

Abstract
Alzheimer’s disease (AD) genetics implies a causal role for innate immune genes, TREM2 and CD33, products that oppose each other in the downstream Syk tyrosine kinase pathway, activating microglial phagocytosis of amyloid (Aβ). We report effects of low (Curc-lo) and high (Curc-hi) doses of curcumin on neuroinflammation in APPsw transgenic mice. Results showed that Curc-lo decreased CD33 and increased TREM2 expression (predicted to decrease AD risk) and also increased TyroBP, which controls a neuroinflammatory gene network implicated in AD as well as phagocytosis markers CD68 and Arg1. Curc-lo coordinately restored tightly correlated relationships between these genes’ expression levels, and decreased expression of genes characteristic of toxic pro-inflammatory M1 microglia (CD11b, iNOS, COX-2, IL1β). In contrast, very high dose curcumin did not show these effects, failed to clear amyloid plaques, and dysregulated gene expression relationships. Curc-lo stimulated microglial migration to and phagocytosis of amyloid plaques both in vivo and in ex vivo assays of sections of human AD brain and of mouse brain. Curcumin also reduced levels of miR-155, a micro-RNA reported to drive a neurodegenerative microglial phenotype. In conditions without amyloid (human microglial cells in vitro, aged wild-type mice), Curc-lo similarly decreased CD33 and increased TREM2. Like curcumin, anti-Aβ antibody (also reported to engage the Syk pathway, increase CD68, and decrease amyloid burden in human and mouse brain) increased TREM2 in APPsw mice and decreased amyloid in human AD sections ex vivo. We conclude that curcumin is an immunomodulatory treatment capable of emulating anti-Aβ vaccine in stimulating phagocytic clearance of amyloid by reducing CD33 and increasing TREM2 and TyroBP, while restoring neuroinflammatory networks implicated in neurodegenerative diseases.
Keywords: Alzheimer’s disease, Amyloid, CD33, Innate immune, Phagocytosis, TREM2, Amyloid vaccine
1. Introduction
In Alzheimer Disease accumulation of beta-amyloid peptide (Aβ) initiates amyloid deposition and a complex inflammatory cascade (Hardy et al., 2014). Although initial Aβ aggregates are effectively cleared by innate immune microglia (Lucin et al., 2013; Rogers et al., 2002), clearance fails with chronic inflammation and the onset of amyloid plaques (Hickman et al., 2008; Krabbe et al., 2013). The pleiotropic roles of microglia, ranging from protective to harmful (Akiyama et al., 2000), cloud our understanding of their role in AD. A simplified classification is often divided into the pro-inflammatory “M1” class or the more phagocytic, resolving “M2” classes although the actual subtypes are far more complex. They can also be defined by function, including morphology, phagocytic phenotype, and more complex marker expression (Cherry et al., 2014; Colton and Wilcock, 2010; Colton, 2009; Van Eldik et al., 2016; Weekman et al., 2014). Aβ aggregates in vitro (Michelucci et al., 2009) and in vivo (Hickman et al., 2008) polarize microglia toward M1 and away from M2 phenotype, which decreases Aβ clearance (Koenigsknecht-Talboo and Landreth, 2005).
One approach to identifying a therapeutic target is to increase expression of selected M2-related microglial genes (Mandrekar-Colucci et al., 2012). Another is to selectively alter innate immune genes linked to increased AD risk, for example, CD33, a sialic acid receptor in microglia (Carrasquillo et al., 2011; Hollingworth et al., 2011; Naj et al., 2011) and Triggering Receptor Expressed on Myeloid cells (TREM2) (Malpass, 2013). These genes are in a network of AD-dysregulated innate immune genes controlled by hub gene TyroBP (DAP12) (Zhang et al., 2013). There are SNPs in human CD33 that either decrease functional CD33 expression and AD risk or reduce both CD33 expression and AD risk; further, microglial CD33 expression is upregulated in APP Tg mice and sporadic AD and associated with increased amyloid burden and microglial activation (Bradshaw et al., 2013; Griciuc et al., 2013; Malik et al., 2013). Therefore, AD therapeutics that limit CD33 expression may prove beneficial.
TREM2 is a classical innate immune system receptor restricted to microglia in the brain (Ulrich et al., 2017; Yeh et al., 2017) (and related peripheral cells) that requires interaction with TyroBP for its signaling through tyrosine kinases. This signaling is opposed by CD33-linked tyrosine phosphatases (Malik et al., 2015) (Fig. 1). TREM2 is highly and persistently upregulated in the M2 “resolution” phase of CNS injury-induced inflammation (Ydens et al., 2012), promoting pro-phagocytic and anti-inflammatory activities (Painter et al., 2015; Takahashi et al., 2007) in amyloid plaque-associated microglia (Frank et al., 2008; Melchior et al., 2010). TREM2-ir plaque-associated microglia in APP/PS1 are reduced with ApoE deletion (Ulrich et al., 2018). While the impact of specifically targeting only TREM2 expression remains controversial (Wang et al., 2015), loss of either TREM2 or TyroBP leads to chronic uncontrolled inflammation (Thrash et al., 2009), and the loss-of-function SNPs decrease functional expression and increase AD risk (Cheng-Hathaway et al., 2018; Guerreiro et al., 2013; Jonsson et al., 2013; Pottier et al., 2016). TREM2 AD-risk SNPs greatly reduce cellular phagocytic activity (Kleinberger et al., 2014).
Fig. 1.

Activation of microglia and phagocytosis controlled by tyrosine phosphorylated AD risk-associated genes (*). Schematic diagram of a microglial cell with the Fc(g)R1 receptor for antibody, opsonized Aβ and parallel signaling receptors: TREM2* and CD11b*, all linked to ITAM domain phosphorylation signaling on TYROBP/DAP12* or FCER1G, that then signal through Syk tyrosine kinase to promote phagocytosis. This ITAM signaling is negatively regulated by CD33* (and other Siglecs with ITIM domains), which couple to SHPS like SHP1 (tyrosine phosphatases). SHP1 dephosphorylates (deactivates) the TYROBP/ITAM-activated, phagocytosis-promoting signaling pathways as shown. The activation of these signaling pathways is represented by phosphorylated tyrosine residues (“PT”). The process of phagocytic engulfment and a mature phagosome fusing with a lysosome (marked by the glycoprotein CD68* (mouse macrosialin) is shown with engulfed Aβ. The classic microglia marker CD68 is a lysosomal innate immune marker of the phagocytic microglial phenotype (reviewed in Zotova et al., 2011). CD11b (ITGAM) is a classic marker of M1 activation that complexes with CD18 (ITGB2), a proximal TyroBP partner. CD11b also serves as a receptor for complement C3b opsonized Aβ aggregates (Wyss-Coray and Mucke, 2002). C1q interacts directly with Aβ, upstream of C3, and its expression is markedly elevated in AD (Yasojima et al., 1999). This figure is adapted from Zhang and Painter (Painter et al., 2015; Zhang et al., 2013).
TREM2 coordinates with microRNA155 in the development of the neurodegenerative activation states, so it is important to assess miR155 when TREM2 is elevated (Krasemann et al., 2017).
Aβ antibody infusions may be a useful intervention for early stage disease (Jack Jr. et al., 2010; Penninkilampi et al., 2017; Rygiel, 2016). Therefore, an alternative immunomodulatory approach to optimize resolution and amyloid clearance could be to coordinate gene expression within the TyroBP network (including increasing TREM2 and decreasing CD33), rather than simply suppressing microglial “activation” (Wyss-Coray and Mucke, 2002) with classic non-steroidal anti-inflammatory drugs (NSAIDs). Although NSAIDs were strongly supported pre-clinically (Lim et al., 2000; McKee et al., 2008; Van Dam et al., 2010; Yan et al., 2003), clinical data have yielded mixed results (Aisen, 2008; Breitner et al., 2011; Pasqualetti et al., 2009; Sonnen et al., 2010; Vlad et al., 2008). Clearance of Aβ with antibody (Lemere and Masliah, 2010) requires TREM2 expression (Xiang et al., 2016). We previously demonstrated that the amyloid binding polyphenol curcumin also reduced amyloid burden (Yang et al., 2005) by immunomodulating microglia (increasing phagocytosis) and reducing pro-inflammatory cytokines (Cole et al., 2004; Cole et al., 2003), similar to other polyphenols (Bickford et al., 2017; Rojanathammanee et al., 2013). It also protects in other models, including against tauopathy (Ma et al., 2013), Aβ toxicity, and Aβ accumulation (Frautschy et al., 2001; Lim et al., 2001). We therefore investigated immune gene expression after administration of curcumin. We used brain tissue in APPsw mice, which had previously been characterized for curcumin reduction in amyloid burden (Yang et al., 2005) (Lim et al., 2001) as well as human and rodent microglial cells to examine curcumin’s immunomodulatory effects on TREM2, CD33, and components of the TyroBP network.
2. Material & methods
2.1. Animals and treatments
All animal experiments complied with the National Institute Health’s guide for Care and Use of Laboratory Animals Publication 8023 and were approved by the institutional animal committees. Tg2576 mice (B6; SJL-Tg (APPSWE)2576Kh) with the APP Swe transgene) were treated with curcumin, as previously described (Lim et al., 2001). Briefly, 10-month old male and female Tg2576 from 12 litters were randomly split between treatment groups. Mice were fed either chow (PMI 5015; Feeds Inc., St. Louis, MO) containing a low dose of curcumin (160 ppm, n = 9, Sigma, St. Louis, MO), a high dose of curcumin (5000 ppm, n = 6), or no drug (n = 8) for 6 months before euthanasia. The low dose was chosen based on the amount of curcumin in turmeric consumed in the Indian diet and our prior study showing amyloid reduction, while the higher dose was based on anti-carcinogenic activity.
To measure mRNA responses to curcumin independent of amyloid, we conducted an acute study feeding curcumin to aged wildtype mice (C57Bl6/J) at 19 months of age. We fed them one of three treatment diets (n = 7 per group, gender-balanced) for 7 days: curcumin at 333 or 1000 ppm, or control diet (PMI 5015 supplemented with vehicle: olive oil). In the acute study we mixed curcumin into powdered chow, first dissolving curcumin into olive oil to ensure homogeneity of dispersion in the chow. At euthanasia, blood was collected and the buffy coat separated. After brain removal, the hippocampus was dissected and used for measuring the mRNAs for TREM2 and CD33.
For the passive immunization study, Tg2576 transgene-positive mice at 19.5 months of age were treated with antibody to Aβ (10G4, anti-Aβ 1–15, an IgG2b isotype, n = 4) or control IgG2b antibody (n = 8). Injections were given biweekly (0.5 mg i.p.), totaling 6 injections over 2.5 months. Mice were euthanized at 22 months of age and brain collected and dissected for measurement of TREM2 mRNA and Aβ.
Since micro-RNA were not available from the Tg2576 experiment and ApoE, Trem2 and miR155 induce a neurodegenerative microglial phenotype, we also examined curcumin’s effect on miR-155 in apoE3–5xFAD mice (n = 5) (human ApoE3 allele targeted replacement (Knouff et al., 1999), either transgenic for 5 FAD mutations in the human APP and PS1 genes (Youmans et al., 2012) (Tg+), or littermates without the 5xFAD transgenes (Tg-)), that were treated for 2 months with 500 ppm curcumin in diet starting at age 13 months.
During treatments with curcumin or immunization, there were no signs of toxicity and there were no differences in body weight between treatment groups.
2.2. Euthanasia and tissue preparation
After a lethal dose of pentobarbital (100 mg/kg, i.p.) and upon deep anesthesia indicated by cessation of toe, corneal and tail reflexes, the chest cavity was opened and mice perfused intracardially with a physiologically isotonic buffer containing 10 mM HEPES, 137 mM NaCl, 4.6 mM KCL, 1.1 mM KH2PO4, 0.6 mM MgSO4, 1.1 mM EDTA, and protease inhibitors (5 mg/mL of leupeptin and aprotinin and 2 mg/mL pepstatin A, pH 7.4).
Brains were bisected and the right hemibrain immersion-fixed in 4% formalin, sucrose cryopreserved, and frozen. Coronal cryostat sections (10 μm) were prepared for immunocytochemistry. The left hemibrain was dissected into major regions (hippocampus and cortex), snap frozen in liquid nitrogen and powderized for measurements of mRNA levels.
2.3. Gene expression mRNA levels
Levels of mRNA for genes were measured after RNA isolation, DNase treatment, and reverse transcription (random primers) using Ambion kits (Life Technologies), and specific gene mRNAs measured by standard Taqman QPCR methods, with gene-specific primers (genes TREM2, TyroBP/DAP12, CD68, Arg1, CD36, CD11b, iNOS, COX-2, C1q, and GAPDH; Life Tech., ABI) run in 384 well plates on an ABI 7900HT thermocycler; levels of gene-specific mRNA were normalized to levels of mRNA for housekeeping gene GAPDH in the same RNA samples. Statistical analyses were performed with SPSS V24 software (IBM Corp.). Differences among means were assessed by ANOVA followed by LSD post-hoc tests. Inclusion of sex in the ANOVA (transgene x sex), showed no sex effect in our cohorts, which were not powered to detect sex effects.
2.4. MiRNA-155 levels
Cortical total RNA including micro-RNA were prepared with MirVana kits (ThermoFisher Sci.) and 10 ng RNA was converted to cDNA using the TaqMan Advanced system (ThermoFisher Sci.). miR155 levels were measured with TaqMan Advanced assay in an ABI7900HT thermocycler, and levels were normalized by the deltadeltaCt method to levels of miR672 in the same samples (duplexed assay).
2.5. Immunohistochemistry (ICC) and staining by Griffonia (Bandeiraea) simplicifolia lectin I isolectin B4 (GS Isolectin B4)
Immunohistochemistry of anti-amyloid-stained brain deposits and other proteins was performed on coronal brain sections from animals treated with low-dose curcumin as described previously (Lim et al., 2001). Briefly, the hemibrain was sectioned from the posterior pole to the anterior margin of the hippocampus. Antigen retrieval was accomplished by incubating sections in an unmasking solution (Vector Laboratories, Burlingame, CA) for 30 s in a pressure cooker, allowed to cool to room temperature, then washed with TBS. Endogenous peroxidase was quenched with 0.3% hydrogen peroxide for 15 min. After blocking with normal serum, sections were incubated overnight at 4 °C (unless otherwise specified) with primary polyclonal antibodies: anti-Aβ (DAE (1:50) to Aβ1–13; anti-CD68 (1:50, sc9139, H255), anti-TREM2 (1:40, sc48765, M-227). The primary antibody to phosphotyrosine (PT, 1:50) was incubated at 37 °C for 40 min, which stains microglia (Frautschy et al., 1998).
The tissue was then incubated with secondary antibodies and developed for 30 min at 37 °C. For confocal microscopy, sections were incubated with fluorescent goat anti-rabbit antibodies (1:1000) and counterstained with DAPI (4–6-diamidino-2-phenylindole-containing medium, Vector Laboratories), while sections for light microscopy were incubated with biotinylated goat anti-mouse antibodies (1:1000) followed by ABC reagent, and then developed with metal enhanced DAΒ (Pierce, Rockford, IL) as previously described (Calon et al., 2004).
For staining the galactosidase resides on microglia, sections were first incubated with blocking solution, followed by the stain GS Isolectin B4 (1:100), then detected with the ABC kit and developed with DAB.
2.6. Ring analysis of CD11c stained microglia
Brain tissue slices from Tg2576 mice were examined for CD11b and CD11c immunoreactivity in proximity to amyloid plaques in the cortex by ring analysis, as previously described (Frautschy et al., 2001; Frautschy et al., 1998; Lim et al., 2001). Briefly, brain sections were taken at 2.5 mm posterior to Bregma ( ± 0.3 mm). Sections were double labeled for Aβ (anti-Aβ 1–13 DAE; blue) and microglia (anti-mouse CD11c (BD Biosciences)) with avidin biotin-peroxidase kit and the peroxidase detector (diaminobenzidine; brown). For acquisition of histological and immunohistochemical images for analysis, the blue slice (filter) of the RGB stack was chosen and density slice threshold selected such that the blue plaque would not be picked up when imaging the brown microglia; all images were captured using the same density slice threshold. Images were acquired at 20×, digitized, and quantitatively analyzed with NIH-Image public domain software. A custom Pascal macro subroutine measured the microglia as a % of total area, in relation to distance from the Aβ-immunoreactive plaque center in each of a series of three concentric circles within and around Aβ-immunoreactive plaques, the ring widths of which correspond to the radius of the Aβ plaque, where Ring 1 defines 2 plaque radius, and Rings 2 and 3 are one and two plaque radii away from the plaque edge.
2.7. High glucose induced inflammation of THP-1 cells (human monocytic cell line)
THP-1 cells were cultured at 37 °C and 5% CO2 in RPMI medium containing 10% FBS and 1% antibiotics. THP-1 cells were seeded in each well of 6-well plate at 2.5 × 105 cells for 24 h and 1 × 105 cells for 72 h treatment. After 24 h, THP-1 cells were pretreated for 1 h with curcumin (1.5 μM, in DMSO 0.1% v/v; see below) or without curcumin (adding the same volume of DMSO), then treated with hyperglycemic high glucose (25 mM) or mannitol (19.5 mM; isomolar control) for 24 h or 72 h. Cells were washed 3 times in PBS, then lysed with lysis buffer that included protease and phosphatase inhibitors (Complete, PhosStop; Roche). Curcumin (Sigma, MW: 368.4) was dissolved in DMSO, vortexed, incubated at 37 °C for 5 min, vortexed again, then aliquoted and frozen at −20 °C, all the while avoiding light. Western blot was used to measure cellular proteins TREM2, CD33, and GAPDH.
2.8. Aβ ELISA
ELISA measurements of protein levels were previously described (Lim et al., 2000). Briefly, Aβ (insoluble) was measured in tissue samples that were first homogenized in 10× vol of TBS, centrifuged (100,000 xg for 20 min at 4 °C) and the TBS-insoluble pellet was dissolved in 70% formic acid (triturated then sonicated for 10 min), followed by neutralization with NaOH and dilution with Tris buffer. The monoclonal 4G8 against Aβ 17–24 (Senentek, Napa, CA) was the capture antibody (3 μg/ml), biotinylated 10G4 against Aβ 1–15 was the detecting antibody, and the reporter system was streptavidin-alkaline phosphatase with AttoPhos (JBL Scientific Inc., San Luis Obispo, CA) as the substrate (excitation 450 nm/emission 580 nm).
2.9. IL1β sandwich ELISA
IL1β in cell culture media was measured using the polyclonal antibody against mouse IL1β (Endogen, Woburn, MA) as the capture antibody and monoclonal anti- mouse IL1β (Endogen) as the detector with the minimum sensitivity for ILβ at 0.5 pg, as previously described (Lim et al., 2000). Statistical analysis of ELISA data was by ANOVA and LSD Fisher for post-hoc analysis of planned comparisons.
2.10. Co-culture of mouse microglia with human AD brain slices
We originally developed the method of co-culturing human AD brain tissue with mouse microglia (Ard et al., 1996), and it has been subsequently used by several groups (Bard et al., 2000; Demattos et al., 2012; Hashioka et al., 2008; Lucin et al., 2013; Ostrowitzki et al., 2012). Culture media used were basal media (BM) (Thermofisher), glial media without serum (GM) (Thermofisher), or glial media with 10% fetal calf serum (GM/FCS).
Human AD brain from fresh frozen autopsy was obtained from the UCLA and USC Brain Banks. Temporal lobe brain was cryosectioned (10 μM slices) and thaw-mounted on PdL coated glass coverslip, that was then placed in a 24 well plate, air-dried for 30 min, then washed twice in BM. Experimental medium (400 μl of GM) containing drugs, e.g. 10G4 antibody against Aβ, control isotype antibody IgG2b, or curcumin, was added and incubated for 1 h at 37 °C in a CO2 incubator. The medium was removed (and saved for later continued treatment after microglial seeding). Mouse microglia (400,000 cells) were incubated in GM/FCS and seeded on top of the human brain slice for 1 h. Then the cultures were washed three times with BM. Mouse primary microglia were derived from brain glial cultures from postnatal day 3 FVB mouse pups as previously described (Saura et al., 2003). The experimental medium containing drugs was returned to each well, and the cultures were maintained for 36 h. The cultures were washed with cold PBS, and harvested in TBS containing a cocktail of protease inhibitors (20 μg/ml each pepstatin A, aprotinin, phosphoramidon, and leupeptin, 0.5 mM PMSF, and 1 mM EGTA), then prepared for either Aβ ELISA or fixed for ICC.
For treatment and control conditions, adjacent AD brain slices were used pairwise, so that Aβ levels were similar within pairs. For statistical analysis, Aβ levels in paired treatment and control slices were analyzed by a non-parametric Mann-Whitney pairwise statistical test.
2.11. Hippocampal slice culture and exogenous amyloid deposition
Organotypic hippocampal slice cultures (OHSC) were prepared from wild-type C57Bl6 mice at 7 days of age and maintained as described (Harris-White et al., 1998; Stoppini et al., 1991; Teter et al., 1999). Briefly, 400 μm slices were maintained in membrane inserts (Costar, Cambridge, MA, 0.4 mm) using media of MEM plus HEPES with the serum substitute TCM (final concentration 2%; ICN Pharmaceuticals, Costa Mesa, CA). Starting on the first day in vitro (0 DIV), cultures were treated with Aβ40 (15 μg/ml) for 4 days. The medium was changed to that without Aβ and cultured for 4 more days, to allow Aβ deposits to develop. At 8 DIV, cultures were treated for 4 days with or without curcumin (at two doses), or the anti-Aβ antibody 10G4 (5 μg/ml), or control isotype antibody IgG2b. The media was changed at day 2 of the treatment period to fresh media containing the respective treatments. The cultures were harvested, extracted with formic acid, and Aβ measured by ELISA. Controls (not shown) were used to show that addition of 10G4 to cultures immediately before formic acid treatment did not interfere with the ELISA, which used 10G4 for detection, presumably because formic acid irreversibly denatured the 10G4 antibody.
2.12. Phagocytosis assay in vitro
BV2 cells were grown to ~50% confluency in 96 well plates (Black, Nunc; parallel cultures in clear plates were also grown to monitor cultures); triplicate wells were treated. Curcumin (1 μM) in 0.05% DMSO was added for 4.5 h, followed by addition of “Bioparticles”, fluorescein-labeled E. coli cells (Vybrant Phagocytosis Assay kit; Molecular Probes), to the media for 3 h. Media is removed and cells were treated with trypan blue for 1 min (which effectively quenches the extracellular Bioparticle fluorescence). Cells were washed 5 times in ice cold Dulbecco’s Modified Eagle’s Medium (without phenol red), then washed once with sodium citrate buffer (pH 5.0) (to further quench extracellular fluorescence); this buffer was removed, then fluorescence was measured at Ex/Em ~480/520 nm in a plate reader. Controls included azide pretreatment which blocked phagocytosis (fluorescence values in azide-treated well were subtracted from all other reading as a measure of background fluorescence).
3. Results
The activation of microglia and associated phagocytosis phenotype is controlled by tyrosine-phosphorylation of AD-associated innate immune risk genes, as illustrated in Fig. 1.
The effects of different doses of diet-administered curcumin, low dose (160 ppm, “Curc-Lo”) and 30-fold higher dose (5000 ppm, “Curc-Hi”), on microglial activation and expression of AD genetic risk factor microglial genes were evaluated in APPSwe (Tg2576) transgenic amyloid plaque-forming mice. Tg2576 mice were fed curcumin doses of 0, 160 or 5000 ppm in PMI 5015 breeder chow from 10 to 16 months of age. We previously reported that these Curc-Lo-treated mice showed decreased inflammation and oxidative damage (e.g. decreased IL1β, GFAP and carbonyl (oxidized) proteins) as well as decreased Aβ levels (decreased amyloid plaque burden, soluble Aβ, and insoluble Aβ) in the brain (Lim et al., 2001). While using an intermediate dosing with 500 ppm from 17 to 22 months (after amyloid accumulation) produced large decreases in IL1β and amyloid burden (Yang et al., 2005), surprisingly, Curc-Hi did not affect insoluble or soluble Aβ levels (Lim et al., 2001),
3.1. Effects of curcumin on brain innate immune gene expression
To evaluate the role of microglial activation state in these differential effects of curcumin dose on inflammation and amyloid, we examined brain cortex expression levels of inflammatory genes implicated in amyloid clearance, including CD36, C1q, innate immune genes
TREM2, TyroBP/DAP12, and CD33 (genetic risk factors for AD), microglia phenotype markers CD11c, CD11b, CD68 and P2ry12, and the brain microglia-specific gene TMEM119 (see Fig. 1). Gene mRNA levels, measured by real time qPCR, were differentially regulated by low and high dose curcumin (Fig. 2A); genes upregulated and downregulated by Curc-Lo are shown in the upper and lower rows, respectively, of Fig. 2A. In addition, to examine coordinated regulation, gene expression ratios and correlations were examined. Details of the statistical analyses are shown in Table 1, and the main findings are summarized in Table 2.
Fig. 2.

Gene mRNA levels differentially regulated and restored by low and/or high dose curcumin. (A) Gene expression mRNA levels were measured by quantitative RT-PCR (Taqman method), and levels were normalized to levels of GAPDH or HPRT in each sample. Each gene’s mRNA level was normalized to the average level in the Tg + Control diet animals, whose expression levels are therefore defined as 1.0 in the bar graph. (B) The ratio of mRNA levels for TREM2 and either CD33, CD11b, or TMEM119 were calculated. Curcumin increased the TREM2:CD33, TREM2:CD11b, and TREM2:TMEM119 gene expression ratios, more so with low dose curcumin than high dose curcumin. (C) TREM2 mRNA levels in hippocampus of C57Bl/6J mice at 19 months treated with curcumin in diet at doses of 333 ppm and 1000 ppm for 7 days. TREM2 levels were normalized to GAPDH mRNA in the same samples. (D) The ratio of mRNA levels of TREM2 to CD33 (normalized to GAPDH) in hippocampus and blood buffy coat of C57Bl/6J mice at 19 months treated with curcumin in diet at doses of 333 ppm and 1000 ppm for 7 days. (E) ApoE3–5XFAD mice (Tg+) with targeted hu ApoE3 and littermate 5xFAD transgene negative (Tg-) (n = 6–10/group) were treated with curcumin at 500 PPM for 2 months starting at 13 months age. Brain cortex was used to measure microRNA155 (normalized to miR672). The FAD-dependent increase in miR155 was effectively reduced by curcumin to control levels. Error bars indicate the SEM; *p < .05; **p < .01; ***p < .005; ****p < .0001. Multivariate analysis was used and post hoc tests included Fisher’s LSD with homogeneity or Games Howell when variances were unequal (p < .05 Levene’s test, Table 1).
Table 1.
Statistics for Fig. 2, Gene mRNA levels differentially regulated and restored by low and high dose curcumin.
| A. CDllb, CDllc, iNOS, C1q, Arg1 (For Fig. 2A). | ||||
|---|---|---|---|---|
| MANOVA F(10,30) = 5.545 |
LSD post-hoc, LEVENE p < .05 |
|||
| Wilks = 0.123, n2 = 0.649, p < .00001 | Control vs Low | Control vs High | High vs Low | |
| logCDllb to control | F(2,19) = 4.087, p = .033 | 0.012 | 0.076 | NS |
| INOS normal control | F(2,19) = 6.950, p = .005 | 0.002 | 0.013 | NS |
| COX2 | F(2,18) =16.580, p = .0001 | 0.001 | 0.001 | NS |
| sqrtC1q to control | F(2,19) = 6.589, p = .007 | 0.050 | 0.112 | 0.002 |
| sqrtCD11c control | F(2,19) = 3.142, p = .066 | 0.021 | NS | 0.155 |
| ARG1 normal control | F(2,19) = 5.979, p = .01 | 0.107 | 0.003 | 0.148 |
| B. TMEM and P2yr12 (For Fig. 2A) | ||||
| MANOVA F(4,52) = 4.654 |
p Values for GAMES HOWELL post-hoc when LEVENE p > .05 |
|||
| Wilks = 0.542 n2 = 0.264 p < .003 | Control/Low | Control/High | High/Low | |
| TMEM normal Ctrl | F(2,27) = 8.057, p = .002 | 0.001 | NS | 0.085 |
| P2ry12 normal to control | F(2,27) = 7.222, p = .003 | 0.011 | NS | 0.008 |
| C. TREM2, CD33, CD68, TYROBP (For Fig. 2A) | ||||
| MANOVA F(8,48)=11.226 |
p Values for GAMES HOWELL post-hoc when Levene p > .05 |
|||
| Wilks = 0.12, n2 = 0.562, p < .0001 | Control/Low | Control/High | High/Low | |
| TREM2 | F(2,27) = 9.975, p = .001 | 0.0001 | NS | 0.002 |
| CD33 | F(2,27) = 10.735, p = .0001 | NS | 0.0001 | 0.095 |
| CD68 | F(2,27) = 9.788, p = .001 | 0.038 | NS | 0.055 |
| TYROBP | F(2,27) = 6.844, p = .004 | 0.049 | NS | 0.05 |
| D. TREM2 ratios (For Fig. 2B) | ||||
| MANOVA F(6,34) = 5.039 |
p values for LSD post-hoc, Levene p < .05 |
|||
| Wilks = 0.280, n2 = 0.471, p < .001 | Control vs Low | Control vs High | High vs Low | |
| TREM2/CD33 | F(2,19) = 19.11, p = .0001 | 0.0001 | 0.001 | 0.048 |
| LOG (TREM2/CD11b) | F(2,19) = 8.678, p = .002 | 0.001 | NS | 0.029 |
| log (TREM2/TMEM119) | F(2,19) = 5.247, p = .015 | 0.005 | NS | 0.044 |
| E. Log Trem-2 mRNA in hippocampus (For Fig. 2C) | ||||
| One Way ANOVA F(2,20) = 3.093, p = .070 |
p Values for LSD post-hoc, Levene p < .05 |
|||
| Control vs Low | Control vs High | High Vs Low | ||
| 0.028 | NS | NS | ||
| F. TREM2/CD33 buffy and hippocampus (hippo) (For Fig. 2D) | ||||
| UNIVARIATE | F(5,37) = 5.025 | p Values for Games Howell post-hoc, Levene p > .05 | ||
| 2-way dose (hi vs lo) × compartment (buffy vs hippo) | p = .013, r2 = 0.250 | Control vs Low | Control vs High | High Vs Low |
| Dose | F(2,37) = 6.538, p = .005 | buffy 0.001 | NS | 0.001 |
| Compartment (buffy vs hippo) | F(2,37) = 0.32, NS | hippo 0.050 | NS | NS |
| Dose* compartment | F(2,37) = 3.082, p = .06 | |||
| G. Log miRNA155 (for Fig. 2E) | ||||
| UNIVARIATE | F(3,28) = 16.482 | p Values for LSD post-hoc, Levene p = .354 | ||
| Two way Diet × FAD | p < .001 | FAD Curc vs FAD Control | Tg- Curc vs Tg-Control | FAD control vs Tg- control |
| Diet | F(1,28) = 18.528, p = .0001 | 0.001 | NS | p < .001 |
| FAD | F(1,28) = 11,225, p = .003 | |||
| Diet × FAD | F(1,28) = 7.624, p = .011 | |||
Table 2.
Summary of the curcumin effects on genes
| Summary of gene effects | Gene | Role | Low curcumin | High curcumin |
|---|---|---|---|---|
| Innate immune, neuroprotective-associated microglial phenotype | TREM2 | Triggering receptor on myeloid cells, central microglial hub gene, regulating tyrosine kinase signaling in microglia; loss of function increases AD risk (Cherry et al., 2014,Kamphuis et al., 2016; Keren-Shaul et al., 2017) | ↑ | ns |
| TyroBP (DAP12) | Central microglial hub gene coding for adapter protein for TREM2 (and Fc receptor) coupling to tyrosine kinase signaling in microglia, with genetic variants that impact AD risk (Forabosco et al., 2013; Hickman et al., 2013; Keren-Shaul et al., 2017; Zhang et al., 2013) | ↑ | ns | |
| Arg1 | M2 marker associated with resolving IL4 mediated Aβ plaque reduction (Gaikwad and Heneka, 2013) | ns | ↓ | |
| Cd11c | (CR4, ITGAX) Innate immune cell-specific integrin (Kamphuis et al., 2016; Keren-Shaul et al., 2017; Krasemann et al.,2017) | ↑ | ns | |
| CD68 | Innate immune microglial lysosomal antigen upregulated in phagocytic cells (Zhao et al., 2016; Zotova et al., 2011) | ↑ | ns | |
| Immunodysregulation, neurodegenerative associated microglial phenotype | Cd11b | (CR3, ITGAM) integrin receptor for complement opsonized Abeta, on innate immune cells, “M1” microglia activation marker, elevated in AD (Gaikwad and Heneka, 2013) | ↓ | ns |
| iNOS* | Enzyme synthesizing NO, M1 microglia activation marker, elevated in AD (Gaikwad and Heneka, 2013) | ↓ | ↓ | |
| C1q | Highly elevated in aging and AD, first step in complement cascade and associated with synaptic stripping (Yasojima et al., 1999) | ↓ | ns | |
| CD33 | Siglec receptor, tyrosine kinase signaling, elevated levels genetically implicated in AD (Bradshaw et al., 2013; Griciuc et al., 2013;Malik et al., 2015) | ns | ↓ | |
| mir155 | Induces neurodegenerative microglia (Krasemann et al., 2017; Ma et al., 2017) | ↓ | – | |
| COX2* | pro-inflammatory enzyme, elevated (neuronal) in AD, affects excitoxicity (Chaudhry et al., 2010) | ↓ | ↓ | |
| Microglial markers | TMEM119 | Transmembrane protein, Microglia -specific marker, elevated in AD (Keren-Shaul et al., 2017; Satoh et al., 2016) | ↓ | ns |
| P2ry12 | Purinergic G-protein coupled receptor for ATP/ADP, on non-activated as well as activated microglia (Keren-Shaul et al., 2017) | ↓ | ns |
Low and high dose have similar effects.
CD11c (an integrin, “Itgam”), has low expression in normal mouse brain, but increased expression is associated with microglial activation in Alzheimer’s disease and in several AD mouse models (Kamphuis et al., 2016; Keren-Shaul et al., 2017; Krasemann et al., 2017). CD11c mRNA was increased nearly 2-fold by Curc-Lo (+98%; p < .05) but less by Curc-Hi (+46%; not significant (n.s.)).
CD11b (an M1 marker (Gaikwad and Heneka, 2013), “Itgax”) was significantly decreased by Curc-Lo (−35%; p < .05) and slightly less so by Curc-Hi (−22%; n.s.).
TREM2, an AD risk gene whose expression is critical for the development of candidate beneficial microglial activation states (Kamphuis et al., 2016; Keren-Shaul et al., 2017) and an M2 marker (Cherry et al., 2015), was significantly increased > 2-fold by Curc-Lo (+109%; p < .0001). In contrast, Curc-Hi did not change TREM2 expression.
CD33 is a siglec receptor with genetically elevated levels in AD. Although Curc-Lo did not decrease CD33 significantly (p = .095), Curc-Hi decreased CD33 by 44%; (p < .0001). While the effect of Curc-Lo (160 ppm) on CD33 was small in this study, a slightly higher dose of curcumin (500 ppm) decreased CD33 more (−33%; p = .06) (data not shown) in the same Tg2576 mice, but on a different base diet; this was associated with a large, significant reduction in plaques and Aβ (measured by ELISA) to a level lower than the level the mice started with at the beginning of treatment (17 months of age, an age that has a large amyloid burden), suggesting this curcumin dose (500 ppm) caused Aβ/amyloid clearance (Yang et al., 2005).
TyroBP (DAP12), a TREM2 signal transduction effector with upregulated expression in the early development of a putative disease restricting microglial phenotype (Keren-Shaul et al., 2017) was significantly increased by Curc-Lo (+87%; p < .05) but not by Curc-Hi.
CD68, a marker of microglial phagocytosis, was increased 2.3-fold by Curc-Lo (+130%; p < .05), but not Curc-Hi. This supports an increased phagocytic phenotype for Curc-Lo but not Curc-Hi.
Arg1 (an M2 marker) (Gaikwad and Heneka, 2013), was not significantly changed by Curc-Lo, but in sharp contrast, Curc-Hi greatly decreased Arg1 (−66%; p < .005).
iNOS (an M1 marker) (Gaikwad and Heneka, 2013) was decreased by Curc-Lo (−53%; p < .01) and less so by Curc-Hi (−38%; p < .05). Further, iNOS and CD11b (both M1 markers) expression levels were correlated in Curc-Lo (see Gene expression correlations, Table 3 below).
Table 3.
Correlations between gene expression (mRNA levels) affected by curcumin. Coordinated changes in gene expression were evaluated by comparing two gene mRNA levels, or marker values [phosphotyrosine (PT) or TREM2:CD33 ratio], in conditions of control diet, Curc-Lo, or Curc-Hi.
| Ratio of genes or markers | ||||||
|---|---|---|---|---|---|---|
| Treatment in Tg2576 | TREM2: TYROBP | TREM2: CD68 | TREM2: PT | iNOS: CD11b | Arg1: (TREM2:CD33) | Arg1: TYROBP |
| Control diet | 0.64*** | 0.77** | n.s. | n.s. | n.s. | n.s. |
| Curcumin-Low (160 ppm) | 0.98** | 0.98** | 0.96** | +0.94** | n.s. | +0.72* |
| Curcumin-High (5000 ppm) | n.s. | n.s. | PT Not determined | n.s. | n.s. | −0.66* |
R2 values and statistical significance are shown in each cell for Pearson, 1-tailed correlations. Not significant (n.s.) p >
p < .05
p < .01
p < .001.
COX-2 (Prostaglandin-endoperoxide synthase 2; PTGS2) is an inducible cyclooxygenase found in neurons, endothelial cells and activated microglia in Alzheimer brain (Chaudhry et al., 2010) and produces prostaglandins in response to its upregulation by inflammation. COX-2 was decreased by Curc-Lo (−66%; p < .0001) and by Curc-Hi (−54%; p < .005).
C1q interacts directly with Aβ, and its expression is markedly elevated in AD (Yasojima et al., 1999). Curc-Lo but not Curc-Hi decreased C1q (−38%; p < .05).
TMEM119, a brain microglia-specific gene whose gene expression reduction is a marker of candidate beneficial disease-associated microglial (“DAM”) phenotype changes in AD and AD mouse models (Keren-Shaul et al., 2017; Satoh et al., 2016) was reduced by Curc-Lo (−68%, p < .005), but a lesser reduction by Curc-Hi was not significant.
P2ry12, a G-protein coupled receptor, whose gene expression reduction marks an early stage of candidate “DAM” beneficial microglial phenotype changes in AD and AD mouse models (Keren-Shaul et al., 2017) was reduced by Curc-Lo (−43%; p = .011), but unchanged by Curc-Hi.
3.2. Gene expression ratios reflect balanced regulation
TREM2:CD33. The ratio of gene expression levels for TREM2:CD33 was increased by curcumin treatments, more so (p < .05) for Curc-Lo (160 ppm) which increased TREM2:CD33 2.4-fold (p < .0001; Fig. 2B) while Curc-Hi increased TREM2:CD33 1.9-fold (p < .01), which was significantly less than the increase by Curc-Lo (p < .05). Further, only Curc-Lo increased the ratio of TREM2 to TMEM119 (p < .005) and TREM2 to CD11b (p < .001) (Fig. 2B), indicating that curcumin was modulating the microglial phenotype, not just increasing microglial numbers. Another relatively low dose of curcumin (500 ppm) in a base diet high in n-6 polyunsaturated fatty acids (Calon et al., 2004; Lim et al., 2005) treated from 17 to 22 months of age also increased the TREM2:CD33 ratio 54% (from 3.5 on the base diet to 5.4 with curcumin (p < .001), data not shown) and was also associated with reductions in amyloid plaques and insoluble Aβ (Yang et al., 2005).
3.3. Gene expression correlations reflect coordinate regulation
Innate immune responses can be coordinately regulated at the gene transcription level, for example, by NFκB, a transcription factor responsive to curcumin or by TyroBP, a master regulator of gene expression changes in AD (Zhang et al., 2013) as well as by PU.1 (Efthymiou and Goate, 2017, see Discussion). Consistent with a coordinated transcriptional impact and tighter control over hub gene expression, Curc-Lo treatment led to more highly correlated expression levels of several hub genes and microglial signaling activation markers (Table 3).
TREM2:TyroBP. TREM2 and TyroBP mRNA levels were significantly correlated in control diet (r2 = 0.64; p < .01). Curc-Lo increased both TREM2 and TyroBP mRNA levels, as well the magnitude of the correlations within each animal (r2 = 0.98; p < .01). In contrast, Curc-Hi treatment eliminated this tight correlation between TREM2 and TyroBP (r2 = 0.23).
TREM2:CD68. Similarly, TREM2 and CD68 mRNA levels were correlated in control diet (r2 = 0.77; p < .01). Curc-Lo increased both TREM2 and CD68 mRNA levels as well as the magnitude of the correlations within each animal (r2 = 0.98; p < .01). In contrast, Curc-Hi treatment eliminated the correlation between TREM2 and CD68 (r2 = 0.30, n.s.).
TREM2:phospho-tyrosine (PT). Increased phosphotyrosine is a marker of microglial activation, reflecting increased phosphorylation of signaling proteins that regulate microglial phenotypes (see Fig. 1). We previously showed that, in the same Tg2576 mice analyzed here, Curc-Lo (160 ppm) increased PT in and around amyloid plaques while decreasing PT in other areas (Frautschy et al., 2001; Lim et al., 2001). We correlated PT levels around plaques with TREM2 mRNA levels in the contralateral hemisphere within each animal. In control animals, TREM2 mRNA and PT around plaques were not correlated (Table 3). In contrast, with Curc-Lo treatment, TREM2 mRNA and PT around plaques were highly correlated (r2 = 0.96; p < .01). This close relationship between TREM2 and PT supports an impact of hub genes on tyrosine phosphorylation in plaque microglia and was further verified by immunocytochemical colocalization (see below, Fig. 3B). A similar effect was seen for TyroBP/DAP12 mRNA levels and PT (data not shown), reflecting the high correlation between TREM2 and TyroBP/DAP12 in Curc-Lo animals (see above).
Fig. 3.

Immunocytochemical colocalization of Aβ and microglial markers in untreated and Curc-Lo (160 ppm) treated Tg2576 mice. Light microscopy images of brain sections from Tg2576 mice fed control diet (control, left columns) or curcumin 160 ppm diet (Curcumin, right columns) (A–C). Sections were stained as follows: DAPI (blue, nuclei). (A) Aβ (blue); PT (brown). Compared to control-treated mice, curcumin increase colocalization of PT (microglia) and Aβ in and around the amyloid plaque. (B) Aβ (blue); CD11c (brown). Curcumin treatment increased the CD11c staining adjacent to the amyloid plaque. (C) In both groups, there was colocalization of Aβ (blue) and CD11b (brown). (D) Graphs represent image analysis of microglia around plaques according to distance from plaque with ring 1 representing area within the Aβ-ir plaque and ring width is plaque radius (see Methods). The majority of microglia inhabit the central portion of plaques (Ring 1), while outer portions of plaques (Rings 2 and 3) contain fewer microglia. Two-way ANOVA was performed (ring * treatment). Both CD11b and CD11c showed significant ring effects for both variables (cell size and total cell area per ring, p < .0001). However, there were only treatment effects for CD11c (cell size: p = .05; total cell area: p = .007). t-tests showed differences between treatments in ring 2 (cell size: p = .003; total area ring: p = .004). Both CD11c variables showed higher values in the Curc-lo group (E), Griffonia simplicifolia isolectin B4 (red); Aβ (green). In control animals, Aβ plaques and lectin microglia show no overlap (yellow). Curcumin increased the overlap between lectin-stained microglia and Aβ in and around plaques in the hippocampus. z-plane analysis (right panel shown; z = 0.05) indicated that the overlap (yellow) occurs in the same optical plane, suggesting internalization of the amyloid by microglia. (F) Aβ (green); PT (red). Curcumin treatment increased the colocalization (yellow) of PT within and around amyloid plaques. (G) Aβ (green); CD68 (red). Curcumin treatment increased the number of CD68 (phagocytic) microglia. The inset shows a magnified view of one plaque with extensive overlap (yellow) of CD68 (lysosome marker in microglia) with amyloid. (H) TREM2 (green); PT (red). This section is a plaque-containing field, adjacent to plaques shown by amyloid staining in Panel E. Curcumin treatment increased TREM2 and PT and increased their co-localization (yellow) within plaques (plaques identified in Panel E). Scale Bar Magnification = 25 μm. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
iNOS:CD11b. iNOS and CD11b (both M1 markers) expression levels were correlated in the Curc-Lo-treated mice (r2 = +0.94; p = .004), but not in the control-treated mice. Curc-Hi treatment tended to reverse the correlation to r2 = −0.50 (n.s., Table 1).
Arg1 mRNA levels were significantly and positively correlated with TyroBP/DAP levels with Curc-Lo treatment but significantly negatively correlated with Curc-Hi treatment (Table 1).
To summarize gene expression changes and correlations, Curc-Lo promoted up-regulation of innate immune hub genes TREM2 and TyroBP and increased their tightly coordinated regulation with peri-plaque microglial tyrosine phosphorylation known to control functional activation. Curc-Lo also led to coordinately regulated TREM2 and CD33, as reflected in their increased ratio, a measure that was useful in describing curcumin effects in aged mice, described below. Curc-Lo upregulated markers related to microglial phagocytosis, CD68 and Arg1, while coordinately down-regulating closely correlated M1 markers CD11b and iNOS.
In contrast to these effects of Curc-Lo, Curc-Hi treatment showed: (1) only some of the immunosuppressive effects seen with Curc-Lo and diminished responses in TREM2, TyroBP, CD68, and Arg1; (2) Curc-Hi expression ratios of TREM2:TyroBP, TREM2:CD68, iNOS:CD11b, and Arg1:TyroBP were no longer correlated and (3) C1q was upregulated- in contrast to suppressed as with Curc-Lo. These findings indicate Curc-Hi may suppress some beneficial microglial activities related to amyloid clearance and control of complement.
3.4. Immunomodulatory effect of curcumin in the absence of amyloid pathology
Because curcumin decreased amyloid burden in Tg2576 mice, and neuroinflammation markers interact with amyloid load, we investigated the impact of curcumin on aging in the absence of amyloid reduction effects. We used aging as a driver of neuroinflammation in wild-type mice and assessed the dose-dependent impact of short-term curcumin on the ratio of TREM2 to CD33 expression in both brain and blood.
C57Bl6/J mice at 19 months of age were treated with curcumin in diet at levels of 333 ppm and 1000 ppm for 7 days. Blood buffy coat and hippocampus were harvested, before and after perfusion, respectively. TREM2 and CD33 mRNA levels were measured and normalized to GAPDH mRNA in the same samples.
In the hippocampus, curcumin at 333 ppm increased TREM2 by 31% (p < .05) but curcumin 1000 ppm did not (11% increase; not significant, Fig. 2C). The TREM2:CD33 ratio was significantly increased by curcumin 333 ppm in both hippocampus (24%; p < .05) and buffy coat (65%; p = .001) (Fig. 2D, Table 3). In contrast to these effects of curcumin at 333 ppm, curcumin at 1000 ppm did not change the TREM2:CD33 ratio in the hippocampus or buffy coat (−36%; n.s.) Fig. 2D).
These results further define the curcumin dose response showing beneficial, coordinated regulation at 333 ppm like those seen at 160 ppm, and lack of these beneficial effects and potential deleterious effects at 1000 ppm like those seen at 5000 ppm. These results show that dose-dependent effects of curcumin impact inflammation induced by aging, like those induced by amyloid in the Tg2576 mice.
Immunomodulatory effects of curcumin could be mediated by its master regulator, microRNA-155 (miR-155) (Krasemann et al., 2017), which has been shown to be regulated by curcumin in vitro (Ma et al., 2017). The miR-155 gene contains NFκB elements (Elton et al., 2013) that could mediate transcriptional control by curcumin’s target, NFκB. To measure miR155 we used another model because there was no remaining RNA left from the Tg2576 curcumin study. We treated apoE3–5xFAD mice (human ApoE3 allele targeted replacement, with (Tg+) or without (Tg-) the transgene of 5 FAD mutations in the human APP and PS1 genes) with curcumin at 500 PPM in diet for 2 months starting at age 13 months. These E3FAD Tg + mice showed a 2-fold increase in miR-155 compared to FAD Tg- littermates (p < .001) (Fig. 2E), coinciding with the extensive amyloid pathology, inflammation, and neurodegeneration (neuritic dystrophy and synapse loss) at this age. The Tg2576 and E3FAD have less neuron loss at the ages examined than the 5xFAD (due to ApoE3 slowing progression), but have similar levels of amyloid and synaptic pathology. Curcumin treatment reduced the level of miR-155 in FAD Tg + to that of the FAD Tg- mice (p < .001).
3.5. Microglial activation responses to amyloid and low dose curcumin
We previously showed that curcumin has different effects on microglia in relation to their proximity to amyloid plaques (Lim et al., 2001). Here we examined plaque-associated microglia, and the co-immunolocalization of microglial marker (GS-Lectin) or amyloid plaques with microglial activation phenotype markers, TREM2, CD68, and phosphotyrosine (PT) (Fig. 3A–H).
Phosphotyrosine, in microglia was decreased by curcumin treatment of rodents (mice and rats) in many brain regions when measured away from amyloid plaques, but was increased in and around amyloid plaques (Frautschy et al., 2001; Lim et al., 2001). This effect is further evaluated in the present study co-localizing PT (DAB, brown) and Aβ (Oregon blue) (Fig. 3A) and confocal imaging (Fig. 3F, green, FITC, Aβ; red, rhodamine, PT), where representative examples of amyloid plaques show PT-stained microglial-like cells clustered around plaques in both control and curcumin-treated mice, but curcumin treatment showed intensely PT-stained microglia within the plaque itself, consistent with our previous findings in curcumin-treated, plaque-bearing mice and rats (Frautschy et al., 2001; Lim et al., 2001).
3.6. Colocalization of plaque-associated PT and TREM2
We hypothesized that if the curcumin-increased PT staining within plaques reflects phosphorylation in protein signaling cascades associated with TREM2 activation, then TREM2 and PT should co-localize within plaques. Using confocal double-labeling, TREM2 and PT co-localized in plaques in Curc-Lo treated mice (Fig. 3H). The fluorescent overlap of TREM2 (green) and PT (red) is shown by yellow fluorescence within plaques that were verified in the adjacent section with staining for amyloid (Fig. 3E). This qualitative protein co-localization supports the quantitative correlation (described above) between elevated TREM2 mRNA levels and plaque-associated PT levels within animals treated with Curc-Lo.
Curc-Lo treatment led to a qualitative increased overlap of CD68 (a marker of phagocytosis) and Aβ/plaques (Fig. 3G) consistent with elevated CD68 mRNA levels. The inset shows a high magnification of one plaque showing CD68 (red) microglial morphology, and distinct overlap (yellow) between CD68 and Aβ (green) (Fig. 3G).
GS Isolectin B4, binds to terminal alpha-D-galactosyl residues of polysaccharides and glycoproteins, specific to microglia (Streit and Kreutzberg, 1987) and strongly labels cells with activated morphology (Taylor et al., 2002). In control animals, amyloid (green) and lectin (red) microglial staining was typically separate with little overlap (yellow) as shown in Fig. 3E (left panel). In contrast, Curc-Lo treated mice (Fig. 3E, right panel), showed abundant co-localization of amyloid and microglia (yellow amorphous structures). Further, isolated Z-planes with confocal microscopy revealed that the colour overlap of amyloid and microglia (overlap is yellow) occurs in the same optical plane, consistent with internalization of the amyloid by microglia (data not shown).
Fig. 3B, C shows images of colocalization of Aβ with either CD11c or CD11b with control or Curc-lo (Fig. 3B, C). CD11b stained microglia that were colocalized with Aβ in both groups. In control mice, there was less staining for CD11c than for CD11b, and the CD11c staining was diffuse. In contrast, CD11c staining in Curc-lo group was prominent in cells and colocalized with Aβ inside and outside of the plaque.
This was quantified by image analysis of CD11c and CD11b immunostaining in spatial relation to plaques, called ring analysis (illustrated in Fig. 3D). Rings 1–3 indicate the cell size and cumulative areas of CD11b-ir or CD11c-ir; Ring 1 is the area of the Aβ-ir plaque. The width of Rings 2 and 3 is the plaque radius (see Methods). In untreated and untreated Tg2576 mice, the majority of CD11b-ir and CD11c-ir microglia inhabit the central portion of plaques (Ring 1), while outer portions of plaques (Rings 2 and 3) contain fewer amounts of microglia.
Two-way ANOVA (ring * treatment) was performed on cell size and total cell area per ring for both CD11b and CD11c. While there were no interactions between treatment and ring, both CD11b and CD11c showed significant ring effects for both variables (cell size and total cell area per ring): cell size (CD11b: F(2,807) = 217.0, p = .0001; CD11c: F(2,1118) = 99.9, p = .0001) and total cell area within ring (CD11b: F(2,807) = 901, p = .0001; CD11c: F(2,118) = 463.2, p = .0001).
While there were no treatment effects with CD11b for any variables, there were treatment effects for CD11c (cell size: F(1,118) = 3.74, p = .05; total cell area: F(1,118) = 7.33; p = .007). T-tests showed differences between treatments in ring 2 (cell size: t(139) = −3.0; p = .003; total area ring: t(138) = −3.6, p = .004.)
Because the in vivo results in Tg2576 mice suggested curcumin altered AD-related innate immune gene expression and function, we next asked whether it might have similar activity with human innate immune cells.
3.7. Curcumin regulation of TREM2 and CD33 protein in human THP-1 monocytic cell line in vitro
THP-1 is a human monocytic cell line that can be activated by high glucose (HG), and the resulting pro-inflammatory phenotype can be suppressed by 1.5 μM curcumin through NF-κB inhibition (Yun et al., 2011). We used this model to test whether this suppression of the inflammatory response by curcumin involves altered TREM2 and CD33 expression. Consistent with a delayed TREM2 response, after 72 h of HG treatment, TREM2 protein was significantly increased by low dose curcumin (0.1 μM), but not high dose curcumin (1.5 μM) (Fig. 4A, B) but TREM2 protein was not modulated in this system at 24 h of HG treatment.
Fig. 4.

Curcumin dose effects on TREM2 and CD33 protein in activated human THP-1 monocyte cells in vitro. Treatment of Thp-1 cells with high glucose (HG) models NFκB-regulated inflammation. Cells were treated for 1 h with 0.1 μM or 1.5 μM curcumin, then treated with HG (or mannitol control, (CNTR)) for 24 or 72 h, then cells were harvested and proteins quantified on Western blot; TREM2 and CD33 protein levels were normalized to GAPDH protein levels within same sample. (A) Western blot of TREM2 and GAPDH. (B) Levels of TREM2 normalized to GAPDH. (C) Western blot of CD33 and GAPDH. (D) Levels of CD33 normalized to GAPDH. ***p < .001. All values represent the average and SEM of triplicate wells.
In contrast, although CD33 protein was modulated in this system at 24 h but not 72 h of HG treatment: at 24 h of HG treatment, CD33 protein was decreased by both low dose curcumin (0.1 μM) and high dose curcumin (1.5 μM) (Fig. 4C, D). These results suggest amyloid-independent, direct time- and dose-dependent effects of curcumin on TREM2 and CD33 protein levels in human innate immune cells resembling the in vivo mRNA expression effects in Tg2576 mouse brain.
3.8. Curcumin stimulation of microglial phagocytosis, migration, and morphology in vitro
While co-cultured rodent microglia on unfixed human AD brain tissue sections fail to clear amyloid plaques (Ard et al., 1996), several groups have observed anti-Aβ antibody stimulation of plaque clearance with this system (Bard et al., 2000; Demattos et al., 2012; Hashioka et al., 2008; Lucin et al., 2013; Ostrowitzki et al., 2012). To examine the effect of curcumin on microglial phagocytosis of amyloid plaques, primary neonatal mouse microglia (400,000 cells) were evenly seeded on human AD plaque-laden cryostat sections (unfixed, serial 10 μm, temporal lobe) and cultured in vitro for 48 h with adjacent sections treated with curcumin (0.1 μM) or vehicle control (ethanol). Microglial responses (migration to plaques and clearance of amyloid, cytokine production) in brain slices and in response to curcumin were examined. Co-localization of Aβ (4G8) and microglia (Ox42) was examined using confocal microscopy. Compared to vehicle treatment (Fig. 5A), curcumin (0.1 μM) treated cultures revealed microglial clustering in and around amyloid plaques (Fig. 5B). Some curcumin-treated microglia were co-localized with Aβ staining as shown by the yellow overlap in Fig. 5C, consistent with microglial internalization of Aβ.
Fig. 5.

Human Alzheimer’s disease autopsied brain sections cultured with mouse microglia and treated with curcumin in vitro. (A-F) Microglia (from neonatal mice; 400,000 cells) were evenly seeded on top of cryostat sections (10 μm thick) of AD human brain with high amyloid plaque burden. Cultures were maintained for 48 h, then harvested for Aβ ELISA or fixed for ICC. (A, B, C) Sections were double-labeled for Aβ (red, 4G8 (Aβ1–13) antibody) and microglia (green, OX42, an antibody to CD11b/c) for confocal microscopy. Compared to vehicle treatment (A), curcumin treatment (0.1 μM) increased clustering of microglial around plaques (B), which suggested that CDllb/c co-localized with Aβ staining (yellow) (C). The magnification scale bar indicates 50 μm. (D) Percentage area stained with OX42 in AD brain sections was calculated using Image J and values for curcumin treatment (0.1 μM) normalized to control. Plaque-containing regions but not non-plaque containing regions or control glass, showed that Curc increased CD11b/c staining (50% increase, t-test **p = .008). (E) The effect of curcumin or Aβ antibody (10G4) in AD brain slices co-cultured with or without primary murine microglia on Aβ levels (measured by ELISA). Adjacent AD brain slices were used pairwise for treatment (treat.) and control (cnt.) conditions, so that Aβ levels were similar across pairs [n = 6 pairs of cultures for curcumin (0.1 μM) vs. vehicle; n = 6 pairs for microglia (+ cells) vs. no microglia (− cells); and n = 3 pairs for 10G4 vs. control antibody (IgG2b)]. Aβ levels for each treatment were normalized to levels in adjacent control slices. Aβ levels in paired treatment and control slices were analyzed by a non-parametric pairwise statistical test. The presence of microglia (+ cells) did not change Aβ levels compared to adjacent slices without microglia (−cells). Curcumin and 10G4 treatment reduced the level of Aβ by 24% (p < .04) and 48% (p = .025), respectively. (F) IL-1β in microglia-conditioned media. Il-1β was measured by a mouse-specific ELISA in media from curcumin (0.1 μM) treated and control (ethanol) treated microglia that were cultured on unfixed AD brain tissue slices. Curcumin treatment (0.1 μM) reduced the level of IL-1β by 61% (***ANOVA p < 1 × 10−6). (G) Ex vivo organotypic hippocampal slice cultures from wild-type mice were exposed to exogenous Aβ40 for 4 days and then treated with curcumin at low dose (5 μM) or high dose (500 μM) or the anti-Aβ antibody 10G4 (5 mg/ml) for 4 more days. Aβ levels in treated slices were normalized to Aβ levels in vehicle treated slices. Aβ levels measured by ELISA were reduced by low dose curcumin by 51% (*p < .05) but were not changed with high dose curcumin, and the anti-Aβ antibody 10G4 reduced Aβ levels by 52% (p < .057). (H) BV2 microglia phagocytosis of fluorescent E. coli particles. Pretreatment of BV2 cells with 1 μM curcumin stimulated phagocytosis by 57% (*p < .05). Independent samples t-test showed a significant difference in values of phagocytosed fluorescent E-coli in cells after treatment with curcumin (mean = 30.25, SD 5.888) versus vehicle (mean = 19.5, SD 2.986); T(6) = −3.56). Error bars indicate the SEM. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Microglial density was quantified by measuring the percentage area of microglia in high plaque-containing grey-matter and low-plaque containing white matter regions of the AD brain sections, by image analysis of OX42+ microglia. In plaque-rich regions, curcumin (0.1 μM) significantly increased the number of microglia by 50% compared to vehicle treatment (t-test p = .008), but curcumin had no impact in non-plaque regions (white matter), or on control glass slides with no AD section (Fig. 5D). Comparing serial sections with phase contrast observations, curcumin appeared to stimulate the seeded microglia to migrate to and cluster on amyloid plaques. Microglial morphology also differed between these culture regions. Microglia on top of the AD brain slices had an amoeboid morphology, compared to microglia on the glass slide outside of the AD brain slice, which had branched pseudopodia (data not shown).
3.9. Curcumin decreased microglial secretion of IL1β
IL1β secreted by microglia, an index of activation state, was measured in media from curcumin-treated and control (ethanol)-treated cocultures of human AD slices with mouse microglia. Consistent with immunomodulatory activity, low-dose curcumin treatment (0.1 μM) decreased the level of IL1β by 61% (*p < 1 × 10−6) (Fig. 5F).
3.10. Curcumin stimulates amyloid beta removal by microglia
In this system, the untreated microglia do not significantly remove/phagocytose amyloid plaques (Ard et al., 1996). After 48 h, Aβ in sections was extracted with formic acid and assayed by ELISA. Cultures without microglia had the same level of Aβ remaining as control cultures with microglia (Fig. 5E, first two bars). Curcumin treatment (0.1 μM) decreased the level of Aβ remaining by 24% (p < .04) (Fig. 5E).
Similar results were obtained in a different system where Aβ40 was exogenously-deposited in ex vivo neonatal organotypic hippocampal slice cultures (OHSC) from C57Bl6 mice with viable endogenous microglia (Harris-White et al., 1998). After Aβ40 was deposited, cultures were treated with low (5 μM) or high (50 μM) doses of curcumin for 4 days. Low dose curcumin, but not high dose curcumin, caused a 51% loss of the previously-deposited exogenous Aβ (p = .053) (Fig. 5G). This ability of low but not high dose curcumin to decrease deposited Aβ in vitro resembles our in vivo data.
3.11. Anti-Aβ antibody stimulates removal of amyloid
In both of these in vitro/ex vivo systems, in which curcumin increased amyloid removal by microglia, treatment with an antibody to Aβ (10G4) also significantly decreased Aβ levels by 48% (p = .025) in human AD slices with mouse microglia (Fig. 5E) and by 52% (p < .05) in OHSC with exogenously-deposited Aβ (Fig. 5G). This is consistent with previous reports of antibody to Aβ mediating microglial removal of amyloid in this ex vivo system (Bard et al., 2000; Demattos et al., 2012; Hashioka et al., 2008; Lucin et al., 2013; Ostrowitzki et al., 2012).
3.12. Gene expression levels in Tg2576 mice at older ages and in response to passive immunization with anti-Aβ antibodies
To further compare anti-Aβ treatment effects with curcumin effects on microglial gene expression, we examined innate immune gene expression levels in mouse brain in vivo. We measured levels of mRNA in Tg2576 mice at 22 months of age and at 22 months of age in response to passive immunization with antibodies to Aβ. Compared to transgene-negative littermates, TREM2 in Tg2576 transgene-carrying mice was increased 2.2-fold (p < .01) at 22 months of age (Fig. 6, left). We have previously shown 10G4 antibody treatment reduces amyloid in Tg2576 mice (Ma et al., 2006, 2007). Passive immunization of 22 month old Tg2576 mice with anti-Aβ (10G4) for 2.5 months caused a 2.4-fold increase in TREM2 mRNA in the brain, compared to control (isotype antibody treated) Tg2576 animals (Fig. 6, right).
Fig. 6.
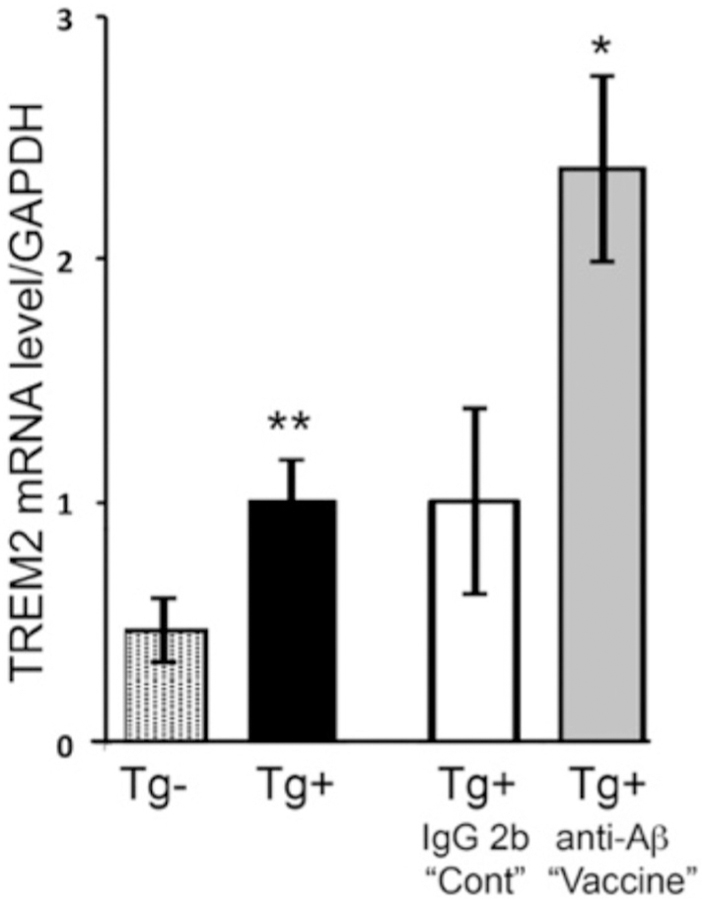
TREM2 gene expression levels in Tg2576 mice at 22 months of age and in response to passive immunization with anti-Aβ antibody. TREM2 mRNA levels were measured in the cortex of Tg2576 mice (Tg+; n = 8) or their non-transgenic littermates (Tg-; n = 8), and levels were normalized to Tg + mice (=1.0) (left two bars)`. TREM2 mRNA was measured in Tg+ mice treated by i.p. passive immunization with antibody to Aβ 10G4 “vaccine” (n = 4), or control antibody IgG2b “cont” (n = 8), and levels were normalized to control treatment Tg+ mice (=1.0) (right two bars). All mRNA levels were initially normalized to the levels of GAPDH mRNA in the same sample. *p < .05; **p < .01. Error bars indicate the SEM.
4. Discussion
Low dose curcumin, that reduces amyloid burden and chronic neuroinflammation (Lim et al., 2001; Yang et al., 2005), coordinately increases expression of innate immune genes TREM2, TyroBP and Arg1 while decreasing, CD11b, iNOS, COX-2, C1q, and the sialic acid-binding “siglec” receptor CD33. Our data is consistent with curcumin playing an immunomodulatory rather than simple immunosuppressive role in correcting dysfunction in the innate immune system.
Reduction in aberrant CD33 expression may facilitate curcumin’s correction of dysregulation in this tightly regulated feedback loop. This is consistent with clinical, genetic and basic research, which suggest that aberrant overexpression or increased functional forms of CD33 impede amyloid clearance (Bradshaw et al., 2013; Griciuc et al., 2013; Guerreiro et al., 2013; Jonsson et al., 2013; Malik et al., 2013, 2015; Pottier et al., 2016). However, curcumin targeting of CD33 alone was not sufficient for correcting immune function in our model since high dose curcumin that reduced CD33 did not reduce amyloid burden. In contrast, the diminished efficacy of high dose curcumin on amyloid clearance paralleled a diminished stimulation of TREM2, TyroBP, Arg1, and CD68, (another index of phagocytosis), supporting a coordinated role of multiple gene products working together in restoring aberrant innate immune function.
Coordinated decreases in CD33 and increases in TREM2 and TyroBP are all predicted to act in concert to increase microglial tyrosine kinase signaling, consistent with TREM2 correlating with increased microglial phosphotyrosine immunoreactivity in the Curc-Lo group. We previously reported that amyloid-reducing doses of Curc were consistently associated with decreased IL1β and microgliosis distal to plaques, but increased PT-labeled microglia within and adjacent to amyloid plaques (Frautschy et al., 2001; Lim et al., 2001). PT is increased by Aβ oligomers in both rodent models and human disease (Dhawan et al., 2012). In the Curc-lo group, plaque-associated PT appeared to be co-localized with TREM2 protein and correlated with increased TREM2 mRNA levels, consistent with Curc-lo increasing the TREM2/TyroBP (DAP12)-coupled tyrosine kinase signaling. Together these findings support a major role for TREM2 working in concert with CD33 and TyroBP mediating the Curc-lo diet impact on increased peri-plaque microglial tyrosine phosphorylation signaling, amyloid reduction and decreased pro-inflammatory IL1β. This is consistent with prior reports, using high-resolution confocal microscopy showing plaque-associated microglia are enriched in TREM2, especially at the leading edges of microglia processes directly contacting amyloid plaques, similar to TYROBP, and phosphorylated tyrosine (Yuan et al., 2016).
In our study, TREM2 increases within the context of other network gene alterations that appear critical to amyloid lowering, clinical and research data present a complex and controversial role of selective genetic alteration of TREM2 expression in AD models which can be both protective and deleterious (Song et al., 2017). For example, 50% TREM2 loss altered microglial morphology but had little effect on plaques (Ulrich et al., 2014) or increased plaques (Cheng-Hathaway et al., 2018), while TREM2 knockout crossed onto mutant APP/PS1 mice decreased Aβ internalization and caused late-stage increases in plaques (Wang et al., 2015). However, subsequent studies found that TREM2 deficiency increases amyloid at later stages and suggests stage-dependent effects that help explain these disparate reports (Jay et al., 2017). Further, the effect of TREM2 over- or under-expression in AD dendritic cells is genetic variant-dependent (Song et al., 2017). TREM2 is focally upregulated along with TyroBP/DAP12 in phagocytic microglia-like cells around plaques (Frank et al., 2008; Melchior et al., 2010), which may occur in invading macrophages (Jay et al., 2015) or resident microglia (Hickman and El Khoury, 2014; Wang et al., 2016). Regardless, lentiviral-directed microglial overexpression of TREM2 decreased amyloid, neurodegeneration, and neuroinflammation in young (Jiang et al., 2014) but not old mice (Jiang et al., 2017). These data show context-dependent effects and support the importance of restoring coordinated feedback interactions within other parts of innate immune network in older subjects with longstanding pathology.
Nevertheless TREM2 overexpression in tau-p301s mice also limited tauopathy, neurodegeneration, and neuroinflammation, while increasing Arg1 and other M2 microglial markers (Jiang et al., 2016) while TREM2 SNPs impact CSF tau, tangles, and AD progression (Cruchaga et al., 2013) arguing for a positive TREM2 role beyond amyloid phagocytosis (Ghosh et al., 2013; Kitazawa et al., 2011; Li et al., 2003).
TREM2 increases in our mice survived normalization to CD11b (a microglial activation marker) arguing against microglial activation and proliferation mediating TREM2 increases. Instead there was evidence of immunomodulation as Curc-lo, (relative to Curc-hi) increased TREM2 relative to CD33 and the effect was independent of amyloid since this was also observed in aged C57Bl6/J wildtype mice. Aging (Flanary et al., 2007) without amyloid drives microglial gene expression changes that include significant reductions in TREM2, and in TyroBP (DAP12), central to a microglial “sensome” hub (Hickman et al., 2013), suggesting curcumin-increased TREM2 in aging brain could be restorative in the context of coordinate increases in TyroBP.
Curcumin increased TREM2:CD33 ratio in the blood buffy coat cells in aged C57Bl/6J (Fig. 2D) which could provide a surrogate biomarker of immunomodulation in human studies. Low dose curcumin but not high dose curcumin also increased TREM2 and decreased CD33 in human THP-1 which is homozygous for the CC CD33 risk allele (Fig. 4).
Taken together, these results show that curcumin’s transcriptional restoration of hub genes central to the innate immune network can occur in the absence of amyloid-driven inflammation in vivo in brain and blood and directly in human monocytic THP-1 and mouse microglial cells.
In vivo upregulation of TyroBP, a master regulator of gene expression changes in AD (Zhang et al., 2013), by curc-lo was closely correlated with increased TREM2 and Arg1. Both these genes are in regulatory networks controlled by PU.1 and NFκB (Numasawa et al., 2011; Satoh et al., 2012) that also include CD33, CD68, and CD11b (Zhao et al., 2016). NFκB activation, driven by IKK phosphorylation (Zhang et al., 2013) is a direct inhibitory target of curcumin (Bharti et al., 2003; Pan et al., 2000). Since increased miR-155 expression is a central drive for “neurodegenerative microglia” linked to deleterious activities, including dystrophic neurite formation (Keren-Shaul et al., 2017), the observed reduction of miR-155 by curcumin should limit a degenerative microglial phenotype and account for curcumin’s rapid amelioration of dystrophic neurites (Garcia-Alloza et al., 2007). In turn, miR-155 is increased by IKK and NFκB explaining miR-155 reduction by curcumin (Ma et al., 2017). Thus, curcumin’s inhibition of NFκB may explain our expression and protective phenotypic changes.
Innate immune cells have the capacity to clear amyloid upon initial exposure to Aβ or inflammatory cytokines (Frautschy et al., 1992; Koenigsknecht and Landreth, 2004; Kopec and Carroll, 2000; Streit et al., 2004a). But, with chronic exposure, the unresolved inflammation represses microglial phagocytic machinery (Floden et al., 2005; Streit et al., 2004b), producing “bystander microglia” (Ard et al., 1996; Chung et al., 1999; Frackowiak et al., 1992; Kloss et al., 2001). Curcumin stimulates new microglial migration (Karlstetter et al., 2011) to plaques that can depend on TREM2 (Cheng-Hathaway et al., 2018; Wang et al., 2016), and stimulates amyloid clearance tracked by multiphoton imaging (Garcia-Alloza et al., 2007). TREM2 AD-risk SNPs greatly reduce cellular phagocytic activity (Kleinberger et al., 2014) (albeit not consistently, Yuan et al., 2016). We show that low dose curcumin increases phagocytosis of BV2 microglia (Fig. 5H), increased expression of CD68, a marker of microglial phagocytosis (Fig. 2A), and increases removal of amyloid in brain slice cultures (Fig. 5). Our data provide a plausible role for increasing coordinated TREM2/TyroBP hub gene expression controlling many genes involved in microglial motility, migration and phagocytosis (Forabosco et al., 2013). Functionally, curcumin stimulated downstream tyrosine phosphorylation illustrated by increases in microglial PT, overlap of PT and TREM2, and overlap of CD68 (a marker of phagocytosis) and TREM2. Increased overlap of Aβ and myeloid cells (TREM2, lectin or CD68) support clearance in vivo analogous to our findings of Aβ reduction and recruitment of microglia to plaques in human AD brain and AD model slices cultured with rodent microglia.
Active or passive anti-Aβ antibodies also act to increase amyloid phagocytosis (Bard et al., 2000; Demattos et al., 2012; Ostrowitzki et al., 2012; Schenk et al., 1999) via microglial recruitment (Koenigsknecht-Talboo et al., 2008) and Fc-Receptor pathway activation (Bacskai et al., 2001; Bard et al., 2003; Bard et al., 2000), with increased CD68 (Zotova et al., 2011). For example, aducanumab, stimulated microglial recruitment and phagocytic clearance of amyloid by mouse microglia in vitro and in AD model mice successfully predicting reduced Aβ plaques with potential cognitive benefits in a clinical trial (Sevigny et al., 2016). TREM2 pathways overlap with this beneficial Fc receptor signaling. Vaccination with Aβ in AD model mice increases TREM2 and amyloid clearance (Fisher et al., 2010), while TREM2 deficiency decreases anti-Aβ-induced amyloid clearance (Xiang et al., 2016). Consistent with this, we found that that anti-Aβ increased TREM2 mRNA in brain, and stimulated amyloid clearance in slices of human AD brain and rodent hippocampus. This is consistent with our hypothesis (Fig. 1) that increased TREM2/TyroBP signaling to Syk could parallel and substitute for the anti-Aβ Fc signaling to Syk. These parallels support curcumin as a low cost, small molecule candidate therapy for AD prevention.
Curcumin is in numerous clinical trials for inflammatory conditions. However, our own and other early AD clinical trials did not achieve therapeutic free curcumin plasma levels (reviewed in Goozee et al., 2016). In animal models, we reported effective dosing provided plasma levels of ~50–200 nM free curcumin. The Curc-Lo group dosing produced brain levels ~0.6 μM, which correlated with decreased brain IL1β and GFAP (Begum et al., 2008). New formulations increase human blood levels of free curcumin to therapeutic levels (Gota et al., 2010), label human retinal amyloid in vivo (Koronyo et al., 2017), decrease plasma Aβ in middle aged subjects (Disilvestro et al., 2012), and improve memory and other cognitive endpoints after short-term treatment in a healthy older population (Cox et al., 2015).
Limitations of the study include that it was not powered to detect sex effects. Also, this study and others’ data show that curcumin has immunomodulatory activity independent of amyloid, we cannot exclude that curcumin bound to amyloid (via the diketone bridge, Yang et al., 2005) also independently alters the inflammatory response; since its metabolite, which binds amyloid with lower infinity, has a reduced effect on clearance of amyloid (Begum et al., 2008).
5. Conclusion
In conclusion, our data highlight a potential key mechanism underlying the actions of curcumin as an immunomodulator of the TREM2-CD33-TyroBP hub, promoting beneficial inflammatory states in the brain and ameliorating Alzheimer’s pathology. Both TREM2 and CD33 are expressed by microglia, among other cells types, and therefore highlight the potential therapeutic relevance of targeting this pathway to modulate a neuroinflammatory outcome. Curcumin stimulates phagocytosis, alters expression of inflammatory cytokines and has been shown to specifically influence pathogenesis in AD models. The data show that the innate immune regulatory hub genes under transcriptional control by AD genes, can be coordinately regulated by curcumin to emulate Aβ immunotherapy and control signaling and phagocytic clearance of amyloid in vitro and in vivo.
Acknowledgments and funding
Funding was supplied R01 AG021975 (SAF), U01 AG028583 (SAF), VA Merit BX001257 (SAF), VA Merit BX000542 (GMC), R21 AG055024 (BT). There are no competing financial interests. We thank Walter Beech and Dr. Fusheng Yang, M.D. for technical support. We acknowledge the Carol Moss Spivak Cell Imaging Facility at the UCLA Semel Institute of Neuroscience for the use of their confocal micro-scope. Human brain tissue was obtained from the AD Research Center Neuropathology Cores NIA grants P50 AG05142 (USC, Dr. Carol Miller), P50 AG16970 (UCLA, Dr. H.V. Vinters). We are grateful for the support from the UCLA Mary S. Easton Alzheimer’s Center and the Oskar Fischer Foundation (Dr. James Truchard, Ph.D).
Footnotes
Declaration of interests
All authors declare no financial interests. SAF and GMC are coin-ventors on a UCLA/VA patent for curcumin formulations.
References
- Aisen PS, 2008. The inflammatory hypothesis of Alzheimer disease: dead or alive? Alzheimer Dis. Assoc. Disord 22, 4–5. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, et al. , 2000. Inflammation and Alzheimer’s disease. Neurobiol. Aging 21, 383–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ard MD, Cole GM, Wei J, Mehrle AP, Fratkin JD, 1996. Scavenging of Alzheimer’s amyloid beta-protein by microglia in culture. J. Neurosci. Res 43, 190–202. [DOI] [PubMed] [Google Scholar]
- Bacskai BJ, Kajdasz ST, Christie RH, Carter C, Games D, Seubert P, Schenk D, Hyman BT, 2001. Imaging of amyloid-ß deposits in brains of living mice permits direct observation of clearance of plaques with immunotherapy. Nat. Med 7, 369–372. [DOI] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke R, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, et al. , 2000. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat. Med 6, 916–919. [DOI] [PubMed] [Google Scholar]
- Bard F, Barbour R, Cannon C, Carretto R, Fox M, Games D, Guido T, Hoenow K, Hu K, Johnson-Wood K, et al. , 2003. Epitope and isotype specificities of antibodies to beta -amyloid peptide for protection against Alzheimer’s disease-like neuropathology. Proc. Natl. Acad. Sci. U. S. A 100, 2023–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begum AN, Jones MR, Lim GP, Morihara T, Kim P, Heath DD, Rock CL, Pruitt MA, Yang F, Hudspeth B, et al. , 2008. Curcumin structure-function, bioavailability, and efficacy in models of neuroinflammation and Alzheimer’s disease. J. Pharmacol. Exp. Ther 326, 196–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharti AC, Donato N, Singh S, Aggarwal BB, 2003. Curcumin (diferuloylmethane) down-regulates the constitutive activation of nuclear factor-kappa B and IkappaBalpha kinase in human multiple myeloma cells, leading to suppression of proliferation and induction of apoptosis. Blood 101, 1053–1062. [DOI] [PubMed] [Google Scholar]
- Bickford PC, Flowers A, Grimmig B, 2017. Aging leads to altered microglial function that reduces brain resiliency increasing vulnerability to neurodegenerative diseases. Exp. Gerontol 94, 4–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw EM, Chibnik LB, Keenan BT, Ottoboni L, Raj T, Tang A, Rosenkrantz LL, Imboywa S, Lee M, Von Korff A, et al. , 2013. CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nat. Neurosci 16, 848–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitner JC, Baker LD, Montine TJ, Meinert CL, Lyketsos CG, Ashe KH, Brandt J, Craft S, Evans DE, Green RC, et al. , 2011. Extended results of the Alzheimer’s disease anti-inflammatory prevention trial. Alzheimers Dement 7, 402–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calon F, Lim GP, Yang F, Morihara T, Teter B, Ubeda O, Rostaing P, Triller A, Salem N Jr., Ashe KH, et al. , 2004. Docosahexaenoic acid protects from dendritic pathology in an Alzheimer’s disease mouse model. Neuron 43, 633–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasquillo MM, Belbin O, Hunter TA, Ma L, Bisceglio GD, Zou F, Crook JE, Pankratz VS, Sando SB, Aasly JO, et al. , 2011. Replication of EPHA1 and CD33 associations with late-onset Alzheimer’s disease: a multi-centre case-control study. Mol. Neurodegener 6, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry U, Zhuang H, Dore S, 2010. Microsomal prostaglandin E synthase-2: cellular distribution and expression in Alzheimer’s disease. Exp. Neurol 223, 359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng-Hathaway PJ, Reed-Geaghan EG, Jay TR, Casali BT, Bemiller SM, Puntambekar SS, von Saucken VE, Williams RY, Karlo JC, Moutinho M, et al. , 2018. The Trem2 R47H variant confers loss-of-function-like phenotypes in Alzheimer’s disease. Mol. Neurodegener 13, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JD, Olschowka JA, O’Banion MK, 2014. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J. Neuroinflammation 11, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JD, Olschowka JA, O’Banion MK, 2015. Arginase 1+ microglia reduce Abeta plaque deposition during IL-1beta-dependent neuroinflammation. J. Neuroinflammation 12, 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung H, Brazil M, Soe T, Maxfield F, 1999. Uptake, degradation, and release of fibrillar and soluble forms of alzheimer’s amyloid beta-peptide by microglial cells. J. Biol. Chem 274, 32301–32308. [DOI] [PubMed] [Google Scholar]
- Cole GM, Yang F, Lim GP, Cummings JL, Masterman DL, Frautschy SA, 2003. A rationale for Curcuminoids for the prevention or treatment of Alzheimer’s disease. Curr. Med. Chem. Immun. Endoc. Metab. Agents 3, 15–25. [Google Scholar]
- Cole GM, Morihara T, Lim GP, Yang F, Begum A, Frautschy SA, 2004. NSAID and antioxidant prevention of Alzheimer’s disease: lessons from in vitro and animal models. Ann. N. Y. Acad. Sci 1035, 68–84. [DOI] [PubMed] [Google Scholar]
- Colton CA, 2009. Heterogeneity of microglial activation in the innate immune response in the brain. J. NeuroImmune Pharmacol 4, 399–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colton C, Wilcock DM, 2010. Assessing activation states in microglia. CNS Neurol. Disord. Drug Targets 9, 174–191. [DOI] [PubMed] [Google Scholar]
- Cox KH, Pipingas A, Scholey AB, 2015. Investigation of the effects of solid lipid curcumin on cognition and mood in a healthy older population. J. Psychopharmacol 29, 642–651. [DOI] [PubMed] [Google Scholar]
- Cruchaga C, Kauwe JS, Harari O, Jin SC, Cai Y, Karch CM, Benitez BA, Jeng AT, Skorupa T, Carrell D, et al. , 2013. GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer’s disease. Neuron 78, 256–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demattos RB, Lu J, Tang Y, Racke MM, Delong CA, Tzaferis JA, Hole JT, Forster BM, McDonnell PC, Liu F, et al. , 2012. A plaque-specific antibody clears existing beta-amyloid plaques in Alzheimer’s disease mice. Neuron 76, 908–920. [DOI] [PubMed] [Google Scholar]
- Dhawan G, Floden AM, Combs CK, 2012. Amyloid-beta oligomers stimulate microglia through a tyrosine kinase dependent mechanism. Neurobiol. Aging 33, 2247–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disilvestro RA, Joseph E, Zhao S, Joshua B, 2012. Diverse effects of a low dose supplement of lipidated curcumin in healthy middle aged people. Nutr. J 11, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efthymiou AG, Goate AM, 2017. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener 12, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elton TS, Selemon H, Elton SM, Parinandi NL, 2013. Regulation of the MIR155 host gene in physiological and pathological processes. Gene 532, 1–12. [DOI] [PubMed] [Google Scholar]
- Fisher Y, Nemirovsky A, Baron R, Monsonego A, 2010. T cells specifically targeted to amyloid plaques enhance plaque clearance in a mouse model of Alzheimer’s disease. PLoS One 5, e10830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanary BE, Sammons NW, Nguyen C, Walker D, Streit WJ, 2007. Evidence that aging and amyloid promote microglial cell senescence. Rejuvenation Res 10, 61–74. [DOI] [PubMed] [Google Scholar]
- Floden AM, Li S, Combs CK, 2005. Beta-amyloid-stimulated microglia induce neuron death via synergistic stimulation of tumor necrosis factor alpha and NMDA receptors. J. Neurosci 25, 2566–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forabosco P, Ramasamy A, Trabzuni D, Walker R, Smith C, Bras J, Levine AP, Hardy J, Pocock JM, Guerreiro R, et al. , 2013. Insights into TREM2 biology by network analysis of human brain gene expression data. Neurobiol. Aging 34, 2699–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frackowiak J, Wisniewski HM, Wegiel J, Merz GS, Iqbal K, Wang KC, 1992. Ultrastructure of the microglia that phagocytose amyloid and the microglia that produce β-amyloid fibrils. Acta Neuropathol (Berl) 84, 225–233. [DOI] [PubMed] [Google Scholar]
- Frank S, Burbach GJ, Bonin M, Walter M, Streit W, Bechmann I, Deller T, 2008. TREM2 is upregulated in amyloid plaque-associated microglia in aged APP23 transgenic mice. Glia 56, 1438–1447. [DOI] [PubMed] [Google Scholar]
- Frautschy S, Cole GM, Baird A, 1992. Phagocytosis and deposition of vascular b-amyloid in rat brains injected with Alzheimer β-amyloid. Am. J. Pathol 140, 1389–1399. [PMC free article] [PubMed] [Google Scholar]
- Frautschy SA, Yang F, Irizarry M, Hyman B, Saido TC, Hsiao K, Cole GM, 1998. Microglial response to amyloid plaques in APPsw transgenic mice. Am. J. Pathol 152, 307–317. [PMC free article] [PubMed] [Google Scholar]
- Frautschy SA, Hu W, Kim P, Miller SA, Chu T, Harris-White ME, Cole GM, 2001. Phenolic anti-inflammatory antioxidant reversal of Abeta-induced cognitive deficits and neuropathology. Neurobiol. Aging 22, 993–1005. [DOI] [PubMed] [Google Scholar]
- Gaikwad SM, Heneka MT, 2013. Studying M1 and M2 states in adult microglia. Methods Mol. Biol 1041, 185–197. [DOI] [PubMed] [Google Scholar]
- Garcia-Alloza M, Borrelli LA, Rozkalne A, Hyman BT, Bacskai BJ, 2007. Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model. J. Neurochem 102, 1095–1104. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Wu MD, Shaftel SS, Kyrkanides S, LaFerla FM, Olschowka JA, O’Banion MK, 2013. Sustained interleukin-1beta overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J. Neurosci 33, 5053–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goozee KG, Shah TM, Sohrabi HR, Rainey-Smith SR, Brown B, Verdile G, Martins RN, 2016. Examining the potential clinical value of curcumin in the prevention and diagnosis of Alzheimer’s disease. Br. J. Nutr 115, 449–465. [DOI] [PubMed] [Google Scholar]
- Gota VS, Maru GB, Soni TG, Gandhi TR, Kochar N, Agarwal MG, 2010. Safety and pharmacokinetics of a solid lipid curcumin particle formulation in osteosarcoma patients and healthy volunteers. J. Agric. Food Chem 58, 2095–2099. [DOI] [PubMed] [Google Scholar]
- Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, Hooli B, Choi SH, Hyman BT, Tanzi RE, 2013. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 78, 631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, et al. , 2013. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med 368, 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Bogdanovic N, Winblad B, Portelius E, Andreasen N, Cedazo-Minguez A, Zetterberg H, 2014. Pathways to Alzheimer’s disease. J. Intern. Med 275, 296–303. [DOI] [PubMed] [Google Scholar]
- Harris-White ME, Frautschy SA, Cole GM, 1998. Methods for evaluating a slice culture model of Alzheimer’s disease. In: Studies of Aging Springer, Berlin, Heidelberg, pp. 55–65. [Google Scholar]
- Hashioka S, Miklossy J, Schwab C, Klegeris A, McGeer PL, 2008. Adhesion of exogenous human microglia and THP-1 cells to amyloid plaques of postmortem Alzheimer’s disease brain. J. Alzheimers Dis 14, 345–352. [DOI] [PubMed] [Google Scholar]
- Hickman SE, El Khoury J, 2014. TREM2 and the neuroimmunology of Alzheimer’s disease. Biochem. Pharmacol 88, 495–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman SE, Allison EK, El Khoury J, 2008. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci 28, 8354–8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, El Khoury J, 2013. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci 16, 1896–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, Abraham R, Hamshere ML, Pahwa JS, Moskvina V, et al. , 2011. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet 43, 429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ, 2010. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol 9, 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML, Xu G, Margevicius D, Karlo JC, Sousa GL, et al. , 2015. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J. Exp. Med 212, 287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay TR, Hirsch AM, Broihier ML, Miller CM, Neilson LE, Ransohoff RM, Lamb BT, Landreth GE, 2017. Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer’s disease. J. Neurosci 37, 637–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T, Tan L, Zhu XC, Zhang QQ, Cao L, Tan MS, Gu LZ, Wang HF, Ding ZZ, Zhang YD, et al. , 2014. Up-regulation of TREM2 ameliorates neuropathology and rescues spatial cognitive impairment in a transgenic mouse model of Alzheimer’s disease. Neuropsychopharmacology 39, 2949–2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T, Zhang YD, Chen Q, Gao Q, Zhu XC, Zhou JS, Shi JQ, Lu H, Tan L, Yu JT, 2016. TREM2 modifies microglial phenotype and provides neuroprotection in P301S tau transgenic mice. Neuropharmacology 105, 196–206. [DOI] [PubMed] [Google Scholar]
- Jiang T, Wan Y, Zhang YD, Zhou JS, Gao Q, Zhu XC, Shi JQ, Lu H, Tan L, Yu JT, 2017. TREM2 overexpression has no improvement on neuropathology and cognitive impairment in aging APPswe/PS1dE9 mice. Mol. Neurobiol 54, 855–865. [DOI] [PubMed] [Google Scholar]
- Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, et al. , 2013. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med 368, 107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphuis W, Kooijman L, Schetters S, Orre M, Hol EM, 2016. Transcriptional profiling of CD11c-positive microglia accumulating around amyloid plaques in a mouse model for Alzheimer’s disease. Biochim. Biophys. Acta 1862, 1847–1860. [DOI] [PubMed] [Google Scholar]
- Karlstetter M, Lippe E, Walczak Y, Moehle C, Aslanidis A, Mirza M, Langmann T, 2011. Curcumin is a potent modulator of microglial gene expression and migration. J. Neuroinflammation 8, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, et al. , 2017. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169 (1276–1290), e17. [DOI] [PubMed] [Google Scholar]
- Kitazawa M, Cheng D, Tsukamoto MR, Koike MA, Wes PD, Vasilevko V, Cribbs DH, LaFerla FM, 2011. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal beta-catenin pathway function in an Alzheimer’s disease model. J. Immunol 187, 6539–6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinberger G, Yamanishi Y, Suarez-Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger-Weinzierl A, Mazaheri F, et al. , 2014. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl. Med 6 243ra86. [DOI] [PubMed] [Google Scholar]
- Kloss CU, Bohatschek M, Kreutzberg GW, Raivich G, 2001. Effect of lipopolysaccharide on the morphology and integrin immunoreactivity of ramified microglia in the mouse brain and in cell culture. Exp. Neurol 168, 32–46. [DOI] [PubMed] [Google Scholar]
- Knouff C, Hinsdale ME, Mezdour H, Altenburg MK, Watanabe M, Quarfordt SH, Sullivan PM, Maeda N, 1999. Apo E structure determines VLDL clearance and atherosclerosis risk in mice. J. Clin. Invest 103, 1579–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenigsknecht J, Landreth G, 2004. Microglial phagocytosis of fibrillar beta-amyloid through a beta1 integrin-dependent mechanism. J. Neurosci 24, 9838–9846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenigsknecht-Talboo J, Landreth GE, 2005. Microglial phagocytosis induced by fibrillar beta-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J. Neurosci 25, 8240–8249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenigsknecht-Talboo J, Meyer-Luehmann M, Parsadanian M, Garcia-Alloza M, Finn MB, Hyman BT, Bacskai BJ, Holtzman DM, 2008. Rapid microglial response around amyloid pathology after systemic anti-Abeta antibody administration in PDAPP mice. J. Neurosci 28, 14156–14164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopec KK, Carroll RT, 2000. Phagocytosis is regulated by nitric oxide in murine microglia. Nitric Oxide 4, 103–111. [DOI] [PubMed] [Google Scholar]
- Koronyo Y, Biggs D, Barron E, Boyer DS, Pearlman JA, Au WJ, Kile SJ, Blanco A, Fuchs DT, Ashfaq A, et al. , 2017. Retinal amyloid pathology and proof-of-concept imaging trial in Alzheimer’s disease. JCI Insight 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krabbe G, Halle A, Matyash V, Rinnenthal JL, Eom GD, Bernhardt U, Miller KR, Prokop S, Kettenmann H, Heppner FL, 2013. Functional impairment of microglia coincides with Beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS One 8, e60921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O’Loughlin E, Xu Y, Fanek Z, et al. , 2017. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47 (566–581), e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemere CA, Masliah E, 2010. Can Alzheimer disease be prevented by amyloid-beta immunotherapy? Nat. Rev. Neurol 6, 108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Liu L, Barger SW, Griffin WS, 2003. Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J. Neurosci 23, 1605–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, Tran T, Ubeda O, Ashe KH, Frautschy SA, et al. , 2000. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer’s disease. J. Neurosci 20, 5709–5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim GP, Chu T, Yang F, Beech W, Frautschy SA, Cole GM, 2001. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J. Neurosci 21, 8370–8377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim GP, Calon F, Morihara T, Yang F, Teter B, Ubeda O, Salem N Jr., Frautschy SA, Cole GM, 2005. A diet enriched with the omega-3 fatty acid docosahexaenoic acid reduces amyloid burden in an aged Alzheimer mouse model. J. Neurosci 25, 3032–3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucin KM, O’Brien CE, Bieri G, Czirr E, Mosher KI, Abbey RJ, Mastroeni DF, Rogers J, Spencer B, Masliah E, et al. , 2013. Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer’s disease. Neuron 79, 873–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma QL, Lim GP, Harris-White ME, Yang F, Ambegaokar SS, Ubeda OJ, Glabe CG, Teter B, Frautschy SA, Cole GM, 2006. Antibodies against beta-amyloid reduce Abeta oligomers, glycogen synthase kinase-3beta activation and tau phosphorylation in vivo and in vitro. J. Neurosci. Res 83, 374–384. [DOI] [PubMed] [Google Scholar]
- Ma QL, Harris-White ME, Ubeda OJ, Simmons M, Beech W, Lim GP, Teter B, Frautschy SA, Cole GM, 2007. Evidence of Abeta- and transgene-dependent defects in ERK-CREB signaling in Alzheimer’s models. J. Neurochem 103, 1594–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma QL, Zuo X, Yang F, Ubeda OJ, Gant DJ, Alaverdyan M, Teng E, Hu S, Chen PP, Maiti P, et al. , 2013. Curcumin suppresses soluble tau dimers and corrects molecular chaperone, synaptic, and behavioral deficits in aged human tau transgenic mice. J. Biol. Chem 288, 4056–4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma F, Liu F, Ding L, You M, Yue H, Zhou Y, Hou Y, 2017. Anti-inflammatory effects of curcumin are associated with down regulating microRNA-155 in LPS-treated macrophages and mice. Pharm. Biol 55, 1263–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik M, Simpson JF, Parikh I, Wilfred BR, Fardo DW, Nelson PT, Estus S, 2013. CD33 Alzheimer’s risk-altering polymorphism, CD33 expression, and exon 2 splicing. J. Neurosci 33, 13320–13325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik M, Parikh I, Vasquez JB, Smith C, Tai L, Bu G, LaDu MJ, Fardo DW, Rebeck GW, Estus S, 2015. Genetics ignite focus on microglial inflammation in Alzheimer’s disease. Mol. Neurodegener 10, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malpass K, 2013. Alzheimer disease: functional dissection of CD33 locus implicates innate immune response in Alzheimer disease pathology. Nat. Rev. Neurol 9, 360. [DOI] [PubMed] [Google Scholar]
- Mandrekar-Colucci S, Karlo JC, Landreth GE, 2012. Mechanisms underlying the rapid peroxisome proliferator-activated receptor-gamma-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer’s disease. J. Neurosci 32, 10117–10128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AC, Carreras I, Hossain L, Ryu H, Klein WL, Oddo S, LaFerla FM, Jenkins BG, Kowall NW, Dedeoglu A, 2008. Ibuprofen reduces Abeta, hyper-phosphorylated tau and memory deficits in Alzheimer mice. Brain Res 1207, 225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchior B, Garcia AE, Hsiung BK, Lo KM, Doose JM, Thrash JC, Stalder AK, Staufenbiel M, Neumann H, Carson MJ, 2010. Dual induction of TREM2 and tolerance-related transcript, Tmem176b, in amyloid transgenic mice: implications for vaccine-based therapies for Alzheimer’s disease. ASN Neuro 2, e00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelucci A, Heurtaux T, Grandbarbe L, Morga E, Heuschling P, 2009. Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: effects of oligomeric and fibrillar amyloid-beta. J. Neuroimmunol 210, 3–12. [DOI] [PubMed] [Google Scholar]
- Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, et al. , 2011. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet 43, 436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numasawa Y, Yamaura C, Ishihara S, Shintani S, Yamazaki M, Tabunoki H, Satoh JI, 2011. Nasu-Hakola disease with a splicing mutation of TREM2 in a Japanese family. Eur. J. Neurol 18, 1179–1183. [DOI] [PubMed] [Google Scholar]
- Ostrowitzki S, Deptula D, Thurfjell L, Barkhof F, Bohrmann B, Brooks DJ, Klunk WE, Ashford E, Yoo K, Xu ZX, et al. , 2012. Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Arch. Neurol 69, 198–207. [DOI] [PubMed] [Google Scholar]
- Painter MM, Atagi Y, Liu CC, Rademakers R, Xu H, Fryer JD, Bu G, 2015. TREM2 in CNS homeostasis and neurodegenerative disease. Mol. Neurodegener 10, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan MH, Lin-Shiau SY, Lin JK, 2000. Comparative studies on the suppression of nitric oxide synthase by curcumin and its hydrogenated metabolites through down-regulation of IkappaB kinase and NFkappa B in macrophages. Biochem. Pharmacol 60, 1665–1676. [DOI] [PubMed] [Google Scholar]
- Pasqualetti P, Bonomini C, Dal Forno G, Paulon L, Sinforiani E, Marra C, Zanetti O, Rossini PM, 2009. A randomized controlled study on effects of ibuprofen on cognitive progression of Alzheimer’s disease. Aging Clin. Exp. Res 21, 102–110. [DOI] [PubMed] [Google Scholar]
- Penninkilampi R, Brothers HM, Eslick GD, 2017. Safety and efficacy of anti-amyloid-beta immunotherapy in Alzheimer’s disease: a systematic review and meta-analysis. J. NeuroImmune Pharmacol 12, 194–203. [DOI] [PubMed] [Google Scholar]
- Pottier C, Ravenscroft TA, Brown PH, Finch NA, Baker M, Parsons M, Asmann YW, Ren Y, Christopher E, Levitch D, et al. , 2016. TYROBP genetic variants in early-onset Alzheimer’s disease. Neurobiol. Aging 48, 222 e9–222 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers J, Strohmeyer R, Kovelowski CJ, Li R, 2002. Microglia and inflammatory mechanisms in the clearance of amyloid beta peptide. Glia 40, 260–269. [DOI] [PubMed] [Google Scholar]
- Rojanathammanee L, Puig KL, Combs CK, 2013. Pomegranate polyphenols and extract inhibit nuclear factor of activated T-cell activity and microglial activation in vitro and in a transgenic mouse model of Alzheimer disease. J. Nutr 143, 597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rygiel K, 2016. Novel strategies for Alzheimer’s disease treatment: an overview of anti-amyloid beta monoclonal antibodies. Indian J. Pharm 48, 629–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh J, Shimamura Y, Tabunoki H, 2012. Gene expression profile of THP-1 monocytes following knockdown of DAP12, a causative gene for Nasu-Hakola disease. Cell. Mol. Neurobiol 32, 337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh J, Kino Y, Asahina N, Takitani M, Miyoshi J, Ishida T, Saito Y, 2016. TMEM119 marks a subset of microglia in the human brain. Neuropathology 36, 39–49. [DOI] [PubMed] [Google Scholar]
- Saura J, Petegnief V, Wu X, Liang Y, Paul SM, 2003. Microglial apolipoprotein E and astroglial apolipoprotein J expression in vitro: opposite effects of lipopolysaccharide. J. Neurochem 85, 1455–1467. [DOI] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, et al. , 1999. Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400, 173–177. [DOI] [PubMed] [Google Scholar]
- Sevigny J, Chiao P, Bussiere T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y, et al. , 2016. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 537, 50–56. [DOI] [PubMed] [Google Scholar]
- Song W, Hooli B, Mullin K, Jin SC, Cella M, Ulland TK, Wang Y, Tanzi RE, Colonna M, 2017. Alzheimer’s disease-associated TREM2 variants exhibit either decreased or increased ligand-dependent activation. Alzheimers Dement 13, 381–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnen JA, Larson EB, Walker RL, Haneuse S, Crane PK, Gray SL, Breitner JC, Montine TJ, 2010. Nonsteroidal anti-inflammatory drugs are associated with increased neuritic plaques. Neurology 75, 1203–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D, 1991. A simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods 37, 173–182. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Kreutzberg GW, 1987. Lectin binding by resting and reactive microglia. J. Neurocytol 16, 249–260. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Mrak RE, Griffin WS, 2004a. Microglia and neuroinflammation: a pathological perspective. J. Neuroinflammation 1, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ, Sammons NW, Kuhns AJ, Sparks DL, 2004b. Dystrophic microglia in the aging human brain. Glia 45, 208–212. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Prinz M, Stagi M, Chechneva O, Neumann H, 2007. TREM2-transduced myeloid precursors mediate nervous tissue debris clearance and facilitate recovery in an animal model of multiple sclerosis. PLoS Med 4, e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DL, Diemel LT, Cuzner ML, Pocock JM, 2002. Activation of group II metabotropic glutamate receptors underlies microglial reactivity and neurotoxicity following stimulation with chromogranin a, a peptide up-regulated in Alzheimer’s disease. J. Neurochem 82, 1179–1191. [DOI] [PubMed] [Google Scholar]
- Teter B, Harris-White ME, Frautschy SA, Cole GM, 1999. Role of apolipoprotein E and estrogen in mossy fiber sprouting in hippocampal slice cultures. Neuroscience 91, 1009–1016. [DOI] [PubMed] [Google Scholar]
- Thrash JC, Torbett BE, Carson MJ, 2009. Developmental regulation of TREM2 and DAP12 expression in the murine CNS: implications for Nasu-Hakola disease. Neurochem. Res 34, 38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich JD, Finn MB, Wang Y, Shen A, Mahan TE, Jiang H, Stewart FR, Piccio L, Colonna M, Holtzman DM, 2014. Altered microglial response to Abeta plaques in APPPS1–21 mice heterozygous for TREM2. Mol. Neurodegener 9, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich JD, Ulland TK, Colonna M, Holtzman DM, 2017. Elucidating the role of TREM2 in Alzheimer’s disease. Neuron 94, 237–248. [DOI] [PubMed] [Google Scholar]
- Ulrich JD, Ulland TK, Mahan TE, Nystrom S, Nilsson KP, Song WM, Zhou Y, Reinartz M, Choi S, Jiang H, et al. , 2018. ApoE facilitates the microglial response to amyloid plaque pathology. J. Exp. Med 215, 1047–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dam D, Coen K, De Deyn PP, 2010. Ibuprofen modifies cognitive disease progression in an Alzheimer’s mouse model. J. Psychopharmacol 24, 383–388. [DOI] [PubMed] [Google Scholar]
- Van Eldik LJ, Carrillo MC, Cole PE, Feuerbach D, Greenberg BD, Hendrix JA, Kennedy M, Kozauer N, Margolin RA, Molinuevo JL, et al. , 2016. The roles of inflammation and immune mechanisms in Alzheimer’s disease. Alzheimers Dement (N Y) 2, 99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlad SC, Miller DR, Kowall NW, Felson DT, 2008. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology 70, 1672–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, et al. , 2015. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 160, 1061–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, Yuan P, Mahan TE, Shi Y, Gilfillan S, et al. , 2016. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med 213, 667–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weekman EM, Sudduth TL, Abner EL, Popa GJ, Mendenhall MD, Brothers HM, Braun K, Greenstein A, Wilcock DM, 2014. Transition from an M1 to a mixed neuroinflammatory phenotype increases amyloid deposition in APP/PS1 transgenic mice. J. Neuroinflammation 11, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Mucke L, 2002. Inflammation in neurodegenerative disease—a double-edged sword. Neuron 35, 419–432. [DOI] [PubMed] [Google Scholar]
- Xiang X, Werner G, Bohrmann B, Liesz A, Mazaheri F, Capell A, Feederle R, Knuesel I, Kleinberger G, Haass C, 2016. TREM2 deficiency reduces the efficacy of immunotherapeutic amyloid clearance. EMBO Mol. Med 8, 992–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q, Zhang J, Liu H, Babu-Khan S, Vassar R, Biere AL, Citron M, Landreth G, 2003. Anti-inflammatory drug therapy alters beta-amyloid processing and deposition in an animal model of Alzheimer’s disease. J. Neurosci 23, 7504–7509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen PP, Kayed R, Glabe CG, Frautschy SA, et al. , 2005. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem 280, 5892–5901. [DOI] [PubMed] [Google Scholar]
- Yasojima K, Schwab C, McGeer EG, McGeer PL, 1999. Up-regulated production and activation of the complement system in Alzheimer’s disease brain. Am. J. Pathol 154, 927–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ydens E, Cauwels A, Asselbergh B, Goethals S, Peeraer L, Lornet G, Almeida-Souza L, Van Ginderachter JA, Timmerman V, Janssens S, 2012. Acute injury in the peripheral nervous system triggers an alternative macrophage response. J. Neuroinflammation 9, 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh FL, Hansen DV, Sheng M, 2017. TREM2, microglia, and neurodegenerative diseases. Trends Mol. Med 23, 512–533. [DOI] [PubMed] [Google Scholar]
- Youmans KL, Tai LM, Nwabuisi-Heath E, Jungbauer L, Kanekiyo T, Gan M, Kim J, Eimer WA, Estus S, Rebeck GW, et al. , 2012. APOE4-specific changes in Abeta accumulation in a new transgenic mouse model of Alzheimer disease. J. Biol. Chem 287, 41774–41786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, Luo W, Colonna M, Baddeley D, Grutzendler J, 2016. TREM2 Haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron 90, 724–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun JM, Jialal I, Devaraj S, 2011. Epigenetic regulation of high glucose-induced proinflammatory cytokine production in monocytes by curcumin. J. Nutr. Biochem 22, 450–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, Zhang C, Xie T, Tran L, Dobrin R, et al. , 2013. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 153, 707–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Jaber V, Lukiw WJ, 2016. Over-expressed pathogenic miRNAs in Alzheimer’s disease (AD) and prion disease (PrD) drive deficits in TREM2-mediated Abeta42 peptide clearance. Front. Aging Neurosci 8, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zotova E, Holmes C, Johnston D, Neal JW, Nicoll JA, Boche D, 2011. Microglial alterations in human Alzheimer’s disease following Abeta42 immunization. Neuropathol. Appl. Neurobiol 37, 513–524. [DOI] [PubMed] [Google Scholar]