

Abstract
Diffuse large B-cell lymphoma (DLBCL) is the major type of aggressive B-cell lymphoma. High-grade B-cell lymphoma (HGBCL) with MYC/BCL2 double-hit (DH) represents a distinct entity with dismal prognosis after standard immunochemotherapy in the current WHO lymphoma classification. However, whether TP53 mutation synergizes with MYC abnormalities (MYC rearrangement and/or Myc protein overexpression) contributing to HGBCL-like biology and prognosis is not well investigated. In this study, patients with DLBCL with MYC/TP53 abnormalities demonstrated poor clinical outcome, high-grade morphology, and distinct gene expression signatures. To identify more effective therapies for this distinctive DLBCL subset, novel MYC/TP53/BCL-2–targeted agents were investigated in DLBCL cells with MYC/TP53 dual alterations or HGBCL-MYC/BCL2-DH. A BET inhibitor INCB057643 effectively inhibited cell viability and induced apoptosis in DLBCL/HGBCL cells regardless of MYC/BCL2/TP53 status. Combining INCB057643 with a MDM2-p53 inhibitor DS3032b significantly enhanced the cytotoxic effects in HGBCL-DH without TP53 mutation, while combining with the BCL-2 inhibitor venetoclax displayed potent therapeutic synergy in DLBCL/HGBCL cells with and without concurrent TP53 mutation. Reverse-phase protein arrays revealed the synergistic molecular actions by INCB057643, DS3032b and venetoclax to induce cell-cycle arrest and apoptosis and to inhibit AKT/MEK/ERK/mTOR pathways, as well as potential drug resistance mechanisms mediated by upregulation of Mcl-1 and RAS/RAF/MEK/ERK pathways. In summary, these findings support subclassification of DLBCL/HGBCL with dual MYC/TP53 alterations, which demonstrates distinct pathobiologic features and dismal survival with standard therapy, therefore requiring additional targeted therapies.
Implications:
The clinical and pharmacologic studies suggest recognizing DLBCL with concomitant TP53 mutation and MYC abnormalities as a distinctive entity necessary for precision oncology practice.
Visual Overview:
http://mcr.aacrjournals.org/content/molcanres/19/2/249/F1.large.jpg.
Graphical Abstract
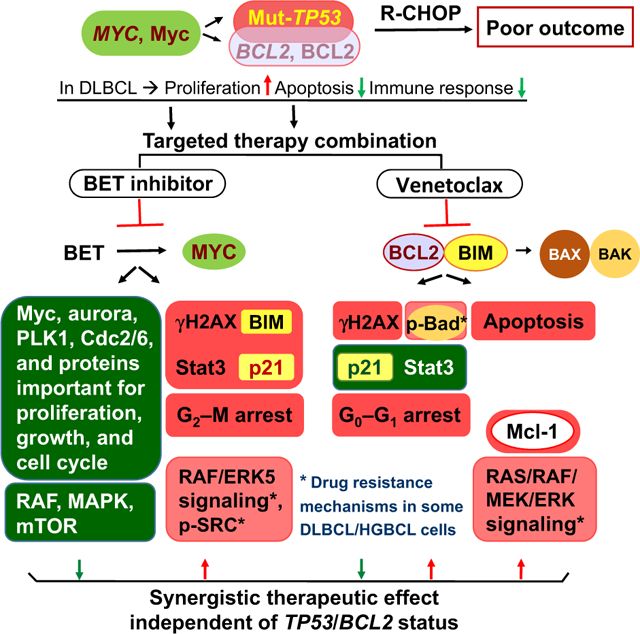
Introduction
Diffuse large B-cell lymphoma (DLBCL) is a morphologically, clinically, and genetically heterogeneous entity, representing about 40% of B-cell lymphomas worldwide (1, 2). Although the 5-year overall survival (OS) with first-line rituximab, cyclophosphamide, hydroxydaunorubicin, vincristine, and prednisone (R-CHOP) has reached 60%, unfortunately, 40% of patients with DLBCL had refractory or relapsed disease (RR-DLBCL) with very poor clinical outcomes (3). The most extensively utilized prognostic markers, including the International Prognostic Index (IPI) and cell-of-origin have limited capability to recognize high-risk patients with RR-DLBCL. Predictive biomarkers and novel therapies for RR-DLBCL have been actively explored with little success.
Myc is a master transcription regulator that modulates cell proliferation, differentiation, survival, and metabolism (4). Because of the pivotally regulatory role of Myc in the cellular signaling circuitry, MYC gene rearrangement (MYC-R) leading to high expression of Myc (Mychigh) is often regarded as a hallmark of malignant diseases and associated with poor prognosis (5, 6). However, MYC alteration (MYC-R or Mychigh) alone is insufficient and requires additional prosurvival factors to cooperatively induce neoplastic transformation, because overexpressed Myc could also induce p53-mediated apoptosis (7). It has been well-established that DLBCL with concurrent MYC and BCL2 and/or BCL6 rearrangements, known as “double-hit” or “triple-hit” lymphoma (DHL/THL) occurring in 5% to 10% of DLBCL, predicts dismal progression-free survival (PFS) and OS (8), and has been recognized as a distinct entity (HGBCL-DH or -TH) in the 2016 World Health Organization classification of lymphoid neoplasms (9, 10). Lymphoma with MYC/BCL-2 double-expression (DE) is more frequent than HGBCL-MYC/BCL2-DH and also associated with poor prognosis (11).
The TP53 gene encodes the p53 tumor suppressor and is the most frequently mutated gene in human malignancies (12). Our studies demonstrated that TP53 mutations occurs in about 22% of DLBCL and predicts significantly poor survival independent of cell-of-origin (13–15). TP53 mutations are associated with overexpression of mutant (Mut) p53 proteins with loss-of-function of wild-type (Wt) p53 and oncogenic gain-of-function (12, 16), predicted significantly worse survival (17). Considering the role of Wt-p53 in Myc-induced apoptosis, it is logical that Mut-TP53 cooperates with MYC abnormalities to confer worse prognosis in DLBCL. This new double-hit concept was suggested by a former study with limited cases showing that MYC-R and a second hit (Mut-TP53 or BCL2-R) depended on each other in predicting poor survival (18). MYC/TP53 dual-alteration was seen in approximately one third of MYC-R and HGBCL-DH patients (18–20). p53 over expression (p53high) concurrent with MYC-R or Mychigh has also been evaluated in previous studies in which the dual abnormality predicted poor OS in DLBCL, regardless of BCL-2 expression status (21, 22).
In this study, we systemically investigated the additive impact of TP53 alterations (Mut-TP53 or p53high) on prognosis and gene expression profiles to that of MYC alteration (MYC-R or Mychigh) in 487 patients with DLBCL and further explored the potential of targeted therapies. To target MYC, we used a novel BET inhibitor INCB057643 (23), because BET inhibitors have shown profound inhibitory effects on MYC expression and tumor growth, and can overcome resistance to venetoclax (BCL-2 inhibitor) in DHL models (24). To target TP53 mutation and mutant p53 expression, which is considered “undruggable” in cancer (12), we tried a MDM2 inhibitor DS-3032b (25, 26) and the BCL-2 inhibitor venetoclax (27, 28), because high BCL-2 expression is frequent in DLBCL mediating resistance to the cytotoxicity of BET inhibitors (29). Reverse-phase protein array (RPPA) was utilized as a proteomics approach to understand the action of these three targeted agents and synergy of combination treatment.
Materials and Methods
Patients and samples
Four hundred and eighty patients with de novo DLBCL treated with standard R-CHOP regimen from collaborative medical centers were studied as a part of the DLBCL Consortium Program. Seven patients with both MYC-R and Mut-TP53, identified through routine clinical practice, were additionally included. The current study was approved by the Institutional Research Board from each of the participating medical centers and performed in accordance with the Declaration of Helsinki.
IHC, FISH, and TP53 mutational analysis
Evaluation of genetic alterations and protein expression has been described previously (11, 14, 17, 30–32). Formalin-fixed paraffin-embedded (FFPE) biopsies were organized prior to R-CHOP treatment and tissue microarray was constructed for study. IHC analysis was performed for the following markers with corresponding antibodies: MYC (clone Y69, Epitomics), BCL-2 (clone 124, DAKO), p53 (clone DO-7, DAKO), and Ki-67 (clone MIB-1, DAKO). The cut-off values were 40% for MYC, 50% for BCL-2, 30% for p53 (average of the two cutoffs in previous studies; refs. 14, 17), and 70% for Ki-67 as previously determined.
FISH analysis was performed on FFPE sections to detect MYC rearrangement using a Vysis LSI MYC dual-color break-apart rearrangement probe (Abbott Molecular) following the manufacturer’s instructions. Images were captured and reviewed using Cytovision software (Applied Imaging). At least 200 tumor cell nuclei were scored and a cutoff of more than 5% of positive cells was used to determine the presence of MYC rearrangement.
TP53 mutations were detected using the AmpliChip p53 Research Test (Roche Molecular Systems), which is a microarray-based assay that detects mutations in exons 2–11 (14).
Gene expression profiling analysis
Gene expression microarray data (GEO accession number GSE#31312; ref. 33) as a part of the DLBCL Consortium Program are available for 440 cases in this study. The microarray data were normalized by the RMA method and differentially expressed genes (DEG) were identified using the Student t test with P value cutoff of 0.001.
Cells and reagents
Two DLBCL-NOS cell lines (GR, HBL1) and six HGBCL-DH cell lines (MZ, TMD8, OCI-LY10, OCI-LY9, RC, and MCA) were kind gifts from MD Anderson Cancer Center Laboratory (Drs. Pham and Ford; refs. 34–36). We performed targeted next-generation sequencing, IHC, and FISH to determine the TP53, MYC, and BCL2 alteration status in all DLBCL cell lines. Cells were cultured in RPMI1640 (CORNING) supplemented with 10% to 15% FBS (Gibco, Life Technologies) with 100 U/mL penicillin and 100 μg/mL streptomycin at 37°C in a humidified atmosphere with 5% CO2. All cultured cell lines were routinely tested for Mycoplasma spp. using a MycoSEQ Mycoplasma Detection kit (Invitrogen). Stocks of authenticated cell lines were cryopreserved, and all cell lines used here were obtained from these authenticated stocks and passaged for <6 months in culture for the performed experiments. INCB057643 and DS3032b were supported separately by Incyte and Daiichi Sankyo under approved material transfer agreement. Venetoclax (ABT-199) was purchased from Selleckchem.
Cell viability assay
The DLBCL/HGBCL cell lines were separately seeded at 5,000 to 10,000 cells per well in 384-well plates. After treatment with single or combination drugs for 72 hours, cell viabilities were determined by the CellTiter-Glo Luminescent Cell Viability Assay, according to the manufacturer’s instructions (Promega). The experiments were performed at least three times with triplicates independently.
Apoptosis assay
Annexin V/PI (eBioscience) dual-staining was utilized to detect apoptotic cells according to the manufacturer’s instructions. Briefly, cells were incubated in a 24-well plate at a density of 5 × 105 cells/well and treated with indicated concentrations of INCB057643 alone or cotreatment with venetoclax or DS3032b for 48 hours. After harvesting and washing, cells were resuspended in 100 μL 1× binding buffer containing 5 μL Annexin V and 5 μL PI solution for 15 minutes in the dark at 4°C and then analyzed by flow cytometry using a FACS canto (BD).
Cell-cycle assay
Cell cycle was assayed by Click-iT EdU Flow Cytometry Assay Kits. According to the manufacturer’s instruction, upon exposure with designated doses of drugs for 24 hours, cells were then exposed to 10 μmol/L EdU incubating another 2 hours in 37°C with 5% CO2 incubator. Next, cells were further washed, fixed, and permeabilized, and followed by staining with the click reaction. Before analyzing by flow cytometer, 5 μg/mL PI was added into the prestained EdU cells. Finally, cells were detected and analyzed using a flow cytometer.
Western blot analysis
Whole-cell extracts were subjected to SDS-PAGE on a 4% to 15% gel (Bio-Rad) and then transferred proteins onto PVDF membranes. After blocking nonspecific binding for 1 hour using 1× TBS 1% Casein blocker (Bio-Rad), the membranes were probed with specific primary antibodies overnight at 4°C and then incubated with a secondary HRP-conjugated mAb (1:5,000, Cell Signaling Technology) for 1 hour at room temperature. Proteins were visualized using an ECL system. Antibodies against Aurora A (rabbit; 1:1,000); p53 (mouse; 1:1,000); p-PLK1 (rabbit; 1:1,000); PLK1 (rabbit; 1:1,000); CDC2, (mouse; 1:1,000); p-CDC2 (rabbit; 1:1,000); Myc (rabbit; 1:1,000); p21 (rabbit; 1:1,000); cleaved-PARP (rabbit; 1:1,000); and Bcl-2 (mouse; 1:1,000) were all purchased from Cell Signaling Technology.
RPPA analysis
RPPA analysis and antibody validation were performed by The MD Anderson Cancer Center Functional Proteomics RPPA Core Facility. Briefly, DLBCL cells were separately seeded at 1 × 106 cells per well in 6-well plates for treating with the vehicle or the drug group for 24 hours. After terminating the treatment, cells were washed twice with ice-cold PBS, respectively. In the following steps, total protein lysates were prepared by suspending lymphoma cells in lysis buffer (1% Triton X-100, 50 mmol/L HEPES, 150 mmol/L NaCl, 1.5 mmol/L MgCl2, 1 mmol/L EGTA, 100 mmol/L NaF, 10 mmol/L NaPPi, 10% glycerol, 1 mmol/L PMSF, 1 mmol/L Na3VO4, and 10 μg/mL aprotinin, pH 7.4). Protein concentration was determined by Quick Start Bradford protein assay and adjusted to a concentration of 1.5 μg/μL. Cell lysate samples were serially diluted 2-fold and arrayed on nitro-cellulose-coated slides (Grace Bio Lab). Total 5808 array spots were arranged on each slide and sample spots were probed with 439 unique antibodies covering major oncogenic and signaling pathways in cancer by tyramide-based signal amplification approach, stained and scanned on Aushon 2470 Arrayer and Huron TissueScope scanner. The signal densities were quantified by Array-Pro-Analyzer (Aushon Bio-Systems), relative protein levels determined by interpolating standard curve, and normalized for protein loading.
RPPA analysis for significantly changed protein expression with drug treatment were by Cluster 3.0 (http://bonsai.hgc.jp/~mdehoon/software/cluster/software.htm) and Java TreeView (http://jtreeview.sourceforge.net) software. Heatmaps for differential expression were generated by R.
Statistical analysis
Kaplan–Meier survival curves and the log-rank (Mantel–Cox) test were used to assess survival differences between groups. Multivariate survival analysis was performed on IBM statistics SPSS 19 using the Cox proportional hazards regression model. Differences with P ≤ 0.05 were considered to be statistically significant. For the experimental results in cell viability and apoptosis, the statistical significance was determined by two-sided t test. *, **, ***, and **** indicate P < 0.05, <0.01, <0.001, and <0.001, respectively.
Results
Clinical and pathologic features associated with MYC/TP53 dual abnormalities
Clinicopathologic and molecular features of the patients included in the study are summarized in Supplementary Table S1. FISH detected MYC-R in 10.9% and AmpliChip detected Mut-TP53 in 22.5% of the cohort. IHC determined Mychigh and p53high in 64.6% and 22.2% of the cohort, respectively. The frequency of cases with Mychigh Mut-TP53, Mychighp53high, MYC-R Mut-TP53, and MYC-R p53high dual-abnormalities was 14.4%, 16.5%, 2.6%, and 4.2%, respectively. With the addition of 7 MYC-R Mut-TP53 cases identified in routine clinical practice, the frequency of MYC-R Mut-TP53 rose to 4.7% (Supplementary Table S2).
By Fisher exact test, cases with Mychigh Mut-TP53, Mychighp53high, or MYC-R p53high compared with corresponding other cases had higher frequencies of MYC/BCL-2-DE (56%, 54%, and 46%, respectively), while MYC-R Mut-TP53 and Mychighp53high cases had higher frequencies of HGBCL-MYC/BCL2-DH (33% and 7.9%, respectively) (Supplementary Table S1). Clinically, Mychighp53high was associated with ≥2 extranodal involvement, IPI >2, and high Ki-67; Mychigh Mut-TP53 was associated with high Ki-67; MYC-R p53high was associated with bulky tumor and GCB subtype; and MYC-R Mut-TP53 dual abnormality was associated with elevated serum LDH level, bulky tumor, ≥2 extranodal involvement, and GCB subtype. All these dual MYC/TP53 alteration subtypes were associated with not achieving complete response to R-CHOP therapy (Supplementary Table S1). Pathologically, 15 patients with MYC-R Mut-TP53 dual abnormality and 20 patients with MYC-R p53high showed morphologic features of HGBCL, similar to HGBCL with MYC/BCL2-DH or HGBCL-NOS. The lymphoma cells were diffuse in pattern and predominantly intermediate in size with brisk mitotic figures and apoptotic bodies. Foci of microscopic necrosis were present. The biopsies showed morphologic features of Burkitt or Burkitt-like lymphoma. Representative images are provided in Supplementary Fig. S1.
Synergistic prognostic effect of Myc overexpression and TP53 alterations
To illustrate the prognostic effect of MYC and TP53 dual abnormalities in DLBCL, we first analyzed the Mychighp53high dual expression. Patients with dual Mychighp53high expression had poor survival (median OS, 38.4 months; median PFS, 30.0 months), which was significantly worse than the Myclowp53low (double-negative) subset (Supplementary Fig. S2, P < 0.0001). The other two subgroups, Mychighp53low (single Mychigh abnormality) and Myclowp53high (single p53high abnormality), had relatively better survival the Mychighp53high dual-abnormality, but not statistically significant. The additive effect of p53high to that of Mychigh was especially significant in the ABC-subtype, whereas was comparably weaker in the GCB-subtype (Supplementary Fig. S2).
Among the 78 patients with dual Mychighp53high, 49 patients had Mut-TP53 (Mychigh Mut-TP53) and 29 patients had Wt-TP53. Different from Mychighp53high, the Mychigh Mut-TP53 dual-abnormality (69 cases) predicted significantly inferior OS and PFS than all the other three Myc/TP53 subgroups irrespective of GCB/ABC subtype (Fig. 1). In the overall cohort, the median OS and PFS of patients with Mychigh Mut-TP53 dual abnormality was 25.8 and 22.4 months, respectively, which were significantly worse compared with those of all other 3 Myc/TP53 groups, Mychigh Wt-TP53 (82.1 and 71.0 months, respectively), Myclow Mut-TP53 (undefined), and Myclow Wt-TP53 (undefined) patients (Fig. 1).
Figure 1.
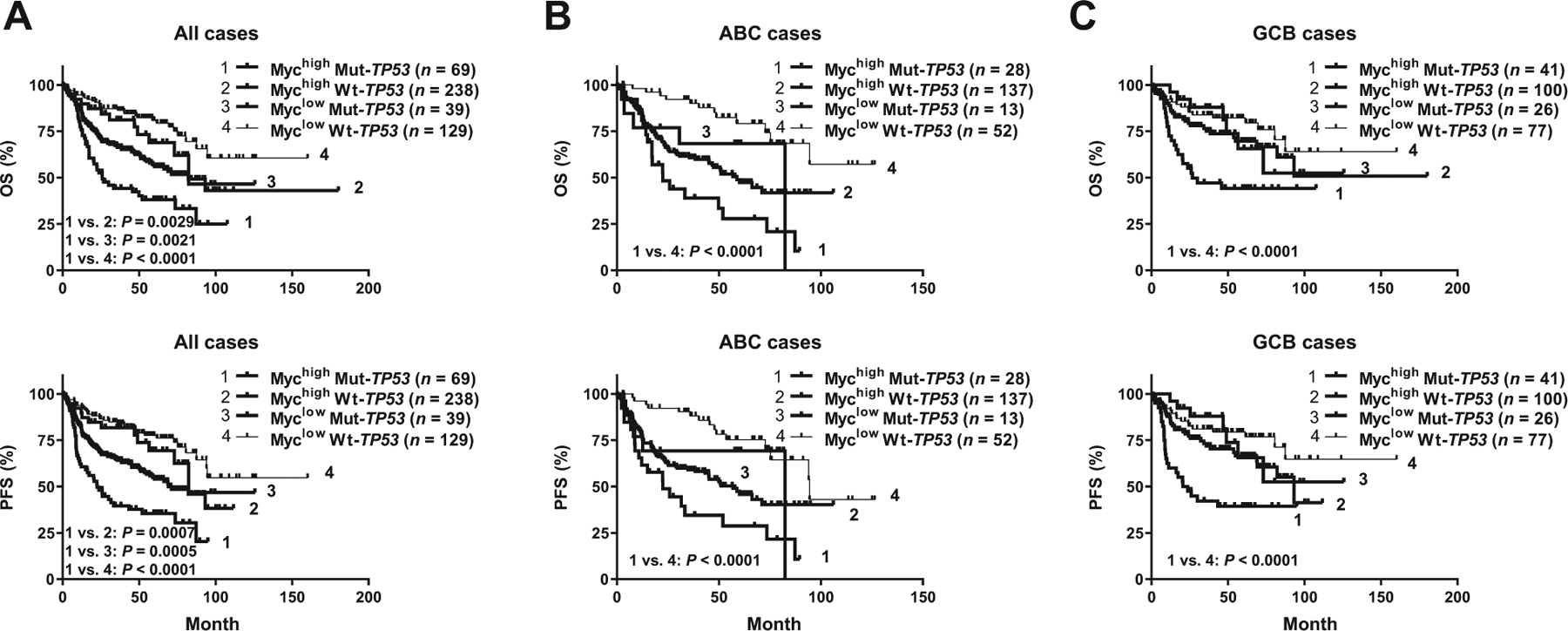
Prognostic effects of TP53 mutation (Mut-TP53) in patients with and without Myc overexpression (Mychigh). Mychigh and Mut-TP53 dual abnormality was associated with significantly inferior OS and PFS in overall cohort (A) and the ABC (B) and GCB (C) subtypes.
Synergistic prognostic effect of MYC-R and TP53 alterations
Next, we examined whether p53 overexpression and TP53 mutation rendered additive negative effects to MYC-R. While dual p53high MYC-R compared with single p53high or MYC-R alteration did not show significantly additive worse impact (Fig. 2A), patients with Mut-TP53 MYC-R dual alterations had significantly worse survival compared with patients with either TP53 mutation or MYC-R alteration alone (Fig. 2B). The median OS and PFS of MYC-R Mut-TP53 patients were strikingly low (9.3 and 8.98 months, respectively). Although TP53 mutation conferred significantly adverse prognostic impact in the absence of MYC-R (Fig. 2C), the Mut-TP53 non-MYC-R group (single TP53 abnormality) had significantly better median OS/PFS (73.1/56.5 months) than the MYC-R Mut-TP53 dual abnormality group (P = 0.0009 and 0.0044, respectively; Fig. 2B–D).
Figure 2.
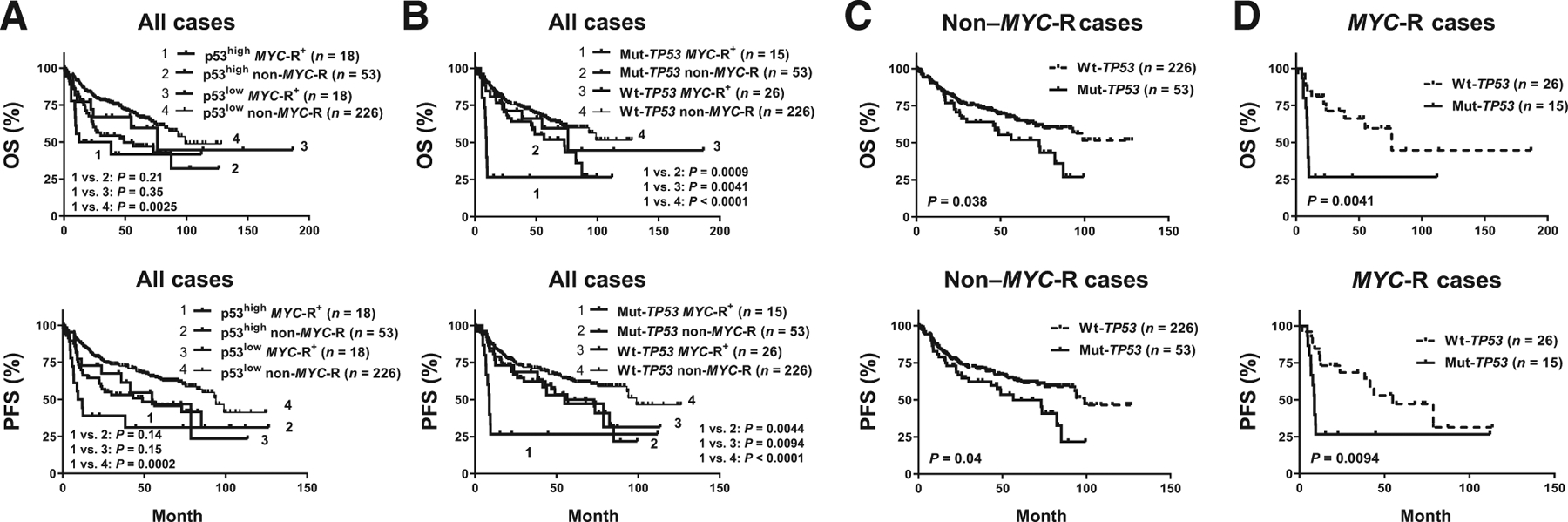
Prognostic effects of TP53 mutation (Mut-TP53) and p53 overexpression (p53high) in patients with MYC rearrangement (MYC-R). A, Patients with DLBCL with concurrent MYC-R and p53high displayed poor OS and PFS but showed little additive negative effect to that of single MYC-R or p53high abnormality. B, Concurrent Mut-TP53 and MYC-R synergistically worsened the OS and PFS in DLBCL. C and D, TP53 mutation had significantly adverse impact independent of MYC-R status.
Supplementary Table S3 summarized the significant prognostic factors by univariate analysis, including B symptoms, high IPI, ABC subtype, and MYC/TP53 alterations. Multivariate analysis showed that MYC-R Mut-TP53 dual abnormality and high IPI still were independent prognostic factors for both OS and PFS.
Synergistic effect of MYC and TP53 alterations on gene expression
Gene expression profiling (GEP) data were analyzed to gain insight into the molecular mechanisms underlying the poor prognosis of Mut-TP53 and MYC dual-abnormalities. We first sought to understand the additive biology effect of Mut-TP53 to MYC-R by identifying significant DEGs between DLBCLs with concurrent MYC-R/Mut-TP53 alterations and DLBCLs with MYC-R alone (MYC-R Wt-TP53). With an FDR threshold of 0.05, 135 DEGs were identified (Supplementary Fig. S3A). Genes downregulated in the Mut-TP53 group include several positive regulators of apoptosis (STK17B, IL24, and RASSF5), ADPRHL2 (which plays key role in DNA damage response), PTEN, WDR81 (negative regulator of PI3K activity), T-cell receptor/CD3 complex and MHC class II molecules. Gene upregulated in the Mut-TP53 group include RAD54B, UNG, DDX11-AS1, and E2F2, which have functions in DNA repair, transcription, and cell-cycle control (Table 1).
Table 1.
Dissecting the role of Mut-TP53, MYC-R, and Mychigh in gene expression profiles.
| Upregulated genes | Downregulated genes |
|---|---|
| Mut-TP53 signature: MYC-R Mut-TP53 vs. MYC-R Wt-TP53 (FDR 0.05) | |
| TEAD4, FAH, EPN2-AS1, SPTBN5, FGF9, SYP, SMIM6, AMBN, SRY, LOC339316, IVL, CASP16, IQGAP3, E2F1, PKD1L1, FRRS1, MT3, C12orf60, C7orf33, CAMP, LOC101928191, PHOX2A, JMJD4, OTOF, LINC00544, GOLGA6A, PTH, ZNF516, LOC101926942, LOC101927043, GFAP, ONECUT2, SLC2A1-AS1, PLCH1, MS4A3, ABCC12, LOC101928921, ZNF560, KIF2B, CDH7, LOC100130155, KCNA6, ANKRD16, BOP1, LRRC3B, LOC101926934, GPR27, FAM19A4, TPTE2P6, CPSF3L, CDK10, POLA2, DDX11-AS1*, E2F2, RAD54B*, UNG*, OGFOD3, CEP70, RTTN, GTF3C4 | ASTE1, NPLOC4, WDR81, PBXIP1, MDFIC, SUCLG1, STK17B, CYTIP, PNRC1, SP140L, PECAM1, CMTM7, C16orf54, FAM65B, BANK1, RASSF5, DOCK8, GVINP1, CYLD, PARP12, LOC100507463, YPEL5, HLA-DRA, HLA-DMA, HLA-DMB, PTPRC, REEP5, CBLB, SNX18, YIPF5, PTEN, WIPF1, PIAS1, CAP1, HIPK3, C16orf72, MGEA5, GLS, CDC42SE1, SP110, CNOT6L, TMEM2, LBH, TMF1, CD3D, TRBC1, GIMAP4, IL6ST, FYB, CD3E, ITM2A, GPRIN3, C5orf20, ZBTB4, CBLN3, PTGDS, TOX4, COMMD9, LIPT1, LYPLAL1, LRP10, SPATA20, ZNF266, CCRL2, ADPRHL2*, SLC41A1, IL24, DCP1B, LRPAP1, DUSP10, SOS1, ACAA1 |
| MYC-R signature: MYC-R Mut-TP53 vs. Non-MYC-R Mut-TP53 (FDR 0.05, fold change >3) | |
| LOC646278, FOXO6, WASF1, LOC100507266, VWCE, NREP. STRBP, NCR3LG1, TERT, ZNF385B, TFDP2, GTF2IRD1, UHRF1*, UNG*, YEATS4*, CDC25A, PAICS, MYC, SNORA71A, ACACA, NUDT11, NTF3, POLR1A, LOC284219, ACP6, PHGDH, IFRD2, FAM27A, CCDC74B-AS1, C16orf89, C7orf33, PHOX2A, RHOV, SPATA3, HK2, SCML2, CCDC77, SLC16A14 | ADPRHL2*, CBLN3, SPATA20, ELF4, HCLS1, PCED1B, APOBEC3G, CCDC92, RASSF5, MBD4*, TRAPPC8, AHNAK, IL10RA, CCND2, ELK3, LOC100507463, CD99, ELL2, PRDM1, RHOG, DRAM2, CCRL2, PARP12, TAX1BP3, MGEA5, KIAA1279, WIPI1, SMAP2, TRIM21, AKR1A1, PNRC1, PARP14, CKLF, REEP5, ZFP36, TMEM2, WSB2, SERPINB1, TNFRSF1B, ARHGAP18, MCL1, SP110, SAMD9L, CAPNS1, ARPC2*, LCP1, EDF1, ASAP1, ANXA4, SCP2, UBB*, SNX3, RNF111, RAP2C, CFLAR, STAT3, IL21R, FNBP1, TNFAIP3, GVINP1, FAS, RNF19A, PTPRC, YPEL5, GPR174, JAK2, IFNGR1, RNASEK, LY96, ANXA7, SH3GLB1, GNG5, ANXA2, PTTG1IP, UBC, CAP1, SH3BGRL3, IL6ST, EPSTI1, PLSCR1, DAPK1, ARL6IP5, HLA-E, HLA-F, HLA-A, HLA-C, HLA-B, SRGN, SAT1, PLA2G7, IL13RA1, FTH1, LYZ, ADAMDEC1, KCTD12, VMP1, CD3D, RAB27A, FYB, GIMAP4, GBP2, GZMK, STAT1, GIMAP2, SLAMF7, ITM2B, UBD, CCL5, CSF2RB, PTGDS, IL18, IL2RG, CD58, ITGAV, HCP5, CTSH, KIAA1551, NFKBIA, CD44, HLA-DMB, HLA-DRA, HLA-DMA, TMSB4X, ALDH2, PEA15, PLEK, ENPP2, TMEM163, HS3ST3A1, ROBO1, CSTA, CHI3L1, FAM129A, MMP9, MDFIC, AHR, BCL2A1, MPEG1, RASSF2*, BIRC3, F8A1, RGS1, CXCL13, MAFF, VOPP1, BASP1, AIM2, FCRL3, CD83 |
| Mychigh signature: Mychigh Mut-TP53 vs. Myclow Mut-TP53 (FDR 0.05, fold change >2) | |
| EHBP1, MIR17HG, NOL11, GAS5, EEF1B2, NOP58, PUS7, CDC7, BZW2, IARS, SNHG1, GCSH, WDR43, MYC, PAICS, DKC1, EBNA1BP2, TMEM97, UHRF1*, TOP2A, PM20D2, HNRNPAB, SRPK1, TOPBP1*, TIPIN, EXO1*, WDR75, IPO4, HDGF, ASF1B, DPY19L2P2, IMPDH2, KIF20A, CBWD5, RIOK1, TRIT1, PRMT3, PPP1R8 | WDTC1, ADCY4, PLD3, ZBTB38, AHNAK, PBXIP1, RNF213, ARL4C, SLFN5, FYB, BCL11B, CD3D, TRBC1, GPR174, EVI2B, CD226, LOC100507463, IL10RA |
Notes: Genes involved in apoptosis, cell death, p53 pathway regulation, or tumor suppression are in bold;
genes involved in DNA repair or DNA damage response are marked by *;
genes indicating impaired immune responses are underlined with straight underlines;
and MIR17HG is underlined with a wavy underline.
To dissect the role of MYC-R in the MYC-R Mut-TP53 dual-abnormality, GEP data were compared between the Mut-TP53 MYC-R and Mut-TP53 non-MYC-R groups (Supplementary Fig. S3B). With an FDR threshold of 0.05, 189 genes were upregulated and 290 genes were downregulated in the MYC-R group. Among these, 187 genes had >3 fold changes in expression; MYC and many genes involved in the cell cycle, biosynthesis, and metabolisms were upregulated, whereas multiple genes involved in DNA repair and/or DNA damage response (MBD4, ARPC2, UBB, and ADPRHL2), cell-cycle arrest (RASSF2), apoptosis (FAS, SH3GLB1, DRAM2, DAPK1, VMP1, SRGN, RASSF2, and RASSF5), degradation, and immune responses (8 HLA/MHC genes and CD3D) were downregulated, consistent with the oncogenic role of MYC. Different from these oncogenic signatures, ZNF385B with role in p53-mediated apoptosis and NCR3LG1, which activates natural killer cells via NCR3 interaction, were upregulated in the MYC-R group, whereas several negative regulators of apoptosis were downregulated, including MCL1, CFLAR, PEA15, TNFRSF1B, BIRC3, BCL2A1, RFFL, and PARP14 (Table 1).
To further understand the synergistic effects of MYC/TP53 alterations, patients with both MYC-R and Mut-TP53 (double-positive) were compared with those with neither of these alterations (double-negative), which showed a more prominent gene signature than when compared with patients with single MYC-R or Mut-TP53 alteration. Largely, the 213 DEGs identified with an FDR threshold of 0.0005 were a combination of the dissected “MYC-R signature” and “Mut-TP53 signature” with improved significance plus a few additional genes, including NLRC5, TINF2, CLIP1, CCR7, and MIR155HG downregulated and BMP3 and DIP2C upregulated in Mut-TP53 MYC-R patients (Supplementary Table S4). Both antiapoptotic genes (MCL1, CFLAR, BCL2A1, HIPK3, and BIRC3) and proapoptotic/autophagy genes were downregulated (FAS, CASP4, SH3GLB1, LITAF, RASSF4, RASSF5, RASSF2, VMP1, and DAPK1), whereas proapoptotic CDKN2A and ZNF385B were upregulated in MYC-R Mut-TP53 patients.
A similar method was used to dissect the additive role of Mychigh in the Mychigh Mut-TP53 dual-abnormality and to understand the combined effects of Mychigh and Mut-TP53. Comparing Mychigh Mut-TP53 (double-positive) with Myclow Mut-TP53 (single-positive) patients identified a Mychigh signature of 56 DEGs with an FDR threshold of 0.05 and a fold change cutoff of >2 (Supplementary Fig. S3C); comparing Mychigh Mut-TP53 with Myclow Wt-TP53 (double-negative) patients identified an integrated “Mychigh Mut-TP53” signature, which significantly enhanced the “Mychigh” signature, 227 DEGs with FDR < 0.0001 (Supplementary Fig. S3D). Compared with the MYC-R and MYC-R Mut-TP53 signatures, the Mychigh and Mychigh Mut-TP53 signatures were more related to DNA replication, cell cycle, mitosis, survival after DNA damage or replication stress, antiapoptosis (NAA40, PEG10, BIRC5, EI24, SNHG1, and HK2), and miRNA (MIR17HG upregulated and MIR155HG downregulated; Table 1; Supplementary Table S4).
Cytotoxicity and proteomic effects of INCB057643 in DLBCL with MYC aberrations
The prognostic and GEP analysis indicated a significant unmet clinical need in DLBCL/HGBCL with concurrent MYC/TP53 alterations. To investigate more effective therapeutic regimens, we first evaluated the cytotoxicity of INCB057643 in 8 MYC-driven (MYC-R or Mychigh) DLBCL cell lines, including 4 lines with Mut-TP53 (2 lines also had BCL2-R) and 4 with Wt-TP53 (all had concurrent BCL2-R; Supplementary Table S5). These cell lines were highly sensitive to INCB057643 treatment with IC50 values below 1 μmol/L, except the GR cell line with IC50 of 3.67 μmol/L (Fig. 3A and B). They all showed increased apoptosis after INCB057643 treatment for 48 hours relative to controls, in a dose-dependent manner (Fig. 3C). Cell-cycle distribution analysis indicated that treatment with INCB057643 for 24 hours induced G2–M phase arrest (Supplementary Fig. S4).
Figure 3.
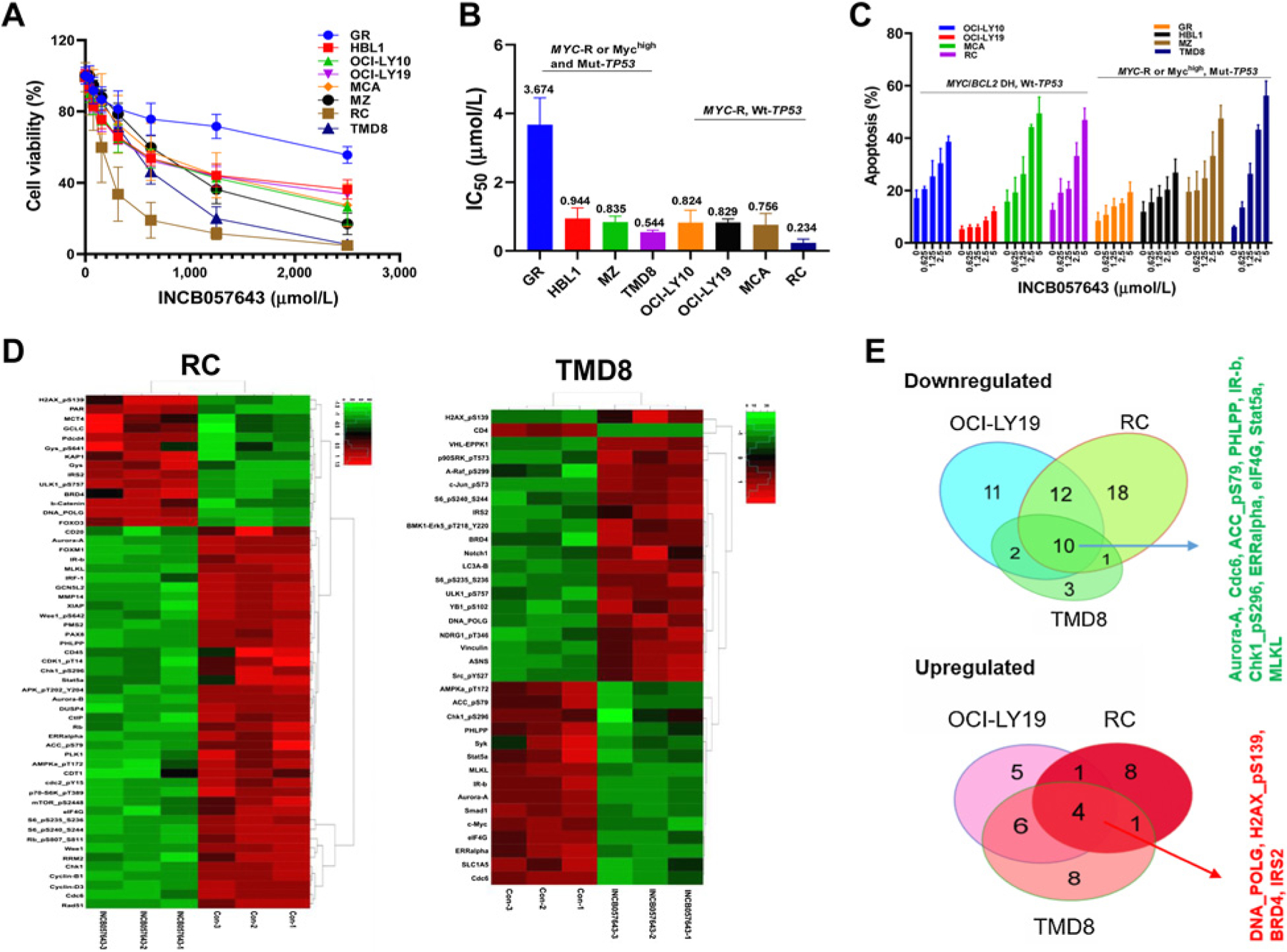
Cytotoxicity and proteomic effects of a BET inhibitor INCB057643 in DLBCL/HGBCL cell lines with MYC rearrangement or Myc overexpression and with or without TP53 mutation. A total of 8 DLBCL/HGBCL cell lines were selected for study, including 4 Wt-TP53 (OCI-LY10, OCI-LY19, MCA, RC) and 4 Mut-TP53 (GR, HBL1, MZ, and TMD8) cell lines. A, Cell viability of lymphoma cells was significantly inhibited after exposure with INCB057643 for 72 hours using CellTiter-Glo 2.0 Assay. B, IC50 values of INCB057643 in 8 DLBCL/HGBCL cell lines as calculated by GraphPad Prism 8. C, Percentage of apoptotic cells by Annexin V/PI double staining flow cytometry analysis after treatment with INCB057643 for 48 hours. Values indicate mean ± SD for at least three independent experiments performed in triplicate. D, Heatmaps illustrating significantly increased or decreased protein expression measured by the RPPA assays after INCB057643 treatment (2.5 μmol/L for 24 hours) in RC and TMD8 cell lines. E, Venn diagrams (right) illustrate the common and unique modulations in three evaluated cell lines (OCI-LY19, RC, and TMD8). RPPA assays were performed for three independent experiments.
RPPA was utilized as a proteomics approach to investigate the biological mechanisms of INCB057643 in 3 HGBCL-DH cell lines, including two with Wt-TP53 (OCI-LY19 and RC) and one with Mut-TP53 (TMD8). After exposure lymphoma cells to INCB057643 for 24 hours, proteins were extracted for proteomic analysis with a panel of 439 antibodies. Normalized expression data went through supervised or unsupervised hierarchical clustering analysis. Ninety proteins were found down- or upregulated >1.32 fold upon INCB057643 treatment in 3 analyzed HGBCL cells (Fig. 3D; Supplementary Fig. S5; Table 2), many of which could be attributable to downregulation of the AKT/mTOR and RAF/MEK/ERK pathways (37). Fourteen proteins were consistently modulated by INBC057643 treatment in all lymphoma lines (Fig. 3E): 10 proteins including Aurora-A and Chk1_pS296 were significantly downregulated, which were involved in mitosis, cell cycle, DNA replication, protein synthesis, metabolism, and growth/survival signaling; 4 proteins, including H2AX_pS139 (involved in double-strand DNA break repair), DNA_POLG, BRD4, and IRS2, were upregulated in the three DLBCL/HGBCL lines. In addition, INBC057643 upregulated Bim (the target of BCL-2 oncogenic activity) and p21 (CDKN1A, involved in p53-mediated DNA damage response) in OCI-LY19 cells, while downregulated Cyclin-B1 and Rb_pS807_S811 in OCI-LY19 and RC cells, CD20 and PMS2 (mismatch repair) in RC cells, and CD4 in TMD8 cells. However, in TMD8 cells Raf_pS299, Erk5_pT218_Y220, p90RSK_pT573, S6_pS235_S236, S6_pS240_S244, and c-Jun_pS73 were upregulated after the treatment, suggesting activation of the RAF/ERK pathway (Table 2).
Table 2.
Proteomic analysis with RPPA for targeted treatment in DLBCL/HGBCL cells
| Decreased protein after treatment | Increased protein after treatment |
|---|---|
| INCB057643 treatment in OCI-LY19, RC, and TMD8 cell lines | |
|
In all cell lines, Aurora-A, Cdc6, ACC_pS79, PHLPP, IR-b, Chk1_pS296, ERRalpha, eIF4G, Stat5a, MLKL; In two cell lines, OCI-LY19, TMD8: Syk, c-Myc; OCI-LY19, RC: Rb_pS807_S811, Wee1, FOXM1, Chk1, Cyclin-B1, Cyclin-D3, MAPK_pT202_Y204, CD45, GCN5L2, mTOR_pS2448, CtIP, IRF-1; RC, TMD8: AMPKa_pT172; In one cell line, OCI-LY19: ACC1, Gab2, Hif-1-alpha, 4E-BP1_pT37_T46, 4E-BP1_pS65, B-Raf_pS445, FASN, Dvl3, E-Cadherin, C-Raf_pS338, C-Raf; RC: S6_pS235_S236, S6_pS240_S244, RRM2, CD20, Rad51, DUSP4, Aurora-B, PAX8, CDK1_pT14, PLK1, Rb, CDC2_pY15, CDT1, MMP14, p70-S6K_pT389, PMS2, XIAP, Wee1_pS642; TMD8: CD4, Smad1, SLC1A5. |
In all cell lines, DNA_POLG, H2AX_pS139, BRD4, IRS2; In two cell lines, OCI-LY19, TMD8: p90RSK_pT573, Vinculin, c-Jun_pS73, NDRG1_pT346, LC3A-B, Notch1; OCI-LY19, RC: PAR; RC, TMD8: ULK1_pS757; In one cell line, OCI-LY19: Stat3, Bim, PDK1_pS241, RIP, p21; RC: FOXO3, KAP1, b-Catenin, GCLC, Gys, Gys_pS641, MCT4, Pdcd4; TMD8: EPPK1, YB1_pS102, ASNS, S6_pS235_S236, S6_pS240_S244, BMK1-Erk5_pT218_Y220, Src_pY527, Raf_pS299. |
| DS3032-b treatment in OCI-LY10, OCI-LY19, MCA, and RC cell lines | |
|
In all cell lines, Cdc6, Chk1, RRM2, Rad51, FOXM1, Wee1; In three cell lines, OCI-LY10, LY19, RC: CDK1_pT14, EPPK1; OCI-LY10, MCA, RC: Rb_pS807_S811; In two cell lines, MCA, RC: Chk1_pS296; OCI-LY10, MCA: S6_pS240_S244, S6_pS235_S236, DNMT1; In one cell line, RC: DUSP4; OCI-LY10: PLK1, p90RSK_pT573, Cyclin-B1, Aurora-A, Aurora-B, c-Myc, Cox-IV, eEF2, MAPK_pT202_Y204; MCA: PHLPP, PAX8, CtIP, ARID1A, Mcl-1, MSH6, Wee1_pS642, XPA. |
In all cell lines, p21, p53, PUMA; In three cell lines, OCI-LY19, RC, MCA: Histone-H3; OCI-LY10, MCA, RC: H2AX_pS139; In two cell lines, OCI-LY10, OCI-LY19: Bax; OCI-LY10, RC: Axl; MCA, RC: Caspase-7-cleaved; In one cell line, OCI-LY10: Src_pY416; OCI-LY19: S6, HMHA1, PAR, Stat3, ATRX; MCA: Caveolin-1, PAI-1, MDM2_pS166; RC: Histone-H3_pS10, Akt_pS473. |
| Venetoclax treatment in OCI-LY19, RC, and TMD8 cell lines | |
|
In two cell lines,
OCI-LY19, RC: p21; RC, TMD8: AMPKa_pT172, ACC_pS79; In one cell line, OCI-LY19: IGFBP2, JNK2; RC: Aurora-A, Cyclin-B1; TMD8: Axl, Cdc6, Chk1_pS296, eEF2K, Myosin-IIa_pS1943, PAR, PARG, PHLPP, Rad51, Stat3, Stat5a, Smad1, Smac. |
In all cell lines, H2AX_pS139, Mcl-1; In two cell lines, OCI-LY19, RC: PAR; OCI-LY19, TMD8: ER-a_pS118; In one cell lines, OCI-LY19: PRAS40_pT246, S6_pS235_S236, S6_pS240_S244; RC: Caspase-7-cleaved; TMD8: H2AX_pS140, A-Raf_pS299, Bad_pS112, CREB_pS133, c-Jun_pS73, HSP27_pS82, p90RSK_pT573. |
Synergistic effects of INCB057643 and DS3032b in HGBCL-DH with Wt-TP53
MDM2 negatively regulates p53 by mediating p53 ubiquitination and degradation. A novel MDM2 inhibitor DS3032b was investigated as monotherapy and in combination with INBC057643 in the 8 MYC-driven lymphoma cells with different TP53 mutation status. Expression of p53 was greatly increased by DS3032b in 4 HGBCL-MYC/BCL2-DH cell lines with Wt-TP53 (OCI-LY10, OCI-L19, MCA, and RC) but not in those with Mut-TP53 (TMD8 and MZ, Supplementary Fig. S6A). In Wt-TP53 cell lines (all HGBCL-DH), DS3032b consistently induced loss of cell viability with IC50 values varying from 62.71 to 237.08 nmol/L and promoted apoptosis in a dose-dependent manner and G0–G1-phase cell-cycle arrest (Supplementary Fig. S6B–S6E). In contrast, DS3032b did not show such therapeutic effects in all the 4 Mut-TP53 cell lines (Supplementary Fig. S6B and S6D).
RPPA demonstrated 19 upregulated and 31 downregulated proteins in four Wt-TP53 cell lines (OCI-LY10, OCI-LY19, MCA, and RC). In all cell lines, DS3032b treatment significantly upregulated p53, PUMA, and p21 expression, whereas downregulated 6 proteins with roles in DNA replication (Cdc6 and RRM2), DNA repair (Rad51), mitosis (FOXM1), and cell-cycle regulation (Chk1 and Wee1) resembling the effect of INCB057643 (Table 2). In one to two cell lines, DS3032b downregulated Myc, Aurora-A, Aurora-B, PLK1, Cyclin-B1, p90RSK_pT573, MAPK_pT202_Y204, MSH6 (mismatch repair), and Mcl-1 (anti-apoptotic), and upregulated Bax (pro-apoptotic), Axl (activating Akt), and Akt_pS473.
DS3032b was then combined with INCB057643 and their synergy was evaluated. In HGBCL-MYC/BCL2-DH cells with Wt-TP53, combination treatment significantly inhibited cell viability and enhanced apoptosis compared with INCB057643 or DS3032b monotherapy (Fig. 4A and B). However, combination with DS3032b did not show additional therapeutic effect in cells with Mut-TP53 (Supplementary Fig. S7).
Figure 4.
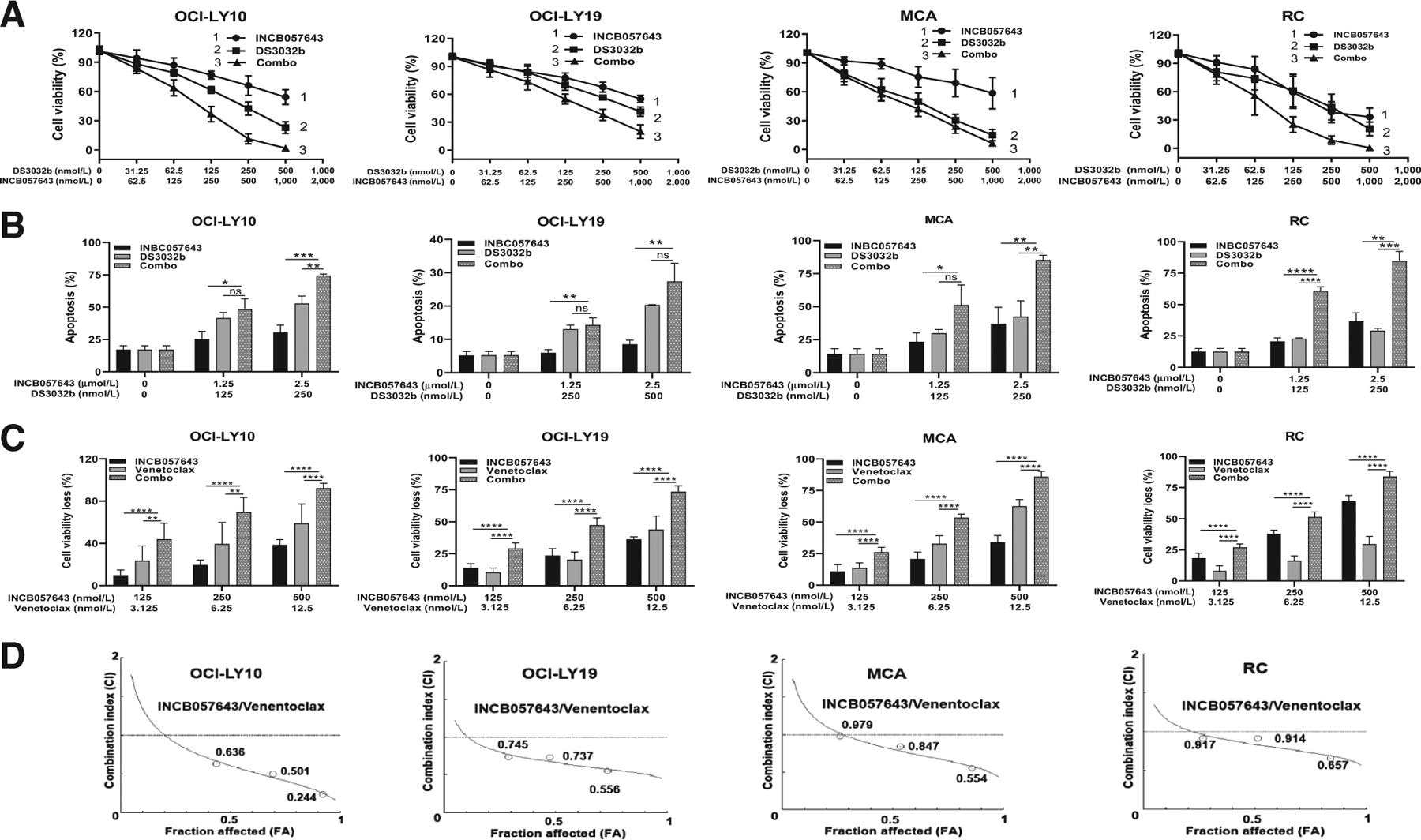
Cotreatment of INCB057643 with DS3032b or venetoclax showed significant and synergistic cytotoxicity on HGBCL-DH cells with Wt-TP53. OCI-LY10, OCI-LY19, MCA, and RC all are HGBCL cells with concurrent MYC-R, BCL2-R, and Wt-TP53 (A) DS3032b combined with INCB057643 induced remarkable cell viability loss in OCI-LY10, OCI-LY19, MCA, and RC cells. B, INCB057643 and DS3032b combination induced higher percentage of apoptosis than single-agent measured in all cells. C, Combined venetoclax and INCB057643 synergistically inhibited cell viability. D, The combination index (CI) of INCB057643 and venetoclax was calculated by CompuSyn software according to the Chou–Talalay method, where CI < 1 indicates synergy. Cell viability was assessed by CellTiter-Glo 2.0 Assay at 72 hours. Apoptosis was determined by Annexin V/PI staining at 48 hours. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant. Values indicate mean ± SD for at least three independent experiments performed in triplicate.
Synergistic effects of INCB057643 and venetoclax in DLBCL/HGBCL regardless of TP53 mutation status
To overcome the therapeutic resistance conferred by TP53 mutation, venetoclax was evaluated as monotherapy and in combination with INCB057643. Venetoclax alone showed cytotoxicity in lymphoma cells regardless of MYC/BCL2/TP53 status, but had more prominent effect in Wt-TP53 than Mut-TP53 cells. RPPA was used for three cell lines (OCI-LY19, RC, and TMD8), and found downregulation of 20 proteins and upregulation of 15 proteins, largely involved in inducing apoptosis and DNA damage response and inhibiting cell-cycle progression (Table 2). Of the 35 venetoclax-modulated proteins, 15 and 11 were modulated also by INCB057643 and DS3032b, respectively, whereas 7 proteins (p21, Mcl-1, Stat3, Axl, S6_pS235_S236, S6_pS240_S244, and p90RSK_pT573) were oppositely modulated by INCB057643 and/or DS3032b. In RC and MCA cells, venetoclax and DS3032b alone (but not INCB057643) significantly upregulated cleaved caspase-7 expression. Interestingly, venetoclax upregulated H2AX_pS139 and Mcl-1 in all cell lines and p21 was downregulated in 2 of 3 RPPA-tested cell lines (OCI-LY19 and RC). In OCI-LY19 and/or TMD8 cells, many proteins involved in the RAS/RAF/MEK/ERK pathway (including the downstream Mcl-1, Bad_pS112, p90RSK_pT573, S6_pS235_S236, S6_pS240_S244, CREB_pS133, and c-Jun_pS73) were upregulated after venetoclax treatment (Table 2).
Remarkably, combined venetoclax and INCB057643 treatment demonstrated synergy and effectively reduced cell viability in MYC-abnormal lymphoma cells regardless of Wt-TP53 (Fig. 4C and D) or Mut-TP53 (Fig. 5A and B) status. The synergism was observed in all tested MYC-driven lymphoma cells with or without Mut-TP53, indicated by combination index <1, which was calculated according to the Chou–Talalay method (Figs. 5B and 4D). All Mut-TP53 cells showed synergistic proapoptotic response to the combination therapy (Fig. 5C). Western blotting confirmed that INCB057643 treatment alone markedly inhibited Myc, Aurora-A, p-PLK1, and p-CDC2 expression in Mut-TP53 cells, whereas p21 expression was enhanced in three of the four Mut-TP53 cell lines (Fig. 5D; Supplementary Fig. S8). Myc, Aurora-A, p-PLK1, and p-CDC2 inhibition were further decreased by combination with venetoclax, although venetoclax alone had limited or no effects on these proteins. Similar to INCB057643, venetoclax also resulted in increased cleaved-PARP in Mut-TP53 cells, and such effects were markedly enhanced with INCB057643 addition (Fig. 5D). BCL-2 expression showed minimal changes with venetoclax or INCB057643 monotherapy and in combination (Supplementary Fig. S8).
Figure 5.
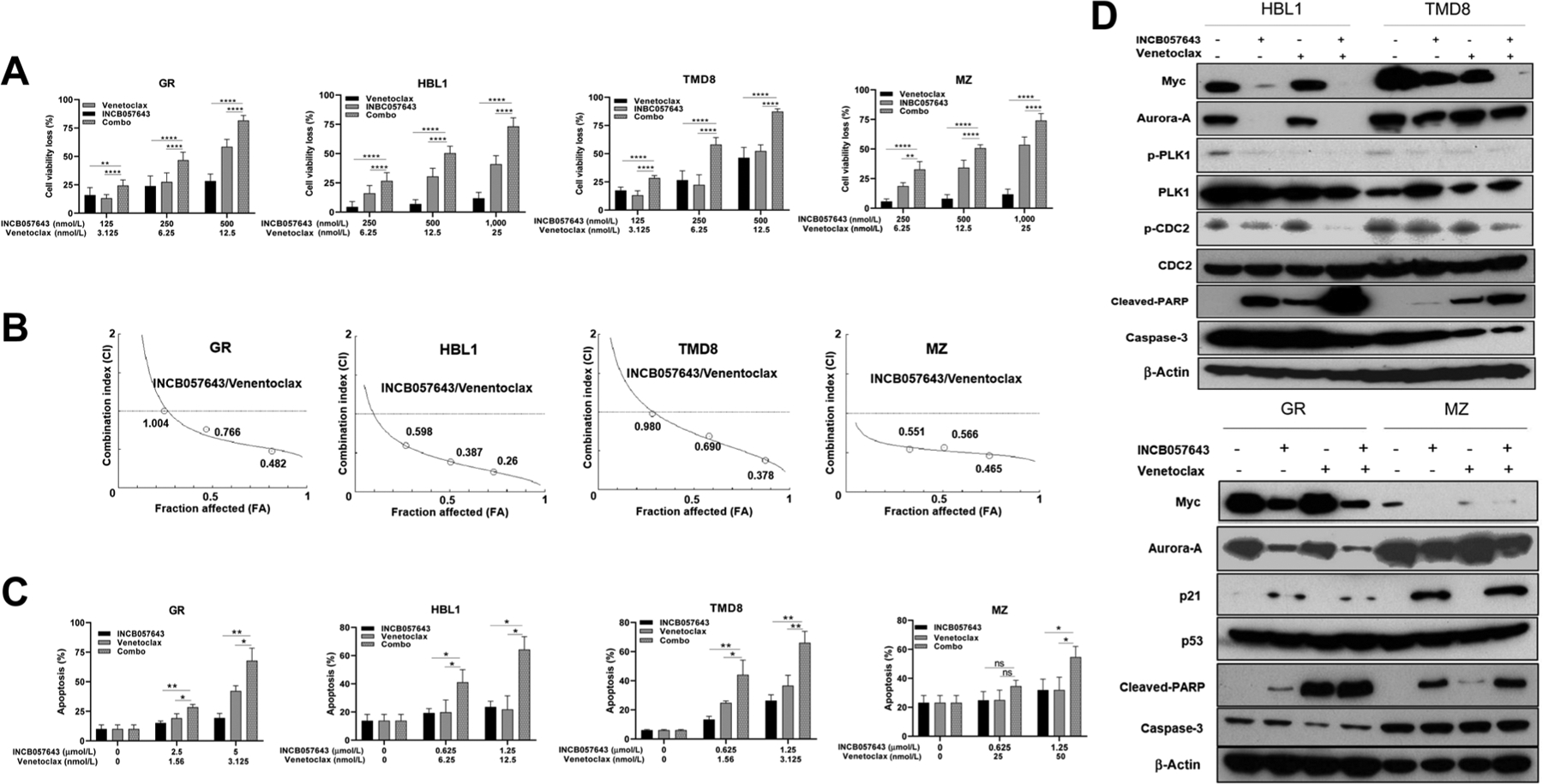
Combination therapy with INCB057643 and venetoclax in DLBCL/HGBCL cells with concurrent MYC alterations and Mut-TP53. A, Cotreatment with venetoclax and INCB057643 synergistically inhibited cell viability in four DLBCL/HGBCL cell lines with concurrent TP53 mutations and MYC-R (MZ and TMD8) or Mychigh (GR and HBL1). B, The combination index (CI) of INCB057643 and venetoclax was calculated by CompuSyn software according to the Chou–Talalay method, where CI < 1 indicates synergy, = 1 is additive, and > 1 is antagonistic effect. C, Apoptosis in GR, HBL1, MZ, and TMD8 was measured by Annexin V/PI staining after treatment with venetoclax, INCB057643 and the combination for 48 hours at designated concentration. The data are presented as mean ± SD for at least three independent experiments performed in triplicate. “ns” indicates not significant; *, P < 0.05; **, P < 0.01; ****, P < 0.0001. D, Western blot analysis to validate RPPA results, including proteins with roles in mitosis and cell growth (Myc, Aurora-A, PLK1, and CDC2), apoptotic indicator cleaved PARP, caspase-3, p53, and p53-targeted p21. β-Actin was probed as loading control. Four DLBCL/HGBCL cell lines with mutant TP53 were exposed to single-agent or combined venetoclax (3.125 nmol/L for GR; 6.25 nmol/L for HBL1; 25 nmol/L for MZ; and 3.125 nmol/L for TMD8) and INCB057643 (2.5 μmol/L for GR; 1.25 μmol/L for HBL1, MZ, and TMD8) treatment for 24 hours. Combined INCB057643 and venetoclax treatment decreased expression of Myc, Aurora-A, and the phosphorylation of downstream targets CDC2 and PLK1, and increased cleaved PARP and p21 expression.
Discussion
We and others have shown that TP53 mutation has significant adverse prognostic effects in DLBCL (13–15), and that aberrant MYC activation due to MYC-R or dysregulated signaling synergizes with oncogenic BCL2-R or BCL-2 overexpression (11, 30–32) owing to the cooperation between cell proliferation driven by MYC and antiapoptosis mediated by BCL-2. Because Myc/MYC-R can biologically induce Wt-p53–mediated apoptosis (38), Mut-TP53 could function as a second-hit synergizing with MYC abnormalities suggested by previous reports (21–23), but this novel double-hit requires validation and refinement. In this study, TP53 and MYC (Myc/MYC) dual alterations were indeed biologically synergistic and predicted worse prognosis in DLBCL than single TP53 or MYC alteration. DLBCL with concurrent MYC-R and TP53 mutation or MYC-R and p53high showed morphologic features of HGBCL, similar to HGBCL-MYC/BCL2-DH and HGBCL-NOS. Furthermore, DLBCL cells with MYC/TP53 dual alterations, biologically resembling HGBCL-MYC/BCL2-DH and MYC/BCL-2-DE, could be effectively treated with combined BET and BCL-2 inhibitors.
Four pathologic dual alterations, including Mychighp53high, Mychigh Mut-TP53, MYC-R p53high, and MYC-R Mut-TP53 were assessed for prognostic significance. These subtypes were associated with unfavorable clinical factors and/or high Ki-67 proliferation index. TP53 mutation rendered more additive negative prognostic effects than p53 overexpression when combined with MYC-R or Mychigh. The MYC-R Mut-TP53 dual abnormality (frequency, 2.6%–4.7%) had the worst adverse prognostic effect. Approximately 33% of MYC-R Mut-TP53 patients harbored MYC/BCL2-DH, accounting for 27% to 42% of HGBCL-MYC/BCL2-DH cases. Compared with MYC-R Mut-TP53, Mychigh Mut-TP53 dual-abnormality occurred more frequently (14.4%). Mychigh Mut-TP53 patients had significantly poorer survival than patients with single-alteration, Mychigh or Mut-TP53. Approximately 56% of Mychigh Mut-TP53 cases displayed MychighBCL-2high phenotype, accounting for approximately 24% of MYC/BCL-2-DE cases.
More effective therapies need to be developed for DLBCL/HGBCL with MYC/TP53 dual alterations or MYC/BCL2-DH with dismal prognosis (39, 40). A number of BET inhibitors targeting MYC expression have been assessed in clinical trials (41, 42). We evaluated the cytotoxicity and proteomic effects of INCB057643, which in a clinical trial showed favorable pharmacokinetic and pharmacodynamics profile allowing maximization of therapeutic index with an optimal and tolerable dose (23). Our data indicated that INCB057643 reduced DLBCL/HGBCL cell viability and enhanced apoptosis in a dose-dependent manner and had efficacy in all DLBCL/HGBCL cell lines regardless of MYC-R, BCL2-R, and TP53 mutation status. Proteomic analysis showed that INCB057643 treatment in DLBCL cells downregulated Myc and molecules that are important for proliferation, cell cycle (e.g., Chk1, Chk1_pS296, Cyclin-B1, Cyclin-D3), mitosis (e.g., Aurora-A/B, Cdc6, Cdc2), metabolism (ACC_pS79, IR-b, ERRalpha, mTOR_pS2448), and protein synthesis (e.g., eIF4G), some of which being part of the MYC GEP signatures (e.g., AURKA, CHEK1) in patients with dual MYC/TP53 alterations, whereas upregulated proteins involved in DNA double-strand break repair (H2AX_pS139), cell-cycle arrest (p21), translation inhibitor (Pdcd4), or apoptosis (Bim, FOXO3) in some cell lines. Previous studies showed that Myc positively regulates AURKA/Aurora-A and Aurora-B expression (43); Aurora-A in turn stabilizes Myc protein mediated via PLK1 and promotes inactivation and degradation of p53, in addition to having a central role in mitotic control (44, 45). The Aurora-A axis has emerged as an attractive target actionable in p53-deficient cancer (44, 46), which may underscore the mechanism of INCB057643 action in addition to Myc inhibition.
However, MYC-R GEP signatures dissected from the MYC-R Mut-TP53 patients showed some proapoptotic components, and by RPPA analysis INCB057643 treatment also decreased expression of PHLPP which promotes apoptosis and MLKL which plays a key role in TNF-induced necroptosis in all MYC-driven DLBCL cell lines, suggesting that therapeutic inhibition of MYC should be combined with proapoptotic agents to maximize the therapeutic effect. We first tried a potent MDM2-p53 inhibitor DS3032b, which is being investigated in phase I/II clinical trials and can re-active TP53 signaling, decrease proliferation, and increase apoptosis in neuroblastoma regardless of the presence of MYCN amplification(25,26).DS3032b was effective in our HGBCL-MYC/BCL2-DH cell lines with Wt-TP53, and the mechanism of action revealed by RPPA analysis included enhancing apoptosis via increasing Wt-p53, Bax, and PUMA proteins whereas decreasing Mcl-1, inducing G0–G1cell-cycle arrest via increasing p21 and H2AX_pS139, and decreasing cell viability via decreasing proteins involved in DNA replication/repair and mitosis (e.g., Cdc6, RRM2, Rad51, FOXM1, Rb_pS807_S811). In one cell line tested, Aurora-A/B were decreased, consistent with the role of p53 in negatively regulating AURKA/Aurora-A expression (47). When combined with INCB057643, DS3032b enhanced the therapeutic potency in all HGBCL-MYC/BCL2-DH cell lines with Wt-TP53, which has significant implications for HGBCL-MYC/BCL2-DH, in that the median OS was less than 2 years with standard therapy (48, 49).
Next, to target TP53 mutation, which is considered “undruggable” in cancer (12), we used venetoclax to counteract BCL-2–mediated antiapoptotic activities in cells with Mut-TP53. Venetoclax has been approved by FDA for treatment of chronic lymphoid leukemia with 17p deletion or TP53 mutation (50). Our in vitro study showed that venetoclax had potent cytotoxic effects in DLBCL/HGBCL cells with more pronounced effect in those with Wt-TP53. Combined venetoclax and INCB056743 treatment showed synergistic cytotoxicity, enhanced Aurora-A inhibition, and cleaved PARP induction in DLBCL with dual Mychigh Mut-TP53 (GR and HBL1) or MYC-R Mut-TP53 (MZ and TMD8, which are also HGBCL-MYC/BCL2-DH). However, these effects need to be confirmed in vivo, as in a phase I/II study, the overall response rate to venetoclax was only 18% in RR-DLBCL (27). Immunotherapy combination may also be necessary, considering downregulation of genes related to MHC and immune responses in patients with dual MYC/Mut-TP53 abnormalities in our GEP analysis.
The molecular mechanisms for the synergy between venetoclax and INCB056743 include enhanced apoptosis due to concurrent inhibition of BCL-2/Bim interaction by venetoclax and Bim upregulation by INCB056743 (in OCI-LY19 cells), consistent with the results for another two BET inhibitors in a recent study (29), as well as a few opposing modulations by these two inhibitors (summarized in Visual Overview). Venetoclax downregulated p21 expression whereas INCB057643 upregulated p21 in several cell lines. In contrast to INCB057643 downregulating a large number of proteins involved in the AKT/mTOR pathway, venetoclax treatment upregulated RAS/RAF/mTOR signaling proteins in a HGBCL-MYC/BCL2-DH (OCI-LY19) cell line and a MYC/BCL2/Mut-TP53-TH (TMD8) cell line, and induced Mcl-1 expression (mTOR regulates Mcl-1 expression) in all tested cell lines, which may represent a potential resistance mechanism for single-agent venetoclax treatment (24). INCB057643 treatment downregulated RAF/mTOR proteins in OCI-LY19, but upregulated RAF/ERK5 signaling proteins in TMD8, suggesting RAF inhibitors can be used in combination therapies for HGBCL-MYC/BCL2/Mut-TP53-TH. The opposing/neutralizing modulations of INCB057643 on AKT/mTOR signaling could also contribute to the synergistic effect of DS3032b and INCB057643 combination in some cell lines, as single-agent DS3032b treatment was associated with increased p-AKT in RC cells and Axl in OCI-LY10 and RC cells.
In conclusion, this study defined a new subset of DLBCL/HGBCL with concomitant MYC/TP53 alterations that exhibits significantly worse survival and unique high-grade morphologic features. Combined MYC/BCL-2–targeted therapy (INCB057643 and venetoclax) could be an alternative therapy for DLBCL with MYC/TP53 dual-abnormality and HGBCL-MYC/BCL2-DH. HGBCL-MYC/BCL2-DH patients without TP53 mutation can also select for combined INCB057643 and DS3032b treatment. Therapeutic synergy may result from concurrent inhibition of mitotic and mTOR pathways and increased proapoptotic activities or proteins, and could be further enhanced by inhibiting the RAS/RAF pathway. These encouraging results provide clinically meaningful and therapeutically important implications for high-grade aggressive B-cell lymphomas.
Supplementary Material
Acknowledgments
This work was supported by NCI/NIH grants 1R01CA233490-01A1, R01CA138688, R01CA187415, and 1RC1CA146299 to K.H. Young and Y. Li. K.H. Young is also supported by The Duke University Institutional Research Grant Award, Hagemeister Lymphoma Foundation, and Incyte Pharmaceutical Corporation.
Authors’ Disclosures
G. Bhagat reports personal fees from Seattle Genetics and Boston Biomedical Inc. outside the submitted work. E.D. Hsi reports other from Eli Lilly and Abbvie, personal fees from Seattle Genetics, Jazz Pharmaceuticals, and Miltenyi Biotec outside the submitted work. B.M. Parsons reports personal fees from Bristol-Myers Squibb, Amgen, and AstraZeneca outside the submitted work. M.A. Piris reports grants from Spanish Association Against Cancer (AECC) during the conduct of the study, grants and personal fees from Takeda, grants from Gilead, and personal fees from EUSA outside the submitted work. J.N. Winter reports grants and personal fees from Merck, Karyopharm, AstraZeneca, Bayer, and Adicet Bio, and grants from Glaxo-Smith-Kline outside the submitted work. No disclosures were reported by the other authors.
Footnotes
Note: Supplementary data for this article are available at Molecular Cancer Research Online (http://mcr.aacrjournals.org/).
References
- 1.Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. N Engl J Med 2018; 378:1396–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med 2018;24:679–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crump M, Neelapu SS, Farooq U, Van Den Neste E, Kuruvilla J, Westin J, et al. Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood 2017;130:1800–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dang CV. MYC on the path to cancer. Cell 2012;149:22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Savage KJ, Johnson NA, Ben-Neriah S, Connors JM, Sehn LH, Farinha P, et al. MYC gene rearrangements are associated with a poor prognosis in diffuse large B-cell lymphoma patients treated with R-CHOP chemotherapy. Blood 2009;114: 3533–7. [DOI] [PubMed] [Google Scholar]
- 6.Battey J, Moulding C, Taub R, Murphy W, Stewart T, Potter H, et al. The human c-myc oncogene: structural consequences of translocation into the IgH locus in Burkitt lymphoma. Cell 1983;34:779–87. [DOI] [PubMed] [Google Scholar]
- 7.McMahon SB. MYC and the control of apoptosis. Cold Spring Harb Perspect Med 2014;4:a014407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Friedberg JW. How I treat double-hit lymphoma. Blood 2017;130:590–6. [DOI] [PubMed] [Google Scholar]
- 9.Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016;127:2375–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ok CY, Medeiros LJ. High-grade B-cell lymphoma: a term re-purposed in the revised WHO classification. Pathology 2020;52:68–77. [DOI] [PubMed] [Google Scholar]
- 11.Hu S, Xu-Monette ZY, Tzankov A, Green T, Wu L, Balasubramanyam A, et al. MYC/BCL2 protein coexpression contributes to the inferior survival of activated B-cell subtype of diffuse large B-cell lymphoma and demonstrates high-risk gene expression signatures: a report from The International DLBCL Rituximab-CHOP Consortium Program. Blood 2013;121:4021–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bykov VJN, Eriksson SE, Bianchi J, Wiman KG. Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer 2018;18:89–102. [DOI] [PubMed] [Google Scholar]
- 13.Young KH, Weisenburger DD, Dave BJ, Smith L, Sanger W, Iqbal J, et al. Mutations in the DNA-binding codons of TP53, which are associated with decreased expression of TRAILreceptor-2, predict for poor survival in diffuse large B-cell lymphoma. Blood 2007;110:4396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu-Monette ZY, Wu L, Visco C, Tai YC, Tzankov A, Liu WM, et al. Mutational profile and prognostic significance of TP53 in diffuse large B-cell lymphoma patients treated with R-CHOP: report from an International DLBCL Rituximab-CHOP Consortium Program Study. Blood 2012;120: 3986–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Young KH, Leroy K, Moller MB, Colleoni GW, Sanchez-Beato M, Kerbauy FR, et al. Structural profiles of TP53 gene mutations predict clinical outcome in diffuse large B-cell lymphoma: an international collaborative study. Blood 2008; 112:3088–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giacomelli AO, Yang X, Lintner RE, McFarland JM, Duby M, Kim J, et al. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat Genet 2018;50:1381–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu-Monette ZY, Moller MB, Tzankov A, Montes-Moreno S, Hu W, Manyam GC, et al. MDM2 phenotypic and genotypic profiling, respective to TP53 genetic status, in diffuse large B-cell lymphoma patients treated with rituximab-CHOP immunochemotherapy: a report from the International DLBCL Rituximab-CHOP Consortium Program. Blood 2013;122:2630–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clipson A, Barrans S, Zeng N, Crouch S, Grigoropoulos NF, Liu H, et al. The prognosis of MYC translocation positive diffuse large B-cell lymphoma depends on the second hit. J Pathol Clin Res 2015;1:125–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gebauer N, Bernard V, Gebauer W, Thorns C, Feller AC, Merz H. TP53 mutations are frequent events in double-hit B-cell lymphomas with MYC and BCL2 but not MYC and BCL6 translocations. Leuk Lymphoma 2015;56:179–85. [DOI] [PubMed] [Google Scholar]
- 20.Schiefer AI, Kornauth C, Simonitsch-Klupp I, Skrabs C, Masel EK, Streubel B, et al. Impact of single or combined genomic alterations of TP53, MYC, and BCL2 on survival of patients with diffuse large B-cell lymphomas: a retrospective cohort study. Medicine 2015;94:e2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang XJ, Medeiros LJ, Bueso-Ramos CE, Tang G, Wang S, Oki Y, et al. P53 expression correlates with poorer survival and augments the negative prognostic effect of MYC rearrangement, expression or concurrent MYC/BCL2 expression in diffuse large B-cell lymphoma. Mod Pathol 2017;30:194–203. [DOI] [PubMed] [Google Scholar]
- 22.Xie Y, Bulbul MA, Ji L, Inouye CM, Groshen SG, Tulpule A, et al. p53 expression is a strong marker of inferior survival in de novo diffuse large B-cell lymphoma and may have enhanced negative effect with MYC coexpression: a single institutional clinicopathologic study. Am J Clin Pathol 2014;141:593–604. [DOI] [PubMed] [Google Scholar]
- 23.Falchook GS, Rosen S, LoRusso PM, Watts JM, Gupta S, Coombs CC, et al. Development of 2 bromodomain and extraterminal inhibitors with distinct pharmacokinetic and pharmacodynamic profiles for the treatment of advanced malignancies. Clin Cancer Res 2020;26:1247–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Esteve-Arenys A, Valero JG, Chamorro-Jorganes A, Gonzalez D, Rodriguez V, Dlouhy I, et al. The BET bromodomain inhibitor CPI203 overcomes resistance to ABT-199 (venetoclax) by downregulation of BFL-1/A1 in in vitro and in vivo models of MYC+/BCL2+ double hit lymphoma. Oncogene 2018;37:1830–44. [DOI] [PubMed] [Google Scholar]
- 25.Arnhold V, Schmelz K, Proba J, Winkler A, Wunschel J, Toedling J, et al. Reactivating TP53 signaling by the novel MDM2 inhibitor DS-3032b as a therapeutic option for high-risk neuroblastoma. Oncotarget 2018;9:2304–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ishizawa J, Nakamaru K, Seki T, Tazaki K, Kojima K, Chachad D, et al. Predictive gene signatures determine tumor sensitivity to MDM2 inhibition. Cancer Res 2018;78:2721–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davids MS, Roberts AW, Seymour JF, Pagel JM, Kahl BS, Wierda WG, et al. Phase I first-in-human study of venetoclax in patients with relapsed or refractory non-Hodgkin lymphoma. J Clin Oncol 2017;35:826–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zelenetz AD, Salles G, Mason KD, Casulo C, Le Gouill S, Sehn LH, et al. Venetoclax plus R- or G-CHOP in non-Hodgkin lymphoma: results from the CAVALLI phase 1b trial. Blood 2019;133:1964–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cummin TEC, Cox KL, Murray TD, Turaj AH, Dunning L, English VL, et al. BET inhibitors synergize with venetoclax to induce apoptosis in MYC-driven lymphomas with high BCL-2 expression. Blood Adv 2020;4:3316–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tzankov A, Xu-Monette ZY, Gerhard M, Visco C, Dirnhofer S, Gisin N, et al. Rearrangements of MYC gene facilitate risk stratification in diffuse large B-cell lymphoma patients treated with rituximab-CHOP. Mod Pathol 2014;27:958–71. [DOI] [PubMed] [Google Scholar]
- 31.Green TM, Young KH, Visco C, Xu-Monette ZY, Orazi A, Go RS, et al. Immunohistochemical double-hit score is a strong predictor of outcome in patients with diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol 2012;30:3460–7. [DOI] [PubMed] [Google Scholar]
- 32.Xu-Monette ZY, Dabaja BS, Wang X, Tu M, Manyam GC, Tzankov A, et al. Clinical features, tumor biology and prognosis associated with MYC rearrangement and overexpression in diffuse large B-cell lymphoma patients treated with rituximab-CHOP. Mod Pathol 2015;28:1555–73. [DOI] [PubMed] [Google Scholar]
- 33.Visco C, Li Y, Xu-Monette ZY, Miranda RN, Green TM, Li Y, et al. Comprehensive gene expression profiling and immunohistochemical studies support application of immunophenotypic algorithm for molecular subtype classification in diffuse large B-cell lymphoma: a report from the International DLBCL Rituximab-CHOP Consortium Program Study. Leukemia 2012;26:2103–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ford RJ, Goodacre A, Ramirez I, Mehta SR, Cabanillas F. Establishment and characterization of human B-cell lymphoma cell lines using B-cell growth factor. Blood 1990;75:1311–8. [PubMed] [Google Scholar]
- 35.Liu W, Chen J, Tamayo AT, Ruan C, Li L, Zhou S, et al. Preclinical efficacy and biological effects of the oral proteasome inhibitor ixazomib in diffuse large B-cell lymphoma. Oncotarget 2018;9:346–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pham LV, Bryant JL, Mendez R, Chen J, Tamayo AT, Xu-Monette ZY, et al. Targeting the hexosamine biosynthetic pathway and O-linked N-acetylglucosamine cycling for therapeutic and imaging capabilities in diffuse large B-cell lymphoma. Oncotarget 2016;7:80599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang J, Xu-Monette ZY, Jabbar KJ, Shen Q, Manyam GC, Tzankov A, et al. AKT hyperactivation and the potential of AKT-targeted therapy in diffuse large B-cell lymphoma. Am J Pathol 2017;187:1700–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bisso A, Sabo A, Amati B. MYC in germinal center-derived lymphomas: mechanisms and therapeutic opportunities. Immunol Rev 2019;288:178–97. [DOI] [PubMed] [Google Scholar]
- 39.Cuccuini W, Briere J, Mounier N, Voelker HU, Rosenwald A, Sundstrom C, et al. MYC+ diffuse large B-cell lymphoma is not salvaged by classical R-ICE or R-DHAP followed by BEAM plus autologous stem cell transplantation. Blood 2012;119:4619–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Herrera AF, Mei M, Low L, Kim HT, Griffin GK, Song JY, et al. Relapsed or refractory double-expressor and double-hit lymphomas have inferior progression-free survival after autologous stem-cell transplantation. J Clin Oncol 2017; 35:24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blum KA, Abramson J, Maris M, Flinn I, Goy A, Mertz J, et al. 410 - A phase I study of CPI-0610, a bromodomain and extra terminal protein (BET) inhibitor in patients with relapsed or refractory lymphoma. Ann Oncol 2018;29:iii7. [Google Scholar]
- 42.Patnaik A, Carvajal RD, Komatsubara KM, Britten CD, Wesolowski R, Michelson G, et al. Phase ib/2a study of PLX51107, a small molecule BET inhibitor, in subjects with advanced hematological malignancies and solid tumors. J Clin Oncol 2018;36:2550. [Google Scholar]
- 43.den Hollander J, Rimpi S, Doherty JR, Rudelius M, Buck A, Hoellein A, et al. Aurora kinases A and B are up-regulated by Myc and are essential for maintenance of the malignant state. Blood 2010;116:1498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murga-Zamalloa C, Inamdar KV, Wilcox RA. The role of aurora A and polo-like kinases in high-risk lymphomas. Blood Adv 2019;3:1778–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sasai K, Treekitkarnmongkol W, Kai K, Katayama H, Sen S. Functional significance of Aurora kinases-p53 protein family interactions in cancer. Front Oncol 2016;6:247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Keen N, Taylor S. Aurora-kinase inhibitors as anticancer agents. Nat Rev Cancer 2004;4:927–36. [DOI] [PubMed] [Google Scholar]
- 47.Wu CC, Yang TY, Yu CT, Phan L, Ivan C, Sood AK, et al. p53 negatively regulates Aurora A via both transcriptional and posttranslational regulation. Cell Cycle 2012;11:3433–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Howlett C, Snedecor SJ, Landsburg DJ, Svoboda J, Chong EA, Schuster SJ, et al. Front-line, dose-escalated immunochemotherapy is associated with a significant progression-free survival advantage in patients with double-hit lymphomas: a systematic review and meta-analysis. Br J Haematol 2015; 170:504–14. [DOI] [PubMed] [Google Scholar]
- 49.Oki Y, Noorani M, Lin P, Davis RE, Neelapu SS, Ma L, et al. Double hit lymphoma: the MD Anderson Cancer Center clinical experience. Br J Haematol 2014;166:891–901. [DOI] [PubMed] [Google Scholar]
- 50.Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med 2016;374:311–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.