to achieve a final concentration of 1, 5, and 15 times the MIC. The first-line TB drug isoniazid at 2 times the MIC (0.08 μg/ml) was used as a positive control. Dimethyl sulfoxide (DMSO) with an equivalent dilution factor at 1 times the MIC of compound 1 was the negative control. All tubes were incubated at 37°C in a shaker incubator (200 rpm). At days 0, 3, 6, and 10, all cultures were diluted and plated onto 7H11 plates for CFU enumeration.
Pharmacokinetic study.
To study the bioavailability of compound 1, 6-week-old female BALB/c mice were assigned to either the intravenous (i.v.) injection or oral (p.o.) administration treatment group. Three mice were used for each treatment. For the i.v. group, the dose was 3 mg/kg and the vehicle contained 75% normal saline, 5% DMSO, 10% Solutol, and 10% ethanol. For the p.o. group, the dose was 10 mg/kg, and the vehicle was a miscible mixture of propylene glycol and Tween 80 at 4:1 (vol/vol). After dosing, retro-orbital blood was collected at 5 min, 15 min, 30 min, 1 h, 2 h, 4 h, 6 h, 8 h, and 24 h. Plasma was separated and stored at −80°C for further analysis using the Sciex 4000 QTRAP liquid chromatography-tandem mass spectrometry (LC-MS/MS) system. Estimation of pharmacokinetic parameters was conducted using noncompartmental analysis with WinNonlin software (Phoenix 6.3).
In vivo efficacy evaluation.
Four-to-6-week-old female BALB/c mice were obtained from Charles River Laboratories. Mice were aerosol infected with M. tuberculosis H37Rv using an inhalation system (Glas-Col Inc., Terre Haute, IN). In order to study the in vivo efficacy of compound 1, two mouse infection and chemotherapy models were carried out. For the acute monotherapy modeling, mice were infected to have a day 1 implantation of 3.2 log10 CFU per lung and treatment initiated at day 1. Groups of five mice were assigned to compound 1 treatment (dosed at 6.25, 12.5, 25, and 50 mg/kg), positive-control (ethambutol at 100 mg/kg), or untreated negative-control groups. After 4 weeks of treatment, mice were sacrificed and lungs were removed, homogenized, and plated onto 7H11 selective agar for CFU enumeration. For the chronic combination therapy modeling, mice were infected to have day 1 implantation of 2.0 log10 CFU per lung. Treatment was initiated on day 28 when CFU reached 7.0 log10 CFU per lung. Mice were randomly grouped to six treatment arms: untreated, compound 1 at 25 mg/kg, rifampin at 10 mg/kg, isoniazid at 10 mg/kg, INH combined with RIF, or compound 1 combined with RIF, with group size of five. After 8 weeks of treatment, mice were sacrificed and lungs were removed, homogenized, and plated onto 7H11 selective agar for CFU enumeration.
Compound 1 was prepared in PG:Tween 80 (4:1), and control compounds were prepared in water. Group size was five mice for all treatments, and all treatments were carried out by oral gavage, once a day and 5 days a week. The Institutional Animal Care and Use Committee of the Johns Hopkins University School of Medicine approved all animal procedures performed in this study.
Data analysis and statistics.
Lung CFU counts (x) were log-transformed (as x + 1) before analysis. Group mean CFU counts were compared using one-way analysis of variance (ANOVA) with Dunnett’s posttest to adjust for multiple comparisons. We used Bliss independence model to examine the synergy of drug combinations. The model is defined by the equation faxy,P = fax + fay − (fax)(fay), where faxy,P is the predicted fraction affected by a combination of drug x and drug y given the experimentally observed fraction affected for drug x (fax) and drug y (fay) individually. The experimentally observed fraction affected by a combination of drug x and drug y (faxy,O) can be compared with the predicted fraction affected. If faxy,O > faxy,P (or faxy,O − faxy,P > 0), the synergy was fulfilled (22). In vivo synergy was tested by using an unpaired t test. All analyses were performed with Prism version 6 (GraphPad, San Diego, CA).
Supplementary Material
ACKNOWLEDGMENTS
This project has been funded in part with federal funds from the National Institutes of Health and National Institute of Allergy and Infectious Diseases, Department of Health and Human Services under grants AI37856 and HL133190. This work has been supported by the National Natural Science Foundation of China (21778019). H.G. acknowledges the ARC Discovery Early Career Researcher Award DE160100482.
REFERENCES
- 1.World Health Organization. 2019. Global tuberculosis report. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.FDA. 2019. FDA news release: FDA approves new drug for treatment-resistant forms of tuberculosis that affects the lungs. FDA, White Oak, MD. [Google Scholar]
- 3.Barry CE, Crick DC, McNeil MR. 2007. Targeting the formation of the cell wall core of M. tuberculosis. Infect Disord Drug Targets 7:182–202. doi: 10.2174/187152607781001808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quémard A. 2016. New insights into the mycolate-containing compound biosynthesis and transport in mycobacteria. Trends Microbiol 24:725–738. doi: 10.1016/j.tim.2016.04.009. [DOI] [PubMed] [Google Scholar]
- 5.Chiaradia L, Lefebvre C, Parra J, Marcoux J, Burlet-Schiltz O, Etienne G, Tropis M, Daffé M. 2017. Dissecting the mycobacterial cell envelope and defining the composition of the native mycomembrane. Sci Rep 7:12807. doi: 10.1038/s41598-017-12718-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Favrot L, Ronning DR. 2012. Targeting the mycobacterial envelope for tuberculosis drug development. Expert Rev Anti Infect Ther 10:1023–1036. doi: 10.1586/eri.12.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen H, Nyantakyi SA, Li M, Gopal P, Aziz DB, Yang T, Moreira W, Gengenbacher M, Dick T, Go ML. 2018. The mycobacterial membrane: a novel target space for anti-tubercular drugs. Front Microbiol 9:1627. doi: 10.3389/fmicb.2018.01627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Portevin D, De Sousa-D'Auria C, Houssin C, Grimaldi C, Chami M, Daffé M, Guilhot C. 2004. A polyketide synthase catalyzes the last condensation step of mycolic acid biosynthesis in mycobacteria and related organisms. Proc Natl Acad Sci U S A 101:314–319. doi: 10.1073/pnas.0305439101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takayama K, Wang C, Besra GS. 2005. Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis. Clin Microbiol Rev 18:81–101. doi: 10.1128/CMR.18.1.81-101.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gavalda S, Léger M, van der Rest B, Stella A, Bardou F, Montrozier H, Chalut C, Burlet-Schiltz O, Marrakchi H, Daffé M, Quémard A. 2009. The Pks13/FadD32 crosstalk for the biosynthesis of mycolic acids in Mycobacterium tuberculosis. J Biol Chem 284:19255–19264. doi: 10.1074/jbc.M109.006940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gavalda S, Bardou F, Laval F, Bon C, Malaga W, Chalut C, Guilhot C, Mourey L, Daffé M, Quémard A. 2014. The polyketide synthase Pks13 catalyzes a novel mechanism of lipid transfer in mycobacteria. Chem Biol 21:1660–1669. doi: 10.1016/j.chembiol.2014.10.011. [DOI] [PubMed] [Google Scholar]
- 12.Ioerger TR, O'Malley T, Liao R, Guinn KM, Hickey MJ, Mohaideen N, Murphy KC, Boshoff HIM, Mizrahi V, Rubin EJ, Sassetti CM, Barry CE, Sherman DR, Parish T, Sacchettini JC. 2013. Identification of new drug targets and resistance mechanisms in Mycobacterium tuberculosis. PLoS One 8:e75245. doi: 10.1371/journal.pone.0075245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilson R, Kumar P, Parashar V, Vilchèze C, Veyron-Churlet R, Freundlich JS, Barnes SW, Walker JR, Szymonifka MJ, Marchiano E, Shenai S, Colangeli R, Jacobs WRJ, Neiditch MB, Kremer L, Alland D. 2013. Antituberculosis thiophenes define a requirement for Pks13 in mycolic acid biosynthesis. Nat Chem Biol 9:499–506. doi: 10.1038/nchembio.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.North EJ, Jackson M, Lee RE. 2014. New approaches to target the mycolic acid biosynthesis pathway for the development of tuberculosis therapeutics. Curr Pharm Des 20:4357–4378. doi: 10.2174/1381612819666131118203641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aggarwal A, Parai MK, Shetty N, Wallis D, Woolhiser L, Hastings C, Dutta NK, Galaviz S, Dhakal RC, Shrestha R, Wakabayashi S, Walpole C, Matthews D, Floyd D, Scullion P, Riley J, Epemolu O, Norval S, Snavely T, Robertson GT, Rubin EJ, Ioerger TR, Sirgel FA, van der Merwe R, van Helden PD, Keller P, Böttger EC, Karakousis PC, Lenaerts AJ, Sacchettini JC. 2017. Development of a novel lead that targets M. tuberculosis polyketide synthase 13. Cell 170:249–259.e25. doi: 10.1016/j.cell.2017.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lehmann J, Cheng T-Y, Aggarwal A, Park AS, Zeiler E, Raju RM, Akopian T, Kandror O, Sacchettini JC, Moody DB, Rubin EJ, Sieber SA. 2018. An antibacterial β-lactone kills Mycobacterium tuberculosis by disrupting mycolic acid biosynthesis. Angew Chem Int Ed Engl 57:348–353. doi: 10.1002/anie.201709365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang W, Lun S, Wang S-H, Jiang X-W, Yang F, Tang J, Manson AL, Earl AM, Gunosewoyo H, Bishai WR, Yu L-F. 2018. Identification of novel coumestan derivatives as polyketide synthase 13 inhibitors against Mycobacterium tuberculosis. J Med Chem 61:791–803. doi: 10.1021/acs.jmedchem.7b01319. [DOI] [PubMed] [Google Scholar]
- 18.Zhang W, Lun S, Liu L-L, Xiao S, Duan G, Gunosewoyo H, Yang F, Tang J, Bishai WR, Yu L-F. 2019. Identification of novel coumestan derivatives as polyketide synthase 13 inhibitors against Mycobacterium tuberculosis. Part II. J Med Chem 62:3575–3589. doi: 10.1021/acs.jmedchem.9b00010. [DOI] [PubMed] [Google Scholar]
- 19.Marrakchi H, Lanéelle M-A, Daffé M. 2014. Mycolic acids: structures, biosynthesis, and beyond. Chem Biol 21:67–85. doi: 10.1016/j.chembiol.2013.11.011. [DOI] [PubMed] [Google Scholar]
- 20.PaweŁczyk J, Kremer L. 2014. The molecular genetics of mycolic acid biosynthesis. Microbiol Spectr 2:MEM2-0003-2013. [DOI] [PubMed] [Google Scholar]
- 21.Lun S, Tasneen R, Chaira T, Stec J, Onajole OK, Yang TJ, Cooper CB, Mdluli K, Converse PJ, Nuermberger EL, Raj VS, Kozikowski A, Bishai WR. 2019. Advancing the therapeutic potential of indoleamides for tuberculosis. Antimicrob Agents Chemother 63:e00343-19. doi: 10.1128/AAC.00343-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laird GM, Bullen CK, Rosenbloom DIS, Martin AR, Hill AL, Durand CM, Siliciano JD, Siliciano RF. 2015. Ex vivo analysis identifies effective HIV-1 latency-reversing drug combinations. J Clin Invest 125:1901–1912. doi: 10.1172/JCI80142. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.